 ,*贵州省农业科学院旱粮研究所, 贵州贵阳 550006
,*贵州省农业科学院旱粮研究所, 贵州贵阳 550006Excavation of main candidate genome regions in Suwan germplasm improvement process of maize
LI Xiu-Shi, WU Xun, WU Wen-Qiang, LIU Peng-Fei, GUO Xiang-Yang, WANG An-Gui, ZHU Yun-Fang, CHEN Ze-Hui,*Upland Crops Institute, Guizhou Academy of Agricultural Sciences, Guiyang 550006, Guizhou, China通讯作者:
收稿日期:2018-06-26接受日期:2018-12-24网络出版日期:2019-01-04
| 基金资助: |
Received:2018-06-26Accepted:2018-12-24Online:2019-01-04
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (3887KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李秀诗, 吴迅, 吴文强, 刘鹏飞, 郭向阳, 王安贵, 祝云芳, 陈泽辉. 玉米Suwan种质改良过程中的关键基因组区段发掘[J]. 作物学报, 2019, 45(4): 568-577. doi:10.3724/SP.J.1006.2019.83052
LI Xiu-Shi, WU Xun, WU Wen-Qiang, LIU Peng-Fei, GUO Xiang-Yang, WANG An-Gui, ZHU Yun-Fang, CHEN Ze-Hui.
近年来, 随着高通量基因型鉴定技术的不断发展, 研究者从分子水平上解析了温带玉米种质的遗传基础。如兰进好等[9]利用黄早四和Mo17构建的F2:3群体, 基于SSR和ALFP标记鉴定出一批控制穗行数、行粒数、百粒重等性状的QTL; Wu等[10]利用包含56,110个SNP标记的基因芯片对367份重要自交系的遗传多样性、群体结构、亲缘关系等进行分析, 并结合全基因组关联分析策略共定位到158个与株型相关性状的SNP位点; Yang等[11]利用B73、SICAU1212及其F2群体, 结合SSR标记基因型进行连锁分析, 共定位到33个QTL与12个农艺性状相关。但是针对Suwan种质的遗传研究较少, 主要集中在表型数据或少数标记的配合力分析、杂种优势分析和群体结构分析等, 所揭示的信息量较为有限。如杨文鹏等[12]利用88个SSR标记对贵州2000年来审定的玉米品种70份亲本材料分析发现, 泰国苏湾热带种质被分为一个亚群, 揭示出贵州以地方亚热带种质和泰国苏湾热带种质为主要杂种优势群的玉米育种模式; 闫飞燕等[13]和番兴明等[14]对热带、亚热带玉米种质群体及自交系的配合力效应和杂种优势分析表明, Suwan 1群体及其衍生自交系具有较高的一般配合力, Suwan 1×Ried是表现较好的杂优模式之一; Zhang等[15]利用MaizeSNP50芯片对西南地区362份玉米自交系的群体结构和遗传多样性进行全基因组关联分析发现, 位于第2染色体130 Mb的一个区域遗传多样性较为丰富, 第7染色体30~120 Mb是S37在西南区玉米育种的一个保守区域; 陈泽辉等[16]利用属Reid自交系和属Tuxpeno自交系构建人工合成群体墨瑞1号, 用属Suwan自交系、Mo17和78599构建人工合成群体苏兰1号, 并利用半同胞相互轮回选择法进行改良, 通过田间鉴定发现Reid-Tuxpeno×Lancaster-Suwan 可作为我国南方玉米杂种优势利用的重要模式之一。然而, 关于Suwan种质改良的遗传基础以及在改良过程中是否存在一些重要遗传区段等研究则属于空白。因此, 本研究利用包含5.6万个标记的MaizeSNP50芯片对Suwan及其衍生群体(苏兰1号)不同改良世代进行基因型鉴定, 基于高密度的基因型鉴定结果分析Suwan及其衍生群体不同改良世代之间的基因组演化特征; 结合全基因组关联分析策略, 初步揭示Suwan种质改良过程中的关键遗传区段并明确其效应, 为玉米Suwan种质改良利用和后续分子标记辅助育种提供参考。
1 材料与方法
1.1 试验材料
以玉米种质群体Suwan 1 (Suwan 1 C10)、苏兰1号的不同改良世代为材料。其中, Suwan 1群体的第11轮(C11)是贵州省农业科学院旱粮研究所1992年从泰国的苏湾农场引进; 而Suwan 1群体的不同改良世代[第10轮(C10)、第12轮(C12)、第13轮(C13)、第15轮(C15)]则是课题组2014年从苏湾农场引进; 苏兰1号(SL1C0)是由苏湾种质与Lancaster种质的优良自交系的人工合成群体, 并通过3次半同胞相互轮回选择法获得SL1C1、SL1C2、SL1C3三轮改良世代[16]。供试材料系谱来源和类群见表1。Table 1
表1
表1供试材料系谱和类群
Table 1
| 序号 No. | 材料 Accession | 系谱 Pedigree | 类群 Group |
|---|---|---|---|
| 1 | Suwan 1 (Suwan 1 C10) | Suwan 1 C9 | Suwan |
