 ,3,*, 韩龙植,1,*
,3,*, 韩龙植,1,*Development of molecular markers polymorphic between Dongxiang wild rice and Geng rice cultivar ‘Nipponbare’
MA Xiao-Ding1, TANG Jiang-Hong2, ZHANG Jia-Ni2, CUI Di1, LI Hui3, LI Mao-Mao,3,*, HAN Long-Zhi,1,*通讯作者:
第一联系人:
收稿日期:2018-07-9接受日期:2018-10-8网络出版日期:2018-11-06
| 基金资助: |
Received:2018-07-9Accepted:2018-10-8Online:2018-11-06
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (3184KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马小定, 唐江红, 张佳妮, 崔迪, 李慧, 黎毛毛, 韩龙植. 东乡野生稻与日本晴多态性标记的开发[J]. 作物学报, 2019, 45(2): 316-321. doi:10.3724/SP.J.1006.2019.82037
MA Xiao-Ding, TANG Jiang-Hong, ZHANG Jia-Ni, CUI Di, LI Hui, LI Mao-Mao, HAN Long-Zhi.
东乡野生稻于1978年在我国江西省东乡县东源乡被发现, 当地称之为“野禾”, 后经水稻和植物学相关专家鉴定, 确定为普通野生稻[1,2], 而且是迄今为止发现的分布于全球最北端(28°14°N)的普通野生稻。东乡野生稻具有丰富的遗传多样性, 而且携带耐寒、耐旱、耐贫瘠、广亲和性、野败育性恢复性、胞质雄性不育、抗病虫和高产等相关基因, 对水稻基础研究和产业发展有着举足轻重的作用, 被誉为“野生植物类的大熊猫”[3]。
分子标记是继形态标记、细胞标记和生化标记后发展起来的一种简单易用的遗传标记。现在水稻研究中被广泛使用的分子标记有简单序列重复(Simple Sequence Repeat, SSR)标记、插入/缺失(Insertion-Deletion, InDel)标记和单核苷酸多态性(Single Nucleotide Polymorphism, SNP)标记。SSR标记是根据广泛存在于基因组序列中的、由1~6个核苷酸为重复单位组成的串联重复序列而设计的分子标记。由于其分布广泛、多态性丰富, 在不同作物功能基因组研究中发挥了重要的作用。InDel标记是指根据相同位点序列由不同数目的核苷酸插入或缺失形成的差异而设计的多态性标记[4]。InDel标记具有基因组差异位点丰富(例如, 日本晴和93-11基因组序列中平均每953 bp就存在1个InDel [5])、染色体位置明确、条带清晰、结果可靠等优点[6,7]。SNP标记是在基因组水平上单个核苷酸的变异引起的DNA序列多态性, 形式包括单碱基的缺失、插入、转换及颠换等。SNP标记遗传稳定性高, 位点丰富且分布广泛, 检测快速并且容易实现自动化分析[8,9,10]。但是目前由于检测方法及成本的限制, 还未被广泛采用。除了上述分子标记外, 近年来由于测序技术的发展和成本的降低, 一些研究机构开始使用全基因组测序技术进行起源进化和功能基因组学的相关研究。全基因组重测序是对已知基因组序列的物种进行不同个体的基因测序, 并在此基础上对个体或者群体进行差异性分析。通过全基因组重测序分析, 可以找到大量的SNP、InDel、结构变异(Structure Variation, SV)和拷贝数变异(Copy Number Variation, CNV)等位点[11]。
前期, 虽然已有一些研究者利用东乡野生稻为供体构建了染色体片段置换系, 但是使用的主要是SSR标记, 而且基因组覆盖不完全[12]。主要是由于东乡野生稻和亚洲栽培稻基因组存在较大的差异, 多态性分子标记数量和质量不佳。本研究以东乡野生稻和日本晴为材料, 通过筛选现有分子标记, 以及利用东乡野生稻基因组重测序数据设计的InDel标记, 形成一套覆盖全基因组的、条带清晰、结果可靠的分子标记, 为东乡野生稻的基础研究和育种利用提供有力的遗传工具。
