 ,*西南大学农学与生物科技学院 / 油菜工程研究中心 / 西南大学现代农业科学研究院, 重庆 400716
,*西南大学农学与生物科技学院 / 油菜工程研究中心 / 西南大学现代农业科学研究院, 重庆 400716Identification and expression analysis of PEBP gene family in oilseed rape
JIAN Hong-Ju**, YANG Bo**, LI Yang-Yang**, YANG Hong**, LIU Lie-Zhao**, XU Xin-Fu**, LI Jia-Na,*College of Agronomy and Biotechnology / Chongqing Engineering Research Center for Rapeseed / Academy of Agricultural Sciences, Southwest University, Chongqing 400715, China通讯作者:
第一联系人:
收稿日期:2018-07-9接受日期:2018-10-8网络出版日期:2018-11-01
| 基金资助: |
Received:2018-07-9Accepted:2018-10-8Online:2018-11-01
| Fund supported: |
作者简介 About authors
荐红举,E-mail:jianhongju1989@126.com。
杨博,E-mail:sheepneck@hotmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (5321KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
荐红举, 杨博, 李阳阳, 杨鸿, 刘列钊, 徐新福, 李加纳. 甘蓝型油菜PEBP基因家族的鉴定与表达分析[J]. 作物学报, 2019, 45(3): 354-364. doi:10.3724/SP.J.1006.2019.84095
JIAN Hong-Ju, YANG Bo, LI Yang-Yang, YANG Hong, LIU Lie-Zhao, XU Xin-Fu, LI Jia-Na.
开花是植物通过有性生殖传递后代的关键前提, 开花时间是影响作物产量和品质最重要的农艺性状之一。相比于其他性状, 开花时间对外界环境更加敏感。因此, 控制开花时间对于提高不同地域的作物产量具有重要意义。截至目前, 有关开花候选基因的解析主要集中在拟南芥等模式植物中。在拟南芥中, 超过300个开花基因得到鉴定[1], 这些基因根据其参与的过程主要分为六大途径, 即春化过程、光周期途径、外界温度、赤霉素途径、自发途径和内在因素[2,3,4]。虽然不同的基因参与不同的开花调控通路, 但各通路被几个关键基因协同调控, 如FT (FLOWERING LOCUS T)、LFY (LEAFY)、FLC (FLOWERING LOCUS C)和SOC1 (SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1)[5]。
FT基因编码一个称为“促花素”的可以长距离移动的信号蛋白, 在开花调控中起着核心调控作用[6]。该基因属于植物磷脂酰乙醇胺结合蛋白PEBP (phosphatidylethanolamine-binding protein)基因, PEBP基因编码蛋白包含一个保守结构域(InterPro: IPR00891)。进化研究表明, 该蛋白家族分为3个亚家族, 即FT、TFL1 (TERMINAL FLOWER 1)和MFT (MOTHER OF FT AND TFL1)。其中, FT主要是诱导开花, TFL1抑制开花, 而MFT主要参与种子发育和萌发过程[7,8,9]。在拟南芥中, PEBP家族包含6个基因成员, 即FT、TFL1、TSF (TWIN SISTER OF FT)、BFT(BROTHER OF FT AND TFL1)、ATC (ARABIDOPSIS THALIANA CENTRORADIALIS)和MFT[10]。其中, FT、TSF和MFT促进开花, TFL1、ATC和BFT抑制开花[11,12,13]。在拟南芥中, 2个FT-like基因, FT和TSF是开花激活因子, 突变该基因将延迟开花[11,14]。在长日照环境中, FT和TSF在叶子韧皮部的伴胞细胞中上调表达[15]。作为ATC的旁系同源基因, TSF在营养生长期的成熟顶端分生组织的内表皮中少量积累, 而开花之后, 其表达量显著下调[16,17]。TFL1蛋白是一个移动信号, 可从顶端分生组织的内表皮移动到外表皮[18]。虽然TFL1和FT在序列上非常相似, 都可以与FD (FLOWERING LOCUS D)互作来调控FD依赖的靶基因, 但在调控开花过程中的作用是相反的[19]。在高盐胁迫中, BFT可以通过调节光周期来适应外界胁迫[20]。MFT特异诱导下胚轴和根的转变, 其突变体表现为在种子萌发过程中对ABA的超敏反应, 而且在萌发的种子中, MFT的表达直接受到ABA-INSENSITIVE3 (ABI3)和ABI5的调控, 同时, MFT通过反馈抑制ABI5的表达来促进胚的发育[21]。
相比于单个PEBP基因成员的研究, 在植物基因组范围内对PEBP的鉴定愈发重要且普遍家族相对较少。比如, 在大豆中有23个PEBP成员得到鉴定, 几乎是拟南芥PEBP基因数目的4倍[22], 其中GmFT2a和GmFT5a通过调控光周期途径来控制开花[23]。在单子叶模式植物水稻中, 有19个PEBP家族得到鉴定, 其中Hd3a (Heading date 3a)和RFT1 (RICE FLOWERING LOCUS T 1)被研究的最为透彻[24]。在短日照环境中, Hd3a促进水稻开花, 而Hd3a的同源基因RFT1主要在Hd3a被抑制时起到控制开花的功能[25]。在玉米的24个PEBP家族中, 只有ZCN8跟拟南芥FT的功能最为相似[26]。
油菜, 作为世界上最重要的油料作物之一, 与模式植物拟南芥具有相似的开花习性。虽然有很多通过基因定位和高通量测序技术研究油菜开花基因或通路的报道, 但是稳定的QTL或者关键调控基因或因子尚未得到解析[27,28]。相比于QTL定位方法筛选候选基因, 以拟南芥基因功能作为参考, 并结合RNA-Seq和分子实验来筛选并鉴定开花相关基因, 然后通过全基因组比较分析作物开花候选基因是非常方便和准确的。这种方式已经在萝卜中得到运用[29]。通过这种方式还可以为进一步的功能鉴定以及开花通路调控因子提供候选基因。PEBP家族成员的进化分析在大豆、棉花、郁金香和小麦等作物中已有报道[10,29-32], 但油菜中尚无有关该基因家族的鉴定、进化和表达分析。本研究利用拟南芥PEBP家族成员的蛋白序列为参考, 通过BlastP方式, 获得甘蓝型油菜以及亲本种白菜和甘蓝的PEBP家族成员, 并分析其进化、选择压力以及组织表达, 可为油菜开花性状候选基因的研究提供理论参考。
1 材料与方法
1.1 序列的获得
从拟南芥数据库(https://www.arabidopsis.org/)中获取拟南芥PEBP基因的蛋白序列, 并以此序列在BRAD数据库(http://brassicadb.org/brad/index. php)中进行BlastP, 阈值E<10-5, 筛选甘蓝型油菜、白菜和甘蓝的同源基因, 并下载相应蛋白和CDS序列。1.2 油菜PEBP基因结构、motif预测以及重复基因检测
根据油菜PEBP基因家族的基因组DNA及CDS序列, 并利用在线工具GSDS (http://gsds.cbi.pku.edu. cn/)分析其基因结构。利用在线工具MEME (http:// meme-suite.org/)预测油菜PEBP基因家族的保守结构元件, 并设置motifs数量为5个, 宽度为6~50。基因结构和motif结果均利用TBtools工具展示。用软件MCScanX 分析26个油菜PEBP基因家族的复制情况, 默认其参数。1.3 序列比对及进化分析
