 ,1,2,3, 杨姗2,3, 崔玉军2,3, 王涛1, 滕越,2,31
,1,2,3, 杨姗2,3, 崔玉军2,3, 王涛1, 滕越,2,31 2
3
Research progress of bacterial minimal genome
Li Jinyu,1,2,3, Yang Shan2,3, Cui Yujun2,3, Wang Tao1, Teng Yue,2,31 2
3
通讯作者: 通讯作者: 滕越,博士,副研究员,研究方向:合成生物学。E-mail:yueteng@sklpb.org
第一联系人:
收稿日期:2020-11-23
| 基金资助: |
Received:2020-11-23
| Fund supported: |

摘要
具有最小基因组的细菌只包含维持自我生命复制所必需的基因,其作为一种潜在的工业生产平台具有诸多优势。由于高通量DNA测序和合成技术的发展,目前已经构建了多种缩减基因组的菌株。本文首先介绍了最小基因组的概念,其次总结了细菌必需基因的相关研究进展,然后梳理了人工缩减与合成微生物基因组的相关工作,最后探讨了在设计和组装基因组的过程中遇到的技术障碍和限制,以期为人工合成基因组的实验与应用提供理论参考。
关键词:
Abstract
Keywords:
PDF (1587KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李金玉, 杨姗, 崔玉军, 王涛, 滕越. 细菌最小基因组研究进展. 遗传[J], 2021, 43(2): 142-159 doi:10.16288/j.yczz.20-301
Li Jinyu.
人们很早就认识到支原体属(Mycoplasma)等物种的基因组比大多数细菌要小得多[1?~3]。虽然最初认为这些小基因组生物代表了生物进化的原始祖先,但到20世纪80年代,基于16S核糖体RNA序列的系统发育研究证实: Mycoplasma[4]以及其他具有小基因组的胞内细菌,如立克次氏体科(Rickettsiaceae)中的一些物种[5],皆来源于具有更大基因组的细菌。20世纪初,基因组测序结果证实了这些发现,并在不同的细菌类群中发现了独立的通过缩减基因组进化的案例,如柔膜菌纲中的Mycoplasma.genitalium[6]、α-变形菌纲中的普氏立克次体(Rickettsia prowazekii)[7]、γ-变形菌纲中的蚜虫内共生菌(Buchnera aphidicola)[8]和螺旋体纲中的伯氏疏螺旋体(Borrelia burgdorferi)[9]。这些微生物的基因组保留了满足其自身在宿主中生存的关键基因,证明不同细菌类群在特定情况下具有丢失多种基因的“能力”,这表明控制基因组缩减的机制和动力可能存在于多数细菌中。
随着全基因组测序技术的发展,通过对几个胞内共生细菌的基因组测序发现存在更小的基因组,这突破了早期测序达到的约500 kb (即500个基因左右)的下限;其中一些基因组还包括其他极端特征,如极快的基因进化、密码子重排和核苷酸组成的极端偏向性等[10,11]。这些变化的主要驱动因素包括较小的种群规模以及无性繁殖背景下的选择、突变和遗传漂变[11?~13]。这类生物不仅编码很少的蛋白质,而且其高度修饰的蛋白质很容易发生错误折叠,需要大量分子伴侣以保证蛋白质具有正常功能[10]。尽管与宿主相互作用的关键基因得以保留,但其基因组通过持续删除特定基因而不断被缩减。共生生物和宿主的共适应可能促进一些基因丢失,但与从线粒体和胞质体向细胞核的基因转移不同,细菌基因向宿主基因组的转移似乎与细菌基因组缩减无关。
20世纪初,伴随重组DNA技术的出现,合成生物学可以设计并构建基于合成遗传回路和通路的新型高潜力生物系统。在合成生物学的“设计-构建-测试-学习”循环中,系统生物学为其设计步骤提供了关于复杂生物过程的完整信息。因此,借助系统生物学和合成生物学,可最小化细菌基因组,使其仅包含细胞复制和产品生产所必需的基因,并以此设计和构建可预测、高效和精于生产的细胞(图1)。本文首先介绍了细菌最小基因组概念,其次总结了必需基因的研究进展,然后回顾了在构建人工缩减与合成基因组方面的研究工作,最后探讨了在设计和构建缩减基因组的过程中遇到的技术障碍和限制,以期为人工合成基因组的实验与应用提供理论参考。
1 最小基因组
1.1 最小基因组的概念
最小基因组被定义为在无外界压力(营养丰富和无应激)条件下足以维持生命活动的基因集。许多实验研究进一步将其定义为在丰富培养基中支持纯种培养的基因集。因为大多数生物在大自然的生态环境中需要额外的基因,所需的基因集会随着环境条件的变化而有所不同。最小基因组概念出现和发展的时间与细菌全基因组测序时代相同,都始于20世纪90年代中期[14]。生物的基因组大小从几十万个碱基对到一千多亿个碱基对不等。其中,布赫纳氏菌(Buchnera spp.)是一种胞内共生菌,与大肠杆菌(Escherichia coli)具有共同祖先;与其祖先相比,基因组缩减达75%,仅为250 kb[15,16],是自然界中基因组大规模缩减的一个明显示例。最小基因组不仅存在于布赫纳氏菌,也存在许多其他共生细菌中。Nasuia deltocephalinicola是一种昆虫共生细菌,具有最小的自我复制基因组(112 kb)[17]。此外,本课题组还聚焦于最近发现的为食液昆虫提供营养的共生细菌中的极端微小基因组,包括来自4个远缘细菌类群的5个独立的极端基因组缩减实例,其基因组大小都不到生殖支原体基因组的一半(甚至不到1/4) (表1)。这些例子显示出源于生态位适应的基因组进化趋势。共生生物不需要环境条件响应相关的基因,因为其宿主提供了稳定的营养供应,并保护其免受恶劣环境变化的影响,因此,在长期进化过程中,这些不必要的基因已经从它们的基因组中移除。许多研究表明,物种种群规模较小、无性繁殖[18?~20]以及细菌所固有的基因删除偏倚[19,21],是基因组缩减的主要原因。由于特定宿主的限制和不同宿主中的菌株之间缺乏重组,导致了高度遗传漂变,使有益但非必需的基因失活和缺失。这种菌群结构的另一个影响是最初发现于B. aphidicola和M. genitalium中的微小基因组基因序列快速进化,及其对蛋白质二级结构稳定性的影响[22,23]。基因组减少的早期阶段以一些较新进化的共生生物为代表,其特征是移动元件的增加,假基因的形成,多个基因组重排和染色体片段的缺失(图2)[24?~26]。然而,在更早期进化的共生体中,如B. aphidicola,移动元件和大多数假基因已被移除。
图1
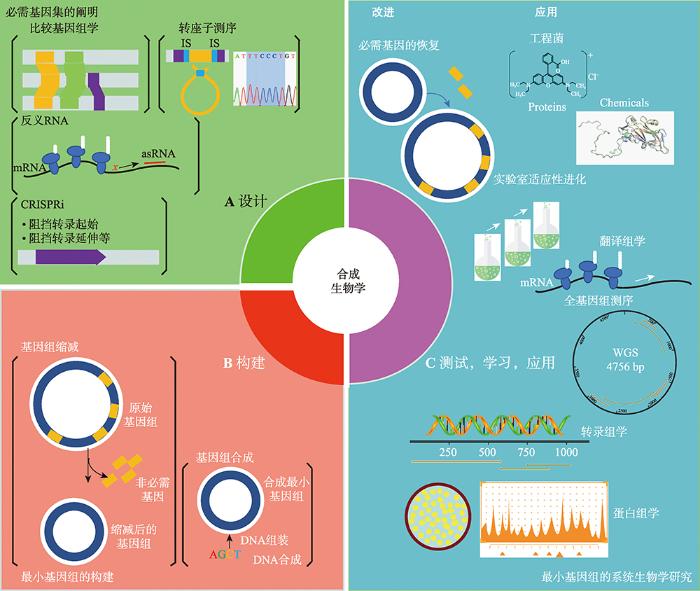 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1设计、构建和测试最小基因组的研究
以合成生物学为基础的最小基因组的构建,包括设计、构建、测试、学习、应用的循环周期。A:通过不同方法确定物种的必需基因集,包括比较基因组学、转座子测序、反义RNA、CRISPRi等。B:构建最小基因组的过程,主要遵循自上而下,自下而上两种原则。C:对构建的最小基因组进行系统生物学研究,以测序技术、组学技术等为手段进行测试、学习、应用。
Fig. 1Design, build and test the minimal genome research
通过对不同生物基因库的计算分析[14,27????~32],以及纯种培养生物的基因诱变破坏实验[33??~36],可对最小基因组的基因完整性进行预测。这种诱变实验只限于可纯种培养的生物,如M. genitalium,它的基因组是迄今为止纯种培养生物中最小的。研究表明,通用基因集很小,且不包括许多已被实验确定为特定生物所必需的基因。因此,不同生物可能以完全不同的方式完成基本生命活动。例如,通过运输而不是从头合成获得所需的化合物,或者使用不相关的基因和不同的途径使特定tRNA装载正确的氨基酸[37]。某些通用或接近通用的基因缺失并不致死[32]。不同的研究预测了不同的最小基因集,虽然这些基因的分布较广泛,但都包括参与细胞基本功能的基因。有些新陈代谢相关基因分布较广泛,在自然环境下对特定生物至关重要,但并非必不可少。
Table 1
表1
表1具有代表性的细菌、共生生物、病毒和细胞器基因组的比较
Table 1
| 微生物 | 分类a | 基因组 大小(bp) | GC含 量(%) | 蛋白质 编码 序列数 | 位于COGs M簇和I簇 的基因数目b | 细胞形态 | 杆状决定基因 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ftsZ | ispA | mreB | rodA | |||||||
| 游离细菌 | ||||||||||
| Escherichia coli | γ-变形菌纲 | 4,639,675 | 50.8 | 4145 | 323 | 杆状[46] | + | + | + | + |
| Bacillus subtilis | 柔膜菌纲 | 4,215,606 | 43.5 | 4176 | 307 | 杆状[46] | + | + | + | + |
| Rickettsia prowazekii | α-变形菌纲 | 1,111,523 | 29.0 | 835 | 100 | 杆状[47] | + | - | + | + |
| Mycobacterium genitalium | Mollicutes | 580,076 | 31.7 | 475 | 17 | '烧瓶形' [48] | + | - | - | - |
| 缩减基因组共生生物 | ||||||||||
| Candidatus Blochmannia floridanus | γ-变形菌纲 | 705,557 | 27.4 | 583 | 77 | 杆状[49] | + | - | + | + |
| Wigglesworthia glossinidia | γ-变形菌纲 | 697,724 | 22.5 | 611 | 85 | 杆状[50] | + | + | + | + |
| Candidatus Baumannia cicadellinicola | γ-变形菌纲 | 686,194 | 33.2 | 595 | 62 | 球状[51] | + | + | + | + |
| Buchnera aphidicola str. APS | γ-变形菌纲 | 640,681 | 26.3 | 564 | 41 | 球状[52] | + | + | - | - |
| Candidatus Moranella endobia' | γ-变形菌纲 | 538,294 | 43.5 | 406 | 36 | 球状[53] | + | - | + | + |
| Buchnera aphidicola str. Cc | γ-变形菌纲 | 416,380 | 20.2 | 357 | 10 | 球状[54] | + | - | - | - |
| 极端缩减基因组共生生物 | ||||||||||
| Candidatus Sulcia muelleri str. GWSS | 黄杆菌纲 | 245,530 | 22.4 | 227 | 5 | 多形性管状[55] | - | - | - | - |
| Candidatus Zinderia insecticola | β-变形菌纲 | 208,564 | 13.5 | 202 | 1 | 多形性斑点[56] | - | - | - | - |
| Candidatus Carsonella ruddii | γ-变形菌纲 | 159,662 | 16.6 | 182 | 1 | 多形性管状[57] | - | - | - | - |
| Candidatus Hodgkinia cicadicola | α-变形菌纲 | 143,795 | 58.4 | 169 | 0 | 多形性管状[58] | - | - | - | - |
| Candidatus Tremblaya princeps | β-变形菌纲 | 138,927 | 58.8 | 121 | 0 | 多形性斑点[53] | - | - | - | - |
| 具有较大基因组的细胞器 | ||||||||||
| Cucurbita pepo | 线粒体 | 982,833 | 42.8 | 38 | NA | NA | - | - | - | - |
