 ,西南大学动物科学技术学院,重庆 402460
,西南大学动物科学技术学院,重庆 402460Progress on meiotic gene expression and epigenetic regulation of male sterility in Dzo cattle
Huiyou Chen, Jianmin Zhang, Baisen Li, Yonglin Deng, Gongwei Zhang,College of Animal Science and Technology , Southwest University, Chongqing 402460, China通讯作者: 张龚炜,博士,副教授,硕士生导师,研究方向:动物遗传育种。E-mail:zgw-vip@163.com
编委: 赵要风
收稿日期:2020-06-15修回日期:2020-07-14网络出版日期:2020-11-20
| 基金资助: |
Received:2020-06-15Revised:2020-07-14Online:2020-11-20
| Fund supported: |
作者简介 About authors
陈会友,在读硕士研究生,专业方向:动物遗传育种。E-mail:

摘要
种间杂交雄性不育是自然界普遍现象,是物种形成生殖隔离的重要方式。犏牛作为牦牛(Bos grunniens)和普通牛(Bos taurus)的种间杂交后代,表现为公犏牛不育,而母犏牛可育,是研究种间杂交雄性不育的良好动物模型。近年来利用分子生物学技术发现犏牛睾丸组织中大量基因表达紊乱。研究表明,DNA甲基化、组蛋白修饰和非编码RNA等表观遗传因素参与精子发生过程。本文从减数分裂相关的基因表达、DNA甲基化、microRNA (miRNA)、PIWI蛋白相互作用的RNA (PIWI-interactingRNA, piRNA)、长链非编码RNA (long non-coding RNA, lncRNA)和组蛋白甲基化修饰等方面总结了犏牛雄性不育的相关研究进展,以期从遗传和表观遗传调控角度更加深入理解犏牛雄性不育的分子机理。
关键词:
Abstract
Interspecific hybrid male sterility is a common occurrence in nature and plays an important role in species reproductive isolation. Dzo (cattle-yak), the offspring of interspecific cross between domestic yak (Bos grunniens) and cattle (Bos taurus), is a unique animal model for investigating interspecific hybrid male sterility. Dzo females are completely fertile while the males are sterile. In recent years, molecular studies have demonstrated that the expressions of genes were dysregulated during meiosis in Dzo testis, as compared to those in cattle or yak. Other studies have revealed that epigenetic factors/events, such as DNA methylation, histone modification and non-coding RNA, are also involved in spermatogenesis. This review summarizes the dysregulation of gene expression, DNA methylation, microRNA (miRNA), PIWI-interacting RNA (piRNA), long non-coding RNA (lncRNA), and histone methylation modification during meiosis in Dzo testis. These results highlighted the potential roles of genetic and epigenetic regulations of meiosis in Dzo testis, thereby providing a more detailed understanding on the molecular mechanisms of interspecific hybrid male sterility.
Keywords:
PDF (650KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈会友, 张建敏, 李柏森, 邓永琳, 张龚炜. 犏牛雄性不育的减数分裂基因表达与表观遗传调控研究进展. 遗传[J], 2020, 42(11): 1081-1092 doi:10.16288/j.yczz.20-176
Huiyou Chen.
牦牛(Bos grunniens)被称为“高原之舟”,是青藏高原地区不可或缺的全能家畜,对高寒、缺氧和强紫外线等恶劣的生态环境条件有极强的适应性,是当地居民不可或缺的生产资料和生活资料。但是牦牛乳、肉用生产性能较低。为了改善牦牛生产性能,利用牦牛和优良肉用、奶用普通牛(Bos taurus)开展种间杂交,F1、F2代犏牛具有明显杂种优势,肉用、奶用均比亲本牦牛有显著提高并能适应高原地区环境气候。但是,F1、F2代犏牛雄性不育,这使得其杂种优势无法通过横交固定,无法通过杂交育种改良牦牛品种,成为阻碍藏区牦牛产业发展的瓶颈问题。种间杂交雄性不育是自然界普遍现象,是物种形成生殖隔离的重要方式。犏牛雄性不育也是探索物种形成生殖隔离的良好动物模型。目前,国内外****已从杂交改良、组织学、内分泌学、生物化学、细胞遗传学和分子生物学等主要领域展开研究[1,2]。最近研究表明,DNA甲基化、组蛋白修饰和非编码RNA等表观遗传因素是调控基因表达的重要因子,并对精子发生过程起关键作用[3]。本文从基因表达、DNA甲基化、组蛋白甲基化修饰和非编码RNA等方面总结了犏牛雄性不育的相关研究进展,以期从遗传和表观遗传调控角度更加深入理解犏牛雄性不育的分子机理。