| 2 | Suwan 1 C11 | Suwan 1 C10 | Suwan |
| 3 | Suwan 1 C12 | Suwan 1 C11 | Suwan |
| 4 | Suwan 1 C13 | Suwan 1 C12 | Suwan |
| 5 | Suwan 1 C15 | Suwan 1 C14 | Suwan |
| 6 | 苏兰1号C0 SL1C0 | Suwan, Lancaster及78599选系 Synthetic populations of Suwan, Lancaster and 78599 selected lines | Suwan-Lancaster |
| 7 | 苏兰1号C1 SL1C1 | 苏兰1号C0 SL1C0 | Suwan-Lancaster |
| 8 | 苏兰1号C2 SL1C2 | 苏兰1号C1 SL1C1 | Suwan-Lancaster |
| 9 | 苏兰1号C3 SL1C3 | 苏兰1号C2 SL1C2 | Suwan-Lancaster |
新窗口打开|下载CSV
1.2 试验方法
1.2.1 田间鉴定 2017年春, 将Suwan 1及4个改良世代、苏兰1号及其3轮改良世代分别在贵州贵阳(26.33°N, 106.64°E)、贵州大方(26.98°N, 105.66°E)和云南罗平(24.78°N, 104°E) 3个不同环境下进行田间鉴定。采用完全随机区组设计, 3次重复, 2行区, 行长5 m, 行距0.7 m, 每行定苗22株。按照常规生产条件进行田间管理。参照石云素等编写的《玉米种质资源描述规范和数据标准》调查表型[17], 即收获后, 从每个小区随机取10个果穗调查穗长、穗行数, 取平均值用于统计分析。1.2.2 基因型鉴定 2016年冬, 在海南省三亚市九所镇贵州南繁基地从Suwan 1及其衍生群体各改良世代中取样。即在玉米大喇叭口时期, 首先依据植株高度将每个群体分为高、中、低3种类型, 从每种类型中取30株幼嫩叶片等量混合和提取DNA, 并采用Illumina公司开发的MaizeSNP50芯片对所取样本进行基因型鉴定, 该芯片包括56,110个SNP标记。参照Illumina 公司提供的操作指南检测基因型。其中DNA提取和基因型鉴定工作均委托北京康普森生物科技有限公司完成。
1.3 表型数据统计分析
采用Microsoft Excel 2007和SAS 9.2软件[18]的PROC UNIVARIATE和PROC GLM程序对田间统计数据进行描述性统计和方差分析等。1.4 候选遗传区段鉴定
基于不同世代的基因型鉴定结果, 通过全基因组比较, 利用GGT32软件鉴定出在各改良世代间稳定遗传和差异的候选区域。1.5 全基因组关联分析
根据最小等位基因频率MAF > 0.05且样本缺失率 < 20%的标准[19], 筛选出43,980个高质量的SNP标记, 利用TASSEL软件的混合线性模型[20]对表型和基因型进行全基因组关联分析。以P < 0.0001为阈值, 鉴定出控制目标性状的关键QTN。在此基础上, 利用生物信息分析手段, 借助公共数据库的定位结果, 初步揭示出目标区段的遗传效应。2 结果与分析
2.1 穗长和穗行数分析
随着改良轮次的增加, 苏兰1号群体的穗长增加, 从改良前的17.67 cm增长到改良后的18.27 cm, 明显高于Suwan 1群体(17.22 cm); 穗行数性状变化较小, 但各世代间存在着明显差异。供试群体在贵阳、大方和罗平3个点的穗长性状的平均值分别为17.56、18.65和17.06 cm, 变异系数分别为6.21%、3.91%和4.92%; 穗行数性状的平均值分别为15.07、14.93和14.45行, 变异系数分别为4.45%、2.82%和4.50% (表2), 说明Suwan 1及其衍生群体各改良世代间穗长和穗行数差异明显。Table 2
表2
表29个供试群体穗长和穗行数
Table 2
| 材料 Accession | 穗长 Ear length | 穗行数 Kernel row number | ||||||
|---|---|---|---|---|---|---|---|---|
| 贵阳 Guiyang | 大方 Dafang | 罗平 Luoping | 平均值 Mean | 贵阳 Guiyang | 大方 Dafang | 罗平 Luoping | 平均值 Mean | |
| 苏兰1号C0 SL1C0 | 17.67 | 18.77 | 17.40 | 17.94 | 15.47 | 14.53 | 14.67 | 14.89 |
| 苏兰1号C1 SL1C1 | 18.13 | 19.33 | 17.17 | 18.21 | 15.33 | 14.73 | 14.27 | 14.78 |
| 苏兰1号C2 SL1C2 | 17.87 | 18.97 | 18.20 | 18.34 | 14.27 | 14.27 | 14.47 | 14.33 |
| 苏兰1号C3 SL1C3 | 18.27 | 19.57 | 17.73 | 18.52 | 15.00 | 15.53 | 15.20 | 15.24 |
| Suwan 1 (Suwan 1 C10) | 17.47 | 18.33 | 16.43 | 17.41 | 15.00 | 14.93 | 14.00 | 14.64 |