| Floydiella terrestris | 叶绿体 | 521,168 | 34.5 | 74 | NA | NA | - | - | - | - |
| Porphyra purpurea | 叶绿体 | 191,028 | 33.0 | 209 | NA | NA | - | - | - | - |
| Reclinomonas americana | 线粒体 | 69,034 | 26.1 | 67 | NA | NA | - | - | - | - |
| 巨大病毒 | ||||||||||
| Acanthamoeba polyphaga mimivirus | 拟菌病毒科 | 1,181,404 | 28.0 | 1262 | NA | NA | NA | NA | NA | NA |
| Cafeteria roenbergensis virus (CroV) | 拟菌病毒科 | 617,453 | 23.4 | 544 | NA | NA | NA | NA | NA | NA |
| Coccolithovirus | 藻类DNA 病毒科 | 407,339 | 40.2 | 472 | NA | NA | NA | NA | NA | NA |
新窗口打开|下载CSV
细菌基因组含有数百万个碱基对。例如,研究最广泛的模式生物大肠杆菌的基因组>5 Mb,包含4000多个基因,其中1000多个为未知功能的基因[38,39]。大肠杆菌可在多种环境下繁殖,如好氧和厌氧,以及不同营养物质、pH值和温度等。大肠杆菌的基因组中有许多基因负责处理环境压力和利用各种营养物质;然而,在环境确定的实验室条件下,不再需要某些应激反应基因;因此许多基因可以被删除,而不会对细胞生长产生负面影响[40]。同时,大肠杆菌基因组还编码了许多实验室培养和工业发酵不需要的基因,这些基因会导致能量和生物质前体的浪费,不必要的基因组片段的复制,以及功能冗余或无用的转录物、蛋白质和代谢物的合成。因此,删除这些不必要基因的生物体或可成为人工条件下产品生产的全新平台[41]。
图2
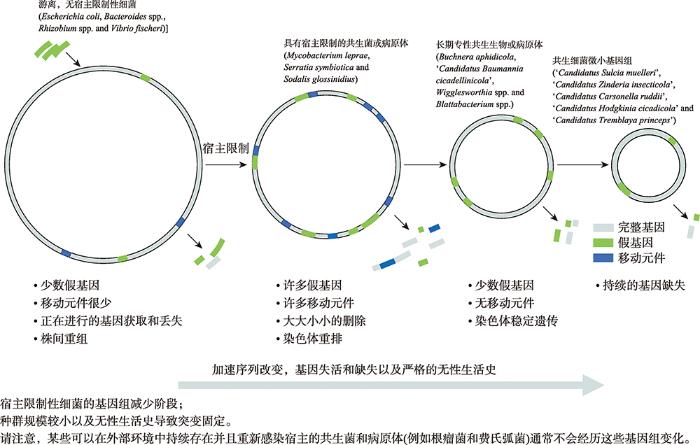 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2细菌基因组的减少阶段及其进化特征
Fig. 2Genome reduction stage of bacteria, and its genome evolution characteristics
1.2 线粒体基因组简化
线粒体比较基因组学使人们了解线粒体祖先的基因组是什么样的,以及它包含的基因是什么。从它所包含的基因组的角度看,线粒体具有毫无疑问的细菌起源,起源于α-变形杆菌(Alpha proteobacteria)[42]。因此,内共生假说认为真核细胞中的线粒体从细菌祖细胞通过共生进化而来。基于系统发育的分析表明,线粒体基因组进化的特征是某些谱系大量扩增,另一些谱系则极度减少和压缩,通过内共生基因转移将线粒体基因组中的许多初始遗传信息重新定位到细胞核[43]。α-变形杆菌基因组之间的比较表明,线粒体的细菌祖先包含约3000~5000个基因,线粒体基因组中直系同源基因的祖先簇的上限约为1700个,这些估计表明,从细菌共生体到原始细胞器的过渡过程中丢失了大约3000个基因[44]。可以预见,基因组学和蛋白质组学数据的不断增长将提供关于线粒体结构和功能的新的数据和见识,并将不断重塑和完善目前的线粒体进化理论。1.3 构建和设计最小基因组
随着基因组学革命以及系统生物学的不断进步,合成生物学在过去10年中飞速发展,尤其是在基于系统和合成生物学的微生物底盘工程和改造领域。在系统生物学中,理想的底盘可代指具有简化的基因组且可以实现全部功能的生物,以及能够更有效地合成所需产物的代谢网络;在合成生物学中,底盘是指通过提供允许其运转的资源来容纳和支持遗传成分的生物。构建合成微生物底盘包括两种方法:自上向下和自下向上。自上而下的方法是通过去除不必要的细胞基因来了解基因组架构并改善其特性的减少基因组大小的策略。基于大规模DNA分析的出现,通过对来自不同生物体的基因组进行比较分析,通常可以揭示对于细胞生命和相似和/或截然不同的代谢途径必不可少的基因。接下来,再通过不同的实验方法来实现缺失。工程设计和修改合成微生物底盘揭示生命的基本原理,是加强其在卫生医药和食品工业中的应用的最佳方法之一。有关这个主题的研究计划已在多个国家获得资助。“最小基因组工厂”(minimum genome factory, MGF)项目于2001年在日本启动,其目标是构建具有较小基因组的微生物以用于工业用途。基于比较大肠杆菌和布赫纳氏菌获得的非必需基因信息,Hiroshi等[45]通过删除大肠杆菌W3110中的大肠杆菌特异性基因构建了大肠杆菌MGF-01,其构建方法与其他缺失方法一致,即连续缺失结合P1转导。简言之,用含有sacB和cat基因的DNA组件同源替换删除靶DNA序列。然后,用另一个DNA组件替换选择标记,用蔗糖进行负筛选获得无标记克隆。最后,在28个P1转导循环中积累了53个缺失,从而使总缺失长度达1.03 Mb。MGF-01的最终生物量是野生型亲本的1.5倍。据称,MGF-01相比亲本菌株葡萄糖利用效率更高,这是因为显著减少了过量的醋酸盐积累(从大肠杆菌W3110中的1.37 g/L降至MGF-01中的0.50 g/L)。此外,与野生型相比,MGF-01的苏氨酸浓度和产量分别提高了2.44倍和1.69倍,醋酸盐副产物则显著降低。MGF-01的这些意想不到的有益特性与其他具有与野生型祖先相似表型的缩减基因组大肠杆菌不同,但需要进一步研究MGF-01的差异性。
2 细菌必需基因
构建最小基因组,首先要阐明维持生命所必需的基因。20世纪90年代,只测得少数微小细菌的全基因组序列。Mushegian和Koonin等[59]对生殖支原体和流感嗜血杆菌(Haemophilus influenza)进行比较,表明这两种细菌都有相对较小的基因组,但表现出完全不同的进化轨迹。根据比较基因组学原则,在多种生物中保守的基因很可能有必需的功能。在生殖支原体和流感嗜血杆菌中,共发现240个直系同源基因;但此分析遗漏了几个编码细胞必需功能的基因,如磷酸甘油酸变位酶和核苷二磷酸激酶基因。这两个物种有不同且不相关的磷酸甘油酸变位酶,因此非同源基因偶尔可以取代古老的基因并破坏其保守性。最终,研究结果证明包括非直系同源基因取代在内,预测共有262个基因构成了两个物种的核心生物功能。尽管这两种细菌和它们的共同祖先之间经历了15亿年的进化,仍有约50%的基因被保存下来。自2000年以来,已获得数万基因组序列。科学家比较了数百个基因组,以确定物种间普遍存在的基因,然而这样的基因并不常见。Brown等[61]比较了45个基因组,发现只有23个保守基因[60];Koonin等[62]报道了普遍存在于100个基因组的63个基因;Charlebois等[63]比较了14个门中的147个原核生物基因组,仅发现34个通用基因。虽然报告的保守基因数目不同,但都这些研究同时证明只有少数保守基因绝对不足以维持生命。因此,比较基因组学对必需基因的理论预测仅限于功能未知、非同源取代和具有数十亿年进化历史的基因。
除了计算预测,研究者还使用实验方法来探究必需基因。最简单的方法是从基因组中删除一个特定的基因位点,观察其致死效应。在迄今为止已知的任何自由生活的生物体中,生殖支原体具有最小的基因组(580 kb,编码517个基因),其中许多基因可能通过转座子作用而失活[64]。在枯草芽孢杆菌(Bacillus subtilis)中,通过对其基因组的79个区域进行随机突变[65],发现只有6个位点是必不可少的,大小约318~526 kb,与生殖支原体基因组大小相似。因此,人们设计了各种方法来确定细菌中的必需基因。如通过重组、转座子插入和反义RNA (asRNA)直接使单个基因失活。通过插入非复制质粒失活枯草芽孢杆菌中的单个基因,Kobayashi等[66]发现4101个基因中只有271个是必需基因。在大肠杆菌中,4288个开放阅读框(open reading frame, ORF)中的3985个可被敲除,表明剩余的303个ORFs对生命至关重要[67]。尽管靶向基因敲除研究提供了基因必需性的直接证据,但该方法耗时长,需要进行数千次删除实验。
为了克服这些限制,可采用基于转座子诱变失活的高通量方法。转座子是一种基因元件,可在基因组内随机移动并插入,破坏基因的正常功能。如果基因组中的必需基因被插入转座子,则此突变体不能存活。利用此特征,可以通过鉴定存活突变体中的转座子插入位点来区分必需和非必需基因。Glass等[68]和Hutchison等[69]分别利用1300和3000个突变体,在生殖支原体中构建了全基因组转座子插入图谱,发现265~382个必需编码序列(coding sequences, CDSs)。由3000个唯一插入位点组成的转座子插入图谱,其平均分辨率约为每200 bp一个插入位点。此分辨率无法检测功能性RNAs (如tRNAs和ncRNAs)等小遗传元件的必需性。此外,一些必需基因在3?端对转座子插入检测具有抗性,因为短截尾或延伸并不影响其功能。为了规避这一限制,研究者发明了一种通过asRNAs使基因失活的方法[70]。与转座子不可逆的基因破坏不同,利用asRNAs可以特异性敲低某个基因。因此,一旦构建出asRNA文库,研究者可以在多种环境条件和迭代过程中评估基因必需性或适合性,而无需重复构建敲除菌株或转座子突变体。
转座子插入位点的鉴定依赖于单个克隆的分离和现有的高通量测序技术的发展;通过与高通量测序技术相结合,可以并行识别多个插入位点。因此,转座子诱变与下一代测序技术(Tn-Seq)相结合,即利用转座子诱变以更高的分辨率检测必需基因。对2×105大肠杆菌转座子突变体文库的统计分析,共鉴定出620个必需基因[71]。Tn-Seq和单个基因敲除研究之间的差异可能源于细胞增殖。一个重要基因,即使不是严格意义上的必需基因,其失活也会导致严重的生长缺陷;在细胞繁殖过程中,这种缺陷可能未被充分表达或逐渐消失。此外,不同的统计临界值和实验条件也可能导致这种差异。
最后,基于成簇规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)引领的技术革命,催化失活Cas9 (dead Cas9, dCas9)可以在转录水平抑制靶基因的表达[72]。由于CRISPR系统的特异性只取决于嵌合单导向RNA (sgRNA)中20 nt的原间隔序列,因此易于通过DNA合成构建大规模全基因组sgRNA文库。利用全基因组CRISPR干扰(CRISPR interference, CRISPRi),通过约59 000个sgRNAs抑制所有基因,在大肠杆菌中鉴定出379个必需基因[73]。利用CRISPRi技术估计的必需基因数量略大于其他方法估计的数量。在细菌中,许多基因是多顺反子转录的,故前导多顺反子的破坏会使操纵子中包含的下游基因失活。因此,即使前导基因是非必需的,其转录抑制也会导致下游必需基因的致死效应,即多顺反子结构导致必需基因的高估。利用目前的高通量DNA合成技术,可以高效合成庞大的sgRNA文库,并将其用于鉴定具有更大基因组的生物(如人类)的必需基因[74,75]。
总之,人们已经采用多种方法来阐明各种生物体中的必需基因。尽管不同方法得到的必需基因的确切数目不同,但500个基因被认为足以维持生命。利用单基因敲除研究、Tn-Seq和CRISPRi直接检查单个基因是否必需无法规避各种技术固有的局限性。因为这些方法依赖于单个基因的移除或失活,而同时失活两个以上的基因从未被测试过。例如,假设两个基因在大肠杆菌中具有相同的基本功能,这两个基因可以单独删除,因为另一个基因可以恢复其功能;但是,两个基因同时失活则产生致死效应。即使在最简单的细菌生殖支原体中,构建双基因敲除库也需要25万个菌株。考虑到构建约4 000个大肠杆菌单基因敲除菌株所需的巨大时间和费用成本[40],需要采用新的技术来探索基因高阶组合的必需性。
3 人工缩减基因组微生物
大肠杆菌是研究最广泛的模式生物,已为其构建了多种缩减基因组,缺失大小从300 kb到1.38 Mb不等(表2)。大肠杆菌K-12菌株的基因组约为4.64 Mb,缺失大小约为原始基因组的6.8%~29.7%。下面简介了一些目前构建的缩减基因组大肠杆菌,还回顾了其他物种中基因组缩减案例。3.1 大肠杆菌CDΔ3456