1 基因表达紊乱与犏牛雄性不育
组织学研究发现,犏牛雄性不育主要表现为精母细胞数量减少,生精小管内极少见精子细胞,表明犏牛雄性不育主要是生精细胞减数分裂过程受阻[4]。减数分裂是有性生殖过程中产生配子的一种特殊分裂方式,是哺乳动物繁衍后代的必须条件。研究人员在减数分裂相关基因上展开大量研究,以探索减数分裂基因表达紊乱与犏牛雄性不育的关系,其研究主要集中在以下蛋白家族(表1)。Table 1
表1
表1犏牛雄性不育相关基因
Table 1
| 基因 | 主要功能 | 参考文献 |
|---|---|---|
| DAZ蛋白家族 | DAZ、DAZL和BOULE蛋白是精子发生的重要调控因子 | [6,26,27] |
| SYCP蛋白家族 | SYCP1、SYCP2、SYCP3和FKBP6是联会复合体的重要组成部分,与精子发生过程中的减数分裂密切相关 | [11~13,28,29] |
| DEAD-box蛋白家族 | DDX4、DDX3Y和DDX25是生殖相关基因,参与广泛RNA代谢过程 | [15,18,29] |
| 减数分裂同源重组相关基因 | DMC1、RAD51、RPA1和BLM是参与哺乳动物减数分裂同源重组修复的关键基因 | [20,21] |
| MSY (Y染色体雄性特异区)相关基因 | Y染色体连锁基因家族(TSPY、PRAMEY等)在精子发生和雄性生育中起重要作用 | [25,30,31] |
| SNRPN | SNRPN (small nuclear ribonucleoprotein polypeptide N)与细胞分化和增殖有关 | [32,33] |
| H19、IGF2 | 印迹基因IGF2 (insulin like growth factor 2)、H19 (H19 imprinted maternally expressed transcript)促进多种细胞的增殖、抑制细胞凋亡 | [33,34] |
| CDC2、CDC25A | CDC2(cell division cycle 2)、CDC25A是减数分裂的两个关键基因 | [35] |
| PIWIL1 | PIWIL1(piwi like RNA-mediated gene silencing 1)在精子发生和转座子控制中起重要作用 | [29,36] |
| STRA8 | STRA8(stimulated by retinoic acid 8)参与调控精原干细胞(SSCs)自我更新与分化 | [37] |
| PRDM9 | PRDM9(PR/SET domain 9)具有PR结构域,和雄性不育相关 | [38] |
| DMRT7 | DMRT7(double sex and mab3-relatated transcrip-tion factor7)缺乏是犏牛雄性不育的一个强有力的候选因素 | [39] |
| ERK1、ERK2 | ERK1 (extracellular signal-regulated kinase 1)、ERK2属于MAPK信号通路 | [40] |
新窗口打开|下载CSV
1.1 DAZ (deleted in azoospermia)蛋白家族
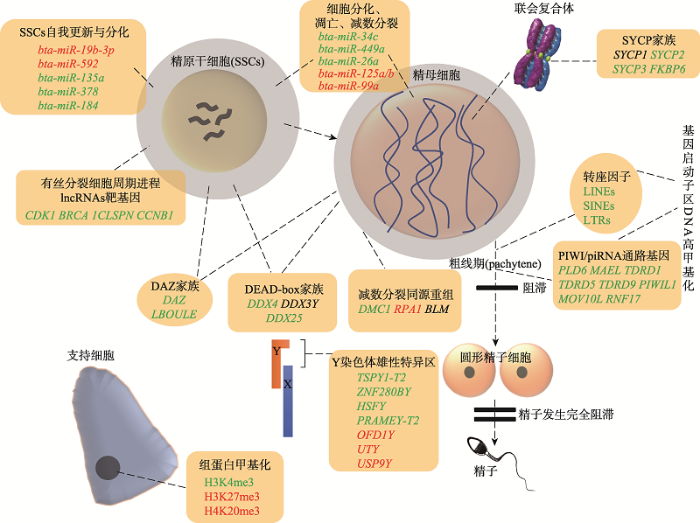
DAZ蛋白家族主要有3个成员,DAZ (the deleted in azoospermia)基因位于Y染色体上,DAZL (deleted in azoospermia like)和BOULE (boule protein)是常染色体基因,这3个基因编码蛋白是RNA结合蛋白,在生殖细胞特异表达,是精子发生过程的主要调控因子。DAZ有4个拷贝,以头对头的形式排列在Y染色体上,基因产物具有RNA结合蛋白特性,在睾丸组织特异性表达,可能与精子生成有关,是决定精子生成的基因[5]。DAZL与DAZ基因同源性约为83%,DAZL在黄牛(Bos taurus domestica)和牦牛睾丸组织中表达,在F1代犏牛睾丸组织不表达,并且DAZL的DNA甲基化程度显著高于黄牛和牦牛[6]。这与小鼠(Mus musculus)DAZL基因敲除后导致精子发生停止或功能异常结果一致[7],说明DAZL基因可能对犏牛雄性不育有重要影响。BOULE是动物精母细胞减数分裂过程中的必需蛋白(图1),与精子发生减数分裂阻滞、雄性不育等密切相关,F1代犏牛BOULE基因表达水平显著低于黄牛,BOULE 5?端DNA甲基化水平极显著高于黄牛和牦牛[8],说明犏牛BOULE基因的高甲基化可能使其mRNA表达下调,对犏牛生精细胞减数分裂、雄性不育有重要影响。
1.2 SYCP (synaptonemal complex protein)蛋白家族
SYCP蛋白家族主要有4个成员,分别是SYCP1 (synaptonemal complex protein 1)、SYCP2、SYCP3和FKBP6 (FKBP prolyl isomerase 6)。它们参与精原细胞的形成,是联会复合体形成的关键蛋白。SYCP1主要在减数分裂前期表达,在同源染色体配对中发挥重要作用,是精子发生过程中所必须的基因。在粗线期和双线期,FKBP6与SYCP1蛋白共同结合于常染色体的联会区域,辅助SYCP1形成横丝(transverse filaments, TF)。SYCP1在犏牛、牦牛和普通牛中都有表达,且差异不显著[9]。SYCP2蛋白能与SYCP3蛋白结合发挥作用。在哺乳动物中,SYCP2和SYCP3是轴向元件(axial element, AE)和侧向元件(lateral elements, LE)形成的主要决定成分[10]。SYCP3蛋白是一个DNA结合蛋白,定位于联会复合体的侧成分,在睾丸中特别是在初级精母细胞中表达(图1),在同源染色体配对中发挥重要作用,是精子发生过程中减数分裂所必须。犏牛睾丸SYCP3、SYCP2表达显著低于牦牛[11],其DNA启动子甲基化水平显著高于牦牛[12]。FKBP6主要在性腺组织的粗线期表达,犏牛睾丸FKBP6表达量显著低于牦牛。因此,SYCP3、SYCP2和FKBP6基因表达紊乱与犏牛雄性不育存在一定联系[13]。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1犏牛睾丸中的生精调控
