0 引言
【研究意义】伴随着耕地面积的不断减少和人口基数的不断壮大,粮食短缺已成为制约中国生态与经济快速发展的重要因素,在水资源严重缺乏的干旱与半干旱地区形势更为严峻[1,2,3],高产育种是解决当前中国粮食短缺的重要途径。谷子是起源于中国的古老栽培作物之一,同时也是中国北方重要的粮食作物[4,5,6]。谷子营养价值丰富,具有适应性广、抗逆性强、耐旱、耐瘠薄等特点,是区域粮食安全的重要保障[6,7]。穗是禾本科作物最重要的生殖器官,也是作物产量的重要来源,因此,穗的发育状况直接影响粮食作物产量,其中谷穗顶端败育现象就是一个影响谷子产量的重要农艺性状。【前人研究进展】作物产量及其构成因子是多基因控制的数量性状,并受基因型和环境的共同影响。而穗是禾本科作物产量的形成器官,因此,穗顶端败育是基因与环境互作的结果[8]。禾本科作物穗顶端败育在玉米中研究较多,且多集中于穗败育与环境因素以及激素水平、营养供应等生理生化指标的相互关系。相关研究表明,环境因素[9,10]、营养供应[11]、内源激素水平[12,13]等都会影响玉米果穗顶端败育,但关于穗顶端败育的遗传规律和分子基础的研究较少。孟昭东等[14]发现玉米中秃尖是质量性状,而秃尖程度是数量性状。才源[15]发现玉米穗秃尖是由2对显性基因控制的,穗秃尖长短由2对主效基因和多个微效基因共同调控,通过SSR分子标记技术,将控制玉米果穗败育的基因定位在玉米第2和第7染色体。McSTEEN等[16]发现了一个叶腋分生组织缺陷型的突变体bif2(barren inflorescence2),研究发现突变基因bif2通过编码丝氨酸/苏氨酸蛋白激酶,调节生长素的运输,最终导致没有小穗和小花,只有裸露的花序轴的突变体性状变异。XIANG等[17]发现一个穗发育异常的突变体lp1,该突变体与sipaa1具有一定的相似性,均表现为株高降低,第一级枝梗和第二级枝梗数减少,谷穗码数大幅减少等,通过对该突变体进行基因挖掘发现了调控突变体株高、穗型的LP1,该基因在拟南芥过表达转基因株系中的ABA敏感性以及耐旱性增强[18]。同时,刁现民研究组已经在谷子中构建了以全基因组测序品种Yugu1为遗传背景且涵盖了谷子株型、叶色、穗发育以及抗病性等多个农艺性状的突变体库,如谷子矮秆突变体sidw3[19]、谷子黄绿叶突变体siygl1[20]、谷子穗发育异常突变体si-spl[21]和sidts1[22]、谷子耐旱耐ABA胁迫突变体siago1b[23]等,利用这些突变体成功克隆得到了谷子穗花粉发育相关基因SiMS1、耐逆相关基因SiAGO1b、叶色基因SiYGL1等。【本研究切入点】前人关于水稻、玉米中籽粒败育的研究难以弥补谷子中该研究缺失的空白,同时玉米果穗、稻穗与谷穗有一定的差异,因此,挖掘谷子穗顶端败育相关基因,探究基因是通过怎样的生理机制调控穗顶端败育。【拟解决的关键问题】本研究通过图位克隆方法对突变基因进行基因定位,寻找Yugu1和sipaa1之间的差异表达基因以及相关生物学通路,找到可能调控穗顶端败育的基因以及调控穗顶端败育的相关生物学通路。1 材料与方法
1.1 试验材料
豫谷一号(Yugu1)是中国华北夏谷区主栽的综合性状优良且最有影响的品种,具有丰产、抗病强、耐旱涝、对光温不敏感、适应性广等特点,且Yugu1全基因组测序已经完成[24]。利用EMS处理Yugu1,获得穗顶端败育突变体,命名为sipaa1。2013年夏,在中国农业科学院作物科学研究所顺义试验站种植突变体sipaa1(母本),分别与SSR41(父本)、Yugu1(父本)进行杂交。2014年,对突变体与Yugu1杂交得到的F1代在北京顺义和海南三亚连续回交两代获得BC1F1,2015年对BC1F1连续自交两代获得回交测序群体BC1F2。2014年,在中国农业科学院北京顺义试验站种植突变体与SSR41杂交得到的F1代,通过对穗部的观察筛选出真杂种,再对真杂种植株进行自交,得到F2代种子。2015年,在同一地点种植F2代群体,在抽穗后对穗顶端败育植株的叶片分单株取样,即为突变基因定位群体,并统计F2代群体中正常性状和突变性状植株的分离比例。所有试验群体的种植均采用条播法,田间管理按照国家谷子品种区试管理要求进行。1.2 定位群体叶片样本的选取
对亲本(sipaa1、SSR41)和F2(sipaa1×SSR41)群体中的隐性单株叶片进行取样,从中共收获592株隐性穗顶端败育突变体株。1.3 sipaa1的初步定位
