 ,1,2
,1,2The osteoporosis susceptible SNP rs4325274 remotely regulates the SOX6 gene through enhancers
Xiaomei Tuo1, Dongli Zhu1,2, Xiaofeng Chen1, Yu Rong1, Yan Guo1, Tielin Yang,1,2通讯作者: 杨铁林,博士,教授,研究方向:生物信息分析及复杂疾病遗传致病机制研究。E-mail:yangtielin@xjtu.edu.cn
责任编辑: 周钢桥
收稿日期:2020-05-26修回日期:2020-09-4网络出版日期:2020-09-20
| 基金资助: |
Received:2020-05-26Revised:2020-09-4Online:2020-09-20
| Fund supported: |
作者简介 About authors
妥晓梅,在读硕士研究生,专业方向:疾病分子遗传机制的基础研究。E-mail:

摘要
骨质疏松症是一种典型的多基因复杂疾病,遗传力高达85%,其发病率已跃居常见疾病的第5位。尽管已经鉴定出大量骨质疏松易感SNP,但大多数SNP位点位于基因组非编码区,且功能机制未知。本研究旨在通过生物信息学分析和功能实验探究骨质疏松非编码功能性易感SNP rs4325274的分子调控机制。首先,通过表观注释发现该SNP所在区域处在增强子上,eQTL和Hi-C分析结果发现SNP调控的潜在靶基因是SOX6;然后,利用多种数据库进行Motif预测,并结合GEO数据库中的ChIP-seq数据分析进行了验证,结果发现转录因子HNF1A更倾向于结合SNP rs4325274-G碱基;进一步通过双荧光素酶报告基因实验验证了该SNP对SOX6基因表达的增强作用;最后,利用shRNA敲低转录因子HNF1A实验,检测靶基因SOX6的表达变化。以上研究结果初步解析了非编码区功能性SNPrs4325274作为增强子远程调控SOX6基因表达的分子机制,为复杂疾病非编码易感SNP的遗传调控研究提供新思路。
关键词:
Abstract
Osteoporosis is a typical polygenic disease, and its heritability is as high as 85%. The incidence of osteoporosis has jumped to the fifth among the common diseases. Although a large number of osteoporosis-susceptible SNPs have been identified, most of them are in the non-coding regions of the genome and the functional mechanisms are unknown. The purpose of this study was to explore the function of non-coding osteoporosis-susceptible SNP rs4325274 and dissect the molecular regulatory mechanisms through integrating bioinformatics analysis and functional experiments. Firstly, we found the SNP rs4325274 resided in a putative enhancer element through functional annotation. eQTL and Hi-C analysis found that the SOX6 gene might be a potential distal target of rs4325274. We conducted the motif prediction using multiple databases and verified the result using ChIP-seq data from GEO database. The result showed that the transcription factor HNF1A could preferentially bind to SNP rs4325274-G allele. We further demonstrated that SNP rs4325274 acted as an enhancer regulating SOX6 gene expression by using dual-luciferase reporter assays. Knockdown of HNF1A decreased the SOX6 gene expression. Taken together, our results uncovered a new mechanism of a non-coding functional SNP rs4325274 as a distal enhancer to modulate SOX6 expression, which provides new insights into deciphering molecular regulatory mechanisms underlying non-coding susceptibility SNPs on complex diseases.
Keywords:
PDF (715KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
妥晓梅, 朱东丽, 陈晓峰, 荣誉, 郭燕, 杨铁林. 骨质疏松易感SNP rs4325274通过增强子远程调控SOX6基因的功能机制研究. 遗传[J], 2020, 42(9): 889-897 doi:10.16288/j.yczz.20-098
Xiaomei Tuo.
骨质疏松症是一种世界范围内流行的全身性骨代谢性疾病,以骨量减少和骨组织微结构损坏为特征[1]。由于其高发病率、高死亡率以及高医疗花费[2,3,4],骨质疏松症已成为当前亟需解决的公共健康问题之一。但至今骨质疏松症的遗传发病机制仍不清楚。因此,开展骨质疏松症的基础研究,深入探索其分子致病机制,对骨质疏松症的预防和临床治疗有着重要意义。
SOX6 (SRY-box transcription factor 6)基因是SRY相关转录因子D亚家族的成员,在软骨细胞分化、软骨形成和软骨内骨形成中起着非常重要的调控作用[5,6]。当SOX6和SOX5基因同时缺失时,小鼠胚胎会发展成严重的泛发性软骨发育不良,在胚胎发育16.5天左右死亡[5]。另外,SOX6和SOX5协同SOX9基因调节软骨中II 型胶原(COL2A1)的表达来控制软骨的重塑,在治疗骨关节炎中软骨退变和软骨损伤修复中具有重要的临床应用价值[7,8]。本课题组前期研究发现SOX6基因参与了软骨形成和成骨的耦合调控作用[9],这提示SOX6基因可能在骨质疏松发生过程起着非常重要的作用。
骨质疏松症的临床诊断和评估主要以骨密度(bone mineral density, BMD)为依据[10],其遗传力高达0.6~0.8[11]。目前,国际上已有多项全基因组关联研究(genome-wide association study, GWAS)发现位于染色体11p15区的SOX6基因内部及上下游区的多个SNPs位点与股骨颈、腰椎和手腕骨密度等骨质疏松症表型显著关联[12,13,14,15]。但对于这些风险SNPs在骨质疏松症发生过程中的功能及作用机制至今尚不清楚。本课题组前期从骨质疏松症遗传因素联合会(genetic factors for osteoporosis consortium, GEFOS)下载和整理了所有与骨质疏松症相关表型的所有GWAS 数据,通过精细定位分析,筛选到SOX6基因上游区域中一个潜在的骨质疏松症易感causal SNP rs4325274位点,已有研究报道了该SNP位点与足跟部骨密度[16]和全身骨密度[17]表现出显著的关联性,P值分别为1.60×10-42和1.73×10-13,但该SNP潜在的功能机制有待进一步研究。本研究将结合生物信息学分析以及功能实验,探究骨质疏松症易感SNP rs4325274调控SOX6基因表达的分子机制,以期为进一步解析骨质疏松症的发生机制提供理论基础。
1 材料与方法
1.1 细胞培养
成骨细胞来源的U2OS细胞,用含有10%胎牛血清,100 U/mL的青霉素和100 μg/mL的链霉素的RPMI-1640培养基在37℃、5%CO2的细胞培养箱中培养。1.2 主要试剂