黄色框内所有基因、miRNAs和组蛋白修饰的符号颜色代表犏牛相对于普通牛和牦牛的差异化程度,红色:上调;绿色:下调;黑色:无差异;:代表来源;
 :代表过程;
:代表过程; :代表范围;
:代表范围; :代表阻滞。
:代表阻滞。1.3 DEAD-box (DEAD-box helicase)蛋白家族
DEAD-box蛋白家族是一个ATP依赖的RNA解旋酶家族,参与多种RNA代谢过程。其中DDX4 (DEAD-box helicase 4)、DDX3Y (DEAD-box helicase 3, Y-linked)和DDX25是与精子发生密切相关的基因。在哺乳动物中,DDX4、DDX3Y和DDX25的缺失或减少会导致不同形式的精子发生障碍。DDX4在哺乳动物生殖系特异表达,作为一种广泛的分子标记物被应用于生殖研究[14]。犏牛睾丸组织DDX4的启动子区甲基化水平显著高于牦牛,其mRNA在犏牛睾丸中的表达量也极显著低于牦牛[15],说明DDX4对犏牛雄性不育存在一定影响。DDX3Y位于牛Y染色体上,在3种牛睾丸组织中mRNA表达量差异不显著[16]。DDX25是已知的唯一由激素调节的RNA解旋酶,DDX25基因敲除小鼠精子发生受阻,致使圆形精子无法继续变形[17]。犏牛睾丸组织中DDX25的基因表达水平也显著低于牦牛[18],可能致使犏牛精子发生受阻。1.4 减数分裂同源重组相关基因
在染色体自我复制过程中可能会出现双链断裂(double strand break, DSB)现象,如果这些断裂未能及时修复就会引起细胞凋亡,修复不准确也会引起基因突变和染色体突变。真核生物对这些断裂的修复有两种机理:非同源末端连接和同源重组,而同源重组是DNA上DSB损伤修复的主要方式,对于保持哺乳动物细胞的基因组完整性十分重要[19]。DMC1 (DNA meiotic recombinase 1)、RPA1 (replication protein A1)和BLM (BLM RecQ like helicase)等是参与哺乳动物减数分裂同源重组修复的关键基因。这些基因的突变、敲除和表达水平的降低均会引起精母细胞减数分裂障碍,最终导致雄性不育。DMC1、RPA1基因在犏牛睾丸组织中的表达水平对比黄牛和牦牛差异显著[20,21,22],且DMC1基因DNA启动子区甲基化水平也差异显著,提示其可能和犏牛雄性不育相关。1.5 Y染色体雄性特异区(male-specific region on the Y, MSY)相关基因
Y染色体是雄性哺乳动物相对于雌性特有的染色体,由常染色体进化而来,和X染色体只有5%的区域相同,该区域是和X染色体同源重组的拟常染色体区,而95%的其他区域则是MSY (图1)。牛Y染色体基因组已经公布(NCBI GenBank accession no. CM001061),通过对牛Y染色体进行测序和注释,牛Y染色体共鉴定出1274个基因,MSY包含28个蛋白编码基因和375个新转录本。利用转录组测序(RNA-seq)技术比较普通牛不同年龄睾丸组织中基因表达模式发现,13个编码基因和220转录本的表达量随睾丸发育显著上调,这表明Y染色体MSY区域的基因参与牛生精过程[23]。MSY相关基因的拷贝数变异(copy number variation, CNV)被证明和雄性生精功能有关[24]。张龚炜等[25]首次对MSY相关基因的CNV进行研究,发现F1、F2代公犏牛MSY相关基因TSPY (testis specific protein, Y-linked)、HSFY (heat shock transcription factor, Y-linked)、PRAMEY (preferentially expressed antigen in melanoma, Y-linked)和ZNF280BY (zinc finger protein 280B, Y-linked)的几何平均拷贝数(the average geometric mean copy number, CN)显著高于普通牛和牦牛,提示犏牛MSY在基因组结构上和牦牛以及普通牛不同,MSY相关基因的CNV可能是犏牛雄性不育的原因。随后详细分析普通牛和牦牛MSY相关基因TSPY、TSPY2、PRAMEY、HSFY和ZNF280BY序列,发现只有TSPY2在牛科是保守的,牦牛缺失TSPY2-T2类型序列,PRAMEY-T2和PRAMEY-T4在牦牛Y染色体上成功扩增,TSPY-T2和ZNF280BY的平均拷贝数在普通牛和牦牛之间差异显著[4],说明普通牛和牦牛MSY存在差异性。犏牛TSPY1-T2、ZNF280BY、HSFY和PRAMEY-T2表达对比牦牛和普通牛显著下调,究其原因可能是犏牛精子发生异常导致无精子生成,提示这些基因主要参与减数分裂后精子形成过程[23,4]。除以上多拷贝基因外,犏牛睾丸组织MSY区域的单拷贝基因UTY (ubiquitously transcribed tetratricopeptide repeat containing, Y-linked)、OFD1Y (oral-facial-digital syndrome 1, Y-linked)和USP9Y (ubiquitin specific peptidase 9, Y-linked)表达对比牦牛和普通牛显著上调。UTY和USP9Y具有组蛋白甲基化和泛素化的功能,提示后续可进一步从组蛋白甲基化和泛素化角度探索犏牛雄性不育的机理[16]。
2 表观遗传与犏牛雄性不育
以DNA甲基化、组蛋白修饰和染色质重塑为特征的表观遗传修饰是包括精子发生在内的许多生物学过程中的重要调节因子[3]。DNA甲基化[41]、组蛋白修饰[42]和非编码RNA[43]作为机体重要的表观遗传修饰类型,是在精子发生过程中调控的关键因素。表观遗传修饰的异常,将使生精过程基因的表达紊乱,进而导致雄性不育。2.1 DNA甲基化与犏牛雄性不育
