 ,昆明理工大学生命科学与技术学院,昆明 650500
,昆明理工大学生命科学与技术学院,昆明 650500Progress on phage genomics of Pseudomonas spp.
Zhiwei Xu, Yunlin Wei, Xiuling Ji,Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming 650500, China通讯作者: 季秀玲,博士,副教授,研究方向:低温微生物。E-mail:jixiuling1023@126.com
编委: 谢建平
收稿日期:2020-04-26修回日期:2020-07-10网络出版日期:2020-08-20
| 基金资助: |
Received:2020-04-26Revised:2020-07-10Online:2020-08-20
| Fund supported: |
作者简介 About authors
徐志伟,在读硕士研究生,专业方向:低温微生物。E-mail:

摘要
假单胞菌属(Pseudomonas spp.)是地球上重要的生态菌群之一,广泛分布于淡水、土壤等生态环境。假单胞菌噬菌体是以假单胞菌为宿主的病毒,不仅影响宿主的生存状况和进化过程,而且在生物物质循环和能量流动中扮演着重要角色。随着基因组测序技术的飞速发展,许多假单胞菌噬菌体的全基因组测序工作已经完成。截至2020年7月,GenBank收录的假单胞菌噬菌体基因组数有247条,占全部病毒基因组(10,069条)的2.45%。由于假单胞菌噬菌体基因组大小差异较大、遗传含量不同、基因组之间相似性较低,因此对假单胞菌噬菌体基因组的研究相对较少。本文主要对假单胞菌噬菌体基因组的特点、遗传多样性和功能基因方面的研究进行了综述,以期为理解细菌和噬菌体的对抗性共进化作用以及噬菌体的遗传进化提供参考。
关键词:
Abstract
Pseudomonas spp. are one of the most important ecological flora on the earth, widely distributed in freshwater, soil and other ecological environments. Pseudomonas phages are viruses hosted by Pseudomonas spp., which not only affect the survival and evolution of the hosts, but also play important roles in biomass circulation and energy flow. With the rapid development of genome sequencing technologies, the whole genome sequences of many Pseudomonas phages have been completed. As of July 2020, 247 Pseudomonas phage genomes were deposited in GenBank, accounting for 2.45% of the total 10,069 viral genomes. The genome sizes of Pseudomonas bacteriophages and the genetic contents are different, and the similarity between genomes is low, so the study on Pseudomonas bacteriophage genomes is relatively less. In this review, we summarize the characteristics, genetic diversity, and functional genes of Pseudomonas bacteriophages genomes in order to provide a reference for understanding the antagonistic coevolution of bacteria and phages and the genetic evolution of phages.
