 ,1, 周一叶2, 曾凡一,1,2
,1, 周一叶2, 曾凡一,1,2Advances in gene therapy for β-thalassemia and hemophilia based on the CRISPR/Cas9 technology
Liwen Bao,1, Yiye Zhou2, Fanyi Zeng,1,2通讯作者: 曾凡一,博士,研究员,研究方向:遗传学。E-mail:fzeng@vip.163.com
编委: 徐湘民
收稿日期:2020-04-21修回日期:2020-08-25网络出版日期:2020-10-20
| 基金资助: |
Received:2020-04-21Revised:2020-08-25Online:2020-10-20
| Fund supported: |
作者简介 About authors
鲍莉雯,硕士研究生,专业方向:生物学。E-mail:

摘要
地中海贫血和血友病是由基因异常引发的常见的遗传性血液病,难以根治且可遗传给下一代,造成严重的家庭和社会负担。基因治疗的出现为遗传性疾病提供了新的治疗方案,但自1990年第1项基因治疗临床试验被批准以来,30年间基因治疗的发展并不乐观。随着基因编辑技术的发展,尤其具有编辑效率高、操作简单、成本低等优势的第三代基因编辑技术CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/ CRISPR-associated protein 9)的发展,基因编辑介导的基因治疗越来越受到关注,有望根治地中海贫血和血友病等遗传性血液病。本文综述了近6年(2014~2020年)基于CRISPR/Cas9技术的β-地中海贫血和血友病基因治疗基础研究进展,总结了基于CRISPR/Cas9技术的基因治疗临床试验概况,并对CRISPR/Cas9技术用于基因治疗存在的问题和可能的解决方案进行探讨,以期为基于CRISPR/Cas9技术的遗传性血液病基因治疗相关研究提供参考。
关键词:
Abstract
Thalassemia and hemophilia are common inherited blood disorders caused by genetic abnormalities. These diseases are difficult to cure and can be inherited to the next generation, causing severe family and social burden. The emergence of gene therapy provides a new treatment for genetic diseases. However, since its first clinical trial in 1990, the development of gene therapy has not been as optimistic in the past three decades as one could hope. The development of gene-editing technology, particularly the third generation gene-editing technology CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9), has given hope in such therapeutic approach for having advantages in high editing efficiency, simple operation, and low cost. Gene editing-mediated gene therapy has thus received increasing attention from the biomedical community. It has shown promises for the treatment of inherited blood disorders, such as thalassemia and hemophilia. This paper reviews the fundamental research progress of gene therapy for β-thalassemia and hemophilia based on CRISPR/Cas9 technology in the past six years. It also summarizes the CRISPR/Cas9-based clinical trials of gene therapy. The problems and possible solutions to this technology for gene therapy are also discussed, thereby providing a reference for the research on gene therapy of inherited blood disorders based on CRISPR/Cas9 technology.
Keywords:
PDF (859KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
鲍莉雯, 周一叶, 曾凡一. 基于CRISPR/Cas9技术的β-地中海贫血和血友病基因治疗研究进展. 遗传[J], 2020, 42(10): 949-964 doi:10.16288/j.yczz.20-110
Liwen Bao.
遗传性血液病是由基因异常引发的血液疾病,研究较为透彻的有地中海贫血(地贫)和血友病。在红细胞中,血红蛋白负责运输氧和二氧化碳,由两条类α珠蛋白链(α链或ξ链)和两条类β珠蛋白链(β链、γ链或ε链)各结合一个血红素组成[1]。珠蛋白基因突变会引起血红蛋白病,包括血红蛋白变异体和地贫,前者受累于血红蛋白结构异常,后者则由于α链或类β链合成数量减少/缺失,致使另一条珠蛋白链含量相对过剩并沉积于红细胞膜,进而引发溶血,严重可导致胎儿流产和患儿死亡[1,2]。根据减少/缺失的珠蛋白链的不同,地贫可分为α-地中海贫血(α-地贫,α链合成减少/缺失)、β-地中海贫血(β-地贫,β链合成减少/缺失)等[3,4]。在我国广西壮族自治区α-地贫发病率高达14.95%,在广州β-地贫发病率高达2.21%[3]。
血友病是由凝血因子基因突变导致相应凝血因子严重缺乏而引起的出血性疾病,其中血友病A和血友病B最常见且均为X连锁隐性遗传病,分别由于凝血因子VIII基因(factor VIII, FVIII)和凝血因子IX基因(factor IX, FIX)突变而引起[5](表1)。
Table 1
表1
表1常见遗传性血液病
Table 1
| 典型疾病 | 致病机制 | 突变基因 | 突变类型 | 参考文献 |
|---|---|---|---|---|
| α-地中海贫血 | α-珠蛋白链合成减少/缺失 | HBA | 大片段缺失 | [1,2] |
| β-地中海贫血 | β-珠蛋白链合成减少/缺失 | HBB | 点突变/移码突变 | [1,4] |
| 镰状细胞贫血 | β-珠蛋白链结构异常 | HBB | 外显子点突变 | [4] |
| 血友病A | 凝血因子VIII缺乏 | FVIII | 内含子倒位 | [5] |
| 血友病B | 凝血因子IX缺乏 | FIX | 点突变 | [5] |
新窗口打开|下载CSV
对于上述遗传性血液病,临床上主要采取长期规范输血、成分输血等方法进行对症治疗[1]。这些短效的对症疗法无法根治遗传性血液病,且长期反复输血会导致铁沉积,还可能引发感染和过敏反应,甚至导致死亡。少部分患者可通过异体骨髓或造血干细胞移植获得长效/永久的治疗效果,但存在供体缺乏和免疫排斥等问题,且费用昂贵。随着现代基因编辑技术的进步,基因治疗成为最具前景的治疗方案,有望根治遗传性血液病。本文从基础研究和临床研究两方面对基于CRISPR/Cas9技术的β-地贫和血友病的基因治疗研究进展进行综述,以期为基于CRISPR/Cas9技术的遗传性血液病基因治疗相关研究提供参考。
1 遗传性血液病基因治疗概况
1990年开展的针对遗传性血液病——重症联合免疫缺陷的基因治疗试验被认为是首次成功的基因治疗临床试验。该研究通过基因增补的方式,利用逆转录病毒将正常的腺苷脱氨酶(adenosine deaminase, ADA)基因体外导入患者T细胞,并随机插入基因组,改造后的T细胞输回患者体内用于治疗[6]。但逆转录病毒介导的基因转导存在安全问题,且淋巴细胞寿命短,不利于发挥长效的治疗效果。此后,多种病毒和非病毒载体被开发,以提高安全性。改造的细胞也能转为无限增殖并分化的造血干细胞和祖细胞(hematopoietic stem and progenitor cell, HSPC),以实现长效治疗。除了体外改造患者细胞的体外途径,基因治疗还可通过体内途径实现[7],即直接将载有目的基因的载体通过静脉输注、肌肉注射等方式输入患者体内,目前血友病的基因治疗主要采取体内途径,比如,通过门静脉输注将载有凝血因子VIII或凝血因子IX基因的肝趋向性腺相关病毒载体(adeno-associated virus, AAV)注入患者体内实现基因治疗[8,9](图1)。但体内途径的基因治疗过程难以控制,出于安全性考虑,以造血干细胞为靶细胞的基因治疗主要采取体外途径。对于β-地贫还可通过调控基因表达的方式进行治疗。上海医学遗传研究所利用羟基脲治疗β+地贫患者(患者能部分合成正常β珠蛋白链),首次发现低剂量的羟基脲可增加正常β链的合成,并显著改善患者表型[10]。此外,由于β-地贫患者体内α链相对过剩,减少α链合成可缓解疾病症状,如利用RNA干扰(RNA interference, RNAi)技术下调α-珠蛋白基因(HBA)表达可缓解β-地贫小鼠模型的贫血症状[11,12]。γ链可在功能上代偿β链的缺失或异常,重激活γ-珠蛋白基因(HBG)表达可改善β-地贫和镰状细胞贫血症状[13,14]。
图1
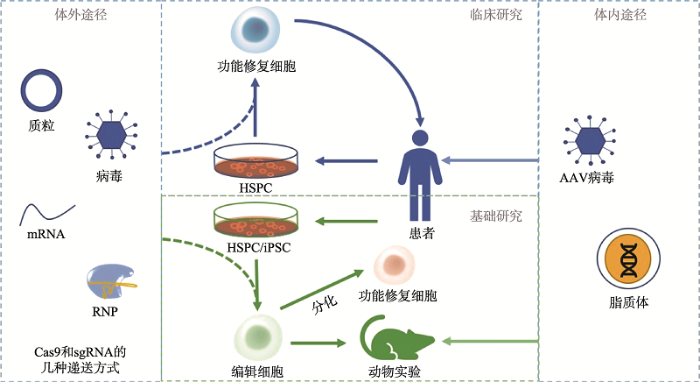 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CRISPR/Cas9用于遗传性血液病基因治疗的研究
sgRNA:向导RNA;RNP:核糖核蛋白复合体;HSPC:造血干细胞或祖细胞;iPSC:诱导多能干细胞。
Fig. 1Strategy on CRISPR/Cas9 for gene therapy of inherited blood disorders
理想的基因治疗是突变基因的原位修复,目前主要通过基因编辑技术实现。经过第一代锌指核酸酶(zinc finger nuclease, ZFN)、第二代转录因子样效应物核酸酶(transcription activator-like effector nuclease, TALEN),基因编辑技术已发展至第三代——CRISPR/Cas9,进一步又发展出单碱基编辑技术(base editing, BE)[15,16]。虽然利用病毒载体增补基因是目前基因治疗临床试验的主流,但基于基因编辑技术的基因治疗临床试验项目正逐年增多,尤其CRISPR/Cas9技术介导的基因治疗临床试验已超过ZFN和TALEN相关临床试验总数。在疾病选择方面,因β-地贫和血友病等致病机制清楚,均为单基因病,且主要突变类型为点突变或移码突变,易于实施基因治疗,成为遗传性血液病基因治疗研究的主要研究对象。
2 基于CRISPR/Cas9技术的遗传性血液病基因治疗策略
CRISPR/Cas9系统由向导RNA (single-guide RNA, sgRNA)和Cas9蛋白组成[17,18,19],与ZFN和TALEN技术类似,CRISPR/Cas9技术同样通过诱导DNA双链断裂,诱发同源重组修复(homology- directed repair, HDR)和/或非同源末端连接(non- homologous end joining, NHEJ)等DNA损伤修复过程,实现基因编辑[19,20,21](图2)。其中同源重组修复需要同源模版,DNA双链断裂按模版精确修复,只在S期晚期和G2期姐妹染色单体存在时起作用;非同源末端连接无需同源模版,无法精确修复DNA双链断裂,修复时在双链断裂处造成核苷酸插入或缺失(insertions and/or deletions, indel),可删除突变位点,或改变关键DNA序列引起基因表达改变,在整个细胞周期均可发生[22,23] (表2)。两种途径的选择主要由同源模版和细胞所处细胞周期决定,难以人为调控。图2
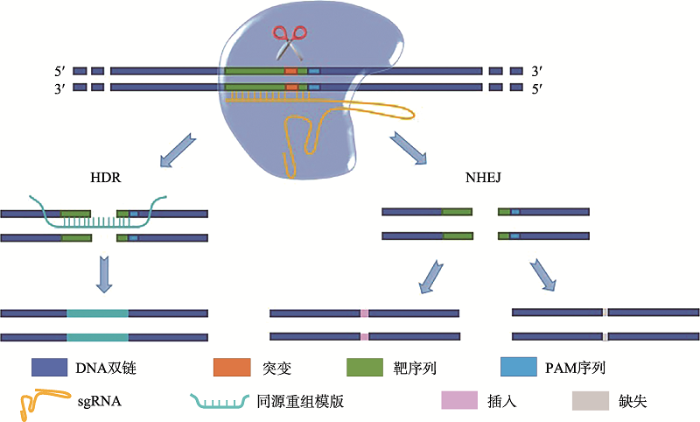 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2CRISPR/Cas9基因编辑原理
HDR:同源重组修复途径;NHEJ:非同源末端连接途径。
Fig. 2Principle of CRISPR/Cas9 gene editing
基于CRISPR/Cas9系统,单碱基编辑技术被开发出来。单碱基编辑技术通过在无核酸酶活性的Cas9蛋白(dCas9)或只有切割DNA单链活性的Cas9蛋白(Cas9-nickase, Cas9n)上融合胞嘧啶脱氨酶或腺嘌呤脱氨酶,在不剪切DNA双链的情况下实现C>T (G>A)或A>G (T>C)的单碱基编辑,不依赖同源重组修复和非同源末端连接等DNA损伤修复过程,无需同源模版便可实现点突变的精确修复,且编辑效率最高可达100%[24,25,26]。
Table 2
表2
表2两种DNA损伤修复途径
Table 2
| 修复途径 | 模版 | 效率 | 作用时期 | 对细胞种类/ 状态依赖性 | 修复结果 | 适用修复突变 | 参考文献 |
|---|---|---|---|---|---|---|---|
| 同源重组修复HDR | 需要 | 低 | S期和G2期 | 高 | 精确修复 | 外显子突变 | [22,23] |
| 非同源末端连接NHEJ | 不需要 | 高 | 整个细胞周期 | 低 | 核苷酸插入/缺失 | 外显子突变 | [22,23] |
| 不可精确修复 | 内含子突变 |
新窗口打开|下载CSV
2.1 原位修复突变基因
2.1.1 CRISPR/Cas9介导的外显子点突变/移码突变的修复外显子点突变/移码突变可导致蛋白质结构或合成数量异常,引发疾病。如我国常见的两种引起β-地贫的β-珠蛋白基因(HBB)突变CD41/42(-TCTT)和CD17(A>T)[27]。由于外显子编码蛋白质,外显子突变需精确修复才可恢复基因功能,因此外显子突变必须依赖同源重组修复途径精确修复,但在造血干细胞和诱导多能干细胞(induced pluripotent stem cell, iPSC)中,同源重组效率低且不稳定(0.045%~ 57%) (表3)[15,28~32]。靶细胞类型、靶细胞所处细胞周期、Cas9蛋白、sgRNA、同源模版以及CRISPR/Cas9系统的递送方式等的差异是造成同源重组效率低且不稳定的可能原因,优化这些参数或可提升同源重组效率。此外,2018年报道的研究利用CRISPR/Cas9技术修复HbE/β-地贫患者(β-珠蛋白等位基因一个带有26号密码子G>A点突变,产生异常β链进而产生异常血红蛋白HbE;另一个带有CD41/42-TCTT缺失,导致β链缺失)来源iPSC中的26号密码子G>A点突变,发现正确修复突变的iPSC经红系分化后β-珠蛋白基因mRNA和蛋白无显著提升,作者分析可能由于β-珠蛋白基因表达所需的转录因子KLF1和BCL11A的表达水平低所致,但遗憾的是该研究未分析修复后细胞产生的β-珠蛋白是否为正常β-珠蛋白[32]。这提示,对基因组的修复可能无法实现转录水平和蛋白水平的修复,导致无效的基因治疗,因此突变修复后的细胞是否能产生正常的转录本并翻译出足够的功能正常的蛋白质也是CRISPR/Cas9用于基因治疗需要考虑的问题。
Table 3
表3
表3CRISPR/Cas9原位修复突变基因的研究
Table 3
| 突变 类型 | 主要疾病 | 策略 | 年份 | 编辑 细胞 | 编辑基因 | 修复/编辑效率 | 脱靶率 | 编辑后体外细胞表型 | 参考文献 |
|---|---|---|---|---|---|---|---|---|---|
| 外显子点突变和移码突变 | β-地贫 镰状细胞贫血 血友病B | HDRa | 2014 | β-地贫iPSC | HBB | -28A/G: 7.8% CD41/42 -TCTT: 9.8% 双位点修复效率: 0 | 未检测到脱靶 | HBB mRNA量升高约16倍 | [15] |
| 2015 | β-地贫iPSC | HBB | 16.67% | 未检测到脱靶 | HBB蛋白量由0上升至正常水平的~60% | [29] | |||
| 2016 | β-地贫iPSC | HBB | 0.045% | 未检测到脱靶 | HBB蛋白量接近正常 | [28] | |||
| 2016 | β-地贫iPSC | HBB | na ??ve iPSC组: 57% primed iPSC组: 32% | na ??ve iPSC: 0 primed iPSC: 5% | 未说明 | [74] | |||
| 2017 | β-地贫iPSC | HBB | 双等位基因修复: 54% 单等位基因修复: 25.5% | 未检测到脱靶 | 有HBB mRNA表达 | [30] | |||
| 2018 | β-地贫iPSC | HBB | 2.9% | 3个克隆中2个检测到HBD点突变 | HBB mRNA和HBB蛋白无明显上升 | [32] | |||