1.3.1 谷子叶片总基因组DNA的提取 采用CTAB法[25]提取谷子叶片基因组总DNA。1.3.2 筛选多态性Indel标记 利用ZHANG等[26]设计的谷子通用Indel 标记,筛选在正常野生型Yugu1和突变体sipaa1之间具有多态性的标记。
1.3.3 混池构建 从592份F2(sipaa1×SSR41)隐性单株中选取30株穗顶端败育突变体叶片,提取总基因组DNA并检测其浓度,将其中浓度相近的DNA等量混合均匀(不少于25株),即为定位群体DNA混池。
1.3.4 突变基因初定位 以定位群体DNA混池模板和两亲本间有多态性的Indel标记进行PCR扩增,利用聚丙烯酰胺凝胶电泳确定突变基因所处的染色体和上下游Indel标记。在上下游Indel标记范围内选取更多具有多态性的标记,利用该标记对592份隐性单株叶片DNA进行PCR扩增,扩增产物经过6%的聚丙烯酰胺凝胶电泳分离,经银染显色后判断其带型。通过对不同Indel标记的重组事件进行统计,确定目标基因所在的精确位置。利用Primer5.0软件设计引物,由生工生物工程(上海)股份有限公司合成,引物序列见电子附表1。
Table S1
附表1
附表1试验所用引物
Table S1All primers involved in the article
| 引物名称 Primer name | 上游引物 Forward primer (5′-3′) | 下游引物 Reverse primer (5′-3′) |
|---|---|---|
| In1-4.73 | GAGCAAGCCTCACG | CCTCGGCATCAGTG |
| In1-6.02 | TGGCATTTCTACACCTGG | ACCTCTATTCGTGCTGATT |
| In1-7.35 | TAGGTAACAACGGGAAGT | AGAAAGAGCAGGACCAT |
| In1-8.77 | GCAGCCATCAGGCAAAT | CAGCCAGACAAGGAAGTAAAA |
| In1-9.11 | GCGTGACGATAAATGAA | ACATAACACCTGCCATT |
| In1-9.123 | GGGCTGTTGGTTTCTTG | TCCTATTCCCACCGTTT |
| In1-9.23 | TCCACGGTTGAAAGAGG | CTGCACGAAGATGACAATAAA |
| In1-9.322 | AGAACATACACCGAGGCT | GGGAGGAGGTTGAAGAA |
| In1-9.333 | CTAAAGCCTACTCCCTC | TCTCCATATCATGCCTC |
| In1-9.36 | ATTACGCAATCGTTCGG | CCATAGGGAGCTGAGGG |
| In1-9.42 | GGTTTATTTGTGCTGCCTGAA | GAAGTGGCGATGGTGGG |
| In1-9.80 | CGAATAGAATGCCCTCAC | AACGCAAACACGATAAGAC |
| In1-10.143 | CAGAGTTTGGTTTGGGTG | GATTGTCGTCGTCTTCG |
| In1-16.2 | GCTAAACGGCAAGA | TACCCGTCACATCG |
| In1-20.60 | CTTCACGGACACCTCATG | GTAGAGTAGCAGGTAGGCAAT |
| In1-24.0 | TATGGCATTTATTAGGAGTC | TGTCTGAACATTGCGTAT |
新窗口打开
1.4 农艺性状调查分析
选取成熟后的野生型Yugu1和突变体sipaa1各5株,调查其株高、穗长、穗粗、每穗粒数、千粒重等重要农艺性状。利用Excel软件整理野生型Yugu1和突变体sipaa1不同农艺性状的测量数据,并进行t检验显著性分析从而计算出P值。在此基础上利用GraphPad Prism 6.07软件进行作图。1.5 突变体sipaa1的转录组测序
利用RNA提取试剂盒(invitrogen)对孕穗期野生型Yugu1与突变体sipaa1的幼穗进行RNA提取,每个样品包含3个生物学重复,并交由北京贝瑞和康生物技术有限公司进行转录组文库构建和RNAseq测序工作。RNAseq数据的生物信息学分析通过百迈克云平台完成(www.biocloud.net)。RNAseq原始数据已经上传至欧洲生物信息研究所核酸序列数据库(www.ebi.ac.uk/ena/submit),ID:PRJEB25199(ERP107094)。2 结果