Keywords:
PDF (1308KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
徐志伟, 魏云林, 季秀玲. 假单胞菌噬菌体基因组学研究进展. 遗传[J], 2020, 42(8): 752-759 doi:10.16288/j.yczz.19-272
Zhiwei Xu.
假单胞菌属(Pseudomonas spp.)是重要的生态菌群之一,包括植物共生体(施氏假单胞菌(Pseudomonas stutzeri)和荧光假单胞菌(Pseudomonas fluorescens)、植物病原体(丁香假单胞菌(Pseudomonas syringae)、人类和动物病原体(铜绿假单胞菌(Pseudomonas aeruginosa))和可用于生物修复的恶臭假单胞菌(Pseudomonas putida)等。假单胞菌对环境具有极强的适应能力,广泛分布于淡水、土壤等生态环境中[1]。
噬菌体是微生物群落的重要组成部分,作为细菌的天敌可以入侵破坏宿主细胞的新陈代谢,对地球生物化学循环和生态系统物质循环产生重要影响。作为病毒的一种,噬菌体具有一般病毒体积微小和结构简单等特性,同时因为体内只含有单一核酸,不具备完整的细胞结构,只能寄生在活菌中,对宿主具有严格的特异性;在调节微生物群落结构、促进生物进化方面扮演着重要作用[2,3]。
假单胞菌噬菌体是以假单胞菌为宿主的病毒,由于其宿主假单胞菌在自然环境中分布广泛,因此易于从自然界中分离得到。对假单胞菌噬菌体基因组的研究揭示了噬菌体基因组的多样性,反映了其宿主假单胞菌的遗传多样性,体现不同假单胞菌噬菌体之间序列的相似性和差异性等。因此,开展假单胞菌噬菌体基因组的研究,对了解噬菌体遗传进化、噬菌体-宿主相互作用的分子机制具有重要的意义[4,5]。结合近年来该领域相关的研究进展,本文主要对假单胞菌噬菌体基因组特点、遗传多样性和功能基因的研究等方面进行综述,以期为进一步研究假单胞菌噬菌体基因组提供参考。
1 假单胞菌噬菌体基因组基本特点
随着DNA测序技术的不断发展,大量噬菌体全基因组被测序。截止2020年7月,GenBank全基因组数据库中提交的假单胞菌噬菌体基因组序列有247条,占全部病毒组数据库(10,069条)的2.45%。综合来看,假单胞菌噬菌体基因组具有如下特点。(1)假单胞菌噬菌体基因组具有嵌合基因结构。这种基因组嵌合结构不是噬菌体群体所特有的,在细菌中也广泛存在,随着可供分析的噬菌体基因组的数量增加,这种现象在噬菌体基因组中表现得更加明显[6]。噬菌体基因组具有嵌合现象最早可以通过异源双链DNA定位法对核苷酸序列比较分析,随后通过对DNA序列的大量解析,可以在两段相应的DNA序列边缘观察到基因组的嵌合结构[7]。嵌合基因结构可以通过高频率水平基因转移(horizontal gene transfer, HGT)得到,每个基因组可以被看作一个可交换的模块(这些模块可以是单个的基因或者基因群,也可以是单个蛋白的功能区域),从而使得噬菌体基因组具有更多的机会去获得所需要的基因库[8]。例如,Amgarten等[9]从堆肥中分离得到3株假单胞菌噬菌体,对其进行了基因组测序、组装和注释;其中ZC03与ZC08两株噬菌体的基因组比对结果显示:它们的基因组序列相似性高达95%;基因的多重比对显示这些基因之间存在共线性关系,为它们属于远源同源基因提供了证据,同时呈现出镶嵌型基因组特点。