1 材料与方法
1.1 研究材料
东乡野生稻取自江西省农业科学院水稻研究所保存的东乡野生稻庵家山居群中的C35单株, 日本晴来源于中国农业科学院作物科学研究所国家种质库。按照常规大田种植方法进行东乡野生稻的种植和栽培稻日本晴育苗移栽。1.2 基因组重测序
分别从3个东乡野生稻植株上取幼嫩的叶片, 然后立即在液氮中冷冻并储存在-80℃冰箱备用。用DNeasy plant mini kit (QIAGEN)试剂盒提取基因组总DNA, 经NanoDrop、Qubit和琼脂糖凝胶检测DNA提取的质量和浓度。5 μg DNA用于北京诺禾致源科技股份有限公司(北京)基因组重测序(Illumina HiSeq2500)。测序下机的原始数据经过滤, 去除接头序列和低质量测序数据获得有效数据。有效测序数据通过BWA软件[13]比对到参考基因组(ftp://ftp.ensemblgenomes.org/pub/plants/release-28/fasta/ oryza_sativa/dna/), SAMTOOLs软件[14]检测长度小于50 bp的小片段InDel, ANNOVAR软件[15]对检测出的InDel注释。1.3 InDel标记的设计
根据东乡野生稻基因组重测序结果, 对比日本晴基因组序列信息(IRGSP-1.0.28版), 以每隔1.5~3.0 Mb的距离在各染色体上查找对应的插入缺失位点, 在位点前后各选择120 bp序列, 利用Primer Premier 5.0设计引物。引物长度一般在19~30 bp之间, Tm值控制在(60±1)℃, 产物设计长度110~180 bp。1.4 DNA提取、PCR扩增及产物检测
分别从东乡野生稻和日本晴植株上取新生的嫩叶, 采用常规CTAB法提取基因组DNA [16]。PCR体系10 μL包含模板DNA 0.5 μL、10×PCR buffer 1.0 μL、10 mmol L-1 dNTPs 0.3 μL、10 μmol L-1正反引物各0.5 μL、Taq DNA聚合酶0.5 U, 用ddH2O补齐至10 μL。PCR程序为94℃ 预变性5 min; 94℃ 30 s, 59℃ 30 s, 72℃ 30 s, 35个循环; 72℃延伸5 min; 16℃保存。
PCR产物加2 μL 6×Loading buffer, 混匀后取3.5 μL于8%聚丙烯酰胺凝胶电泳槽中, 180 V稳压电压10 min后调电压至250 V, 再电泳大约70 min, 电泳结束后用银染法染胶显色[17]。
2 结果与分析
2.1 多态性分子标记开发
针对东乡野生稻、日本晴和东乡野生稻/日本晴杂合体(将两者DNA等量混合, 模拟杂合体基因组DNA)样本, 利用实验室现有的661对SSR标记引物和356对InDel标记引物进行多态性检测, 获得有多态性的标记81 个, 包括42个SSR标记和39个InDel标记, 多态率为8%。为了获得更多的多态性标记, 对东乡野生稻进行了基因组重测序, 通过与对照基因组比对, 分别在染色体基因上游、外显子、内含子和基因下游区共检测到373,501个InDel。根据此结果, 设计了217对InDel标记引物, 经过检测, 其中122个标记显示出多态性, 多态率为56% (图1)。综合以前和本次设计的引物, 共获得203个东乡野生稻与日本晴间有多态的分子标记。每条染色体上分子标记数目为9~30个, 平均间隔为1.2~2.6 Mb (表1和附表1)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1多态性分子标记筛选
1: 东乡野生稻; 2: 日本晴; 3: 东乡野生稻/日本晴杂合体; M: 标准分子量。
Fig. 1Screening of polymorphic molecular markers
1: Dongxiang wild rice; 2: Nipponbare; 3: artificial F1 hybrid between Dongxiang wild rice and Nipponbare; M: DNA size marker.