2002年有两例关于缩减基因组大肠杆菌的报道。其中之一是Yu等[76]报道的CDΔ3456,缺失片段大小超过300 kb。该菌株构建过程中,位于Cre位点特异性重组酶识别的两个loxP位点之间的一个较大的基因组区域被删除。使用转座子将loxP位点预先插入到基因组中的任意位置。具体方法是,用两种抗生素抗性基因盒构建两个转座子突变库,并定位所有转座子插入位点。筛选转座子分别定位在目标缺失区域一端的两个突变体,其中包含两个不同的标记基因,并通过P1转导将其基因组融合。使用两种不同的抗生素抗性基因标记来筛选成功转导的细胞。然后,融合基因组中的两个loxP位点通过Cre重组酶重组,产生较大的靶向缺失。由此构建了6个缺失菌株,并进一步利用P1转导将多个缺失累积到一个基因组中。在此过程中,某些基因组区域可以单独从基因组中删除,但是特定的基因组合不能同时删除,这种关系被称为合成致死,其原因可能是存在基因重复或直系同源物[77]。当两个基因的一个拷贝被删除时,另一个拷贝可以维持生物体正常功能,而双突变体则不能。为了避免合成致死组合,4个区域被整合在CDΔ3456中,该菌株缺少287个ORFs,其中包含179个未知基因,以及与组氨酸生物合成、菌毛和数个转运蛋白相关的基因,最终获得克隆的生长速率与亲本大肠杆菌相当。Table 2
表2
表2缩减基因组大肠杆菌及其特征
Table 2
| 原始菌株 | 缩减基因组菌株名称 | 原始基因组大小(Mb) | 缩减量(%) | 特征 |
|---|---|---|---|---|
| Escherichia coli str. K-12 substr. MG1655 | CDΔ3456 | 4.64 | 313 kb (6.8) | 生长速度正常 |
| Escherichia coli str. K-12 substr. MG1655 | MDS12 | 4.64 | 376 kb (8.1) | 生长速度正常,细菌生长密度提高10% |
| Escherichia coli str. K-12 substr. MG1655 | MDS42 | 4.64 | 708 kb (15.3) | 生长速度正常,更高的转化效率 |
| Escherichia coli str. K-12 substr. MG1655 | MS56 | 4.64 | 1068 kb (23.0) | 生长速度正常,遗传稳定性提高 |
| Escherichia coli str. K-12 substr. MG1655 | Δ16 | 4.64 | 1377 kb (29.7) | 较低的生长速度,异常的拟核结构 |
| Escherichia coli str. K-12 substr. W3110 | MGF-01 | 4.65 | 1030 kb (22.2) | 细菌生长密度提高50%,更高的苏氨酸产量 |
| Bacillus subtilis str. 168 | Δ6 | 4.22 | 320 kb (7.7) | 生长速度正常 |
| Bacillus subtilis str. 168 | PG10 | 4.22 | 1456 kb (34.5) | 较低的生长速度 |
| Bacillus subtilis str. 168 | PG38 | 4.22 | 1535 kb (36.4) | 较低的生长速度 |
| Bacillus subtilis str. 168 | MGB469 | 4.22 | 469 kb (11.1) | 生长速度正常 |
| Bacillus subtilis str. 168 | MG1M | 4.22 | 991 kb (23.5) | 生长速度正常 |
| Bacillus subtilis str. 168 | MBG874 | 4.22 | 874 kb (20.7) | 较低的生长速度,更高的蛋白产量 |
| Streptomyces avermitilis | SUKA17 | 9.03 | 1674 kb (18.5) | 更高的抗生素产量 |
| Schizosaccharomyces pombe | A8 | 12.6 | 657 kb (5.2) | 更高的蛋白产量 |
新窗口打开|下载CSV
3.2 大肠杆菌MS56及其近缘菌株
2002年报道了缩减基因组大肠杆菌MDS12,其缺少大肠杆菌K-12菌株的12个K-岛[78]。K-岛是K-12通过水平基因转移获得的基因组区域。K-岛含有非必需基因,如原噬菌体和转座子。利用I-SceI巨核酶和双链断裂修复系统,用无痕删除法将12个K-岛依次删除。具体方法是,将含有氯霉素抗性基因的DNA组件引入大肠杆菌MG1655,以取代同源靶区。虽然靶标被删除,但下一轮删除需要去除抗性基因,因此采用I-SceI将该基因删除。之后,双链断裂被RecA修复,形成无痕缺失株。经过12次迭代删除和P1转导,删除长度合计为376 kb,包含409个ORFs。由于被删除的基因没有必需功能,MDS12的生长速度和DNA转化效率与原始菌株无差异。MDS12的最终细胞密度比野生型大肠杆菌高约10%。在MDS12中,节省的能量和物质可以转化为生物量,显示出缩减基因组的优势。
4年后,Pósfai等[79]报告了大肠杆菌MDS41、42和43,它们是MDS12的后代,含有额外的缺失(与野生型相比,总缺失为663~708 kb)。MDS42不含任何可转位和插入序列(insertion sequence, IS)元件。据报道,在大肠杆菌中,20%~25%的突变与IS元件有关[79]。MDS42未显示IS介导的基因失活。利用该特性,MDS42可稳定复制携带毒性ORFs的质粒DNA,且基因组构成稳定。大肠杆菌有一个沉默的bgl操作子,能够利用水杨苷;因此,通常条件下,大肠杆菌不利用水杨苷作为其唯一碳源。当使用水杨苷作为唯一碳源培养时,MDS41的bgl操纵子激活率低于MG1655 (8%)。此外,MDS42产生的苏氨酸比野生型大肠杆菌多80%以上[80]。
成功构建MDS菌株之后,Park等[81]在MDS42中又引入了14个缺失,以此构建大肠杆菌MS56。除MDS42的缺失外,还删除了非必需基因和遗传元件,如氢化酶、菌毛样黏附素和厌氧呼吸酶,基因组缩减总量达1.07 Mb。编码抑制大肠杆菌生长的人蛋白异源基因可在MS56中成功表达,不会有任何IS介导的失活,而正常含IS的大肠杆菌中会迅速发生失活。
3.3 大肠杆菌Δ16
Hashimoto等[82]构建了大肠杆菌Δ16,其基因组缩减量为1.377 Mb (占其原始基因组的29.7%),是报道的最大缩减量。与其他缩减基因组一样,Δ16的构建方法也是同源重组连续缺失结合P1转导。Hashimoto等[82]采用正筛选和负筛选两种方法进行基因删除。首先,构建靶区序列末端同源DNA组件。该盒还含有三个基因:氯霉素乙酰转移酶(cat)、rpsL和sacB。将此DNA组件导入后,用氯霉素选择目标基因组区域被此组件取代的克隆。然后,用新的同源DNA组件取代该序列,接着用rpsL和sacB基因进行负筛选,使细胞对链霉素和蔗糖敏感。通过P1转导将16个无痕缺失进行组合,获得最终菌株Δ16,累计缺失1.377 Mb。在连续缺失过程中,倍增时间从MG1655的26.2 min依次增加到Δ16的45.4 min,细胞形状变长变宽,拟核呈不规则分布,其复制与细胞分裂不同步。3.4 枯草芽孢杆菌PG38及其近缘菌株
枯草芽孢杆菌是研究最广泛的革兰氏阳性菌之一,因其具有蛋白分泌系统而成为各种蛋白的优良生产宿主。Westers等[83]通过去除6个基因组位点,包括聚酮、蛋白质抗生素生物合成、原噬菌体和原噬菌体样元件相关基因,构建了基因组缩减枯草芽孢杆菌Δ6。这些位点共包含332个基因(320 kb)。通过使用整合质粒pG + host4整合并切除选择标记实现基因删除[84]。基因组缩减枯草芽孢杆菌Δ6在细胞生理上没有明显变化。与亲本菌株相比,该菌株的生长速度、葡萄糖/醋酸盐代谢通量、异源蛋白分泌和生物量均相同。出乎意料的是,尽管没有删除与细胞运动相关的基因,Δ6在琼脂糖平板上显示出细胞运动性增加。基因组缩减菌株偶尔会出现此类意外表型,说明对即使是微小细菌的基因组理解也是有限的[85]。枯草芽孢杆菌Δ6基因组被进一步缩减,构建形成PG10和PG38。Reu等[86]构建了两个独立的基因组缩减菌株PG10和PG38,分别包含88和94个迭代缺失,缺失部分包含产孢、运动、抗生素合成和次生代谢相关的非必需基因。在连续缺失过程中,中间菌株逐渐丧失DNA整合所需的感受态。因此,在基因组中引入了额外的感受态蛋白(ComK和ComS),以有效导入缺失所需的DNA。两种衍生菌株的生长速度较慢(倍增时间延长约40%),细胞形态呈长丝状。虽然这两个菌株的生长速度都有所降低,与之前认为细胞存活所需的基因组相比,它们的基因组缩减比例是迄今为止最大的(1.46和1.54 Mb;超过了它们原始基因组的1/3)。
3.5 枯草芽孢杆菌MGB874及其近缘菌株
Ara等[87]基于枯草芽孢杆菌168构建了一株缩减基因组枯草芽孢杆菌MGB469。缩减菌株缺少9个与原噬菌体和原噬菌体样元件相关的基因组位点和2个抗生素合成基因(巴斯他汀和聚酮)。首先从枯草芽孢杆菌的基因组中逐一删除这些位点,以确认其不包含必需基因。然后,依次合并删除,使删除区域达469 kb,由此得到菌株MGB469,其生长速度和蛋白质产率与原始菌株无差别,从而达到进一步缩减基因组的目的。该文作者发现一个可以增加蛋白质产率的基因组缩减位点,最终构建出比原始菌株具有更高生产率的基因组缩减菌株。从MGB469中进一步删除单独缺失时增加纤维素酶产率的6个基因组位点,构建了总基因组缩减991 kb的菌株MG1M。然而,MG1M中纤维素酶和蛋白酶的生产没有发生明显的变化。由于枯草芽孢杆菌MGB469和MG1M的表型与野生型相当,并且在进一步基因组缩减后没有产生有益表型,Morimoto等[88]重新利用中间菌株MGB469构建具有优势特征的新型基因组缩减枯草芽孢杆菌。该文作者测试了包括原噬菌体和次级代谢基因在内的74个基因组区域的缺失。在63个可发生缺失的区域中,11个连续缺失被引入MGB469,从而得到缺失长度为874 kb的枯草芽孢杆菌MGB874。虽然MGB874具有正常的细胞形态和拟核结构,但其生长速度降低至野生型枯草芽孢杆菌的70%。与枯草芽孢杆菌的其他缩减基因组不同,MGB874产生的纤维素酶和蛋白酶比枯草芽孢杆菌168分别提高1.7倍和2.5倍。转录组研究发现了许多转录组水平的基因表达变化,如产孢、降解酶分泌和σ因子,推测是其生产率提高的原因。MGB874证明具有缩减基因组的菌株可作为工业蛋白生产的优良宿主。
3.6 阿维链霉菌(Streptomyces avermitilis) SUKA17
放线菌门中的链霉菌属是一类工业和临床应用中重要的放线菌,已知其可产生抗生素等大多数具有生物活性的次级代谢产物。链霉菌属基因组在细菌中相对较大,其染色体DNA呈线性而非环状。基于应用最广的工业菌种之一阿维链霉菌,Komatsu等[89,90]构建了菌株SUKA17。阿维链霉菌中含有20多个次级代谢物生物合成基因簇,主要位于基因组的末端,称为亚端粒区。对灰色链霉菌(Streptomyces griseus)、天蓝色链霉菌(Streptomyces coelicolor)和阿维链霉菌进行比较基因组学研究,揭示了位于基因组中心的必需核心基因。使用同源重组和Cre介导的重组,从阿维链霉菌中删除亚端粒区主要次级代谢相关基因(包括萜烯代谢)。与大肠杆菌和枯草芽孢杆菌不同,阿维链霉菌不易发生发重组。因此,两个loxP位点首先通过同源重组从环状DNA模板导入到基因组中。然后,通过Cre介导重组删除基因组中大片段目标位点。该缺失包括1272个ORFs的基因组区域,总长度为1.67 Mb。由于其基因组较简单,无需承担不必要的次生代谢物合成,含有异源基因簇的SUKA17比原生灰色链球菌和棒状链霉菌(Streptomyces clavuligerus)产生更多的链霉素和头霉素C。4 合成基因组学
4.1 基于合成基因组学的微生物设计与改造