DNA甲基化是在甲基转移酶(DNA methyltransferase, DNMT)催化作用下,以S-腺苷甲硫氨酸(S-Adenosylmethionine, SAM)作为甲基供体,通过共价键结合的方式使基因组CpG二核苷酸中胞嘧啶5号位碳原子获得一个甲基基团的化学修饰过程[44]。DNA甲基化能引起染色质结构、DNA构象、DNA稳定性及DNA与蛋白质相互作用方式的改变,从而调控基因表达[45]。DNA甲基化可能通过调节雄性生殖细胞的增殖和分化而发挥关键作用[46],是雄性不育的一个重要影响因素。在雄性不育模型中观察到的异常DNA甲基化模式可能是精原细胞的再甲基化失败或精母细胞、精子细胞和成熟精子细胞的甲基化状态维持不变所致[47]。由于启动子DNA甲基化通常抑制基因转录,在精子发生过程中DNA甲基化的紊乱导致了生精基因的表达紊乱,和雄性不育高度相关[45]。在犏牛中,研究发现PIWIL1、DAZL和FKBP6基因启动子区域甲基化水平升高导致基因表达下调,进而影响犏牛生殖[48,49,50]。最近利用全基因组甲基化测序技术发现启动子高甲基化基因在配子产生、piRNA (非编码RNA的一种)代谢过程和染色质结构的DNA甲基化过程中显著富集,表明启动子高甲基化和piRNA途径等表观遗传紊乱可能和犏牛雄性不育高度相关[29]。犏牛睾丸中PIWI/piRNA通路基因启动子发生DNA超甲基化,使PIWIL1、DDX4、PLD6 (phospholipase D family member 6)、MAEL (maelstrom spermatogenic transposon silencer)、FKBP6、TDRD1 (tudor domain containing 1)和TDRD5等基因表达下调,并导致犏牛精子发生过程中粗线期piRNA的产生降低,同时还发现转座因子LINEs (long interspersed nuclear elements)、SINEs (short interspersed nuclear elements)和LTRs (long terminal repeats)在犏牛睾丸中高甲基化(图1)。因此,DNA高甲基化和piRNA生成途径中断是导致生殖细胞发育不成功的原因之一,这可能导致犏牛雄性不育[29]。2.2 非编码RNA与犏牛雄性不育
随着基因组研究的深入,以前普遍认为不编码蛋白质的非编码RNA被证明其在转录后具有基因调控的作用。非编码RNA主要包括lncRNA、核糖体RNA (rRNA)、转运RNA (tRNA)、核小RNA (small nuclearRNA, snRNA)、核仁小RNA (small nucleolar RNA, snoRNA)、miRNA和piRNA等多种已知功能的RNA,及未知功能的RNA。这些RNA的共同特点是都能从基因组上转录而来,但是不翻译成蛋白,在RNA水平上就能行使各自的生物学功能。例如piRNA主要在睾丸组织中表达,其在转录后水平调控动物生殖系统[51,52,53]。2.2.1 PIWI/piRNA途径与犏牛雄性不育
piRNA一般长约24~35 nt,通过与AGO蛋白家族相互作用形成piRNA沉默复合体(piRNA-induced silencing complex, pi-RISC)来调控基因重复序列及转座子等基因元件的活性[54],主要影响动物生殖[51]。小鼠减数分裂时期piRNAs主要分前粗线期piRNAs (26~28 nt)和粗线期piRNAs (~30 nt)[55]。前粗线期piRNAs主要在细线期精母细胞中发现,来源于转座子元件[56,57]。粗线期piRNAs起源于基因组不同区域的piRNA簇,并与粗线期精母细胞中的PIWIL1和PIWIL2结合维持到圆形精子细胞阶段,是成年小鼠睾丸中的主要piRNAs,约占总piRNAs的95%[58]。在小鼠中,编码PIWI蛋白的PIWIL1/MIWI、PIWIL2/MILI和PIWIL4/MIWI2基因以及其他协助piRNA产生或功能表达所必需的蛋白是PIWI/piRAN通路的重要组成部分,它们共同维持小鼠的精子发生。一旦piRNA通路基因发生突变,都会导致雄性不育[59,60]。例如,转录因子A-MYB/MYBL1 (myeloblastosis oncogene-like 1)被证明直接识别上游DNA元件来启动粗线期piRNAs的转录[55],生成的piRNA前体转录本被输送到核外细胞质,随后又被PIWI蛋白结构域修剪成前体piRNA,经过一系列修剪后,由核酸外切酶(PARN like, ribonuclease domain containing 1, PNLDC1)[61]对piRNA3?端进一步修剪成成熟大小,最终piRNA3?端被2?-O-甲基转移酶(HEN methyltransferase 1, HENMT1)修饰产生成熟的piRNA[62] (图2)。如前所述,犏牛睾丸中PIWI/piRNA通路相关基因(PIWIL1、DDX4、PLD6、MAEL、FKBP6、TDRD1和TDRD5)启动子超甲基化抑制基因表达,使piRNA生成显著降低[29],表明PIWI/piRNA通路参与犏牛雄性不育的过程。
2.2.2 miRNA与犏牛雄性不育
miRNA是一种长度约为22 nt并高度保守的内源性非编码小分子RNA,虽然不编码蛋白质但具有调控功能。miRNA的生物学功能主要体现在对其靶基因的转录后水平调控,主要有靶标mRNA的降解和mRNA翻译抑制2种方式,它们在精子发生过程中以相对特异性方式表达,在雄性动物生殖健康中起着至关重要的作用[63]。例如,miRNA参与调控睾丸支持细胞的增殖和粘附,一旦支持细胞增殖和粘附功能异常,将会导致精子发生受阻[64]。因此,miRNAs的失调被认为是雄性不育的分子基础,这些分子的异常表达模式可以遗传给后代[65]。研究表明,miRNA在哺乳动物精子发生的不同过程中起着重要的调节作用:miR-20和miR-106a通过靶向STAT3 (signal transducer and activator of transcription 3)和CCND1(Cyclin D1)在转录后水平促进小鼠 SSCs的更新[66],miR-221/222通过抑制KIT (KIT proto-oncogene, receptor tyrosine kinase)的表达在维持精原细胞的未分化状态中发挥关键作用[67]。研究表明,小鼠粗线期精母细胞中存在许多来自X染色体的miRNAs,这些miRNAs可能导致性染色体减数分裂失活[68]。徐传飞[69]通过对小RNA测序发现61个miRNA在犏牛与牦牛睾丸组织之间差异表达,其中bta-miR-19b-3p、bta-miR-592、bta-miR-135a、bta-miR-378和bta-miR-184参与SSCs自我更新以及分化过程;bta-miR-34c和bta-miR-449a参与精子发生过程中减数分离起始过程,说明miRNA在犏牛生精过程中具有影响力。廖珂[70]发现涉及细胞增殖、凋亡过程的miRNA表达在犏牛和普通牛间也存在差异,如bta-miR-26a、bta-miR-125a/b和bta-miR-99a等。徐传飞[71]对差异miRNAs的靶基因进行GO分析和KEGG分析,结果显示bta-miR-34c靶向的CDK2 (cyclin dependent kinase 2)、CDK4和CDK6主要参与细胞分化、增殖以及凋亡等通路。