Table 1
表1
表1各染色体多态性标记分布
Table 1
| 染色体 Chromosome | 染色体长度 Chromosome length (kb) | 标记数量 Number of marker | 平均标记间隔 Mean interval (kb) |
|---|---|---|---|
| 1 | 43270 | 23 | 1881 |
| 2 | 35937 | 22 | 1633 |
| 3 | 36413 | 30 | 1213 |
| 4 | 35502 | 17 | 2088 |
| 5 | 29958 | 16 | 1872 |
| 6 | 31248 | 18 | 1736 |
| 7 | 29697 | 16 | 1856 |
| 8 | 28443 | 13 | 2187 |
| 9 | 23012 | 14 | 1643 |
| 10 | 23207 | 9 | 2578 |
| 11 | 29021 | 13 | 2232 |
| 12 | 27531 | 12 | 2294 |
新窗口打开|下载CSV
为了方便研究者直观地查看标记在染色体上的分布, 利用MapDraw (2.1)软件, 以标记的物理位置为参数制作了203个多态性标记在染色体上的分布图谱(图2)。由图谱可知, 标记在染色体上的分布比较均匀, 基本完全覆盖了12条水稻染色体。
图2
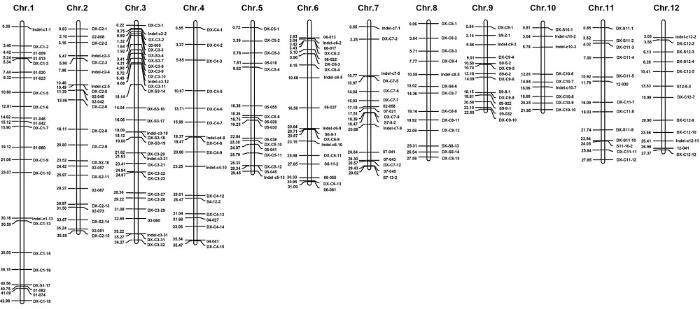 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2多态性分子标记在染色体上的分布图谱
每条染色体左边数字表示引物的物理位置(单位Mb)。
Fig. 2Distribution of polymorphic molecular marker loci on rice chromosomes
The numbers to the left of each chromosome indicate the physical locations of the marker loci in Mb.
2.2 多态性分子标记的应用
为检验所开发的多态性分子标记在其他亚洲栽培稻中的应用价值, 随机选取了59个多态性分子标记, 以5个籼稻和5个粳稻品种为样本, 以东乡野生稻和日本晴为对照检测(图 3)发现, 在粳稻品种中有84.7%的基因型与日本晴一致, 11.6%的基因型与东乡野生稻一致; 在籼稻品种中有73.9%的基因型与东乡野生稻一致, 13.5%的基因型与日本晴一致。而在检测到的异于对照基因型数量中, 籼稻(10.9%)要远远高于粳稻品种(1.7%)(表2和附表2)。因此, 如果利用本套多态性分子标记进行基因型分析, 其在分析东乡野生稻/粳稻杂交后代群体基因型上的应用价值高于分析东乡野生稻/籼稻杂交后代群体。图3
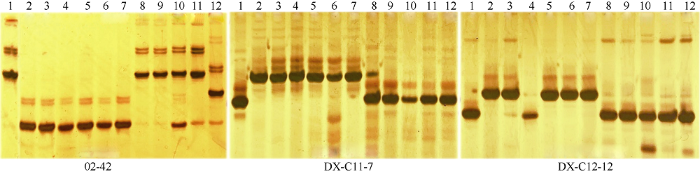 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3多态性分子标记在籼粳亚种中的应用
1: 东乡野生稻; 2: 日本晴; 3: 辽盐241; 4: 松粳8; 5: 吉粳88; 6: 淮稻9号; 7: 合系6号; 8: 黄花占; 9: 白玉丝苗; 10: 扬稻6号; 11: 湘晚籼12号; 12: 鄂中5号。
Fig. 3Genotyping of Xian and Geng cultivars by polymorphic molecular markers
1: Dongxiang wild rice; 2: Nipponbare; 3: Liaoyan 241; 4: Songgeng 8; 5: Jigeng 88; 6: Huaidao 9; 7: Hexi 6; 8: Huanghuazhan; 9: Baiyusimiao; 10: Yangdao 6; 11: Xiangwanxian 12; 12: Ezhong 5.
