 ,1,2, 戴勇,3
,1,2, 戴勇,3Analysis of transcription factors in accessible open chromatin in the 18-trisomy syndrome based on single cell ATAC sequencing technique
Xiaofen Qiu1,2,3, Dong’e Tang3, Haiyan Yu3, Qiuyan Liao3, Zhiyang Hu3, Jun Zhou3, Xin Zhao3, Huiyan He3, Zhuojian Liang3, Chengming Xu2, Ming Yang,1,2, Yong Dai,3通讯作者: 戴勇,博士,主任医师,教授,研究方向:医学遗传和产前诊断。E-mail:daiyong22@aliyun.com杨明,主任医师,学士,硕士生导师,研究方向:传染病学。E-mail:yangming181@yeah.net
编委: 吴旭东
收稿日期:2020-11-8修回日期:2020-12-24网络出版日期:2021-01-20
| 基金资助: |
Received:2020-11-8Revised:2020-12-24Online:2021-01-20
| Fund supported: |
作者简介 About authors
邱晓芬,在读硕士研究生,专业方向:生物学。E-mail:

摘要
18-三体综合征是常见的常染色体非整倍体疾病之一,长期以来人们对18-三体综合征疾病的临床表型(如智力障碍、心脏、肾脏异常等)发生及发展相关的基因调控知之甚少。为探索影响该疾病表型的调控因子,本研究利用单细胞ATAC测序技术,对18-三体综合征和对照组的脐带血单个核细胞的开放性染色质区域的转录因子进行分析。捕获11,611个细胞构建的单细胞文库鉴定得到7种主要免疫细胞群,细胞数量统计的结果提示18-三体综合征的免疫系统异常。通过分析开放性染色质区域,筛选得到14个转录因子(P<0.05,|FC|>1.2)。采用实时荧光定量PCR验证其中4个转录因子(TEAD1、TEAD2、TEAD4、Twist2)的相对表达量的结果符合预期。上述研究结果表明这4个转录因子可能与18-三体综合征患者心脏和骨骼发育的异常相关,为18-三体综合征表型的发生及发展的机制研究提供候选分子。
关键词:
Abstract
Trisomy 18 syndrome is one of the most common autosomal aneuploidy disorders. Little is known about the genetic regulation leading to the clinical phenotypes associated with the occurrence and development of trisomy 18 syndrome disorders (e.g., mental retardation, cardiac and renal abnormalities). To explore the regulatory factors that influence the phenotypes of the disease, this study used single-cell ATAC sequencing to analyze transcription factors in the accessibility chromatin regions of the single-nucleus cells of the cord blood from 18-trisomy syndrome and control subjects. A single-cell library constructed by capturing 11,611 cells identified seven major immune cell populations, and the results of cell number statistics suggested the presence of abnormalities in the immune system of 18-trisomy syndrome patients. Fourteen transcription factors (P<0.05, |FC|>1.2) were identified by analyzed accessibility chromatin regions. The relative expression levels of four of these transcription factors (TEAD1, TEAD2, TEAD4, Twist2) were confirmed using real-time quantitative fluorescence PCR. In conjunction with information from the literature, this study suggests that these four transcription factors may be associated with abnormalities in cardiac and skeletal development in patients with the 18-trisomy syndrome, thereby providing candidate molecules for mechanistic studies on the occurrence and development of the 18-trisomy syndrome phenotypes.
Keywords:
PDF (1429KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
邱晓芬, 汤冬娥, 虞海燕, 廖秋燕, 胡芷洋, 周俊, 赵鑫, 何慧燕, 梁灼健, 许承明, 杨明, 戴勇. 基于单细胞ATAC测序技术对18-三体综合征染色质开放性区域转录因子的分析. 遗传[J], 2021, 43(1): 74-83 doi:10.16288/j.yczz.20-283
Xiaofen Qiu.
18-三体综合征(trisomy 18 syndrome)于1960年首次被遗传学研究者爱德华(Edwards JH)等[1]和史密斯(Smith DW)等[2]报道,因此又常被称为爱德华兹综合征(Edwards syndrome,ES),由于当时染色体鉴定技术尚不完善,第3条18号染色体被认为是17号染色体。ES是由于存在额外的18号染色体而导致的一系列不同身体器官和系统异常的疾病,根据患者核型的不同可分为游离型(症状典型,占90%以上)、嵌合型和易位型,活产率1/6000~1/8000,总体患病率1/2500~1/2600[3],活产率远低于总体患病率的原因是ES有很高的胎儿丢失率和死产率[4],同时早产率高于平均水平[5]。活产儿约50%在出生一周内死亡,其余大多数在一年内死亡,死亡通常是由于中枢性呼吸暂停,上呼吸道阻塞,呼吸功能不全,心力衰竭等综合因素;活产患儿平均寿命为10天[6]。ES的临床特征包括严重的和轻微的异常,产前和出生后生长缺陷,明显的精神和认知障碍。典型的轻微异常包括特征性的头面部特征(后枕部突出,耳位低,耳廓形状异常,眼裂短,小颌),拳头紧握,压指,指甲发育不良,踇趾短,胸骨短,腹股沟疝,骨盆小。最常见的重大畸形是心脏和肾脏畸形,心脏室间隔缺损,马蹄肾,肾盂缺水[3]。