利用Clustal W软件对获取的拟南芥、甘蓝型油菜、白菜和甘蓝基因组内的PEBP的序列进行完全比对。用MEGA 6.0软件, 通过邻接法(Neighbor Joining, NJ)构建基因分子进化树, 设BootStrap抽样次数为1000, 默认其余参数, 并用软件Figtree展示。1.4 选择压力分析
用MEGA 6.0内置的Muscle对拟南芥和甘蓝型油菜之间的共线性PEBP基因对做codon比对[33], 然后用软件KaKs_Calculator 2.0计算共线性基因之间的选择压力[34]。1.5 甘蓝型油菜PEBP家族成员的组织表达分析
利用课题组前期完成的不同发育时期的‘中双11’共111个组织器官的转录组数据(PRJNA358784) 分析甘蓝型油菜PEBP的表达情况, 样品详细信息见附表1。对各成员的表达量取log2 FPKM后利用MeV 4.9.0 (http://en.bio-soft.net/chip/MeV.html)绘制热图展示。Supplementarey table 1
附表1
附表1‘中双11’的111个样品信息
Supplementarey table 1
| 编号 Code | 生育期 Growth stage | 器官 Organ | 取样时间 Treatment time | 编号 Code | 生育期 Growth stage | 器官 Organ | 取样时间 Treatment time | ||
|---|---|---|---|---|---|---|---|---|---|
| Ro_24h | 种子萌发 | 胚根 | 萌发后24 h | Se_7d | 青荚期 | 种子 | 花后7 d | ||
| Ro_48h | 种子萌发 | 胚根 | 萌发后48 h | Se_10d | 青荚期 | 种子 | 花后10 d | ||
| Ro_72h | 种子萌发 | 胚根 | 萌发后72 h | Se_13d | 青荚期 | 种子 | 花后13 d | ||
| Hy_24h | 种子萌发 | 下胚轴 | 萌发后24 h | Se_19d | 青荚期 | 种子 | 花后19 d | ||
| Hy_48h | 种子萌发 | 下胚轴 | 萌发后48 h | Se_21d | 青荚期 | 种子 | 花后21 d | ||
| Hy_72h | 种子萌发 | 下胚轴 | 萌发后72 h | Se_24d | 青荚期 | 种子 | 花后24 d | ||
| Co_24h | 种子萌发 | 子叶 | 萌发后24 h | Se_27d | 青荚期 | 种子 | 花后27 d | ||
| Co_48h | 种子萌发 | 子叶 | 萌发后48 h | Se_30d | 青荚期 | 种子 | 花后30 d | ||
| Co_72h | 种子萌发 | 子叶 | 萌发后72 h | Se_35d | 青荚期 | 种子 | 花后35 d | ||
| GS_12h | 种子萌发 | 整个种子 | 萌发后12 h | Se_40d | 青荚期 | 种子 | 花后40 d | ||
| GS_24h | 种子萌发 | 整个种子 | 萌发后24 h | Se_43d | 青荚期 | 种子 | 花后43 d | ||
| Ro_s | 苗期-温室 | 根 | Se_46d | 青荚期 | 种子 | 花后46 d | |||
| Ro_s_f | 苗期-大田 | 根 | Se_49d | 青荚期 | 种子 | 花后49 d | |||
| Ro_b | 蕾期 | 根 | Em_19d | 青荚期 | 种胚 | 花后19 d | |||
| Ro_i | 初花期 | 根 | Em_21d | 青荚期 | 种胚 | 花后21 d | |||
| Ro_f | 盛花期 | 根 | Em_24d | 青荚期 | 种胚 | 花后24 d | |||
| St_b | 蕾期 | 茎秆 | Em_27d | 青荚期 | 种胚 | 花后27 d | |||
| St_i | 初花期 | 茎秆 | Em_30d | 青荚期 | 种胚 | 花后30 d | |||
| St_f | 盛花期 | 茎秆 | Em_35d | 青荚期 | 种胚 | 花后35 d | |||
| St_24d | 青荚期 | 茎秆 | 花后24 d | Em_40d | 成熟期 | 种胚 | 花后40 d | ||
| St_50d | 成熟期 | 茎秆 | 花后50 d | Em_43d | 成熟期 | 种胚 | 花后43 d | ||
| Le_s | 苗期-温室 | 叶片 | Em_46d | 成熟期 | 种胚 | 花后46 d | |||
| Le_s_f | 苗期-大田 | 叶片 | Em_49d | 成熟期 | 种胚 | 花后49 d | |||
| LeY_b | 蕾期 | 幼叶 | Ra_40d | 成熟期 | 胚芽 | 花后40 d | |||
| LeY_i | 初花期 | 幼叶 | Ra_43d | 成熟期 | 胚芽 | 花后43 d | |||
| LeY_f | 盛花期 | 幼叶 | Ra_46d | 成熟期 | 胚芽 | 花后46 d | |||
| LeY_10d | 青荚期 | 幼叶 | 花后10 d | Ra_49d | 成熟期 | 胚芽 | 花后49 d | ||
| LeY_24d | 青荚期 | 幼叶 | 花后24 d | SC_19d | 青荚期 | 种皮 | 花后19 d | ||
| LeY_30d | 青荚期 | 幼叶 | 花后30 d | SC_21d | 青荚期 | 种皮 | 花后21 d | ||
| LeO_b | 蕾期 | 成熟叶片 | SC_27d | 青荚期 | 种皮 | 花后27 d | |||
| LeO_i | 初花期 | 成熟叶片 | SC_30d | 青荚期 | 种皮 | 花后30 d | |||
| LeO_f | 盛花期 | 成熟叶片 | SC_35d | 青荚期 | 种皮 | 花后35 d | |||
| LeO_10d | 青荚期 | 成熟叶片 | 花后10 d | SC_40d | 青荚期 | 种皮 | 花后40 d | ||
| LeO_24d | 青荚期 | 成熟叶片 | 花后24 d | SC_43d | 成熟期 | 种皮 | 花后43 d | ||
| LeO_30d | 青荚期 | 成熟叶片 | 花后30 d | En_21d | 青荚期 | 内种皮 | 花后21 d | ||
| Bu_b | 蕾期 | 蕾 | En_24d | 青荚期 | 内种皮 | 花后24 d | |||
| Ao_i | 初花期 | 花柄 | Ep_24d | 青荚期 | 外种皮 | 花后24 d | |||
| Ao_f | 盛花期 | 花柄 | Ep_30d | 青荚期 | 外种皮 | 花后30 d | |||
| Cal_i | 初花期 | 萼片 | Fu_27d | 青荚期 | 珠柄 | 花后27 d | |||
| Cal_f | 盛花期 | 萼片 | Fu_35d | 青荚期 | 珠柄 | 花后35 d | |||
| Pe_i | 初花期 | 花瓣 | SP_3d | 青荚期 | 角果皮 | 花后3 d | |||
| Pe_f | 盛花期 | 花瓣 | SP_5d | 青荚期 | 角果皮 | 花后3 d | |||
| Pi_f_un | 盛花期 | 雌蕊-未授粉 | SP_7d | 青荚期 | 角果皮 | 花后7 d | |||
| Pi_i | 初花期 | 雌蕊 | SP_10d | 青荚期 | 角果皮 | 花后10 d | |||
| Pi_f | 盛花期 | 雌蕊 | SP_13d | 青荚期 | 角果皮 | 花后13 d | |||
| Sta_i | 初花期 | 雄蕊 | SP_16d | 青荚期 | 角果皮 | ||||
| Sta_f | 盛花期 | 雄蕊 | SP_19d | 青荚期 | 角果皮 | 花后19 d | |||
| At_i | 初花期 | 花药 | SP_21d | 青荚期 | 角果皮 | 花后21 d | |||
