 ,1, 储文1, 梅德圣1, 成洪涛1, 朱琳琳2, 付丽1, 胡琼1, 刘佳,1,*
,1, 储文1, 梅德圣1, 成洪涛1, 朱琳琳2, 付丽1, 胡琼1, 刘佳,1,*Quantitative trait loci mapping for branch angle and candidate gene screening in Brassica napus L.
WANG Wen-Xiang,1, CHU Wen1, MEI De-Sheng1, CHENG Hong-Tao1, ZHU Lin-Lin2, FU Li1, HU Qiong1, LIU Jia,1,*1 2
通讯作者:
第一联系人:
收稿日期:2018-03-22接受日期:2018-08-20网络出版日期:2018-09-09
| 基金资助: |
Received:2018-03-22Accepted:2018-08-20Online:2018-09-09
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2166KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
汪文祥, 储文, 梅德圣, 成洪涛, 朱琳琳, 付丽, 胡琼, 刘佳. 基于SNP遗传图谱定位甘蓝型油菜分枝角度QTL[J]. 作物学报, 2019, 45(1): 37-45. doi:10.3724/SP.J.1006.2019.84042
WANG Wen-Xiang, CHU Wen, MEI De-Sheng, CHENG Hong-Tao, ZHU Lin-Lin, FU Li, HU Qiong, LIU Jia.
油菜是我国重要的油料作物, 每年提供450万吨左右食用油, 占全国植物油总产的40%以上[1]。现阶段, 油菜单产水平低和机械化生产普及率不高是限制我国油菜产业发展的两大瓶颈。为增加单产、提高油菜机械化生产效率, 改良现有品种的株型、提高油菜的种植密度是一条有效途径。改良油菜株型不仅可以促进光能和肥料利用率, 提高品种高密度耐性, 降低病害, 增加群体产量[2], 还有利于增强抗倒性和机械化生产, 通过资源高效化利用实现低耗高产[3]。
随着甘蓝型油菜全基因组测序的完成[4,5]、高通量SNP芯片技术的完善和分析成本的下降, 油菜60K SNP芯片分型分析成为油菜遗传研究的重要工具[6,7,8,9,10,11]。近几年, 利用油菜60K SNP芯片对油菜产量及品质等性状的遗传定位做了大量的研究[12,13,14]。Liu等[6]利用油菜芯片对RIL群体进行分型, 对种子颜色、木质素、纤维素和半纤维素等性状进行QTL定位。Sun等[15]利用520份油菜资源群体对株高性状进行全基因组关联分析, 鉴定到68个显著的SNP位点, 超过70%的位点与已发表的9个遗传群体中重合。Fu等[16]利用DH群体和永久F2群体对角果长和千粒重性状进行联合QTL分析, 将A9染色体的角果长和千粒重区间缩小到1.14 Mb内。油菜SNP芯片技术的利用, 提高了遗传图谱构建精密度, 为油菜数量性状的研究提供更方便、快捷的途径。
油菜分枝角度是构建理想株型的重要因素, 紧凑型株型有利于密植, 有利于提高群体的产量和抗倒伏性, 减少病害[17,18,19,20,21,22,23,24]。Liu等[18]最早利用143份自然群体对分枝角度进行全基因组关联分析, 获得6个显著关联的基因组区域, 鉴定出其中4个区域内候选基因。Sun等[19]利用520份自然群体进行全基因组关联分析, 共获得52个分枝角度相关位点, 总共可解释表型变异的51.1%, 并对52个位点进行了候选基因分析。Wang等[20]利用分枝角差异显著亲本构建F2群体, 鉴定F2代所有单株的分枝角表型, 挑选极端单株进行混池, 利用BSA-seq方法确定A6染色体定位区间内生长素合成相关基因BnaYUCCA6a为候选基因。Shen等[22]利用包含208个系的DH群体定位了17个油菜分枝角度QTL, 3个主效的QTL各能解释约10%表型变异, 并在QTL区间获得27个候选基因。上述研究表明, 油菜分枝角度受多基因控制, 油菜分枝角度性状的遗传解析对油菜品种的遗传改良具有重要意义。
本研究利用油菜60K SNP芯片构建高密度遗传连锁图谱, 对不同部位油菜分枝角度性状进行QTL定位分析。收集整理前人研究油菜分枝角度基因信息, 并利用甘蓝型油菜基因组序列, 根据QTL区间物理位置及拟南芥功能基因组信息筛选可能的候选基因, 为明确关键QTL和克隆候选基因奠定基础。
1 材料与方法
1.1 材料
以分枝角度小的甘蓝型油菜品系1019B和分枝角度大的R2为亲本, 手工配置F1, 通过小孢子培养获得DH群体。选取其中163个DH系进行SNP标记分型, 用以构建高密度SNP遗传连锁图谱。所有材料均由中国农业科学院油料作物研究所提供及保存。1.2 田间试验
2013年9月与2014年9月, 将亲本及DH系群体种植于中国农业科学院油料作物研究所阳逻试验基地, 分别记录为WH2014和WH2015。随机区组设计2个重复, 每个小区3行, 每行18株, 行距0.33 m, 株距0.10 m。田间管理同常规生产, 确保所有样本的外部生长环境一致, 待成熟后统计数据。1.3 性状考察
油菜成熟后, 从每个株系选取正常生长的5株, 剪取连有油菜上部第一分枝(顶枝)和基部第一分枝(基枝)的茎段, 通过数字图像采集法[17]获取顶端分枝角和基部分枝角图像文件, 将其导入AutoCAD软件, 利用角度工具标注角度, 记录到Microsoft Excel文档中。1.4 遗传连锁图谱及QTL分析
利用油菜60K Illumina Infinium SNP芯片对亲本(1019B和R2)及DH群体进行SNP基因分型。采用GenomeStudio (Illumina公司)软件分析SNP基因型, 排除低于0.05的最小基因型频率(minor allele frequency, MAF), SNP得率(call frequency)小于80%, 筛选在亲本间具有多态性的标记, 利用SNP芯片序列信息和法国公布的甘蓝型油菜品种“Darmor-Bzh”的基因组序列信息进行BlastN比对, E-value阈值为1×10-15, 获得唯一位置的SNP标记信息, 最终获得9521个高质量SNP标记用于后续分析。利用MSTmap软件[25]和JoinMap 4.0软件[26]构建遗传图谱, 通过两两标记之间最小重组频率计算每个连锁群上标记顺序, 构建高密度bin-map遗传图谱。利用Windows QTL Cartographer 2.5软件[27]中复合区间作图(Composite Interval Mapping, CIM)法对2个环境下基部分枝角和顶端分枝角进行单环境QTL检测。运行CIM时, 选用1 cM的步长(walking speed), 同时设置5个标记作为余因子, 采用模型6, 对每个性状分别进行1000次排列测验(permutation test), 显著水平0.05来判断是否存在QTL。运行结果同时给出性状QTL的加性效应和表型贡献率。以q加顶端分枝角度英文缩写“顶端分”或基部分枝角度英文缩写“基部分”, 再加上染色体编号及QTL序号命名QTL。在染色体相同的位置重复的QTL, 且加性效应方向一致, 认为是同一QTL。采用MapChart 2.3[28]绘制QTL定位遗传连锁图。
1.5 候选基因筛选
为了筛选出分枝角度相关的候选基因, 基于油菜基因组和拟南芥基因组序列进化的同源性[29], 在甘蓝型油菜基因组[4]上查询检测到的QTL置信区间对应的序列, 然后与Liu等[15]、Sun等[25]、Li等[21]和Shen等[22]搜索出的拟南芥分枝角度相关基因进行BlastN比对, 将E值设定为1×10-20, 最后筛选出每个QTL置信区间内匹配E值小于阈值的候选基因。2 结果与分析
2.1 双亲及DH群体分枝角度表型变异
在两年的环境中, 2个亲本的顶端分枝角和基部分枝角差异均极显著(表1)。1019B顶端分枝角和基部分枝角均较小, R2顶端分枝角和基部分枝角均较大。DH群体顶端分枝角和基部分枝角均呈连续性分布, 其中一些株系表现出明显的超亲分离现象, 平均变异幅度分别为22.98°~63.99°和20.76°~50.22° (图1), 平均值介于两亲本之间; 顶端分枝角和基部分枝角的变异系数都大于10, 说明顶端分枝角和基部分枝角都具有较大的改良潜力。Table 1
表1
表1亲本及DH群体分枝角度性状两年的表型分析
Table 1
| 年份 Year | 性状 Trait | 亲本 Parent | Pt-test | DH群体 DH population | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1019B (o) | R2 (o) | 范围 Range (o) | 均值 Mean (o) | 标准差 SD | 变异系数 CV | 偏度 Skewness | 峰度 Kurtosis | |||
| 2014 | BBA | 28.13±5.45 | 42.34±4.43 | 6.09E-03 | 26.17-50.22 | 36.46 | 5.23 | 14.35 | -0.777 | -0.085 |