上面讨论的缩减基因组都是使用自上而下的方式对基因组进行缩减而构建的。现今,研究者已实现从头合成病毒、噬菌体和细菌基因组。借助基因组编辑和合成工具,可以进行全基因组水平上进行工程设计。研究者已实现设计并构建基于合成遗传回路和通路的新型高潜力生物系统。借助系统生物学和合成生物学而发展的合成基因组学,可最小化细菌基因组,从头开始合成基因组可以对基因组结构和功能进行前所未有的修饰,获得对生命基本原理的新见解并致力于设计有价值的应用,例如设计并构建可预测的、高效的或是精于生产的细胞(图3)。自1995年关于生殖支原体基因组大小为525个基因的第一份报道以来,该细菌和该属的其他成员已成为探索赋予生命最少数量的细胞成分的最有用的试验台。事实上,到目前为止,某些支原体菌株已成为我们最了解的生物系统,其所有组成部分及其相互作用的各个方面都经过了非常详细的探索和量化。里程碑式的工作包括通过转座子诱变检查每个基因的必要性,实现支原体基因组的完全化学合成等。
支原体是小基因组细菌,其将成为探索生命所需的最低限度的基因组大小的理想平台。Gibson等[91]构建的支原体JCVI-syn1.0含有一个完全化学合成的基因组,即略加修饰的1.08 Mb蕈状支原体(Mycoplasma mycoides)基因组的复制体。通过转座子诱变阐明蕈状支原体的必需基因后,合成了最小基因组蕈状支原体JCVI-syn3.0[92] (包含473个基因,总长度为531 kb)。进一步的实验验证表明,基于分子生物学的系统知识以及有限的转座子诱变数据相结合的初始设计未能产生活细胞。随后,改良的转座子诱变方法揭示了稳健生长所需的一类准必需基因,这解释了其最初设计的失败。最后,研究者通过设计,合成和测试的3个循环(保留了基本必需的基因)产生了JCVI-syn3.0,相对于其亲本菌株而言,基因组的净基因组大小减少了约50% (从约1079 kb减少到531 kb),并将基本生物学信息精简到了473个基因,其基因组比自然界中任何自主复制的细胞都要小。虽然JCVI-syn3.0的倍增时间是JCVI-syn1.0的3倍(~180 min),但远小于具有天然最小基因组的生殖支原体(~16 h)。尽管全基因组合成需要漫长的时间和巨大的费用[93],但无疑是基因组缩减的一种极有吸引力的替代方法。
图3
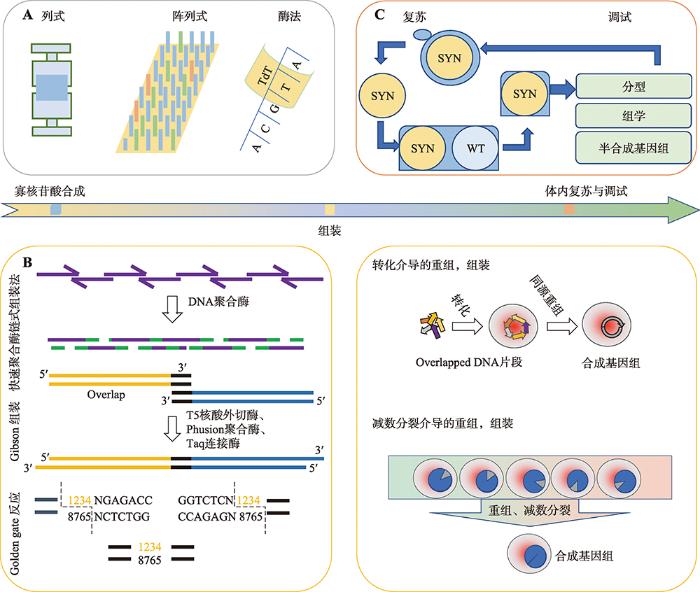 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3合成基因组的过程:从设计序列到体外组装到体内复苏
A:在体外进行寡核苷酸的合成。常用方法包括列式合成、阵列式合成、酶法合成等。B:通过PCA、Gibson组装等进行体外组装,或是导入细胞内进行由转化以及减数分裂介导的重组、组装。C:最后进行分型、调试。
Fig. 3The process of synthesizing genome research: from design sequence to in vitro assembly to in vivo recovery
JCVI-syn3.0是一个多功能平台,可用于研究生活的核心功能并探索全基因组设计。最小细胞的概念乍看之下似乎很简单,但基于实验的实践验证却变得更加复杂。除了必需和非必需基因外,还有许多准必需基因,它们对生存力不是绝对关键,但对于强劲的生长却是必需的。因此,在最小化基因组的过程中,需要在基因组大小和生长速率之间进行权衡。JCVI-syn3.0近似为最小细胞基因组,为最小基因组与实验生物可行生长速率间的折中,它保留了几乎所有与大分子合成和加工有关的基因。出乎意料的是,它还包含149个生物学功能未知的基因,表明存在着生命必需的未发现功能。
4.2 大肠杆菌基因组密码子的简化
大自然使用64个密码子编码来自基因组的蛋白质合成,并从多达6个同义词中选择1个有义密码子编码每个氨基酸。同义密码子选择具有多种重要作用,且许多同义替换是有害的。合成基因组不仅模仿模板DNA,而且允许遗传密码重编程。 Lajoie等[94]将80个大肠杆菌菌株中42个高度表达的必需基因中13个稀有密码子的所有实例与编码相同氨基酸的密码子交换,显示了在活细胞中以全基因组规模进行编码的可行性。生命科学的技术进步加快了我们处理基因组编码信息的能力,包括接合组装基因组改造(conjugative assembly genome engineering, CAGE)技术等。通过定义的同义密码子对目标密码子进行全基因组取代,可以减少用于编码规范氨基酸的密码子数量,最后,通过重新编码18 214个密码子,创建了一个具有61个密码子,基因组大小为4 Mb的大肠杆菌变体,其利用59个密码子编码20个氨基酸,并能够删除以前必不可少的转移RNA[95]。使用减少数量的有义密码子(59)编码20个必需氨基酸产生的生物体表明,生命可以在减少数量的同义有义密码子下运转。研究者开发的将设计的基因组分为多个片段、部分,通过Rexer、定向缀合等方法进行融合,最后进行无缝和强大的集成来实现设计的策略,为将来的基因组合成提供了一个蓝图。
4.3 组装技术
作为合成生物学的基石,DNA组装过程允许使用被定义的、标准化的、特征明确的组件构建新颖的生物学系统。实际上是将多个DNA片段首尾相连的一种物理方式,从而创建目标高阶装配,然后将其连接到载体。采用限制酶切消化和逐个元素克隆的传统技术既耗时又低廉成本。因此,人们正在付出巨大的努力来开发更好的克隆策略和DNA组装技术,以使多基因系统能够被更快,更有效地构建。这样,构建具有复杂遗传功能的菌株将变得更加容易。这些新技术还有助于提高重组菌株的活力、可转化性或是稳定性。DNA组装技术的加速使用使的得研究人员能够进行更复杂的合成项目,常见的组装方法除了上面提到的Gibson 组装,还有Golden Gate组装(GG)。Golden Gate模块化克隆系统利用II型限制酶建立了标准化且可互换的DNA部分文库,随后将其一步一步组装到一个预先设计的4nt悬臂支架。目前,Golden Gate组装已成为许多基因组编辑试剂盒的核
心,许多实验室已经具备在单个反应中按定义的顺序装配许多不同零件的能力。GG的优点之一是它允许组合组装,可用于高效地生成库。此外,使用GG的启动子改组策略用于筛选表达的每个转录单位的最佳启动子-基因对,然后使用最佳启动子组合工程改造脂质过量生产菌株。
5 缩减基因组的障碍与挑战
解析突变和选择对细菌GC含量的不同影响一直是个难题。1962年,研究者提出一个模型,将基因组中GC平均含量描述为由(G或C)→(A或T)和(A或T)→(G或C)碱基替换率差异所驱动的严格中性突变的过程函数。之后,人们将细菌类群之间基因组GC含量的变异归因于谱系特异性突变模式和对各种全基因组特性的选择[96]。最近发表的两篇论文指出存在一种固有且普遍的(G或C)→(A或T)突变偏倚,并表明有利于高GC含量的选择过程是决定细菌基因组碱基组成的主要因素[97,98]。缩减的细菌基因组往往具有更高的AT含量,且有时AT含量会急剧增加。具有最高AT偏倚的细胞基因组来自共生菌‘Candidatus Zinderia insecticola’ (β-变形菌,基因组为209 kb,GC含量为13.5%[99])和‘Candidatus. Carsonella ruddii’ (γ-变形菌,GC含量为16.6%[100])。已提出几种假设来解释严格内共生菌的AT偏倚,包括选择或群体遗传结合突变模式变化[101?~103]。微小基因组倾向于删除许多参与DNA修复的基因,这可能导致更多的A或T突变,因为脱氧核糖核酸损伤,如胞嘧啶脱氨和鸟嘌呤氧化,往往会导致(G或C)→(A或T)[104]。穆勒棘轮效应(Muller’s ratchet)和纯化选择压力减轻将产生更多轻微有害突变,此类突变偏向A或T突变,并在种群中得到固定。这几种因素的共同作用使基因组GC平均含量移向AT含量较高的新平衡点。
值得注意的是,两个最小的细菌基因组,a. Tremblaya princeps[105] (基因组GC含量为58.8%,四重简并密码子第三位的GC含量为66.6%)和Ca. Hodgkinia cicadicola[105] (α-变形菌,基因组大小为144 kb,基因组GC含量为58.4%,四重简并密码子第三位的GC含量为62.5%),打破了极端缩减基因组和低GC含量之间原本普遍的联系。在Ca. Hodgkinia cicadicola中,缩减基因组表现为偏向GC的突变压力,因为在蛋白质序列水平上,四重简并密码子第三位几乎没有选择压力[106]。由此提出一种假设,即在基因组缩减的过程中,Ca. Hodgkinia cicadicola以某种方式维持了自由生活α-变形菌中的高GC含量突变偏倚。通过后续研究Ca. Hodgkinia cicadicola和Ca. Tremblaya princeps的突变方向,可以确定它们的GC突变偏倚是否具有普遍性,或它们是否像大多数(或所有)其他细菌一样,存在AT突变偏倚[97,98]。因此,其独特基因组组成的一个最明显的解释是全基因组范围内偏向GC的选择压力。
在细胞和细胞器基因组中,多种遗传密码独立于“通用”密码[107],UGA从终止密码子重新分配到色氨酸密码子是最常见的密码变化之一。在细菌中,这种密码子改变发现于柔膜菌纲的一个谱系中[108],这也是直到最近唯一报道的细菌编码重组事件。对来自昆虫共生细菌的微小基因组进行测序,发现了这种终止子-色氨酸重编码的两个新案例:Ca. Hodgkinia cicadicola[107]和Ca. Zinderia insecticola[99]。这是柔膜菌纲谱系以外的细菌中唯一发现的密码子重新分配的两个案例,而Ca. Hodgkinia cicadicola是唯一已知的在高GC含量的基因组中发生UGA密码子重新分配的案例。这一发现,以及基于几种线粒体基因组对密码子重新分配机制的分析[109],似乎与广泛引用的观点不符,即低GC含量是密码子重新分配的先决条件(“密码子捕获”假说)[110?~112]。McCutcheon等[106]设想了一种基于“模糊翻译”[109]的机制,其中tRNATrp突变允许色氨酸(UGG)和终止(UGA)密码子的混杂解码,从而允许通过持续的基因组缩减移除识别UGA密码子的肽链释放因子2 (RF-2;由prfB编码)。在此假设中,UGA密码子重新分配是对关键基因prfB缺失的一种遗传共适应,而不是为了使基因组(或翻译)更加精简的适应性事件[113],也不是通过GC含量偏倚导致的密码子丢失和重新分配的完全中性事件。
大部分缩减基因组菌株的生长速度与其原始菌株相当。其中一些菌株的生长速率和最终生物量甚至高于其亲本菌株。此外,缩减基因组还具有诸如更高的转化效率[79]和生物化学法生产[85]等有利特性。少数情况下,尽管与生长、细胞周期和形态相关的
基因未发生改变,但缩减基因组表现出意想不到的表型,如生长迟缓和异常细胞形态。最近一项研究提出了一种实验室适应进化(ALE)技术,以改善缩减基因组大肠杆菌的生长表型[80]。对进化菌株的多组学分析表明,不平衡代谢通过ALE重组代谢扰动,诱导生长延迟和转录组及翻译组重构。上述研究说明我们对基因功能、代谢和基因组的认识还不全面,亟需对细菌基因组进行更全面的研究以填补我们知识空白,解答基因组缩减生物体所呈现的表型特征。
6 结语与展望
回顾全文,首先介绍了基于各种方法阐明必需基因集的研究,并依此设计最小(或缩减)基因组。通过比较多个基因组对必需基因进行计算研究,表明只有不到300个基因是细菌生存复制必需的。然而,这种比较低估了必需基因的数量,因为基因的功能可以被其他基因取代。利用转座子诱变、敲除、asRNA和CRISPRi技术进行实验评估,可揭示必需基因的直接和精确信息。但是,由于上述实验方法使基因破坏或失活,所以无法研究基因失活组合(如合成基因致死),需要开发更精细的方法以阐明基因之间相互关联的上位相互作用。然后,介绍了以前构建的缩减基因组的特点及其构建方法。科学家们试图通过缩减已有基因组,使之成为最小基因组。现有的研究结果证明,缩减基因组是一把“双刃剑”,一方面,基因组缩减菌株可保留原始菌株优势特性,甚至还具有诸如更高的转化效率和生物化学法生产等有利特性;另一方面,仍会出现意想不到的表型,如生长迟缓和异常细胞形态等。这说明我们对基因组、基因功能和代谢的认识还不全面,还需要对细菌基因组进行更全面的研究以填补知识空白。
现今,高速发展的生物技术使设计基因网络,生物合成途径乃至整个基因组的构建成为可能,将遗传学和基因组学领域从描述性应用转移到了合成应用。在合成小型病毒基因组之后,DNA组装和重写技术的进步使细菌基因组(例如生殖支原体)的层次化合成成为可能[114],并且通过将密码子的数量从64个减少到57个来重新编码大肠杆菌基因组。目前,合成基因组学已经发展到合成整个真核基因组的地步。合成酵母基因组计划(Sc2.0)正在进行中,旨在重写所有16个酿酒酵母染色体;到2018年,已经设计并合成了6.5条染色体。使用自下而上的装配并应用全基因组的改变将增进对基因组结构和功能的理解。这种方法不仅将为真核染色体的系统研究提供平台,还将产生可能适用于医学和工业应用的各种“简化”菌株。Sc2.0的目的是设计和完全化学合成16条染色体,这些染色体包含来自啤酒酵母的1250万个碱基和一个带有所有tRNA基因的“新染色体”(