| 2019 | 镰状细胞贫血HSPC | HBB | 24.5±7.6% | Cas9: 0.1%~36.7% HiFi Cas9d: ≤0.23% | HbA水平由5.3±1%提升至25.3±13.9% | [31] | |||
| 内含子点突变 | β-地贫 | HDR | 2018 | β-地贫HSC | HBB | 8% | 未检测到脱靶 | 未说明 | [33] |
| NHEJb | 2019 | β-地贫HSPC | HBB | indel产生率: 93% | 未说明 | HbA分数从36.4%上升至75.6% | [35] | ||
| 内含子倒位 | 血友病A | NAHRc | 2015 | 血友病A iPSC | FVIII | 3.7% | 未检测到脱靶 | F8 mRNA量由0上升至正常水平的50%~120% | [36] |
新窗口打开|下载CSV
2.1.2 CRISPR/Cas9介导的内含子点突变的修复
内含子点突变可能导致mRNA剪接异常,影响蛋白质翻译,从而引发疾病。例如,导致β-地贫的β-珠蛋白基因IVS2-654 (C>T)突变,造成β-珠蛋白mRNA前体剪接异常,产生的异常mRNA (254 bp)较正常mRNA (181 bp)多出73 bp的内含子序列,进而影响β-珠蛋白的合成[11]。内含子不编码蛋白,内含子突变只要不影响mRNA前体的剪接,便不影响基因功能,因此无需精确修复,同源重组修复和非同源末端连接两种DNA双链断裂修复途径都可恢复内含子突变基因的功能[33,34,35]。通过非同源末端连接途径修复内含子突变附近的DNA双链断裂可高效删除内含子突变(效率可达98%) (表3),恢复基因功能[34,35]。但非同源末端连接途径产生的插入或缺失具有异质性,这些插入或缺失的产生是否会引入新的突变,造成基因表达和蛋白功能的异常,值得进一步研究。
2.1.3 CRISPR/Cas9介导的内含子倒位的修复
内含子倒位是引起重型血友病A的常见突变类型,约50%重型血友病A患者因凝血因子VIII基因1号或22号内含子倒位破坏了基因编码区,进而导致凝血因子VIII转录本的缺失,无法合成凝血因子VIII而致病[36]。非等位同源重组(non-allelic homologous recombination, NAHR)是导致凝血因子VIII基因内含子倒位突变的原因[36]。非等位同源重组由基因组中与同源模版序列类似的非同源序列(非同源模版)介导,是引起DNA重排的关键机制之一[37,38]。凝血因子VIII基因含有的两个1号内含子同系物(int1h-1、int1h-2)和3个22号内含子同系物(int22h-1、int22h-2、int22h-3)可作为非同源模版诱发凝血因子VIII基因上的DNA双链断裂经非等位同源重组途径错误修复,产生内含子倒位[36]。
研究表明模拟这一过程可修复倒位突变[39]。通过在倒位片段的两端设计向导RNA,引导Cas9切割倒位片段两端,诱发非等位同源重组,使倒位片段再次发生倒位,实现患者iPSC中内含子倒位的修复,基因修复的细胞分化后凝血因子VIII基因表达量由0上升至正常水平的50%~120%,但修复效率只有3.7%[36]。这可能由于细胞中存在一系列抑制非等位同源重组的调控机制[40](表3)。
2.1.4 单碱基编辑技术介导的原位修复
对于单碱基突变引起的遗传性血液病,除了上述利用CRISPR/Cas9技术进行原位修复,还可通过单碱基编辑技术实现突变的精确修复。对β-珠蛋白基因上-28(A>G)突变进行单碱基编辑,结果表明患者来源的造血干细胞中有36.4%的突变实现精确修复(C>T),56.4%的突变被错误编辑(C>G/A),3.6%的突变未被编辑[41]。说明在患者造血干细胞中单碱基编辑技术介导的精确修复效率有待提升。此外单碱基编辑技术还存在脱靶率高的问题,研究表明单碱基编辑系统可造成基因组水平和转录组水平的大范围脱靶[24]。在HEK293T细胞系中单碱基编辑系统BE3可导致近100%的RNA单核苷酸变异[42]。向脱氨酶中引入点突变可能是减少脱靶的有效方法[42]。
2.2 调控基因表达
α链和β链数量不平衡是导致地贫的直接原因,β-地贫患者的β链合成减少或缺失导致α链相对过剩,减少α链合成可缓解疾病症状[12,43]。对α-珠蛋白的顺式调控元件进行基因编辑,可调控α-珠蛋白的表达量。研究表明利用CRISPR/Cas9敲除β-地贫患者造血干细胞中α-珠蛋白基因增强子MCS-R2的核心元件,可降低α链的合成水平[44]。γ链是胎儿血红蛋白HbF (fetal hemoglobin, α2γ2)的组成部分,在胎儿时期高表达[43]。出生后,γ链逐渐被β链取代,人体内主要血红蛋白转变为HbA (adult hemoglobin, α2β2)[43] (图3)。重激活γ-珠蛋白基因,产生γ链与α链结合形成HbF,可代替HbA发挥功能,缓解α链相对过剩导致的地贫症状和β链结构异常导致的镰状细胞贫血症状[45,46,47,48]。
图3
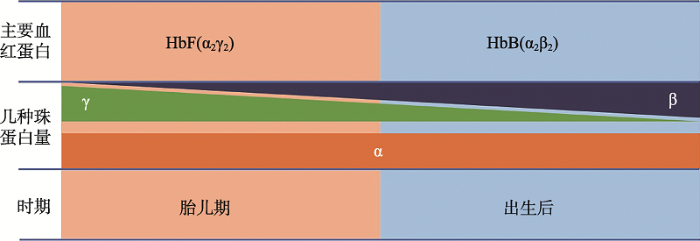 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3血红蛋白演变示意图
Fig. 3Schematic diagram of hemoglobin developmental switches
γ-珠蛋白基因表达受转录因子、启动子、增强子等多种元件调控。其中转录抑制因子BCL11A等是成人红系细胞中γ-珠蛋白基因低表达的重要影响因素[46,49]。BCL11A可以结合γ-珠蛋白基因的近端启动子,并抑制γ-珠蛋白基因表达。由于BCL11A在淋巴细胞发育中有重要调控作用,敲除造血干细胞中的BCL11A虽可显著提升其分化的红系细胞的γ-珠蛋白基因的表达水平,但会影响淋巴细胞的成熟[50,51]。进一步研究发现BCL11A存在一个红系特异增强子,在造血干细胞中敲除该增强子的DNA酶I超敏位点(DNase I hypersensitive site, DHS) +58可特异下调红系细胞中BCL11A的表达,而不影响其他类型细胞,且效果与直接敲除BCL11A相似[45,49]。利用CRISPR/Cas9在β-地贫和镰状细胞贫血患者的造血干细胞和祖细胞中敲除此位点,其分化后的红系细胞可产生治疗水平的胎儿血红蛋白[45](表4)。基于这一思路,相关临床试验已展开(表5)。
Table 4
表4
表4基因表达调控: CRISPR/Cas9体外重激活HBG的基础研究
Table 4
| 年份 | 编辑细胞 | 编辑方式 | 编辑效率 | 脱靶率 | 编辑后体外细胞表型 | 参考文献 |
|---|---|---|---|---|---|---|
| 2015 | 正常人HSPC | 破坏BCL11A红系特异增强子 | N/A | N/A | HBG表达水平上升近4倍,分化后HbF+细胞占比升高近2倍 | [49] |
| 2018 | HUDEP-2a | 破坏HBG近端启动子上BCL11A和ZBTB7A结合位点 | N/A | N/A | mRNA水平: γ/(γ+β)%比值由不到5%上升至~70% 蛋白水平: HbF水平由0上升至58.7±9.6% | [47] |
| 2018 | SCD HSPCb | 敲除13.6kb长的可能的γ-珠蛋白基因抑制因子区域 | 缺失: 34.1% 倒位: 28.1% | 0~4.6% | γ链/类β链的比值较对照上升近2.5倍 0%氧气条件下,镰状细胞数由~65%下降至~30% | [48] |
| 2018 | SCD CD34+ 细胞c | 敲除HRId | N/A | N/A | mRNA水平: HBG/(HBG+HBB)%比值由3.2%上升至24% 蛋白水平: HbF上升近4倍 细胞水平: HbF+细胞占比升高近2倍 | [53] |
| 2019 | β-地贫HSPC | 破坏BCL11A红系特异增强子 | 平均84.4% | 未检测 到脱靶 | HbF/(HbF+HbA+HbA2)%比值由~30%上升至~70% | [45] |
| 2019 | HUDEP-2 | 破坏HBG近端启动子上BCL11A结合位点 | N/Ae | N/A | mRNA水平: γ/(γ+β)%比值由~10%上升至~60% 蛋白水平: HbF/(HbF+HbA)%比值由零上升至~40% | [46] |
| 2020 | β-地贫HSPC | 破坏HBG近端启动子上BCL11A结合位点 | 平均85% | 未检测 到脱靶 | γ-珠蛋白的mRNA水平可达α-珠蛋白的126% | [52] |
新窗口打开|下载CSV
此外,重激活γ-珠蛋白基因还可采取其他方法,如破坏γ-珠蛋白基因近端启动子上转录抑制因子BCL11A或ZBTB7A的结合位点[46],其中BCL11A结合位点(TGACCA:HBG启动子的-114至-119)是基因编辑的理想靶点,在β-地贫患者的造血干细胞和祖细胞中利用CRISPR/Cas9破坏该位点,平均编辑效率可达85%,编辑后的造血干细胞和祖细胞经红系分化,γ-珠蛋白的mRNA可达α-珠蛋白的126%,或利用单碱基编辑器(hAPOBEC3A-Cas9n, hA3A-BE3)破坏该位点,可使γ-珠蛋白mRNA与α-珠蛋白mRNA的比值由~6.8%升至~44.2%[52];模拟引起遗传性胎儿血红蛋白持续存在综合症的-113A>G突变,在γ-珠蛋白基因近端启动子上创造新的转录激活因子GATA1结合位点[47];敲除β-珠蛋白基因簇上可能的γ-珠蛋白基因抑制因子区域[48];下调血红素调节抑制因子(heme-regulated inhibitor, HRI)进而引起BCL11A表达下调[53]等(表4)。
Table 5
表5
表5基于CRISPR/Cas9技术的基因治疗临床试验
Table 5
| NCT编号t | 国家 | 治疗疾病 | 治疗途径 | 编辑细胞 | 编辑方式 | 试验阶段 | 状态 |
|---|---|---|---|---|---|---|---|
| NCT03745287 | 美国、比利时、加拿大、德国和意大利 | 镰状细胞贫血 | 体外 | 自体CD34+ HSPCa | 破坏BCL11Ab的红系特异增强子 | 临床I/II期 | 招募中 |
| NCT03655678 | 美国、加拿大、德国、意大利和英国 | 输血依赖型β-地贫(非β0/β0) | 体外 | 自体CD34+ HSPC | 破坏BCL11A的红系特异增强子 | 临床I/II期 | 招募中 |
| NCT04205435 | 中国 | 重型β地贫 | 体外 | 自体HSC | 修复HBB IVS2-654(C>T)突变 | 临床I/II期 | 未开始招募 |
| NCT03728322 | N/As | 地贫 | 体外 | 自体HSC | HBB基因突变的原位修复 | 早期I期临床 | 未开始招募 |
| NCT04037566 | 中国 | CD19+白血病/淋巴瘤 | 体外 | 自体T细胞 | 敲除靶向CD19c的CAR-Td细胞内源性HPK1e | 临床I期 | 招募中 |
| NCT04035434 | 美国、澳大利亚和德国 | CD19+ B细胞恶性肿瘤 | 体外 | 异体T细胞 | 敲入靶向CD19的CAR并敲除TCRf和MHC Ig | 临床I/II期 | 招募中 |
| NCT03166878 | 中国 | CD19+ B细胞白血病/淋巴瘤 | 体外 | 异体T细胞 | 敲除靶向CD19的CAR-T细胞内源性TCR和B2Mh | 临床I/II期 | 招募中 |
| NCT03398967 | 中国 | 复发难治性B细胞恶性肿瘤 | 体外 | 异体T细胞 | N/A | 临床I/II期 | 招募中 |
| NCT03690011 | 美国 | T细胞白血病或淋巴瘤 | 体外 | 自体T细胞 | 敲除靶向CD7i和CD28j的CAR-T细胞内源性CD7 | 临床I期 | 未开始招募 |
| NCT04244656 | 美国、澳大利亚和西班牙 | 多发性骨髓瘤 | 体外 | 异体T细胞 | 敲入靶向BCMAk的CAR并敲除TCR和MHC I | 临床I期 | 招募中 |
| NCT03399448 | 美国 | 多发性骨髓瘤 黑色素瘤 滑膜肉瘤 黏液样/圆形细胞脂肪肉瘤 | 体外 | 自体T细胞 | 敲除靶向 NY-ESO-1l阳性癌细胞的T细胞内源性TCR和 PDCD1m | 临床I期 | 终止 |
| NCT03044743 | 中国 | EBVn阳性IV期胃癌 EBV阳性鼻咽癌 EBV阳性淋巴瘤 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | 临床I/II期 | 招募中 |
| NCT03747965 | 中国 | 间皮素阳性实体瘤 | 体外 | 靶向间皮素的CAR-T细胞 | 敲除靶向间皮素的CAR-T细胞内源性 PDCD1 | 临床I期 | 招募中 |
| NCT03545815 | 中国 | 间皮素阳性实体瘤 | 体外 | 靶向间皮素的CAR-T细胞 | 敲除靶向间皮素的CAR-T细胞内源性 PDCD1和TCR | 临床I期 | 招募中 |
| NCT04426669 | 美国 | 转移性胃肠上皮癌 | 体外 | 自体肿瘤浸润性淋巴细胞 | 破坏肿瘤浸润性淋巴细胞CISHo基因 | 临床I/II期 | 招募中 |
| NCT03081715 | 中国 | 食管癌 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | N/A | 完成 |
| NCT02863913 | 中国 | 浸润性膀胱癌IV期 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | 临床I期 | 撤回 |
| NCT04438083 | 澳大利亚 | 肾细胞癌 | 体外 | 异体T细胞 | 敲入靶向CD70的CAR并敲除TCR和MHC I | 临床I期 | 招募中 |
| NCT02867332 | N/A | 转移性肾细胞癌 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | 临床I期 | 撤回 |
| NCT编号t | 国家 | 治疗疾病 | 治疗途径 | 编辑细胞 | 编辑方式 | 试验阶段 | 状态 |
| NCT02867345 | 中国 | 激素难治性前列腺癌 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | N/A | 撤回 |
| NCT02793856 | 中国 | 转移性非小细胞肺癌 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | 临床I期 | 未开始招募 |
| NCT04417764 | 中国 | 晚期肝癌 | 体外 | 自体T细胞 | 敲除T细胞内源性PDCD1 | 临床I期 | 招募中 |
| NCT03057912 | 中国 | 人乳头瘤病毒相关恶性肿瘤 | 阴道栓剂 | 人乳头瘤病毒 | 破坏人乳头瘤病毒E6/E7基因 | 临床I期 | 未知 |
| NCT03164135 | 中国 | HIV-1感染合并有血液肿瘤 | 体外 | 异体CD34+ HSPC | 敲除CCR5p基因 | N/A | 招募中 |
| NCT03855631 | 法国 | 歌舞伎综合征1 | 细胞水平研究 | 自体成纤维细胞 | 表观修饰提升野生KMT2Dq表达 | N/A | 未开始招募 |
| NCT03872479 | 美国 | Leber先天性黑蒙症10型 | 体内(视网膜下注射) | N/A | 破坏CEP290 IVS26 A>G突变r | 临床I/II期 | 招募中 |
新窗口打开|下载CSV
3 CRISPR/Cas9治疗策略与其他基因疗法的比较
对于遗传性血液病,目前利用病毒载体增补基因的基因治疗方法仍是临床试验的主流,尤其安全性更高、包装能力更强的慢病毒载体应用最为广泛。2018年报道的临床试验利用慢病毒载体将β-珠蛋白基因导入患者造血干细胞,对22例输血依赖型地贫患者进行基因治疗,其中12例非β0/β0基因型患者停止接受红细胞输入,另9例β0/β0基因型或双IVS1-110突变患者年输血量中位值降低了73%[54],证实了通过增补基因治疗β-地贫的可行性。但基因的随机整合存在插入突变等安全问题[55],因此与整合型病毒介导的基因增补相比,CRISPR/Cas9技术能直接靶向突变位点,目的性强。采用非整合型病毒可降低随机整合引起的安全问题,如腺相关病毒,一般在胞内以附加体形式存在,基本不整合到基因组中,但其导入基因的长期表达效果不理想[56]。将腺相关病毒载体注射到体内增补凝血因子VIII或凝血因子IX基因是血友病主要的基因治疗方法[9]。对于血友病B,2017年报道的临床试验中凝血因子IX平均促凝活性水平可达33.7±18.5%,持续时间超1年[9,57];另一项研究中凝血因子IX表达时间可达8年甚至更久[9]。在血友病A基因治疗临床试验中,凝血因子VIII促凝活性水平可达52.3%,表达时间可达3年[9]。但随着细胞分裂,非整合型的腺相关病毒载体可能被丢失[56]。理想状态下,CRISPR/Cas9系统只需短暂存在于待编辑的细胞中便可实现永久的靶向基因修复。有动物实验通过颞静脉注射,将载有CRISPR/Cas9基因编辑系统的腺相关病毒载体注入凝血因子IX敲除的小鼠模型中,以实现人凝血因子IX的敲入[58]。治疗后,小鼠体内人凝血因子IX活性达正常水平的120%~ 160%,并至少可持续32周[58]。目前尚无基于CRISPR/ Cas9技术的血友病基因治疗相关临床试验。