| TBA | 36.32±5.46 | 46.65±4.08 | 1.47E-02 | 22.98-56.37 | 37.74 | 5.41 | 14.33 | 0.515 | 0.080 | |
| 2015 | BBA | 27.90±3.50 | 37.72±2.25 | 1.44E-05 | 20.76-45.64 | 32.68 | 5.54 | 16.94 | -0.201 | 0.150 |
| TBA | 38.75±6.30 | 47.66±6.10 | 2.46E-03 | 26.30-63.99 | 40.43 | 6.70 | 16.57 | 1.364 | 0.625 | |
新窗口打开|下载CSV
图1
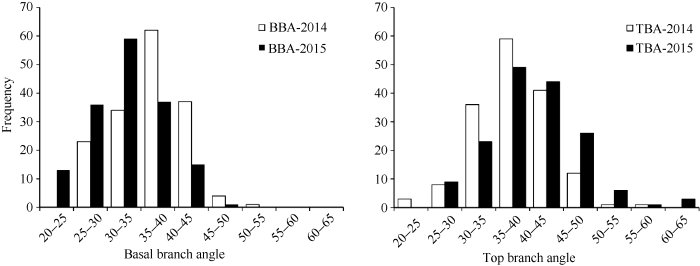 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1油菜1019B×R2 DH群体分枝角度性状(BBA和TBA)两年的频率分布图
Fig. 1Frequency distribution of branch angle (BBA and TBA) in 1019B×R2 DH population planted in two environments
相关性分析表明, 在2014年和2015年的2个环境中顶端分枝角和基部分枝角表现均呈极显著正相关(表2), 相关系数分别为0.362和0.411, 说明油菜分枝角性状遗传稳定, 但受一定环境影响。在2个生长周期里, 顶端分枝角和基部分枝角呈极显著正相关, 相关系数分别为0.542和0.586, 说明油菜顶端分枝角和基部分枝角相关性较强, 具有一致性。
Table 2
表2
表2在2014年和2015年甘蓝型油菜DH群体分枝角度性状的相关系数
Table 2
| 2015BBA | 2014TBA | 2015TBA | |
|---|---|---|---|
| 2014BBA | 0.411** | 0.542** | 0.353** |
| 2015BBA | 0.432** | 0.586** | |
| 2014TBA | 0.362** |
新窗口打开|下载CSV
2.2 高密度遗传图构建
利用油菜SNP芯片, 检测DH群体的基因型, 共得到9521个高质量多态性SNP标记。基于这些SNP标记, 利用MSTmap软件构建DH群体的重组区块图谱共得到1442个Bin, 并利用这些Bin构建的遗传连锁图谱总长度为2544.07 cM, 标记间的平均距离1.76 cM, 标记间最大距离为30.59 cM, 最小距离0.06 cM; 其中A10染色体上标记间的平均遗传距离最小(1.03 cM), C09染色体上标记间平均遗传距离最大(3.73 cM)(图2)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2SNP标记簇在连锁群上分布图
纵坐标显示甘蓝型油菜基因组19个连锁群中的每一个SNP簇的遗传距离。
Fig. 2Overview of genome-wide SNP density in the bin map of DH population
The ordinate shows the genetic distance along each of the 19 linkage groups corresponding to B. napus genome.
2.3 油菜分枝角度QTL分析
利用软件Windows QTL Cartographer 2.5对顶端分枝角和基部分枝角性状进行QTL分析, 共检测到17个QTL, 分布于A01、A02、A03、A06、A09、C02、C03、C04、C06和C08染色体上, 单个QTL可解释的表型变异为6.39%~21.78% (表3和图3)。其中检测到6个基部分枝角QTL, 单个QTL解释的表型变异为8.07%~15.10%; 检测到11个顶端分枝角QTL, 单个QTL解释的表型变异为6.39%~21.78%。在2年内重复检测到基部分枝角性状显著效应的QTL qBBA.A03.1和qBBA.A03.2, 与在一个环境中检测到的顶端分枝角性状QTL (qTBA.A03.1)在同一个置信区间, 在3个环境中解释的表型变异为8.28%~11.79%, 加性效应值为1.74~2.03。另外, 在同一环境中检测到顶端分枝角和基部分枝角共同的QTL (qBBA.C08.1和qTBA.C08.1), 解释表型变异8.78%~8.85%。Table 3
表3
表32个环境中检测到的顶端分枝角和基部分枝角QTL
Table 3
| 年份 Year | 性状 Trait | 数量性状位点 QTL | 染色体 Chromosome | 位置 Position | 置信区间 Confidence interval | 阈值 LOD | 加性效应 Additive | 贡献率 R2(%) |
|---|---|---|---|---|---|---|---|---|
| 2014 | BBA | qBBA.A02.1 | A02 | 4.0 | 0-24.0 | 3.7 | 2.15 | 15.10 |
| qBBA.A03.1 | A03 | 11.1 | 7.9-15.0 | 2.8 | 1.74 | 9.31 | ||
| qBBA.C08.1 | C08 | 8.4 | 0-11.4 | 2.8 | -1.94 | 8.85 | ||
| 2015 | BBA | qBBA.A01.1 | A01 | 34.0 | 32.5-35.4 | 2.8 | 1.70 | 8.07 |
| qBBA.A03.2 | A03 | 11.8 | 10.2-14.9 | 3.9 | 2.03 | 11.79 | ||
| qBBA.C03.1 | C03 | 101.7 | 99.7-103.6 | 3.9 | -2.12 | 12.66 | ||
| 2014 | TBA | qTBA.A06.1 | A06 | 90.4 | 87.2-91.4 | 2.9 | -1.44 | 8.20 |
| qTBA.A09.1 | A09 | 87.0 | 82.0-90.2 | 6.9 | -2.35 | 21.78 | ||
| qTBA.C03.1 | C03 | 146.0 | 144.2-148.5 | 4.3 | -1.79 | 12.73 | ||
| qTBA.C08.1 | C08 | 8.4 | 0-11.4 | 3.0 | -1.69 | 8.78 | ||
| 2015 | TBA | qTBA.A03.1 | A03 | 17.6 | 14.9-18.8 | 0.2 | 1.85 | 8.28 |
| qTBA.A09.2 | A09 | 3.1 | 0.9-4.2 | 4.1 | -2.14 | 11.08 | ||
| qTBA.C02.1 | C02 | 47.3 | 45.8-49.2 | 2.8 | 1.61 | 6.39 | ||
| qTBA.C02.2 | C02 | 54.8 | 53.4-57.5 | 3.2 | 1.83 | 8.07 | ||
| qTBA.C02.3 | C02 | 65.5 | 64.5-71.6 | 2.8 | 1.65 | 6.43 | ||
| qTBA.C04.1 | C04 | 1.6 | 0-5.7 | 3.9 | -2.13 | 10.24 | ||
| qTBA.C06.1 | C06 | 80.3 | 79.7-92.2 | 4.1 | 5.20 | 11.00 |
新窗口打开|下载CSV
图3
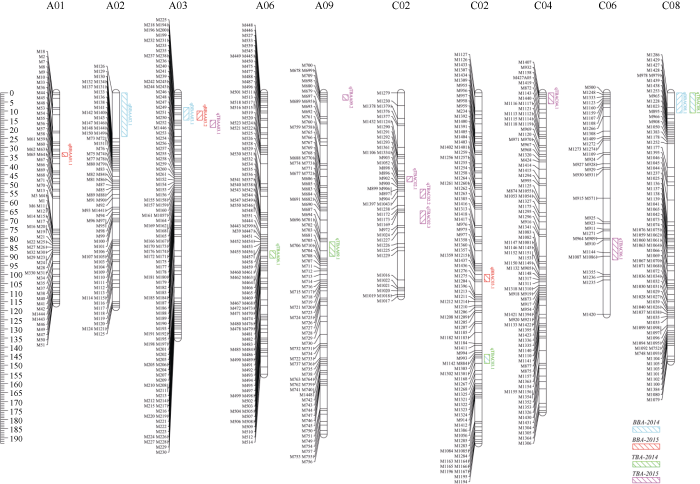 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘蓝型油菜顶端分枝角和基部分枝角QTL在连锁群上分布图
Fig. 3Putative QTLs of basal branch angle and top branch angle on the genetic map
2.4 分枝角度候选基因筛选
将17个QTL置信区间序列与前人报道的181个分枝角度相关基因分别比对, 在其中6个QTL区间内检测到12个候选基因。在两年均检测到的QTL (qBBA.A03.1、qBBA.A03.2和qTBA.A03.1)置信区间内发现生长素转运相关的基因VAMP714, 其物理位置离整合后的QTL峰值非常近, 该基因也存在于Sun等[19]定位的分枝角度QTL区间内。在基部分枝角度QTL (qBBA.A01.1)置信区间内筛选到生长素相关的基因ARF16; 在顶端分枝角度2个QTL (qTBA.A09.2和 qTBA.C06.1)置信区间内分别筛选出2个分枝角度候选基因。另外, 在顶端分枝角度QTL (qTBA.C03.1)置信区间内筛选到4个生长素相关候选基因; 与控制水稻分蘖角基因TAC1同源的基因也在顶端分枝角的QTL (qTBA.C04.1)区间内检测到(表4)。Table 4
表4
表4在甘蓝型油菜分枝角QTL置信区间比对获得的候选基因
Table 4
| 性状 Trait | 名称 Name | 物理区间 Physical interval | 预测基因 Gene prediction | 拟南芥相关基因 Related genes in A. thaliana | 参考文献Reference | |
|---|---|---|---|---|---|---|