从小型病毒基因组的合成到酵母染色体Sc2.0,从罕见的密码子替代到全基因组重新编码,合成基因组学的可行性和能力已得到一次又一次的证明。Sc2.0着重介绍了用于生产非天然药物和工业化合物(如青蒿素)的酵母“底盘”的开发。基于合成染色体,可以将多种异源途径直接整合到酵母基因组中。合成基因组学还使改组基因组以快速生成新的基因组并筛选所需的特性成为可能。
最后,合成最小基因组是合成基因组学领域的一个里程碑,高通量基因合成技术正在持续发展过程中,凭借其已被证实的优势,使得最小基因组生物成为科学、工业和许多其他应用的理想生物平台,相信随着系统和合成生物学前沿技术的不断发展,终将解决目前存在的许多障碍和挑战。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nrn3214URLPMID:22498897 [本文引用: 1]

The brain is expensive, incurring high material and metabolic costs for its size--relative to the size of the body--and many aspects of brain network organization can be mostly explained by a parsimonious drive to minimize these costs. However, brain networks or connectomes also have high topological efficiency, robustness, modularity and a 'rich club' of connector hubs. Many of these and other advantageous topological properties will probably entail a wiring-cost premium. We propose that brain organization is shaped by an economic trade-off between minimizing costs and allowing the emergence of adaptively valuable topological patterns of anatomical or functional connectivity between multiple neuronal populations. This process of negotiating, and re-negotiating, trade-offs between wiring cost and topological value continues over long (decades) and short (millisecond) timescales as brain networks evolve, grow and adapt to changing cognitive demands. An economical analysis of neuropsychiatric disorders highlights the vulnerability of the more costly elements of brain networks to pathological attack or abnormal development.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/s0168-9525(01)02447-7URLPMID:11585665 [本文引用: 1]
Although bacteria increase their DNA content through horizontal transfer and gene duplication, their genomes remain small and, in particular, lack nonfunctional sequences. This pattern is most readily explained by a pervasive bias towards higher numbers of deletions than insertions. When selection is not strong enough to maintain them, genes are lost in large deletions or inactivated and subsequently eroded. Gene inactivation and loss are particularly apparent in obligate parasites and symbionts, in which dramatic reductions in genome size can result not from selection to lose DNA, but from decreased selection to maintain gene functionality. Here we discuss the evidence showing that deletional bias is a major force that shapes bacterial genomes.
[本文引用: 1]
John Wiley, Chapman & Hall,
[本文引用: 2]
DOI:10.1038/nrmicro2670URLPMID:22064560 [本文引用: 1]
Since 2006, numerous cases of bacterial symbionts with extraordinarily small genomes have been reported. These organisms represent independent lineages from diverse bacterial groups. They have diminutive gene sets that rival some mitochondria and chloroplasts in terms of gene numbers and lack genes that are considered to be essential in other bacteria. These symbionts have numerous features in common, such as extraordinarily fast protein evolution and a high abundance of chaperones. Together, these features point to highly degenerate genomes that retain only the most essential functions, often including a considerable fraction of genes that serve the hosts. These discoveries have implications for the concept of minimal genomes, the origins of cellular organelles, and studies of symbiosis and host-associated microbiota.
[本文引用: 1]
DOI:10.1093/gbe/evt118URLPMID:23918810 [本文引用: 1]
Many insects rely on bacterial symbionts with tiny genomes specialized for provisioning nutrients lacking in host diets. Xylem sap and phloem sap are both deficient as insect diets, but differ dramatically in nutrient content, potentially affecting symbiont genome evolution. For sap-feeding insects, sequenced symbiont genomes are available only for phloem-feeding examples from the suborder Sternorrhyncha and xylem-feeding examples from the suborder Auchenorrhyncha, confounding comparisons. We sequenced genomes of the obligate symbionts, Sulcia muelleri and Nasuia deltocephalinicola, of the phloem-feeding pest insect, Macrosteles quadrilineatus (Auchenorrhyncha: Cicadellidae). Our results reveal that Nasuia-ALF has the smallest bacterial genome yet sequenced (112 kb), and that the Sulcia-ALF genome (190 kb) is smaller than that of Sulcia in other insect lineages. Together, these symbionts retain the capability to synthesize the 10 essential amino acids, as observed for several symbiont pairs from xylem-feeding Auchenorrhyncha. Nasuia retains genes enabling synthesis of two amino acids, DNA replication, transcription, and translation. Both symbionts have lost genes underlying ATP synthesis through oxidative phosphorylation, possibly as a consequence of the enriched sugar content of phloem. Shared genomic features, including reassignment of the UGA codon from Stop to tryptophan, and phylogenetic results suggest that Nasuia-ALF is most closely related to Zinderia, the betaproteobacterial symbiont of spittlebugs. Thus, Nasuia/Zinderia and Sulcia likely represent ancient associates that have co-resided in hosts since the divergence of leafhoppers and spittlebugs >200 Ma, and possibly since the origin of the Auchenorrhyncha, >260 Ma.