ES是染色体非整倍性疾病,目前无法根治,主要依靠产前筛查进行预防,对于已出生的患儿,可采用外科手术等进行延长寿命的干预措施[7]。超声波检查,人类染色体鉴定技术和高通量测序等技术的发展有效地增加了对胎儿染色体异常疾病的早期筛查力度,对预防出生缺陷具有重要意义。FitzPatrick等[8]将来自21-三体综合征和13-三体综合征病例羊水样本原代培养细胞的mRNA与来自正常细胞的mRNA的cDNA阵列进行对比杂交,当阵列cDNA按染色体位点分组时,可以清楚地识别出相关三体染色体显示出最显著的转录失调,与正常细胞相比,三体染色体上基因的平均转录水平仅提高了1.1倍。他们的数据显示大多数(>95%)>±2SD异常表达的基因没有定位到三体染色体上,并且在13-三体综合征中比21-三体综合征中发生了显著的差异表达更普遍。这暗示着基因组调控的机制可能比以往认为的更远距离上起作用,即一个染色体的增加可能会影响其他染色体基因的转录水平,三体染色体上转录因子的差异表达将产生全基因组转录失调的影响,他们得出一个结论:三体染色体上基因的略微上调导致了继发性的、广泛性的和更极端的转录失调,这种错误调控的程度可能决定了表型的严重程度。因此,探究18-三体综合征染色质的开放性区域的转录因子,不仅可以加强对18-三体综合征临床表型的发生和发展中调控因子变化的理解,而且也可为疾病的诊治提供潜在的候选分子。
随着单细胞测序技术的发展,近几年相继出现单细胞转录组测序技术、单细胞染色质可及性测序等技术。单细胞测序技术克服了传统的高通量测序技术对细胞的平均水平的基因表达情况进行分析的局限性。2015年出现了基于转座酶的高通量染色质可及性单细胞测序法scATAC-seq (single cell assay for transposase accessible chromatin using sequencing)[9],该方法建库过程不包含读段的长度筛选,可以同时检测开放性DNA区域和相应的转录因子,是基于Tn5转座酶可及染色质的开放性区域的单细胞测序的高通量测序技术。传统测序建库的过程包括DNA片段化、末端修复、接头连接、文库扩增、多次纯化分选等步骤,耗时长,但是将Tn5转座酶用于测序文库构建时,可将DNA片段化、末端修复、接头连接等多步反应转变为一步反应,缩短建库时间。染色质的可及性,也称为染色质的可接近性,是指细胞核内一些参与DNA复制或转录的大分子能够与染色质中DNA所能发生物理接触的程度,由核小体或其他染色质结合因子在染色质上的占据情况决定,也可以通过染色质中DNA对DNase的敏感性评估[10]。不同类型的细胞或在不同的生理条件或外界刺激下,细胞核中的染色质会呈现出不同的结构和状态,并且在发育期间对外部刺激的响应表现为动态变化,这些差异或动态变化的状态的表现形式之一就是染色质可及性的变化[11]。表观遗传调控的重要机制之一是通过改变染色质可及性来调控基因表达的,简而言之,染色质可及性的改变对基因的表达起着重要的调控作用。
为了定位细胞水平上与18-三体综合发生相关的调控因子,揭示相关的转录因子,本研究利用scATAC-seq技术对18-三体综合征的脐带血单个核细胞以及对照组的脐带血单个核细胞进行无细胞差异的测序及分析,探索性的对18-三体综合征的转录前调控水平进行初步的构建,揭示疾病相关活性基因的定位以及细胞特异性的相关基因作用程度。
1 材料与方法
1.1 人脐带血单个核细胞提取
选取深圳市人民医院接诊的经人类染色体核型鉴定的脐带血进行研究。纳入本次研究的实验组(N=1)材料的人类染色体G显带鉴定结果为18-三体综合征,核型结果是:47,XY,+18;对照组(N=1)材料的人类染色体G显带鉴定结果为:46,XY。2~3 mL脐带血于EDTA抗凝管中,采用淋巴细胞分离液提取脐带血单个核细胞保存备用,检测细胞活性大于80%后进行单细胞文库构建。本研究由深圳市人民医院医学伦理委员会批准,且所有孕妇签署了知情同意书。1.2 scATAC-seq文库构建及细胞类型聚类与鉴定
ScATAC-seq使用10×微流控测序平台。本研究样本在10× Chromium平台上生成脐带血单个核细胞scATAC-seq数据的所有步骤,包括细胞核提取和悬浮、文库构建、仪器和测序设置,都遵循官方推荐,可在此下载:Table 1
表1
表1qPCR的反应体系
Table 1
| 试剂 | 剂量(μL) |
|---|---|
| 2 × ChamQ Universal SYBR qPCR Master Mix | 10 |
| Primer F | 0.4 |
| Primer R | 0.4 |
| cDNA | 0.3 |
| H2O | 8.7 |
| 合计 | 20 |
新窗口打开|下载CSV
1.3 实时荧光定量PCR检测差异表达的转录因子
为了验证这些转录因子是否在疾病组中是否差异表达,采用实时荧光定量PCR (quantitative real- time PCR, qPCR)检测TEAD1、TEAD2、TEAD4和Twist2这4个转录因子在疾病组和对照组中的表达量,设置3个实验重复。步骤如下:(1) Trizol法提取细胞悬液的RNA;(2)使用提取的RNA合成cDNA:采用反转录试剂VAZYME R222-01进行cDNA逆转录合成,冰上配制逆转录体系(20 μL):RNA加1 pg~ 1 μg,5×HiScript?II qRT SuperMixa加2 μL,加入RNase-free H2O将体系的体积配至20 μL,50℃温育15 min,85℃高温灭活5 s,收集反转录的cDNA,用于荧光定量检测;(3)以反转录的cDNA为模板,分别加入引物T-β-actin,xw-0000291,caspase-3,Bcl-2,Beclin1,Bax进行相对定量分析,反应体系如表1,将96-PCR板置于Realtime PCR仪(Biometra Tone)上进行PCR反应。所有的指标均按以下程序进行:95℃,30秒;40个PCR循环(95℃,5 s;60℃,34 s (收集荧光))。为了建立PCR产物的熔解曲线,扩增反应结束后,按95℃,15 s;60℃,60 s;95℃,15 s进行反应;并从60℃缓慢加热到99℃ (仪器自动进行Ramp Rate为0.05 ℃/s),反应的引物序列见表2。各样品的目的基因和管家基因分别进行Realtime PCR反应,根据测得的各样品目的基因和管家基因的Ct值进行ddct法的相对定量分析。Table 2
表2
表2qPCR的引物序列
Table 2
| 名称 | 序列(5'→ 3') | 碱基数 |
|---|---|---|
| H-TEAD1-F | GCCACTGCCATTCATAACAAGC | 22 |
| H-TEAD1-R | CCTGGCTGCCCTGTTTGAATC | 21 |
| H-TEAD2-F | CCATTCTCACAGACACCGTTCAC | 23 |
| H-TEAD2-R | TCCACGAAGGCTGAGAACTCTAC | 23 |
| H-TEAD4-F | GACACGTACAACAAGCACCTG | 21 |
| H-TEAD4-R | CCGTTCGAAGAGATCCTTGAGTC | 23 |
| H-Twist2-F | CCCTCTGACAAGCTGAGCAAG | 21 |
| H-Twist2-R | CATGCGCCACACGGAGAAG | 19 |
新窗口打开|下载CSV
2 结果与分析
2.1 聚类鉴定得到7类主要免疫细胞群