菌株感受态细胞E.coli DH5α购自北京天根生化技术有限公司;双荧光素酶报告基因载体PGL-3 Basic、内参海肾载体phRL和ViaFect转染试剂均购自美国Promega公司;定点突变试剂盒购自北京天恩泽技术有限公司;限制性内切酶,DNA ligation Mix,Ex Taq DNA 聚合酶均购自日本TaKaRa公司;胶纯化回收试剂盒购自上海GENEray公司;血液/组织/细胞基因组DNA提取试剂盒,无内毒素质粒小量中提提取试剂盒均购自北京天根生化技术有限公司。
1.3 表观功能注释
利用ANNOVAR软件注释causal SNP rs4325274的基因组位置,利用WashU Epigenome Brower (1.4 顺式表达数量性状(expression quantitative trait locus, eQTL)分析
利用福明翰心脏研究(Framingham Heart Study, FHS)中的全血eQTL数据(1.5 高通量染色质互作(high-throughput chromatin interaction analysis, Hi-C)分析
结合本实验室测得成骨细胞的Hi-C数据、ChIP-seq测序数据(doi:1.6 双荧光素酶报告基因实验
1.6.1 野生型和突变型两种荧光素酶重组载体构建构建含SOX6启动子片段的PGL3荧光素酶报告载体,正确载体命名为SOX6 promoter-Luc+。然后,构建含SNP rs4325274片段的SOX6 promoter-Luc+重组载体。NCBI(
Table 1
表1
表1实验所用引物序列
Table 1
| 引物名称 | 引物序列(5?→3?) |
|---|---|
| SOX6启动子扩增引物 | 上游:CCCAAGCTTATGGTGCCGATAGACTTGCC(Hind III) |
| 下游:CCGCTCGAGCATCTTATGGGTTCCACGCCT(Xho I) | |
| SNP扩增引物 | 上游:CGACGCGTAGAAGAGAAACGAGGTGTTGGT(Mlu I) |
| 下游:CCGCTCGAGACAAGTTTAGTGGGACAGGGTT(Xho I) | |
| SNP突变引物 | 上游:GGGATCCTGCTTACTTACTATCTTTGGTCTTAAACTAGACC |
| 下游:TAAGTAAGCAGGATCCCGATATCAAGTACCAGG | |
| SNP分型引物 | 上游:AAGAGAAACGAGGTGTTGGTGAT |
| 下游:GGTATCTTCCGTGCAGTTTTGG | |
| SOX6qRT-PCR引物 | 上游:AGAACGCGCTTTGAGAATTT |
| 下游:GCCCAGTTTTCCATCTTCAT | |
| HNF1AqRT-PCR引物[18] | 上游:CCCCAGATTCAGGATCAGACA |
| 下游:CCATCATGTTCCATTTTTCGC |
新窗口打开|下载CSV
1.6.2 报告基因载体的转染及荧光素酶活性检测
分别将构建好的SOX6 promoter-Luc+、rs4325274- C-SOX6 promoter-Luc+和rs4325274-G-SOX6 promoter- Luc+荧光素酶报告基因载体与内参海肾质粒(phRL) 共转染至成骨系细胞U2OS中,根据Dual Luciferase Reporter Assay System操作说明书,利用化学发光仪检测rs4325274两种碱基型增强子对SOX6基因启动子的调控活性,以确定其是否对SOX6基因有增强子作用。
1.7 Motif预测分析
从meme suite网站(1.8 转录因子shRNA敲低实验
首先在U2OS细胞中,对SNP rs4325274进行了分型,利用血液/组织/细胞基因组DNA提取试剂盒提取U2OS细胞的基因组DNA作为模板进行PCR扩增,将得到的PCR产物送北京擎科生物科技有限公司进行测序检测SNP基因型。本研究选取文献[19]已报道的HNF1A-shRNA寡核苷酸序列为shRNA序列:HNF1A-shRNA1:5?-GGCAGAAGAACCCTAGCAA-3?,HNF1A-shRNA2:5?-GGTCTTCACCTCAGACACT-3?,sh-NC:5?-GTTCTCCGAACGTGTCACGT-3?,构建到miR-30骨架上,经Xho I和EcoR I双酶切克隆到pcDNA3.1表达载体上,酶切及测序鉴定验证,即构建好sh-HNF1A-1,sh-HNF1A-2和sh-NC (空白对照)表达质粒。然后用ViaFect转染试剂分别转染U2OS细胞。转染48h后,提取细胞总RNA,反转录,用实时荧光定量PCR(qRT-PCR)检测HNF1A的敲低情况及SOX6基因mRNA水平的表达情况。qRT-PCR检测引物序列见表1。
2 结果与分析
2.1 表观注释提示SNP rs4325274处于成骨细胞相关增强子上
利用ENCODE数据库里的成骨细胞及GM12878的ChIP-seq数据对筛选出的SNP rs4325274所在区域进行表观注释,利用WashU可视化工具作图,发现SNP rs4325274周围富集了多个具有较强的激活型组蛋白标记,如H3K4me1、H3K4me3、H3K27ac,以及转录激活因子P300和DNase I 超敏感位点(DHS) (图1),提示SNP rs4325274处于增强子元件内,初步推断该SNP所在区域可能是一个增强子。图1
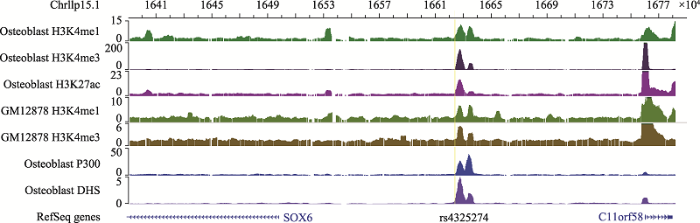 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1SNP rs4325274组蛋白注释结果(采用Wash U基因组浏览器可视化)
Fig. 1SNP rs4325274 histone annotation results (visualized using Wash U Genome browser)
2.2 eQTL和Hi-C分析发现易感SNP rs4325274作用的靶基因可能是SOX6基因
研究发现,基因组可折叠成环使得直线物理位置较远的增强子和靶基因的启动子在三维结构上接近,进而发挥调控作用[20,21]。为了证明易感SNP rs4325274与靶基因之间的关系,利用FHS中5257个人的全血eQTL数据进行了eQTL分析,结果发现SNP rs4325274 (P = 2.06×10-12)与SOX6基因之间的关系较显著。利用本课题组前期对成骨细胞高分辨率的Hi-C测序数据及搭建好的Hi-C分析流程,数据分辨率为2 kb,发现非编码区SNP rs4325274与SOX6基因存在远程互作(图2)。eQTL分析和Hi-C分析结果表明,易感SNP rs4325274所调控的靶基因可能是SOX6基因。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Hi-C数据分析结果示意图
Fig. 2Schematic diagram of the results of Hi-C data analysis
2.3 rs4325274对SOX6基因表达具有增强子调控活性
分别将包含rs4325274(C/G)不同等位基因的片段(约0.8 kb)及SOX6基因启动子区片段(约1.2 kb)克隆到荧光素酶报告基因载体(PGL3-Basic)上,与内参海肾质粒(phRL)共转染至U2OS细胞中,检测其荧光素酶活性。结果发现,相比只含SOX6基因启动子的载体,含SNP不同等位基因的重组载体表达活性显著增强,且含有G碱基的载体表达活性相比C碱基的载体表达活性显著增强(图3),这进一步证实了SNP rs4325274所在的片段是作为一个增强子来调控SOX6基因表达,而且不同等位基因调控活性有差异,其中G等位基因的调控活性更高。图3
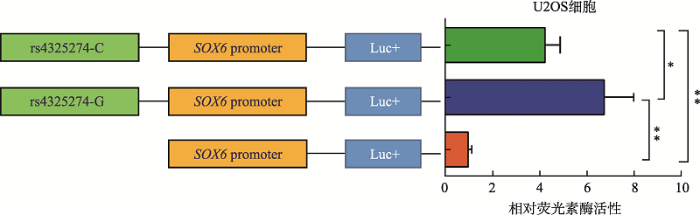 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3双荧光素酶报告基因活性检测rs4325274的增强子活性结果
数据为均值 ± 标准差;*:P<0.05;**:P< 0.01。
Fig. 3Enhancer activity of rs4325274 detected by dual-luciferase reporter gene activity assays
2.4 Motif预测发现rs4325274结合转录因子HNF1A
根据JASPAR 2018、HOCOMOCOv11、SwissRegulon和TRANSFAC四个权威数据库对SNP rs4325274 (G/C)序列进行转录因子预测,结果同时在SwissRegulon和TRANSFAC数据库中发现SNP rs4325274都结合转录因子HNF1A,而且转录因子HNF1A更倾向于结合rs4325274-G等位基因(图4)。因此,推测SNP rs4325274通过不同基因型结合转录因子的活性对增强子具有潜在的激活作用。图4