URLPMID:11028996 [本文引用: 1]
DOI:10.1126/science.1132493URLPMID:17158324 [本文引用: 2]
Biological organisms perform complex information processing and control tasks using sophisticated biochemical circuits, yet the engineering of such circuits remains ineffective compared with that of electronic circuits. To systematically create complex yet reliable circuits, electrical engineers use digital logic, wherein gates and subcircuits are composed modularly and signal restoration prevents signal degradation. We report the design and experimental implementation of DNA-based digital logic circuits. We demonstrate AND, OR, and NOT gates, signal restoration, amplification, feedback, and cascading. Gate design and circuit construction is modular. The gates use single-stranded nucleic acids as inputs and outputs, and the mechanism relies exclusively on sequence recognition and strand displacement. Biological nucleic acids such as microRNAs can serve as inputs, suggesting applications in biotechnology and bioengineering.
[本文引用: 1]
[C].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:20419221 [本文引用: 1]
URLPMID:11127835 [本文引用: 1]
[本文引用: 1]
URLPMID:20493680 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
Education (ICCSE),
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:30698741 [本文引用: 1]
URLPMID:16397293 [本文引用: 1]
URLPMID:16738554 [本文引用: 2]
DOI:10.1111/j.1574-6976.2008.00151.xURLPMID:19067748 [本文引用: 1]
Recent technical and conceptual advances in the biological sciences opened the possibility of the construction of newly designed cells. In this paper we review the state of the art of cell engineering in the context of genome research, paying particular attention to what we can learn on naturally reduced genomes from either symbiotic or free living bacteria. Different minimal hypothetically viable cells can be defined on the basis of several computational and experimental approaches. Projects aiming at simplifying living cells converge with efforts to make synthetic genomes for minimal cells. The panorama of this particular view of synthetic biology lead us to consider the use of defined minimal cells to be applied in biomedical, bioremediation, or bioenergy application by taking advantage of existing naturally minimized cells.
URLPMID:17483224 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/dnares/dsn019URLPMID:18753290 [本文引用: 1]
Escherichia coli has dispensable genome regions and eliminating them may improve cell use by reducing unnecessary metabolic pathways and complex regulatory networks. Although several strains with reduced genomes have already been constructed, there have been no reports of strains constructed with deletions assayed for influence on growth. To retain robust growth and fundamental metabolic pathways, the growth of each deletion strain and combination effects of deletions were checked using M9 minimal medium. Then a new strain, MGF-01, with a 1 Mb reduced genome was constructed by integrating deletions that did not affect growth. MGF-01 grew as well as the wild type in the exponential phase and continued growing after the wild type had entered the stationary phase. The final cell density of MGF-01 was 1.5 times higher than that of the wild-type strain. Using MGF-01 as a production host, a 2.4-fold increase in l-threonine production was achieved.
[本文引用: 2]
URLPMID:6783534 [本文引用: 1]
Secondary chicken embryo fibroblasts infected in suspension with the Breinl strain of Rickettsia prowazekii and grown in monolayer culture were examined by both transmission and scanning electron microscopy at specific intervals after infection to study the effects of prolonged intracellular growth on the fine structure of the host cell and the rickettsiae. Cytopathological changes in the infected host cells were not apparent until late in the intracellular growth cycle when the cells began to rupture as a result of a large rickettsial burden. The only recognizable changes in heavily infected cells before lysis were the condensation of the intercristal matrix of some mitochondria and the apparent dissociation of ribosomes from the rough-surfaced endoplasmic reticulum. Although the effects of intracellular growth of rickettsiae on the fine structure of the host cell were rather unremarkable when compared with those imposed by Rickettsia rickettsii in a similar cell system, noticeable morphological changes in the rickettsiae were recognized during the intracellular growth cycle. These changes first became apparent about 40 h postinfection and consisted primarily of an increased electron density of the rickettsiae, the appearance of numerous vacuoles in the rickettsial cytoplasm, and a slight reduction in size of the rickettsiae. Changes of this nature may reflect transitional phases of growth characteristically seen in free-living bacterial cell systems.
URLPMID:6112607 [本文引用: 1]
URLPMID:8866472 [本文引用: 1]
URLPMID:7547309 [本文引用: 1]
URLPMID:12558594 [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1021/ac00133a015URLPMID:2953269 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:12618371 [本文引用: 1]
URLPMID:15035042 [本文引用: 1]
DOI:10.1101/gr.3024704URLPMID:15574825 [本文引用: 1]
The genomic core concept has found several uses in comparative and evolutionary genomics. Defined as the set of all genes common to (ubiquitous among) all genomes in a phylogenetically coherent group, core size decreases as the number and phylogenetic diversity of the relevant group increases. Here, we focus on methods for defining the size and composition of the core of all genes shared by sequenced genomes of prokaryotes (Bacteria and Archaea). There are few (almost certainly less than 50) genes shared by all of the 147 genomes compared, surely insufficient to conduct all essential functions. Sequencing and annotation errors are responsible for the apparent absence of some genes, while very limited but genuine disappearances (from just one or a few genomes) can account for several others. Core size will continue to decrease as more genome sequences appear, unless the requirement for ubiquity is relaxed. Such relaxation seems consistent with any reasonable biological purpose for seeking a core, but it renders the problem of definition more problematic. We propose an alternative approach (the phylogenetically balanced core), which preserves some of the biological utility of the core concept. Cores, however delimited, preferentially contain informational rather than operational genes; we present a new hypothesis for why this might be so.
[本文引用: 1]
DOI:10.1016/0014-5793(95)00233-yURLPMID:7729508 [本文引用: 1]
The number of indispensable chromosomal loci for a bacterium, Bacillus subtilis was estimated. Seventy-nine randomly selected chromosomal loci were investigated by mutagenesis. Mutation at only six loci rendered B. subtilis unable to form colonies. In contrast, mutants for the rest of the 73 loci retained the ability to form colonies. Mutant B. subtilis with multiple-fold mutations of those dispensable loci (7-, 12- or 33-fold) were not impaired in their ability to form colonies on nutritionally adequate medium, indicating that up to 33 dispensable loci were simultaneously abolished. Given the statistical analyses for the frequency of indispensable loci (6 out of 79), total indispensable genetic material would be included within about 562 kbp. The hypothetical minimum genome size lies in the range of those currently determined smallest genomes for bacteria.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:11567142 [本文引用: 1]
DOI:10.1128/jb.185.19.5673-5684.2003URLPMID:13129938 [本文引用: 1]
Defining the gene products that play an essential role in an organism's functional repertoire is vital to understanding the system level organization of living cells. We used a genetic footprinting technique for a genome-wide assessment of genes required for robust aerobic growth of Escherichia coli in rich media. We identified 620 genes as essential and 3,126 genes as dispensable for growth under these conditions. Functional context analysis of these data allows individual functional assignments to be refined. Evolutionary context analysis demonstrates a significant tendency of essential E. coli genes to be preserved throughout the bacterial kingdom. Projection of these data over metabolic subsystems reveals topologic modules with essential and evolutionarily preserved enzymes with reduced capacity for error tolerance.
DOI:10.1016/j.cell.2013.02.022URL [本文引用: 1]
Targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering cellular systems. Here, we develop a method for controlling gene expression based on Cas9, an RNA-guided DNA endonuclease from a type II CRISPR system. We show that a catalytically dead Cas9 lacking endonuclease activity, when coexpressed with a guide RNA, generates a DNA recognition complex that can specifically interfere with transcriptional elongation, RNA polymerase binding, or transcription factor binding. This system, which we call CRISPR interference (CRISPRi), can efficiently repress expression of targeted genes in Escherichia coli, with no detectable off-target effects. CRISPRi can be used to repress multiple target genes simultaneously, and its effects are reversible. We also show evidence that the system can be adapted for gene repression in mammalian cells. This RNA-guided DNA recognition platform provides a simple approach for selectively perturbing gene expression on a genome-wide scale.
DOI:10.1371/journal.pgen.1007749URLPMID:30403660 [本文引用: 1]
High-throughput genetic screens are powerful methods to identify genes linked to a given phenotype. The catalytic null mutant of the Cas9 RNA-guided nuclease (dCas9) can be conveniently used to silence genes of interest in a method also known as CRISPRi. Here, we report a genome-wide CRISPR-dCas9 screen using a starting pool of ~ 92,000 sgRNAs which target random positions in the chromosome of E. coli. To benchmark our method, we first investigate its utility to predict gene essentiality in the genome of E. coli during growth in rich medium. We could identify 79% of the genes previously reported as essential and demonstrate the non-essentiality of some genes annotated as essential. In addition, we took advantage of the intermediate repression levels obtained when targeting the template strand of genes to show that cells are very sensitive to the expression level of a limited set of essential genes. Our data can be visualized on CRISPRbrowser, a custom web interface available at crispr.pasteur.fr. We then apply the screen to discover E. coli genes required by phages lambda, T4 and 186 to kill their host, highlighting the involvement of diverse host pathways in the infection process of the three tested phages. We also identify colanic acid capsule synthesis as a shared resistance mechanism to all three phages. Finally, using a plasmid packaging system and a transduction assay, we identify genes required for the formation of functional lambda capsids, thus covering the entire phage cycle. This study demonstrates the usefulness and convenience of pooled genome-wide CRISPR-dCas9 screens in bacteria and paves the way for their broader use as a powerful tool in bacterial genomics.
DOI:10.1126/science.1247005URLPMID:24336571 [本文引用: 1]
The simplicity of programming the CRISPR (clustered regularly interspaced short palindromic repeats)-associated nuclease Cas9 to modify specific genomic loci suggests a new way to interrogate gene function on a genome-wide scale. We show that lentiviral delivery of a genome-scale CRISPR-Cas9 knockout (GeCKO) library targeting 18,080 genes with 64,751 unique guide sequences enables both negative and positive selection screening in human cells. First, we used the GeCKO library to identify genes essential for cell viability in cancer and pluripotent stem cells. Next, in a melanoma model, we screened for genes whose loss is involved in resistance to vemurafenib, a therapeutic RAF inhibitor. Our highest-ranking candidates include previously validated genes NF1 and MED12, as well as novel hits NF2, CUL3, TADA2B, and TADA1. We observe a high level of consistency between independent guide RNAs targeting the same gene and a high rate of hit confirmation, demonstrating the promise of genome-scale screening with Cas9.
[本文引用: 1]
DOI:10.1007/978-1-59745-321-9_17URLPMID:18392973 [本文引用: 1]
Efficient genome-engineering tools have been developed for use in whole-genome essentiality studies. In this chapter, we describe a powerful genomic deletion tool, the Tn5-targeted Cre/loxP excision system, for determining genetic essentiality and minimizing bacterial genomes on a genome-wide scale. This tool is based on the Tn5 transposition system, phage P1 transduction, and the Cre/loxP excision system. We have generated two large pools of independent transposon insertion mutants in Escherichia coli using random transposition of two modified Tn5 transposons (TnKloxP and TnCloxP) with two different selection markers, kanamycin-resistance gene (Km(R)) or chloramphenicol-resistance gene (Cm(R)), and a loxP site. Transposon integration sites are identified by direct genome sequencing of the genomic DNA. By combining a mapped transposon mutation from each of the mutant pools into the same chromosome using phage P1 transduction and then excising the nonessential genomic regions flanked by the two loxP sites using Cre-mediated loxP recombination, we can obtain numerous E. coli deletion strains from which nonessential regions of the genome are deleted. In addition to the combinatorial deletion of the E. coli genomic regions, we can create a cumulative E. coli deletion strain from which all the individual deleted regions are excised. This process will eventually yield an E. coli strain in which the genome is reduced in size and contains only regions that are essential for viability.