2.1 突变体sipaa1表型特征及主要农艺性状分析
与野生型相比,突变体sipaa1的穗部顶端在穗发育早期就已出现了明显的败育现象,一二级枝梗数显著锐减,花器官异常发育(图1)。野生型株高139.00 cm,较突变体株高(146.80 cm)略有升高,差异不显著(图1-A、图2-A);野生型叶长45.33 cm,较突变体(40.50 cm)降低10.66%(图2-B);野生型叶宽3.54 mm,较突变体(3.36 mm)降低了5.06%,差异不显著(图2-C)。对穗部主要性状进行统计发现,野生型穗长为23.67 cm,较突变体(20.98 mm)略有降低,二者差异不显著(图1-B、图2-E);野生型Yugu1穗粗30.64 mm,较突变体(25.70 mm)降低了16.12%(图1-B、图2-F);野生型谷码数为104.20,较突变体(70.25)减少了32.58%(图1-D、图2-H);野生型千粒重为2.64 g,较突变体(3.12 g)增加18.18%(图2-J)。总之,与野生型Yugu1相比,突变体sipaa1的变化主要集中在穗部性状,如穗粗、码数、千粒重等,降幅均在10% 以上,而株高、叶宽等性状则没有明显差异。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1野生型(Yugu1)和突变体(sipaa1)成熟期表型比较
A:成熟期的植株;B—E:成熟期的穗
-->Fig. 1Phenotype of the mutant and its wild-type (Yugu1) at mature stage
A: Whole plant phenotype; B-E: The panicle at mature stage
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2野生型Yugu1和突变体sipaa1主要农艺性状的比较
A:株高;B:叶长;C:叶宽;D:茎节数;E:穗长;F:穗粗;G:单株穗重;H:谷码数;I:单穗粒重;J:千粒重
-->Fig.2Comparison of major agronomic traits between the sipaa1 and its wild-type parent Yugu1
A: Plant height; B: Leaf length; C: Leaf width; D: Number of stem nodes; E: Panicle length; F: Panicle width; G: Panicle weights per plant; H: Spikelet number; I: Grain weight per panicle; J: 1000-grain weight
-->
2.2 突变体的突变基因定位
突变体sipaa1连续多代自交能够保持稳定遗传,且sipaa1×SSR41的子一代(F1)谷穗正常发育,子二代(F2)开始出现性状分离。经统计发现,F2中出现1 783株正常株,592株突变株,其分离比符合3﹕1,经卡方(χ2)检验,χ2=0.0068<3.841(χ20.05(1)=3.841),表明该突变由隐性单基因控制。在谷子9条染色体上均匀选取了180对Indel引物,检测其多态性,最终找到76个可用的Indel标记。利用这76个标记分别扩增父本SSR41、母本sipaa1和隐性混池DNA,发现在第1染色体上有一个与目标基因紧密连锁且在父母本间有显著多态性的Indel标记In1-8.77(图3、表1)。进一步在In1-8.77标记的上下游寻找标记,最后选出6个可用的多态性标记,分别为In1-6.02、In1-7.35、In1-9.8、In1-10.143、In1-16.2和In1-24.0(表1)。用两端的标记In1-6.02、In1-24.0以及In1-8.77分别检测收取的592株突变体叶片的DNA,根据电泳检测结果,将突变基因定位于Indel 1-8.77与Indel 1-9.8之间。同时将其中显示H带(混池DNA既有父本带又有母本带)的单株挑选出来,共计80株。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3sipaa1×SSR41群体连锁标记检测
♂:父本SSR41;♀:母本sipaa1;P:F2隐性单株混池