Pauline等[10]从铜绿假单胞菌转座噬菌体中分离得到噬菌体D3112,并对D3112的全基因组进行了测序;基因序列与Mu样噬菌体和前噬菌体具有48%的相似性,同时尾蛋白基因与大肠杆菌(Escherichia coli)的原噬菌体和金黄色葡萄球菌(Staphylococcus aureus)噬菌体Phi12中的原噬菌体序列高度相似;系统发育分析揭示了假单胞噬菌体D3112基因组的高度镶嵌结构,并证明了遗传物质的水平交换在噬菌体进化中发挥了重要作用。嵌合基因结构是噬菌体在漫长的进化中不断选择或者淘汰重组子的结果,噬菌体基因组的嵌合结构有益于研究基因的进化历程。
(2)假单胞菌噬菌体具有比宿主菌更低的G+C含量,基因组之间遗传差异较大。Denver等[11]对130个假单胞菌噬菌体基因组进行测序分析,发现这些假单胞菌噬菌体基因组具有多种G+C含量,最低的G+C含量为37%。Wittmann等[12]从废水处理厂分离得到两株噬菌体JWAlpha和JWDelta,基因组测序分析发现JWAlpha和JWDelta的G+C含量分别为54.4%和54.2%,明显低于宿主菌的G+C含量(65%~66%);其他N4样噬菌体基因组也存在G+C含量明显低于宿主菌的现象。铜绿假单胞菌噬菌体D3112的G+C平均含量为64.34%,而其宿主铜绿假单胞菌的G+C含量为67% (图1)[10],两者之间含量非常接近;D3112基因组中ORF1与ORF2的G+C含量更低,分别为56%和53%,还有一些ORF的G+C含量在60%左右;表明这些基因来自于低G+C含量的生物,揭示了噬菌体从其他细菌宿主中转移的现象,导致假单胞菌噬菌体的基因组在遗传进化过程中差异性较大。假单胞菌噬菌体基因组中低水平的G+C含量,也是宿主菌长期适应不同环境而产生的一种进化。
图1
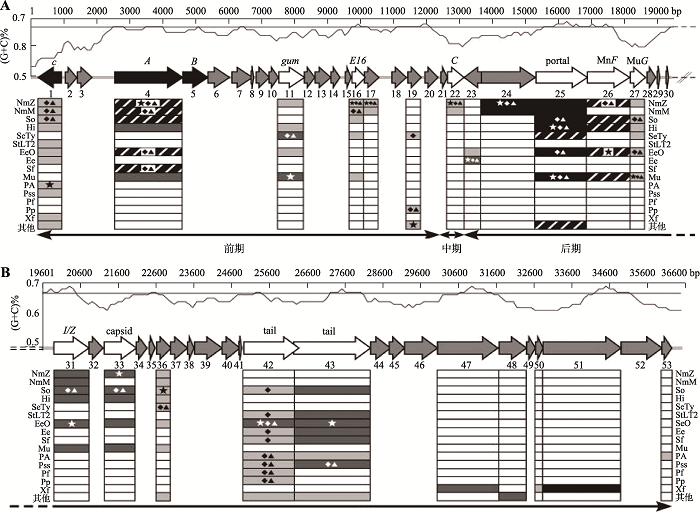 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1铜绿假单胞菌与噬菌体D3112的遗传图谱
A:铜绿假单胞菌遗传图;B:噬菌体D3112遗传图。遗传图谱上方是G+C含量的图形表示,与基因图谱按比例绘制。中间部分显示了D3112预测的ORF及其方向,由实心箭头表示。每个箭头下方的数字对应于ORF编号,每个箭头上方的名称标识具有高度相似性的已知蛋白质。黑色箭头A、B、c表示3个先前测序的基因,白色箭头表示与已知蛋白质相似的ORF,灰色箭头表示未知的ORF。左侧方框表示与噬菌体D3112宿主高度相似的细菌,阴影表示不同程度的相似性。NmZ:脑膜炎奈瑟球菌Z2491;NmM:脑膜炎奈瑟球菌MC58;So:S. oneidensis;Hi:流感嗜血杆菌;SeTy:肠炎沙门氏菌;StLT2:肠炎链球菌LT2;EcO:大肠杆菌O157:H7;Ec:大肠杆菌;Sf:弗氏志贺菌;Mu:肠噬菌体Mu;PA:铜绿假单胞菌;Pss:丁香属;Pf:荧光假单胞菌;Pp:恶臭假单胞菌;Xf:X. fastidiosa。框内的星号表示基于期望值与D3112 ORF具有最高相似性的物种,菱形显示通过贝叶斯方法与D3112 ORF最密切相关的物种,而三角形显示通过邻点连接法与D3112 ORF最密切相关的物种。
Fig. 1Genetic maps of Pseudomonas aeruginosa and bacteriophage D3112