通过10× Genomics平台的微流控系统对实验组(ES)和对照组(NC)进行scATAC-seq。在去除低质量和不合格的细胞后,一共捕获了11,611个细胞,其中ES组5296个细胞,NC组6315个细胞。通过对单细胞测序得到的数据进行聚类分析后得到7个不同类别的类群。对这些细胞类群进行无抗体的细胞标记基因鉴定,确定了7类主要的免疫细胞,然后用已知的细胞标记基因进行鉴定[14,15]。用CD3D和IL7R鉴定T细胞(T),用MS4A1、CD79A、CD79B鉴定B细胞(B),用GZMB和NKG7鉴定自然杀伤细胞(natural killer cell, NK),用CD83和IL3RA鉴定树突状细胞(dendritic cell, DC),用CD14、CD36、CD68鉴定单核细胞(monocyte),用CD27和CD4鉴定CD4+T细胞,用CD8A、CD8B、CD3E鉴定CD8+T细胞(图1),每种颜色峰的高度代表对应细胞类型染色质的开放程度。统计各细胞群数量的结果显示,ES组中,T细胞,NK细胞和DC细胞的细胞数量显著减少,B细胞和CD4+T细胞的细胞数量显著增加(图2)。研究结果提示,18-三体综合征患儿在子宫内免疫系统发育异常。让人兴奋的是,早在1994年,就有研究人员[16]研究了18-三体综合胎儿血液的免疫系统,在妊娠20~36周时通过脐带穿刺术从8个18-三体综合征的胎中获得的胎儿血液中淋巴细胞亚群,使用流式细胞仪分析并计数,得出结论:与染色体正常胎儿的相比,在18-三体综合征中,平均T细胞和NK细胞计数显著降低。图1
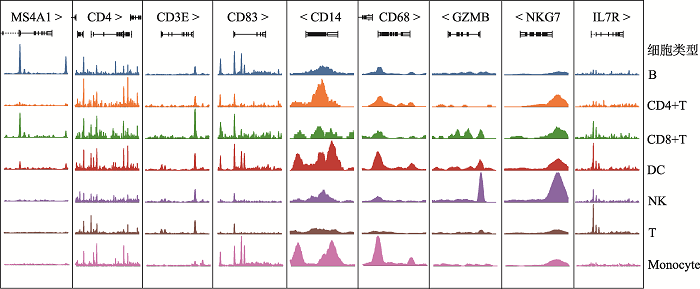 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1部分特异性细胞标记基因对应染色质可及性开放程度
Fig. 1Part of the specific cell marker genes correspond to chromatin accessibility
图2
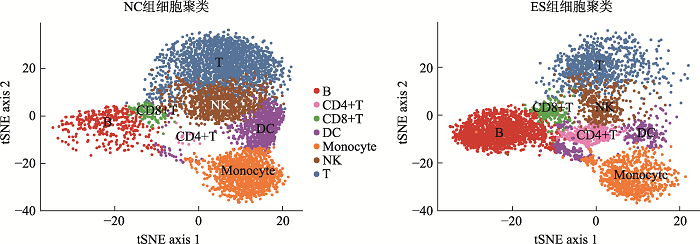 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2脐带血单个核细胞t-SNE聚类与鉴定
Fig. 2T-SNE clustering and identification of cord blood mononuclear cells
2.2 可及性位点差异转录因子
在细胞层面上对18-三体综合征进行了差异可及性位点的motif分析,motif是转录因子结合的基序,通过motif可以找到结合该motif的转录因子。以P<0.05, |FC|>1.2为筛选阈值,其中B细胞筛选得到11个转录因子:Esrra、PBX1、TEAD1、TEAD2、TEAD4、Twist2、HOXC12、ZNF410、SREBF1、FOSL2::JUN、FOS::JUNB;单核细胞(Monocyte)筛选得到7个转录因子:Esrra、PBX1、TEAD1、TEAD2、HOXC12、TBX20、ZNF410。T细胞筛选得到6个因子:Esrra、PBX1、TEAD1、TEAD2、TEAD4、MYBL1。自然杀伤细胞(NK)筛选得到1个差异转录因子:SREBF2;树突细胞群(DC) 1个差异转录因子:HOXC12;CD8+T细胞群筛选得到1个转录因子:TEAD1;CD4+T细胞群没有得到筛选到转录因子(图4)。通过整理,一共得到筛选得到14个的转录因子:Esrra、PBX1、TEAD1、TEAD2、TEAD4、Twist2、HOXC12、ZNF410、SREBF1、FOSL2::JUN、FOS::JUNB、SREBF2、TBX20、MYBL1。随后我们对不同细胞类群的差异转录因子调控的基因进行了GO功能分析。图5展示了B细胞群差异转录因子调控基因功能分析的结果,其他细胞群的结果见附加材料(附图1~5)。结果显示在已鉴定到的7类主要免疫细胞的主要生物进程功能是转录调控,信号转导;细胞组分功能主要集中在细胞核、细胞膜和细胞质;分子功能主要是蛋白质结合、金属离子结合、DNA结合。图3
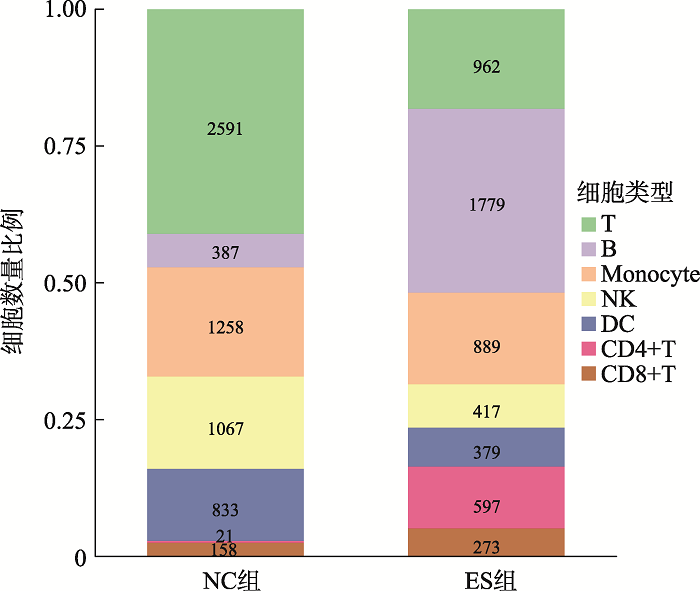 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图37类细胞群数量及比例
Fig. 3The number and proportion of 7 cell groups
图4
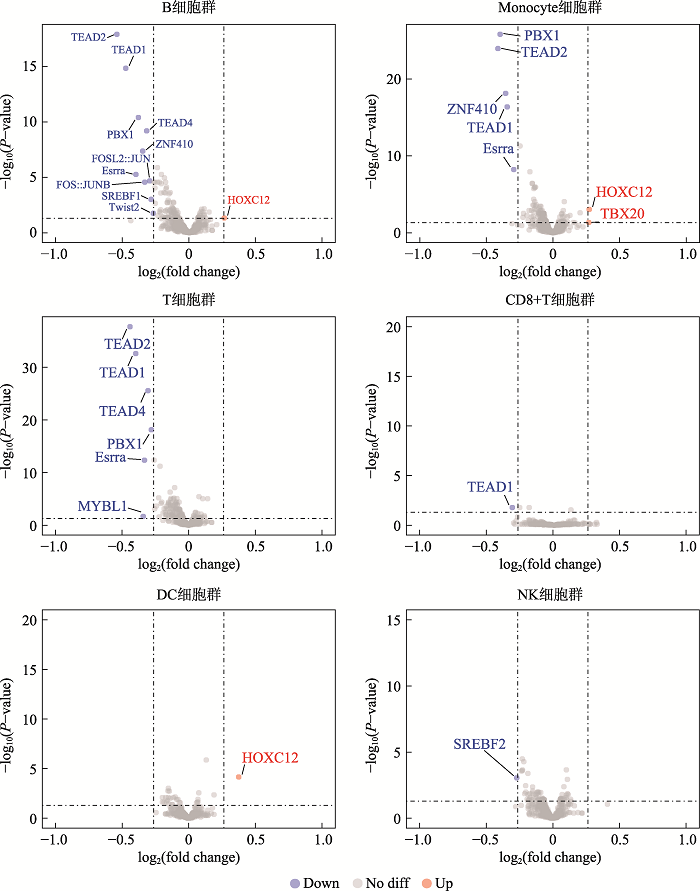 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同细胞群转录因子差异表达火山图
Fig. 4Differential expression of transcription factors in different cell populations
图5
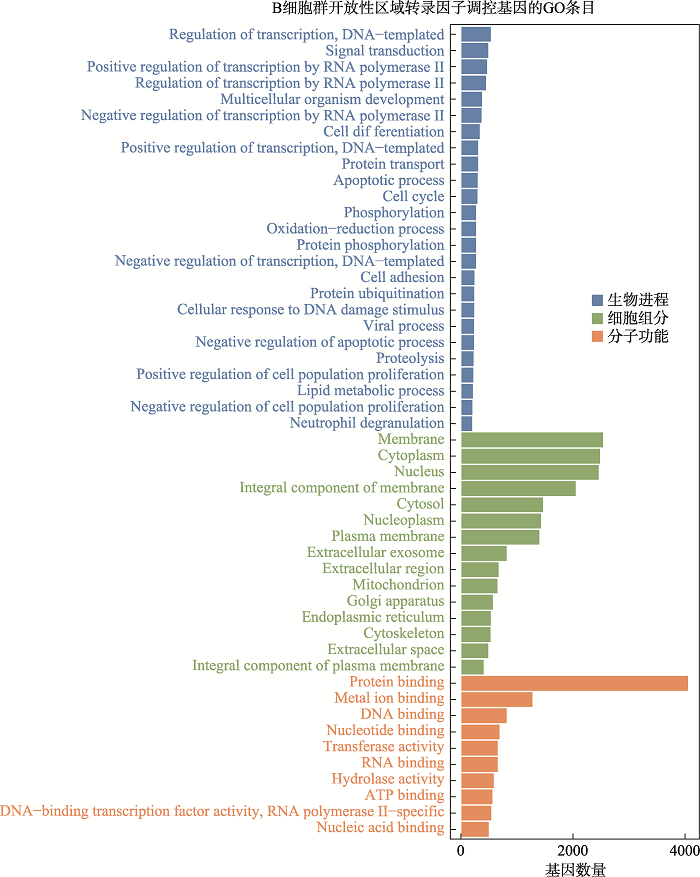 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5B细胞开放性区域差异转录因子调控基因GO分析
Fig. 5GO analysis of genes regulated by differential transcription factors in accessible chromatin region of B cells
2.3 转录因子qPCR相对表达量
qPCR实验验证了4个转录因子相对表达量的结果(图6),4个实验组(ES)的转录因子(TEAD1、TEAD2、TEAD4、Twist2)的相对表达量低于对照组(NC),符合实验预期的结果。图6
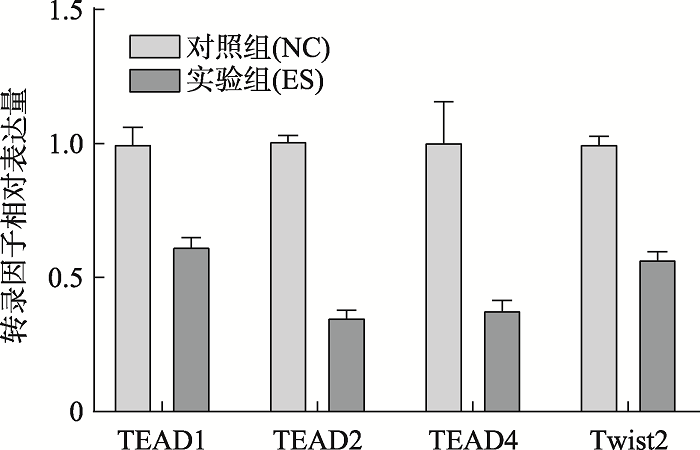 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6脐带血单个核细胞转录因子qPCR相对表达量
Fig. 6Relative expression of transcription factor qPCR in mononuclear cells of cord blood
3 讨论
单细胞ATAC-seq技术能在单细胞水平上捕获每种细胞类型的开放性染色质区域,实现无标记鉴定细胞类型特异性的顺式和反式调控元件和与疾病相关转录因子活性的定位。本研究得到了疾病组和对照组的单细胞文库,鉴定得到7类主要的免疫细胞群,筛选得到14个转录因子,选取了4个通过qPCR验证的转录因子进行讨论。本研究的不足是样本数量不足。采用单细胞测序技术,得出7种主要的免疫细胞数量比例的结果以及差异转录因子,对18-三体综合征疾病的认识有意义。已有研究者通过对18-三体综合征胎儿的脐带血进行流式细胞仪分析后报道[16]18-三体综合征胎儿宫内时期的免疫系统的异常情况。本次研究通过单细胞测序技术,进行无抗体的细胞群鉴定,发现了18-三体综合征胎儿脐带血单个核细胞比例与对照组的差异。随后我们发现了疾病组差异表达的转录因子,验证了其中4个(TEAD1、TEAD2、TEAD4、Twist2)转录因子的相对表达量与实验结果相符。
转录增强关联域(transcriptional enhanced associate domain, TEAD)蛋白家族由4个旁系转录因子组成,其功能是调节基因表达,以响应Hippo信号通路,Hippo信号通路是调控器官发育、细胞生长、增殖和组织稳态和再生的重要转录信号通路[17,18,19]。2019年Akerberg BN等研究发现TEAD1是心脏转录调控网络的核心组成部分,控制心脏调控区域和心肌细胞特异性基因功能[20]。Joshi S等[21]的研究表明TEAD因子在肌细胞分化中具有特定作用,TEAD1是已知的Hippo信号转导的转录因子,参与心脏发育。Wen T等[22] 的研究揭示了TEAD1在小鼠心血管发育中的关键作用,并确定了TEAD1在遗传调节层次的上游起作用,以促成平滑肌收缩。Osman I等[23] 的研究表明TEAD1通过转录诱导SLC1A5促进血管平滑肌细胞增殖,从而激活mTORC1信号传导并促进新血管内膜形成。同时,Liu R等[24]的研究表明TEAD1在维持正常的成人心脏功能中具有非常重要的作用。