| 基因名 Gene name | 登录号 Accession number | |||||
| BBA | qBBA.A01.1 | 2,992,973-3,391,055 | BnaA01g06910D | ARF16 | AT4G30080 | Shen et al.[22] |
| qBBA.A03.1 | 3,256,156-4,877,684 | BnaA03g08500D | VAMP714 | AT5G22360 | Sun et al.[19] | |
| TBA | qTBA.A09.2 | 963,282-1,890,151 | BnaA09g02390D | ABCB19/PGP19 | AT3G28860 | Sun et al.[19] |
| BnaA09g02480D | EXPA5 | AT3G29030 | Sun et al.[19] | |||
| qTBA.C03.1 | 26,692 780-33,111,760 | BnaC03g46000D | GH3-10, DFL2 | AT4G03400 | Shen et al.[22] | |
| BnaC03g46010D | GH3-10, DFL2 | AT4G03400 | Shen et al.[22] | |||
| BnaC03g46450D | SAUR8 | AT2G16580 | Shen et al.[22] | |||
| BnaC03g46960D | SAUR42 | AT2G28085 | Shen et al.[22] | |||
| qTBA.C04.1 | 1563-990,575 | BnaC04g00310D | WRK23 | AT2G47260 | Li et al.[21] | |
| BnaC04g00780D | TAC1 | AT2G46640 | Li et al.[21] | |||
| qTBA.C06.1 | 27,360,890-32,410,130 | BnaC06g29230D | IAR1 | AT1G68100 | Li et al.[21] | |
| BnaC06g31170D | IAR4 | AT1G24180 | Li et al.[21] | |||
新窗口打开|下载CSV
3 讨论
株型紧凑和抗倒伏是实现油菜全程机械化栽培的重要性状。本研究通过高通量SNP标记构建高密度遗传图谱, 鉴定获得了油菜分枝角的QTL及两侧与性状连锁的标记, 为油菜分枝角度的分子标记辅助育种提供了依据。我们前期研究发现, 油菜从基部到顶部的分枝角度并不完全一样, 有逐渐增大的趋势[30]。不同材料间从下到上分枝角度变化具有同样的趋势, 这是由油菜多分枝生长发育习性决定的。本研究同时调查顶部和基部的分枝角度, 可以对这一性状进行更全面的分析。本研究获得的油菜分枝角度QTL多数与已研究报道的QTL[18,19,20,21,22]相吻合, 还鉴定到一些新的QTL; 2个环境重复检测到位于A03连锁群的QTL效应稳定, 与前人利用关联分析方法获得的peak-SNP (Bn-A03-p4342338)位点相同。顶端分枝角度和基部分枝角在同一环境中均在C08连锁群定位到重合的QTL, 并且表型数据表明顶端分枝角和基部分枝角具有显著相关性, 说明可能存在同时控制油菜上、下分枝角度的基因。随着高通量测序技术的发展, 油菜参考基因组信息的公布[4,5], 油菜基因组上大量的SSR、Indel、SNP等标记被发掘出来[31,32]。SNP标记因为数量多、分布广, 在基因组中密度更高和分布更均匀, 已实现SNP基因型分型的高通量、快速和自动化检测, 使不同研究者间的数据整合分析成为现实[33]。本研究构建的高密度遗传图谱, 标记间平均距离1.76 cM, 是较精密的甘蓝型油菜遗传图谱之一。利用该图谱获得的分枝角性状相关SNP标记, 有助于进一步发展加密QTL区间的分子标记, 用于进一步精细定位和功能基因克隆。
利用油菜基因组序列信息及SNP标记的侧翼序列, 从分枝角度的QTL置信区段内, 借助和拟南芥基因组间同源区间序列比对, 筛选出12个油菜分枝角候选基因。其中坐落于A03染色体上的候选基因, 与拟南芥的VAMP714基因同源。据拟南芥基因的功能注释, VAMP714基因正向调节生长素, 在生长素的极性运输中通过正确的PIN蛋白分泌与定位来实现其功能, 在植物分枝数或分生组织的建成中发挥着重要作用[34]。位于C04染色体的候选基因BnaC04g00780D, 该基因的拟南芥同源基因是TAC1。TAC1是控制水稻分蘖角度增加的主效基因, 具有显性效应[35]; 在玉米[36]、小麦和桃树[37]中也已分别克隆和验证了TAC1基因的功能。因此, TAC1类基因在植物中具有广泛的调节侧生分枝水平生长的功能[34]。在定位到的分枝角度QTL区间内还鉴定到生长激素相关的基因ARF16、GH3、SAUR和IAR。此外, 本研究还在QTL区间内发现许多未知功能的候选基因, 可能存在油菜特有的分枝角度的功能基因, 有待进一步挖掘。
4 结论
本研究构建了包含19条染色体的油菜高密度遗传图谱, 该图谱含9521个多态性标记, 经过整合为1442个簇, 相邻簇之间平均距离为1.76 cM。定位到位于A01、A02、A03、A06、A09、C02、C03、C04、C06和C08染色体上的17个分枝角度QTL, 其中位于A03染色体上的QTL能在两年环境下被重复检测到, 贡献率为9.31%~11.80%, 在A03染色体上重复检测到的QTL 置信区间获得1个与分枝角度相关的候选基因VAMP714, 在C04染色体上的QTL置信区间获得1个控制水稻分蘖角度的候选基因TAC1; 分别在染色体A01、A09、C03和C06上QTL中检测到生长激素相关基因ARF16、GH3、SAUR和IAR。本研究为进一步鉴定和克隆油菜分枝角度功能基因奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/srep21625URLPMID:26880301 [本文引用: 1]

An optimized plant architecture (PA) is fundamental for high-yield breeding but the genetic control of the important trait is largely unknown in rapeseed. Here plant architecture factors (PAFs) were proposed to consist of main inflorescence length proportion (MILP), branch height proportion (BHP), and branch segment proportion (BSP). Comparison of different genotypes in a DH population grown in diverse environments showed that an optimized PAF performance with MILP and BHP between 0.3–0.4 was important for high yield potential. In total, 163 unique quantitative trait loci (QTLs) for PA- and plant yield (PY)-related traits were mapped onto a high-density genetic map. Furthermore, 190 PA-related candidate genes for 91 unique PA QTLs and 2350 PY epistatic interaction loci-pairs were identified, which explain 2.8–51.8% and 5.2–23.6% of phenotypic variation, respectively. Three gene categories, transcription factor, auxin/IAA, and gibberellin, comprise the largest proportions of candidate genes for PA-related QTLs. The effectiveness of QTL candidate genes prediction was demonstrated by cloning of three candidate genes, Bna.A02.CLV2, Bna.A09.SLY2, and Bna.C07.AHK4. The study thus outlines a gene network for control of PA-related traits and provides novel information for understanding the establishment of ideal PA and for developing effective breeding strategies for yield improvement in rapeseed and other crops.