(3)假单胞菌噬菌体基因组密度大、非编码区域少。假单胞菌噬菌体基因组中,平均每600 bp长度含有一个假定基因[13]。从基因密度来看:编码区堆积紧密,基因之间的间隙少,编码潜力很高。例如,假单胞菌噬菌体PaP1基因组由91,715个碱基对(bp)组成,G+C含量为49.36%。基因组中总共预测到541个ORF,只有约13.8%属于非编码区[14]。从噬菌体PaP1 (GenBank ID:HQ832595)的基因组注释结果来看(图2),由于ORF之间紧密地结合在一起,导致基因之间的间隔很小,显示编码区的基因密度很高。18株铜绿假单胞菌噬菌体基因组图谱分析表明:基因组中编码区排列紧凑,功能基因之间间隙较少每个铜绿假单胞菌噬菌体基因组的平均基因编码潜力为93.0%,每1 kb约有1.5个基因[15]。对于PB1样假单胞菌噬菌体,其基因组的密度很大,完整基因组中包含九个重叠群,长度范围从65,762 bp到66,283 bp,每个基因组包含了85~89个注释基因[16]。假单胞菌噬菌体PFP1的基因组总长度为40,914 bp,包含45个预测的ORF (图3),ORF长度在117~ 3996 bp之间,功能区之间排列紧凑,平均约900 bp序列含有一个基因;说明噬菌体对基因组的利用很充分,非编码区域很少[17]。假单胞菌噬菌体PaP3的基因组中平均约600 bp分布一个基因,而在真核生物大麦基因组中平均每20 kb序列含有一个基因;拟南芥(Arabidopsis thaliana)基因组中平均每4.8 kb片段含有一个基因;人类基因组共30亿碱基对,编码蛋白的碱基仅约3~5万个左右,即平均约60 kb序列存在一个基因[18]。因此,物种进化越高级,其基因密度越小,非编码序列越多。例如,人类基因组约95%的序列都属于非编码序列,表明噬菌体作为古老的生物在基因序列上具有更强的利用能力。
图2
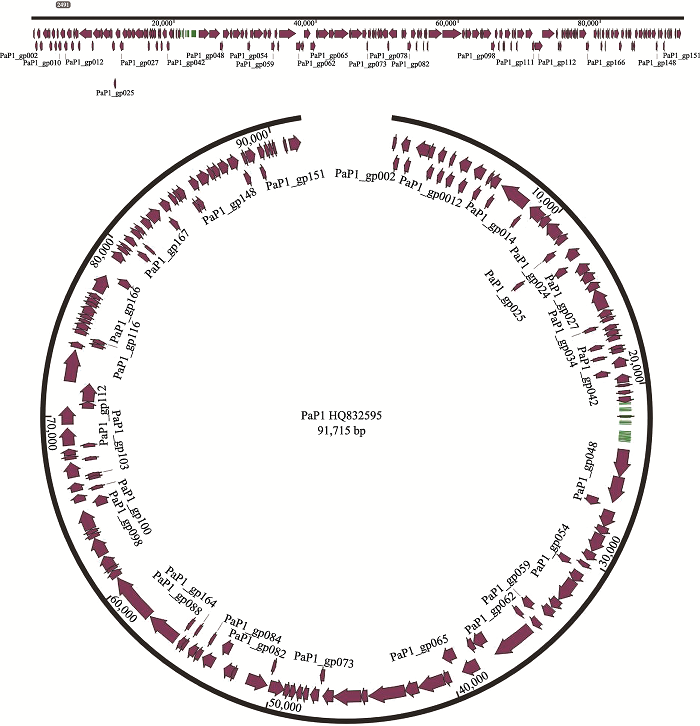 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2假单胞菌噬菌体PaP1基因组 基因组细节可参考文献[14]。
Fig. 2Genome of Pseudomonas phage PaP1
图3
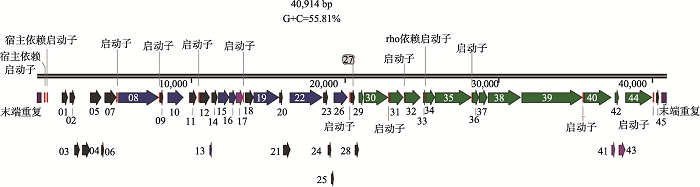 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3假单胞菌噬菌体PFP1的基因组图
图上的数字代
Fig. 3Genomic map of the Pseudomonas phage PFP1
2 假单胞菌噬菌体基因组遗传多样性
噬菌体是迄今为止生物圈中最丰富的生命实体,包含大量的、新的遗传信息。假单胞菌噬菌体种群具有显著的遗传多样性,然而这些噬菌体基因组之间的序列相似性却很小[19]。例如,新发现的一株铜绿假单胞菌噬菌体Podovirius04,其基因组序列与目前已报道的Podovirius噬菌体相比,相似性都很低[20]。Carson等[21]对6种铜绿假单胞菌基因组进行测序分析,并与已知的铜绿假单胞菌双链DNA噬菌体的基因组序列进行比较,表明这6种铜绿假单胞菌噬菌体基因组序列差异较大,相似性极低。Kwan等[15]对18株铜绿假单胞菌噬菌体基因组进行分析,揭示了编码蛋白质组相关基因的多样性;通过序列比较分析和ORF图谱分析表明:宿主菌同属,大多数噬菌体基因组之间相似性很低。但是,基因组测序也发现:相同宿主菌的噬菌体之间,基因组具有潜在相似性。例如,铜绿假单胞菌噬菌体ZC03和ZC08的宿主菌相同,均为铜绿假单胞菌;噬菌体ZC03和ZC08的基因组很相似,不但具有36个相似性极高的同源基团,还均携带特殊的tRNA基因[9]。铜绿假单胞菌噬菌体PA11与噬菌体04,基因组序列相似性为95%;噬菌体04基因组编码55种ORF,与噬菌体PA11中的48个蛋白质基因有40%~100%的相似性,与另外7个蛋白质基因有27%~40%的相似性[22]。徐彬等[23]从环境污水中分离到一株铜绿假单胞菌噬菌体D204,基因组测序和比对发现D204基因组与铜绿假单胞菌噬菌体PA11的基因组具有83%的序列相似性;在D204基因组片段上,一些特殊位置的编码基因还与铜绿假单胞菌噬菌体PaeP、PaP3及PaP4的编码基因类似。假单胞菌噬菌体基因组之间具有多样性,同时各基因组之间又存在潜在相似性,从而导致假单胞菌噬菌体种群之间具有显著的遗传多样性。3 假单胞菌噬菌体基因组中功能基因分析