-->Fig. 3Linked markers results of sipaa1×SSR41 population
♂: SSR41; ♀: sipaa1; P: DNA pool of recessive individuals of F2 population
-->
Table 1
表1
表1部分隐性单株Indel标记检测结果
Table 1Test results of Indel markers in partial recessive individual plants
| 株号 Code | Indel标记 Indel markers | |||||||
|---|---|---|---|---|---|---|---|---|
| In1-4.73 | In1-6.02 | In1-7.35 | In1-7.41 | In1-8.77 | In1-16.2 | 1-20.6 | 1-24.0 | |
| 6 | H | H | S | S | S | H | H | H |
| 8 | H | H | S | S | S | S | S | S |
| 9 | H | H | S | S | S | S | S | H |
| 11 | H | H | H | H | S | S | S | S |
| 16 | H | H | S | S | S | S | S | H |
| 18 | H | H | H | S | S | S | S | S |
| 22 | H | H | S | S | S | S | S | S |
| 24 | H | H | S | S | S | S | S | H |
| 26 | H | H | S | S | S | S | S | S |
| 27 | H | H | H | H | S | S | S | S |
| 31 | H | H | S | S | S | S | S | S |
| 33 | H | H | H | S | S | S | S | S |
| 34 | H | H | S | S | S | S | S | H |
| 35 | H | H | H | H | S | S | S | S |
| 38 | H | H | S | S | S | S | S | S |
| 45 | H | H | S | S | S | S | S | S |
| 50 | H | H | H | H | S | S | S | S |
| 54 | H | H | H | H | S | S | S | S |
| 55 | H | H | H | H | S | S | S | S |
| 60 | H | H | H | H | S | S | S | S |
| 62 | H | H | H | H | S | H | H | H |
| 65 | H | H | H | H | S | S | S | S |
| 66 | H | H | H | H | S | S | S | S |
新窗口打开
为了进一步缩小区间,在In1-8.77与In1-9.8之间设计20个Indel标记,经检测发现其中7个标记在父本(SSR41)和母本(sipaa1)之间有多态性,分别为In1-9.11、Ind1-9.123、Inl1-9.23、In1-9.322、In1-9.333、In1-9.36和In1-9.42。以7对多态性标记进行PCR检测上述80株隐性突变体单株的带型,将突变基因定位在In1-9.23与In1-9.322之间约100 kb范围内(图3和图4)。继续在该范围内设计多态性引物,结果发现,在592株突变体F2隐性单株后代中没有了交换株,难以再进一步缩小区间,因此,最终定位目的基因在In1-9.23与In1-9.322区间内。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4穗顶端败育突变基因sipaa1的定位
-->Fig.4Gene mapping of the panicle apical abortion mutant sipaa1
-->
2.3 突变体sipaa1差异表达基因鉴定