DOI:10.1109/83.760334URLPMID:18444901 [本文引用: 1]
Higher plants display a variety of architectures that are defined by the degree of branching, internodal elongation, and shoot determinancy. Studies on the model plants of Arabidopsis thaliana and tomato and on crop plants such as rice and maize have greatly strengthened our understanding on the molecular genetic bases of plant architecture, one of the hottest areas in plant developmental biology. The identification of mutants that are defective in plant architecture and characterization of the corresponding and related genes will eventually enable us to elucidate the molecular mechanisms underlying plant architecture. The achievements made so far in studying plant architecture have already allowed us to pave a way for optimizing the plant architecture of crops by molecular design and improving grain productivity.
[本文引用: 3]
DOI:10.1111/tpj.13669URLPMID:28849613 [本文引用: 2]
react-text: 64 Improving the trait based the results from GWAS /react-text react-text: 65 /react-text
DOI:10.1371/journal.pone.0083052URLPMID:3873396 [本文引用: 2]
A high density genetic linkage map for the complex allotetraploid crop species Brassica napus (oilseed rape) was constructed in a late-generation recombinant inbred line (RIL) population, using genome-wide single nucleotide polymorphism (SNP) markers assayed by the Brassica 60 K Infinium BeadChip Array. The linkage map contains 9164 SNP markers covering 1832.9 cM. 1232 bins account for 7648 of the markers. A subset of 2795 SNP markers, with an average distance of 0.66 cM between adjacent markers, was applied for QTL mapping of seed colour and the cell wall fiber components acid detergent lignin (ADL), cellulose and hemicellulose. After phenotypic analyses across four different environments a total of 11 QTL were detected for seed colour and fiber traits. The high-density map considerably improved QTL resolution compared to the previous low-density maps. A previously identified major QTL with very high effects on seed colour and ADL was pinpointed to a narrow genome interval on chromosome A09, while a minor QTL explaining 8.1% to 14.1% of variation for ADL was detected on chromosome C05. Five and three QTL accounting for 4.7% to 21.9% and 7.3% to 16.9% of the phenotypic variation for cellulose and hemicellulose, respectively, were also detected. To our knowledge this is the first description of QTL for seed cellulose and hemicellulose in B. napus, representing interesting new targets for improving oil content. The high density SNP genetic map enables navigation from interesting B. napus QTL to Brassica genome sequences, giving useful new information for understanding the genetics of key seed quality traits in rapeseed.
DOI:10.1007/s00122-014-2343-6URLPMID:24947439 [本文引用: 1]
Abstract KEY MESSAGE: Considerable genome variation had been incorporated within rapeseed breeding programs over past decades. In past decades, there have been substantial changes in phenotypic properties of rapeseed as a result of extensive breeding effort. Uncovering the underlying patterns of allelic variation in the context of genome organisation would provide knowledge to guide future genetic improvement. We assessed genome-wide genetic changes, including population structure, genetic relatedness, the extent of linkage disequilibrium, nucleotide diversity and genetic differentiation based on F ST outlier detection, for a panel of 472 Brassica napus inbred accessions using a 60 k Brassica Infinium庐 SNP array. We found genetic diversity varied in different sub-groups. Moreover, the genetic diversity increased from 1950 to 1980 and then remained at a similar level in China and Europe. We also found ~6-10 % genomic regions revealed high F ST values. Some QTLs previously associated with important agronomic traits overlapped with these regions. Overall, the B. napus C genome was found to have more high F ST signals than the A genome, and we concluded that the C genome may contribute more valuable alleles to generate elite traits. The results of this study indicate that considerable genome variation had been incorporated within rapeseed breeding programs over past decades. These results also contribute to understanding the impact of rapeseed improvement on available genome variation and the potential for dissecting complex agronomic traits.
DOI:10.7505/j.issn.1007-9084.2014.06.001URL [本文引用: 1]
Plant height is an important factor affecting the yield of oilseed rape. In order to find the markers linked with plant height and to increase the efficiency of plant height improvement by molecular marker-assisted selection, identification of quantitative trait loci (QTL) with single environment and multi-environment detection methods for plant height was conducted in a recombinant inbred lines (RILs) population derived from a cross between 888-5 (a dwarf line) and M083 (a tall line) by employing the newly developed Brassica napus 60K SNP array. The results indicated that a total of 27 putative QTLs for plant height in the 4 environments were detected on the chromosomes A2, A4, A5, A6, A9, A10, C3 and C7, each accounting for 0.70% - 26.10% of the phenotypic variation. Among them, 6 QTLs showed interaction with the environment, and their effect values in the 4 environments were different, but their directions were consistent. QTL qPHE2-4 which had the largest effect in the 27 QTLs was detected in 2 environments, explaining 17.96%-26.10% of the phenotypic variation, and 2 linkage SNP markers in this QTL region had less than 0.2 cM distance from the peak of the QTL hese QTLs provided useful information for improving the plant height in oilseed rape breeding.
DOI:10.7505/j.issn.1007-9084.2014.06.001URL [本文引用: 1]
Plant height is an important factor affecting the yield of oilseed rape. In order to find the markers linked with plant height and to increase the efficiency of plant height improvement by molecular marker-assisted selection, identification of quantitative trait loci (QTL) with single environment and multi-environment detection methods for plant height was conducted in a recombinant inbred lines (RILs) population derived from a cross between 888-5 (a dwarf line) and M083 (a tall line) by employing the newly developed Brassica napus 60K SNP array. The results indicated that a total of 27 putative QTLs for plant height in the 4 environments were detected on the chromosomes A2, A4, A5, A6, A9, A10, C3 and C7, each accounting for 0.70% - 26.10% of the phenotypic variation. Among them, 6 QTLs showed interaction with the environment, and their effect values in the 4 environments were different, but their directions were consistent. QTL qPHE2-4 which had the largest effect in the 27 QTLs was detected in 2 environments, explaining 17.96%-26.10% of the phenotypic variation, and 2 linkage SNP markers in this QTL region had less than 0.2 cM distance from the peak of the QTL hese QTLs provided useful information for improving the plant height in oilseed rape breeding.
DOI:10.1093/dnares/dsv035PMID:26659471 [本文引用: 1]
Flowering time adaptation is a major breeding goal in the allopolyploid species Brassica napus. To investigate the genetic architecture of flowering time, a genome-wide association study (GWAS) of flowering time was conducted with a diversity panel comprising 523 B. napus cultivars and inbred lines grown in eight different environments. Genotyping was performed with a Brassica 60K Illumina Infinium SNP array. A total of 41 single-nucleotide polymorphisms (SNPs) distributed on 14 chromosomes were found to be associated with flowering time, and 12 SNPs located in the confidence intervals of quantitative trait loci (QTL) identified in previous researches based on linkage analyses. Twenty-five candidate genes were orthologous to Arabidopsis thaliana flowering genes. To further our understanding of the genetic factors influencing flowering time in different environments, GWAS was performed on two derived traits, environment sensitivity and temperature sensitivity. The most significant SNPs were found near Bn-scaff_16362_1-p380982, just 13 kb away from BnaC09g41990D, which is orthologous to A. thaliana CONSTANS (CO), an important gene in the photoperiod flowering pathway. These results provide new insights into the genetic control of flowering time in B. napus and indicate that GWAS is an effective method by which to reveal natural variations of complex traits in B. napus.<br>
DOI:10.1016/j.plantsci.2015.05.012URLPMID:26566834 [本文引用: 1]
Crop plant architecture plays a highly important role in its agronomic performance. Plant height (PH) and primary branch number (PB) are two major factors that affect the plant architecture of rapeseed (Brassica napus). Previous studies have shown that these two traits are controlled by multiple quantitative trait loci (QTL); however, QTLs have not been delimited to regions less than 10cM. Genome-wide association study (GWAS) is a highly efficient approach for identifying genetic loci controlling traits at relatively high resolution. In this study, variations in PH and PB of a panel of 472 rapeseed accessions that had previously been analyzed by a 60k SNP array were investigated for three consecutive years and studied by GWAS. Eight QTLs on chromosome A03, A05, A07 and C07 were identified for PH, and five QTLs on A01, A03, A07 and C07 were identified for PB. Although most QTLs have been detected in previous studies based on linkage analyses, the two QTLs of PH on A05 and the QTL of PB on C07 were novel. In the genomic regions close to the GWAS peaks, orthologs of the genes involved in flower development, phytohormone biosynthesis, metabolism and signaling in Arabidopsis were identified.