与其他基因编辑技术相比CRISPR/Cas9技术主要胜在应用面广、易操作、效率高。不同于ZFN和TALEN需通过合成特殊蛋白识别靶DNA,CRISPR/ Cas9只需借助一段22nt的与DNA碱基互补配对的向导RNA,避免了复杂且昂贵的蛋白设计合成过程,且向导RNA设计合成灵活简便,CRISPR/Cas9系统装配简单,使得CRISPR/Cas9技术迅速得到广泛应用,包括用于基因治疗[59](表6)。
单碱基编辑系统对靶位点的识别机制与CRISPR/ Cas9相同,不同的是,CRISPR/Cas9介导的基因编辑依赖同源重组修复和非同源末端连接等DNA损伤修复途径对DNA双链断裂的修复[16]。同源重组修复虽能精确修复突变但效率低,非同源末端连接会造成核苷酸插入或缺失,可能引入新的突变[16]。单碱基编辑技术不造成DNA双链断裂,其介导的基因编辑不依赖同源重组修复和非同源末端连接等DNA损伤修复过程,不会造成核苷酸插入或缺失,且对单个碱基的精确编辑效率高,可达100%[16,25,26]。但单碱基编辑系统只能实现单个碱基的编辑,而CRISPR/Cas9除可实现单个碱基的编辑外,还可实现DNA大片段的插入或敲除。
4 基于CRISPR/Cas9技术的基因治疗临床研究
2016年中国开展了世界首个基于CRISPR/Cas9技术针对转移性非小细胞肺癌的基因治疗临床试验[60]。截止至2020年6月,基于CRISPR/Cas9技术的基因治疗临床试验共26项(表5),涉及遗传性血液病、血液肿瘤、实体瘤、感染性疾病和罕见病。对于血液肿瘤和实体瘤,CRISPR/Cas9主要用于编辑嵌合抗原受体(chimeric antigen receptor, CAR) T细胞(CAR-T cell)和患者自体T细胞,以提高治疗的安全性和有效性。2020年5月上述首个基于CRISPR/ Cas9技术的基因治疗临床试验(NCT02793856)公布了最新结果[61]。该研究利用CRISPR/Cas9技术体外破坏患者自体T细胞的PDCD1基因,以激活T细胞杀伤癌细胞,二代测序结果表明编辑效率中位数为5.81% (范围0.42%~24.85%),脱靶率中位数为0.05% (范围0~0.25%)[61]。12名晚期非小细胞肺癌患者接受基因编辑T细胞治疗,无患者获得病情的部分缓解,2名患者获得病情稳定,未见严重不良反应[61]。另一项临床试验针对癌症-睾丸抗原NY-ESO-1和/或LAGE-1阳性肿瘤(NCT03399448),通过慢病毒载体将靶向NY-ESO-1和LAGE-1的T细胞抗原受体(T cell receptor, TCR)编码序列导入患者自体T细胞,使之靶向NY-ESO-1和/或LAGE-1阳性肿瘤细胞,同时利用CRISPR/Cas9技术敲除T细胞内源TCR和PDCD1,以增强T细胞对肿瘤的杀伤力[62]。2名难治性晚期骨髓瘤患者和1名难治性静态肉瘤患者接受基因编辑T细胞输注后,2名患者获得病情稳定,未见细胞因子释放综合征或细胞输注引起的明显副作用[62]。感染性疾病包括人乳头瘤病毒感染和艾滋病。对于前者,研究利用CRISPR/Cas9靶向破坏人乳头瘤病毒的E6和E7基因,使病毒丧失感染能力。相反,艾滋病的治疗试图利用CRISPR/Cas9靶向破坏编码人类免疫缺陷病毒1型(human immunodeficiency virus-1, HIV-1)主要受体蛋白的CCR5基因,使HIV-1无法感染淋巴细胞。2019年该临床试验(NCT03164135)公布研究结果,1名合并有急性淋巴细胞白血病的艾滋病患者接受CCR5基因敲除的异体CD34+造血干细胞和祖细胞输注后急性淋巴白血病得到完全缓解,携带CCR5突变的造血干细胞和祖细胞在患者体内可长期存活达19个月,但不足以治愈HIV-1感染导致的艾滋病,研究未见基因编辑相关的不良反应[63]。上述临床研究结果显示了基于CRISPR/Cas9技术的基因治疗的可行性及安全性,但有效性有待提升。罕见病有歌舞伎综合征1和Leber先天性黑蒙10型(Leber congenital amaurosis type 10, LCA-10),其中针对Leber先天性黑蒙10型的临床试验是第一项也是目前唯一一项CRISPR/Cas9介导的体内途径的基因治疗临床试验[64]。Table 6
表6
表6基因编辑技术对比
Table 6
| 基因编辑技术 | |||
|---|---|---|---|
| ZFN | TALEN | CRISPR/Cas9 | |
| 识别模式 | 蛋白-DNA | 蛋白-DNA | RNA-DNA |
| 识别序列长度(nt) | 18~36 | 30~40 | 22 |
| 操作 | 复杂 | 复杂 | 简单 |
| 效率 | 低 | 低 | 较高 |
| 脱靶率 | 较低 | 低 | 较高 |
| 成本 | 高 | 高 | 较低 |
新窗口打开|下载CSV
总体来看,基于CRISPR/Cas9技术的基因治疗临床试验以体外途径为主,主要集中于癌肿治疗(20项),可能由于癌肿往往严重威胁患者生命,癌肿的治疗是目前临床上迫切需要解决的问题。从疾病发生系统来说,则主要集中于血液系统疾病(13项),这可能得益于造血干细胞移植的成功和CAR-T细胞疗法在血液肿瘤中取得了较好的临床效果[65]。在遗传性血液病方面,目前临床研究数相对较少(4项),可能由于遗传性血液病目前已有疗法有一定效果,且CRISPR/Cas9系统存在的缺陷暂未很好解决,目前相关临床研究结果尚未发布。
5 结语和展望
CRISPR/Cas9技术走向临床展现了其用于基因治疗的良好前景,但其存在的问题仍不容忽视。(1)安全性问题。研究表明CRISPR/Cas9技术存在严重脱靶效应,脱靶率可达靶向编辑效率的5.6%~125%[66]。最新研究报道Cas9蛋白可能多次切割DNA,造成百万碱基级的染色体末端大片段缺失[67]。(2)精确编辑效率低。(3)编辑产物异质性高。DNA双链断裂经非同源末端连接修复时可产生40种[35],甚至更多的基因型,导致基因编辑产物异质性高,难以进行质控。针对这些问题,可从以下几个方面进行研究和解决。
(1)改造递送系统提高编辑效率,降低脱靶率。质粒、病毒、mRNA和核糖核蛋白复合体(ribonucleoprotein, RNP)是依次开发出的四种CRISPR/Cas9递送系统[68](图1)。最新的核糖核蛋白复合体由向导RNA与Cas9蛋白体外孵育组装而成,进入细胞后发挥短时的基因编辑功能,有较高的基因编辑效率和更低的细胞毒性和脱靶率,更适用于基因治疗,但其转导效率有待进一步提升[45,68,69]。上述3项临床研究中(NCT02793856、NCT03399448、NCT03164135)有两项(NCT03399448、NCT03164135)使用核糖核蛋白复合体递送系统。
(2)改造CRISPR/Cas9系统提高编辑效率,降低脱靶率。在改造向导RNA方面,对于同一靶点不同向导RNA介导的基因编辑效率和脱靶率差异显著[70],目前主要通过实验筛选的方法,筛出效率最高、脱靶率最低的向导RNA进行后续实验。针对人和小鼠基因组,科学家基于大量实验开发了向导RNA库,用于评估向导RNA的编辑效果[70];对于mRNA和核糖核蛋白复合体递送系统,向导RNA的稳定性对基因编辑效率影响较大,化学修饰向导RNA可提升向导RNA稳定性,防止其过快降解,提升基因编辑效率,将插入或缺失(indel)率从2.4%提升至83.3%,同源重组效率从~15%提升至50%[71]。在改造Cas9蛋白方面,改造Cas9蛋白的氨基酸组成可提高靶向编辑率:总编辑率的比值,如p.R691A单点突变的HiFi Cas9,其总编辑效率与野生型Cas9相近,但on-target效率可达总编辑效率的99%,显著高于野生型的28%~72%,且HiFi Cas9较野生型Cas9脱靶率降低近20倍[72];在Cas9蛋白上融合核定位序列,辅助Cas9蛋白入核,也可提升基因编辑效率[45]。
(3)阐明DNA损伤修复机制。CRISPR/Cas9介导的基因编辑依赖DNA损伤修复机制,目前主要考虑同源重组和非同源末端连接。研究表明,微同源末端连接(microhomology-mediated end joining, MMEJ)是DNA损伤修复的又一重要途径,为基因编辑介导的基因治疗策略提供了更多可能[73]。
(4)找寻合适的基因编辑靶点。基因编辑安全实施的关键是找到特异且易编辑的靶点,而找寻靶点的基础是阐明疾病致病机制,使得基因治疗靶点不限于突变位点,而是可以拓展到突变基因的上下游或替代/补偿途径去寻找最合适的编辑靶点,比如,对于β-地贫和镰状细胞贫血,可通过重激活内源γ-珠蛋白基因,缓解疾病症状。该方法不受突变位点影响,适用范围广,为其他有类似调节机制的疾病提供了基因治疗的新思路。基因调控元件作为编辑靶点是近两年的研究趋势,越来越多的研究表明对启动子、增强子等进行编辑可有效调控基因表达,尤其对于谱系特异的调控元件,安全性更高。
(5)发展造血干细胞相关技术。血液病的基因治疗离不开干细胞技术的发展。目前血液病治疗和临床研究主要采用造血干细胞和祖细胞及其分化出的T淋巴细胞。造血干细胞和祖细胞的应用仍存在难以体外增殖、体外培养体系不成熟、已有分子标记特异性不强、同源重组修复效率低且不稳定等问题。基因编辑细胞移植至免疫缺陷小鼠后编辑效率降低可能就由于其中混有未编辑的造血干细胞和祖细胞[45],而目前基因编辑效率难以实现100%,也难以分选出正确编辑的造血干细胞和祖细胞。但造血干细胞和祖细胞安全性好,移植技术成熟,是基因治疗靶细胞的首选。
(6)发展诱导多能干细胞相关技术。2006年诱导多能干细胞(iPSC)的问世为遗传病基因治疗研究提供了良好的细胞模型,患者来源的iPSC与患者遗传背景一致,无伦理问题,且可在体外大量扩增。研究发现,CRISPR/Cas9在na ??ve iPSC中的编辑效率高出primed iPSC近两倍[74];在iPSC培养物中添加小分子化合物,同源重组修复效率可从6%提升至54%,同时抑制非同源末端连接的发生,这些研究可为造血干细胞和祖细胞基因编辑提供指导[30]。iPSC具有分化为三胚层细胞的潜能,但未分化的iPSC有致瘤风险,无法直接用于基因治疗[75]。将其分化为造血干细胞等成体干细胞或可解决此问题。目前将iPSC分化为造血干细胞和祖细胞的分化体系尚不成熟,分化出的造血干细胞在免疫缺陷小鼠中难以长期植入[76],基因组不稳定,易发生白血病转化[77]。此外,虽然实验室中突变修复后的β-地贫患者来源iPSC红系分化效率提高且分化所得红系细胞β-珠蛋白表达提升[15,28,29],但多数研究中突变未修复iPSC和突变修复iPSC来源的红系细胞均主要表达γ-珠蛋白[15,29,78],难以模拟正常人红系细胞主要表达β-珠蛋白的真实情况。
(7)其他。目前CRISPR/Cas9用于基因治疗的技术标准、临床疗效、不良反应等尚不明确,需待进一步的临床研究。基因编辑技术用于基因治疗的相关法律法规也有待进一步完善。
综上所述,基于CRISPR/Cas9技术的基因治疗研究主要集中于血液系统疾病,包括血液肿瘤和遗传性血液病,尤其适用于主要由单基因点突变或移码突变造成的β-地贫等疾病,最安全有效的细胞是患者自体造血干细胞和祖细胞,更安全的途径是体外途径。在体外造血干细胞和祖细胞处于G0期,主要采取非同源末端连接的方式修复DNA双链断裂,因此CRISPR/Cas9虽可修复多种类型突变,但更适用于内含子突变的删除和基因表达调控,尤其利用CRISPR/Cas9重激活γ-珠蛋白基因表达的基因治疗方式,已开始临床研究。但通过非同源末端连接的方式实现基因编辑终究无法精确修复基因突变,潜在的风险无法评估,因此最理想最希望实现的仍是通过同源重组修复精确修复基因突变,提高同源重组修复效率应是今后CRISPR/Cas9用于疾病治疗的重点研究方向。此外,单碱基编辑技术能实现高效且精确的单碱基编辑,在单碱基突变引起的疾病中有巨大的应用前景,可与CRISPR/Cas9技术互为补充。目前单碱基编辑技术进入临床要解决的首要问题亦是脱靶率高。相信随着上述问题的解决,基于CRISPR技术的基因治疗会成为安全且高效的基因治疗方法,为治愈遗传性疾病带来希望。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/S0140-6736(17)31822-6URLPMID:28774421 [本文引用: 3]

Inherited haemoglobin disorders, including thalassaemia and sickle-cell disease, are the most common monogenic diseases worldwide. Several clinical forms of alpha-thalassaemia and beta-thalassaemia, including the co-inheritance of beta-thalassaemia with haemoglobin E resulting in haemoglobin E/beta-thalassaemia, have been described. The disease hallmarks include imbalance in the alpha/beta-globin chain ratio, ineffective erythropoiesis, chronic haemolytic anaemia, compensatory haemopoietic expansion, hypercoagulability, and increased intestinal iron absorption. The complications of iron overload, arising from transfusions that represent the basis of disease management in most patients with severe thalassaemia, might further complicate the clinical phenotype. These pathophysiological mechanisms lead to an array of clinical manifestations involving numerous organ systems. Conventional management primarily relies on transfusion and iron-chelation therapy, as well as splenectomy in specific cases. An increased understanding of the molecular and pathogenic factors that govern the disease process have suggested routes for the development of new therapeutic approaches that address the underlying chain imbalance, ineffective erythropoiesis, and iron dysregulation, with several agents being evaluated in preclinical models and clinical trials.
DOI:10.1016/j.bcmd.2017.09.004URLPMID:29032940 [本文引用: 1]
alpha-Thalassemia is an inherited, autosomal recessive, disorder characterized by a microcytic hypochromic anemia. It is one of the most common monogenic gene disorders in the world population. The clinical severity varies from almost asymptomatic, to mild microcytic hypochromic, and to a lethal hemolytic condition, called Hb Bart's Hydrops Foetalis Syndrome. The molecular basis are usually deletions and less frequently, point mutations affecting the expression of one or more of the duplicated alpha-genes. The clinical variation and increase in disease severity is directly related to the decreased expression of one, two, three or four copies of the alpha-globin genes. Deletions and point mutations in the alpha-globin genes and their regulatory elements have been studied extensively in carriers and patients and these studies have given insight into the alpha-globin genes are regulated. By looking at naturally occurring deletions and point mutations, our knowledge of globin-gene regulation and expression will continue to increase and will lead to new targets of therapy.