对突变体sipaa1与野生型Yugu1的转录组数据进行了差异表达基因(DEGs,differential expressed genes)鉴定(图5)。通过统计Mapped Reads在基因组的CDS(编码区)、5’UTR区(前导序列区)、3’UTR区(尾随序列区)、Intron(内含子区)、Intergenic(基因间区)等区域的分布分别为79%、3%、13%、2%和3%,大部分的Mapped Reads集中在基因的CDS区(图5-A),测序结果符合转录组特征。图5-B为突变体sipaa1和Yugu1各3个生物学重复样品间的相关性。相关系数的大小代表样品之间表达模式的相似度。相似度越高(越接近1),样品之间差异越小;同理,相似度越低(越接近0),样品之间差异越大。野生型Yugu1 3个重复样品WT.1、WT.2和WT.3间的相关系数约为0.92,突变体间相关系数约为0.96,说明突变体与野生型的生物学重复样本之间具有较高的相关性;而突变体sipaa1与野生型Yugu1的相关系数介于0.78—0.81,即突变体和野生型间的差异显著,表明RNAseq的样本选择合理且试验数据可重复性及一致性好(图5-C)。在突变体sipaa1与野生型Yugu1之间存在3 275个差异基因,上调基因2 768个,上调基因远多于下调基因(507个),约为其5.5倍(图5-D)。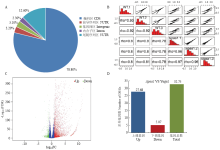 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5突变体sipaa1与野生型Yugu1的差异基因鉴定
A:Reads在基因组上的分布;B:样本相关性;C:基因表达量火山图;D:差异表达基因数
-->Fig. 5Identification of DEGs between the sipaa1 and its wild-type Yugu1
A: The distribution of reads in the genome; B: Sample correlation; C: Volcano plot of gene expression; D: Number of DEGs
-->
2.4 差异表达基因的GO分类与富集分析
将突变体sipaa1的RNAseq数据与谷子蛋白组进行BLASTP比对,并对DEGs进行功能GO注释(图6),根据GO注释结果,将差异表达基因归属于细胞组分(cellular component,CC)、分子功能(molecular function,MF)和生物过程(biological process,BP)三大生物学通路,且各自的差异表达基因通路分别有142、299和1 092条。以校正后的P-Value(FDR)进行筛选(FDR≤0.001)发现,细胞组分、分子功能和生物过程三类注释得到的差异表达基因通路分别有15、5和99条。根据各通路差异基因的富集程度及其层级关系,从中选出了3个具有代表性的细胞组分注释和15个具有代表性的生物过程的注释。由图中可以看出,差异基因主要富集在生物及非生物胁迫的响应(response to water deprivation、defense response to bacterium)、激素响应(response to jasmonic acid stimulus、salicylic acid biosynthetic process、response to abscisic acid stimulus、salicylic acid mediated signaling pathway)、细胞程序性死亡(regulation of programmed cell death)以及转录因子活性(transcription factor activity)这四类通路上。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6突变体sipaa1与野生型Yugu1差异基因GO功能富集分析
-->Fig. 6GO function enrichment analysis of DEGs between the sipaa1 and its wild-type parent Yugu1
-->
2.5 差异表达基因的KEGG分类与富集分析