| Suwan 1 C11 | 17.67 | 18.23 | 17.03 | 17.64 | 15.40 | 14.93 | 14.13 | 14.82 |
| Suwan 1 C12 | 16.87 | 17.97 | 16.33 | 17.06 | 15.13 | 15.87 | 14.60 | 15.20 |
| Suwan 1 C13 | 16.53 | 18.17 | 16.20 | 16.97 | 15.40 | 14.93 | 14.20 | 14.84 |
| Suwan 1 C15 | 17.60 | 18.53 | 17.03 | 17.72 | 14.60 | 14.60 | 14.53 | 14.58 |
| 平均值Mean | 17.56 | 18.65 | 17.06 | 17.76 | 15.07 | 14.93 | 14.45 | 14.81 |
| 标准差Standard deviation | 1.09 | 0.73 | 0.84 | 1.11 | 0.67 | 0.57 | 0.65 | 0.68 |
| 变异系数Coefficient of variation (%) | 6.21 | 3.91 | 4.92 | 6.25 | 4.45 | 3.82 | 4.50 | 4.59 |
新窗口打开|下载CSV
2.2 基因组特征
在Suwan 1群体的不同改良世代中, 共鉴定出9个稳定遗传片段和5个差异遗传片段(图1)。说明Suwan 1群体在改良过程中, 保持着群体内丰富的遗传变异, 其不同改良世代间基因发生交换重组的频率较低, 仅出现5个差异遗传片段, 其中4个出现在第11轮改良世代(Suwan 1 C11), 1个出现在第15轮改良世代(Suwan 1 C15), 其原因是Suwan 1 C11的引入时间较早, 与其他改良世代批次不同, 受到环境驯化和选择发生基因漂移, 而且这9个稳定遗传片段可能在保持Suwan群体优势抗性、适应性等热带种质特征方面发挥作用。而其中的5个差异遗传区段可能是Suwan种质改良过程中的重组热点区域, 可为后续的种质改良提供依据。图1
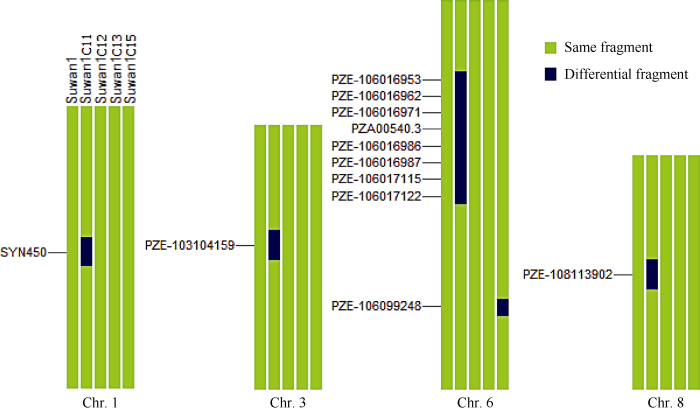 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Suwan 1群体不同改良世代间的基因组特征
特异性SNP标记在染色体上的物理位置见附
Fig. 1Genomic characteristics of Suwan 1 in different improved generations
The physical location of the specific SNP markers on chromosome is shown in the Supplementary table 1.
Table 1
表1
表11 附Suwan 1群体不同改良世代的特异性标记
Table 1
| SNP | 染色体 Chr. | 物理位置 Position | SNP | 染色体 Chr. | 物理位置 Position |
|---|---|---|---|---|---|
| SYN450 | 1 | 35968064 | PZE-106016986 | 6 | 32497838 |
| PZE-103104159 | 3 | 164159285 | PZE-106016987 | 6 | 32498979 |
| PZE-106016953 | 6 | 32495316 | PZE-106017115 | 6 | 32905813 |
| PZE-106016962 | 6 | 32495710 | PZE-106017122 | 6 | 32908494 |
| PZE-106016971 | 6 | 32496071 | PZE-106099248 | 6 | 152896893 |
| PZA00540.3 | 6 | 32496071 | PZE-108113902 | 8 | 163858777 |
新窗口打开|下载CSV
在苏兰1号C0群体不同改良世代中, 共鉴定出26个稳定遗传片段和18个差异遗传片段(图2), 分别位于第1、第2、第3、第4、第5、第6、第7、第9染色体上。其中, 第2、第5、第6染色体上的8个差异遗传片段在其改良世代间稳定出现, 以第2染色体上的多态性最高。说明苏兰1号群体在改良过程中, 不同改良世代间保持着群体内丰富的遗传变异, 少数基因发生交换重组, 获得18个差异遗传片段, 而且这些差异遗传区段可能与苏兰1号群体改良世代间秃尖长、行粒数等产量性状特征方面发挥作用, 可为后续利用分子标记辅助育种提供重要依据。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2苏兰1号群体不同改良世代间的基因组特征
特异性SNP标记在染色体上的物理位置见附
Fig. 2Genomic characteristics of Suwan-Lancaster1 in different improved generations
The physical location of the specific SNP markers on chromosome is shown in Supplementary table 2.