| At_f | 盛花期 | 花药 | SP_24d | 青荚期 | 角果皮 | 花后24 d | |||
| Cap_i | 初花期 | 花丝 | SP_27d | 青荚期 | 角果皮 | 花后27 d | |||
| Cap_f | 盛花期 | 花丝 | SP_30d | 青荚期 | 角果皮 | 花后30 d | |||
| IT_b | 蕾期 | 主序顶端 | SP_35d | 青荚期 | 角果皮 | 花后35 d | |||
| IT_i | 初花期 | 主序顶端 | SP_40d | 成熟期 | 角果皮 | 花后40 d | |||
| IT_f | 盛花期 | 主序顶端 | SP_43d | 成熟期 | 角果皮 | 花后43 d | |||
| Se_3d | 青荚期 | 种子 | 花后3 d | SP_46d | 成熟期 | 角果皮 | 花后46 d | ||
| Se_5d | 青荚期 | 种子 | 花后3 d | ||||||
新窗口打开|下载CSV
2 结果与分析
2.1 甘蓝型油菜、白菜和甘蓝PEBP基因家族成员筛选
为筛选并鉴定甘蓝型油菜及亲本种白菜和甘蓝基因组内PEBP基因成员, 利用拟南芥的6个PEBP蛋白序列在BRAD网站上进行BlastP分析(E<10-5), 分别获得26、12和11个甘蓝型油菜、白菜和甘蓝PEBP基因成员(表1)。值得一提的是, 在甘蓝基因组内没有发现TSF亚家族同源基因, 可能在进化过程中被丢失。Table 1
表1
表1利用BlastP方法获得油菜、白菜、甘蓝和拟南芥PEBP同源基因
Table 1
| AGI ID | 油菜同源基因 B. napus homologs | 白菜或甘蓝同源基因 B. rapa or B. oleracea homologs | 基因模型 Gene models |
|---|---|---|---|
| AT5G62040 | BnaA06g21490D | Bra010052 | BROTHER OF FT AND TFL1 (BFT) |
| AT5G62040 | BnaC03g52010D | Bol012573 | BROTHER OF FT AND TFL1 (BFT) |
| AT5G62040 | -— | Bol015337 | BROTHER OF FT AND TFL1 (BFT) |
| AT2G27550 | BnaA03g22340D | Bra000506 | centroradialis (ATC) |
| AT2G27550 | BnaA04g15930D | Bra034357 | centroradialis (ATC) |
| AT2G27550 | BnaA07g13290D | Bra012010 | centroradialis (ATC) |
| AT2G27550 | BnaC01g41050D | -— | centroradialis (ATC) |
| AT2G27550 | BnaC03g47080D | Bol026421 | centroradialis (ATC) |
| AT2G27550 | BnaC03g64560D | -— | centroradialis (ATC) |
| AT2G27550 | BnaC04g39220D | Bol032920 | centroradialis (ATC) |
| AT2G27550 | BnaC04g16750D | -— | centroradialis (ATC) |
| AT1G65480 | BnaA02g12130D | Bra022475 | FLOWERING LOCUS T (FT) |
| AT1G65480 | -— | Bra004117 | FLOWERING LOCUS T (FT) |
| AT1G65480 | BnaC02g45250D | Bol017639 | FLOWERING LOCUS T (FT) |
| AT1G65480 | BnaC04g14850D | Bol039209 | FLOWERING LOCUS T (FT) |
| AT1G18100 | BnaA09g44700D | Bra031016 | MOTHER OF FT AND TFL1 (MFT) |
| AT1G18100 | BnaA06g12320D | Bra025930 | MOTHER OF FT AND TFL1 (MFT) |
| AT1G18100 | BnaC05g50120D | Bol009744 | MOTHER OF FT AND TFL1 (MFT) |
| AT1G18100 | BnaC08g37400D | Bol030802 | MOTHER OF FT AND TFL1 (MFT) |
| AT5G03840 | BnaA10g26300D | Bra009508 | TERMINAL FLOWER 1 (TFL1) |
| AT5G03840 | BnaAnng00810D | Bra028815 | TERMINAL FLOWER 1 (TFL1) |
| AT5G03840 | -— | Bra005783 | TERMINAL FLOWER 1 (TFL1) |
| AT5G03840 | BnaC02g02900D | Bol005471 | TERMINAL FLOWER 1 (TFL1) |
| AT5G03840 | BnaC03g01440D | Bol010027 | TERMINAL FLOWER 1 (TFL1) |
| AT5G03840 | BnaCnng10680D | Bol015337 | TERMINAL FLOWER 1 (TFL1) |
| AT4G20370 | BnaA07g25310D | Bra022475 | TWIN SISTER OF FT (TSF) |
| AT4G20370 | BnaA07g33120D | Bra015710 | TWIN SISTER OF FT (TSF) |
| AT4G20370 | BnaC02g23820D | -— | TWIN SISTER OF FT (TSF) |
| AT4G20370 | BnaC06g27090D | -— | TWIN SISTER OF FT (TSF) |
新窗口打开|下载CSV
2.2 基因结构与motif预测
基因结构决定其功能, 为进一步分析油菜PEBP基因家族的功能, 利用其CDS和基因组DNA序列分析其外显子-内含子结构, 并按照其进化树顺序展示(图1)。除了BnaC04g39220D (ATC)和BnaC06g27090D (TSF)含有3个外显子和2个内含子之外, 其余PEBP基因家族成员均含有4个外显子和3个内含子。Motif预测结果表明, Motif-1和Motif-2是PEBP基因家族的特征性motif, 除BnaC04g39220D (ATC)和BnaC06g 27090D (TSF)只含有Motif-1和Motif-2外, 其余成员还含有Motif-3、Motif-4和Motif-5, 说明Motif-3、Motif-4和Motif-5的功能也相当保守(图2)。图1
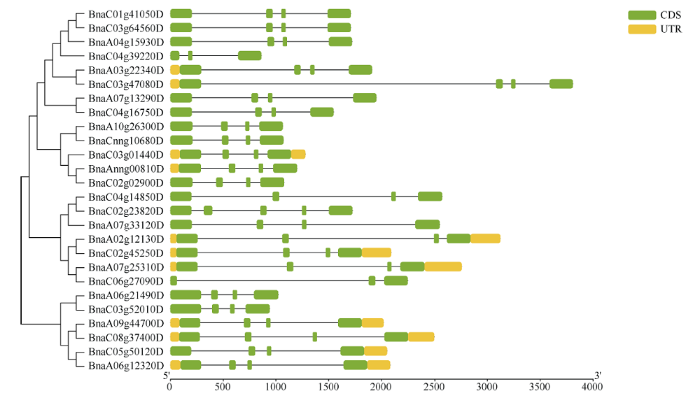 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1油菜PEBP进化树与基因结构
Fig. 1Phylogenetic tree and gene structures of PEBP in B. napus
图2
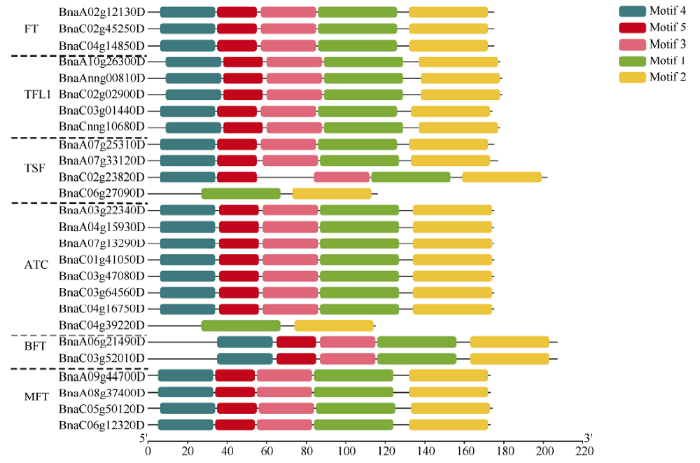 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2油菜PEBP基因家族motif预测
Fig. 2Motif prediction of PEBP genes in B. napus