利用KOBAS软件对差异表达基因的KEGG代谢途径进行分析,选取其中富集程度最高的前20条pathway(图7)。从图中可以看出,差异表达基因(differential expresed genes,DEGs)大多存在于植物激素信号转导、植物-病原互作、苯丙素的生物合成、淀粉和蔗糖代谢四大通路中,分别有56、49、48和43个;其次主要集中于糖酵解和糖异生作用、半胱氨酸和甲硫氨酸代谢、类胡萝卜素的生物合成、苯丙氨酸代谢等,在次生物质代谢通路,如类黄酮生物合成、泛醌和其他萜类醌生物合成、角质,木栓质和蜡质生物合成等中的差异表达基因也有富集,而次生代谢产物对植物胁迫响应和抵抗病原侵袭有重要作用。由此推断,很可能是突变体sipaa1中生物或非生物胁迫引发细胞程序化死亡,最终导致了穗顶端败育的表型。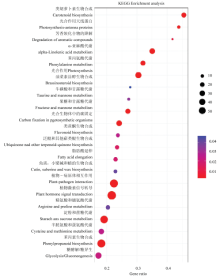 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7突变体sipaa1与野生型Yugu1差异基因KEGG pathway Terms富集图
该图表示差异基因富集程度最高的前20条pathway,横轴表示富集的程度(用P-Value表示,该值越小表示富集程度越高),纵轴表示富集的KEGG Pathway Term。圆点大小表示该通路包含的差异基因数目
-->Fig.7KEGG pathway Terms of DEGs between the sipaa1 and its wild-type parent Yugu1
This figure represents the top 20 pathways with the highest degree of DGEs. The horizontal axis indicates the level of enrichment of the DGEs (the smaller the P-Value is, the higher the enrichment level is). The vertical axis represents the GO Term of the enrichment. The dot size indicates the number of DGEs of the pathway
-->
2.6 候选基因在谷子中的表达与分析
根据Phytozome的对比结果,在定位区间In1-9.23与In1-9.322之间约100 kb范围内确定了6个穗部高度表达的候选基因,对6个候选基因的基因位置、基因长度、基因功能以及其在拟南芥和水稻中的同源基因进行了注释(表2)。同时,对6个候选基因在谷子不同组织部位(包括萌芽期的幼苗、成苗期的叶、茎、根、穗)中的表达量进行了统计(图8),结果发现,与同一植株的其他组织相比,Seita.1G106700、Seita.1G106800、Seita.1G107000和Seita.1G107500 4个候选基因在穗部的表达量最高,Seita.1G107200候选基因在萌芽期的幼苗中表达量最高,叶片中几乎不表达,而Seita.1G107600候选基因在萌芽期的幼苗、根、穗、茎中表达量则比较相近。同时统计了候选基因在Yugu1和sipaa1中的表达量,用FPKM值表示(图9)。候选基因Seita.1G107200的表达量在本次测序中没有检测到。候选基因Seita.1G106700在Yugu1和sipaa1中的表达差异不显著,Seita.1G106800差异显著,而Seita.1G107000、Seita.1G107500与Seita.1G107600则表现为极显著。Table 2
表2
表2候选基因功能分析
Table 2Function analysis of candidate genes
| 基因 Gene | 位置 Location (bp) | 长度 Length (kb) | 拟南芥ID Homologous gene in Ath | 水稻ID Homologous gene in Osa | 基因功能注释 Functional annotation |
|---|---|---|---|---|---|
| Seita.1G106700 | Chr_1:9232806..9236753 | 3948 | AT2G45690 | Os02g03070 | 过氧化物酶体 Peroxin-16 (PEX16) |
| Seita.1G106800 | Chr_1:9237950..9241496 | 3547 | AT1G20930 | Os02g03060 | 细胞周期依赖型蛋白激酶2 Cyclin-dependent kinase 2 (CDK2) |
| Seita.1G107000 | Chr_1:9259107..9261004 | 1898 | AT5G52040 | Os02g03040 | 丝氨酸、苏氨酸剪接因子 Splicing factor, arginine/serine-rich 4/5/6 (SFRS4_5_6) |