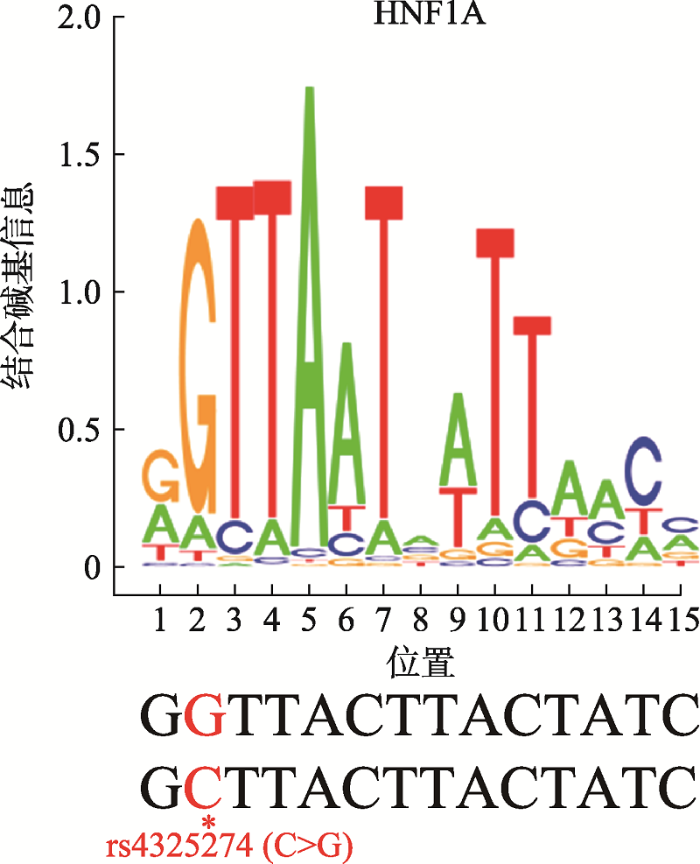 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4转录因子HNF1A结合DNA序列和SNP位置
*:SNP rs4325274位点。
Fig. 4Transcription factor HNF1A binding motif and SNP location
为了验证rs4325274可能与转录因子HNF1A结合,从GEO数据库中获得转录因子HNF1A的ChIP-seq数据(NO:GSM2534454),利用IGV (Integrative Genomics Viewer)工具可视化作图,发现rs4325274位于ChIP信号富集区(图5),这一结果进一步说明了rs4325274可能会结合转录因子HNF1A。
图5
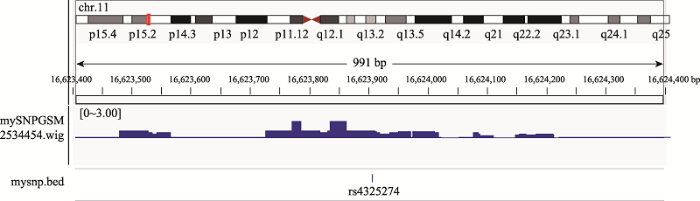 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5ChIP-seq数据分析结果(采用IGV工具可视化)
Fig. 5ChIP-seq data analysis results (visualized using IGV tool)
2.5 HNFIA敲低后SOX6表达降低
对SNP rs4325274在U2OS细胞中进行了分型,分型结果显示rs4325274在U2OS细胞中是杂合型(G/C)(图6),确保了后续敲低实验的进行。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6rs4325274在U2OS细胞中的分型结果
Fig. 6Genotyping results of rs4325274 in U2OS cells
为进一步探究SNPrs4325274是否通过影响转录因子结合机制调控靶基因SOX6的表达,本研究构建了HNF1A-shRNA敲低载体,转染至U2OS细胞,利用qRT-PCR检测SOX6基因的表达情况。结果表明,与阴性对照shRNA转染细胞相比,在转录因子HNF1A敲低的U2OS细胞中,SOX6基因的表达显著降低(图7)。这一结果提示HNF1A可能是SNP rs4325274调控机制中的潜在调节因子,通过目标SNP-转录因子-靶基因的调控机制,进而导致疾病的发生。转录因子HNF1A与SNP rs4325274等位基因的特异性结合需进一步实验去验证。
图7
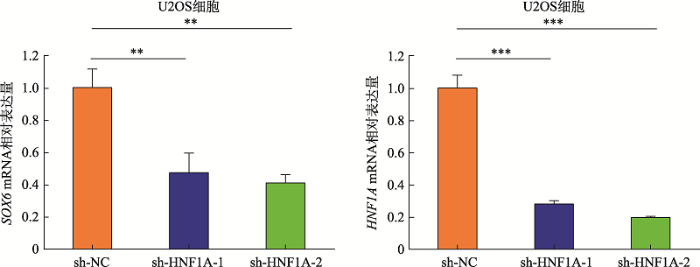 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7在U2OS细胞中敲低HNFIA对SOX6表达的影响
数据为均值 ± 标准差;***:P<0.001;**:P<0.01。
Fig. 7Effects of HNFIA knockdown on SOX6 expression in U2OS cells
3 讨论
骨质疏松症是一种受多基因调控的复杂疾病[11],其遗传力高达85%[22]。随着基因组技术的发展,大规模GWAS的开展,已经成功鉴定了大量与骨质疏松症骨密度相关联的易感变异位点。目前,破译这些易感位点影响疾病发生的功能机制,进而为临床转化提供潜在治疗靶点,是后GWAS时代的研究热点和难点[23]。近年来,随着ENCODE[24,25]和Roadmap[26]计划结果的陆续公布,提供了大量人类基因组上的各种表观调控信息,这为解析非编码区疾病位点的功能提供了新的契机。本研究利用ENCODE数据对骨质疏松易感SNP rs4325274进行表观注释,发现SNP rs4325274位于增强子区域。为进一步证明易感SNP rs4325274与靶基因之间的关系,利用eQTL分析和Hi-C分析发现易感SNP rs4325274所调控的靶基因可能是SOX6基因。在此基础上通过功能实验证实该SNP确实对SOX6基因表达有增强子调控活性,并发现rs4325274-G等位基因表达活性比rs4325274-C等位基因显著增强。
研究表明,SNP影响疾病易感性的机制之一就是通过影响转录因子与DNA的结合调控基因表达[27],在骨质疏松症发生机制中已有相关研究报道。Xiao等[28]研究证明转录因子CDX1特异性结合rs9547970的主等位基因A而非等位基因G,通过该机制调控POSTN基因的转录活性,从而影响骨形成。MPP7基因上的rs4317882与骨密度关联,该研究者[29]同时又证实了转录因子GATA2可特异性结合MPP7 rs4317882的风险等位基因A而非等位基因G。本课题组前期在解析骨质疏松症热点区域13q14.11中易感SNP位点的机制时,证明rs9533090-C等位基因可以大量招募激活型转录因子NFIC,提高其增强子活性,从而增强骨质疏松明星基因RANKL表达的机制[30]。
为进一步探究易感SNP rs4325274位点调控SOX6基因表达的机制,本研究利用多种数据库进行了Motif预测,发现SNP rs4325274结合的转录因子HNF1A,并结合ChIP-seq数据分析进行验证,发现rs4325274位于转录因子ChIP信号富集区,进一步利用shRNA干扰转录因子HNF1A,发现在HNF1A基因敲低的细胞中,SOX6基因的表达显著降低。由此推测骨质疏松易感SNP rs4325274可能通过影响与转录因子HNF1A的特异性结合来调节SOX6基因表达。后续本课题组会进一步通过染色质互作(chromosome conformation capture, 3C)实验和染色质免疫共沉淀(chromatin immunoprecipitation assay, ChIP)等实验来深入探究该SNP位点与转录因子及靶基因SOX6基因之间的作用机制,并在细胞水平和动物模型深入探究SOX6基因在骨质疏松症发病中的真正作用机制。
综上所述,本研究初步解析了非编码区功能性SNPrs4325274作为增强子远程调控SOX6基因表达的分子机制。研究结果将有助于为复杂疾病非编码易感SNP的遗传调控研究提供新思路,并为骨质疏松症的药物开发和治疗提供潜在的药物靶点。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:16568580 [本文引用: 1]

Osteoporosis is a systemic disease characterized by decrease in bone mass per unit volume, compromised bone strength, which predisposes the affected bone to fracture. This is currently one of the leading causes of morbidity and mortality among elderly over the world. In general, osteoporosis is a silent and progressive disorder that is often brought to attention of the patients or physician only after a fracture. The aetiology of osteoporosis is multifactorial and is related to two main processes: acquisition of peak bone density that occurs at the end of the third decade and loss of bone at menopause, going on to old age. The cardinal features of osteoporosis are pain, fracture and deformity. Bone mineral density measurement is the most reliable diagnostic tool in the early stage of osteoporosis. Management of osteoporosis involves prevention and treatment. The best treatment for osteoporosis is prevention. The risk of osteoporosis can be reduced by increasing peak bone mass or by decreasing the bone loss. It needs to be emphasized that bone mineral density (BMD) peaks at about age 35 and then begins to slowly decline with significant acceleration after menopause.Therefore, the most logical and cost-effective preventive strategies are to encourage young women to stop smoking and avoid excessive use of alcohol. They should also be counseled to exercise regularly and consume adequate amounts of calcium and vitamin D.