DOI:10.3389/fpls.2016.01058URLPMID:4954820 [本文引用: 1]
The majority of rapeseed cultivars shatter seeds upon maturity especially under hot-dry and windy conditions, reducing yield and gross margin return to growers. Here, we identified quantitative trait loci (QTL) for resistance to pod shatter in an unstructured diverse panel of 143 rapeseed accessions, and two structured populations derived from bi-parental doubled haploid (DH) and inter-mated (IF2) crosses derived from R1 (resistant to pod shattering) and R2 (prone to pod shattering) accessions. Genome-wide association analysis identified six significant QTL for resistance to pod shatter located on chromosomes A01, A06, A07, A09, C02, and C05. Two of the QTL,qSRI.A09delimited with the SNP marker Bn-A09-p30171993 (A09) andqSRI.A06delimited with the SNP marker Bn-A06-p115948 (A06) could be repeatedly detected across environments in a diversity panel, DH and IF2populations, suggesting that at least two loci on chromosomes A06 and A09 were the main contributors to pod shatter resistance in Chinese germplasm. Significant SNP markers identified in this study especially those that appeared repeatedly across environments provide a cost-effective and an efficient method for introgression and pyramiding of favorable alleles for pod shatter resistance via marker-assisted selection in rapeseed improvement programs.
DOI:10.1186/s12864-015-1607-0URLPMID:25962630 [本文引用: 1]
Background Harvest index (HI), the ratio of grain yield to total biomass, is considered as a measure of biological success in partitioning assimilated photosynthate to the harvestable product. While crop production can be dramatically improved by increasing HI, the underlying molecular genetic mechanism of HI in rapeseed remains to be shown. Results In this study, we examined the genetic architecture of HI using 35,791 high-throughput single nucleotide polymorphisms (SNPs) genotyped by the Illumina BrassicaSNP60 Bead Chip in an association panel with 155 accessions. Five traits including plant height (PH), branch number (BN), biomass yield per plant (BY), harvest index (HI) and seed yield per plant (SY), were phenotyped in four environments. HI was found to be strongly positively correlated with SY, but negatively or not strongly correlated with PH. Model comparisons revealed that the A???D test (ADGWAS model) could perfectly balance false positives and statistical power for HI and associated traits. A total of nine SNPs on the C genome were identified to be significantly associated with HI, and five of them were identified to be simultaneously associated with HI and SY. These nine SNPs explained 3.42 % of the phenotypic variance in HI. Conclusions Our results showed that HI is a complex polygenic phenomenon that is strongly influenced by both environmental and genotype factors. The implications of these results are that HI can be increased by decreasing PH or reducing inefficient transport from pods to seeds in rapeseed. The results from this association mapping study can contribute to a better understanding of natural variations of HI, and facilitate marker-based breeding for HI.
DOI:10.3389/fpls.2017.00206URLPMID:5309214 [本文引用: 1]
Yield is one of the most important yet complex crop traits. To improve our understanding of the genetic basis of yield establishment, and to identify candidate genes responsible for yield improvement inBrassica napus, we performed genome-wide association studies (GWAS) for seven yield-determining traits [main inflorescence pod number (MIPN), branch pod number (BPN), pod number per plant (PNP), seed number per pod (SPP), thousand seed weight, main inflorescence yield (MIY), and branch yield], using data from 520 diverseB. napusaccessions from two different yield environments. In total, we detected 128 significant single nucleotide polymorphisms (SNPs), 93 of which were revealed as novel by integrative analysis. A combination of GWAS and transcriptome sequencing on 21 haplotype blocks from samples pooled by four extremely high-yielding or low-yielding accessions revealed the differential expression of 14 crucial candiate genes (such asBna.MYB83, Bna.SPL5, andBna.ROP3) associated with multiple traits or containing multiple SNPs associated with the same trait. Functional annotation and expression pattern analyses further demonstrated that these 14 candiate genes might be important in developmental processes and biomass accumulation, thus affecting the yield establishment ofB. napus. These results provide valuable information for understanding the genetic mechanisms underlying the establishment of high yield inB. napus, and lay the foundation for developing high-yieldingB. napusvarieties.
DOI:10.1007/s00122-016-2697-zURLPMID:26912143 [本文引用: 1]
A set of additive loci for seed oil content were identified using association mapping and one of the novel loci on02the chromosome A5 was validated by linkage mapping.Increasing seed oil content is on
DOI:10.3389/fpls.2016.01102URLPMID:4961929 [本文引用: 2]
Plant height is a key morphological trait of rapeseed. In this study, we measured plant height of a rapeseed population across six environments. This population contains 476 inbred lines representing the major Chinese rapeseed genepool and 44 lines from other countries. The 60KBrassicaInfinium03 SNP array was utilized to genotype the association panel. A genome-wide association study (GWAS) was performed via three methods, including a robust, novel, nonparametric Anderson–Darling (A–D) test. Consequently, 68 loci were identified as significantly associated with plant height (P< 5.22 × 10615), and more than 70% of the loci (48) overlapped the confidence intervals of reported QTLs from nine mapping populations. Moreover, 24 GWAS loci were detected with selective sweep signals, which reflected the signatures of historical semi-dwarf breeding. In the linkage disequilibrium (LD) decay range up—and downstream of 65 loci (r2> 0.1), we found plausible candidates orthologous to the documentedArabidopsisgenes involved in height regulation. One significant association found by GWAS colocalized with the established height locusBnRGAin rapeseed. Our results provide insights into the genetic basis of plant height in rapeseed and may facilitate marker-based breeding.
DOI:10.1038/srep14407URLPMID:4585775 [本文引用: 1]
Abstract Silique length (SL) and seed weight (SW) are important yield-associated traits in rapeseed (Brassica napus). Although many quantitative trait loci (QTL) for SL and SW have been identified in B. napus, comparative analysis for those QTL is seldom performed. In the present study, 20 and 21 QTL for SL and SW were identified in doubled haploid (DH) and DH-derived reconstructed F2 populations in rapeseed, explaining 55.1-74.3% and 24.4-62.9% of the phenotypic variation across three years, respectively. Of which, 17 QTL with partially or completely overlapped confidence interval on chromosome A09, were homologous with two overlapped QTL on chromosome C08 by aligning QTL confidence intervals with the reference genomes of Brassica crops. By high density selective genotyping of DH lines with extreme phenotypes, using a Brassica single-nucleotide polymorphism (SNP) array, the QTL on chromosome A09 was narrowed, and aligned into 1.14-Mb region from 30.84 to 31.98090009Mb on chromosome R09 of B. rapa and 1.05-Mb region from 27.21 to 28.26090009Mb on chromosome A09 of B. napus. The alignment of QTL with Brassica reference genomes revealed homologous QTL on A09 and C08 for SL. The narrowed QTL region provides clues for gene cloning and breeding cultivars by marker-assisted selection.