Table 2
表2
表22 苏兰1号群体不同改良世代的特异性标记
Table 2
| SNP | 染色体 Chr. | 物理位置 Position | SNP | 染色体 Chr. | 物理位置 Position |
|---|---|---|---|---|---|
| PZE-101205609 | 1 | 253344590 | PZE-102156731 | 2 | 203902578 |
| PZE-101205965 | 1 | 253895960 | SYN15645 | 3 | 182660027 |
| SYN6838 | 1 | 297376850 | PZE-105016506 | 4 | 7204051 |
| PZE-103008521 | 2 | 4666469 | PZE-106033525 | 5 | 76988770 |
| PZE-102060229 | 2 | 38551101 | SYN25006 | 6 | 8192709 |
| PZE-102060230 | 2 | 39031517 | PZE-107126153 | 7 | 163083361 |
| PZE-102061400 | 2 | 39748797 | PZE-109019803 | 7 | 20222259 |
| SYN26925 | 2 | 200353907 | PZE-109051268 | 9 | 85880947 |
| SYN26929 | 2 | 200359094 | PZE-109051619 | 9 | 86414796 |
| SYN10568 | 2 | 200723525 | PZE-109052137 | 9 | 86974491 |
| SYN35589 | 2 | 201246108 |
新窗口打开|下载CSV
基因组信息比对发现, Suwan种质改良形成苏兰1号群体的过程中, 共有53个区段来自Suwan群体和43个区段来自Lancaster种质(图3), 其中有35个Lancaster来源区段在苏兰1号C0群体不同改良世代间稳定出现, 分别位于第1、第3、第4、第5染色体, 说明这些区域发生交换重组的频率较高, 多态性较丰富。
图3
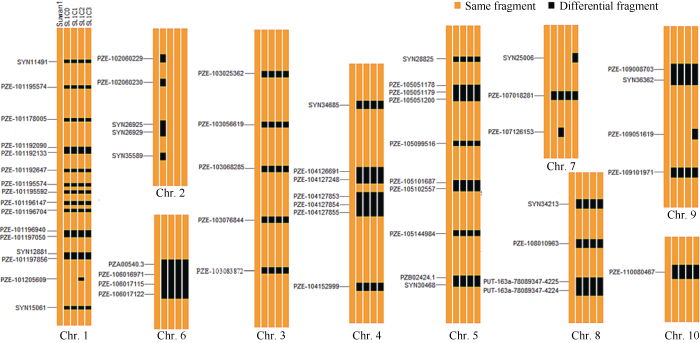 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Suwan 1与苏兰1号不同改良世代间的基因组特征
特异性SNP标记在染色体上的物理位置见附
Fig. 3Genomic characteristics between Suwan 1 and Suwan-Lancaster 1 in different improved generations
The physical location of the specific SNP markers on chromosome is shown in Supplementary table 3.
Table 3
表3
表33 Suwan 1与苏兰1号不同改良世代间的特异性标记
Table 3
| SNP | 染色体 Chr. | 物理位置 Position | SNP | 染色体 Chr. | 物理位置 Position |
|---|---|---|---|---|---|
| SYN11491 | 1 | 3683507 | PZE-104127854 | 4 | 212788677 |
| PZE-101054452 | 1 | 38617583 | PZE-104127855 | 4 | 212788991 |
| PZE-101178005 | 1 | 222370910 | PZE-104152999 | 4 | 243521349 |
| PZE-101192090 | 1 | 237838603 | SYN28825 | 5 | 7319620 |
| PZE-101192133 | 1 | 237900682 | PZE-105051178 | 5 | 43960922 |
| PZE-101192647 | 1 | 238680213 | PZE-105051179 | 5 | 43961044 |
| PZE-101195574 | 1 | 242361972 | PZE-105051200 | 5 | 43966297 |
| PZE-101195592 | 1 | 242398910 | PZE-105099516 | 5 | 146942464 |
| PZE-101196147 | 1 | 243255282 | PZE-105101687 | 5 | 152095801 |
| PZE-101196704 | 1 | 244083020 | PZE-105102557 | 5 | 154315332 |
| PZE-101196940 | 1 | 244512792 | PZE-105144984 | 5 | 197943057 |
| PZE-101197050 | 1 | 244678601 | PZB02424.1 | 5 | 199531408 |
| SYN12881 | 1 | 245242156 | SYN30468 | 5 | 199531778 |