为检测油菜PEBP基因家族的扩增情况, 检测了26个成员复制情况, 发现超过76.9%的成员都属于片段重复, 说明片段复制在油菜PEBP家族扩增中起着重要作用。
2.3 序列比对与进化树构建
利用Clustal W软件将4个物种PEBP蛋白序列进行多序列比对, 并导入DNAMAN中展示(图3)。用MEGA6.0软件, 通过邻接法(Neighbor Joining, NJ) 构建基因分子进化树(图4), 表明该基因可分为3个亚家族, 第一亚家族包括ATC(13)和TFL1(11), 第二亚家族包括FT(6)和TSF(6), 第三亚家族包括BFT(5)和MFT(8)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3油菜与拟南芥PEBP蛋白序列多重比对
Fig. 3Multiple alignment of PEBP protein sequences between B. napus and A. thaliana
图4
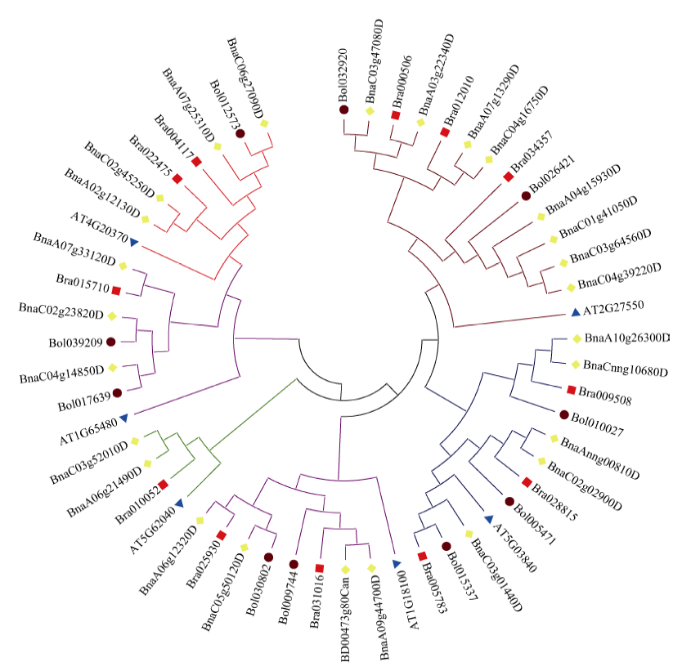 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4油菜、白菜、甘蓝和拟南芥PEBP基因家族进化分析
Fig. 4Phylogenetic analysis of PEBP gene family in B. napus, B. rapa, B. oleracea, and A. thaliana
2.4 选择压力分析
在进化分析中, 常用Ka/Ks或dN/dS来计算某基因是否受到自然选择作用。如果Ka/Ks>1, 认为有正选择效应; 如果Ka/Ks=1, 认为存在中性选择; 如果Ka/Ks<1, 则认为有纯化选择作用。以拟南芥PEBP家族基因为参照估算进化过程中甘蓝型油菜PEBP基因家族的选择压力(表2)。表明, 油菜PEBP基因家族的Ka/Ks均小于1, 即受到纯化选择, 其中TSF和FT亚家族的Ka/Ks较大, ATC和MFT亚家族的Ka/Ks较小, 表明TSF和FT亚家族进化更保守。Table 2
表2
表2油菜和拟南芥PEBP同源基因的Ka/Ks
Table 2
| 油菜基因编号 Gene ID in B. napus | 拟南芥基因编号 Gene ID in A. thaliana | 基因模型 Gene model | 非同义突变频率 Ka | 同义突变频率 Ks | Ka/Ks |
|---|---|---|---|---|---|
| BnaA02g12130D | AT1G65480 | FT | 0.09 | 0.32 | 0.27 |
| BnaC02g45250D | AT1G65480 | FT | 0.08 | 0.35 | 0.24 |
| BnaC04g14850D | AT1G65480 | FT | 0.11 | 0.80 | 0.14 |
| BnaA10g26300D | AT5G03840 | TFL1 | 0.08 | 0.37 | 0.23 |
| BnaAnng00810D | AT5G03840 | TFL1 | 0.05 | 0.39 | 0.14 |
| BnaC02g02900D | AT5G03840 | TFL1 | 0.05 | 0.39 | 0.14 |
| BnaC03g01440D | AT5G03840 | TFL1 | 0.03 | 0.41 | 0.07 |
| BnaCnng10680D | AT5G03840 | TFL1 | 0.08 | 0.35 | 0.22 |
| BnaA07g25310D | AT4G20370 | TSF | 0.10 | 0.80 | 0.12 |
| BnaA07g33120D | AT4G20370 | TSF | 0.09 | 0.45 | 0.21 |
| BnaC02g23820D | AT4G20370 | TSF | 0.13 | 0.45 | 0.30 |
| BnaC06g27090D | AT4G20370 | TSF | 0.23 | 0.88 | 0.26 |
| BnaA03g22340D | AT2G27550 | ATC | 0.02 | 0.31 | 0.07 |
| BnaA04g15930D | AT2G27550 | ATC | 0.03 | 0.33 | 0.08 |
| BnaA07g13290D | AT2G27550 | ATC | 0.03 | 0.49 | 0.06 |
| BnaC01g41050D | AT2G27550 | ATC | 0.03 | 0.33 | 0.08 |
| BnaC03g47080D | AT2G27550 | ATC | 0.03 | 0.31 | 0.10 |
| BnaC03g64560D | AT2G27550 | ATC | 0.03 | 0.33 | 0.08 |
| BnaC04g16750D | AT2G27550 | ATC | 0.03 | 0.52 | 0.07 |
| BnaC04g39220D | AT2G27550 | ATC | 0.06 | 0.39 | 0.15 |
| BnaA06g21490D | AT5G62040 | BFT | 0.05 | 0.31 | 0.17 |
| BnaC03g52010D | AT5G62040 | BFT | 0.05 | 0.31 | 0.18 |
| BnaA09g44700D | AT1G18100 | MFT | 0.02 | 0.40 | 0.05 |
| BnaC08g37400D | AT1G18100 | MFT | 0.02 | 0.45 | 0.05 |
| BnaC05g50120D | AT1G18100 | MFT | 0.03 | 0.37 | 0.08 |
| BnaA06g12320D | AT1G18100 | MFT | 0.03 | 0.37 | 0.09 |
新窗口打开|下载CSV
2.5 甘蓝型油菜PEBP基因家族成员的组织表达分析
为解析甘蓝型油菜PEBP基因家族的功能, 利用本课题组前期完成的不同发育时期的中双11材料的共111个组织器官的转录组数据分析甘蓝型油菜PEBP基因家族的表达情况(图5)。3个FT基因成员中, BnaC04g14850D在各组织器官中表达量均较低或者不表达, 其余两个成员在开花后茎秆、叶片和发育的角果皮中具有较高的表达量, 其他组织器官表达量均较低; 4个TFL1成员中, 除BnaC03g 01440D在花器官中具有一定表达之外, 其他成员在所有检测样品中几乎均不表达; 4个TSF成员中, BnaA07g25310D和BnaC06g27090D在茎秆、叶片、蕾、雄蕊、花药、花丝以及不同发育时期的种子中有一定表达, 其余样品表达量均较低, 剩余2个成员在所有检测样品中几乎均不表达; 8个油菜ATC成员中, 除了BnaA03g22340D和BnaA07g 13290D在胚根和下胚轴中有表达之外, 其余成员均不表达; 2个BFT中, BnaC03g52010D在不同发育时期的叶子中表达, 其余组织器官中不表达, BnaA06g21490D在所有检测的样品中均不表达或者表达量很低; 4个MFT成员均在发育的种子、种胚、胚芽和种皮中特异表达, 其他组织器官中不表达。以上结果表明, 油菜PEBP家族具有较高的组织表达特性, 部分同源基因功能可能发生分化。图5
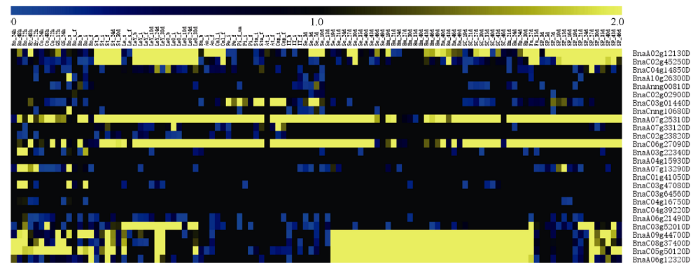 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5油菜PEBP家族成员的组织表达分析
横坐标样品名称详见附
Fig. 5Tissues-specific analysis of PEBP gene members in B. napus
Samples were listed in Supplementary table 1. Blue shows low expression levels and yellow shows high expression levels.