Table 2
表2
表2多态性标记籼梗亚种基因型鉴定一致性分析
Table 2
| 亚种 Subspecies | 鉴定总数 Number of identification | 东乡野生稻基因型数目 Number of Dongxiang wild rice genotype | 日本晴基因型数目 Number of Nipponbare genotype | 杂合基因型数目 Number of heterozygous genotype | 其他基因型数目 Number of others genotype |
|---|---|---|---|---|---|
| 粳 | 295 (5×59) | 34 (11.6%) | 250 (84.7%) | 6 (2.0%) | 5 (1.7%) |
| 籼 | 295 (5×59) | 218 (73.9%) | 40 (13.5%) | 5 (1.7%) | 32 (10.9%) |
新窗口打开|下载CSV
3 讨论
分子标记是现代遗传学和分子生物学研究最常用的技术之一, 随着技术的发展, 分子标记的种类也在不断发展, 例如SNP、CNV标记等。东乡野生稻做为一种普通野生稻, 与亚洲栽培稻基因组具有较大的差异。直接利用常规的SSR标记以及基于籼稻与粳稻设计的InDel标记来研究东乡野生稻, 或者开展分子标记辅助选择育种具有一定的局限性。例如, 本研究直接筛选常用的SSR和InDel标记, 仅仅只有8%的引物存在多态性。因此, 开发一套覆盖东乡野生稻全基因组的, 并且与亚洲栽培稻间存在多态性的分子标记至关重要。本研究在筛选现有分子标记的基础上, 利用东乡野生稻基因组重测序信息设计的InDel标记, 共获得了203个多态性分子标记, 均匀分布于12条染色体上, 标记平均间隔1.9 Mb, 可以满足基因初定位和标记辅助选择育种的需要。分子标记的开发基本上都是基于2个或者多个特定样本, 因此获得的分子标记的应用范围具有一定的局限性。本研究获得的203个多态性分子标记是基于东乡野生稻和日本晴开发的, 通过对5个籼稻和5个粳稻品种的检验, 发现其在粳稻品种中检测的基因型84.7%与日本晴一致, 而在籼稻品种中的基因型73.9%与东乡野生稻一致。如果利用该套分子标记检测东乡野生稻与籼稻杂交的后代, 可能会有大约74%的分子标记不能表现出多态性; 而在检测东乡野生稻与粳稻杂交后代中, 大约会有85%的标记表现出多态性。因此, 该套分子标记在分析东乡野生稻与粳稻杂交后代群体的基因型上具有较大的应用价值。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops. org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http:// c.wanfangdata.com.cn/Periodical-zuowxb.aspx。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]

正 近一、二年,我省鄱阳湖南侧东乡一带,先后传说发现野生稻。为了发掘祖国的野生稻资源,我们在1978年11月上旬前往东乡县,对野生稻的形态特征及其自然繁殖地段的地理生态环境、气候因素等情况,作了初步考查,并采回了样本,供进一步考察和研究应用。现将东乡野生稻(暂名)的调查资料简报如下:
URL [本文引用: 1]
正 近一、二年,我省鄱阳湖南侧东乡一带,先后传说发现野生稻。为了发掘祖国的野生稻资源,我们在1978年11月上旬前往东乡县,对野生稻的形态特征及其自然繁殖地段的地理生态环境、气候因素等情况,作了初步考查,并采回了样本,供进一步考察和研究应用。现将东乡野生稻(暂名)的调查资料简报如下:
[本文引用: 1]
[本文引用: 1]
DOI:10.3969/j.issn.1005-4944.2012.01.004URL [本文引用: 1]
东乡野生稻是20世纪70年代发现的分布于我国最北的野生稻种。东乡野生稻具有抗寒、耐旱、耐瘠、抗病虫等多种优异抗性基因,价值十分珍贵,被誉为“野生植物大熊猫”、“比大熊猫更应得到保护的物种”。笔者在调查研究的基础上,筒述了东乡野生稻的发现;分析了东乡野生稻的价值,认为东乡野生稻的价值体现在4个方面——珍贵的农业文化遗产、优异的植物抗性基因、特有的水稻育种材料、难得的生态旅游资源;最后,指出了当前东乡野生稻濒危现状,提出了抢救、保护东乡野生稻的具体对策和措施。
DOI:10.3969/j.issn.1005-4944.2012.01.004URL [本文引用: 1]
东乡野生稻是20世纪70年代发现的分布于我国最北的野生稻种。东乡野生稻具有抗寒、耐旱、耐瘠、抗病虫等多种优异抗性基因,价值十分珍贵,被誉为“野生植物大熊猫”、“比大熊猫更应得到保护的物种”。笔者在调查研究的基础上,筒述了东乡野生稻的发现;分析了东乡野生稻的价值,认为东乡野生稻的价值体现在4个方面——珍贵的农业文化遗产、优异的植物抗性基因、特有的水稻育种材料、难得的生态旅游资源;最后,指出了当前东乡野生稻濒危现状,提出了抢救、保护东乡野生稻的具体对策和措施。
DOI:10.16590/j.cnki.1001-4705.2017.09.047URL [本文引用: 1]
InDel (insertion-deletion)分子标记是指根据在近缘物种或同一物种不同个体间基因组同一位点的序列发生不同大小DNA片段的插入或缺失(insertion-deletion)而设计的多态性引物.它具有分布密度大、准确性高,重演性好的特点,已广泛应用于水稻的遗传分析和分子辅助育种研究中.本研究对InDel分子标记的开发利用进行了综述,并着重总结了其在水稻的籼粳分化、遗传多样性分析、基因定位以及功能标记开发等方面的应用进展,旨在为今后更好地开展水稻MAS育种研究提供参考.