图2
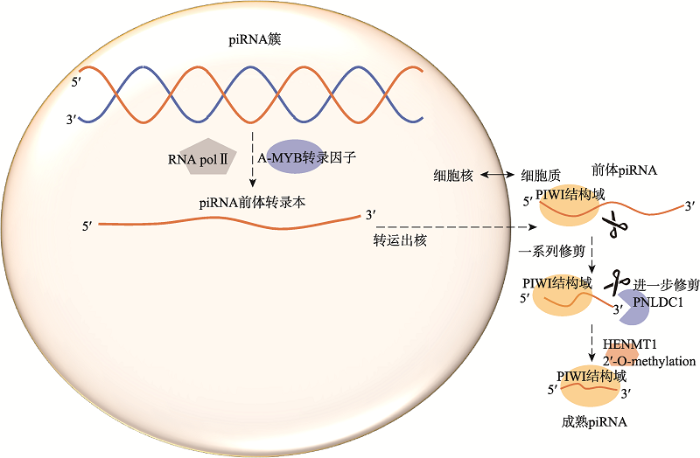 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2粗线期piRNA的生物发生
Fig. 2Biogenesis of pachytene piRNA
2.2.3 lncRNA与犏牛雄性不育
lncRNA长度一般大于200 nt,主要与染色质修饰蛋白、RNA结合蛋白、小RNA和其他lncRNA相互作用调节各种生理过程。lncRNA的功能大致可分为3种调控模式:竞争者(competitor)、激活者(activator)和前体(precursor)[72]。首先,作为竞争者,lncRNAs可以与某些DNA结合蛋白结合,从而抑制其与靶DNA的结合[73]。例如,一些lncRNAs可以与DNA甲基转移酶1 (DNA methyltransferase 1,DNMT1)结合,从而阻止DNMT1与靶DNA结合[74]。因此,DNA靶区域的甲基化状态会受到影响,导致靶基因的转录激活。其次,与竞争者相反,lncRNAs还可以作为招募者,通过将表观遗传修饰因子招募到特定的靶位点来启动表观遗传修饰,从而加强DNA甲基化或组蛋白修饰[75,76]。第三,lncRNAs可以被某些核糖核酸酶(RNase)如Dicer消化,产生小的非编码RNA,如HongrES2。HongrES2是一种附睾特异的1.6 kb mRNA样前体,能产生类似miRNA的小RNA,通过形成类似miRNA的小RNA,HongrES2下调CES7/CES5A(carboxylesterase 5A)蛋白的表达,从而影响精子获能[77]。在沉默牛的lncRNA H19 (H19母系印迹表达转录本)后,生精小管中的细胞数量有减少的趋势,这影响了IGF-1R (insulin-like growth factor I receptor)在支持细胞和生精细胞中的表达。IGF-1维持多种类型干细胞的存活,在雄性生殖系中具有重要功能[78]。近期,阿果约达等[79]通过RNA-seq技术从犏牛、牦牛和普通牛中筛选出6178个差异显著lncRNA转录本,候选靶基因2676个,最终筛选出犏牛不育相关基因PTGDS (prostaglandin D2 synthase)、IGF2 (insulin like growth factor 2)和MEST (mesoderm specific transcript)等,这些差异靶基因与犏牛雄性不育相关。伍仕鑫[80]通过分离出犏牛、牦牛睾丸高纯度精原细胞进行lncRNA的RNA-seq、GO和KEGG富集分析后,发现差异lncRNAs的靶基因CDK1 (Cyclin-dependent kinase 1)、BRCA1 (BRCA1 DNA Repair Associated)、CLSPN (Claspin)和CCNB1 (G2/mitotic-specific cyclin-B1)等主要参与犏牛生精过程中细胞分裂的起始(图1),细胞周期负性调控和相变调控,细胞周期进程的检控,DNA复制的损伤检测及修复,同源重组和细胞的内吞、程序性死亡、生长、分化和凋亡,以及细胞物质代谢等重要的信号通路和生物学过程。这些结果提示lncRNA可以作为犏牛雄性不育的重点研究方向。
2.3 组蛋白甲基化与犏牛雄性不育
组蛋白甲基化是发生在精氨酸和赖氨酸上的共价修饰,是影响基因活性的一种表观遗传机制,其功能主要体现在异染色质形成、基因印记、X染色体失活和转录调控等方面[81]。它由组蛋白甲基转移酶(histone methyltransferase, HMT)调节,可在精氨酸和赖氨酸残基中添加或移除甲基[82,83],所以组蛋白赖氨酸甲基化修饰与基因的活化或抑制有关。通常认为,组蛋白H3K4﹑H3K36和H3K79的甲基化与转录活化基因有关,而H3K9﹑H3K27和H4K20的甲基化抑制基因表达[84,85,86]。例如MLL5/KMT2E (lysine methyltransferase 2E)催化组蛋白H3K4二甲基化(H3K4me2),是形成顶体所必须的组蛋白甲基转移酶,MLL5基因敲除的小鼠是不育的[87]。而H3K9的去甲基化在减数分裂末期对于精子发生的完成至关重要,否则会抑制鱼精蛋白1 (protamine 1, PRM1)和过渡性蛋白1 (transition protein 1, TNP1)表达,进而导致染色质凝集和不育[88]。由此可见,组蛋白甲基转移酶在精子发生中发挥着重要作用。犏牛支持细胞中组蛋白H3K4三甲基化(H3K4me3)缺失,H3K27me3和H4K20me3显著富集(图1),H3K4me3、H3K9me1、H3K9me3和H4K20me3在犏牛精母细胞减数分裂染色体中的水平和定位存在显著差异,这些结果提示了组蛋白甲基化在精子发生和犏牛雄性不育中的潜在作用[89]。3 结语与展望
近年来分子生物学兴起,特别是组学技术的发展,可以从全基因组水平分析基因表达、组蛋白甲基化、DNA甲基化和非编码RNA等与犏牛雄性不育的关系,为理解犏牛雄性不育的分子机理提供了新的角度与见解。但要阐述犏牛雄性不育的分子机制,以下几个方面需要引起特别注意:(1)由于睾丸组织细胞异质性和精子发生的动态与连续性,基于睾丸组织的组学研究难以明确具体发生紊乱的细胞类型及发育阶段;(2)由于精子发生细胞无法建立体外培养体系,对候选基因的功能验证需要借助小鼠敲除/敲入体系;(3)前期研究主要关注生精相关细胞的表达调控关系,忽略了支持细胞等睾丸微环境对精子发生的调控作用[64,89];(4)各种表观遗传因素表现出细胞类型特异性和生精阶段特异性的表达调控特性,阐述不同表观遗传因素的动态调控网络是今后一个主要方向,如piRNAs主要在哺乳动物粗线期精母细胞表达[29,58],miRNA参与调控支持细胞和精原细胞[64,69]。综上所述,表观遗传调控在犏牛雄性不育过程中起到极为关键的作用。今后研究可重点关注犏牛睾丸支持细胞和粗线期以前阶段生精细胞,DNA甲基化、非编码RNA和组蛋白甲基化等表观遗传角度是研究犏牛雄性不育的良好切入点。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 2]
.