3 讨论
随着白菜、甘蓝和甘蓝型油菜基因组数据的释放, 芸薹属植物的遗传和分子功能研究进入一个新的时代。有关油菜产量性状如千粒重、农艺性状如株高以及抗性性状如核盘菌抗性的研究已经有大量报道, 而且利用分子标记技术对开花时间等数量性状QTL的定位同样有很多研究。PEBP家族功能非常保守而且在动植物中参与多种生物学过程[35]。其家族成员的功能在拟南芥、水稻、葡萄等作物中有一定研究, 但油菜中除一例报道外[36], 对该基因家族的功能特点研究相对较少。基于同源比对的方法, 我们在油菜基因组内筛选出26个PEBP基因家族成员, 分别包括2个BFT、8个ATC、3个FT、4个MFT、5个TFL1和4个TSF。进化树表明, 共包含3个亚家族, 其中ATC和TFL1亚家族, 为开花抑制因子; FT和TSF亚家族, 为开花促进因子; BFT和MFT属于第三亚家族。结构分析表明, 大分部成员含有4个外显子和3个内含子, 表明油菜PEBP基因家族进化非常保守。Motif预测表明Motif-1和-2是PEBP基因家族特征性基序。TSF和FT亚家族的Ka/Ks较大, ATC和MFT亚家族的Ka/Ks较小, 表明TSF和FT亚家族进化更保守。
PEBP家族成员的组织器官时空表达特性在拟南芥、葡萄和水稻等作物中已有报道[37,38,39]。但油菜中尚无有该基因的组织表达特性的研究。在本研究中, 3个FT基因成员除BnaC04g14850D在各组织器官中表达量均较低或者不表达, 其余两个成员在开花后器官如茎秆、叶片和发育的角果皮中具有较高的表达量, 其他组织器官中表达量均较低, 这与拟南芥中的表达模式一致[40]。4个TFL1成员中, 只有BnaC03g01440D在花器官中具有一定表达, 其他成员均不表达, 而棉花GhTFL1a和GhTFL1c主要在根中特异表达[30]。作为开花抑制因子, 油菜ATC基因仅在子叶和萌发的根中表达, 这与拟南芥ATC基因的表达模式一致。5个TFL1基因成员中, 只有BnaC03g01440D在花器官中有表达, 而其他成员均不表达, 表明BnaC03g01440D为TFL1的主要关键成员, 该基因与ATC功能类似, 都抑制植物开花[41]。与TFL1有功能冗余的2个油菜BFT基因, 只有BnaC03g52010D在不同发育时期的叶子中表达[42]。MFT基因主要在种子中表达, 且对ABA有响应[21], 油菜的4个MFT基因成员在不同发育时期的种子中均有较高水平表达。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1093/nar/gkv1054URLPMID:26476447 [本文引用: 1]

Flowering is a hot topic in Plant Biology and important progress has been made inArabidopsis thalianatoward unraveling the genetic networks involved. The increasing complexity and the explosion of literature however require development of new tools for information management and update. We therefore created an evolutive and interactive database of flowering time genes, named FLOR-ID (Flowering-Interactive Database), which is freely accessible athttp://www.flor-id.org. The hand-curated database contains information on 306 genes and links to 1595 publications gathering the work of >4500 authors. Gene/protein functions and interactions within the flowering pathways were inferred from the analysis of related publications, included in the database and translated into interactive manually drawn snapshots.
.
DOI:10.1007/s00018-011-0673-yURLPMID:21611891 [本文引用: 1]
Plants undergo a major physiological change as they transition from vegetative growth to reproductive development. This transition is a result of responses to various endogenous and exogenous signals that later integrate to result in flowering. Five genetically defined pathways have been identified that control flowering. The vernalization pathway refers to the acceleration of flowering on exposure to a long period of cold. The photoperiod pathway refers to regulation of flowering in response to day length and quality of light perceived. The gibberellin pathway refers to the requirement of gibberellic acid for normal flowering patterns. The autonomous pathway refers to endogenous regulators that are independent of the photoperiod and gibberellin pathways. Most recently, an endogenous pathway that adds plant age to the control of flowering time has been described. The molecular mechanisms of these pathways have been studied extensively in Arabidopsis thaliana and several other flowering plants.
.
DOI:10.1002/(ISSN)1521-1878URL [本文引用: 1]
.
DOI:10.1016/j.tplants.2006.06.006URLPMID:16843035 [本文引用: 1]
In wild and cultivated annual plant species, flowering time is an important life-history trait that coordinates the life cycle with local environmental conditions. Extensive studies on the genetic basis of flowering time in the model species Arabidopsis thaliana have revealed a complex genetic network that can detect environmental and internal signals. Based on this knowledge and on known pleiotropic effects associated with flowering time genes, we suggest that a natural shift towards an early-flowering life cycle might involve only particular functional regions in a limited number of genes. Our predictions are supported by genetic theories of adaptation and by recent data about genes associated with natural variation. We analyse the extent to which these predictions can also apply to crop species.
.
[本文引用: 1]
.
DOI:10.1109/TASC.2007.898014URLPMID:18444908 [本文引用: 1]
The transition from vegetative to reproductive growth is controlled by day length in many plant species. Day length is perceived in leaves and induces a systemic signal, called florigen, that moves through the phloem to the shoot apex. At the shoot apical meristem (SAM), florigen causes changes in gene expression that reprogram the SAM to form flowers instead of leaves. Analysis of flowering of Arabidopsis thaliana placed the CONSTANS/FLOWERING LOCUS T (CO/FT) module at the core of a pathway that promotes flowering in response to changes in day length. We describe progress in defining the molecular mechanisms that activate this module in response to changing day length and the increasing evidence that FT protein is a major component of florigen. Finally, we discuss conservation of FT function in other species and how variation in its regulation could generate different flowering behaviors.
DOI:10.1104/pp.108.132134URLPMID:19168644 [本文引用: 1]
Five barley (Hordeum vulgare) PEBP (for phosphatidylethanolamine-binding protein) genes were analyzed to clarify their functional roles in flowering using trans genie, expression, and quantitative trait locus analyses. Introduction of HvTFL1 and HvMFT1 into rice (Oryza sativa) plants did not result in any changes in flowering, suggesting that these two genes have functions distinct from flowering. Overexpression of HvFT1, HvFT2, and HvFT3 in rice resulted in early heading, indicating that these FT-like genes can act as promoters of the floral transition. HvFTl transgenic plants showed the most robust flowering initiation. In barley, HvFT1 was expressed at the time of shoot meristem phase transition. These results suggest that HvFT1 is the key gene responsible for flowering in the barley FT-like gene family. HvFT2 transgenic plants also showed robust flowering initiation, but HvFT1 was expressed only under short-day (SD) conditions during the phase transition, suggesting that its role is limited to specific photoperiodic conditions in barley. Flowering activity in HvFT3 transgenic rice was not as strong and was modulated by the photoperiod. These results suggest that HvFT3 functions in flowering promotion but that its effect is indirect. HvFT3 expression was observed in Morex, a barley cultivar carrying a dominant allele of Ppd-H2, a major quantitative trait locus for flowering under SD conditions, although no expression was detected in Steptoe, a cultivar carrying ppd-H2. HvFT3 was expressed in Morex under both long-day and SD conditions, although its expression was increased under SD conditions. HvFT3 was mapped to chromosome 1HL, the same chromosome that carries Ppd-H2. Genomic sequence analyses revealed that Morex possesses an intact HvFT3 gene, whereas most of this gene has been lost in Steptoe. These data strongly suggest that HvFT3 may be identical to Ppd-H2.