假单胞菌噬菌体基因组上常见的功能基因包括编码裂解蛋白、调节蛋白、尾部蛋白、衣壳蛋白、DNA聚合酶和DNA解旋酶等。随着对假单胞菌噬菌体基因组研究的不断深入,可以把功能基因分为以下几类:第一类:与噬菌体DNA复制相关的酶。噬菌体C11的基因组中发现了与噬菌体DNA复制有关的酶,包括ORF 101、ORF 102、ORF 109、ORF 123、ORF 125、ORF 126。0RF 101和ORF 102分别编码DNA解旋酶(DNA primase/helicase)和DNA聚合酶(DNA polymerase)[19]。噬菌体ZC01与YuA具有直系同源性。因此DNA解旋酶、RecD样蛋白脱氧脲苷酸羟甲基转移酶和DNA聚合酶A存在于多个直系同源基因中[9,24]。
第二类:与噬菌体核衣壳蛋白形成相关的酶。如在噬菌体Ardmore中,ORF115 (卷尺蛋白)可决定噬菌体尾部的长度,Che8噬菌体的GP116与TM4噬菌体的GP119和Che9d噬菌体的GP119,是尾部亚基的组成部分。此外,还有与噬菌体DNA包装相关的基因,如Ardmore噬菌体的ORF11和ORF12,其中ORF12编码末端酶[25]。
第三类:转录调节功能因子。铜绿假单胞菌基因组中的前噬菌体预测显示,前噬菌体存在于少数假单胞菌菌株中,如NCGM2.S1和VRFPA04噬菌体。在前噬菌体中鉴定出了编码毒力相关蛋白和转录调节因子的几种基因,如ACG 06-06430[26]。
除了上述常见的功能基因外,假单胞菌噬菌体基因组上还存在抗CRISPR-Cas基因。CRISPR-Cas系统是原核生物中最广泛的噬菌体抗性机制之一,它是成簇的、有规律间隔的短回文重复序列,可以对外来入侵的噬菌体基因进行切割[27,28]。April等[29]发现铜绿假单胞菌噬菌体基因组中存在抗CRISPR基因,编码铜绿假单胞菌IF CRISPR-CAS系统的蛋白抑制剂,为进一步探究噬菌体与宿主菌之间对抗性共进化机制提供了新的研究思路。
假单胞菌噬菌体影响假单胞菌致病性有3个关键因素:生物膜的形成、毒力和抗生素抗性。据报道溶源性噬菌体Pf4在释放细胞外DNA(eDNA)过程中扮演重要作用,该酶稳定了生物膜基质并降低其对抗生素的渗透性。另一种溶源性噬菌体D3112可以阻止细菌菌毛的形成,导致细菌粘附力下降,抑制生物膜形成[30]。相反,噬菌体抗性突变体通常是由于假单胞菌的脂多糖(LPS)的改变引起的,这些与生物膜的形成和毒力的变化有关。噬菌体还可以通过产生特定的毒力因子来增加毒力,例如外毒素(如霍乱毒素)和多糖(如藻酸盐);因此噬菌体对环境假单胞菌的致病性具有复杂的影响作用[31,32]。假单胞菌噬菌体基因组包含许多未确定的功能基因,并且大部分由功能基因预测的蛋白质生物学功能也无法确定,目前尚无能够进行匹配的数据库,因此假单胞菌噬菌体基因组中有很多尚未发现的新功能基因。
4 结语与展望
近年来,噬菌体基因组学研究取得了巨大进展。高通量测序技术的快速发展为获得更多新噬菌体基因组带来了极大的便利,促进了噬菌体基因组学相关研究的不断发展。噬菌体是宿主遗传进化的主要动力,直接影响致病性假单胞菌宿主在生态环境中的作用[33]。假单胞菌及其噬菌体同时也是研究病毒与宿主之间相互作用分子机制的理想模型。对噬菌体与假单胞菌相互作用的研究体现了这些生物实体在生物技术应用方面的潜力。噬菌体蛋白在感染周期的每个阶段都具有潜在的生物技术应用,病毒相关宿主受体的特异性使它们成为快速鉴定病原体的理想工具。一些噬菌体转录或复制相关的酶,也已经成为分子实验室中常用的工具[34]。假单胞菌噬菌体还可以作为控制细菌感染的新手段,将假单胞菌噬菌体作为抗生素替代品或者直接利用噬菌体产生的裂解酶作为抗菌剂。此外,假单胞菌噬菌体基因组功能区的注释,有利于形成一套新的基因工具,为合成生物学家提供了新思路。随着对假单胞菌噬菌体基因组的不断研究,利用其与宿主菌的共同进化作用开发新型药物,为抗生素耐药菌的治疗带来希望。因此,开展假单胞菌噬菌体基因组的研究将具有非常重要的现实意义,不仅有助于拓展我们对噬菌体生物学的了解,而且有助于优化并利用这些宝贵的生物有机体。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/j.1574-6976.2011.00269.xURLPMID:21361996 [本文引用: 1]

Members of the genus Pseudomonas inhabit a wide variety of environments, which is reflected in their versatile metabolic capacity and broad potential for adaptation to fluctuating environmental conditions. Here, we examine and compare the genomes of a range of Pseudomonas spp. encompassing plant, insect and human pathogens, and environmental saprophytes. In addition to a large number of allelic differences of common genes that confer regulatory and metabolic flexibility, genome analysis suggests that many other factors contribute to the diversity and adaptability of Pseudomonas spp. Horizontal gene transfer has impacted the capability of pathogenic Pseudomonas spp. in terms of disease severity (Pseudomonas aeruginosa) and specificity (Pseudomonas syringae). Genome rearrangements likely contribute to adaptation, and a considerable complement of unique genes undoubtedly contributes to strain- and species-specific activities by as yet unknown mechanisms. Because of the lack of conserved phenotypic differences, the classification of the genus has long been contentious. DNA hybridization and genome-based analyses show close relationships among members of P. aeruginosa, but that isolates within the Pseudomonas fluorescens and P. syringae species are less closely related and may constitute different species. Collectively, genome sequences of Pseudomonas spp. have provided insights into pathogenesis and the genetic basis for diversity and adaptation.