URLPMID:31103297 [本文引用: 3]
.
[本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 2]
.
[本文引用: 2]
[本文引用: 2]
.
URLPMID:21035437 [本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 2]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
URLPMID:12947387 [本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 2]
[本文引用: 2]
64,
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 1]
.
DOI:10.2527/jas.2015-9983URLPMID:27135999 [本文引用: 2]

Crossbreeding between cattle () and yak () exhibits significant hybrid advantages in milk yield and meat production. By contrast, cattle-yak F hybrid bulls are sterile. Copy number variations (CNV) of multicopy gene families in male-specific regions of the mammalian Y chromosome (MSY) affect human and animal fertility. The present study investigated CNV of (), (), (), and () in 5 yak breed bulls ( = 63), cattle-yak F ( = 22) and F ( = 2) hybrid bulls, and Chinese Yellow (CY) cattle bulls ( = 10) by quantitative real-time PCR. showed restricted amplification in yak bulls in that the average geometric mean copy number (CN) was estimated to be 4 copies. The most compelling finding is that there is a tremendous expansion of CN in F hybrids (385 copies; 95% confidence interval [CI] = 351-421) and F hybrids (356 copies) compared with the male parent breed CY cattle (142 copies; 95% CI = 95-211). Copy numbers of and were also extensively expanded on the Y chromosome in yak and CY cattle bulls. The geometric mean CN of and were estimated to be 123 (95% CI = 114-132) and 250 copies (95% CI = 233-268) in yak bulls and 71 (95% CI = 61-82) and 133 (95% CI = 107-164) copies in CY cattle, respectively. Yak and CY cattle have 2 copies of the gene on the Y chromosome. Similarly to gene, the F and F hybrid bulls have higher CN of , , and than CY cattle ( < 0.01). These results indicated that the MSY of yak and cattle-yak crossbred hybrids was fundamentally different from cattle MSY in the context of genomic organization. Based on the model of cattle-yak F and F hybrid bull sterility, the CNV of may serve as a potential risk factor for crossbred bull ( x ) infertility. To our knowledge, this is the first study to examine differences in multicopy genes in MSY between yak and cattle-yak bulls.
.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1371/journal.pone.0128250URLPMID:26030766 [本文引用: 1]
Non-primate mammals have two deleted azoospermia (DAZ) family genes, DAZL and Boule; genes in this family encode RNA-binding proteins essential for male fertility in diverse animals. Testicular DAZL transcription is regulated by epigenetic factors such as DNA methylation. However, nothing is known about the epigenetic regulation of Boule. Here, we explored the role of DNA methylation in the regulation of the bovine Boule (bBoule) gene. We found that a long CpG island (CGI) in the bBoule promoter was hypermethylated in the testes of cattle-yak hybrids with low bBoule expression, whereas cattle had relatively low methylation levels (P < 0.01), and there was no difference in the methylation level in the short CGI of the gene body between cattle and cattle-yak hybrids (P > 0.05). We identified a 107 bp proximal core promoter region of bBoule. Intriguingly, the differences in the methylation level between cattle and cattle-yak hybrids were larger in the core promoter than outside the core promoter. An in vitro methylation assay showed that the core promoter activity of bBoule decreased significantly after M.SssI methylase treatment (P < 0.01). We also observed dramatically increased bBoule transcription in bovine mammary epithelial cells (BMECs) after treatment with the methyltransferase inhibitor 5-Aza-dC. Taken together, our results establish that methylation status of the core promoter might be involved in testicular bBoule transcription, and may provide new insight into the epigenetic regulation of DAZ family genes and clinical insights regarding male infertility.
.
[本文引用: 1]
.