[本文引用: 1]
[本文引用: 1]
DOI:10.1155/2015/874361URLPMID:4576011 [本文引用: 2]
Early flowering is an important trait influencing grain yield and quality in wheat (Triticum aestivum L.) and barley (Hordeum vulgare L.) in short-season cropping regions. However, due to large and complex genomes of these species, direct identification of flowering genes and their molecular characterization remain challenging. Here, we used a bioinformatic approach to predict flowering-related genes in wheat and barley from 190 known Arabidopsis (Arabidopsis thaliana (L.) Heynh.) flowering genes. We identified 900 and 275 putative orthologs in wheat and barley, respectively. The annotated flowering-related genes were clustered into 144 orthologous groups with one-to-one, one-to-many, many-to-one, and many-to-many orthology relationships. Our approach was further validated by domain and phylogenetic analyses of flowering-related proteins and comparative analysis of publicly available microarray data sets for in silico expression profiling of flowering-related genes in 13 different developmental stages of wheat and barley. These further analyses showed that orthologous gene pairs in three critical flowering gene families (PEBP, MADS, and BBX) exhibited similar expression patterns among 13 developmental stages in wheat and barley, suggesting similar functions among the orthologous genes with sequence and expression similarities. The predicted candidate flowering genes can be confirmed and incorporated into molecular breeding for early flowering wheat and barley in short-season cropping regions.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.286.5446.1962URL [本文引用: 1]
DOI:10.1111/j.1365-313X.2009.03986.xURLPMID:19656342 [本文引用: 1]
Flowering is controlled by a network of pathways that converge to regulate a small number of floral integrator genes. We studied the interactions in Arabidopsis between three of these integrators, FLOWERING LOCUS T (FT), TWIN SISTER OF FT (TSF) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), as well as their repression by the MADS box transcription factor SHORT VEGETATIVE PHASE (SVP). FT is a mobile signal transmitted from the leaf to the meristem to initiate flowering. Using mRNA null alleles, we show that FT and the closely related TSF are not essential for flowering, but that the double mutant is photoperiod-insensitive. Inactivation of both genes also fully suppresses the early-flowering phenotype caused by over-expression of CONSTANS (CO), a transcriptional regulator in the photoperiod pathway. In addition, we demonstrate that TSF and FT have similar biochemical functions by showing that they interact in yeast with the same bZIP transcription factors. Expression of FT or TSF from promoters specific for phloem companion cells drives early flowering of the double mutant, so no expression of either gene is required in the meristem. Furthermore, TSF, like FT, is repressed by SVP, but the triple mutant svp-41 ft-10 tsf-1 expresses SOC1 in the meristem sooner and flowers earlier than ft-10 tsf-1. Thus we distinguish the functions of SVP in repressing FT and TSF in the leaf and SOC1 in the meristem. In addition, a time course of in situ hybridizations suggested that repression of SVP and activation of SOC1 proceed simultaneously in the meristem. These observations clarify the relationships between these early regulators of the floral transition, and further emphasize the relatedness of mechanisms acting in the leaf and meristem to control flowering time.
.
DOI:10.1126/science.275.5296.80URLPMID:8974397 [本文引用: 1]
Flowering plants exhibit one of two types of inflorescence architecture: indeterminate, in which the inflorescence grows indefinitely, or determinate, in which a terminal flower is produced. The indeterminate condition is thought to have evolved from the determinate many times, independently. In two mutants in distantly related species, terminal flower 1 in Arabidopsis and centroradialis in Antirrhinum, inflorescences that are normally indeterminate are converted to a determinate architecture. The Antirrhinum gene CEN-TRORADIALIS (CEN) and the Arabidopsis gene TERMINAL FLOWER 1 (TFL1) were shown to be homologous, which suggests that a common mechanism underlies indeterminacy in these plants. However, unlike CEN, TFL1 is also expressed during the vegetative phase, where it delays the commitment to inflorescence development and thus affects the timing of the formation of the inflorescence meristem as well as its identity.
.
URL [本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.111.088641URL [本文引用: 1]
TERMINAL FLOWER1 (TFL1) is a key regulator of flowering time and the development of the inflorescence meristem in Arabidopsis thaliana. TFL1 and FLOWERING LOCUS T (FT) have highly conserved amino acid sequences but opposite functions. For example, FT promotes flowering and TFL1 represses it; FT-overexpressing plants and TFL1 loss-of-function mutants have a similar phenotype production of terminal flowers in the shoot apex. FT is believed to function in a transcriptional activator complex by interacting with FD. Here, we demonstrate that TFL1 is involved in the transcriptional repression of genes that are activated by FT. We analyzed transgenic plants overexpressing TFL1 fused to a transcriptional repressor domain (TFL1-SRDX) or an activator domain (TFL1-VP16). Plants carrying 35S:TFL1-SRDX showed delayed flowering similar to 35S:TFL1 plants, and plants carrying 35S:TFL1-VP16 showed an early flowering phenotype and produced terminal flowers. Furthermore, the tfl1 and 35S:TFL1-VP16 plant phenotypes were strongly suppressed by the fd mutation, and TFL1 interacted with FD in the cell nucleus, as shown by bimolecular fluorescence complementation experiments. We conclude that TFL1 negatively modulates the FD-dependent transcription of target genes to fine-tune flowering time and the development of the inflorescence meristem.
.
DOI:10.1093/mp/sst114URLPMID:23935007 [本文引用: 1]
Soil salinity is one of the most serious agricultural problems that significantly reduce crop yields in the arid and semi-arid regions. It influences various phases of plant growth and developmental processes, such as seed germination, leaf and stem growth, and reproductive propagation. Salt stress delays the onset of flowering in many plant species. We have previously reported that the Arabidopsis BROTHER OF FT AND TFL1 (BFT) acts as a floral repressor under salt stress. However, the molecular mechanisms underlying the BFT function in the salt regulation of flowering induction is unknown. In this work, we found that BFT delays flowering under high salinity by competing with FLOWERING LOCUS T (FT) for binding to the FD transcription factor. The flowering time of FD-deficient fd-2 mutant was insensitive to high salinity. BFT interacts with FD in the nucleus via the C-terminal domain of FD, which is also required for the interaction of FD with FT, and interferes with the FT–FD interaction. These observations indicate that BFT constitutes a distinct salt stress signaling pathway that modulates the function of the FT–FD module and possibly provides an adaptation strategy that fine-tunes photoperiodic flowering under high salinity. Under high salinity, the floral repressor BFT competes with FT for FD binding and thus interferes with the FT–FD function, resulting in delayed flowering. The BFT-mediated salt stress signaling scheme provides an adaptation strategy by fine-tuning photoperiodic flowering under high salinity.
DOI:10.4161/psb.5.10.13161URLPMID:20551347 [本文引用: 2]
Brassinosteroids (BRs) are a family of plant steroid hormones that play diverse roles in many aspects of plant growth and development. For example, BRs promote seed germination by counteracting the inhibitory effect of ABA and regulate plant reproductive development, thus affecting seed yield. We have recently reported that MOTHER OF FT AND TFL1 (MFT) regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Here, we show that MFT function is also relevant to the BR signaling pathway. In mft loss-of-function mutants, the application of BR could not fully antagonize the inhibitory effect of exogenous ABA on seed germination, suggesting that BR promotes seed germination against ABA partly through MFT. In addition, mft enhances the low-fertility phenotype of det2 in which BR biosynthesis is blocked. This phenotype, together with the observation that MFT is expressed in gametophytes and developing seeds, suggests that MFT and BR play redundant roles in regulating fertility. Therefore, these results suggest that MFT affects seed germination and fertility relevant to the BR signaling pathway.
.
DOI:10.1105/tpc.114.135103URLPMID:25663621 [本文引用: 1]
Gene duplication provides resources for novel gene functions. Identification of the amino acids responsible for functional conservation and divergence of duplicated genes will strengthen our understanding of their evolutionary course. Here, we conducted a systemic functional investigation of phosphatidylethanolamine binding proteins (PEBPs) in soybean (Glycine max) and Arabidopsis thaliana. Our results demonstrated that after the ancestral duplication, the lineage of the common ancestor of the FLOWERING LOCUS T (FT) and TERMINAL FLOWER1 (TFL1) subfamilies functionally diverged from the MOTHER OF FT AND TFL1 (MFT) subfamily to activate flowering and repress flowering, respectively. They also underwent further specialization after subsequent duplications. Although the functional divergence increased with duplication age, we observed rapid functional divergence for a few pairs of young duplicates in soybean. Association analysis between amino acids and functional variations identified critical amino acid residues that led to functional differences in PEBP members. Using transgenic analysis, we validated a subset of these differences. We report clear experimental evidence for the functional evolution of the PEBPs in the MFT, FT, and TFL1 subfamilies, which predate the origin of angiosperms. Our results highlight the role of amino acid divergence in driving evolutionary novelty after duplication.