DOI:10.16590/j.cnki.1001-4705.2017.09.047URL [本文引用: 1]
InDel (insertion-deletion)分子标记是指根据在近缘物种或同一物种不同个体间基因组同一位点的序列发生不同大小DNA片段的插入或缺失(insertion-deletion)而设计的多态性引物.它具有分布密度大、准确性高,重演性好的特点,已广泛应用于水稻的遗传分析和分子辅助育种研究中.本研究对InDel分子标记的开发利用进行了综述,并着重总结了其在水稻的籼粳分化、遗传多样性分析、基因定位以及功能标记开发等方面的应用进展,旨在为今后更好地开展水稻MAS育种研究提供参考.
DOI:10.1104/pp.103.038463URLPMID:15266053 [本文引用: 1]
DNA polymorphism is the basis to develop molecular markers that are widely used in genetic mapping today. A genome-wide rice (Oryza sativa) DNA polymorphism database has been constructed in this work using the genomes of Nipponbare, a cultivar of japonica, and 93-11, a cultivar of indica. This database contains 1,703,176 single nucleotide polymorphisms (SNPs) and 479,406 Insertion/Deletions (InDels), approximately one SNP every 268 bp and one InDel every 953 bp in rice genome. Both SNPs and InDels in the database were experimentally validated. Of 109 randomly selected SNPs, 107 SNPs (98.2%) are accurate. PCR analysis indicated that 90% (97 of 108) of InDels in the database could be used as molecular markers, and 68% to 89% of the 97 InDel markers have polymorphisms between other indica cultivars (Guang-lu-ai 4 and Long-te-pu B) and japonica cultivars (Zhong-hua 11 and 9522). This suggests that this database can be used not only for Nipponbare and 93-11, but also for other japonica and indica cultivars. While validating InDel polymorphisms in the database, a set of InDel markers with each chromosome 3 to 5 marker was developed. These markers are inexpensive and easy to use, and can be used for any combination of japonica and indica cultivars used in this work. This rice DNA polymorphism database will be a valuable resource and important tool for map-based cloning of rice gene, as well as in other various research on rice (http://shenghuan.shnu.edu.cn/ricemarker).
DOI:10.3969/j.issn.1672-416X.2005.05.024URL [本文引用: 1]
为评价插入缺失(InDel)标记在水稻分子育种中的应用价值,本研究用日本晴和9311序列筛选得到遍布每条染色体的20对InDel标记和53对SSR标记,分析46份粳稻和47份籼稻的遗传多样性。研究表明这些InDel标记在籼粳亚种间具有很高的多态性,亚种内多态性较低,平均多态性水平显著小于SSR标记,InDel标记具有数量多,扩增产物稳定和易于检测等优点,将InDel标记应用到遗传图谱构建、基因定位分析和标记辅助选择中可以加速研究进程。
DOI:10.3969/j.issn.1672-416X.2005.05.024URL [本文引用: 1]
为评价插入缺失(InDel)标记在水稻分子育种中的应用价值,本研究用日本晴和9311序列筛选得到遍布每条染色体的20对InDel标记和53对SSR标记,分析46份粳稻和47份籼稻的遗传多样性。研究表明这些InDel标记在籼粳亚种间具有很高的多态性,亚种内多态性较低,平均多态性水平显著小于SSR标记,InDel标记具有数量多,扩增产物稳定和易于检测等优点,将InDel标记应用到遗传图谱构建、基因定位分析和标记辅助选择中可以加速研究进程。
DOI:10.3724/SP.J.1006.2016.00932URL [本文引用: 1]
插入/缺失(InDel)分子 标记具有使用简单,结果清晰可靠的优点。本研究通过比对粳稻品种日本晴和籼稻品种93-11的基因组序列,在全基因组范围内设计了634对InDel候选 标记,通过PCR检测比较2种粳稻(日本晴和台中65)和2种籼稻(93-11和黄华占)的多态性,发现295对标记在2种籼稻间及2种粳稻间均带型一 致,而在籼、粳亚种间有多态性,因此这套295对标记可以在涉及籼粳亚种的基因定位和分子育种中应用。
DOI:10.3724/SP.J.1006.2016.00932URL [本文引用: 1]
插入/缺失(InDel)分子 标记具有使用简单,结果清晰可靠的优点。本研究通过比对粳稻品种日本晴和籼稻品种93-11的基因组序列,在全基因组范围内设计了634对InDel候选 标记,通过PCR检测比较2种粳稻(日本晴和台中65)和2种籼稻(93-11和黄华占)的多态性,发现295对标记在2种籼稻间及2种粳稻间均带型一 致,而在籼、粳亚种间有多态性,因此这套295对标记可以在涉及籼粳亚种的基因定位和分子育种中应用。
DOI:10.1051/gse:19980406URLPMID:9685323 [本文引用: 1]
An efficient strategy to develop a dense set of single-nucleotide polymorphism (SNP) markers is to take advantage of the human genome sequencing effort currently under way. Our approach is based on the fact that bacterial artificial chromosomes (BACs) and P1-based artificial chromosomes (PACs) used in long-range sequencing projects come from diploid libraries. If the overlapping clones sequenced are from different lineages, one is comparing the sequences from 2 homologous chromosomes in the overlapping region. We have analyzed in detail every SNP identified while sequencing three sets of overlapping clones found on chromosome 5p15.2, 7q21-7q22, and 13q12-13q13. In the 200.6 kb of DNA sequence analyzed in these overlaps, 153 SNPs were identified. Computer analysis for repetitive elements and suitability for STS development yielded 44 STSs containing 