DOI:10.7505/j.issn.1007-9084.2015.04.020URL [本文引用: 2]
以数字图像处理技术为基础,建立了一套油菜株型角度测量方法,并根据分枝角度将22份材料分为紧凑型(小于30°)3份,中间型(30°~40°)8份和松散型(大于40°)11份;根据角果着生状态将22份材料分为平生型(趋近于90°)3份和斜生型(小于80°)19份。对其中5份材料进行数字图像处理测量值(x)和传统的手工方法测量值(y)比较,经显著性t测验和方差分析,测定结果符合曲线为y=1.019 1x-1.363 2,相关系数为0.991 9。可见,这一套数字图像技术具有较高的重复性、精确性,更具有较好的可操作性,可为油菜分枝角度和角果着生角度的遗传研究提供技术支撑。
DOI:10.7505/j.issn.1007-9084.2015.04.020URL [本文引用: 2]
以数字图像处理技术为基础,建立了一套油菜株型角度测量方法,并根据分枝角度将22份材料分为紧凑型(小于30°)3份,中间型(30°~40°)8份和松散型(大于40°)11份;根据角果着生状态将22份材料分为平生型(趋近于90°)3份和斜生型(小于80°)19份。对其中5份材料进行数字图像处理测量值(x)和传统的手工方法测量值(y)比较,经显著性t测验和方差分析,测定结果符合曲线为y=1.019 1x-1.363 2,相关系数为0.991 9。可见,这一套数字图像技术具有较高的重复性、精确性,更具有较好的可操作性,可为油菜分枝角度和角果着生角度的遗传研究提供技术支撑。
DOI:10.3389/fpls.2016.00021URLPMID:26870051 [本文引用: 3]
Changes in the rapeseed branch angle alter plant architecture, allowing more efficient light capture as planting density increases. In this study, a natural population of rapeseed was grown in three environments and evaluated for branch angle trait to characterize their phenotypic patterns and genotype with a 60K Brassica Infinium SNP array. Significant phenotypic variation was observed from 20 to 70 degrees. As a result, 25 significant quantitative trait loci (QTL) associated with branch angle were identified on chromosomes A2, A3, A7, C3, C5 and C7 by the MLM model in TASSEL 4.0. Orthologs of the functional candidate genes involved in branch angle were identified. Among the key QTL, the peak SNPs were close to the key orthologous genes BnaA.Lazy1 and BnaC.Lazy1 on A3 and C3 homologous genome blocks. With the exception of Lazy (LA) orthologous genes, SQUMOSA PROMOTER BINDING PROTEIN LIKE 14 (SPL14) and an auxin-responsive GRETCHEN HAGEN 3 (GH3) genes from Arabidopsis thaliana were identified close to two clusters of SNPs on the A7 and C7 chromosomes.These findings on multiple novel loci and candidate genes of branch angle will be useful for further understanding and genetic improvement of plant architecture in rapeseed.
DOI:10.1038/srep33673URLPMID:5028734 [本文引用: 7]
The rapeseed branch angle is an important morphological trait because an adequate branch angle enables more efficient light capture under high planting densities. Here, we report that the average angle of the five top branches provides a reliable representation of the average angle of all branches. Statistical analyses revealed a significantly positive correlation between the branch angle and multiple plant-type and yield-related traits. The 60 BrassicaInfinium single nucleotide polymorphism (SNP) array was utilized to genotype an association panel with 520 diverse accessions. A genome-wide association study was performed to determine the genetic architecture of branch angle, and 56 loci were identified as being significantly associated with the branch angle trait via three models, including a robust, novel, nonparametric Anderson-Darling (A-D) test. Moreover, these loci explained 51.1% of the phenotypic variation when a simple additive model was applied. Within the linkage disequilibrium (LD) decay ranges of 53 loci, we observed plausible candidates orthologous to documentedArabidopsisgenes, such asLAZY1,SGR2,SGR4,SGR8,SGR9,PIN3,PIN7,CRK5,TIR1, andAPD7. These results provide insight into the genetic basis of the branch angle trait in rapeseed and might facilitate marker-based breeding for improvements in plant architecture.
[本文引用: 3]
DOI:10.3389/fpls.2017.01054URLPMID:5474488 [本文引用: 7]
Plant architecture is vital not only for crop yield, but also for field management, such as mechanical harvesting. The branch angle is one of the key factors determining plant architecture. With the aim of revealing the genetic control underlying branch angle in rapeseed (Brassica napusL.), the positional variation of branch angles on individual plants was evaluated, and the branch angle increased with the elevation of branch position. Furthermore, three middle branches of individual plants were selected to measure the branch angle because they exhibited the most representative phenotypic values. An association panel with 472 diverse accessions was estimated for branch angle trait in six environments and genotyped with a 60KBrassicaInfinium SNP array. As a result of association mapping, 46 and 38 significantly-associated loci were detected using a mixed linear model (MLM) and a multi-locus random-SNP-effect mixed linear model (MRMLM), which explained up to 62.2 and 66.2% of the cumulative phenotypic variation, respectively. Numerous highly-promising candidate genes were identified by annotating againstArabidopsis thalianahomologous, including some first found in rapeseed, such asTAC1, SGR1, SGR3, andSGR5. These findings reveal the genetic control underlying branch angle and provide insight into genetic improvements that are possible in the plant architecture of rapeseed.
DOI:10.1007/s00122-017-2986-1URLPMID:28942459 [本文引用: 9]
A high-density SNP map was constructed and several novel QTL for branch angle across six environments inBrassica napuswere identified.Branch angle is a major determinant for the ideotype of a plant,
.
URL [本文引用: 1]