| Seita.1G107200 | Chr_1:9264861..9265325 | 465 | AT2G46600 | Os02g03020 | EF手型结构域钙结合蛋白 Calcium-binding EF-hand family protein |
| Seita.1G107500 | Chr_1:9301930..9307080 | 5151 | AT5G42480 | Os02g03000 | 分子伴侣DnaJ结构域超家族蛋白 Chaperone DnaJ-domain superfamily protein |
| Seita.1G107600 | Chr_1:9307612..9310924 | 3313 | AT1G66070 | Os02g02990 | 翻译起始因子3亚基J(EIF3J) Translation initiation factor 3 subunit J (EIF3J) |
新窗口打开
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图86个候选基因在植物不同组织部位的表达量
-->Fig. 8Expression of six candidate genes in different tissues of plants
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图9候选基因在Yugu1和sipaa1中的表达量
-->Fig. 9Expression of candidate genes between the sipaa1 and its wild-type parent Yugu1
-->
结合基因功能注释、候选基因在谷子不同组织部位表达量以及候选基因在Yugu1和sipaa1之间的表达差异结果进行综合分析发现,Seita.1G106700在穗部高表达时会生产较多过氧化物酶体,过氧化物酶体能够将光合作用的副产物乙醇酸氧化为过氧化氢,大量过氧化氢在植物体内累积导致细胞程序性死亡,表现出谷穗败育的性状。而Seita.1G107200是一种钙结合EF手型家族蛋白基因,该基因编码形成的蛋白质多参与植物体内胁迫反应,例如钙依赖蛋白激酶 CDPKs即为典型的EF手型家族蛋白,有研究证实该蛋白参与多种光、干旱等环境胁迫。这与RNAseq功能富集结果相符合。因此,预测候选基因Seita.1G106700和Seita.1G107200可能与突变体sipaa1穗顶端败育的形成有关。
3 讨论
3.1 穗顶端败育突变体sipaa1的表型比较分析
谷子的穗是一个由三级分枝及其上着生的小穗构成的顶生圆锥花序;每个小穗由两朵小花组成,第一朵小花败育,第二朵小花正常发育结实[27]。而突变体sipaa1的穗顶端一级枝梗(分枝、谷码)数显著减少,第二小花不发育或发育不完全,导致谷码数大幅减少,单穗粒重显著降低。与谷子相比,玉米败育的研究更为透彻。玉米败育包括花败育和粒败育两类。花败育包括败育花和未受精花,籽粒败育包括未灌浆型败育粒和已灌浆型败育粒[28]。按照该分类标准,突变体sipaa1在幼穗分化早期就已出现了顶端穗败育的表型,生长锥发育异常导致穗顶端一级枝梗数大幅减少,小穗原基难以形成正常的小穗,因此,sipaa1的败育可归为花不发育或者发育不全导致的穗顶端败育。一般来说,幼穗分化、以及籽粒形成始于穗的中下部,以后由此处向上或向下同时进行,最后在顶部结束。顶部的小花或受精胚常因外界环境条件不适或自身遗传因素的影响造成养分供应不足从而败育,在谷穗顶端形成秃尖。3.2 RNAseq分析表明突变体sipaa1中致变基因与胁迫响应及细胞死亡有关
据报道,穗顶端败育是遗传因素和植物生理作用二者互作的结果。因此,突变体sipaa1穗顶端败育既是其自身遗传作用的结果,也是外界环境作用的结果[8]。徐云姬等[13]发现玉米中脱落酸(ABA)、吲哚乙酸(IAA)等与籽粒发育呈正相关,赤霉素(GA3)为负相关。张秀梅等[29]发现BR(表油菜素内酯)与玉米败育呈显著负相关。本研究中,GO功能富集中差异表达基因主要在JA和ABA等激素应答通路中富集。同时,GO和KEGG分析表明差异表达基因富集于干旱胁迫或植物激素信号转导,这与前人的研究结果基本一致。也有研究指出,植物在干旱条件下代谢异常,导致植物中可利用的蔗糖含量减少,这种外部伤害会激活衰老基因导致细胞程序化死亡,进而导致籽粒败育[30]。由此可以推断,该突变体穗顶端败育与胁迫响应及激素信号转导相关。高学曾等[31]发现与正常籽粒相比,败育玉米籽粒中的过氧化氢酶及其同工酶表现出更高的活性。高素伟[32]通过DAB检测发现过氧化氢的累积可能导致穗发育受阻,显示小穗败育可能是细胞程序性死亡的一种表现,本研究RNAseq的数据分析结果也符合这一猜想。因此,对过氧化氢与籽粒败育关系的研究有待进一步深入。3.3 候选基因初步探索