Twist2是Twist子家族的高度保守成员,负责间质细胞谱系中发育程序的转录调控[25]。碱性螺旋-环-螺旋(Basic helix-loop-helix protein, b HLH)家族成员Twist2对间质细胞系的发生和发育起转录调节作用,直接或间接机制发挥分子开关功能,从而激活或抑制靶基因,Twist2对骨骼发育存在影响[26]。Liu N等[27]通过在小鼠中表达Twist2转录因子的祖细胞的谱系追踪,发现了位于成年骨骼肌基底层之外的肌源系,并发现Twist2祖细胞在肌肉再生过程中对IIb/x型肌纤维有重要贡献。同时,2019年Albizua I等[28]对18-三体综合征患者的基因组表达谱分析后得出结论:SHOX2、TBX4、ALX3、ALX4和Twist1等关键转录因子在18-三体相关骨骼发育中起关键作用,他们的研究结果中报告了Twist家族的转录因子Twist1表达降低的小鼠表现出多指畸形。2010年,Koide等[29]对18-三体综合征和对照组的羊水上清提取总RNA,发现与35个基因与肾上腺发育有关的基因的显著下调,其中17个是焦点基因。
本研究在单细胞染色质可及性的水平上揭示免疫细胞类群异常的转录因子,特别是经过实验证实下调表达4个的转录因子(TEAD1、TEAD2、TEAD4、Twist2)。TEAD (TEAD1、TEAD2、TEAD4)转录因子家族与成肌细胞分化有关,特别是TEAD1与心脏,心血管发育等有关,的下调表达可能会影响18-三体综合征患者心脏的正常的发育调控;Twist2是可能导致18-三体综合征的骨骼异常的转录因子。揭示18-三体综合征表型发生和发展的机制,仍有更远的路要走。
附录
附加材料详见电子版附图1
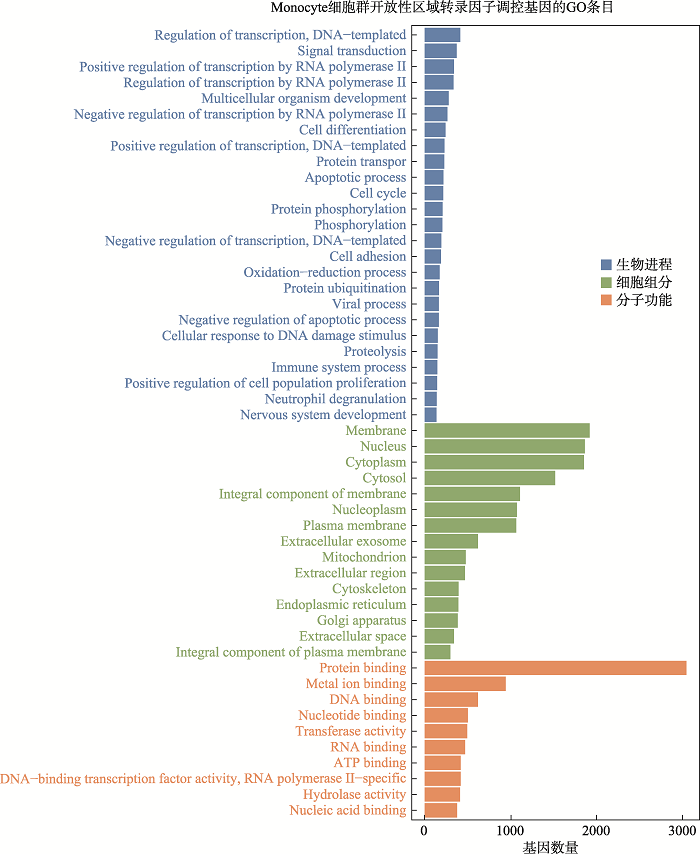 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1Monocytes开放性区域差异转录因子调控基因GO分析
Suppl fig. 1GO analysis of genes regulated by differential transcription factors in accessible chromatin region of monocytes
附图2
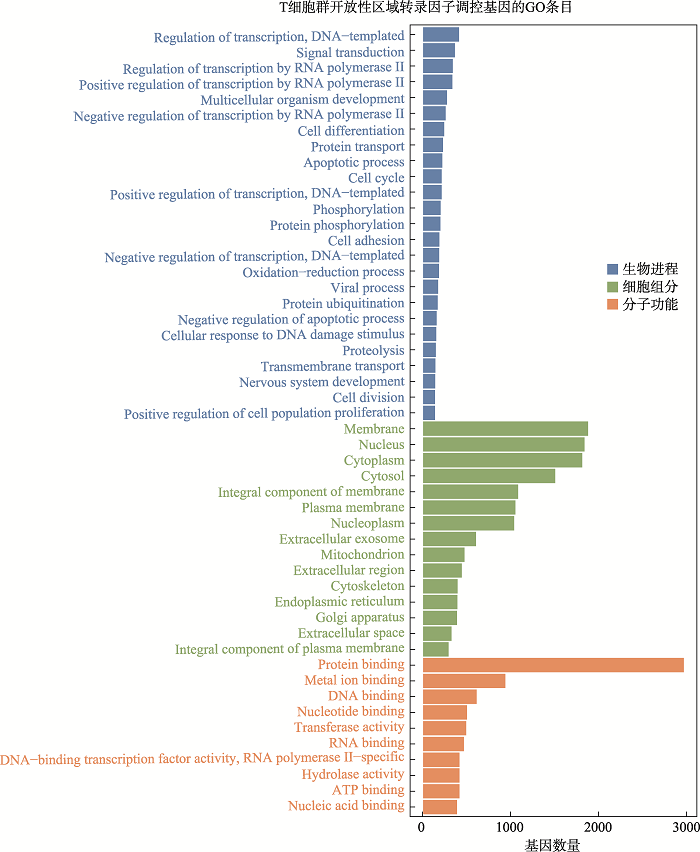 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图2T细胞群开放性区域差异转录因子调控基因GO分析
Suppl fig. 2GO analysis of genes regulated by differential transcription factors in accessible chromatin region of T cells
附图3
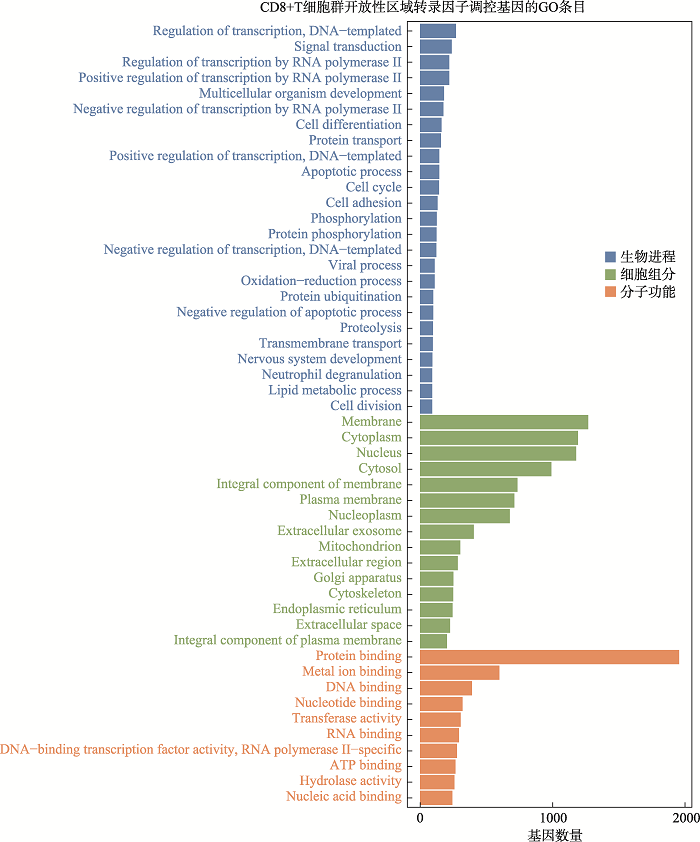 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图3CD8+T细胞群开放性区域差异转录因子调控基因GO分析
Suppl fig. 3GO analysis of genes regulated by differential transcription factors in accessible chromatin region of CD8+T cells
附图4
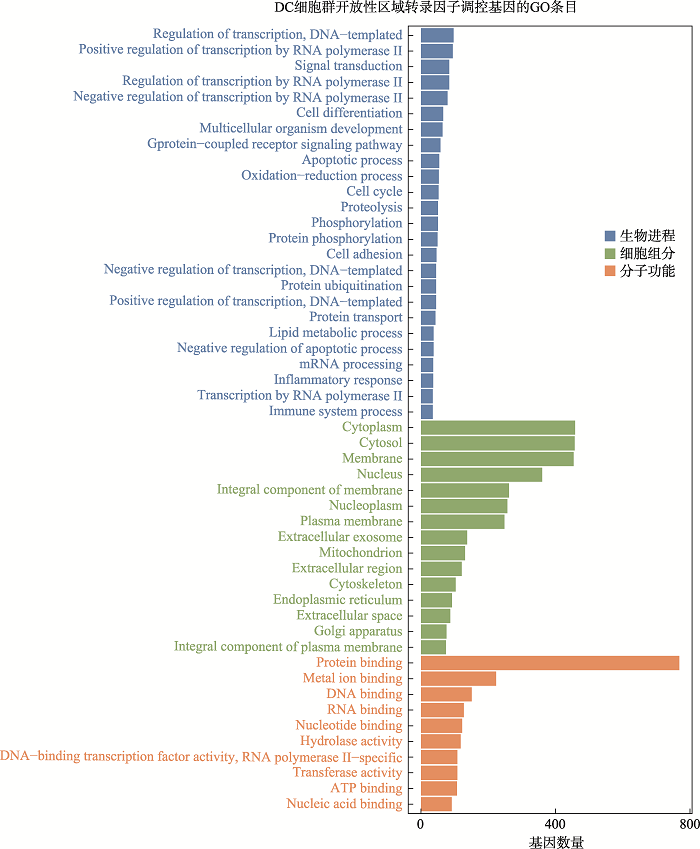 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图4DC细胞群开放性区域差异转录因子调控基因GO分析
Suppl fig. 4GO analysis of genes regulated by differential transcription factors in accessible chromatin region of DC
附图5
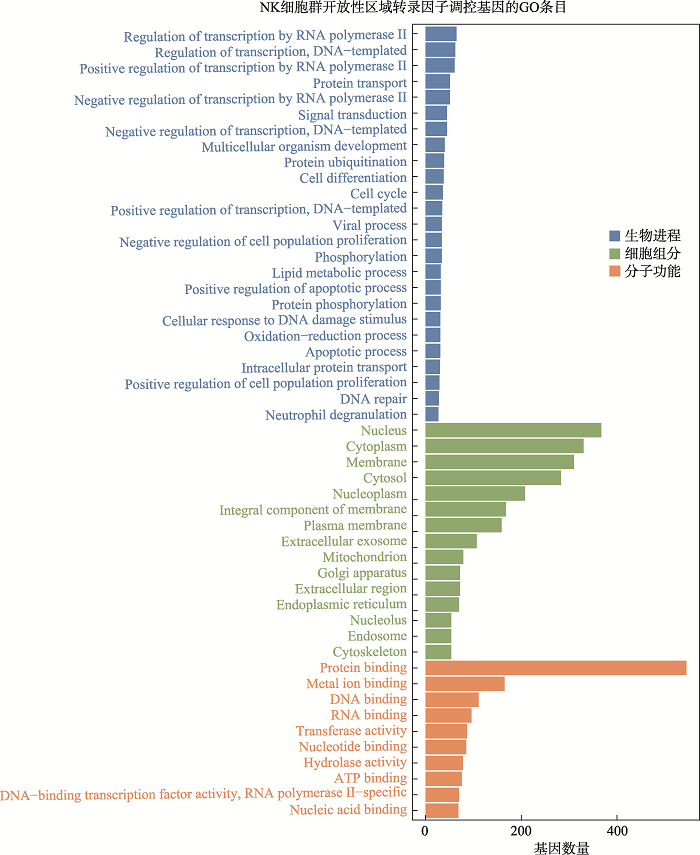 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图5NK细胞群开放性区域差异转录因子调控基因GO分析
Suppl fig. 5GO analysis of genes regulated by differential transcription factors in accessible chromatin region of NK
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/s0140-6736(60)90675-9URLPMID:13819419 [本文引用: 1]
DOI:10.1016/s0022-3476(60)80241-7URLPMID:13831938 [本文引用: 1]
DOI:10.1186/1750-1172-7-81URLPMID:23088440 [本文引用: 2]

The trisomy 18 syndrome, also known as Edwards syndrome, is a common chromosomal disorder due to the presence of an extra chromosome 18, either full, mosaic trisomy, or partial trisomy 18q. The condition is the second most common autosomal trisomy syndrome after trisomy 21. The live born prevalence is estimated as 1/6,000-1/8,000, but the overall prevalence is higher (1/2500-1/2600) due to the high frequency of fetal loss and pregnancy termination after prenatal diagnosis. The prevalence of trisomy 18 rises with the increasing maternal age. The recurrence risk for a family with a child with full trisomy 18 is about 1%. Currently most cases of trisomy 18 are prenatally diagnosed, based on screening by maternal age, maternal serum marker screening, or detection of sonographic abnormalities (e.g., increased nuchal translucency thickness, growth retardation, choroid plexus cyst, overlapping of fingers, and congenital heart defects ). The recognizable syndrome pattern consists of major and minor anomalies, prenatal and postnatal growth deficiency, an increased risk of neonatal and infant mortality, and marked psychomotor and cognitive disability. Typical minor anomalies include characteristic craniofacial features, clenched fist with overriding fingers, small fingernails, underdeveloped thumbs, and short sternum. The presence of major malformations is common, and the most frequent are heart and kidney anomalies. Feeding problems occur consistently and may require enteral nutrition. Despite the well known infant mortality, approximately 50% of babies with trisomy 18 live longer than 1 week and about 5-10% of children beyond the first year. The major causes of death include central apnea, cardiac failure due to cardiac malformations, respiratory insufficiency due to hypoventilation, aspiration, or upper airway obstruction and, likely, the combination of these and other factors (including decisions regarding aggressive care). Upper airway obstruction is likely more common than previously realized and should be investigated when full care is opted by the family and medical team. The complexity and the severity of the clinical presentation at birth and the high neonatal and infant mortality make the perinatal and neonatal management of babies with trisomy 18 particularly challenging, controversial, and unique among multiple congenital anomaly syndromes. Health supervision should be diligent, especially in the first 12 months of life, and can require multiple pediatric and specialist evaluations.
DOI:10.1002/ajmg.a.38123URLPMID:28328132 [本文引用: 1]
Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13) both have high natural fetal loss rates. The aim of this study was to provide estimates of these fetal loss rates by single gestational week of age using data from the National Down Syndrome Cytogenetic Register. Data from all pregnancies with Edwards or Patau syndrome that were prenatally detected in England and Wales from 2004 to 2014 was analyzed using Kaplan-Meier survival estimates. Pregnancies were entered into the analysis at the time of gestation at diagnosis, and were considered
DOI:10.1002/ajmg.a.31241URLPMID:16652360 [本文引用: 1]
To investigate the pregnancy outcome of fetuses affected with trisomy 18, we analyzed 63 cases diagnosed at our hospital from January 1993 to December 2004. Twenty-nine were males and 34 were females. Fifty-eight were prenatally diagnosed, and in 16 (27.6%) of them intrauterine fetal death (IUFD) occurred between 28 weeks and 41 weeks gestation (34.6 +/- 3.9 weeks, Mean +/- SD). Ten (17.2%) fetuses died during labor and their age ranged from 30 weeks to 40 weeks of gestation. The total number of cases ending in fetal demise was 26 (44.8%) and the mean gestational age at the time of fetal demise was 35.0 +/- 3.6 weeks (Mean +/- SD). All liveborn infants (n = 36) were born after 31 weeks gestation. In our study the preterm birth ratio for trisomy 18 is 34.8%, which is much higher than the ratio for the general population. Females are more likely than males to be long-term survivors. These data are helpful in the counseling of parents faced with the difficult decision of whether or not to continue a pregnancy with a fetus affected with trisomy 18.