68 SNPs for further study. All 68 SNPs were confirmed to be present in at least one of the three (Caucasian, African-American, Hispanic) populations studied. Furthermore, 42 of the SNPs tested (62%) were informative in at least one population, 32 (47%) were informative in two or more populations, and 23 (34%) were informative in all three populations. These results clearly indicate that developing SNP markers from overlapping genomic sequence is highly efficient and cost effective, requiring only the two simple steps of developing STSs around the known SNPs and characterizing them in the appropriate populations.
DOI:10.3969/j.issn.1000-6850.2012.12.028URL [本文引用: 1]
SNP (single nucleotide polymorphism) marker, as a molecular marker of the third generation, plays an important role in molecular genetics, pharmacogenetics, forensic medicine, diagnosis and treatment of diseases recently. In this paper, the definition and characteristics of SNP molecular marker were both introduced. And the application profiles of Bin map, high throughput SNP-chip classification and population evolution research on the basis of SNP molecular marker was presented respectively. Finally, the problems of SNP molecular marker at the present stage were proposed, and the prospects of SNP molecular marker was predicted as well.
DOI:10.3969/j.issn.1000-6850.2012.12.028URL [本文引用: 1]
SNP (single nucleotide polymorphism) marker, as a molecular marker of the third generation, plays an important role in molecular genetics, pharmacogenetics, forensic medicine, diagnosis and treatment of diseases recently. In this paper, the definition and characteristics of SNP molecular marker were both introduced. And the application profiles of Bin map, high throughput SNP-chip classification and population evolution research on the basis of SNP molecular marker was presented respectively. Finally, the problems of SNP molecular marker at the present stage were proposed, and the prospects of SNP molecular marker was predicted as well.
DOI:10.1371/journal.pone.0017595URL [本文引用: 1]
DOI:10.1101/gr.114876.110URLPMID:21324876 [本文引用: 1]
Copy number variation (CNV) in the genome is a complex phenomenon, and not completely understood. We have developed a method, CNVnator, for CNV discovery and genotyping from read-depth (RD) analysis of personal genome sequencing. Our method is based on combining the established mean-shift approach with additional refinements (multiple-bandwidth partitioning and GC correction) to broaden the range of discovered CNVs. We calibrated CNVnator using the extensive validation performed by the 1000 Genomes Project. Because of this, we could use CNVnator for CNV discovery and genotyping in a population and characterization of atypical CNVs, such as de novo and multi-allelic events. Overall, for CNVs accessible by RD, CNVnator has high sensitivity (86%-96%), low false-discovery rate (3%-20%), high genotyping accuracy (93%-95%), and high resolution in breakpoint discovery (<200 bp in 90% of cases with high sequencing coverage). Furthermore, CNVnator is complementary in a straightforward way to split-read and read-pair approaches: It misses CNVs created by retrotransposable elements, but more than half of the validated CNVs that it identifies are not detected by split-read or read-pair. By genotyping CNVs in the CEPH, Yoruba, and Chinese-Japanese populations, we estimated that at least 11% of all CNV loci involve complex, multi-allelic events, a considerably higher estimate than reported earlier. Moreover, among these events, we observed cases with allele distribution strongly deviating from Hardy-Weinberg equilibrium, possibly implying selection on certain complex loci. Finally, by combining discovery and genotyping, we identified six potential de novo CNVs in two family trios.