[本文引用: 1]
DOI:10.2174/0929867324666170413100136URLPMID:28412903 [本文引用: 1]
Bacteriophages (phages or bacterial viruses) are the most abundant biological entities in our planet; their influence reaches far beyond the microorganisms they parasitize. Phages are present in every environment and shape up every bacterial population in both active and passive ways. They participate in the circulation of organic matter and drive the evolution of microorganisms by horizontal gene transfer at unprecedented scales. The mass flow of genetic information in the microbial world influences the biosphere and poses challenges for science and medicine. The genetic flow, however, depends on the fate of the viral DNA injected into the bacterial cell. The archetypal notion of phages only engaging in predatorprey relationships is slowly fading. Because of their varied development cycles, environmental conditions, and the diversity of microorganisms they parasitize, phages form a dense and highly complex web of dependencies, which has important consequences for life on Earth. The sophisticated phage-bacteria interplay includes both aggressive action (bacterial lysis) and
DOI:10.1038/s41586-019-1662-9URLPMID:31645729 [本文引用: 1]
About half of all bacteria carry genes for CRISPR-Cas adaptive immune systems(1), which provide immunological memory by inserting short DNA sequences from phage and other parasitic DNA elements into CRISPR loci on the host genome(2). Whereas CRISPR loci evolve rapidly in natural environments(3,4), bacterial species typically evolve phage resistance by the mutation or loss of phage receptors under laboratory conditions(5,6). Here we report how this discrepancy may in part be explained by differences in the biotic complexity of in vitro and natural environments(7,8). Specifically, by using the opportunistic pathogen Pseudomonas aeruginosa and its phage DMS3vir, we show that coexistence with other human pathogens amplifies the fitness trade-offs associated with the mutation of phage receptors, and therefore tips the balance in favour of the evolution of CRISPR-based resistance. We also demonstrate that this has important knock-on effects for the virulence of P. aeruginosa, which became attenuated only if the bacteria evolved surface-based resistance. Our data reveal that the biotic complexity of microbial communities in natural environments is an important driver of the evolution of CRISPR-Cas adaptive immunity, with key implications for bacterial fitness and virulence.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.virol.2008.04.038URLPMID:18519145 [本文引用: 1]
The virulent Pseudomonas aeruginosa bacteriophage LUZ24 (45,625 bp) was isolated from hospital sewage. It belongs to the family of the Podoviridae, and carries a bidirectionally transcribed dsDNA genome delineated by two direct terminal repeats of 184 bp. In vitro transcriptional analysis identified seven sigma(70) promoters, revealing a bias towards stronger promoter strength in the late genomic region. Reverse transcription demonstrated in vivo splicing of a 668 bp Group I intron embedded inside the DNA polymerase gene. Using mass spectrometry, nine structural proteins were identified as part of the phage particle. The lytic characteristics of LUZ24 are evaluated against its genomic content, which displays an overall 71% sequence similarity to the temperate phage PaP3.
DOI:10.1128/JB.01441-07URLPMID:18065532 [本文引用: 1]
Pseudomonas aeruginosa phage YuA (Siphoviridae) was isolated from a pond near Moscow, Russia. It has an elongated head, encapsulating a circularly permuted genome of 58,663 bp, and a flexible, noncontractile tail, which is terminally and subterminally decorated with short fibers. The YuA genome is neither Mu- nor lambda-like and encodes 78 gene products that cluster in three major regions involved in (i) DNA metabolism and replication, (ii) host interaction, and (iii) phage particle formation and host lysis. At the protein level, YuA displays significant homology with phages M6, phiJL001, 73, B3, DMS3, and D3112. Eighteen YuA proteins were identified as part of the phage particle by mass spectrometry analysis. Five different bacterial promoters were experimentally identified using a promoter trap assay, three of which have a sigma54-specific binding site and regulate transcription in the genome region involved in phage particle formation and host lysis. The dependency of these promoters on the host sigma54 factor was confirmed by analysis of an rpoN mutant strain of P. aeruginosa PAO1. At the DNA level, YuA is 91% identical to the recently (July 2007) annotated phage M6 of the Lindberg typing set. Despite this level of DNA homology throughout the genome, both phages combined have 15 unique genes that do not occur in the other phage. The genome organization of both phages differs substantially from those of the other known Pseudomonas-infecting Siphoviridae, delineating them as a distinct genus within this family.