DOI:10.1016/j.febslet.2010.11.024URLPMID:21094158 [本文引用: 1]
Synthetic lethality occurs when the simultaneous perturbation of two genes results in cellular or organismal death. Synthetic lethality also occurs between genes and small molecules, and can be used to elucidate the mechanism of action of drugs. This area has recently attracted attention because of the prospect of a new generation of anti-cancer drugs. Based on studies ranging from yeast to human cells, this review provides an overview of the general principles that underlie synthetic lethality and relates them to its utility for identifying gene function, drug action and cancer therapy. It also identifies the latest strategies for the large-scale mapping of synthetic lethalities in human cells which bring us closer to the generation of comprehensive human genetic interaction maps.
DOI:10.1101/gr.217202URLPMID:11932248 [本文引用: 1]
Our goal is to construct an improved Escherichia coli to serve both as a better model organism and as a more useful technological tool for genome science. We developed techniques for precise genomic surgery and applied them to deleting the largest K-islands of E. coli, identified by comparative genomics as recent horizontal acquisitions to the genome. They are loaded with cryptic prophages, transposons, damaged genes, and genes of unknown function. Our method leaves no scars or markers behind and can be applied sequentially. Twelve K-islands were successfully deleted, resulting in an 8.1% reduced genome size, a 9.3% reduction of gene count, and elimination of 24 of the 44 transposable elements of E. coli. These are particularly detrimental because they can mutagenize the genome or transpose into clones being propagated for sequencing, as happened in 18 places of the draft human genome sequence. We found no change in the growth rate on minimal medium, confirming the nonessential nature of these islands. This demonstration of feasibility opens the way for constructing a maximally reduced strain, which will provide a clean background for functional genomics studies, a more efficient background for use in biotechnology applications, and a unique tool for studies of genome stability and evolution.
.
URLPMID:16645050 [本文引用: 3]
DOI:10.1186/1475-2859-8-2URLPMID:19128451 [本文引用: 2]
BACKGROUND: Deletion of large blocks of nonessential genes that are not needed for metabolic pathways of interest can reduce the production of unwanted by-products, increase genome stability, and streamline metabolism without physiological compromise. Researchers have recently constructed a reduced-genome Escherichia coli strain MDS42 that lacks 14.3% of its chromosome. RESULTS: Here we describe the reengineering of the MDS42 genome to increase the production of the essential amino acid L-threonine. To this end, we over-expressed a feedback-resistant threonine operon (thrA*BC), deleted the genes that encode threonine dehydrogenase (tdh) and threonine transporters (tdcC and sstT), and introduced a mutant threonine exporter (rhtA23) in MDS42. The resulting strain, MDS-205, shows an ~83% increase in L-threonine production when cells are grown by flask fermentation, compared to a wild-type E. coli strain MG1655 engineered with the same threonine-specific modifications described above. And transcriptional analysis revealed the effect of the deletion of non-essential genes on the central metabolism and threonine pathways in MDS-205. CONCLUSION: This result demonstrates that the elimination of genes unnecessary for cell growth can increase the productivity of an industrial strain, most likely by reducing the metabolic burden and improving the metabolic efficiency of cells.
DOI:10.1007/s00253-014-5739-yURLPMID:24752842 [本文引用: 1]
The genomic stability and integrity of host strains are critical for the production of recombinant proteins in biotechnology. Bacterial genomes contain numerous jumping genetic elements, the insertion sequences (ISs) that cause a variety of genetic rearrangements, resulting in adverse effects such as genome and recombinant plasmid instability. To minimize the harmful effects of ISs on the expression of recombinant proteins in Escherichia coli, we developed an IS-free, minimized E. coli strain (MS56) in which about 23 % of the genome, including all ISs and many unnecessary genes, was removed. Here, we compared the expression profiles of recombinant proteins such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and bone morphogenetic protein-2 (BMP2) in MG1655 and MS56. Hopping of ISs (IS1, IS3, or IS5) into the TRAIL and BMP2 genes occurred at the rate of ~10(-8)/gene/h in MG1655 whereas such events were not observed in MS56. Even though IS hopping occurred very rarely (10(-8)/gene/h), cells containing the IS-inserted TRAIL and BMP2 plasmids became dominant (~52 % of the total population) 28 h after fermentation began due to their growth advantage over cells containing intact plasmids, significantly reducing recombinant protein production in batch fermentation. Our findings clearly indicate that IS hopping is detrimental to the industrial production of recombinant proteins, emphasizing the importance of the development of IS-free host strains.
DOI:10.1111/j.1365-2958.2004.04386.xURLPMID:15612923 [本文引用: 2]
The minimization of a genome is necessary to identify experimentally the minimal gene set that contains only those genes that are essential and sufficient to sustain a functioning cell. Recent developments in genetic techniques have made it possible to generate bacteria with a markedly reduced genome. We developed a simple system for formation of markerless chromosomal deletions, and constructed and characterized a series of large-scale chromosomal deletion mutants of Escherichia coli that lack between 2.4 and 29.7% of the parental chromosome. Combining deletion mutations changes cell length and width, and the mutant cells with larger deletions were even longer and wider than the parental cells. The nucleoid organization of the mutants is also changed: the nucleoids occur as multiple small nucleoids and are localized peripherally near the envelope. Inhibition of translation causes them to condense into one or two packed nucleoids, suggesting that the coupling of transcription and translation of membrane proteins peripherally localizes chromosomes. Because these phenotypes are similar to those of spherical cells, those may be a consequence of the morphological change. Based on the nucleoid localization observed with these mutants, we discuss the cellular nucleoid dynamics.
DOI:10.1093/molbev/msg219URLPMID:12949151 [本文引用: 1]
Bacterial genomes contain 250 to 500 essential genes, as suggested by single gene disruptions and theoretical considerations. If this view is correct, the remaining nonessential genes of an organism, such as Bacillus subtilis, have been acquired during evolution in its perpetually changing ecological niches. Notably, approximately 47% of the approximately 4,100 genes of B. subtilis belong to paralogous gene families in which several members have overlapping functions. Thus, essential gene functions will outnumber essential genes. To answer the question to what extent the most recently acquired DNA contributes to the life of B. subtilis under standard laboratory growth conditions, we initiated a
DOI:10.1128/jb.175.11.3628-3635.1993URLPMID:8501066 [本文引用: 1]
A system for high-efficiency single- and double-crossover homologous integration in gram-positive bacteria has been developed, with Lactococcus lactis as a model system. The system is based on a thermosensitive broad-host-range rolling-circle plasmid, pG+host5, which contains a pBR322 replicon for propagation in Escherichia coli at 37 degrees C. A nested set of L. lactis chromosomal fragments cloned onto pG+host5 were used to show that the single-crossover integration frequency was logarithmically proportional to the length of homology for DNA fragments between 0.35 and 2.5 kb. Using random chromosomal 1-kb fragments, we showed that homologous integration can occur along the entire chromosome. We made use of the reported stimulatory effect of rolling-circle replication on intramolecular recombination to develop a protocol for gene replacement. Cultures were first maintained at 37 degrees C to select for a bacterial population enriched for plasmid integrants; activation of the integrated rolling-circle plasmid by a temperature shift to 28 degrees C resulted in efficient plasmid excision by homologous recombination and replacement of a chromosomal gene by the plasmid-carried modified copy. More than 50% of cells underwent replacement recombination when selection was applied for the replacing gene. Between 1 and 40% of cells underwent replacement recombination when no selection was applied. Chromosomal insertions and deletions were obtained in this way. These results show that gene replacement can be obtained at an extremely high efficiency by making use of the thermosensitive rolling-circle nature of the delivery vector. This procedure is applicable to numerous gram-positive bacteria.
.
DOI:10.1038/s41467-019-08888-6URLPMID:30804335 [本文引用: 2]
Synthetic biology aims to design and construct bacterial genomes harboring the minimum number of genes required for self-replicable life. However, the genome-reduced bacteria often show impaired growth under laboratory conditions that cannot be understood based on the removed genes. The unexpected phenotypes highlight our limited understanding of bacterial genomes. Here, we deploy adaptive laboratory evolution (ALE) to re-optimize growth performance of a genome-reduced strain. The basis for suboptimal growth is the imbalanced metabolism that is rewired during ALE. The metabolic rewiring is globally orchestrated by mutations in rpoD altering promoter binding of RNA polymerase. Lastly, the evolved strain has no translational buffering capacity, enabling effective translation of abundant mRNAs. Multi-omic analysis of the evolved strain reveals transcriptome- and translatome-wide remodeling that orchestrate metabolism and growth. These results reveal that failure of prediction may not be associated with understanding individual genes, but rather from insufficient understanding of the strain's systems biology.
DOI:10.1101/gr.215293.116URLPMID:27965289 [本文引用: 1]
Understanding cellular life requires a comprehensive knowledge of the essential cellular functions, the components involved, and their interactions. Minimized genomes are an important tool to gain this knowledge. We have constructed strains of the model bacterium, Bacillus subtilis, whose genomes have been reduced by approximately 36%. These strains are fully viable, and their growth rates in complex medium are comparable to those of wild type strains. An in-depth multi-omics analysis of the genome reduced strains revealed how the deletions affect the transcription regulatory network of the cell, translation resource allocation, and metabolism. A comparison of gene counts and resource allocation demonstrates drastic differences in the two parameters, with 50% of the genes using as little as 10% of translation capacity, whereas the 6% essential genes require 57% of the translation resources. Taken together, the results are a valuable resource on gene dispensability in B. subtilis, and they suggest the roads to further genome reduction to approach the final aim of a minimal cell in which all functions are understood.
DOI:10.1042/BA20060111URLPMID:17115975 [本文引用: 1]
In 1997, the complete genomic DNA sequence of Bacillus subtilis (4.2 Mbp) was determined and 4100 genes were identified [Kunst, Ogasawara, Moszer, Albertini, Alloni, Azevedo, Bertero, Bessieres, Bolotin, Borchert, S. et al. (1997) Nature 90, 249-256]. In addition, B. subtilis, which shows an excellent ability to secrete proteins (enzymes) and antibiotics in large quantities outside the cell, plays an important role in industrial and medical fields. It is necessary to clarify the genes involved in the production of compounds by understanding the network of these 4100 genes and the proceeding analysis of genes of unknown functions. In promoting such a study, it is expected that the regulatory system of B. subtilis can be simplified by the creation of a Bacillus strain with a reduced genome by discriminating genes unnecessary for the production of proteins from essential genes, and deleting as many of these unnecessary genes as possible, which may help to understand this complex network of genes. We have previously distinguished essential and non-essential genes by evaluating the growth and enzyme-producing properties of strains of B. subtilis in which about 3000 genes (except 271 essential genes) have been disrupted or deleted singly, and have successfully utilized the findings from these studies in creating the MG1M strain with an approx. 1 Mbp deletion by serially deleting 17 unnecessary regions from the genome. This strain showed slightly reduced growth in enzyme-production medium, but no marked morphological changes. Moreover, we confirmed that the MG1M strain had cellulase and protease productivity comparable with that of the B. subtilis 168 strain, thus demonstrating that genome reduction does not contribute to a negative influence on enzyme productivity.
DOI:10.1093/dnares/dsn002URLPMID:18334513 [本文引用: 1]
The emerging field of synthetic genomics is expected to facilitate the generation of microorganisms with the potential to achieve a sustainable society. One approach towards this goal is the reduction of microbial genomes by rationally designed deletions to create simplified cells with predictable behavior that act as a platform to build in various genetic systems for specific purposes. We report a novel Bacillus subtilis strain, MBG874, depleted of 874 kb (20%) of the genomic sequence. When compared with wild-type cells, the regulatory network of gene expression of the mutant strain is reorganized after entry into the transition state due to the synergistic effect of multiple deletions, and productivity of extracellular cellulase and protease from transformed plasmids harboring the corresponding genes is remarkably enhanced. To our knowledge, this is the first report demonstrating that genome reduction actually contributes to the creation of bacterial cells with a practical application in industry. Further systematic analysis of changes in the transcriptional regulatory network of MGB874 cells in relation to protein productivity should facilitate the generation of improved B. subtilis cells as hosts of industrial protein production.