DOI:10.1371/journal.pone.0097669URLPMID:24845624 [本文引用: 1]
FLOWERING LOCUS T (FT) is the key flowering integrator in Arabidopsis (Arabidopsis thaliana), and its homologs encode florigens in many plant species regardless of their photoperiodic response. Two FT homologs, GmFT2a and GmFT5a, are involved in photoperiod-regulated flowering and coordinately control flowering in soybean. However, the molecular and genetic understanding of the roles played by GmFT2a and GmFT5a in photoperiod-regulated flowering in soybean is very limited. In this study, we demonstrated that GmFT2a and GmFT5a were able to promote early flowering in soybean by overexpressing these two genes in the soybean cultivar Williams 82 under noninductive long-day (LD) conditions. The soybean homologs of several floral identity genes, such as GmAP1, GmSOC1 and GmLFY, were significantly upregulated by GmFT2a and GmFT5a in a redundant and differential pattern. A bZIP transcription factor, GmFDL19, was identified as interacting with both GmFT2a and GmFT5a, and this interaction was confirmed by yeast two-hybridization and bimolecular fluorescence complementation (BiFC). The overexpression of GmFDL19 in soybean caused early flowering, and the transcription levels of the flowering identity genes were also upregulated by GmFDL19, as was consistent with the upregulation of GmFT2a and GmFT5a. The transcription of GmFDL19 was also induced by GmFT2a. The results of the electrophoretic mobility shift assay (EMSA) indicated that GmFDL19 was able to bind with the cis-elements in the promoter of GmAP1a. Taken together, our results suggest that GmFT2a and GmFT5a redundantly and differentially control photoperiod-regulated flowering in soybean through both physical interaction with and transcriptional upregulation of the bZIP transcription factor GmFDL19, thereby inducing the expression of floral identity genes.
DOI:10.1007/s00239-004-0179-4URLPMID:16170456 [本文引用: 1]
The TFL1 and FT genes, which are key genes in the control of flowering time in Arabidopsis thaliana, belong to a small multigene family characterized by a specific phosphatidylethanolamine-binding protein domain, termed the PEBP gene family. Several PEBP genes are found in dicots and monocots, and act on the control of flowering time. We investigated the evolution of the PEBP gene family in cereals. First, taking advantage of the complete rice genome sequence and EST databases, we found 19 PEBP genes in this species, 6 of which were not previously described. Ten genes correspond to five pairs of paralogs mapped on known duplicated regions of the rice genome. Phylogenetic analysis of Arabidopsis and rice genes indicates that the PEBP gene family consists of three main homology classes (the so-called TFL1-LIKE, MFT-LIKE, and FT-LIKE subfamilies), in which gene duplication and/or loss occurred independently in Arabidopsis and rice. Second, phylogenetic analyses of genomic and EST sequences from five cereal species indicate that the three subfamilies of PEBP genes have been conserved in cereals. The tree structure suggests that the ancestral grass genome had at least two MFT-like genes, two TFL1-like genes, and eight FT-like genes. A phylogenomic approach leads to some hypotheses about conservation of gene function within the subfamilies.
[本文引用: 1]
DOI:10.1105/tpc.110.081406URLPMID:21441432 [本文引用: 1]
The mobile floral-promoting signal, florigen, is thought to consist of, in part, the FT protein named after the Arabidopsis thaliana gene FLOWERING LOCUS T. FT is transcribed and translated in leaves and its protein moves via the phloem to the shoot apical meristem where it promotes the transition from vegetative to reproductive development. In our search for a maize FT-like floral activator(s), seven Zea mays CENTRORADIALIS (ZCN) genes encoding FT homologous proteins were studied. ZCN8 stood out as the only ZCN having the requisite characteristics for possessing florigenic activity. In photoperiod sensitive tropical lines, ZCN8 transcripts were strongly upregulated in a diurnal manner under floral-inductive short days. In day-neutral temperate lines, ZCN8 mRNA level was independent of daylength and displayed only a weak cycling pattern. ZCN8 is normally expressed in leaf phloem, but ectopic expression of ZCN8 in vegetative stage shoot apices induced early flowering in transgenic plants. Silencing of ZCN8 by artificial microRNA resulted in late flowering. ZCN8 was placed downstream of indeterminatel and upstream of delayed floweringi, two other floral activator genes. We propose a flowering model linking photoperiod sensitivity of tropical maize to diurnal regulation of ZCN8.
.
DOI:10.1111/pce.12644URLPMID:26428711 [本文引用: 1]
Abstract Optimum flowering time is the key to maximize canola production in order to meet global demand of vegetable oil, biodiesel and canola-meal. We reveal extensive variation in flowering time across diverse genotypes of canola under field, glasshouse and controlled environmental conditions. We conduct a genome-wide association study and identify 69 single nucleotide polymorphism (SNP) markers associated with flowering time, which are repeatedly detected across experiments. Several associated SNPs occur in clusters across the canola genome; seven of them were detected within 20 b regions of a priori candidate genes; FLOWERING LOCUS T , FRUITFUL, FLOWERING LOCUS C, CONSTANS, FRIGIDA, PHYTOCHROME B and an additional five SNPs were localized within 14 b of a previously identified quantitative trait loci for flowering time. Expression analyses showed that among FLC paralogs, BnFLC.A2 accounts for ~23% of natural variation in diverse accessions. Genome-wide association analysis for FLC expression levels mapped not only BnFLC.C2 but also other loci that contribute to variation in FLC expression. In addition to revealing the complex genetic architecture of flowering time variation, we demonstrate that the identified SNPs can be modelled to predict flowering time in diverse canola germplasm accurately and hence are suitable for genomic selection of adaptative traits in canola improvement programmes.
DOI:10.1093/dnares/dsv035URL [本文引用: 1]
Key message: A set of additive loci for seed oil content were identified using association mapping and one of the novel loci on the chromosome A5 was validated by linkage mapping. Increasing seed oil content is one of the most important goals in the breeding of oilseed crops including Brassica napus, yet the genetic basis for variations in this important trait remains unclear. By genome-wide... [Show full abstract]
URL [本文引用: 2]
Anthocyanins are widely distributed water-soluble phytochemical pigments belonging to the flavonoid group. To date, limited knowledge is available about the regulatory roles of miRNAs in anthocyanin biosynthesis in plants. To identify the miRNAs associated with anthocyanin biosynthesis in radish, five small RNA (sRNA) libraries constructed from 'Xinlimei' radish roots at 11, 21, 44, 56 and 73... [Show full abstract]
DOI:10.1371/journal.pone.0161080URLPMID:725 [本文引用: 1]
Upland cotton (Gossypium hirsutumL.) is a naturally occurring photoperiod-sensitive perennial plant species. However, sensitivity to the day length was lost during domestication. The phosphatidylethanolamine-binding protein (PEBP) gene family, of which three subclades have been identified in angiosperms, functions to promote and suppress flowering in photoperiod pathway. Recent evidence indicates that PEBP family genes play an important role in generating mobile flowering signals. We isolated homologues of thePEBPgene family in upland cotton and examined their regulation and function. NinePEBP-like genes were cloned and phylogenetic analysis indicated the genes belonged to four subclades (FT,MFT,TFL1andPEBP). CottonPEBP-like genes showed distinct expression patterns in relation to different cotton genotypes, photoperiod responsive and cultivar maturity. TheGhFTgene expression of a semi-wild race of upland cotton were strongly induced under short day condition, whereas theGhPEBP2gene expression was induced under long days. We also elucidated that GhFT but not GhPEBP2 interacted with FD-like bZIP transcription factor GhFD and promote flowering under both long- and short-day conditions. The present result indicated thatGhPEBP-like genes may perform different functions. This work corroborates the involvement ofPEBP-like genes in photoperiod response and regulation of flowering time in different cotton genotypes, and contributes to an improved understanding of the function ofPEBP-like genes in cotton.