DOI:10.1016/j.bone.2005.11.024URLPMID:16455317 [本文引用: 1]
It is estimated that over 200 million people worldwide have osteoporosis. The prevalence of osteoporosis is continuing to escalate with the increasingly elderly population. The major complication of osteoporosis is an increase in fragility fractures leading to morbidity, mortality, and decreased quality of life. In the European Union, in 2000, the number of osteoporotic fractures was estimated at 3.79 million. A baseline fracture is a very strong predictor of further fractures with 20% of patients experiencing a second fracture within the first year. The costs to health care services are already considerable and, on current trends, are predicted to double by 2050. The direct costs of osteoporotic fractures to the health services in the European Union in the year 2000 were estimated at 32 billion Euros. Guidelines for the diagnosis and treatment of osteoporosis are available in many countries; however, implementation is generally poor despite the availability of treatments with proven efficacy. Programs to increase awareness of osteoporosis and its outcomes are necessary for healthcare specialists and the general public. Earlier diagnosis and intervention prior to the first fracture are highly desirable.
[本文引用: 1]
URLPMID:14984358 [本文引用: 1]
Osteoporosis is: (1) Underrated. Currently costs about 7 billion dollars annually in Australia. Has high morbidity and 2-3-fold increase in risk of death after any major osteoporotic fracture. Genetic factors contribute highly to risk, modified by lifestyle and hormonal factors. (2) Underdiagnosed. Bone density is a good predictor of subsequent risk. Anyone with a low-trauma fracture has osteoporosis unless proven otherwise. Every individual with a low trauma fracture should be investigated for exclusion of underlying osteoporosis and considered for effective treatment to reduce future fracture risk. More than 75% of women and about 90% of men with a high likelihood of osteoporosis are not investigated. (3) Undertreated. More than 75% of those affected are not treated. Effective treatments (eg, hormone replacement therapy, selective oestrogen receptor modifiers and bisphosphonates) reduce fracture risk by 30%-60%. Simple measures like vitamin D and calcium supplementation and use of hip protectors can reduce hip fractures, particularly in institutionalised and housebound elderly people
DOI:10.1016/s1534-5807(01)00003-xURLPMID:11702786 [本文引用: 2]
L-Sox5 and Sox6 are highly identical Sry-related transcription factors coexpressed in cartilage. Whereas Sox5 and Sox6 single null mice are born with mild skeletal abnormalities, Sox5; Sox6 double null fetuses die with a severe, generalized chondrodysplasia. In these double mutants, chondroblasts poorly differentiate. They express the genes for all essential cartilage extracellular matrix components at low or undetectable levels and initiate proliferation after a long delay. All cartilages are thus extracellular matrix deficient and remain rudimentary. While chondroblasts in the center of cartilages ultimately activate prehypertrophic chondrocyte markers, epiphyseal chondroblasts ectopically activate hypertrophic chondrocyte markers. Thick intramembranous bone collars develop, but the formation of cartilage growth plates and endochondral bones is disrupted. L-Sox5 and Sox6 are thus redundant, potent enhancers of chondroblast functions, thereby essential for endochondral skeleton formation.