DOI:10.1136/jmg.24.10.578URLPMID:3500312 [本文引用: 2]
A large scale survey of haemoglobinopathies and thalassaemia has been carried out in China, involving 900,000 people in 28 provinces. It has resulted in the finding of many new variants and some interesting cases of thalassaemia, and in a study on the chemical structure of abnormal haemoglobins and DNA analysis of thalassaemia. We report here data on haemoglobin disorders in the Chinese, mainly the characterisation of the geographical distribution of haemoglobin variants, the analysis of globin genes of alpha, beta, gamma, or delta beta thalassaemia, and the progress in prenatal diagnosis of alpha and beta thalassaemia conducted in the authors' laboratory.
[本文引用: 1]
[本文引用: 1]
DOI:10.1055/s-0033-1353996URLPMID:24014073 [本文引用: 1]
Hemophilia A and B are traditionally considered clinically indistinguishable; however, differences in bleeding frequency, clinical scores, use of prophylaxis, and need for orthopedic surgery have been reported, suggesting that the bleeding tendency associated with factor IX deficiency may be less severe with consequent better outcomes in the long term.Hemophilia A and B show their own peculiar aspects, not only in terms of epidemiological and clinical features, including inhibitor incidence and associated symptoms, but also with respect to molecular defects. The type of factor VIII/IX mutation is a major determinant of the bleeding tendency as well as of the risk of inhibitor formation; thus, there is a biological plausibility behind the different clinical expression of these two forms of congenital hemophilia. The distinction of various bleeding phenotypes in hemophilia has considerable therapeutic implications; therefore, further research in this field is required to optimize treatment regimens.
DOI:10.1126/science.270.5235.475URLPMID:7570001 [本文引用: 1]
In 1990, a clinical trial was started using retroviral-mediated transfer of the adenosine deaminase (ADA) gene into the T cells of two children with severe combined immunodeficiency (ADA- SCID). The number of blood T cells normalized as did many cellular and humoral immune responses. Gene treatment ended after 2 years, but integrated vector and ADA gene expression in T cells persisted. Although many components remain to be perfected, it is concluded here that gene therapy can be a safe and effective addition to treatment for some patients with this severe immunodeficiency disease.
[本文引用: 1]
[本文引用: 1]
DOI:10.1089/hum.2017.167URLPMID:28835123 [本文引用: 1]
Gene therapy provides hope for a cure for patients with hemophilia by establishing continuous endogenous expression of factor VIII or factor IX following transfer of a functional gene copy to replace the hemophilic patient's own defective gene. Hemophilia may be considered a
DOI:10.1111/hae.13816URLPMID:31282050 [本文引用: 5]
Gene therapy is rapidly becoming a new therapeutic strategy for haemophilia A and B treatment. In the 1990s, studies in animal models showed that adeno-associated vectors (AAV) exhibited an efficient expression of factor IX (FIX). In the first clinical trial in patients with haemophilia B, therapeutic levels of FIX were documented but the expression remained only for few weeks. Subsequently, improvements in vector design, such as the use of different AAV serotypes, the development of the self-complementary vector, the engineering of the transgene with codon optimization and liver-specific expression cassette resulted in circulating FIX level between 2% and 5% for long-lasting period. Recently, a natural gain of function FIX variant (Padua) inserted in the F9 cDNA improved the expression of FIX achieving a level of more than 30% resulting in cessation of infusions and in a greatly reduction of bleeding events. Encouraging clinical progresses have been also obtained from trials of gene therapy for haemophilia A. Transgene expression persisted for three years with circulating FVIII activity levels of 52.3% in patients treated with AAV vector containing a codon-optimized F8 cDNA. A complication, reported in both clinical trials for haemophilia A and B, was the elevation of liver enzymes, which resolved with steroid treatment in a large group of patients. However, to date, the pathophysiological mechanism for the liver toxicity remains still unclear. Clinical trials with adeno-associated vectors have documented a significant success for haemophilia gene therapy demonstrating potential to transform haemophilia treatment offering hope for a long-term expression.
URLPMID:7646994 [本文引用: 1]
DOI:10.1007/s12185-010-0727-1URL [本文引用: 2]
Although the therapeutic efficacy of beta(654)-thalassaemia treatment using a combination of RNAi and antisense RNA to balance the synthesis of alpha- and beta-globin chains has been demonstrated previously, and the safety of lentiviral delivery remains unclear. Herein, we used the same beta(654)-thalassaemia mouse model to develop a therapy involving direct delivery of siRNA and antisense RNA plasmids via intravenous injection to simultaneously knock down alpha-globin transcript levels and restore correct beta-globin splicing. The amount of alpha-globin mRNAs in siRNA-treated MEL cells decreased significantly, and the properly spliced beta-globin mRNA was restored in HeLa beta(654) cells transfected with pcDNA-antisense plasmid. Furthermore, treatment of beta(654)-thalassaemic mice with siRNA and antisense RNA plasmids resulted in significant reduction of poikilocytosis and reticulocyte counts in blood samples, decreased nucleated cell populations in bone marrow, and reduced intrasinusoidal extramedullary haematopoiesis loci and iron accumulation in liver. RT-PCR analysis revealed that treatment resulted in down-regulation of alpha-globin mRNA synthesis by similar to 50% along with an increase in the presence of normally spliced beta-globin transcripts, indicating that the phenotypic changes observed in beta(654)-thalassaemic mice following treatment resulted from restoration of the balance of alpha/beta-globin biosynthesis.
DOI:10.1093/hmg/ddm218URLPMID:17716993 [本文引用: 2]
The beta-thalassemia is associated with abnormality in beta-globin gene, leading to imbalanced synthesis of alpha-/beta-globin chains. Consequently, the excessive free alpha-globin chains precipitate to the erythrocyte membrane, resulting in hemolytic anemia. We have explored post-transcriptional strategies aiming at alpha-globin reduction and beta-globin enrichment on beta(654) (Hbb(th-4)/Hbb(+)) mouse, carrying a human splicing-deficient beta-globin allele (Hbb(th-4)). Lentiviral vectors of short hairpin RNA (shRNA) targeting alpha-globin and/or antisense RNA facilitating beta-globin correct splicing were microinjected into beta(654) single-cell embryos. Three transgenic strains were generated, as alpha(i)-Hbb(th-4)/Hbb(+)(shRNA), beta(a)-Hbb(th-4)/Hbb(+)(antisense) and alpha(i)beta(a)-Hbb(th-4)/Hbb(+)(both shRNA and antisense). Without notable abnormalities, all the founders and their offsprings showed sustained amelioration of hematologic parameters, ineffective erythropoiesis and extramedullary hematopoiesis. Augmented effects appeared in alpha(i)beta(a)-Hbb(th-4)/Hbb(+), which correlated with a better-balanced alpha-/beta-globin mRNA level. Among the transgenic mice integrated with shRNA and antisense RNA, one homozygous mouse (Hbb(th-4)/Hbb(th-4)) had been viable, and the 3-week survival rate for heterozygotes (Hbb(th-4)/Hbb(+)) was 97%, compared with 45.4% for untreated. Our data have demonstrated the feasibility of techniques for beta-thalassemia therapy by balancing the synthesis of alpha-/beta-globin chains.