油菜是我国重要的油料作物之一,如何最大程度的提高油菜的产量和实现油菜的机械化生产是油菜育种和栽培的主要目标。通过对油菜株型相关性状的遗传规律和QTL定位进行研究分析,可以为这个目标的实现提供了理论依据。本文采用松散型甘蓝型油菜S33B和紧凑型甘蓝型油菜86-5-10-17组合杂交所得的F2群体为对象,探讨了与油菜机械化收获紧密相关的包括株高、分枝高度、分枝数和分枝角度4个性状的发育动态及遗传规律,并在构建遗传连锁图谱的基础上进行了4个性状的QTL定位。主要研究结果如下: 1.根据分枝角度的大小将甘蓝型油菜的株型分为紧凑型、中间型和分散型。这三种类型油菜的株高、分枝高度和分枝数的发育动态都不相同,但是每个性状的发育动态变化趋势几乎是一致的。从初花期到盛花期是油菜株高、分枝高度和分枝数快速生长期,在这四个生育期内株高、分枝数都是中间型株型松散型株型紧凑型株型;分枝高度则是松散株型中间型株型紧凑型株型。 2.在紧凑型株型中,第一分枝角度和第三分枝角都呈先缓下降后上升最后快速下降的趋势,第二分枝角度呈先上升后下降的趋势,第四分枝角度在整个生育期内都是呈直线缓慢下降的趋势;在中间型油菜株型中,第二分枝角度和第四分枝角度变化趋势是一致的,都是先下降后上升的的趋势,第一分枝角度呈先下降后上升最后再下降的趋势,第三分枝角度呈不断上升趋势;在松散型油菜株型中,第一个分枝角度,在整个生育期内都呈不断增加趋势,第二个分枝角度呈先下降后上升的趋势,从第三个分枝角呈先缓慢下降后上升的趋势第四分枝角度呈先下降后上升的趋势。 3.甘蓝型油菜株高和分枝数的F1正反交均值都超过了亲本,说明株高和分枝数F1具有很好的杂种优势;分枝角度的F1正反交均值都介于双亲之间,都偏向于亲本S33B,说明控制分枝角度的基因是以加性效应为主,有部分显性效应,分散型对紧凑型为部分显性;分枝高度的F1正交均值比亲本的要大,F1反交均值介于双亲之间,偏向于亲本86-5-10-17,说明分枝高度可能存在一定的胞质效应。 4.甘蓝型油菜的株高符合两对主基因控制的等显性模型(B-6模型),这两个主基因的显性效应相等并与相应的加性效应也相等,无上位性效应存在;分枝高度是一对主基因控制的加性和部分显性或超显性模型(A-1模型):分枝数符合两对主基因控制的等加性模型(B-4模型),两对主基因的加性效应相等且无显性效应和上位性效应。分枝角度平均数符合加性-显性-上位性模型(B-1模型),有两个主基因控制的株高,分枝高度,分枝数,和分枝角度的主基因遗传率分别为:44.71%,64.43%,4.55%,60.52%。多基因遗传率分别为:1.36%,8.34%,0.56%,0.95%。都以主基因遗传为主。 5.用223个SSR多态性标记构建了包含了13个连锁群163个标记位点的F2家系群体连锁图谱,有60个标记位点没有进入任何连锁群,占总数的26.9%。该遗传连锁图谱总长度为906.986cM。最大的连锁群有63个标记,最小的只有2个标记,标记间平均距离为12.286cM。其中超过100cM的有3个连锁群,分别是LG1、LG2和LG8。其中LG1距离最长,为177.222cM,LG13距离最短,仅为9.481cM。平均间距最大的为LG11,达30.968cM;平均间距最小的为LG1,只有2.858cM。 6.共检测到11个与这四个株型性状相关的QTLs,分别位于LG1、LG2、 LG3、LG5、LG8连锁群上,且都具有较大的遗传效率。其中株高4个,分别位于LG1、LG2连锁群上,可解释表型变异的8.54%-17.04%;分枝高度5个,分别位于LG3、LG5、LG8连锁群上,可解释表型变异的3.92%-21.82%;分枝数1个,位于LG3连锁群上,可解释表型变异的11.08%;分枝角度1个,位于LG1连锁群上,可解释表型变异的14.16%。只有分枝高度的第二个QTL、分枝数和分枝角度QTL的增效基因都来源于松散型亲本S33B,其余QTL的增效基因都来源于紧凑型亲本86-5-10-17。油菜株型的初步定位结果为该基因的精细定位、分子标记辅助育种和基因克隆功能分析等奠定基础。
URL [本文引用: 1]
油菜是我国重要的油料作物之一,如何最大程度的提高油菜的产量和实现油菜的机械化生产是油菜育种和栽培的主要目标。通过对油菜株型相关性状的遗传规律和QTL定位进行研究分析,可以为这个目标的实现提供了理论依据。本文采用松散型甘蓝型油菜S33B和紧凑型甘蓝型油菜86-5-10-17组合杂交所得的F2群体为对象,探讨了与油菜机械化收获紧密相关的包括株高、分枝高度、分枝数和分枝角度4个性状的发育动态及遗传规律,并在构建遗传连锁图谱的基础上进行了4个性状的QTL定位。主要研究结果如下: 1.根据分枝角度的大小将甘蓝型油菜的株型分为紧凑型、中间型和分散型。这三种类型油菜的株高、分枝高度和分枝数的发育动态都不相同,但是每个性状的发育动态变化趋势几乎是一致的。从初花期到盛花期是油菜株高、分枝高度和分枝数快速生长期,在这四个生育期内株高、分枝数都是中间型株型松散型株型紧凑型株型;分枝高度则是松散株型中间型株型紧凑型株型。 2.在紧凑型株型中,第一分枝角度和第三分枝角都呈先缓下降后上升最后快速下降的趋势,第二分枝角度呈先上升后下降的趋势,第四分枝角度在整个生育期内都是呈直线缓慢下降的趋势;在中间型油菜株型中,第二分枝角度和第四分枝角度变化趋势是一致的,都是先下降后上升的的趋势,第一分枝角度呈先下降后上升最后再下降的趋势,第三分枝角度呈不断上升趋势;在松散型油菜株型中,第一个分枝角度,在整个生育期内都呈不断增加趋势,第二个分枝角度呈先下降后上升的趋势,从第三个分枝角呈先缓慢下降后上升的趋势第四分枝角度呈先下降后上升的趋势。 3.甘蓝型油菜株高和分枝数的F1正反交均值都超过了亲本,说明株高和分枝数F1具有很好的杂种优势;分枝角度的F1正反交均值都介于双亲之间,都偏向于亲本S33B,说明控制分枝角度的基因是以加性效应为主,有部分显性效应,分散型对紧凑型为部分显性;分枝高度的F1正交均值比亲本的要大,F1反交均值介于双亲之间,偏向于亲本86-5-10-17,说明分枝高度可能存在一定的胞质效应。 4.甘蓝型油菜的株高符合两对主基因控制的等显性模型(B-6模型),这两个主基因的显性效应相等并与相应的加性效应也相等,无上位性效应存在;分枝高度是一对主基因控制的加性和部分显性或超显性模型(A-1模型):分枝数符合两对主基因控制的等加性模型(B-4模型),两对主基因的加性效应相等且无显性效应和上位性效应。分枝角度平均数符合加性-显性-上位性模型(B-1模型),有两个主基因控制的株高,分枝高度,分枝数,和分枝角度的主基因遗传率分别为:44.71%,64.43%,4.55%,60.52%。多基因遗传率分别为:1.36%,8.34%,0.56%,0.95%。都以主基因遗传为主。 5.用223个SSR多态性标记构建了包含了13个连锁群163个标记位点的F2家系群体连锁图谱,有60个标记位点没有进入任何连锁群,占总数的26.9%。该遗传连锁图谱总长度为906.986cM。最大的连锁群有63个标记,最小的只有2个标记,标记间平均距离为12.286cM。其中超过100cM的有3个连锁群,分别是LG1、LG2和LG8。其中LG1距离最长,为177.222cM,LG13距离最短,仅为9.481cM。平均间距最大的为LG11,达30.968cM;平均间距最小的为LG1,只有2.858cM。 6.共检测到11个与这四个株型性状相关的QTLs,分别位于LG1、LG2、 LG3、LG5、LG8连锁群上,且都具有较大的遗传效率。其中株高4个,分别位于LG1、LG2连锁群上,可解释表型变异的8.54%-17.04%;分枝高度5个,分别位于LG3、LG5、LG8连锁群上,可解释表型变异的3.92%-21.82%;分枝数1个,位于LG3连锁群上,可解释表型变异的11.08%;分枝角度1个,位于LG1连锁群上,可解释表型变异的14.16%。只有分枝高度的第二个QTL、分枝数和分枝角度QTL的增效基因都来源于松散型亲本S33B,其余QTL的增效基因都来源于紧凑型亲本86-5-10-17。油菜株型的初步定位结果为该基因的精细定位、分子标记辅助育种和基因克隆功能分析等奠定基础。
DOI:10.3724/SP.J.1006.2016.01103URL [本文引用: 1]
分枝角度是油菜株型重要性状,是油菜品种高产及适合机械化收获理想株型的基本组成之一。为明确油菜分枝角度的遗传,本研究选用油菜分枝角度大的松散型材料6098B和分枝角度小的紧凑型材料Purler配制杂交组合,采用主基因+多基因混合遗传模型方法对该组合6世代(P1、P2、F1、F2、BCP1和BCP2)的分枝角度进行了遗传分析。结果表明,上部第一分枝(顶枝)和基部第一分枝(基枝)角度的最适合遗传模型均为D-0(1对加性-显性主基因+加性-显性-上位性多基因)。顶枝角的主基因加性效应值为4.939o,显性效应值为–4.156o,主基因遗传率在BCP1、BCP2和F2中分别是34.08%、1.40%和14.99%,多基因遗传率分别为24.43%、61.72%和63.98%;而基枝角的主基因加性效应值为2.217o,显性效应值为–1.941o,主基因遗传率在BCP1、BCP2和F2中分别是7.86%、1.24%和4.84%,多基因遗传率分别为66.46%、58.49%和73.96%。结果发现油菜分枝角度明显存在主效基因,为油菜分枝角度的遗传改良奠定了基础。
DOI:10.3724/SP.J.1006.2016.01103URL [本文引用: 1]
分枝角度是油菜株型重要性状,是油菜品种高产及适合机械化收获理想株型的基本组成之一。为明确油菜分枝角度的遗传,本研究选用油菜分枝角度大的松散型材料6098B和分枝角度小的紧凑型材料Purler配制杂交组合,采用主基因+多基因混合遗传模型方法对该组合6世代(P1、P2、F1、F2、BCP1和BCP2)的分枝角度进行了遗传分析。结果表明,上部第一分枝(顶枝)和基部第一分枝(基枝)角度的最适合遗传模型均为D-0(1对加性-显性主基因+加性-显性-上位性多基因)。顶枝角的主基因加性效应值为4.939o,显性效应值为–4.156o,主基因遗传率在BCP1、BCP2和F2中分别是34.08%、1.40%和14.99%,多基因遗传率分别为24.43%、61.72%和63.98%;而基枝角的主基因加性效应值为2.217o,显性效应值为–1.941o,主基因遗传率在BCP1、BCP2和F2中分别是7.86%、1.24%和4.84%,多基因遗传率分别为66.46%、58.49%和73.96%。结果发现油菜分枝角度明显存在主效基因,为油菜分枝角度的遗传改良奠定了基础。
DOI:10.1371/journal.pgen.1000212URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1093/jhered/93.1.77URLPMID:12011185 [本文引用: 1]
Provides information on MapChart software designed for the graphical representation of linkage maps and quantitative trait loci. Features of the software; Details of how the software works; Discussion on data and formatting of the software.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/dnares/dst040URLPMID:3989493 [本文引用: 1]
<p id="p-3">Although much research has been conducted, the pattern of microsatellite distribution has remained ambiguous, and the development/utilization of microsatellite markers has still been limited/inefficient in Brassica, due to the lack of genome sequences. In view of this, we conducted genome-wide microsatellite characterization and marker development in three recently sequenced Brassica crops: Brassica rapa, Brassica oleracea and Brassica napus. The analysed microsatellite characteristics of these Brassica species were highly similar or almost identical, which suggests that the pattern of microsatellite distribution is likely conservative in Brassica. The genomic distribution of microsatellites was highly non-uniform and positively or negatively correlated with genes or transposable elements, respectively. Of the total of 115 869, 185 662 and 356 522 simple sequence repeat (SSR) markers developed with high frequencies (408.2, 343.8 and 356.2 per Mb or one every 2.45, 2.91 and 2.81 kb, respectively), most represented new SSR markers, the majority had determined physical positions, and a large number were genic or putative single-locus SSR markers. We also constructed a comprehensive database for the newly developed SSR markers, which was integrated with public Brassica SSR markers and annotated genome components. The genome-wide SSR markers developed in this study provide a useful tool to extend the annotated genome resources of sequenced Brassica species to genetic study/breeding in different Brassica species.