DOI:10.1083/jcb.200312045URLPMID:14993235 [本文引用: 1]
Sox5 and Sox6 encode Sry-related transcription factors that redundantly promote early chondroblast differentiation. Using mouse embryos with three or four null alleles of Sox5 and Sox6, we show that they are also essential and redundant in major steps of growth plate chondrocyte differentiation. Sox5 and Sox6 promote the development of a highly proliferating pool of chondroblasts between the epiphyses and metaphyses of future long bones. This pool is the likely cellular source of growth plates. Sox5 and Sox6 permit formation of growth plate columnar zones by keeping chondroblasts proliferating and by delaying chondrocyte prehypertrophy. They allow induction of chondrocyte hypertrophy and permit formation of prehypertrophic and hypertrophic zones by delaying chondrocyte terminal differentiation induced by ossification fronts. They act, at least in part, by down-regulating Ihh signaling, Fgfr3, and Runx2 and by up-regulating Bmp6. In conclusion, Sox5 and Sox6 are needed for the establishment of multilayered growth plates, and thereby for proper and timely development of endochondral bones.
DOI:10.1007/s00109-011-0842-3URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1359/JBMR.040132URLPMID:15125790 [本文引用: 1]
UNLABELLED: BMD is a primary predictor of osteoporotic fracture, and its genetic determination is still unclear. This study showed that the correlation between BMD at different skeletal sites is caused by an underlying genetic structure of common genetic effects. In addition to possible shared (pleiotropic) genetic and environmental effects, each of the BMD variables may also be determined by site-specific genetic factors. INTRODUCTION: BMD is a primary predictor of osteoporotic fracture and a key phenotype for the genetic study of osteoporosis. The interindividual variation in BMD measured at a given skeletal site is largely regulated by genetic factors. A strong phenotypic covariation exists for BMD at different skeletal sites. This study tests the hypothesis that the covariation is in fact caused by an underlying genetic structure of common genetic effects and that, in addition to possible shared (pleiotropic) genetic effects, each of the BMD variables may also be determined by site-specific genetic factors MATERIALS AND METHODS: A bivariate complex segregation analysis as implemented in statistical package PAP was conducted to explore various models of pleiotropic genetic and environmental transmission in lumbar spine and femoral neck BMD, as well as in compact and spongious segments of hand phalanges. The BMD was obtained in three ethnically, culturally, and socially heterogeneous samples of white pedigrees, with 2549 individuals between 18 and 100 years of age, from Australia, Europe, and North America. RESULTS AND CONCLUSIONS: The genetic correlation between BMD measures ranged between 0.50 +/- 0.09 and 0.79 +/- 0.04 in the three samples. In each sample, the model incorporated a major locus pleiotropic effect, and residual correlation was found to be the most parsimonious model. Estimated parameters from the model indicated a significant pleiotropic major gene effect on both lumbar spine and femoral neck BMD, with the existence of a significant residual correlation (0.51 +/- 0.07 to 0.66 +/- 0.04). These results suggest that the covariation in BMD at different skeletal sites, and between mostly compact versus mostly trabecular bone, was largely determined by common genetic factors that are pleiotropic or in close linkage and linkage disequilibirum, while at the same time, exhibiting considerable evidence of shared environmental effects. The results, for the first time, suggest that the possibility of pleiotropic genetic effect may be controlled by a major genetic locus. Identification of the major locus could open new opportunity to understanding the liability and pathogenic processes in which they are involved in the determination of fracture risk.
DOI:10.1210/edrv.23.3.0464URLPMID:12050122 [本文引用: 2]
Osteoporosis is a common multifactorial disorder of reduced bone mass. The disorder in its most common form is generalized, affecting the elderly, both sexes, and all racial groups. Multiple environmental factors are involved in the pathogenesis. Genes also play a major role as reflected by heritability of many components of bone strength. Quantitative phenotypes in bone strength in the normal population do not conform to a monogenetic mode of inheritance. The common form of osteoporosis is generally considered to be a polygenic disorder arising from the interaction of common polymorphic alleles at quantitative trait loci, with multiple environmental factors. Finding the susceptibility genes underlying osteoporosis requires identifying specific alleles that coinherit with key heritable phenotypes in bone strength. Because of the close correspondence among mammalian genomes, identification of the genes underlying bone strength in mammals such as the mouse is likely to be of major assistance in human studies. Identification of susceptibility genes for osteoporosis is one of several important approaches toward the long-term goal of understanding the molecular biology of the normal variation in bone strength and how it may be modified to prevent osteoporosis. As with all genetic studies in humans, these scientific advances will need to be made in an environment of legal and ethical safeguards that are acceptable to the general public.
DOI:10.1038/ng.446URLPMID:19801982 [本文引用: 1]
Bone mineral density (BMD) is a heritable complex trait used in the clinical diagnosis of osteoporosis and the assessment of fracture risk. We performed meta-analysis of five genome-wide association studies of femoral neck and lumbar spine BMD in 19,195 subjects of Northern European descent. We identified 20 BMD loci that reached genome-wide significance (GWS; P < 5 x 10(-8)), of which 13 map to regions not previously associated with this trait: 1p31.3 (GPR177), 2p21 (SPTBN1), 3p22 (CTNNB1), 4q21.1 (MEPE), 5q14 (MEF2C), 7p14 (STARD3NL), 7q21.3 (FLJ42280), 11p11.2 (LRP4, ARHGAP1, F2), 11p14.1 (DCDC5), 11p15 (SOX6), 16q24 (FOXL1), 17q21 (HDAC5) and 17q12 (CRHR1). The meta-analysis also confirmed at GWS level seven known BMD loci on 1p36 (ZBTB40), 6q25 (ESR1), 8q24 (TNFRSF11B), 11q13.4 (LRP5), 12q13 (SP7), 13q14 (TNFSF11) and 18q21 (TNFRSF11A). The many SNPs associated with BMD map to genes in signaling pathways with relevance to bone metabolism and highlight the complex genetic architecture that underlies osteoporosis and variation in BMD.