DOI:10.1542/peds.111.4.777URLPMID:12671111 [本文引用: 1]
OBJECTIVE: Although trisomy 13 and trisomy 18 are generally considered to be lethal, long-term survival of patients has been reported. We sought to evaluate mortality in people with trisomy 13 or 18 using 2 population-based strategies. METHODS: In the first analysis, infants who had trisomy 13 or 18 and were born during 1968-1999 were identified using the Metropolitan Atlanta Congenital Defects Program, a population-based birth defects surveillance system. Dates of death were documented using hospital records, Georgia vital records, and the National Death Index. In the second analysis, we used the Multiple-Cause Mortality Files compiled from US death certificates from 1979 through 1997. Using these 2 analyses, we examined median survival time or median age at death, survival beyond 1 year of age, and factors associated with longer survival. RESULTS: Using Metropolitan Atlanta Congenital Defects Program, we identified 70 liveborn infants with trisomy 13 and 114 liveborn infants with trisomy 18. Median survival time was 7 days (95% confidence interval [CI]: 3-15) for people with trisomy 13 and 14.5 days (95% CI: 8-28) for people with trisomy 18. For each condition, 91% of infants died within the first year. Neither race nor gender affected survival for trisomy 13, but for trisomy 18, girls and infants of races other than white seemed to survive longer. The presence of a heart defect did not seem to affect survival for either condition. Using MCMF, we identified 5515 people with trisomy 13 and 8750 people with trisomy 18 listed on their death certificates. Median ages at death for people with trisomy 13 and trisomy 18 both were 10 days; 5.6% of people with trisomy 13 and 5.6% of people with trisomy 18 died at age 1 year or greater. Race and gender seemed to affect survival in both conditions, with girls and blacks showing higher median ages at death. CONCLUSIONS: Although survival is greatly affected by trisomy 13 and trisomy 18, 5% to 10% of people with these conditions survive beyond the first year of life. These population-based data are useful to clinicians who care for patients with these trisomies or counsel families with infants or fetuses who have a diagnosis of trisomy 13 or 18.
DOI:10.1001/jama.2016.9819URLPMID:27458947 [本文引用: 1]
IMPORTANCE: Trisomy 13 and 18 are genetic diagnoses with characteristic physical features, organ anomalies, and neurodevelopmental disability. Most children with these disorders die shortly after birth, although limited data suggest some children survive longer. Surgeries are controversial, and little evidence is available about outcomes. OBJECTIVE: To describe survival and utilization of any type of surgery among children with trisomy 13 and 18 born over a 21-year period in Ontario, Canada. DESIGN, SETTING, AND PARTICIPANTS: This retrospective cohort study used linked health administrative databases to identify children born in Ontario between April 1, 1991, and March 31, 2012, with a diagnosis code for trisomy 13 or 18 on a hospital record in the first year of life. Survival was calculated from birth and death dates; children living on March 31, 2013, were censored at their last clinical encounter. EXPOSURES: All procedures classified as occurring in an operating room through March 31, 2013, were categorized as major, intermediate, or minor surgeries. MAIN OUTCOMES AND MEASURES: Survival and surgical procedure utilization. RESULTS: The cohorts included 174 children with trisomy 13 (mean [SD] birth weight, 2.5 [0.7] kg; 98 [56.3%] female); and 254 children with trisomy 18 (mean birth weight, 1.8 [0.7] kg; 157 [61.8%] female), with follow-up times of 0 to more than 7000 days. Median (interquartile range [IQR]) survival times were 12.5 (2-195) days for trisomy 13 and 9 (2-92) days for trisomy 18. Mean 1-year survival for trisomy 13 was 19.8% (95% CI, 14.2%-26.1%) and 12.6% (95% CI, 8.9%-17.1%) for trisomy 18. Ten-year survival for trisomy 13 was 12.9% (95% CI, 8.4%-18.5%) and 9.8% (95% CI, 6.4%-14.0%) for trisomy 18. Survival did not change over the study period. Forty-one children (23.6%) with trisomy 13 and 35 children (13.8%) with trisomy 18 underwent surgeries, ranging from myringotomy to complex cardiac repair. Median age at first surgery for trisomy 13 was 92 (IQR, 30.5-384.5) days and for trisomy 18, it was 205.5 (IQR, 20.0-518.0) days. Kaplan-Meier curves showed 1-year survival after first surgery of 70.7% (95% CI, 54.3%-82.2%; n = 23) for trisomy 13 and 68.6% (95% CI, 50.5%-81.2%; n = 29) for trisomy 18. CONCLUSIONS AND RELEVANCE: Among children born with trisomy 13 or 18 in Ontario, early mortality was the most common outcome, but 10% to 13% survived for 10 years. Among children who underwent surgical interventions, 1-year survival was high.
DOI:10.1093/hmg/11.26.3249URLPMID:12471051 [本文引用: 1]
We present transcriptome analyses of primary cultures of human fetal cells from pregnancies affected with trisomy 21 (t21) and trisomy 13 (t13). Pooled mRNA samples from t21 and t13 cases were used for comparative hybridizations to cDNA arrays with pooled mRNA from normal cells. When the array cDNAs were grouped by chromosomal location the relevant trisomic chromosome could be clearly identified as showing the most significant misregulation. The average level of transcription on the trisomic chromosome was increased only approximately 1.1-fold compared to normal cells on array analysis. Since the karyotype could be accurately predicted by the transcriptome this could provide a novel method of detecting aneusomy of unknown position. Subsequent analysis of individuals cases demonstrated that variation in transcriptional profiles between samples within each class made transcriptional karyotyping difficult without pooling or the use of arrays with a higher proportion of all human cDNAs. Interestingly, consistent differences in the relative expression levels between chromosomes were detected suggesting that genomic control mechanisms may act over larger distances than previously thought. Most (>95%) >+/-2 SD misregulated genes did not map to the trisomic chromosome and significant misregulation was more common in t13 than t21. These data support a model of a subtle primary upregulation of genes on the trisomic chromosome resulting in a secondary, generalized and more extreme transcriptional misregulation. It seems likely that the degree of this misregulation determines the severity of the phenotype in most aneuploidy.
DOI:10.1038/nature14590URLPMID:26083756 [本文引用: 1]
Cell-to-cell variation is a universal feature of life that affects a wide range of biological phenomena, from developmental plasticity to tumour heterogeneity. Although recent advances have improved our ability to document cellular phenotypic variation, the fundamental mechanisms that generate variability from identical DNA sequences remain elusive. Here we reveal the landscape and principles of mammalian DNA regulatory variation by developing a robust method for mapping the accessible genome of individual cells by assay for transposase-accessible chromatin using sequencing (ATAC-seq) integrated into a programmable microfluidics platform. Single-cell ATAC-seq (scATAC-seq) maps from hundreds of single cells in aggregate closely resemble accessibility profiles from tens of millions of cells and provide insights into cell-to-cell variation. Accessibility variance is systematically associated with specific trans-factors and cis-elements, and we discover combinations of trans-factors associated with either induction or suppression of cell-to-cell variability. We further identify sets of trans-factors associated with cell-type-specific accessibility variance across eight cell types. Targeted perturbations of cell cycle or transcription factor signalling evoke stimulus-specific changes in this observed variability. The pattern of accessibility variation in cis across the genome recapitulates chromosome compartments de novo, linking single-cell accessibility variation to three-dimensional genome organization. Single-cell analysis of DNA accessibility provides new insight into cellular variation of the 'regulome'.
DOI:10.1146/annurev.genom.7.080505.115613URLPMID:16756479 [本文引用: 1]
Centromeres are the elements of chromosomes that assemble the proteinaceous kinetochore, maintain sister chromatid cohesion, regulate chromosome attachment to the spindle, and direct chromosome movement during cell division. Although the functions of centromeres and the proteins that contribute to their complex structure and function are conserved in eukaryotes, centromeric DNA diverges rapidly. Human centromeres are particularly complicated. Here, we review studies on the organization of homogeneous arrays of chromosome-specific alpha-satellite repeats and evolutionary links among eukaryotic centromeric sequences. We also discuss epigenetic mechanisms of centromere identity that confer structural and functional features of the centromere through DNA-protein interactions and post-translational modifications, producing centromere-specific chromatin signatures. The assembly and organization of human centromeres, the contributions of satellite DNA to centromere identity and diversity, and the mechanism whereby centromeres are distinguished from the rest of the genome reflect ongoing puzzles in chromosome biology.