在Indel1-9.23与Indel1-9.322之间约100 kb范围的定位区间内找到12个候选基因,通过分析候选基因在谷子不同组织部位的表达量差异,发现其中6个候选基因在突变体sipaa1穗部的表达量明显高于根、茎、叶等其他组织器官,分别为Seita.1G106700、Seita.1G106800、Seita.1G107000、Seita.1G107200、Seita.1G107500、Seita.1G107600。在数据库中对候选基因进行查找发现,Seita.1G106700编码过氧化物酶体的膜周边或整合蛋白(peroxin-16,PEX16),该基因在拟南芥中的同源基因为AT2G45690且已被克隆,其缺失突变体种胚中没有正常的过氧化物酶体,导致种子油脂和蛋白质含量将低,种子皱缩等现象,因此又被称为SSE1(shrunken seed protein)[33,34]。Seita.1G106800为细胞周期蛋白依赖性激酶2(Cyclin dependent kinase 2,CDK2),在水稻中的同源基因是LOC_Os02g03060,推测其可能编码细胞周期蛋白依赖性激酶A2(cyclin-dependent kinase A-2,CDKA2),属于细胞周期蛋白依赖性激酶,该类基因控制细胞周期各个环节的起始,从而调控胚和胚乳的发育。Seita.1G107000是精氨酸/丝氨酸富集剪接因子4/5/6(splicing factor, arginine/serine-rich 4/5/6,SFRS4_5_6),参与RNA前体剪接过程;在拟南芥中的同源基因是AT2G46610,具有4个转录本Seita.1G107000.1、Seita.1G107000.2、Seita.1G107000.3和Seita.1G107000.4,该基因编码RRM型结构域的RNA结合家族蛋白(RRM/RBD/RNP RNA-binding family proteins,RRM RBPs),在植物开花时间转换、花发育、ABA信号通路、逆境响应、生物节律与染色质修饰等生长发育过程中起重要作用[35]。Seita.1G107200编码EF手型钙结构域结合蛋白(EF- HAND CALCIUM-BINDING DOMAIN CONTAINING PROTEIN),该基因在水稻中的同源基因是LOC_Os01g57470,属于EF手型家族蛋白,该类基因参与植物体内钙离子信号转导,调控植物的花发育(成花诱导、花芽分化和开花调控)、有性生殖(花粉萌发和花粉管生长)、逆境生理等[36]。该基因的功能与突变体顶端败育的性状可能有一定的相关性。Seita.1G107500在拟南芥中的同源基因是AT5G42480,该基因编码分子伴侣J-蛋白,该类蛋白参与植物重力信号过程和对外界多种刺激的响应[37]。Seita.1G107600编码转录起始因子3J(translation initiation factor 3 subunit J,EIF3J),属于转录起始因子家族,调节植物转录起始。综合图位克隆、RNAseq以及候选区间基因基因功能分析结果,Seita.1G106700和Seita.1G107200相关研究有待进一步深入。4 结论
与Yugu1相比,突变体sipaa1穗顶端较早的出现了败育的表型,一、二级枝梗数骤减,谷码数、千粒重等穗部性状有显著差异。谷子穗顶端败育由隐性单基因控制,并将控制穗败育表型的候选基因定位在谷子第1染色体标记In1-9.23与In1-9.322之间约100 kb范围内,在该定位区间内的候选基因Seita.1G106700和Seita.1G107200参与了激素、干旱胁迫以及过氧化氢毒害等生理生化反应,而外界胁迫和过氧化氢累积与细胞程序性死亡密切相关。因此,谷子顶端小穗败育可能是细胞程序性死亡的一种典型表现。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}