DOI:10.1371/journal.pgen.1000977URLPMID:20548944 [本文引用: 1]
Osteoporosis is a complex disorder and commonly leads to fractures in elderly persons. Genome-wide association studies (GWAS) have become an unbiased approach to identify variations in the genome that potentially affect health. However, the genetic variants identified so far only explain a small proportion of the heritability for complex traits. Due to the modest genetic effect size and inadequate power, true association signals may not be revealed based on a stringent genome-wide significance threshold. Here, we take advantage of SNP and transcript arrays and integrate GWAS and expression signature profiling relevant to the skeletal system in cellular and animal models to prioritize the discovery of novel candidate genes for osteoporosis-related traits, including bone mineral density (BMD) at the lumbar spine (LS) and femoral neck (FN), as well as geometric indices of the hip (femoral neck-shaft angle, NSA; femoral neck length, NL; and narrow-neck width, NW). A two-stage meta-analysis of GWAS from 7,633 Caucasian women and 3,657 men, revealed three novel loci associated with osteoporosis-related traits, including chromosome 1p13.2 (RAP1A, p = 3.6x10(-8)), 2q11.2 (TBC1D8), and 18q11.2 (OSBPL1A), and confirmed a previously reported region near TNFRSF11B/OPG gene. We also prioritized 16 suggestive genome-wide significant candidate genes based on their potential involvement in skeletal metabolism. Among them, 3 candidate genes were associated with BMD in women. Notably, 2 out of these 3 genes (GPR177, p = 2.6x10(-13); SOX6, p = 6.4x10(-10)) associated with BMD in women have been successfully replicated in a large-scale meta-analysis of BMD, but none of the non-prioritized candidates (associated with BMD) did. Our results support the concept of our prioritization strategy. In the absence of direct biological support for identified genes, we highlighted the efficiency of subsequent functional characterization using publicly available expression profiling relevant to the skeletal system in cellular or whole animal models to prioritize candidate genes for further functional validation.
DOI:10.1007/s11427-010-4056-7URLPMID:21104366 [本文引用: 1]
Osteoporosis is a highly heritable common bone disease leading to fractures that severely impair the life quality of patients. Wrist fractures caused by osteoporosis are largely due to the scarcity of wrist bone mass. Here we report the results of a genome-wide association study (GWAS) of wrist bone mineral density (BMD). We examined approximately 500000 SNP markers in 1000 unrelated homogeneous Caucasian subjects and found a novel allelic association with wrist BMD at rs11023787 in the SOX6 (SRY (sex determining region Y)-box 6) gene (P=9.00x10(-5)). Subjects carrying the C allele of rs11023787 in SOX6 had significantly higher mean wrist BMD values than those with the T allele (0.485:0.462 g cm(-2) for C allele vs. T allele carriers). For validation, we performed SOX6 association for BMD in an independent Chinese sample and found that SNP rs11023787 was significantly associated with wrist BMD in the Chinese sample (P=6.41x10(-3)). Meta-analyses of the GWAS scan and the replication studies yielded P-values of 5.20x10(-6) for rs11023787. Results of this study, together with the functional relevance of SOX6 in cartilage formation, support the SOX6 gene as an important gene for BMD variation.
DOI:10.1155/2017/5831020URLPMID:28840121 [本文引用: 1]
To identify genetic variants influencing bone mineral density (BMD) in the Mexican-Mestizo population, we performed a GWAS for femoral neck (FN) and lumbar spine (LS) in Mexican-Mestizo postmenopausal women. In the discovery sample, 300,000 SNPs were genotyped in a cohort of 411 postmenopausal women and seven SNPs were analyzed in the replication cohort (n = 420). The combined results of a meta-analysis from the discovery and replication samples identified two loci, RMND1 (rs6904364, P = 2.77 x 10(-4)) and CCDC170 (rs17081341, P = 1.62 x 10(-5)), associated with FN BMD. We also compared our results with those of the Genetic Factors for Osteoporosis (GEFOS) Consortium meta-analysis. The comparison revealed two loci previously reported in the GEFOS meta-analysis: SOX6 (rs7128738) and PKDCC (rs11887431) associated with FN and LS BMD, respectively, in our study population. Interestingly, rs17081341 rare in Caucasians (minor allele frequency < 0.03) was found in high frequency in our population, which suggests that this association could be specific to non-Caucasian populations. In conclusion, the first pilot Mexican GWA study of BMD confirmed previously identified loci and also demonstrated the importance of studying variability in diverse populations and/or specific populations.
DOI:10.1038/s41588-018-0302-xURLPMID:30598549 [本文引用: 1]
Osteoporosis is a common aging-related disease diagnosed primarily using bone mineral density (BMD). We assessed genetic determinants of BMD as estimated by heel quantitative ultrasound in 426,824 individuals, identifying 518 genome-wide significant loci (301 novel), explaining 20% of its variance. We identified 13 bone fracture loci, all associated with estimated BMD (eBMD), in ~1.2 million individuals. We then identified target genes enriched for genes known to influence bone density and strength (maximum odds ratio (OR) = 58, P = 1 x 10(-75)) from cell-specific features, including chromatin conformation and accessible chromatin sites. We next performed rapid-throughput skeletal phenotyping of 126 knockout mice with disruptions in predicted target genes and found an increased abnormal skeletal phenotype frequency compared to 526 unselected lines (P < 0.0001). In-depth analysis of one gene, DAAM2, showed a disproportionate decrease in bone strength relative to mineralization. This genetic atlas provides evidence linking associated SNPs to causal genes, offers new insight into osteoporosis pathophysiology, and highlights opportunities for drug development.
DOI:10.1016/j.ajhg.2017.12.005URLPMID:29304378 [本文引用: 1]
Bone mineral density (BMD) assessed by DXA is used to evaluate bone health. In children, total body (TB) measurements are commonly used; in older individuals, BMD at the lumbar spine (LS) and femoral neck (FN) is used to diagnose osteoporosis. To date, genetic variants in more than 60 loci have been identified as associated with BMD. To investigate the genetic determinants of TB-BMD variation along the life course and test for age-specific effects, we performed a meta-analysis of 30 genome-wide association studies (GWASs) of TB-BMD including 66,628 individuals overall and divided across five age strata, each spanning 15 years. We identified variants associated with TB-BMD at 80 loci, of which 36 have not been previously identified; overall, they explain approximately 10% of the TB-BMD variance when combining all age groups and influence the risk of fracture. Pathway and enrichment analysis of the association signals showed clustering within gene sets implicated in the regulation of cell growth and SMAD proteins, overexpressed in the musculoskeletal system, and enriched in enhancer and promoter regions. These findings reveal TB-BMD as a relevant trait for genetic studies of osteoporosis, enabling the identification of variants and pathways influencing different bone compartments. Only variants in ESR1 and close proximity to RANKL showed a clear effect dependency on age. This most likely indicates that the majority of genetic variants identified influence BMD early in life and that their effect can be captured throughout the life course.