DOI:10.1101/gr.180646.114URLPMID:25373146 [本文引用: 1]
Mitosis entails global alterations to chromosome structure and nuclear architecture, concomitant with transient silencing of transcription. How cells transmit transcriptional states through mitosis remains incompletely understood. While many nuclear factors dissociate from mitotic chromosomes, the observation that certain nuclear factors and chromatin features remain associated with individual loci during mitosis originated the hypothesis that such mitotically retained molecular signatures could provide transcriptional memory through mitosis. To understand the role of chromatin structure in mitotic memory, we performed the first genome-wide comparison of DNase I sensitivity of chromatin in mitosis and interphase, using a murine erythroblast model. Despite chromosome condensation during mitosis visible by microscopy, the landscape of chromatin accessibility at the macromolecular level is largely unaltered. However, mitotic chromatin accessibility is locally dynamic, with individual loci maintaining none, some, or all of their interphase accessibility. Mitotic reduction in accessibility occurs primarily within narrow, highly DNase hypersensitive sites that frequently coincide with transcription factor binding sites, whereas broader domains of moderate accessibility tend to be more stable. In mitosis, proximal promoters generally maintain their accessibility more strongly, whereas distal regulatory elements tend to lose accessibility. Large domains of DNA hypomethylation mark a subset of promoters that retain accessibility during mitosis and across many cell types in interphase. Erythroid transcription factor GATA1 exerts site-specific changes in interphase accessibility that are most pronounced at distal regulatory elements, but has little influence on mitotic accessibility. We conclude that features of open chromatin are remarkably stable through mitosis, but are modulated at the level of individual genes and regulatory elements.
DOI:10.1142/S0219720016500232URLPMID:27427382 [本文引用: 1]
We propose a new method to visualize gene expression experiments inspired by the latent semantic indexing technique originally proposed in the textual analysis context. By using the correspondence word-gene document-experiment, we define an asymmetric similarity measure of association for genes that accounts for potential hierarchies in the data, the key to obtain meaningful gene mappings. We use the polar decomposition to obtain the sources of asymmetry of the similarity matrix, which are later combined with previous knowledge. Genetic classes of genes are identified by means of a mixture model applied in the genes latent space. We describe the steps of the procedure and we show its utility in the Human Cancer dataset.
DOI:10.1093/nar/gkh012URLPMID:14681366 [本文引用: 1]
The analysis of regulatory regions in genome sequences is strongly based on the detection of potential transcription factor binding sites. The preferred models for representation of transcription factor binding specificity have been termed position-specific scoring matrices. JASPAR is an open-access database of annotated, high-quality, matrix-based transcription factor binding site profiles for multicellular eukaryotes. The profiles were derived exclusively from sets of nucleotide sequences experimentally demonstrated to bind transcription factors. The database is complemented by a web interface for browsing, searching and subset selection, an online sequence analysis utility and a suite of programming tools for genome-wide and comparative genomic analysis of regulatory regions. JASPAR is available at http://jaspar. cgb.ki.se.
DOI:10.1038/s41467-017-02289-3URLPMID:29230012 [本文引用: 1]
As interactions between the immune system and tumour cells are governed by a complex network of cell-cell interactions, knowing the specific immune cell composition of a solid tumour may be essential to predict a patient's response to immunotherapy. Here, we analyse in depth how to derive the cellular composition of a solid tumour from bulk gene expression data by mathematical deconvolution, using indication-specific and cell type-specific reference gene expression profiles (RGEPs) from tumour-derived single-cell RNA sequencing data. We demonstrate that tumour-derived RGEPs are essential for the successful deconvolution and that RGEPs from peripheral blood are insufficient. We distinguish nine major cell types, as well as three T cell subtypes. Using the tumour-derived RGEPs, we can estimate the content of many tumours associated immune and stromal cell types, their therapeutically relevant ratios, as well as an improved gene expression profile of the malignant cells.
DOI:10.1093/nar/gky007URLPMID:29361178 [本文引用: 1]
Droplet based single cell transcriptomics has recently enabled parallel screening of tens of thousands of single cells. Clustering methods that scale for such high dimensional data without compromising accuracy are scarce. We exploit Locality Sensitive Hashing, an approximate nearest neighbour search technique to develop a de novo clustering algorithm for large-scale single cell data. On a number of real datasets, dropClust outperformed the existing best practice methods in terms of execution time, clustering accuracy and detectability of minor cell sub-types.
DOI:10.1002/pd.1970140403URLPMID:8066033 [本文引用: 2]
Flow cytometry was used to enumerate the lymphocyte subpopulations in fetal blood obtained by cordocentesis from eight trisomy 18 fetuses at 20-36 weeks' gestation. Compared with values in chromosomally normal fetuses, in trisomy 18 the mean T- and natural killer (NK) cell counts were significantly lower (t = -7.63, P < 0.001 and t = -3.58, P < 0.01, respectively); the mean B-cell count was not significantly different (t = -1.32). These findings demonstrate that in trisomy 18 there is abnormal intrauterine development of the immune system.
DOI:10.1016/j.tibs.2017.09.003URLPMID:28964625 [本文引用: 1]
The TEAD transcription factor family is best known for transcriptional output of the Hippo signaling pathway and has been implicated in processes such as development, cell growth and proliferation, tissue homeostasis, and regeneration. Our understanding of the functional importance of TEADs has increased dramatically since its initial discovery three decades ago. The majority of our knowledge of TEADs is in the context of Hippo signaling as nuclear DNA-binding proteins passively activated by Yes-associated protein (YAP) and transcriptional activator with PDZ-binding domain (TAZ), transcription coactivators downstream of the Hippo pathway. However, recent studies suggest that TEAD itself is actively regulated. Here, we highlight evidence demonstrating Hippo-independent regulation of TEADs and the potential impacts these studies may have on new cancer therapeutics.
DOI:10.1038/nchembio.2036URLPMID:26900866 [本文引用: 1]
TEA domain (TEAD) transcription factors bind to the coactivators YAP and TAZ and regulate the transcriptional output of the Hippo pathway, playing critical roles in organ size control and tumorigenesis. Protein S-palmitoylation attaches a fatty acid, palmitate, to cysteine residues and regulates protein trafficking, membrane localization and signaling activities. Using activity-based chemical probes, we discovered that human TEADs possess intrinsic palmitoylating enzyme-like activities and undergo autopalmitoylation at evolutionarily conserved cysteine residues under physiological conditions. We determined the crystal structures of lipid-bound TEADs and found that the lipid chain of palmitate inserts into a conserved deep hydrophobic pocket. Strikingly, palmitoylation did not alter TEAD's localization, but it was required for TEAD's binding to YAP and TAZ and was dispensable for its binding to the Vgll4 tumor suppressor. Moreover, palmitoylation-deficient TEAD mutants impaired TAZ-mediated muscle differentiation in vitro and tissue overgrowth mediated by the Drosophila YAP homolog Yorkie in vivo. Our study directly links autopalmitoylation to the transcriptional regulation of the Hippo pathway.
DOI:10.1016/j.str.2015.11.005URLPMID:26724994 [本文引用: 1]
The Hippo signaling pathway is responsible for regulating the function of TEAD family transcription factors in metazoans. TEADs, with their co-activators YAP/TAZ, are critical for controlling cell differentiation and organ size through their transcriptional activation of genes involved in cell growth and proliferation. Dysregulation of the Hippo pathway has been implicated in multiple forms of cancer. Here, we identify a novel form of regulation of TEAD family proteins. We show that human TEADs are palmitoylated at a universally conserved cysteine, and report the crystal structures of the human TEAD2 and TEAD3 YAP-binding domains in their palmitoylated forms. These structures show a palmitate bound within a highly conserved hydrophobic cavity at each protein's core. Our findings also demonstrate that this modification is required for proper TEAD folding and stability, indicating a potential new avenue for pharmacologically regulating the Hippo pathway through the modulation of TEAD palmitoylation.
DOI:10.1038/s41467-019-12812-3URLPMID:31659164 [本文引用: 1]
Mapping the chromatin occupancy of transcription factors (TFs) is a key step in deciphering developmental transcriptional programs. Here we use biotinylated knockin alleles of seven key cardiac TFs (GATA4, NKX2-5, MEF2A, MEF2C, SRF, TBX5, TEAD1) to sensitively and reproducibly map their genome-wide occupancy in the fetal and adult mouse heart. These maps show that TF occupancy is dynamic between developmental stages and that multiple TFs often collaboratively occupy the same chromatin region through indirect cooperativity. Multi-TF regions exhibit features of functional regulatory elements, including evolutionary conservation, chromatin accessibility, and activity in transcriptional enhancer assays. H3K27ac, a feature of many enhancers, incompletely overlaps multi-TF regions, and multi-TF regions lacking H3K27ac retain conservation and enhancer activity. TEAD1 is a core component of the cardiac transcriptional network, co-occupying cardiac regulatory regions and controlling cardiomyocyte-specific gene functions. Our study provides a resource for deciphering the cardiac transcriptional regulatory network and gaining insights into the molecular mechanisms governing heart development.