| PZE-101197856 | 1 | 245308899 | PZA00540.3 | 6 | 32496071 |
| SYN15061 | 1 | 261370341 | PZE-106016971 | 6 | 32496071 |
| PZE-102060229 | 2 | 38551101 | PZE-106017115 | 6 | 32905813 |
| PZE-102060230 | 2 | 39031517 | PZE-106017122 | 6 | 32908494 |
| SYN26925 | 2 | 200353907 | SYN25006 | 7 | 8192709 |
| SYN26929 | 2 | 200359094 | PZE-107018281 | 7 | 15776884 |
| SYN35589 | 2 | 201246108 | PZE-107126153 | 7 | 163083361 |
| PZE-103025362 | 3 | 17738644 | SYN34213 | 8 | 5090046 |
| PZE-103056619 | 3 | 68433447 | PZE-108010963 | 8 | 11645850 |
| PZE-103068285 | 3 | 108233400 | PUT-163a-78089347-4225 | 8 | 169951365 |
| PZE-103076844 | 3 | 123683053 | PUT-163a-78089347-4224 | 8 | 169951478 |
| PZE-103083872 | 3 | 135004208 | PZE-109008703 | 9 | 9247324 |
| SYN34685 | 4 | 202660380 | SYN36362 | 9 | 9247324 |
| PZE-104126691 | 4 | 210495187 | PZE-109051619 | 9 | 86414796 |
| PZE-104127248 | 4 | 211708774 | PZE-109101971 | 9 | 141650742 |
| PZE-104127853 | 4 | 212785374 | PZE-110080467 | 10 | 134211006 |
新窗口打开|下载CSV
2.3 全基因组关联分析
共发掘出16个与穗行数显著关联(P<0.0001)的QTN (表3和图4), 分别位于第2、第3、第5、第6、第7、第8、第9染色体上, 其中SYN25713和SYN36577分别位于苏兰1号群体的第4染色体Lancaster特异性遗传区段212.79~243.52 Mb和第7染色体8.19~15.78 Mb范围内(附表3)。Table 3
表3
表3穗行数显著关联的SNP位点
Table 3
| SNP | 染色体 Chr. | 物理位置 Position | P值 P-value (×10-4) | 最小等位基因频率 Minimum allele frequency |
|---|---|---|---|---|
| PZE-102049428 | 2 | 27586143 | 0.83 | 0.22 |
| SYN29936 | 3 | 214728752 | 0.79 | 0.53 |
| SYN25713 | 4 | 218657640 | 0.50 | 0.39 |
| PZE-104012465 | 4 | 10671690 | 0.95 | 0.39 |
| PZE-105068275 | 5 | 70167652 | 0.38 | 0.50 |
| SYN35408 | 5 | 64502995 | 0.64 | 0.53 |
| ZM013904-0312 | 5 | 64757602 | 0.64 | 0.53 |
| PZE-105060180 | 5 | 58448124 | 0.80 | 0.44 |
| PZE-106105143 | 6 | 155807238 | 0.29 | 0.42 |
| PUT-163a-60355888-2779 | 6 | 30864359 | 0.32 | 0.61 |
| PUT-163a-60355888-2773 | 6 | 30864423 | 0.32 | 0.61 |
| SYN36577 | 7 | 9216854 | 0.53 | 0.67 |
| SYN36527 | 8 | 166695438 | 0.72 | 0.17 |
| SYN36532 | 8 | 166695690 | 0.72 | 0.83 |
| PZE-108065598 | 8 | 115805296 | 0.89 | 0.33 |
| PZE-109004718 | 9 | 5289730 | 0.54 | 0.31 |
新窗口打开|下载CSV
图4
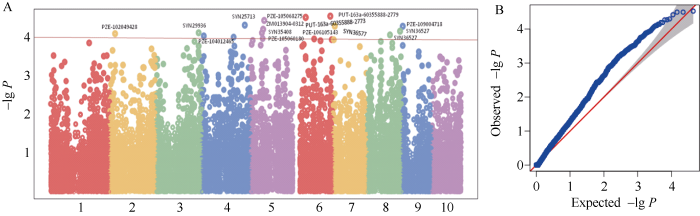 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4穗行数相关QTNA和B分别代表曼哈顿图和QQ图。A and B represent Manhattan plot and QQ plot, respectively.