DOI:10.1016/j.omtm.2018.12.004URLPMID:30603654 [本文引用: 1]
Hemoglobinopathies, including sickle cell disease and thalassemia, are among the most common inherited genetic diseases worldwide. Due to the relative ease of isolating and genetically modifying hematopoietic stem and progenitor cells, recent gene editing and gene therapy strategies have progressed to clinical trials with promising outcomes; however, challenges remain and necessitate the continued exploration of new gene engineering and cell transplantation protocols. Current gene engineering strategies aim at reactivating the expression of the fetal gamma-globin genes in adult erythroid cells. The gamma-globin proteins exhibit anti-sickling properties and can functionally replace adult beta-globin. Here, we describe and compare the current genetic engineering procedures that may develop into safe and efficient therapies for hemoglobinopathies in the near future.
DOI:10.1111/ejh.12182URL [本文引用: 1]
ObjectiveTo assess the efficacy and safety of combined hydroxyurea (HU) and recombinant human erythropoietin (rHuEPO) in -thalassemia intermedia (TI) patients compared with single HU therapy.
MethodsAn interventional prospective randomized study registered in the ClinicalTrials.gov (NCT01624038) was performed on 80 TI patients (18yr) divided into group A (40 patients received combined HU and rHuEPO) and group B (40 patients received single HU therapy). Baseline serum EPO levels were measured, and both groups were followed up for a mean period of 1yr with regular assessment of transfusion requirements, blood pressure, ferritin, liver and renal functions, hemoglobin, and HbF. Quality of life (QoL) was assessed at the start and end of the study.
ResultsTransfusion frequency and index were significantly decreased, while QoL was increased in group A compared with group B where 85% of patients showed improvement on combined therapy compared with 50% of patients on HU. Hemoglobin and HbF were significantly increased in both TI groups; however, this was more evident in group A than in group B. Also, 37.5% of patients in group A became transfusion-independent compared with 15% in group B. EPO levels were negatively related to increments of hemoglobin and HbF. Splenectomized patients and those with initial HbF% >40% had the best response to combined therapy. No serious adverse events necessitating discontinuation of therapy in both groups.
ConclusionsHU was effective in management of TI; however, combination with rHuEPO gave a superior therapeutic effect resulting in the best clinical and hematological responses without adverse events.
[本文引用: 4]
[本文引用: 4]
[本文引用: 4]
DOI:10.1038/nature09523URLPMID:21048762 [本文引用: 1]
Bacteria and Archaea have developed several defence strategies against foreign nucleic acids such as viral genomes and plasmids. Among them, clustered regularly interspaced short palindromic repeats (CRISPR) loci together with cas (CRISPR-associated) genes form the CRISPR/Cas immune system, which involves partially palindromic repeats separated by short stretches of DNA called spacers, acquired from extrachromosomal elements. It was recently demonstrated that these variable loci can incorporate spacers from infecting bacteriophages and then provide immunity against subsequent bacteriophage infections in a sequence-specific manner. Here we show that the Streptococcus thermophilus CRISPR1/Cas system can also naturally acquire spacers from a self-replicating plasmid containing an antibiotic-resistance gene, leading to plasmid loss. Acquired spacers that match antibiotic-resistance genes provide a novel means to naturally select bacteria that cannot uptake and disseminate such genes. We also provide in vivo evidence that the CRISPR1/Cas system specifically cleaves plasmid and bacteriophage double-stranded DNA within the proto-spacer, at specific sites. Our data show that the CRISPR/Cas immune system is remarkably adapted to cleave invading DNA rapidly and has the potential for exploitation to generate safer microbial strains.
DOI:10.1038/nature09886URLPMID:21455174 [本文引用: 1]
CRISPR/Cas systems constitute a widespread class of immunity systems that protect bacteria and archaea against phages and plasmids, and commonly use repeat/spacer-derived short crRNAs to silence foreign nucleic acids in a sequence-specific manner. Although the maturation of crRNAs represents a key event in CRISPR activation, the responsible endoribonucleases (CasE, Cas6, Csy4) are missing in many CRISPR/Cas subtypes. Here, differential RNA sequencing of the human pathogen Streptococcus pyogenes uncovered tracrRNA, a trans-encoded small RNA with 24-nucleotide complementarity to the repeat regions of crRNA precursor transcripts. We show that tracrRNA directs the maturation of crRNAs by the activities of the widely conserved endogenous RNase III and the CRISPR-associated Csn1 protein; all these components are essential to protect S. pyogenes against prophage-derived DNA. Our study reveals a novel pathway of small guide RNA maturation and the first example of a host factor (RNase III) required for bacterial RNA-mediated immunity against invaders.
DOI:10.1126/science.1225829URLPMID:22745249 [本文引用: 2]
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
[本文引用: 1]
DOI:10.1080/21655979.2016.1189039URLPMID:27340770 [本文引用: 1]
CRISPR (Clustered Regularly-Interspaced Short Palindromic Repeats)-Cas9 (CRISPR associated protein 9) has rapidly become the most promising genome editing tool with great potential to revolutionize medicine. Through guidance of a 20 nucleotide RNA (gRNA), CRISPR-Cas9 finds and cuts target protospacer DNA precisely 3 base pairs upstream of a PAM (Protospacer Adjacent Motif). The broken DNA ends are repaired by either NHEJ (Non-Homologous End Joining) resulting in small indels, or by HDR (Homology Directed Repair) for precise gene or nucleotide replacement. Theoretically, CRISPR-Cas9 could be used to modify any genomic sequences, thereby providing a simple, easy, and cost effective means of genome wide gene editing. However, the off-target activity of CRISPR-Cas9 that cuts DNA sites with imperfect matches with gRNA have been of significant concern because clinical applications require 100% accuracy. Additionally, CRISPR-Cas9 has unpredictable efficiency among different DNA target sites and the PAM requirements greatly restrict its genome editing frequency. A large number of efforts have been made to address these impeding issues, but much more is needed to fully realize the medical potential of CRISPR-Cas9. In this article, we summarize the existing problems and current advances of the CRISPR-Cas9 technology and provide perspectives for the ultimate perfection of Cas9-mediated genome editing.
URLPMID:28512351 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1007/s13238-017-0418-2URLPMID:28585179 [本文引用: 2]
Targeted point mutagenesis through homologous recombination has been widely used in genetic studies and holds considerable promise for repairing disease-causing mutations in patients. However, problems such as mosaicism and low mutagenesis efficiency continue to pose challenges to clinical application of such approaches. Recently, a base editor (BE) system built on cytidine (C) deaminase and CRISPR/Cas9 technology was developed as an alternative method for targeted point mutagenesis in plant, yeast, and human cells. Base editors convert C in the deamination window to thymidine (T) efficiently, however, it remains unclear whether targeted base editing in mouse embryos is feasible. In this report, we generated a modified high-fidelity version of base editor 2 (HF2-BE2), and investigated its base editing efficacy in mouse embryos. We found that HF2-BE2 could convert C to T efficiently, with up to 100% biallelic mutation efficiency in mouse embryos. Unlike BE3, HF2-BE2 could convert C to T on both the target and non-target strand, expanding the editing scope of base editors. Surprisingly, we found HF2-BE2 could also deaminate C that was proximal to the gRNA-binding region. Taken together, our work demonstrates the feasibility of generating point mutations in mouse by base editing, and underscores the need to carefully optimize base editing systems in order to eliminate proximal-site deamination.
DOI:10.1038/nbt.3816URLPMID:28244995 [本文引用: 2]
Base editors (BEs) composed of a cytidine deaminase fused to CRISPR-Cas9 convert cytidine to uridine, leading to single-base-pair substitutions in eukaryotic cells. We delivered BE mRNA or ribonucleoproteins targeting the Dmd or Tyr gene via electroporation or microinjection into mouse zygotes. F0 mice showed nonsense mutations with an efficiency of 44-57% and allelic frequencies of up to 100%, demonstrating an efficient method to generate mice with targeted point mutations.
DOI:10.1038/s41598-017-00967-2URLPMID:28424478 [本文引用: 1]
Comprehensive data regarding the epidemiology and prevalence of thalassemia in mainland China are lacking. To assess the prevalence of thalassemia, we performed a meta-analysis including 16 articles published from 1981 to 2015. The overall prevalence of alpha-thalassemia, beta-thalassemia and alpha + beta-thalassemia was 7.88%, 2.21% and 0.48%, respectively. Trends in thalassemia prevalence in mainland China were not steady; a prevalence map based on a geographic information system (GIS) showed that the geographic distribution of thalassemia was highest in the south of China and decreased from south to north. Additionally, the most common alpha- and beta-globin gene mutation was --(SEA) and CD41/42, respectively. The current study provides valuable information regarding epidemiology and intervention and supports the planning, implementation and management of prevention programmes for public health.
DOI:10.1074/jbc.M116.719237URL [本文引用: 2]
DOI:10.1089/scd.2014.0347URLPMID:25517294 [本文引用: 2]
The generation of beta-thalassemia (beta-Thal) patient-specific induced pluripotent stem cells (iPSCs), subsequent homologous recombination-based gene correction of disease-causing mutations/deletions in the beta-globin gene (HBB), and their derived hematopoietic stem cell (HSC) transplantation offers an ideal therapeutic solution for treating this disease. However, the hematopoietic differentiation efficiency of gene-corrected beta-Thal iPSCs has not been well evaluated in the previous studies. In this study, we used the latest gene-editing tool, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9), to correct beta-Thal iPSCs; gene-corrected cells exhibit normal karyotypes and full pluripotency as human embryonic stem cells (hESCs) showed no off-targeting effects. Then, we evaluated the differentiation efficiency of the gene-corrected beta-Thal iPSCs. We found that during hematopoietic differentiation, gene-corrected beta-Thal iPSCs showed an increased embryoid body ratio and various hematopoietic progenitor cell percentages. More importantly, the gene-corrected beta-Thal iPSC lines restored HBB expression and reduced reactive oxygen species production compared with the uncorrected group. Our study suggested that hematopoietic differentiation efficiency of beta-Thal iPSCs was greatly improved once corrected by the CRISPR/Cas9 system, and the information gained from our study would greatly promote the clinical application of beta-Thal iPSC-derived HSCs in transplantation.
DOI:10.1016/j.omtn.2016.11.010URLPMID:28325300 [本文引用: 1]
Monogenic disorders (MGDs), which are caused by single gene mutations, have a serious effect on human health. Among these, beta-thalassemia (beta-thal) represents one of the most common hereditary hematological diseases caused by mutations in the human hemoglobin beta (HBB) gene. The technologies of induced pluripotent stem cells (iPSCs) and genetic correction provide insights into the treatments for MGDs, including beta-thal. However, traditional approaches for correcting mutations have a low efficiency and leave a residual footprint, which leads to some safety concerns in clinical applications. As a proof of concept, we utilized single-strand oligodeoxynucleotides (ssODNs), high-fidelity CRISPR/Cas9 nuclease, and small molecules to achieve a seamless correction of the beta-41/42 (TCTT) deletion mutation in beta thalassemia patient-specific iPSCs with remarkable efficiency. Additionally, off-target analysis and whole-exome sequencing results revealed that corrected cells exhibited a minimal mutational load and no off-target mutagenesis. When differentiated into hematopoietic progenitor cells (HPCs) and then further to erythroblasts, the genetically corrected cells expressed normal beta-globin transcripts. Our studies provide the most efficient and safe approach for the genetic correction of the beta-41/42 (TCTT) deletion in iPSCs for further potential cell therapy of beta-thal, which represents a potential therapeutic avenue for the gene correction of MGD-associated mutants in patient-specific iPSCs.
DOI:10.1093/nar/gkz475URLPMID:31147717
Sickle cell disease (SCD) is a monogenic disorder that affects millions worldwide. Allogeneic hematopoietic stem cell transplantation is the only available cure. Here, we demonstrate the use of CRISPR/Cas9 and a short single-stranded oligonucleotide template to correct the sickle mutation in the beta-globin gene in hematopoietic stem and progenitor cells (HSPCs) from peripheral blood or bone marrow of patients with SCD, with 24.5 +/- 7.6% efficiency without selection. Erythrocytes derived from gene-edited cells showed a marked reduction of sickle cells, with the level of normal hemoglobin (HbA) increased to 25.3 +/- 13.9%. Gene-corrected SCD HSPCs retained the ability to engraft when transplanted into non-obese diabetic (NOD)-SCID-gamma (NSG) mice with detectable levels of gene correction 16-19 weeks post-transplantation. We show that, by using a high-fidelity SpyCas9 that maintained the same level of on-target gene modification, the off-target effects including chromosomal rearrangements were significantly reduced. Taken together, our results demonstrate efficient gene correction of the sickle mutation in both peripheral blood and bone marrow-derived SCD HSPCs, a significant reduction in sickling of red blood cells, engraftment of gene-edited SCD HSPCs in vivo and the importance of reducing off-target effects; all are essential for moving genome editing based SCD treatment into clinical practice.
DOI:10.1186/s13287-018-0779-3URL [本文引用: 2]
DOI:10.1186/s40348-018-0086-1URLPMID:30430274 [本文引用: 1]
BACKGROUND: beta-Thalassemia is an inherited hematological disorder caused by mutations in the human hemoglobin beta (HBB) gene that reduce or abrogate beta-globin expression. Although lentiviral-mediated expression of beta-globin and autologous transplantation is a promising therapeutic approach, the risk of insertional mutagenesis or low transgene expression is apparent. However, targeted gene correction of HBB mutations with programmable nucleases such as CRISPR/Cas9, TALENs, and ZFNs with non-viral repair templates ensures a higher safety profile and endogenous expression control. METHODS: We have compared three different gene-editing tools (CRISPR/Cas9, TALENs, and ZFNs) for their targeting efficiency of the HBB gene locus. As a proof of concept, we studied the personalized gene-correction therapy for a common beta-thalassemia splicing variant HBB(IVS1-110) using Cas9 mRNA and several optimally designed single-stranded oligonucleotide (ssODN) donors in K562 and CD34(+) hematopoietic stem cells (HSCs). RESULTS: Our results exhibited that indel frequency of CRISPR/Cas9 was superior to TALENs and ZFNs (P < 0.0001). Our designed sgRNA targeting the site of HBB(IVS1-110) mutation showed indels in both K562 cells (up to 77%) and CD34(+) hematopoietic stem cells-HSCs (up to 87%). The absolute quantification by next-generation sequencing showed that up to 8% site-specific insertion of the NheI tag was achieved using Cas9 mRNA and a chemically modified ssODN in CD34(+) HSCs. CONCLUSION: Our approach provides guidance on non-viral gene correction in CD34(+) HSCs using Cas9 mRNA and chemically modified ssODN. However, further optimization is needed to increase the homology directed repair (HDR) to attain a real clinical benefit for beta-thalassemia.