DOI:10.7666/d.y558395URL [本文引用: 1]
普通野生稻是栽培稻的祖先种,从野生稻中发掘栽培稻中已丢失或削弱了的优异基因已成为当前水稻育种和资源研究的热点之一。本研究选用世界上生境最北(28°14′N)、地下茎能够耐-12.8℃严冬,具有强耐旱性且米质优良的江西东乡普通野生稻(简称东乡普野,下同)为供体亲本,以曾多年在云南创亩产吨粮记录的优良品种“桂朝2号”为轮回亲本构建一套高代回交群体,用AB-QTL分析法对株高、单株产量等7个性状进行QTL定位。为了全面发掘东乡普野中优良基因,在桂朝2号遗传背景下构建一套渗入系并绘出图示基因型。同时对东乡普野渗入片段大小、数目和染色体分布等因子作了分析。主要结果归纳如下: 1.通过对BC_4F_1的基因型分析和BC_4F_2群体株高、单株产量及产量构成因素的调查,采用AB-QTL分析法共定位了63个影响7个农艺性状的QTL。在第2和11染色体上定位了2个高产QTL(来自东乡普野),它们分别使桂朝2号单株产量提高24.96%和22.54%。其中第2染色体上的高产QTL(qGY2-1)贡献率高达17%,是一个主效基因。此基因在其它研究中未见报道。 2.借助分子标记辅助选择,构建了159个以桂朝2号为遗传背景,东乡普野为供体的野栽渗入系。该套渗入系共覆盖东乡普野基因组的67.5%。其中有23个东乡普野单片段纯合渗入系和14个东乡普野单片段杂合渗入系。 3.根据159个渗入系的基因型数据,采用GGT软件绘出了渗入系的图示基因型,同时获得了两亲本纯合及杂合区段在渗入系基因组中的比例及长度。 4.以株高QTL-NIL为材料,通过渗入系作图分析,初步定位了两个影响株高的主效QTL,并对这两个QTL的表型效应作了分析。 5.以159个渗入系为研究对象,对东乡普野渗入片段大小、数目和染色体位置等因子分析结果表明,大多数东乡普野渗入片段的小于10cM,且多为单标记渗入;每个渗入系包含的纯合和杂合渗入片段数在0—8个和0—7个之间,平均为3.08个渗入片段;渗入片段在染色体上的位置也不是随机分布的,其中以染色体端部和近端部(定义为从末端向内15cM)为主,占总片段数的71%。 6.研究表明渗入片段数在不同的染色体间存在差异,其中以第1染色体最高,第7染色体最低,它们的渗入片段数分别为87和14个。此外,渗入片段在不同染色体上的覆盖率也存在差异,以第2染色体最高和第3染色体最低,覆盖率分别为91.3%和27.4%。
DOI:10.7666/d.y558395URL [本文引用: 1]
普通野生稻是栽培稻的祖先种,从野生稻中发掘栽培稻中已丢失或削弱了的优异基因已成为当前水稻育种和资源研究的热点之一。本研究选用世界上生境最北(28°14′N)、地下茎能够耐-12.8℃严冬,具有强耐旱性且米质优良的江西东乡普通野生稻(简称东乡普野,下同)为供体亲本,以曾多年在云南创亩产吨粮记录的优良品种“桂朝2号”为轮回亲本构建一套高代回交群体,用AB-QTL分析法对株高、单株产量等7个性状进行QTL定位。为了全面发掘东乡普野中优良基因,在桂朝2号遗传背景下构建一套渗入系并绘出图示基因型。同时对东乡普野渗入片段大小、数目和染色体分布等因子作了分析。主要结果归纳如下: 1.通过对BC_4F_1的基因型分析和BC_4F_2群体株高、单株产量及产量构成因素的调查,采用AB-QTL分析法共定位了63个影响7个农艺性状的QTL。在第2和11染色体上定位了2个高产QTL(来自东乡普野),它们分别使桂朝2号单株产量提高24.96%和22.54%。其中第2染色体上的高产QTL(qGY2-1)贡献率高达17%,是一个主效基因。此基因在其它研究中未见报道。 2.借助分子标记辅助选择,构建了159个以桂朝2号为遗传背景,东乡普野为供体的野栽渗入系。该套渗入系共覆盖东乡普野基因组的67.5%。其中有23个东乡普野单片段纯合渗入系和14个东乡普野单片段杂合渗入系。 3.根据159个渗入系的基因型数据,采用GGT软件绘出了渗入系的图示基因型,同时获得了两亲本纯合及杂合区段在渗入系基因组中的比例及长度。 4.以株高QTL-NIL为材料,通过渗入系作图分析,初步定位了两个影响株高的主效QTL,并对这两个QTL的表型效应作了分析。 5.以159个渗入系为研究对象,对东乡普野渗入片段大小、数目和染色体位置等因子分析结果表明,大多数东乡普野渗入片段的小于10cM,且多为单标记渗入;每个渗入系包含的纯合和杂合渗入片段数在0—8个和0—7个之间,平均为3.08个渗入片段;渗入片段在染色体上的位置也不是随机分布的,其中以染色体端部和近端部(定义为从末端向内15cM)为主,占总片段数的71%。 6.研究表明渗入片段数在不同的染色体间存在差异,其中以第1染色体最高,第7染色体最低,它们的渗入片段数分别为87和14个。此外,渗入片段在不同染色体上的覆盖率也存在差异,以第2染色体最高和第3染色体最低,覆盖率分别为91.3%和27.4%。
DOI:10.1093/bioinformatics/btp324URLPMID:19451168 [本文引用: 1]
The enormous amount of short reads generated by the new DNA sequencing technologies call for the development of fast and accurate read alignment programs. A first generation of hash table-based methods has been developed, including MAQ, which is accurate, feature rich and fast enough to align short reads from a single individual. However, MAQ does not support gapped alignment for single-end reads, which makes it unsuitable for alignment of longer reads where indels may occur frequently. The speed of MAQ is also a concern when the alignment is scaled up to the resequencing of hundreds of individuals. We implemented Burrows-Wheeler Alignment tool (BWA), a new read alignment package that is based on backward search with Burrows-Wheeler Transform (BWT), to efficiently align short sequencing reads against a large reference sequence such as the human genome, allowing mismatches and gaps. BWA supports both base space reads, e.g. from Illumina sequencing machines, and color space reads from AB SOLiD machines. Evaluations on both simulated and real data suggest that BWA is approximately 10-20x faster than MAQ, while achieving similar accuracy. In addition, BWA outputs alignment in the new standard SAM (Sequence Alignment/Map) format. Variant calling and other downstream analyses after the alignment can be achieved with the open source SAMtools software package. http://maq.sourceforge.net.
DOI:10.1046/j.1440-1665.1999.0178e.xURLPMID:19505943 [本文引用: 1]
The Sequence Alignment/Map (SAM) format is a generic alignment format for storing read alignments against reference sequences, supporting short and long reads (up to 128 Mbp) produced by different sequencing platforms. It is flexible in style, compact in size, efficient in random access and is the format in which alignments from the 1000 Genomes Project are released. SAMtools implements various utilities for post-processing alignments in the SAM format, such as indexing, variant caller and alignment viewer, and thus provides universal tools for processing read alignments. http://samtools.sourceforge.net.
DOI:10.1093/nar/gkq603URL [本文引用: 1]
DOI:10.1093/nar/8.19.4321URL [本文引用: 1]
DOI:10.1016/0003-2697(91)90120-IURLPMID:1716076 [本文引用: 1]
Abstract The photochemically derived silver stain of nucleic acids in polyacrylamide gels originally described by Merril et al. (1981, Science 211, 1437-1438) was modified to reduce unspecific background staining and increase sensitivity (down to 1 pg/mm2 band cross-section). Detection limits for double-stranded DNA fragments from HaeIII endonuclease digests of phage phi X174 were maintained despite eliminating oxidation pretreatment of fixed gels and reducing silver nitrate concentration. Preexposure to formaldehyde during silver impregnation enhanced sensitivity and the inclusion of the silver-complexing agent sodium thiosulphate in the image developer decreased background staining. Higher formaldehyde concentration during image development resulted in darker bands with good contrast. The procedure almost halves the number of steps, solutions and experimental time required and can be used for the staining of DNA fragments in polyacrylamide gels bound to a polyester backing film by controlling temperature during image development. We have applied this improved staining procedure for the routine analysis of complex DNA profiles generated by DNA amplification fingerprinting (DAF).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}