Fig. 4Kernel rows per ear related QTN
穗长关联分析结果显示,共发掘出13个与穗长显著关联(P < 0.001)的SNP位点(表4和图5),分别位于第1、第2、第5、第7、第8、第9染色体上,其中PZE-105143697 (196993540)位于苏兰1号群体的第5染色体Lancaster特异性遗传区段157.32~ 197.94 Mb范围内(附表3)。
Table 4
表4
表4穗长显著关联的SNP位点
Table 4
| SNP | 染色体 Chr. | 物理位置 Position | P值 P-value (×10-4) | 最小等位基因频率 Minimum allele frequency |
|---|---|---|---|---|
| SYN28790 | 1 | 198085193 | 3.55 | 0.56 |
| PZE-102089207 | 2 | 89322082 | 3.55 | 0.83 |
| PZE-102089216 | 2 | 89350485 | 7.42 | 0.17 |
| PZE-102094273 | 2 | 103594813 | 7.42 | 0.28 |
| PZE-103146876 | 3 | 199472604 | 5.85 | 0.64 |
| PZE-105143697 | 5 | 196993540 | 6.06 | 0.22 |
| SYN2938 | 5 | 212502760 | 2.47 | 0.44 |
| PZE-105181391 | 5 | 215066204 | 8.77 | 0.22 |
| SYN19052 | 7 | 125475003 | 6.64 | 0.78 |
| SYN10053 | 8 | 1816317 | 7.98 | 0.39 |
| PZA-000908002 | 8 | 99959553 | 2.84 | 0.44 |
| PZE-108076469 | 8 | 130589109 | 3.55 | 0.83 |
| PZE-109008801 | 9 | 9401106 | 7.42 | 0.44 |
新窗口打开|下载CSV
图5
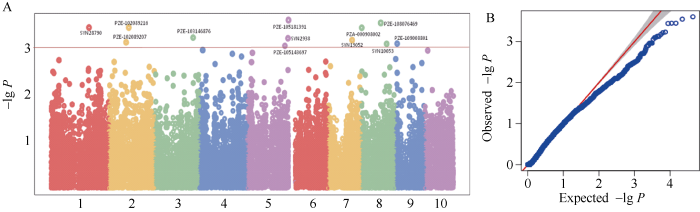 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5穗长相关QTNA和B分别代表曼哈顿图和QQ图。A and B represent Manhattan plot and QQ plot, respectively.
Fig. 5Ear length related QTN
3 讨论
3.1 Suwan种质改良过程中的一些重要区段
群体改良是一种打破有利基因与不利基因连锁, 使有利基因频率得到提高, 从而改良玉米育种材料的过程[1,21]。如雍红军等[22]以辽旅综、中综3号、BSCB1等10个玉米群体改良杂交种吉单261的育种利用发现, 供试群体具有改良杂交种性状的利用价值, 能提高杂交种产量和穗行数的潜力; 贵州省农业科学院将Lancaster种质导入Suwan种质形成苏兰1号群体, 与墨瑞1号群体进行半同胞相互轮回选择改良后, 形成新一轮改良群体组配群体杂交组合, 通过田间鉴定表明, 墨瑞1号C1×苏兰1号C1比墨瑞1号C0×苏兰1号C0的产量提高7.84%, 群体间的平均杂种优势也从改良前的9.86%提高到改良后的16.29%[16]。李芦江等[23]研究5轮控制双亲混合选择对2个玉米人工合成窄基群体P3C0和P4C0的改良效果发现, 控制双亲混合选择对群体单株产量和主要构成性状及其一般配合力(GCA)改良效果明显, 多数性状及其GCA均大于C0, 但是当选择响应到达最大以后, 持续改良会使群体的选择增益下降, 甚至出现负增益。说明群体在改良过程中, 其农艺性状会随着改良轮次的增加呈现出不同的改良效应。本研究结果表明, 群体在改良过程中, 保持着群体内丰富的遗传变异, 不同改良世代间有少数基因发生交换重组, 其中在Suwan 1群体的改良世代中仅获得5个差异遗传片段, 苏兰1号C0群体改良世代中共获得18个差异遗传区段, 有8个在其不同改良世代间稳定出现。另外, Suwan种质改良形成苏兰1号群体的过程中, 共获得53个来自Suwan群体的稳定遗传区段和43个来自Lancaster的差异遗传区段, 而且有35个差异遗传片段在苏兰1号C0群体及其改良世代间稳定出现。说明这些Suwan来源的稳定遗传区段可能在保持Suwan种质优势性状方面发挥作用, 所以在育种选择中被保留下来, 而Lancaster来源的差异遗传区段则可能在改良Suwan种质生育期长、株高等性状缺点方面起作用。这些研究结果从分子水平上解释了苏兰1号群体在育种和生产中应用与Suwan种质存在差异的原因, 也为后续Suwan 1及其衍生群体(苏兰1号)的改良利用提供了一定的理论支撑。
3.2 Suwan群体改良过程中候选区段的效应
本研究共发掘出16个与穗行数显著关联的QTN位点, 分别位于第2、第3、第5、第6、第7、第8、第9染色体上, 其中有2个位点(SYN25713和SYN36577)位于苏兰1号群体的Lancaster来源的遗传区段内; 发掘出13个与穗长显著相关联的QTN位点, 分别位于第1、第2、第5、第7、第8、第9染色体上, 有1个位点(PZE-105143697)位于Lancaster来源的特异性遗传区段内, 研究这些位点对理解玉米Suwan种质遗传机制有重要作用。与前人研究结果的比较发现, Lu等[24]利用B73/丹340的F2群体进行QTL定位, 在第4染色体上检测到qERN4a (umc1294-bnlg1265)和qERN4c (umc2188-umc1101), 与本文第4染色体上发掘出的2个穗行数QTN (PZE-104012465和SYN25713)位于同一位置, 这一结果与Yan等[25]的定位结果相一致。王辉等[26]利用郑58/HD568构建重组自交系群体进行QTL定位, 检测到qEL3 (198.52~201.32 Mb)和qERN4 (211.08~ 219.99 Mb)与本文在第3染色体发掘出的穗长SNP标记PZE-103146876 (199472604)和第4染色体发掘出的穗行数SNP标记 SYN25713 (218657640)位于同一染色体区段的位点; Zhou等[27]利用掖478/ SL17-1构建群体进行QTL定位, 在第7染色体上定位到多效性qEL7.02 (umc1393-bnlg657)与本文在第7染色体发掘出的穗长SNP标记SYN19052 (125475003)所在标记区间一致。说明在这些区段内含有控制相关性状的基因位点且可靠性较高, 该区域在玉米产量相关性状改良过程中起到一定作用, 可以作为今后精细定位的候选区域。4 结论
在Suwan种质改良形成苏兰1号群体的过程中, 共发现53个Suwan来源的稳定遗传区段和43个Lancaster来源的差异遗传区段, 这些区段内存在控制穗行数和穗长的QTN, 对于保持各自优势性状具有重要意义。该研究结果将为Suwan种质改良过程中的优势基因聚合提供很好的借鉴。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
DOI:10.2135/cropsci1992.0011183X003200040010xURL