[本文引用: 1]
DOI:10.1042/BA20060106URLPMID:17300221 [本文引用: 1]
Various systems for the production of useful proteins have been developed using the fission yeast Schizosaccharomyces pombe as a host, and some are now being used commercially. It is necessary, however, to improve the system further for the production of low-cost chemicals and commodities, so that the host becomes more economical and productive and can be widely used for the production of different molecules. We hypothesized that many S. pombe genes are not necessary under nutrient-rich growth conditions; or rather, they serve only to waste energy when seen from the viewpoint of protein production, because their products are necessary only for adaptation to different environments. Thus we have tried to create S. pombe mutants that are dedicated to heterologous protein production by deleting as many non-essential genes as possible. Putative essential genes were mapped using the genome information of S. pombe. The transcriptome of gene disruptants was analysed using microarrays and, using this system, a new promoter was identified. The method (called the Latour method) has been developed to delete efficiently a large region from the chromosome, resulting in the establishment of mutant strains lacking approx. 500 kb of genetic material. New experimental strains auxotrophic for six nutrients were established that were conveniently used for co-expression of proteins using multiple plasmids. An efficient transformation method has also been developed that is useful for investigating heterologous protein production in a variety of strains. Incidentally, in heterologous protein production systems, products are often degraded, leading to a decline in production efficiency. Thus, to examine heterologous protein production, we created 52 S. pombe mutant strains in each of which a single protease gene was destroyed. We also successfully constructed strains in which multiple protease genes were disrupted. As a result, it was shown that the production of a model protein, human growth hormone, was increased in this strain. Furthermore, we obtained many strains that lacked genes related to glucose metabolism, intracellular transport or biosynthesis of sugar chains. The present minireview covers the results of functional analysis of these strains. By preparing strains in which large chromosomal regions have been deleted and then combining strains defective in various functional genes, the establishment of effective hosts will become possible.
DOI:10.1126/science.1190719URLPMID:20488990 [本文引用: 1]
We report the design, synthesis, and assembly of the 1.08-mega-base pair Mycoplasma mycoides JCVI-syn1.0 genome starting from digitized genome sequence information and its transplantation into a M. capricolum recipient cell to create new M. mycoides cells that are controlled only by the synthetic chromosome. The only DNA in the cells is the designed synthetic DNA sequence, including
DOI:10.4161/bbug.1.4.12465URLPMID:21327053 [本文引用: 1]
DOI:10.1126/science.1241460URL [本文引用: 1]
Engineering radically altered genetic codes will allow for genomically recoded organisms that have expanded chemical capabilities and are isolated from nature. We have previously reassigned the translation function of the UAG stop codon; however, reassigning sense codons poses a greater challenge because such codons are more prevalent, and their usage regulates gene expression in ways that are difficult to predict. To assess the feasibility of radically altering the genetic code, we selected a panel of 42 highly expressed essential genes for modification. Across 80 Escherichia coli strains, we removed all instances of 13 rare codons from these genes and attempted to shuffle all remaining codons. Our results suggest that the genome-wide removal of 13 codons is feasible; however, several genome design constraints were apparent, underscoring the importance of a strategy that rapidly prototypes and tests many designs in small pieces.
[本文引用: 1]
DOI:10.1016/j.neunet.2011.11.001URLPMID:22154354 [本文引用: 1]
This paper introduces a process to compute optical flow using an asynchronous event-based retina at high speed and low computational load. A new generation of artificial vision sensors has now started to rely on biologically inspired designs for light acquisition. Biological retinas, and their artificial counterparts, are totally asynchronous and data driven and rely on a paradigm of light acquisition radically different from most of the currently used frame-grabber technologies. This paper introduces a framework for processing visual data using asynchronous event-based acquisition, providing a method for the evaluation of optical flow. The paper shows that current limitations of optical flow computation can be overcome by using event-based visual acquisition, where high data sparseness and high temporal resolution permit the computation of optical flow with micro-second accuracy and at very low computational cost.
DOI:10.1109/TVCG.2012.225URLPMID:26357175 [本文引用: 2]
Event sequence data is common in many domains, ranging from electronic medical records (EMRs) to sports events. Moreover, such sequences often result in measurable outcomes (e.g., life or death, win or loss). Collections of event sequences can be aggregated together to form event progression pathways. These pathways can then be connected with outcomes to model how alternative chains of events may lead to different results. This paper describes the Outflow visualization technique, designed to (1) aggregate multiple event sequences, (2) display the aggregate pathways through different event states with timing and cardinality, (3) summarize the pathways' corresponding outcomes, and (4) allow users to explore external factors that correlate with specific pathway state transitions. Results from a user study with twelve participants show that users were able to learn how to use Outflow easily with limited training and perform a range of tasks both accurately and rapidly.
DOI:10.1109/TNNLS.2011.2180025URLPMID:24808513 [本文引用: 2]
We present a novel event-based stereo matching algorithm that exploits the asynchronous visual events from a pair of silicon retinas. Unlike conventional frame-based cameras, recent artificial retinas transmit their outputs as a continuous stream of asynchronous temporal events, in a manner similar to the output cells of the biological retina. Our algorithm uses the timing information carried by this representation in addressing the stereo-matching problem on moving objects. Using the high temporal resolution of the acquired data stream for the dynamic vision sensor, we show that matching on the timing of the visual events provides a new solution to the real-time computation of 3-D objects when combined with geometric constraints using the distance to the epipolar lines. The proposed algorithm is able to filter out incorrect matches and to accurately reconstruct the depth of moving objects despite the low spatial resolution of the sensor. This brief sets up the principles for further event-based vision processing and demonstrates the importance of dynamic information and spike timing in processing asynchronous streams of visual events.
DOI:10.1038/srep40703URLPMID:28079187 [本文引用: 2]
Stereo vision is an important feature that enables machine vision systems to perceive their environment in 3D. While machine vision has spawned a variety of software algorithms to solve the stereo-correspondence problem, their implementation and integration in small, fast, and efficient hardware vision systems remains a difficult challenge. Recent advances made in neuromorphic engineering offer a possible solution to this problem, with the use of a new class of event-based vision sensors and neural processing devices inspired by the organizing principles of the brain. Here we propose a radically novel model that solves the stereo-correspondence problem with a spiking neural network that can be directly implemented with massively parallel, compact, low-latency and low-power neuromorphic engineering devices. We validate the model with experimental results, highlighting features that are in agreement with both computational neuroscience stereo vision theories and experimental findings. We demonstrate its features with a prototype neuromorphic hardware system and provide testable predictions on the role of spike-based representations and temporal dynamics in biological stereo vision processing systems.
[本文引用: 1]
[本文引用: 1]
DOI:10.1162/08997660152002852URLPMID:11387046 [本文引用: 1]
It is often supposed that the messages sent to the visual cortex by the retinal ganglion cells are encoded by the mean firing rates observed on spike trains generated with a Poisson process. Using an information transmission approach, we evaluate the performances of two such codes, one based on the spike count and the other on the mean interspike interval, and compare the results with a rank order code, where the first ganglion cells to emit a spike are given a maximal weight. Our results show that the rate codes are far from optimal for fast information transmission and that the temporal structure of the spike train can be efficiently used to maximize the information transfer rate under conditions where each cell needs to fire only one spike.
DOI:10.1038/35035038URLPMID:11028996 [本文引用: 1]
Recent work has demonstrated the self-assembly of designed periodic two-dimensional arrays composed of DNA tiles, in which the intermolecular contacts are directed by 'sticky' ends. In a mathematical context, aperiodic mosaics may be formed by the self-assembly of 'Wang' tiles, a process that emulates the operation of a Turing machine. Macroscopic self-assembly has been used to perform computations; there is also a logical equivalence between DNA sticky ends and Wang tile edges. This suggests that the self-assembly of DNA-based tiles could be used to perform DNA-based computation. Algorithmic aperiodic self-assembly requires greater fidelity than periodic self-assembly, because correct tiles must compete with partially correct tiles. Here we report a one-dimensional algorithmic self-assembly of DNA triple-crossover molecules that can be used to execute four steps of a logical (cumulative XOR) operation on a string of binary bits.
DOI:10.3389/fnins.2016.00405URLPMID:27630540 [本文引用: 1]
DOI:10.1093/nar/gks230URLPMID:22416064
Isothermal nucleic acid amplification is becoming increasingly important for molecular diagnostics. Therefore, new computational tools are needed to facilitate assay design. In the isothermal EXPonential Amplification Reaction (EXPAR), template sequences with similar thermodynamic characteristics perform very differently. To understand what causes this variability, we characterized the performance of 384 template sequences, and used this data to develop two computational methods to predict EXPAR template performance based on sequence: a position weight matrix approach with support vector machine classifier, and RELIEF attribute evaluation with Naive Bayes classification. The methods identified well and poorly performing EXPAR templates with 67-70% sensitivity and 77-80% specificity. We combined these methods into a computational tool that can accelerate new assay design by ruling out likely poor performers. Furthermore, our data suggest that variability in template performance is linked to specific sequence motifs. Cytidine, a pyrimidine base, is over-represented in certain positions of well-performing templates. Guanosine and adenosine, both purine bases, are over-represented in similar regions of poorly performing templates, frequently as GA or AG dimers. Since polymerases have a higher affinity for purine oligonucleotides, polymerase binding to GA-rich regions of a single-stranded DNA template may promote non-specific amplification in EXPAR and other nucleic acid amplification reactions.
DOI:10.1177/0278364913491297URL [本文引用: 2]
We present a novel dataset captured from a VW station wagon for use in mobile robotics and autonomous driving research. In total, we recorded 6 hours of traffic scenarios at 10-100 Hz using a variety of sensor modalities such as high-resolution color and grayscale stereo cameras, a Velodyne 3D laser scanner and a high-precision GPS/IMU inertial navigation system. The scenarios are diverse, capturing real-world traffic situations, and range from freeways over rural areas to inner-city scenes with many static and dynamic objects. Our data is calibrated, synchronized and timestamped, and we provide the rectified and raw image sequences. Our dataset also contains object labels in the form of 3D tracklets, and we provide online benchmarks for stereo, optical flow, object detection and other tasks. This paper describes our recording platform, the data format and the utilities that we provide.
DOI:10.3389/fnins.2016.00049URLPMID:26941595 [本文引用: 2]
Standardized benchmarks in Computer Vision have greatly contributed to the advance of approaches to many problems in the field. If we want to enhance the visibility of event-driven vision and increase its impact, we will need benchmarks that allow comparison among different neuromorphic methods as well as comparison to Computer Vision conventional approaches. We present datasets to evaluate the accuracy of frame-free and frame-based approaches for tasks of visual navigation. Similar to conventional Computer Vision datasets, we provide synthetic and real scenes, with the synthetic data created with graphics packages, and the real data recorded using a mobile robotic platform carrying a dynamic and active pixel vision sensor (DAVIS) and an RGB+Depth sensor. For both datasets the cameras move with a rigid motion in a static scene, and the data includes the images, events, optic flow, 3D camera motion, and the depth of the scene, along with calibration procedures. Finally, we also provide simulated event data generated synthetically from well-known frame-based optical flow datasets.
DOI:10.1126/science.7973651URLPMID:7973651 [本文引用: 1]
The tools of molecular biology were used to solve an instance of the directed Hamiltonian path problem. A small graph was encoded in molecules of DNA, and the
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nbt0708-771URLPMID:18612298 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}