DOI:10.1186/s13045-015-0143-3URLPMID:25953102 [本文引用: 1]
BACKGROUND: The therapeutic efficacy of arsenic trioxide (As2O3) in acute myeloid leukemia (AML) is modest, which is partly related to its limited intracellular uptake into the leukemic cells. As2O3 enters cells via the transmembrane protein aquaglyceroporin 9 (AQP9). Azacytidine, a demethylating agent that is approved for the treatment of AML, has been shown to have synergistic effect with As2O3. We tested the hypothesis that azacytidine might up-regulate AQP9 and enhances As2O3-mediated cytotoxicity in AML. METHODS: Arsenic-induced cytotoxicity, the expression of AQP9, and the intracellular uptake of As2O3 were determined in AML cell lines and primary AML cells with or without azacytidine pre-treatment. The mechanism of AQP9 up-regulation was then investigated by examining the expression of transcription factors for AQP9 gene and the methylation status of their gene promoters. RESULTS: As2O3-induced cytotoxicity in AML cell lines was significantly enhanced after azacytidine pre-treatment as a result of AQP9 up-regulation, leading to increased arsenic uptake and hence intracellular concentration. Blocking AQP9-mediated As2O3 uptake with mercury chloride abrogated the sensitization effect of azacytidine. AQP9 promoter does not contain CpG islands. Instead, azacytidine pre-treatment led to increased expression of HNF1A, a transcription activator of AQP9, through demethylation of HNF1A promoter. HNF1 knockdown abrogated azacytidine-induced AQP9 up-regulation and almost completely blocked intracellular As2O3 entry, confirming that azacytidine enhanced As2O3-mediated cell death via up-regulation of HNF1A and hence increased AQP9 and As2O3 intracellular concentration. Azacytidine sensitization to As2O3 treatment was re-capitulated also in primary AML samples. Finally, azacytidine did not enhance arsenic toxicity in a liver cell line, where HNF1A was largely unmethylated. CONCLUSIONS: Azacytidine sensitizes AML cells to As2O3 treatment, and our results provide proof-of-principle evidence that pharmacological up-regulation of AQP9 potentially expands the therapeutic spectrum of As2O3. Further clinical trial should evaluate the efficacy of azacytidine in combination with As2O3 in the treatment of AML.
DOI:10.1002/hep.23362URLPMID:20041408 [本文引用: 1]
UNLABELLED: Hepatocellular adenomas (HCAs) are benign liver tumors that usually develop in women who are taking oral contraceptives. Among these tumors, biallelic inactivating mutations of the hepatocyte nuclear factor 1alpha (HNF1A) transcription factor have been frequently identified and in rare cases of hepatocellular carcinomas developed in noncirrhotic liver. Because HNF1A meets the genetic criteria of a tumor suppressor gene, we aimed to elucidate the tumorigenic mechanisms related to HNF1alpha inactivation in hepatocytes. We searched for signaling pathways aberrantly activated in human HNF1A-mutated HCA (H-HCA) using a genome-wide transcriptome analysis comparing five H-HCA with four normal livers. We validated the main pathways by quantitative reverse transcription polymerase chain reaction (RT-PCR) and western blotting in a large series of samples. Then, we assessed the role of HNF1alpha in the observed deregulations in hepatocellular cell models (HepG2 and Hep3B) by silencing its endogenous expression using small interfering RNA. Along with the previously described induction of glycolysis and lipogenesis, H-HCA also displayed overexpression of several genes encoding growth factor receptors, components of the translation machinery, cell cycle, and angiogenesis regulators, with, in particular, activation of the mammalian target of rapamycin (mTOR) pathway. Moreover, estradiol detoxification activities were shut down, suggesting a hypersensitivity of H-HCA to estrogenic stimulation. In the cell model, inhibition of HNF1alpha recapitulated most of these identified transcriptional deregulations, demonstrating that they were related to HNF1alpha inhibition. CONCLUSION: H-HCA showed a combination of alterations related to HNF1alpha inactivation that may cooperate to promote tumor development. Interestingly, mTOR appears as a potential new attractive therapeutic target for treatment of this group of HCAs.
DOI:10.1038/nrm.2016.138URLPMID:27826147 [本文引用: 1]
Genetic variation associated with disease often appears in non-coding parts of the genome. Understanding the mechanisms by which this phenomenon leads to disease is necessary to translate results from genetic association studies to the clinic. Assigning function to this type of variation is notoriously difficult because the human genome harbours a complex regulatory landscape with a dizzying array of transcriptional regulatory sequences, such as enhancers that have unpredictable, promiscuous and context-dependent behaviour. In this Review, we discuss how technological advances have provided increasingly detailed information on genome folding; for example, genome folding forms loops that bring enhancers and target genes into close proximity. We also now know that enhancers function within topologically associated domains, which are structural and functional units of chromosomes. Studying disease-associated mutations and chromosomal rearrangements in the context of the 3D genome will enable the identification of dysregulated target genes and aid the progression from descriptive genetic association results to discovering molecular mechanisms underlying disease.
[本文引用: 1]
DOI:10.1016/j.mce.2015.12.021URLPMID:26747728 [本文引用: 1]
Bone mineral density (BMD) is a quantitative traits used as a surrogate phenotype for the diagnosis of osteoporosis, a common metabolic disorder characterized by increased fracture risk as a result of a decreased bone mass and deterioration of the microarchitecture of the bone. Normal variation in BMD is determined by both environmental and genetic factors. According to heritability studies, 50-85% of the variance in BMD is controlled by genetic factors which are mostly polygenic. In contrast to the complex etiology of osteoporosis, there are disorders with deviating BMD values caused by one mutation with a large impact. These mutations can result in monogenic bone disorders with either an extreme high (sclerosteosis, Van Buchem disease, osteopetrosis, high bone mass phenotype) or low BMD (osteogenesis imperfecta, juvenile osteoporosis, primary osteoporosis). Identification of the disease causing genes, increased the knowledge on the regulation of BMD and highlighted important signaling pathways and novel therapeutic targets such as sclerostin, RANKL and cathepsin K. Genetic variation in genes involved in these pathways are often also involved in the regulation of normal variation in BMD and osteoporosis susceptibility. In the last decades, identification of genetic factors regulating BMD has proven to be a challenge. Several approaches have been tested such as linkage studies and candidate and genome wide association studies. Although, throughout the years, technological developments made it possible to study increasing numbers of genetic variants in populations with increasing sample sizes at the same time, only a small fraction of the genetic impact can yet be explained. In order to elucidate the missing heritability, the focus shifted to studying the role of rare variants, copy number variations and epigenetic influences. This review summarizes the genetic cause of different monogenic bone disorders with deviating BMD and the knowledge on genetic factors explaining normal variation in BMD and osteoporosis risk.