Knowledge about the combining ability and heterotic patterns among CIMMYT's maize (Zea mays L.) germplasm is essential for hybrid development work at CIMMYT, as well as at other national research programs using CIMMYT germplasm. This study was conducted to determine the heterosis and combining ability among CIMMYT's subtropical and temperate early-maturity maize germplasm. A seven-parent diallel involving two populations and five gene pools was made. The parents and 21 crosses were evaluated in 17 temperate and 5 subtropical environments during 1985-1986. Average yield across temperate environments (4.35 Mg ha-1) was comparable to that obtained in subtropical environments (4.59 Mg ha-1). Highest yield in subtropical environments was recorded by Population 48 x Pool 27 (5.42 Mg ha-1), with a high-parent heterosis of 9.9%. Maximum high-parent heterosis was observed in Population 46 x Pool 30 (13%), which yielded 5.17 Mg ha-1. Under temperate environments, the highest-yielding comb
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12864-016-3041-3URLPMID:5007717 [本文引用: 1]
Maize breeding germplasm used in Southwest China has high complexity because of the diverse ecological features of this area. In this study, the population structure, genetic diversity, and linkage disequilibrium decay distance of 362 important inbred lines collected from the breeding program of Southwest China were characterized using the MaizeSNP50 BeadChip with 56,110 single nucleotide polymorphisms (SNPs). With respect to population structure, two (Tropical and Temperate), three (Tropical, Stiff Stalk and non-Stiff Stalk), four [Tropical, group A germplasm derived from modern U.S. hybrids (PA), group B germplasm derived from modern U.S. hybrids (PB) and Reid] and six (Tropical, PB, Reid, Iowa Stiff Stalk Synthetic, PA and North) subgroups were identified. With increasing K value, the Temperate group showed pronounced hierarchical structure with division into further subgroups. The Genetic Diversity of each group was also estimated, and the Tropical group was more diverse than the Temperate group. Seven low-genetic-diversity and one high-genetic-diversity regions were collectively identified in the Temperate, Tropical groups, and the entire panel. SNPs with significant variation in allele frequency between the Tropical and Temperate groups were also evaluated. Among them, a region located at 13002Mb on Chromosome 2 showed the highest genetic diversity, including both number of SNPs with significant variation and the ratio of significant SNPs to total SNPs. Linkage disequilibrium decay distance in the Temperate group was greater (2.5–302Mb) than that in the entire panel (0.5–0.7502Mb) and the Tropical group (0.25–0.502Mb). A large region at 30–12002Mb of Chromosome 7 was concluded to be a region conserved during the breeding process by comparison between S37, which was considered a representative tropical line in Southwest China, and its 30 most similar derived lines. For the panel covered most of widely used inbred lines in Southwest China, this work representatively not only illustrates the foundation and evolution trend of maize breeding resource as a theoretical reference for the improvement of heterosis, but also provides plenty of information for genetic researches such as genome-wide association study and marker-assisted selection in the future. The online version of this article (doi:10.1186/s12864-016-3041-3) contains supplementary material, which is available to authorized users.