DOI:10.1111/cpr.12491URLPMID:30070404 [本文引用: 2]
OBJECTIVES: This study explored whether TALENs-mediated non-homologous end joining (NHEJ) targeting the mutation site can correct the aberrant beta-globin RNA splicing, and ameliorate the beta-thalassaemia phenotype in beta(654) mice. MATERIAL AND METHODS: TALENs vectors targeted to the human beta-globin gene (HBB) IVS2-654C >T mutation in a mouse model were constructed and selected to generate double heterozygous TALENs(+) /beta(654) mice. The gene editing and off-target effects were analysed by sequencing analysis. beta-globin expression was identified by RT-PCR and Western blot analysis. Various clinical indices including haematologic parameters and tissue pathology were examined to determine the therapeutic effect in these TALENs(+) /beta(654) mice. RESULTS: Sequencing analysis revealed that the HBB IVS2-654C >T point mutation was deleted in over 50% of the TALENs(+) /beta(654) mice tested, and off-target effects were not detected. RT-PCR and Western blot analysis confirmed the expression of normal beta-globin in TALENs(+) /beta(654) mice. The haematologic parameters were significantly improved as compared with their affected littermates. The proportion of nucleated cells in bone marrow was considerably decreased, splenomegaly with extramedullary haematopoiesis was reduced, and significant decreases in iron deposition were seen in spleen and liver of the TALENs(+) /beta(654) mice. CONCLUSION: These results suggest effective treatment of the anaemia phenotype in TALENs(+) /beta(654) mice following deletion of the mutation site by TALENs, demonstrating a simple and straightforward strategy for gene therapy of beta(654) -thalassaemia in the future.
DOI:10.1182/blood-2019-01-895094URLPMID:30704988 [本文引用: 3]
The thalassemias are compelling targets for therapeutic genome editing in part because monoallelic correction of a subset of hematopoietic stem cells (HSCs) would be sufficient for enduring disease amelioration. A primary challenge is the development of efficient repair strategies that are effective in HSCs. Here, we demonstrate that allelic disruption of aberrant splice sites, one of the major classes of thalassemia mutations, is a robust approach to restore gene function. We target the IVS1-110G>A mutation using Cas9 ribonucleoprotein (RNP) and the IVS2-654C>T mutation by Cas12a/Cpf1 RNP in primary CD34(+) hematopoietic stem and progenitor cells (HSPCs) from beta-thalassemia patients. Each of these nuclease complexes achieves high efficiency and penetrance of therapeutic edits. Erythroid progeny of edited patient HSPCs show reversal of aberrant splicing and restoration of beta-globin expression. This strategy could enable correction of a substantial fraction of transfusion-dependent beta-thalassemia genotypes with currently available gene-editing technology.
DOI:10.1016/j.stem.2015.07.001URLPMID:26212079 [本文引用: 4]
Hemophilia A is an X-linked genetic disorder caused by mutations in the F8 gene, which encodes the blood coagulation factor VIII. Almost half of all severe hemophilia A cases result from two gross (140-kbp or 600-kbp) chromosomal inversions that involve introns 1 and 22 of the F8 gene, respectively. We derived induced pluripotent stem cells (iPSCs) from patients with these inversion genotypes and used CRISPR-Cas9 nucleases to revert these chromosomal segments back to the WT situation. We isolated inversion-corrected iPSCs with frequencies of up to 6.7% without detectable off-target mutations based on whole-genome sequencing or targeted deep sequencing. Endothelial cells differentiated from corrected iPSCs expressed the F8 gene and functionally rescued factor VIII deficiency in an otherwise lethal mouse model of hemophilia. Our results therefore provide a proof of principle for functional correction of large chromosomal rearrangements in patient-derived iPSCs and suggest potential therapeutic applications.
DOI:10.1093/hmg/ddu533URLPMID:25324539 [本文引用: 1]
Non-allelic homologous recombination (NAHR) is one of the key mechanisms of DNA rearrangement. NAHR occurring between direct homologous repeats can generate genomic copy number variation (CNV) and make significant contributions to both genome evolution and human diseases such as cancer. Intriguingly, previous observations on the rare CNVs at certain genomic disorder loci suggested that NAHR frequency could be dependent on homology properties. However, such a correlation remains unclear at the other NAHR-mediated CNV loci, especially the common CNVs in human populations. Different from the rare CNVs associated with genomic disorders, it is challenging to identify de novo NAHR events at common CNV loci. Therefore, our previously proposed statistic M was employed in estimating relative mutation rate for the NAHR-mediated CNVs in human populations. By utilizing generalized regression neural network and principal component analysis in studying 4330 CNVs ascertained in 3 HapMap populations, we identified the CNVs mediated by NAHR between paired segmental duplications (SDs) and further revealed the correlations between SD properties and NAHR probability. SD length and inter-SD distance were shown to make major contributions to the occurrence of NAHR, whereas chromosomal position and sequence similarity of paired SDs are also involved in NAHR. An integrated effect of SD properties on NAHR frequency was revealed for the common CNVs in human populations. These observations can be well explained by ectopic synapsis in NAHR together with our proposed model of chromosomal compression/extension/looping (CCEL) for homology mis-pairing. Our findings showed the important roles of SDs in NAHR and human genomic evolution.
DOI:10.1038/nrm2849URLPMID:20164840 [本文引用: 1]
Meiotic recombination, which promotes proper homologous chromosome segregation at the first meiotic division, normally occurs between allelic sequences on homologues. However, recombination can also take place between non-allelic DNA segments that share high sequence identity. Such non-allelic homologous recombination (NAHR) can markedly alter genome architecture during gametogenesis by generating chromosomal rearrangements. Indeed, NAHR-mediated deletions, duplications, inversions and other alterations have been implicated in numerous human genetic disorders. Studies in yeast have provided insights into the molecular mechanisms of meiotic NAHR as well as the cellular strategies that limit it.
DOI:10.1073/pnas.1323941111URL [本文引用: 1]
DOI:10.3109/10409238.2012.675644URLPMID:22494239 [本文引用: 1]
Repetitive DNA is present in the eukaryotic genome in the form of segmental duplications, tandem and interspersed repeats, and satellites. Repetitive sequences can be beneficial by serving specific cellular functions (e.g. centromeric and telomeric DNA) and by providing a rapid means for adaptive evolution. However, such elements are also substrates for deleterious chromosomal rearrangements that affect fitness and promote human disease. Recent studies analyzing the role of nuclear organization in DNA repair and factors that suppress non-allelic homologous recombination (NAHR) have provided insights into how genome stability is maintained in eukaryotes. In this review, we outline the types of repetitive sequences seen in eukaryotic genomes and how recombination mechanisms are regulated at the DNA sequence, cell organization, chromatin structure, and cell cycle control levels to prevent chromosomal rearrangements involving these sequences.
DOI:10.1038/s41591-020-0790-yURLPMID:32284612 [本文引用: 1]
Base editing by nucleotide deaminases linked to programmable DNA-binding proteins represents a promising approach to permanently remedy blood disorders, although its application in engrafting hematopoietic stem cells (HSCs) remains unexplored. In this study, we purified A3A (N57Q)-BE3 base editor for ribonucleoprotein (RNP) electroporation of human-peripheral-blood-mobilized CD34(+) hematopoietic stem and progenitor cells (HSPCs). We observed frequent on-target cytosine base edits at the BCL11A erythroid enhancer at +58 with few indels. Fetal hemoglobin (HbF) induction in erythroid progeny after base editing or nuclease editing was similar. A single therapeutic base edit of the BCL11A enhancer prevented sickling and ameliorated globin chain imbalance in erythroid progeny from sickle cell disease and beta-thalassemia patient-derived HSPCs, respectively. Moreover, efficient multiplex editing could be achieved with combined disruption of the BCL11A erythroid enhancer and correction of the HBB -28A>G promoter mutation. Finally, base edits could be produced in multilineage-repopulating self-renewing human HSCs with high frequency as assayed in primary and secondary recipient animals resulting in potent HbF induction in vivo. Together, these results demonstrate the potential of RNP base editing of human HSPCs as a feasible alternative to nuclease editing for HSC-targeted therapeutic genome modification.
DOI:10.1038/s41586-019-1314-0URLPMID:31181567 [本文引用: 2]
Recently developed DNA base editing methods enable the direct generation of desired point mutations in genomic DNA without generating any double-strand breaks(1-3), but the issue of off-target edits has limited the application of these methods. Although several previous studies have evaluated off-target mutations in genomic DNA(4-8), it is now clear that the deaminases that are integral to commonly used DNA base editors often bind to RNA(9-13). For example, the cytosine deaminase APOBEC1-which is used in cytosine base editors (CBEs)-targets both DNA and RNA(12), and the adenine deaminase TadA-which is used in adenine base editors (ABEs)-induces site-specific inosine formation on RNA(9,11). However, any potential RNA mutations caused by DNA base editors have not been evaluated. Adeno-associated viruses are the most common delivery system for gene therapies that involve DNA editing; these viruses can sustain long-term gene expression in vivo, so the extent of potential RNA mutations induced by DNA base editors is of great concern(14-16). Here we quantitatively evaluated RNA single nucleotide variations (SNVs) that were induced by CBEs or ABEs. Both the cytosine base editor BE3 and the adenine base editor ABE7.10 generated tens of thousands of off-target RNA SNVs. Subsequently, by engineering deaminases, we found that three CBE variants and one ABE variant showed a reduction in off-target RNA SNVs to the baseline while maintaining efficient DNA on-target activity. This study reveals a previously overlooked aspect of off-target effects in DNA editing and also demonstrates that such effects can be eliminated by engineering deaminases.
DOI:10.1016/j.bcmd.2017.06.001URLPMID:28651846 [本文引用: 3]
The remarkable phenotypic diversity of beta thalassemia that range from severe anemia and transfusion-dependency, to a clinically asymptomatic state exemplifies how a spectrum of disease severity can be generated in single gene disorders. While the genetic basis for beta thalassemia, and how severity of the anemia could be modified at different levels of its pathophysiology have been well documented, therapy remains largely supportive with bone marrow transplant being the only cure. Identification of the genetic variants modifying fetal hemoglobin (HbF) production in combination with alpha globin genotype provide some prediction of disease severity for beta thalassemia but generation of a personalized genetic risk score to inform prognosis and guide management requires a larger panel of genetic modifiers yet to be discovered. Nonetheless, genetic studies have been successful in characterizing the key variants and pathways involved in HbF regulation, providing new therapeutic targets for HbF reactivation. BCL11A has been established as a quantitative repressor, and progress has been made in manipulating its expression using genomic and gene-editing approaches for therapeutic benefits. Recent discoveries and understanding in the mechanisms associated with ineffective and abnormal erythropoiesis have also provided additional therapeutic targets, a couple of which are currently being tested in clinical trials.
DOI:10.1038/s41467-017-00479-7URLPMID:28871148 [本文引用: 1]
beta-Thalassemia is one of the most common inherited anemias, with no effective cure for most patients. The pathophysiology reflects an imbalance between alpha- and beta-globin chains with an excess of free alpha-globin chains causing ineffective erythropoiesis and hemolysis. When alpha-thalassemia is co-inherited with beta-thalassemia, excess free alpha-globin chains are reduced significantly ameliorating the clinical severity. Here we demonstrate the use of CRISPR/Cas9 genome editing of primary human hematopoietic stem/progenitor (CD34+) cells to emulate a natural mutation, which deletes the MCS-R2 alpha-globin enhancer and causes alpha-thalassemia. When edited CD34+ cells are differentiated into erythroid cells, we observe the expected reduction in alpha-globin expression and a correction of the pathologic globin chain imbalance in cells from patients with beta-thalassemia. Xenograft assays show that a proportion of the edited CD34+ cells are long-term repopulating hematopoietic stem cells, demonstrating the potential of this approach for translation into a therapy for beta-thalassemia.beta-thalassemia is characterised by the presence of an excess of alpha-globin chains, which contribute to erythrocyte pathology. Here the authors use CRISP/Cas9 to reduce alpha-globin expression in hematopoietic precursors, and show effectiveness in xenograft assays in mice.
DOI:10.1038/s41591-019-0401-yURLPMID:30911135 [本文引用: 6]
Re-expression of the paralogous gamma-globin genes (HBG1/2) could be a universal strategy to ameliorate the severe beta-globin disorders sickle cell disease (SCD) and beta-thalassemia by induction of fetal hemoglobin (HbF, alpha2gamma2)(1). Previously, we and others have shown that core sequences at the BCL11A erythroid enhancer are required for repression of HbF in adult-stage erythroid cells but are dispensable in non-erythroid cells(2-6). CRISPR-Cas9-mediated gene modification has demonstrated variable efficiency, specificity, and persistence in hematopoietic stem cells (HSCs). Here, we demonstrate that Cas9:sgRNA ribonucleoprotein (RNP)-mediated cleavage within a GATA1 binding site at the +58 BCL11A erythroid enhancer results in highly penetrant disruption of this motif, reduction of BCL11A expression, and induction of fetal gamma-globin. We optimize conditions for selection-free on-target editing in patient-derived HSCs as a nearly complete reaction lacking detectable genotoxicity or deleterious impact on stem cell function. HSCs preferentially undergo non-homologous compared with microhomology-mediated end joining repair. Erythroid progeny of edited engrafting SCD HSCs express therapeutic levels of HbF and resist sickling, while those from patients with beta-thalassemia show restored globin chain balance. Non-homologous end joining repair-based BCL11A enhancer editing approaching complete allelic disruption in HSCs is a practicable therapeutic strategy to produce durable HbF induction.