[本文引用: 1]
.
DOI:10.1093/molbev/mst197URL [本文引用: 1]
.
URLPMID:2702329 [本文引用: 1]
Background Over the past two decades, there have been several approximate methods that adopt different mutation models and used for estimating nonsynonymous and synonymous substitution rates (Ka and Ks) based on protein-coding sequences across species or even different evolutionary lineages. Among them, MYN method (a Modified version of Yang- Nielsen method) considers three major dynamic features of evolving DNA sequences???bias in transition/transversion rate, nucleotide frequency, and unequal transitional substitution but leaves out another important feature unequal substitution rates among different sites or nucleotide positions. Results We incorporated a new feature for analyzing evolving DNA sequences???unequal substitution rates among different sites???into MYN method, and proposed a modified version, namely ?? (gamma)-MYN, based on an assumption that the evolutionary rate at each site follows a mode of ??-distribution. We applied ??-MYN to analyze the key estimator of selective pressure ?? (Ka/Ks) and other relevant parameters in comparison to two other related methods, YN and MYN, and found that neglecting the variation of substitution rates among different sites may lead to biased estimations of ??. Our new method appears to have minimal deviations when relevant parameters vary within normal ranges defined by empirical data. Conclusion Our results indicate that unequal substitution rates among different sites have variable influences on ?? under different evolutionary rates while both transition/transversion rate ratio and unequal nucleotide frequencies affect Ka and Ks thus selective pressure ??. Reviewers This paper was reviewed by Kateryna Makova, David A. Liberles (nominated by David H Ardell), Zhaolei Zhang (nominated by Mark Gerstein), and Shamil Sunyaev.
DOI:10.1104/pp.107.109538URLPMID:17993543 [本文引用: 1]
The phosphatidylethanolamine-binding proteins (PEBPs) represent an ancient protein family found across the biosphere. In animals they are known to act as kinase and serine protease inhibitors controlling cell growth and differentiation. In plants the most extensively studied PEBP genes, the Arabidopsis (Arabidopsis thaliana) FLOWERING LOCUS T (FT) and TERMINAL FLOWER1 (TFL1) genes, function, respectively, as a promoter and a repressor of the floral transition. Twenty-five maize (Zea mays) genes that encode PEBP-like proteins, likely the entire gene family, were identified and named Zea mays CENTRORADIALIS (ZCN), after the first described plant PEBP gene from Antirrhinum. The maize family is expanded relative to eudicots (typically six to eight genes) and rice (Oryza sativa; 19 genes). Genomic structures, map locations, and syntenous relationships with rice were determined for 24 of the maize ZCN genes. Phylogenetic analysis assigned the maize ZCN proteins to three major subfamilies: TFL1-like (six members), MOTHER OF FT AND TFL1-like (three), and FT-like (15). Expression analysis demonstrated transcription for at least 21 ZCN genes, many with developmentally specific patterns and some having alternatively spliced transcripts. Expression patterns and protein structural analysis identified maize candidates likely having conserved gene function of TFL1. Expression patterns and interaction of the ZCN8 protein with the floral activator DLF1 in the yeast (Saccharomyces cerevisiae) two-hybrid assay strongly supports that ZCN8 plays an orthologous FT function in maize. The expression of other ZCN genes in roots, kernels, and flowers implies their involvement in diverse developmental processes.
DOI:10.3389/fpls.2014.00282URLPMID:4060206 [本文引用: 1]
Rapeseed (Brassica napus L.) is grown in different geographical regions of the world. It is adapted to different environments by modification of flowering time and requirement for cold. A broad variation exists from very early-flowering spring-type to late-flowering winter cultivars which only flower after exposure to an extended cold period. B. napus is an allopolyploid species which resulted from the hybridization between B. rapa and B. oleracea. In Arabidopsis thaliana, the PEBP-domain genes FLOWERING LOCUS-T (FT) and TERMINAL FLOWER-1 (TFL1) are important integrators of different flowering pathways. Six FT and four TFL1 paralogs have been identified in B. napus. However, their role in flowering time control is unknown. We identified EMS mutants of the B. napus winter-type inbreed line Express 617. In total, 103 mutant alleles have been determined for BnC6FTb, BnC6FTa, and BnTFL1-2 paralogs. We chose three non-sense and 15 missense mutant lines (M3) which were grown in the greenhouse. Although only two out of 6 FT paralogs were mutated, 6 out of 8 BnC6FTb mutant lines flowered later as the control, whereas all five BnC6FTa mutant lines started flowering as the non-mutated parent. Mutations within the BnTFL1-2 paralog had no large effects on flowering time but on yield components. F1 hybrids between BnTFL1-2 mutants and non-mutated parents had increased seed number per pod and total seeds per plant suggesting that heterozygous mutations in a TFL1 paralog may impact heterosis in rapeseed. We demonstrate that single point-mutations in BnFT and BnTFL1 paralogs have effects on flowering time despite the redundancy of the rapeseed genome. Moreover, our results suggest pleiotropic effects of BnTFL1 paralogs beyond the regulation of flowering time.
DOI:10.1007/s11103-006-9113-zURLPMID:17160562 [本文引用: 1]
http://link.springer.com/article/10.1007%2Fs11103-006-9113-z
[本文引用: 1]
DOI:10.1007/s11103-009-9478-xURLPMID:19288213 [本文引用: 1]
Angiosperm genes sharing a conserved phosphatidylethanolamine-binding (PEPB) domain have been shown to be involved in the control of shoot meristem identity and flowering time. The family is divided into three subfamilies, FT -like, TFL1 -like and MFT -like. This study is focused on the evolution of the MFT -like clade, suggested to be ancestral to the two other clades. We report that the bryophyte Physcomitrella patens and the lycopod Selaginella moellendorfii contain four and two MFT -like genes respectively. Neither species have any FT or TFL1 -like genes. Furthermore, we have identified a new subclade of MFT-like genes in Angiosperms. Quantitative expression analysis of MFT -like genes in Physcomitrella patens reveals that the expression patterns are circadian and reaches maximum in gametangia and sporophytes. Our data suggest that the occurrence FT and TFL1 -like genes, is associated with the evolution of seed plants. Expression data for Physcomitrella MFT-like genes implicates an involvement in the development of reproductive tissues in the moss.
.
DOI:10.1126/science.286.5446.1960URL [本文引用: 1]
DOI:10.1093/jxb/erv247URLPMID:4507777 [本文引用: 1]
Plants carefully control where and when flowers are made through activators and repressors. We show that spatially the shoot meristem is key in responding to the repressors of flowering TFL1. Models for the control of above-ground plant architectures show how meristems can be programmed to be either shoots or flowers. Molecular, genetic, transgenic, and mathematical studies have greatly refined these models, suggesting that the phase of the shoot reflects different genes contributing to its repression of flowering, its vegetativeness (‘veg’), before activators promote flower development. Key elements of how the repressor of flowering and shoot meristem gene TFL1 acts have now been tested, by changing its spatiotemporal pattern. It is shown that TFL1 can act outside of its normal expression domain in leaf primordia or floral meristems to repress flower identity. These data show how the timing and spatial pattern of TFL1 expression affect overall plant architecture. This reveals that the underlying pattern of TFL1 interactors is complex and that they may be spatially more widespread than TFL1 itself, which is confined to shoots. However, the data show that while TFL1 and floral genes can both act and compete in the same meristem, it appears that the main shoot meristem is more sensitive to TFL1 rather than floral genes. This spatial analysis therefore reveals how a difference in response helps maintain the ‘veg’ state of the shoot meristem.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}