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-013-2246-yURLPMID:24343198 [本文引用: 1]
To investigate the genetic structure of Chinese maize germplasm, the MaizeSNP50 BeadChip with 56,110 single nucleotide polymorphisms (SNPs) was used to genotype a collection of 367 inbred lines widely used in maize breeding of China. A total of 41,819 informative SNPs with minor allele number of more than 0.05 were used to estimate the genetic diversity, relatedness, and linkage disequilibrium (LD) decay. Totally 1,015 SNPs evenly distributed in the genome were selected randomly to evaluate the population structure of these accessions. Results showed that two main groups could be determined i.e., the introduced germplasm and the local germplasm. Further, five subgroups corresponding to different heterotic groups, that is, Reid Yellow Dent (Reid), Lancaster Sure Crop (Lancaster), P group (P), Tang Sipingtou (TSPT), and Tem-tropic I group (Tem-tropic I), were determined. The genetic diversity of within subgroups was highest in the Tem-Tropic I and lowest in the P. Most lines in this panel showed limited relatedness with each other. Comparisons of gene diversity showed that there existed some conservative genetic regions in specific subgroups across the ten chromosomes, i.e., seven in the Lancaster, seven in the Reid, six in the TSPT, five in the P, and two in the Tem-Tropical I. In addition, the results also revealed that there existed fifteen conservative regions transmitted from Huangzaosi, an important foundation parent, to its descendants. These are important for further studies since the outcomes may provide clues to understand why Huangzaosi could become a foundation parent in Chinese maize breeding. For the panel of 367 elite lines, average LD distance was 391kb and varied among different chromosomes as well as in different genomic regions of one chromosome. This analysis uncovered a high natural genetic diversity in the elite maize inbred set, suggesting that the panel can be used in association study, esp. for temperate regions.
DOI:10.1093/bioinformatics/btm308URLPMID:20320202020202020202020 [本文引用: 1]
Summary: Association analyses that exploit the natural diversity of a genome to map at very high resolutions are becoming increasingly important. In most studies, however, researchers must contend with the confounding effects of both population and family structure. TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) implements general linear model and mixed linear model approaches for controlling population and family structure. For result interpretation, the program allows for linkage disequilibrium statistics to be calculated and visualized graphically. Database browsing and data importation is facilitated by integrated middleware. Other features include analyzing insertions/deletions, calculating diversity statistics, integration of phenotypic and genotypic data, imputing missing data and calculating principal components. Availability: The TASSEL executable, user manual, example data sets and tutorial document are freely available at http://www.maizegenetics.net/tassel. The source code for TASSEL can be found at http://sourceforge.net/projects/tassel. Contact: pjb39@cornell.edu
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11032-010-9468-3URL [本文引用: 1]
Genetic factors controlling quantitative inheritance of grain yield and its components have been intensively investigated during recent decades using diverse populations in maize (Zea mays L.). Notwithstanding this, quantitative trait loci (QTL) for kernel row number (KRN) with large and consistent effect have not been identified. In this study, a linkage map of 150 simple sequence repeat (SSR) loci was constructed by using a population of 500 F2 individuals derived from a cross between elite inbreds Ye478 and Dan340. The linkage map spanned a total of 1478 cM with an average interval of 10.0 cM. A total of 397 F2:3 lines were evaluated across seven diverse environments for mapping QTL for KRN. Some QTL for grain yield and its components had previously been confirmed with this population across environments. A total of 13 QTL for KRN were identified, with each QTL explaining from 3.0 to 17.9% of phenotypic variance. The gene action for KRN was mainly additive to partial dominance. A large-effect QTL (qkrn7) with partial dominance effect accounting for 17.9% of the phenotypic variation for KRN was identified on chromosome 7 near marker umc1865 with consistent gene effect across seven diverse environments. This study has laid a foundation for map-based cloning of this major QTL and for developing molecular markers for marker-assisted selection of high KRN.
DOI:10.1007/s10681-005-9060-9URL [本文引用: 1]
The aim of this investigation was to map quantitative trait loci (QTL) associated with grain yield and yield components in maize and to analyze the role of epistasis in controlling these traits. An F 2:3 population from an elite hybrid (Zong387-1) was used to evaluate grain yield and yield components in two locations (Wuhan and Xiangfan, China) using a randomized complete-block design. The mapping population included 266 F 2:3 family lines. A genetic linkage map containing 150 simple sequence repeats and 24 restriction fragment length polymorphism markers was constructed, spanning a total of 2531.6 cM with an average interval of 14.5 cM. A logarithm-of-odds threshold of 2.8 was used as the criterion to confirm the presence of one QTL after 1000 permutations. Twenty-nine QTL were detected for four yield traits, with 11 of them detected simultaneously in both locations. Single QTL contribution to phenotypic variations ranged from 3.7% to 16.8%. Additive, partial dominance, dominance, and overdominance effects were all identified for investigated traits. A greater proportion of overdominance effects was always observed for traits that exhibited higher levels of heterosis. At the P 0.005 level with 1000 random permutations, 175 and 315 significant digenic interactions were detected in two locations for four yield traits using all possible locus pairs of molecular markers. Twenty-four significant digenic interactions were simultaneously detected for four yield traits at both locations. All three possible digenic interaction types were observed for investigated traits. Each of the interactions accounted for only a small proportion of the phenotypic variation, with an average of 4.0% for single interaction. Most interactions (74.9%) occurred among marker loci, in which significant effects were not detected by single-locus analysis. Some QTL (52.2%) detected by single-locus analysis were involved in epistatic interactions. These results demonstrate that digenic interactions at the two-locus level might play an important role in the genetic basis of maize heterosis.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}