DOI:10.1371/journal.pgen.1006600URLPMID:28178271 [本文引用: 1]
The TEAD family of transcription factors (TEAD1-4) bind the MCAT element in the regulatory elements of both growth promoting and myogenic differentiation genes. Defining TEAD transcription factor function in myogenesis has proved elusive due to overlapping expression of family members and their functional redundancy. We show that silencing of either Tead1, Tead2 or Tead4 did not effect primary myoblast (PM) differentiation, but that their simultaneous knockdown strongly impaired differentiation. In contrast, Tead1 or Tead4 silencing impaired C2C12 differentiation showing their different contributions in PMs and C2C12 cells. Chromatin immunoprecipitation identified enhancers associated with myogenic genes bound by combinations of Tead4, Myod1 or Myog. Tead4 regulated distinct gene sets in C2C12 cells and PMs involving both activation of the myogenic program and repression of growth and signaling pathways. ChIP-seq from mature mouse muscle fibres in vivo identified a set of highly transcribed muscle cell-identity genes and sites bound by Tead1 and Tead4. Although inactivation of Tead4 in mature muscle fibres caused no obvious phenotype under normal conditions, notexin-induced muscle regeneration was delayed in Tead4 mutants suggesting an important role in myogenic differentiation in vivo. By combining knockdown in cell models in vitro with Tead4 inactivation in muscle in vivo, we provide the first comprehensive description of the specific and redundant roles of Tead factors in myogenic differentiation.
DOI:10.1038/s41418-019-0335-4URLPMID:31024075 [本文引用: 1]
TEAD1 (TEA domain transcription factor 1), a transcription factor known for the functional output of Hippo signaling, is important for tumorigenesis. However, the role of TEAD1 in the development of vascular smooth muscle cell (VSMC) is unknown. To investigate cell-specific role of Tead1, we generated cardiomyocyte (CMC) and VSMC-specific Tead1 knockout mice. We found CMC/VSMC-specific deletion of Tead1 led to embryonic lethality by E14.5 in mice due to hypoplastic cardiac and vascular walls, as a result of impaired CMC and VSMC proliferation. Whole transcriptome analysis revealed that deletion of Tead1 in CMCs/VSMCs downregulated expression of muscle contractile genes and key transcription factors including Pitx2c and myocardin. In vitro studies demonstrated that PITX2c and myocardin rescued TEAD1-dependent defects in VSMC differentiation. We further identified Pitx2c as a novel transcriptional target of TEAD1, and PITX2c exhibited functional synergy with myocardin by directly interacting with myocardin, leading to augment the differentiation of VSMC. In summary, our study reveals a critical role of Tead1 in cardiovascular development in mice, but also identifies a novel regulatory mechanism, whereby Tead1 functions upstream of the genetic regulatory hierarchy for establishing smooth muscle contractile phenotype.
DOI:10.1161/CIRCRESAHA.118.314187URLPMID:30801233 [本文引用: 1]
RATIONALE: TEAD (TEA domain transcription factor) 1-a major effector of the Hippo signaling pathway-acts as an oncoprotein in a variety of tumors. However, the function of TEAD1 in vascular smooth muscle cells (VSMCs) remains unclear. OBJECTIVE: To assess the role of TEAD1 in vascular injury-induced smooth muscle proliferation and delineate the mechanisms underlying its action. METHODS AND RESULTS: We found that TEAD1 expression is enhanced in mouse femoral artery after wire injury and correlates with the activation of mTORC1 (mechanistic target of rapamycin complex 1) signaling in vivo. Using an inducible smooth muscle-specific Tead1 KO (knockout) mouse model, we found that specific deletion of Tead1 in adult VSMCs is sufficient to attenuate arterial injury-induced neointima formation due to inhibition of mTORC1 activation and VSMC proliferation. Furthermore, we found that TEAD1 plays a unique role in VSMCs, where it not only downregulates VSMC differentiation markers but also activates mTORC1 signaling, leading to enhanced VSMC proliferation. Using whole-transcriptome sequencing analysis, we identified Slc1a5 (solute carrier family 1 member 5)-a key glutamine transporter-as a novel TEAD1 target gene. SLC1A5 overexpression mimicked TEAD1 in promoting mTORC1 activation and VSMC proliferation. Moreover, depletion of SLC1A5 by silencing RNA or blocking SLC1A5-mediated glutamine uptake attenuated TEAD1-dependent mTORC1 activation and VSMC proliferation. CONCLUSIONS: Our study unravels a novel mechanism by which TEAD1 promotes VSMC proliferation via transcriptional induction of SLC1A5, thereby activating mTORC1 signaling and promoting neointima formation.
DOI:10.1172/jci.insight.93343URL [本文引用: 1]
DOI:10.1093/nar/gkq890URLPMID:20935057 [本文引用: 1]
Twist1 and Twist2 are highly conserved members of the Twist subfamily of bHLH proteins responsible for the transcriptional regulation of the developmental programs in mesenchymal cell lineages. The regulation of such processes requires that Twist1 and Twist2 function as molecular switches to activate and repress target genes by employing several direct and indirect mechanisms. Modes of action by these proteins include direct DNA binding to conserved E-box sequences and recruitment of coactivators or repressors, sequestration of E-protein modulators, and interruption of proper activator/repressor function through protein-protein interactions. Regulatory outcomes of Twist1 and Twist2 are themselves controlled by spatial-temporal expression, phosphoregulation, dimer choice and cellular localization. Although these two proteins are highly conserved and exhibit similar functions in vitro, emerging literature have demonstrated different roles in vivo. The involvement of Twist1 and Twist2 in a broad spectrum of regulatory pathways highlights the importance of understanding their roles in normal development, homeostasis and disease. Here we focus on the mechanistic models of transcriptional regulation and summarize the similarities and differences between Twist1 and Twist2 in the context of myogenesis, osteogenesis, immune system development and cancer.
[本文引用: 1]
DOI:10.1038/ncb3477URLPMID:28218909 [本文引用: 1]
Skeletal muscle possesses remarkable regenerative potential due to satellite cells, an injury-responsive stem cell population located beneath the muscle basal lamina that expresses Pax7. By lineage tracing of progenitor cells expressing the Twist2 (Tw2) transcription factor in mice, we discovered a myogenic lineage that resides outside the basal lamina of adult skeletal muscle. Tw2(+) progenitors are molecularly and anatomically distinct from satellite cells, are highly myogenic in vitro, and can fuse with themselves and with satellite cells. Tw2(+) progenitors contribute specifically to type IIb/x myofibres during adulthood and muscle regeneration, and their genetic ablation causes wasting of type IIb myofibres. We show that Tw2 expression maintains progenitor cells in an undifferentiated state that is poised to initiate myogenesis in response to appropriate cues that extinguish Tw2 expression. Tw2-expressing myogenic progenitors represent a previously unrecognized, fibre-type-specific stem cell involved in postnatal muscle growth and regeneration.
DOI:10.1093/hmg/ddz279URLPMID:31813999 [本文引用: 1]
Trisomy 18, sometimes called Edwards syndrome, occurs in about 1 in 6000 live births and causes multiple birth defects in affected infants. The extra copy of chromosome 18 causes the altered expression of many genes and leads to severe skeletal, cardiovascular and neurological systems malformations as well as other medical problems. Due to the low rate of survival and the massive genetic imbalance, little research has been aimed at understanding the molecular consequences of trisomy 18 or considering potential therapeutic approaches. Our research is the first study to characterize whole-genome expression in fibroblast cells obtained from two patients with trisomy 18 and two matched controls, with follow-up expression confirmation studies on six independent controls. We show a detailed analysis of the most highly dysregulated genes on chromosome 18 and those genome-wide. The identified effector genes and the dysregulated downstream pathways provide hints of possible genotype-phenotype relationships to some of the most common symptoms observed in trisomy 18. We also provide a possible explanation for the sex-specific differences in survival, a unique characteristic of trisomy 18. Our analysis of genome-wide expression data moves us closer to understanding the molecular consequences of the second most common human autosomal trisomy of infants who survive to term. These insights might also translate to the understanding of the etiology of associated birth defects and medical conditions among those with trisomy 18.
DOI:10.1007/s00439-010-0923-3URLPMID:21152935 [本文引用: 1]
Trisomy 18 is a common human aneuploidy that is associated with significant perinatal mortality. Unlike the well-characterized

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}