DOI:10.1007/s00122-016-2849-1URLPMID:28220206 [本文引用: 1]
The Brassica napus 60K Illumina Infinium SNP array has had huge international uptake in the rapeseed community due to the revolutionary speed of acquisition and ease of analysis of this high-throughput genotyping data, particularly when coupled with the newly available reference genome sequence. However, further utilization of this valuable resource can be optimized by better understanding the promises and pitfalls of SNP arrays. We outline how best to analyze Brassica SNP marker array data for diverse applications, including linkage and association mapping, genetic diversity and genomic introgression studies. We present data on which SNPs are locus-specific in winter, semi-winter and spring B. napus germplasm pools, rather than amplifying both an A-genome and a C-genome locus or multiple loci. Common issues that arise when analyzing array data will be discussed, particularly those unique to SNP markers and how to deal with these for practical applications in Brassica breeding applications.
.
URL [本文引用: 2]
An activation tagging screen was carried out to identify gain-of-function mutants showing potential auxin defects, with the aim of identifying the new genes regulating plant development. The conjoined (cnj) mutant isolated from the activation tagged screen exhibits duplication of the seedling axis. Sequencing the insertion locus revealed that the activation tag was positioned between ATP-binding protein and vesicle associated membrane protein genes. Cloned mutant locus and expression studies indicated that the vesicle associated membrane protein gene is upregulated in the activation tagging line. The aim of this work is to investigate the function of the vesicle associated membrane protein gene (AtVAMP714), which is a member of an R-SNARE protein family. To determine the expression pattern of AtVAMP714, proAtVAMP714::GUS expression in seedlings was examined. GUS activity was observed in the hypocotyl and roots and the strongest expression was observed in the root vascular tissues. The AtVAMP714 gene is positively auxin regulated. VAMP714::GFP fusion protein localized to Golgi vesicles suggesting it may be involved in the Golgi secretory pathway. Auxin transport levels in roots and shoots found to be greatly reduced in AtVAMP714 overexpressors compared to the wild type plants. Quantitative real-time RT-PCR showed that transcript levels of IAA1 and IAA2 were significantly reduced in AtVAMP714 overexpressors. Immunolocalization of PIN1 and PIN2 showed strong defects in localization. To characterize the development role of VAMP714, SALK null mutants of the AtVAMP714 gene were identified. The mutant phenotype showed an abnormal branching and short root phenotype and PIN1, PIN2 and PIN4 transcript levels were significantly reduced. Dominant negative transgenics of AtVAMP714 were also created as a method of knocking out the function of the protein and F2 generation plants were analysed for developmental defects. Consistent with the above results the dominant negative transgenics showed a short root phenotype with dwarfed, branchy shoots. PIN1 and PIN2 proteins were mislocalized in dominant negative transgenics. The results presented provide evidence for a role of AtVAMP714 in auxin transport through a requirement for correct PIN protein secretion and localization.
DOI:10.1111/j.1365-313X.2007.03284.xURLPMID:17908158 [本文引用: 1]
A critical step during rice ( Oryza sativa ) cultivation is dense planting: a wider tiller angle will increase leaf shade and decrease photosynthesis efficiency, whereas a narrower tiller angle makes for more efficient plant architecture. The molecular basis of tiller angle remains unknown. This research demonstrates that tiller angle is controlled by a major quantitative trait locus, TAC1 ( Tiller Angle Control 1 ). TAC1 was mapped to a 35-kb region on chromosome 9 using a large F 2 population from crosses between an indica rice, IR24, which displays a relatively spread-out plant architecture, and an introgressed line, IL55, derived from japonica rice Asominori, which displays a compact plant architecture with extremely erect tillers. Genetic complementation further identified the TAC1 gene, which harbors three introns in its coding region and a fourth 1.5-kb intron in the 3'-untranslated region. A mutation in the 3'-splicing site of this 1.5-kb intron from 'AGGA' to 'GGGA' decreases the level of tac1 , resulting in a compact plant architecture with a tiller angle close to zero. Further sequence verification of the mutation in the 3'-splicing site of the 1.5-kb intron revealed that the tac1 mutation 'GGGA' was present in 88 compact japonica rice accessions and TAC1 with 'AGGA' was present in 21 wild rice accessions and 43 indica rice accessions, all with the spread-out form, indicating that tac1 had been extensively utilized in densely planted rice grown in high-latitude temperate areas and at high altitudes where japonica rice varieties are widely cultivated.
DOI:10.1371/journal.pone.0020621URLPMID:3110200 [本文引用: 1]
Modifying plant architecture to increase photosynthesis efficiency and reduce shade avoidance response is very important for further yield improvement when crops are grown in high density. Identification of alleles controlling leaf angle in maize is needed to provide insight into molecular mechanism of leaf development and achieving ideal plant architecture to improve grain yield. The gene cloning was done by using comparative genomics, and then performing real-time polymerase chain reaction (RT-PCR) analysis to assay gene expression. The gene function was validated by sequence dissimilarity analysis and QTL mapping using a functional cleaved amplified polymorphism (CAP). The leaf angle is controlled by a major quantitative trait locus,ZmTAC1(Zea maysL. Leaf Angle Control 1).ZmTAC1has 4 exons encoding a protein with 263 amino acids, and its domains are the same as those of the riceOsTAC1protein.ZmTAC1was found to be located in the region of qLA2 by using the CAP marker and the F2:3families from the cross between Yu82 and Shen137. Real-time PCR analysis revealedZmTAC1expression was the highest in the leaf-sheath pulvinus, less in the leaf and shoot apical meristem, and the lowest in the root. A nucleotide difference in the 5′-untranslated region (UTR) between the compact inbred line Yu82 (“CTCC”) and the expanded inbred line Shen137 (“CCCC”) influences the expression level ofZmTAC1, further controlling the size of the leaf angle. Sequence verification of the change in the 5′-UTR revealedZmTAC1with “CTCC” was present in 13 compact inbred lines andZmTAC1with “CCCC” was present in 18 expanded inbred lines, indicatingZmTAC1had been extensively utilized in breeding with regard to the improvement of the maize plant architecture.
DOI:10.1111/tpj.12234URLPMID:23663106 [本文引用: 1]
Trees are capable of tremendous architectural plasticity, allowing them to maximize their light exposure under highly competitive environments. One key component of tree architecture is the branch angle, yet little is known about the molecular basis for the spatial patterning of branches in trees. Here, we report the identification of a candidate gene for the br mutation in Prunus persica (peach) associated with vertically oriented growth of branches, referred to as pillar' or broomy'. Ppa010082, annotated as hypothetical protein in the peach genome sequence, was identified as a candidate gene for br using a next generation sequence-based mapping approach. Sequence similarity searches identified rice TAC1 (tiller angle control 1) as a putative ortholog, and we thus named it PpeTAC1. In monocots, TAC1 is known to lead to less compact growth by increasing the tiller angle. In Arabidopsis, an attac1 mutant showed more vertical branch growth angles, suggesting that the gene functions universally to promote the horizontal growth of branches. TAC1 genes belong to a gene family (here named IGT for a shared conserved motif) found in all plant genomes, consisting of two clades: one containing TAC1-like genes; the other containing LAZY1, which contains an EAR motif, and promotes vertical shoot growth in Oryza sativa (rice) and Arabidopsis through influencing polar auxin transport. The data suggest that IGT genes are ancient, and play conserved roles in determining shoot growth angles in plants. Understanding how IGT genes modulate branch angles will provide insights into how different architectural growth habits evolved in terrestrial plants.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}