DOI:10.1016/j.jgg.2015.02.001URLPMID:25819085 [本文引用: 1]
Genome-wide association studies (GWASs) have identified thousands of genes and genetic variants (mainly SNPs) that contribute to complex diseases in humans. Functional characterization and mechanistic elucidation of these SNPs and genes action are the next major challenge. It has been well established that SNPs altering the amino acids of protein-coding genes can drastically impact protein function, and play an important role in molecular pathogenesis. Functions of regulatory SNPs can be complex and elusive, and involve gene expression regulation through the effect on RNA splicing, transcription factor binding, DNA methylation and miRNA recruitment. In the present review, we summarize the recent progress in our understanding of functional consequences of GWAS-associated non-coding regulatory SNPs, and discuss the application of systems genetics and network biology in the interpretation of GWAS findings.
DOI:10.1038/nature11247URL [本文引用: 1]
The human genome encodes the blueprint of life, but the function of the vast majority of its nearly three billion bases is unknown. The Encyclopedia of DNA Elements (ENCODE) project has systematically mapped regions of transcription, transcription factor association, chromatin structure and histone modification. These data enabled us to assign biochemical functions for 80% of the genome, in particular outside of the well-studied protein-coding regions. Many discovered candidate regulatory elements are physically associated with one another and with expressed genes, providing new insights into the mechanisms of gene regulation. The newly identified elements also show a statistical correspondence to sequence variants linked to human disease, and can thereby guide interpretation of this variation. Overall, the project provides new insights into the organization and regulation of our genes and genome, and is an expansive resource of functional annotations for biomedical research.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nbt1010-1045URLPMID:20944595 [本文引用: 1]
The NIH Roadmap Epigenomics Mapping Consortium aims to produce a public resource of epigenomic maps for stem cells and primary ex vivo tissues selected to represent the normal counterparts of tissues and organ systems frequently involved in human disease.
DOI:10.1126/science.1242463URL [本文引用: 1]
DNA sequence variation has been associated with quantitative changes in molecular phenotypes such as gene expression, but its impact on chromatin states is poorly characterized. To understand the interplay between chromatin and genetic control of gene regulation, we quantified allelic variability in transcription factor binding, histone modifications, and gene expression within humans. We found abundant allelic specificity in chromatin and extensive local, short-range, and long-range allelic coordination among the studied molecular phenotypes. We observed genetic influence on most of these phenotypes, with histone modifications exhibiting strong context-dependent behavior. Our results implicate transcription factors as primary mediators of sequence-specific regulation of gene expression programs, with histone modifications frequently reflecting the primary regulatory event.
[本文引用: 1]
DOI:10.1093/hmg/ddr586URL [本文引用: 1]
Our previous genome-wide association study (GWAS) in a Hong Kong Southern Chinese population with extreme bone mineral density (BMD) scores revealed suggestive association with MPP7, which ranked second after JAG1 as a candidate gene for BMD. To follow-up this suggestive signal, we replicated the top single-nucleotide polymorphism rs4317882 of MPP7 in three additional independent Asian-descent samples (n 2684). The association of rs4317882 reached the genome-wide significance in the meta-analysis of all available subjects (P-meta 4.58 10(8), n 4204). Site heterogeneity was observed, with a larger effect on spine than hip BMD. Further functional studies in a zebrafish model revealed that vertebral bone mass was lower in an mpp7 knock-down model compared with the wide-type (P 9.64 10(4), n 21). In addition, MPP7 was found to have constitutive expression in human bone-derived cells during osteogenesis. Immunostaining of murine MC3T3-E1 cells revealed that the Mpp7 protein is localized in the plasma membrane and intracytoplasmic compartment of osteoblasts. In an assessment of the function of identified variants, an electrophoretic mobility shift assay demonstrated the binding of transcriptional factor GATA2 to the risk allele oA' but not the oG' allele of rs4317882. An mRNA expression study in human peripheral blood mononuclear cells confirmed that the low BMD-related allele oA' of rs4317882 was associated with lower MPP7 expression (P 9.07 10(3), n 135). Our data suggest a genetic and functional association of MPP7 with BMD variation.
DOI:10.1002/jbmr.3419URLPMID:29528523 [本文引用: 1]
RANKL is a key regulator involved in bone metabolism, and a drug target for osteoporosis. The clinical diagnosis and assessment of osteoporosis are mainly based on bone mineral density (BMD). Previous powerful genomewide association studies (GWASs) have identified multiple intergenic single-nucleotide polymorphisms (SNPs) located over 100 kb upstream of RANKL and 65 kb downstream of AKAP11 at 13q14.11 for osteoporosis. Whether these SNPs exert their roles on osteoporosis through RANKL is unknown. In this study, we conducted integrative analyses combining expression quantitative trait locus (eQTL), genomic chromatin interaction (high-throughput chromosome conformation capture [Hi-C]), epigenetic annotation, and a series of functional assays. The eQTL analysis identified six potential functional SNPs (rs9533090, rs9594738, r8001611, rs9533094, rs9533095, and rs9594759) exclusively correlated with RANKL gene expression (p < 0.001) at 13q14.11. Co-localization analyses suggested that eQTL signal for RANKL and BMD-GWAS signal shared the same causal variants. Hi-C analysis and functional annotation further validated that the first five osteoporosis SNPs are located in a super-enhancer region to regulate the expression of RANKL via long-range chromosomal interaction. Particularly, dual-luciferase assay showed that the region harboring rs9533090 in the super-enhancer has the strongest enhancer activity, and rs9533090 is an allele-specific regulatory SNP. Furthermore, deletion of the region harboring rs9533090 using CRISPR/Cas9 genome editing significantly reduced RANKL expression in both mRNA level and protein level. Finally, we found that the rs9533090-C robustly recruits transcription factor NFIC, which efficiently elevates the enhancer activity and increases the RANKL expression. In summary, we provided a feasible method to identify regulatory noncoding SNPs to distally regulate their target gene underlying the pathogenesis of osteoporosis by using bioinformatics data analyses and experimental validation. Our findings would be a potential and promising therapeutic target for precision medicine in osteoporosis. (c) 2018 American Society for Bone and Mineral Research.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}