DOI:10.1038/s41588-018-0085-0URLPMID:29610478 [本文引用: 3]
beta-hemoglobinopathies such as sickle cell disease (SCD) and beta-thalassemia result from mutations in the adult HBB (beta-globin) gene. Reactivating the developmentally silenced fetal HBG1 and HBG2 (gamma-globin) genes is a therapeutic goal for treating SCD and beta-thalassemia (1) . Some forms of hereditary persistence of fetal hemoglobin (HPFH), a rare benign condition in which individuals express the gamma-globin gene throughout adulthood, are caused by point mutations in the gamma-globin gene promoter at regions residing ~115 and 200 bp upstream of the transcription start site. We found that the major fetal globin gene repressors BCL11A and ZBTB7A (also known as LRF) directly bound to the sites at -115 and -200 bp, respectively. Furthermore, introduction of naturally occurring HPFH-associated mutations into erythroid cells by CRISPR-Cas9 disrupted repressor binding and raised gamma-globin gene expression. These findings clarify how these HPFH-associated mutations operate and demonstrate that BCL11A and ZBTB7A are major direct repressors of the fetal globin gene.
DOI:10.1182/blood-2018-07-863951URLPMID:30617196 [本文引用: 2]
beta-hemoglobinopathies, such as sickle cell disease and beta-thalassemia, result from mutations in the adult beta-globin gene. Reactivating the developmentally silenced fetal gamma-globin gene elevates fetal hemoglobin levels and ameliorates symptoms of beta-hemoglobinopathies. The continued expression of fetal gamma-globin into adulthood occurs naturally in a genetic condition termed hereditary persistence of fetal hemoglobin (HPFH). Point mutations in the fetal gamma-globin proximal promoter can cause HPFH. The -113A>G HPFH mutation falls within the -115 cluster of HPFH mutations, a binding site for the fetal globin repressor BCL11A. We demonstrate that the -113A>G HPFH mutation, unlike other mutations in the cluster, does not disrupt BCL11A binding but rather creates a de novo binding site for the transcriptional activator GATA1. Introduction of the -113A>G HPFH mutation into erythroid cells using the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) system increases GATA1 binding and elevates fetal globin levels. These results reveal the mechanism by which the -113A>G HPFH mutation elevates fetal globin and demonstrate the sensitivity of the fetal globin promoter to point mutations that often disrupt repressor binding sites but here create a de novo site for an erythroid activator.
DOI:10.1182/blood-2017-10-811505URLPMID:29519807 [本文引用: 2]
Naturally occurring, large deletions in the beta-globin locus result in hereditary persistence of fetal hemoglobin, a condition that mitigates the clinical severity of sickle cell disease (SCD) and beta-thalassemia. We designed a clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) (CRISPR/Cas9) strategy to disrupt a 13.6-kb genomic region encompassing the delta- and beta-globin genes and a putative gamma-delta intergenic fetal hemoglobin (HbF) silencer. Disruption of just the putative HbF silencer results in a mild increase in gamma-globin expression, whereas deletion or inversion of a 13.6-kb region causes a robust reactivation of HbF synthesis in adult erythroblasts that is associated with epigenetic modifications and changes in chromatin contacts within the beta-globin locus. In primary SCD patient-derived hematopoietic stem/progenitor cells, targeting the 13.6-kb region results in a high proportion of gamma-globin expression in erythroblasts, increased HbF synthesis, and amelioration of the sickling cell phenotype. Overall, this study provides clues for a potential CRISPR/Cas9 genome editing approach to the therapy of beta-hemoglobinopathies.
DOI:10.1038/nature15521URLPMID:26375006 [本文引用: 2]
Enhancers, critical determinants of cellular identity, are commonly recognized by correlative chromatin marks and gain-of-function potential, although only loss-of-function studies can demonstrate their requirement in the native genomic context. Previously, we identified an erythroid enhancer of human BCL11A, subject to common genetic variation associated with the fetal haemoglobin level, the mouse orthologue of which is necessary for erythroid BCL11A expression. Here we develop pooled clustered regularly interspaced palindromic repeat (CRISPR)-Cas9 guide RNA libraries to perform in situ saturating mutagenesis of the human and mouse enhancers. This approach reveals critical minimal features and discrete vulnerabilities of these enhancers. Despite conserved function of the composite enhancers, their architecture diverges. The crucial human sequences appear to be primate-specific. Through editing of primary human progenitors and mouse transgenesis, we validate the BCL11A erythroid enhancer as a target for fetal haemoglobin reinduction. The detailed enhancer map will inform therapeutic genome editing, and the screening approach described here is generally applicable to functional interrogation of non-coding genomic elements.
DOI:10.1038/ni925URLPMID:12717432 [本文引用: 1]
Bcl11a (also called Evi9) functions as a myeloid or B cell proto-oncogene in mice and humans, respectively. Here we show that Bcl11a is essential for postnatal development and normal lymphopoiesis. Bcl11a mutant embryos lack B cells and have alterations in several types of T cells. Phenotypic and expression studies show that Bcl11a functions upstream of the transcription factors Ebf1 and Pax5 in the B cell pathway. Transplantation studies show that these defects in Bcl11a mutant mice are intrinsic to fetal liver precursor cells. Mice transplanted with Bcl11a-deficient cells died from T cell leukemia derived from the host. Thus, Bcl11a may also function as a non-autonomous T cell tumor suppressor gene.
DOI:10.1080/03630269.2018.1515774URLPMID:30821197 [本文引用: 1]
Fetal hemoglobin (Hb F, alpha2gamma2) is a potent genetic modifier of the severity of beta-thalassemia (beta-thal) and sickle cell anemia. Differences in the levels of HbF that persist into adulthood affect the severity of sickle cell disease and the beta-thal syndromes. B-cell lymphoma 11 A (BCL11A) is a potent silencer of HbF. Here, we reactivated gamma-globin expression by down-regulating BCL11A to alleviate anemia in the beta-thal major (beta-TM) patients. BCL11A were down-regulated by lentiviral RNAi (RNA interference) in the K562 cell line and an in vitro culture model of human erythropoiesis in which erythroblasts are derived from the normal donor mononuclear cells (MNC) or beta-TM MNC. The expression of gamma-globin were analyzed by qPCR (quantitative real-time polymerase chain reaction) and Western blot techniques. Our data showed that down-regulation of BCL11A induces gamma-globin production in the K562 cell line and human erythrocytes from normal donors and beta-TM donors, without altering erythroid maturation. This is the first report on gamma-globin induction by down-regulation of BCL11A in human erythroblasts derived from beta-TM.
DOI:10.1038/s41422-019-0267-zURLPMID:31911671 [本文引用: 1]
DOI:10.1126/science.aao0932URLPMID:30026227 [本文引用: 1]
Increasing fetal hemoglobin (HbF) levels in adult red blood cells provides clinical benefit to patients with sickle cell disease and some forms of beta-thalassemia. To identify potentially druggable HbF regulators in adult human erythroid cells, we employed a protein kinase domain-focused CRISPR-Cas9-based genetic screen with a newly optimized single-guide RNA scaffold. The screen uncovered the heme-regulated inhibitor HRI (also known as EIF2AK1), an erythroid-specific kinase that controls protein translation, as an HbF repressor. HRI depletion markedly increased HbF production in a specific manner and reduced sickling in cultured erythroid cells. Diminished expression of the HbF repressor BCL11A accounted in large part for the effects of HRI depletion. Taken together, these results suggest HRI as a potential therapeutic target for hemoglobinopathies.
DOI:10.1056/NEJMoa1705342URLPMID:29669226 [本文引用: 1]
BACKGROUND: Donor availability and transplantation-related risks limit the broad use of allogeneic hematopoietic-cell transplantation in patients with transfusion-dependent beta-thalassemia. After previously establishing that lentiviral transfer of a marked beta-globin (beta(A-T87Q)) gene could substitute for long-term red-cell transfusions in a patient with beta-thalassemia, we wanted to evaluate the safety and efficacy of such gene therapy in patients with transfusion-dependent beta-thalassemia. METHODS: In two phase 1-2 studies, we obtained mobilized autologous CD34+ cells from 22 patients (12 to 35 years of age) with transfusion-dependent beta-thalassemia and transduced the cells ex vivo with LentiGlobin BB305 vector, which encodes adult hemoglobin (HbA) with a T87Q amino acid substitution (HbA(T87Q)). The cells were then reinfused after the patients had undergone myeloablative busulfan conditioning. We subsequently monitored adverse events, vector integration, and levels of replication-competent lentivirus. Efficacy assessments included levels of total hemoglobin and HbA(T87Q), transfusion requirements, and average vector copy number. RESULTS: At a median of 26 months (range, 15 to 42) after infusion of the gene-modified cells, all but 1 of the 13 patients who had a non-beta(0)/beta(0) genotype had stopped receiving red-cell transfusions; the levels of HbA(T87Q) ranged from 3.4 to 10.0 g per deciliter, and the levels of total hemoglobin ranged from 8.2 to 13.7 g per deciliter. Correction of biologic markers of dyserythropoiesis was achieved in evaluated patients with hemoglobin levels near normal ranges. In 9 patients with a beta(0)/beta(0) genotype or two copies of the IVS1-110 mutation, the median annualized transfusion volume was decreased by 73%, and red-cell transfusions were discontinued in 3 patients. Treatment-related adverse events were typical of those associated with autologous stem-cell transplantation. No clonal dominance related to vector integration was observed. CONCLUSIONS: Gene therapy with autologous CD34+ cells transduced with the BB305 vector reduced or eliminated the need for long-term red-cell transfusions in 22 patients with severe beta-thalassemia without serious adverse events related to the drug product. (Funded by Bluebird Bio and others; HGB-204 and HGB-205 ClinicalTrials.gov numbers, NCT01745120 and NCT02151526 .).
DOI:10.1002/cpmo.58URLPMID:30485696 [本文引用: 1]
Viral vectors are a promising tool for effective delivery of genetic material into cells. They take advantage of the natural ability of a virus to deliver a genetic payload into cells while being genetically modified such that their ability to replicate is crippled or removed. Here, an updated overview of routinely used viral vectors, including adeno-associated viruses (AAV), retroviruses/lentiviruses, and adenoviruses (Ads), is provided, as well as perspectives on their advantages and disadvantages in research and gene therapy. (c) 2018 by John Wiley & Sons, Inc.
DOI:10.1038/mt.2008.72URLPMID:18414478 [本文引用: 2]
Recombinant adeno-associated viral (rAAV) vectors have shown promise for use in liver-targeted gene delivery, but their effects have not been extensively investigated in the immature liver. Understanding the impact of liver growth on the efficacy of transduction is essential, because many monogenic liver diseases that are amenable to gene therapy will require treatment early in life. Here we show that rAAV2/8 transduces the neonatal mouse liver with high efficiency. With just one doubling in liver weight, however, there is a rapid reduction in vector genome numbers, irrespective of form, and the loss of episomal vector is almost complete by 2 weeks. Stable transgene expression is observed in a small percentage of hepatocytes, often in two- to eight-cell clusters, suggestive of genomic integration. Delivery at serially older ages was associated with progressively improved episome persistence and transgene expression. Vector re-administration was possible following initial neonatal administration, albeit at reduced efficacy because of an anticapsid humoral immune response. We also found that intraperitoneal (i.p.) delivery of rAAV2/8 was highly effective at all ages, and that promoter selection is the critical determinant of the intensity and pattern of transgene expression across the hepatic lobule. We conclude that successful use of rAAV to treat liver disease in early childhood will require optimally efficient vector constructs and probable re-administration.
DOI:10.1056/NEJMoa1708538URLPMID:29211678 [本文引用: 1]
BACKGROUND: The prevention of bleeding with adequately sustained levels of clotting factor, after a single therapeutic intervention and without the need for further medical intervention, represents an important goal in the treatment of hemophilia. METHODS: We infused a single-stranded adeno-associated viral (AAV) vector consisting of a bioengineered capsid, liver-specific promoter and factor IX Padua (factor IX-R338L) transgene at a dose of 5x10(11) vector genomes per kilogram of body weight in 10 men with hemophilia B who had factor IX coagulant activity of 2% or less of the normal value. Laboratory values, bleeding frequency, and consumption of factor IX concentrate were prospectively evaluated after vector infusion and were compared with baseline values. RESULTS: No serious adverse events occurred during or after vector infusion. Vector-derived factor IX coagulant activity was sustained in all the participants, with a mean (+/-SD) steady-state factor IX coagulant activity of 33.7+/-18.5% (range, 14 to 81). On cumulative follow-up of 492 weeks among all the participants (range of follow-up in individual participants, 28 to 78 weeks), the annualized bleeding rate was significantly reduced (mean rate, 11.1 events per year [range, 0 to 48] before vector administration vs. 0.4 events per year [range, 0 to 4] after administration; P=0.02), as was factor use (mean dose, 2908 IU per kilogram [range, 0 to 8090] before vector administration vs. 49.3 IU per kilogram [range, 0 to 376] after administration; P=0.004). A total of 8 of 10 participants did not use factor, and 9 of 10 did not have bleeds after vector administration. An asymptomatic increase in liver-enzyme levels developed in 2 participants and resolved with short-term prednisone treatment. One participant, who had substantial, advanced arthropathy at baseline, administered factor for bleeding but overall used 91% less factor than before vector infusion. CONCLUSIONS: We found sustained therapeutic expression of factor IX coagulant activity after gene transfer in 10 participants with hemophilia who received the same vector dose. Transgene-derived factor IX coagulant activity enabled the termination of baseline prophylaxis and the near elimination of bleeding and factor use. (Funded by Spark Therapeutics and Pfizer; ClinicalTrials.gov number, NCT02484092 .).
DOI:10.1182/blood.2019000790URLPMID:30975639 [本文引用: 2]
Many genetic diseases, including hemophilia, require long-term therapeutic effects. Despite the initial success of liver-directed adeno-associated virus (AAV) gene therapy for hemophilia in clinical trials, long-term sustained therapeutic effects have yet to be seen. One explanation for the gradual decline of efficacy over time is that the nonintegrating AAV vector genome could be lost during cell division during hepatocyte turnover, albeit at a slow pace in adults. Readministering the same vector is challenging as a result of the AAV-neutralizing antibodies elicited by the initial treatment. Here, we investigated the use of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated homology-directed gene targeting for sustained treatment of hemophilia B. We developed a donor vector containing a promoterless partial human factor IX (FIX) complementary DNA carrying the hyperactive FIX Padua mutation. A single injection of dual AAV vectors in newborn and adult FIX-knockout (FIX-KO) mice led to stable expression of FIX at or above the normal levels for 8 months. Eight weeks after the vector treatment, we subjected a subgroup of newborn and adult treated FIX-KO mice to a two-thirds partial hepatectomy; all of these animals survived the procedure without any complications or interventions. FIX levels persisted at similar levels for 24 weeks after partial hepatectomy, indicating stable genomic targeting. Our results lend support for the use of a CRISPR/Cas9 approach to achieve lifelong expression of therapeutic proteins.