DOI:10.1080/15592294.2020.1738026URLPMID:32141383 [本文引用: 7]
Hybrid male sterility (HMS) is a postzygotic reproductive isolation mechanism that enforces speciation. A bovine example of HMS is the yattle (also called dzo), an interspecies hybrid of taurine cattle (Bos taurus) and yak (Bos grunniens). The molecular mechanisms underlying HMS of yattle are not well understood. Epigenetic modifications of DNA methylation and P-element induced wimpy testis (PIWI)-interacting RNA (piRNAs) are important regulators in spermatogenesis. In this study, we investigated DNA methylation patterns and piRNA expression in adult testes in hybrid infertile yattle bulls and fertile cattle and yak bulls using whole genome bisulphite-seq and small RNA-seq. Promoter hypermethylation in yattle were associated with DNA methylation involved in gamete generation, piRNA metabolic processes, spermatogenesis, and spermatid development (P < 2.6 x 10(-5)). Male infertility in yattle was associated with the promoter hypermethylation-associated silencing of PIWI/piRNA pathway genes including PIWIL1, DDX4, PLD6, MAEL, FKBP6, TDRD1 and TDRD5. The downstream effects of silencing these genes were diminished production of 29- to 31- nucleotide pachytene piRNAs in yattle testes. Hypermethylation events at transposable element loci (LINEs, SINEs, and LTRs) were found in yattle. LINE-derived prepachytene piRNAs increased and SINE-derived prepachytene piRNAs were reduced in yattle testes. Our data suggests that DNA methylation affects the PIWI/piRNA pathway and is involved in gene expression and pachytene piRNA production during spermatogenesis in bovine HMS. DNA hypermethylation and disruption of piRNA production contributed to unsuccessful germ cell development that may drive bovine HMS.
.
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1530/REP-16-0620URLPMID:27965401 [本文引用: 1]
PIWI proteins and their associated piRNAs have been the focus of intensive research in the past decade; therefore, their participation in the maintenance of genomic integrity during spermatogenesis has been well established. Recent studies have suggested important roles for the PIWI/piRNA system outside of gametogenesis, based on the presence of piRNAs and PIWI proteins in several somatic tissues, cancers, and the early embryo. Here, we investigated the small RNA complement present in bovine gonads, gametes, and embryos through next-generation sequencing. A distinct piRNA population was present in the testis as expected. However, we also found a large population of slightly shorter, 24-27 nt piRNA-like RNA (pilRNAs) in pools of oocytes and zygotes. These oocyte and embryo pilRNAs exhibited many of the canonical characteristics of piRNAs including a 1U bias, the presence of a 'ping-pong' signature, genomic clustering, and transposable element targeting. Some of the major transposons targeted by oocyte and zygote pilRNA were from the LINE RTE and ERV1 classes. We also identified pools of pilRNA potentially derived from, or targeted at, specific mRNA sequences. We compared the frequency of these gene-associated pilRNAs to the fold change in the expression of respective mRNAs from two previously reported transcriptome datasets. We observed significant negative correlations between the number of pilRNAs targeting mRNAs, and their fold change in expression between the 4-8 cell and 8-16 cell stages. Together, these results represent one of the first characterizations of the PIWI/piRNA pathway in the translational bovine model, and in the novel context of embryogenesis.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
.
URLPMID:27659720 [本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 2]
.
URLPMID:25105055 [本文引用: 1]
.
URLPMID:21747415 [本文引用: 1]
.
URLPMID:23830763 [本文引用: 1]
.
URLPMID:21724343 [本文引用: 1]
.
URLPMID:27658575 [本文引用: 1]
.
URLPMID:30446728 [本文引用: 2]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1016/j.molcel.2013.02.016URLPMID:23523368 [本文引用: 2]
Animal germ cells produce PIWI-interacting RNAs (piRNAs), small silencing RNAs that suppress transposons and enable gamete maturation. Mammalian transposon-silencing piRNAs accumulate early in spermatogenesis, whereas pachytene piRNAs are produced later during postnatal spermatogenesis and account for >95% of all piRNAs in the adult mouse testis. Mutants defective for pachytene piRNA pathway proteins fail to produce mature sperm, but neither the piRNA precursor transcripts nor the trigger for pachytene piRNA production is known. Here, we show that the transcription factor A-MYB initiates pachytene piRNA production. A-MYB drives transcription of both pachytene piRNA precursor RNAs and the mRNAs for core piRNA biogenesis factors including MIWI, the protein through which pachytene piRNAs function. A-MYB regulation of piRNA pathway proteins and piRNA genes creates a coherent feedforward loop that ensures the robust accumulation of pachytene piRNAs. This regulatory circuit, which can be detected in rooster testes, likely predates the divergence of birds and mammals.
.
[本文引用: 1]
.
URLPMID:18922463 [本文引用: 1]
.
URLPMID:16751776 [本文引用: 2]
.
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 1]
.
[本文引用: 3]
[本文引用: 3]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
.
DOI:10.1093/nar/gkl096URLPMID:16582102 [本文引用: 1]
MicroRNAs (miRNAs), which are non-coding RNAs 18-25 nt in length, regulate a variety of biological processes, including vertebrate development. To identify new species of miRNA and to simultaneously obtain a comprehensive quantitative profile of small RNA expression in mouse embryos, we used the massively parallel signature sequencing technology that potentially identifies virtually all of the small RNAs in a sample. This approach allowed us to detect a total of 390 miRNAs, including 195 known miRNAs covering approximately 80% of previously registered mouse miRNAs as well as 195 new miRNAs, which are so far unknown in mouse. Some of these miRNAs showed temporal expression profiles during prenatal development (E9.5, E10.5 and E11.5). Several miRNAs were positioned in polycistron clusters, including one particular large transcription unit consisting of 16 known and 23 new miRNAs. Our results indicate existence of a significant number of new miRNAs expressed at specific stages of mammalian embryonic development and which were not detected by earlier methods.
.
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.7150/ijbs.38232URLPMID:31929752 [本文引用: 1]
The male infertility of cattleyak resulted from spermatogenic arrest has greatly restricted the effective utilization of the heterosis from crossbreeding of cattle and yak. Based on our previous studies, the significant divergences of the transcriptomic and proteomic sequencing between yak and cattleyak prompt us to investigate the critical roles of microRNAs in post-transcriptional regulation of gene expression during spermatogenesis. TUNEL-POD analysis presented sharply decreased spermatogenic cell types and the increased apoptotic spermatogonia in cattleyak. The STA-PUT velocity sedimentation was employed to obtain spermatogonia and spermatocytes from cattle, yak and cattleyak and these spermatogenic cells were verified by the morphological and phenotypic identification. MicroRNA microarray showed that 27 differentially expressed miRNAs were simultaneously identified both in cattleyak vs cattle and in cattleyak vs yak comparisons. Further analysis revealed that the down-regulation of bta-let-7 families, bta-miR-125 and bta-miR-23a might impair the RA-induced differentiation of spermatogonia. Target gene analysis for differentially expressed miRNAs revealed that miRNAs targeted major players involved in vesicle-mediated transport, regulation of protein kinase activity and Pathways in cancer. In addition, spermatogonia transfection analysis revealed that the down-regulation of bta-miR-449a in the cattleyak might block the transition of male germ cells from the mitotic cycle to the meiotic program. The present study provided valuable information for future elucidating the regulatory roles of miRNAs involved in spermatogenic arrest of cattleyak.