DOI:10.3892/ijo.2018.4434URLPMID:29901119 [本文引用: 1]
One of the fundamental discoveries in the field of biology is the ability to modulate the genome and to monitor the functional outputs derived from genomic alterations. In order to unravel new therapeutic options, scientists had initially focused on inducing genetic alterations in primary cells, in established cancer cell lines and mouse models using either RNA interference or cDNA overexpression or various programmable nucleases [zinc finger nucleases (ZNF), transcription activator-like effector nucleases (TALEN)]. Even though a huge volume of data was produced, its use was neither cheap nor accurate. Therefore, the clustered regularly interspaced short palindromic repeats (CRISPR) system was evidenced to be the next step in genome engineering tools. CRISPR-associated protein 9 (Cas9)-mediated genetic perturbation is simple, precise and highly efficient, empowering researchers to apply this method to immortalized cancerous cell lines, primary cells derived from mouse and human origins, xenografts, induced pluripotent stem cells, organoid cultures, as well as the generation of genetically engineered animal models. In this review, we assess the development of the CRISPR system and its therapeutic applications to a wide range of complex diseases (particularly distinct tumors), aiming at personalized therapy. Special emphasis is given to organoids and CRISPR screens in the design of innovative therapeutic approaches. Overall, the CRISPR system is regarded as an eminent genome engineering tool in therapeutics. We envision a new era in cancer biology during which the CRISPR-based genome engineering toolbox will serve as the fundamental conduit between the bench and the bedside; nonetheless, certain obstacles need to be addressed, such as the eradication of side-effects, maximization of efficiency, the assurance of delivery and the elimination of immunogenicity.
URLPMID:27882996 [本文引用: 1]
DOI:10.1038/s41591-020-0840-5URLPMID:32341578 [本文引用: 3]
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 editing of immune checkpoint genes could improve the efficacy of T cell therapy, but the first necessary undertaking is to understand the safety and feasibility. Here, we report results from a first-in-human phase I clinical trial of CRISPR-Cas9 PD-1-edited T cells in patients with advanced non-small-cell lung cancer (ClinicalTrials.gov NCT02793856). Primary endpoints were safety and feasibility, and the secondary endpoint was efficacy. The exploratory objectives included tracking of edited T cells. All prespecified endpoints were met. PD-1-edited T cells were manufactured ex vivo by cotransfection using electroporation of Cas9 and single guide RNA plasmids. A total of 22 patients were enrolled; 17 had sufficient edited T cells for infusion, and 12 were able to receive treatment. All treatment-related adverse events were grade 1/2. Edited T cells were detectable in peripheral blood after infusion. The median progression-free survival was 7.7 weeks (95% confidence interval, 6.9 to 8.5 weeks) and median overall survival was 42.6 weeks (95% confidence interval, 10.3-74.9 weeks). The median mutation frequency of off-target events was 0.05% (range, 0-0.25%) at 18 candidate sites by next generation sequencing. We conclude that clinical application of CRISPR-Cas9 gene-edited T cells is generally safe and feasible. Future trials should use superior gene editing approaches to improve therapeutic efficacy.
URLPMID:32108095 [本文引用: 2]
DOI:10.1056/NEJMoa1817426URLPMID:31509667 [本文引用: 1]
The safety of CRISPR (clustered regularly interspaced short palindromic repeats)-based genome editing in the context of human gene therapy is largely unknown. CCR5 is a reasonable but not absolutely protective target for a cure of human immunodeficiency virus type 1 (HIV-1) infection, because CCR5-null blood cells are largely resistant to HIV-1 entry. We transplanted CRISPR-edited CCR5-ablated hematopoietic stem and progenitor cells (HSPCs) into a patient with HIV-1 infection and acute lymphoblastic leukemia. The acute lymphoblastic leukemia was in complete remission with full donor chimerism, and donor cells carrying the ablated CCR5 persisted for more than 19 months without gene editing-related adverse events. The percentage of CD4+ cells with CCR5 ablation increased by a small degree during a period of antiretroviral-therapy interruption. Although we achieved successful transplantation and long-term engraftment of CRISPR-edited HSPCs, the percentage of CCR5 disruption in lymphocytes was only approximately 5%, which indicates the need for further research into this approach. (Funded by the Beijing Municipal Science and Technology Commission and others; ClinicalTrials.gov number, NCT03164135.).
URLPMID:32528163 [本文引用: 1]
DOI:10.1182/blood-2016-02-629063URLPMID:27207800 [本文引用: 1]
Adoptive transfer of T cells genetically modified to express chimeric antigen receptors (CARs) targeting CD19 has produced impressive results in treating patients with B-cell malignancies. Although these CAR-modified T cells target the same antigen, the designs of CARs vary as well as several key aspects of the clinical trials in which these CARs have been studied. It is unclear whether these differences have any impact on clinical outcome and treatment-related toxicities. Herein, we review clinical results reflecting the investigational use of CD19-targeted CAR T-cell therapeutics in patients with B-cell hematologic malignancies, in light of differences in CAR design and production, and outline the limitations inherent in comparing outcomes between studies.
DOI:10.1038/nbt.2623URLPMID:23792628 [本文引用: 1]
Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) have rapidly emerged as a facile and efficient platform for genome editing. Here, we use a human cell-based reporter assay to characterize off-target cleavage of CRISPR-associated (Cas)9-based RGNs. We find that single and double mismatches are tolerated to varying degrees depending on their position along the guide RNA (gRNA)-DNA interface. We also readily detected off-target alterations induced by four out of six RGNs targeted to endogenous loci in human cells by examination of partially mismatched sites. The off-target sites we identified harbored up to five mismatches and many were mutagenized with frequencies comparable to (or higher than) those observed at the intended on-target site. Our work demonstrates that RGNs can be highly active even with imperfectly matched RNA-DNA interfaces in human cells, a finding that might confound their use in research and therapeutic applications.
DOI:10.1038/s41467-019-09006-2URLPMID:30850590 [本文引用: 1]
CRISPR-Cas9 is a promising technology for genome editing. Here we use Cas9 nuclease-induced double-strand break DNA (DSB) at the UROS locus to model and correct congenital erythropoietic porphyria. We demonstrate that homology-directed repair is rare compared with NHEJ pathway leading to on-target indels and causing unwanted dysfunctional protein. Moreover, we describe unexpected chromosomal truncations resulting from only one Cas9 nuclease-induced DSB in cell lines and primary cells by a p53-dependent mechanism. Altogether, these side effects may limit the promising perspectives of the CRISPR-Cas9 nuclease system for disease modeling and gene therapy. We show that the single nickase approach could be safer since it prevents on- and off-target indels and chromosomal truncations. These results demonstrate that the single nickase and not the nuclease approach is preferable, not only for modeling disease but also and more importantly for the safe management of future CRISPR-Cas9-mediated gene therapies.
URLPMID:30424953 [本文引用: 2]
URLPMID:27820943 [本文引用: 1]
DOI:10.1038/nbt.3437URLPMID:26780180 [本文引用: 2]
CRISPR-Cas9-based genetic screens are a powerful new tool in biology. By simply altering the sequence of the single-guide RNA (sgRNA), one can reprogram Cas9 to target different sites in the genome with relative ease, but the on-target activity and off-target effects of individual sgRNAs can vary widely. Here, we use recently devised sgRNA design rules to create human and mouse genome-wide libraries, perform positive and negative selection screens and observe that the use of these rules produced improved results. Additionally, we profile the off-target activity of thousands of sgRNAs and develop a metric to predict off-target sites. We incorporate these findings from large-scale, empirical data to improve our computational design rules and create optimized sgRNA libraries that maximize on-target activity and minimize off-target effects to enable more effective and efficient genetic screens and genome engineering.
DOI:10.1038/nbt.3290URLPMID:26121415 [本文引用: 1]
CRISPR-Cas-mediated genome editing relies on guide RNAs that direct site-specific DNA cleavage facilitated by the Cas endonuclease. Here we report that chemical alterations to synthesized single guide RNAs (sgRNAs) enhance genome editing efficiency in human primary T cells and CD34(+) hematopoietic stem and progenitor cells. Co-delivering chemically modified sgRNAs with Cas9 mRNA or protein is an efficient RNA- or ribonucleoprotein (RNP)-based delivery method for the CRISPR-Cas system, without the toxicity associated with DNA delivery. This approach is a simple and effective way to streamline the development of genome editing with the potential to accelerate a wide array of biotechnological and therapeutic applications of the CRISPR-Cas technology.
DOI:10.1038/s41591-018-0137-0URLPMID:30082871 [本文引用: 1]
Translation of the CRISPR-Cas9 system to human therapeutics holds high promise. However, specificity remains a concern especially when modifying stem cell populations. We show that existing rationally engineered Cas9 high-fidelity variants have reduced on-target activity when using the therapeutically relevant ribonucleoprotein (RNP) delivery method. Therefore, we devised an unbiased bacterial screen to isolate variants that retain activity in the RNP format. Introduction of a single point mutation, p.R691A, in Cas9 (high-fidelity (HiFi) Cas9) retained the high on-target activity of Cas9 while reducing off-target editing. HiFi Cas9 induces robust AAV6-mediated gene targeting at five therapeutically relevant loci (HBB, IL2RG, CCR5, HEXB, and TRAC) in human CD34(+) hematopoietic stem and progenitor cells (HSPCs) as well as primary T cells. We also show that HiFi Cas9 mediates high-level correction of the sickle cell disease (SCD)-causing p.E6V mutation in HSPCs derived from patients with SCD. We anticipate that HiFi Cas9 will have wide utility for both basic science and therapeutic genome-editing applications.
URLPMID:30405244 [本文引用: 1]
DOI:10.5966/sctm.2015-0157erratumURL [本文引用: 1]
DOI:10.7150/ijms.21666URLPMID:29333086 [本文引用: 1]
Results obtained from completed and on-going clinical studies indicate huge therapeutic potential of stem cell-based therapy in the treatment of degenerative, autoimmune and genetic disorders. However, clinical application of stem cells raises numerous ethical and safety concerns. In this review, we provide an overview of the most important ethical issues in stem cell therapy, as a contribution to the controversial debate about their clinical usage in regenerative and transplantation medicine. We describe ethical challenges regarding human embryonic stem cell (hESC) research, emphasizing that ethical dilemma involving the destruction of a human embryo is a major factor that may have limited the development of hESC-based clinical therapies. With previous derivation of induced pluripotent stem cells (iPSCs) this problem has been overcome, however current perspectives regarding clinical translation of iPSCs still remain. Unlimited differentiation potential of iPSCs which can be used in human reproductive cloning, as a risk for generation of genetically engineered human embryos and human-animal chimeras, is major ethical issue, while undesired differentiation and malignant transformation are major safety issues. Although clinical application of mesenchymal stem cells (MSCs) has shown beneficial effects in the therapy of autoimmune and chronic inflammatory diseases, the ability to promote tumor growth and metastasis and overestimated therapeutic potential of MSCs still provide concerns for the field of regenerative medicine. This review offers stem cell scientists, clinicians and patient's useful information and could be used as a starting point for more in-depth analysis of ethical and safety issues related to clinical application of stem cells.
DOI:10.1007/s10565-016-9377-2URLPMID:28039590 [本文引用: 1]
Ten years have passed since the first publication announcing the generation of induced pluripotent stem cells (iPSCs). Issues related to ethics, immune rejection, and cell availability seemed to be solved following this breakthrough. The development of iPSC technology allows advances in in vitro cell differentiation for cell therapy purpose and other clinical applications. This review provides a perspective on the iPSC potential for cell therapies, particularly for hematological applications. We discuss the advances in in vitro hematopoietic differentiation, the possibilities to employ iPSC in hematology studies, and their potential clinical application in hematologic diseases. The generation of red blood cells and functional T cells and the genome editing technology applied to mutation correction are also covered. We highlight some of the requirements and obstacles to be overcome before translating these cells from research to the clinic, for instance, iPSC variability, genotoxicity, the differentiation process, and engraftment. Also, we evaluate the patent landscape and compile the clinical trials in the field of pluripotent stem cells. Currently, we know much more about iPSC than in 2006, but there are still challenges that must be solved. A greater understanding of molecular mechanisms underlying the generation of hematopoietic stem cells is necessary to produce suitable and transplantable hematopoietic stem progenitor cells from iPSC.
URLPMID:29386396 [本文引用: 1]
DOI:10.1002/stem.2517URLPMID:27739611 [本文引用: 1]
Human embryonic stem (ES) cells and induced pluripotent stem (iPS) cells represent an ideal source for in vitro modeling of erythropoiesis and a potential alternative source for red blood cell transfusions. However, iPS cell-derived erythroid cells predominantly produce epsilon- and gamma-globin without beta-globin production. We recently demonstrated that ES cell-derived sacs (ES sacs), known to express hemangioblast markers, allow for efficient erythroid cell generation with beta-globin production. In this study, we generated several iPS cell lines derived from bone marrow stromal cells (MSCs) and peripheral blood erythroid progenitors (EPs) from sickle cell disease patients, and evaluated hematopoietic stem/progenitor cell (HSPC) generation after iPS sac induction as well as subsequent erythroid differentiation. MSC-derived iPS sacs yielded greater amounts of immature hematopoietic progenitors (VEGFR2 + GPA-), definitive HSPCs (CD34 + CD45+), and megakaryoerythroid progenitors (GPA + CD41a+), as compared to EP-derived iPS sacs. Erythroid differentiation from MSC-derived iPS sacs resulted in greater amounts of erythroid cells (GPA+) and higher beta-globin (and betaS-globin) expression, comparable to ES sac-derived cells. These data demonstrate that human MSC-derived iPS sacs allow for more efficient erythroid cell generation with higher beta-globin production, likely due to heightened emergence of immature progenitors. Our findings should be important for iPS cell-derived erythroid cell generation. Stem Cells 2017;35:586-596.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}