.
[本文引用: 1]
.
URLPMID:21642992 [本文引用: 1]
.
URLPMID:24107992 [本文引用: 1]
.
URLPMID:24089468 [本文引用: 1]
.
DOI:10.1016/j.cell.2013.01.003URLPMID:23352431 [本文引用: 1]
Long noncoding RNAs (lncRNAs) are often expressed in a development-specific manner, yet little is known about their roles in lineage commitment. Here, we identified Braveheart (Bvht), a heart-associated lncRNA in mouse. Using multiple embryonic stem cell (ESC) differentiation strategies, we show that Bvht is required for progression of nascent mesoderm toward a cardiac fate. We find that Bvht is necessary for activation of a core cardiovascular gene network and functions upstream of mesoderm posterior 1 (MesP1), a master regulator of a common multipotent cardiovascular progenitor. We also show that Bvht interacts with SUZ12, a component of polycomb-repressive complex 2 (PRC2), during cardiomyocyte differentiation, suggesting that Bvht mediates epigenetic regulation of cardiac commitment. Finally, we demonstrate a role for Bvht in maintaining cardiac fate in neonatal cardiomyocytes. Together, our work provides evidence for a long noncoding RNA with critical roles in the establishment of the cardiovascular lineage during mammalian development.
.
[本文引用: 1]
.
URLPMID:30069947 [本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1101/gad.1110503URLPMID:12897054 [本文引用: 1]
On the histone H3 tail, Lys 9 and Lys 27 are both methylation sites associated with epigenetic repression, and reside within a highly related sequence motif ARKS. Here we show that the chromodomain proteins Polycomb (Pc) and HP1 (heterochromatin protein 1) are highly discriminatory for binding to these sites in vivo and in vitro. In Drosophila S2 cells, and on polytene chromosomes, methyl-Lys 27 and Pc are both excluded from areas that are enriched in methyl-Lys 9 and HP1. Swapping of the chromodomain regions of Pc and HP1 is sufficient for switching the nuclear localization patterns of these factors, indicating a role for their chromodomains in both target site binding and discrimination. To better understand the molecular basis for the selection of methyl-lysine binding sites, we solved the 1.8 A structure of the Pc chromodomain in complex with a H3 peptide bearing trimethyl-Lys 27, and compared it with our previously determined structure of the HP1 chromodomain in complex with a H3 peptide bearing trimethyl-Lys 9. The Pc chromodomain distinguishes its methylation target on the H3 tail via an extended recognition groove that binds five additional residues preceding the ARKS motif.
.
[本文引用: 1]
.
DOI:10.1111/j.1365-2605.2008.00872.xURLPMID:18298569 [本文引用: 1]
During the elongating spermatid stage of spermatogenesis, there is a step-wise replacement of nuclear histones with protamines 1 and 2. In fertile men, the ratio of protamine 1/protamine 2 (P1/P2) is within the narrow range of 0.8-1.2. Ratios above or below that range are associated with infertility, exhibiting a wide range of defects including decreased sperm counts, morphology, fertilization ability, and embryo implantation capacity. In this review, we highlight studies evaluating potential causes of abnormal protamine expression, including the sequencing of genes relevant to protamine expression in both affected patients and controls. While the variants of the protamine genes themselves do not appear to be responsible for most observed defects, variants of the Contrin gene, a transcription factor and translation repressor, appear to be contributory to some cases of abnormal expression. Additionally, we explore the potential effects of abnormal protamine replacement on the epigenome of human sperm. Ongoing studies are evaluating the role of retained histones and DNA methylation in sperm, which may be affected in sperm with aberrant protamine replacement. This important area of epigenetic research has profound clinical implications.
.
DOI:10.1016/j.molcel.2011.06.015URLPMID:21726805 [本文引用: 1]
In this issue of Molecular Cell, Wu et al. (2011) reveal that ubiquitylation of histone 2B lysine 34 stimulates histone methyltransferase activity on nucleosomes, a finding with implications for the general mechanism by which monoubiquitylation may influence subsequent modification activities.
.
[本文引用: 1]
.
URLPMID:22069496 [本文引用: 1]
.
DOI:10.1038/nature06236URLPMID:17943087 [本文引用: 1]
Recent studies indicate that, similar to other covalent modifications, histone lysine methylation is subject to enzyme-catalysed reversion. So far, LSD1 (also known as AOF2) and the jumonji C (JmjC)-domain-containing proteins have been shown to possess histone demethylase activity. LSD1 catalyses removal of H3K4me2/H3K4me1 through a flavin-adenine-dinucleotide-dependent oxidation reaction. In contrast, JmjC-domain-containing proteins remove methyl groups from histones through a hydroxylation reaction that requires alpha-ketoglutarate and Fe(II) as cofactors. Although an increasing number of histone demethylases have been identified and biochemically characterized, their biological functions, particularly in the context of an animal model, are poorly characterized. Here we use a loss-of-function approach to demonstrate that the mouse H3K9me2/1-specific demethylase JHDM2A (JmjC-domain-containing histone demethylase 2A, also known as JMJD1A) is essential for spermatogenesis. We show that Jhdm2a-deficient mice exhibit post-meiotic chromatin condensation defects, and that JHDM2A directly binds to and controls the expression of transition nuclear protein 1 (Tnp1) and protamine 1 (Prm1) genes, the products of which are required for packaging and condensation of sperm chromatin. Thus, our work uncovers a role for JHDM2A in spermatogenesis and reveals transition nuclear protein and protamine genes as direct targets of JHDM2A.
.
[本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}