DOI:10.1371/journal.pgen.0020092URLPMID:16789831 [本文引用: 1]
Bacteriophages are the most abundant forms of life in the biosphere and carry genomes characterized by high genetic diversity and mosaic architectures. The complete sequences of 30 mycobacteriophage genomes show them collectively to encode 101 tRNAs, three tmRNAs, and 3,357 proteins belonging to 1,536
DOI:10.1186/s12864-017-3729-zURLPMID:28472930 [本文引用: 3]
BACKGROUND: Among viruses, bacteriophages are a group of special interest due to their capacity of infecting bacteria that are important for biotechnology and human health. Composting is a microbial-driven process in which complex organic matter is converted into humus-like substances. In thermophilic composting, the degradation activity is carried out primarily by bacteria and little is known about the presence and role of bacteriophages in this process. RESULTS: Using Pseudomonas aeruginosa as host, we isolated three new phages from a composting operation at the Sao Paulo Zoo Park (Brazil). One of the isolated phages is similar to Pseudomonas phage Ab18 and belongs to the Siphoviridae YuA-like viral genus. The other two isolated phages are similar to each other and present genomes sharing low similarity with phage genomes in public databases; we therefore hypothesize that they belong to a new genus in the Podoviridae family. Detailed genomic descriptions and comparisons of the three phages are presented, as well as two new clusters of phage genomes in the Viral Orthologous Clusters database of large DNA viruses. We found sequences encoding homing endonucleases that disrupt a putative ribonucleotide reductase gene and an RNA polymerase subunit 2 gene in two of the phages. These findings provide insights about the evolution of two-subunits RNA polymerases and the possible role of homing endonucleases in this process. Infection tests on 30 different strains of bacteria reveal a narrow host range for the three phages, restricted to P. aeruginosa PA14 and three other P. aeruginosa clinical isolates. Biofilm dissolution assays suggest that these phages could be promising antimicrobial agents against P. aeruginosa PA14 infections. Analyses on composting metagenomic and metatranscriptomic data indicate association between abundance variations in both phage and host populations in the environment. CONCLUSION: The results about the newly discovered and described phages contribute to the understanding of tailed bacteriophage diversity, evolution, and role in the complex composting environment.
DOI:10.1128/jb.186.2.400-410.2004URLPMID:14702309 [本文引用: 2]
Bacteriophage D3112 represents one of two distinct groups of transposable phage found in the clinically relevant, opportunistic pathogen Pseudomonas aeruginosa. To further our understanding of transposable phage in P. aeruginosa, we have sequenced the complete genome of D3112. The genome is 37,611 bp, with an overall G+C content of 65%. We have identified 53 potential open reading frames, including three genes (the c repressor gene and early genes A and B) that have been previously characterized and sequenced. The organization of the putative coding regions corresponds to published genetic and transcriptional maps and is very similar to that of enterobacteriophage Mu. In contrast, the International Committee on Taxonomy of Viruses has classified D3112 as a lambda-like phage on the basis of its morphology. Similarity-based analyses identified 27 open reading frames with significant matches to proteins in the NCBI databases. Forty-eight percent of these were similar to Mu-like phage and prophage sequences, including proteins responsible for transposition, transcriptional regulation, virion morphogenesis, and capsid formation. The tail proteins were highly similar to prophage sequences in Escherichia coli and phage Phi12 from Staphylococcus aureus, while proteins at the right end were highly similar to proteins in Xylella fastidiosa. We performed phylogenetic analyses to understand the evolutionary relationships of D3112 with respect to Mu-like versus lambda-like bacteriophages. Different results were obtained from similarity-based versus phylogenetic analyses in some instances. Overall, our findings reveal a highly mosaic structure and suggest that extensive horizontal exchange of genetic material played an important role in the evolution of D3112.
DOI:10.3389/fmicb.2018.01456URLPMID:30022972 [本文引用: 1]
Bacteria of the genus Pseudomonas are genetically diverse and ubiquitous in the environment. Like other bacteria, those of the genus Pseudomonas are susceptible to bacteriophages which can significantly affect their host in many ways, ranging from cell lysis to major changes in morphology and virulence. Insights into phage genomes, evolution, and functional relationships with their hosts have the potential to contribute to a broader understanding of Pseudomonas biology, and the development of novel phage therapy strategies. Here we provide a broad-based comparative and evolutionary analysis of 130 complete Pseudomonas phage genome sequences available in online databases. We discovered extensive variation in genome size (ranging from 3 to 316 kb), G + C percentage (ranging from 37 to 66%), and overall gene content (ranging from 81-96% of genome space). Based on overall nucleotide similarity and the numbers of shared gene products, 100 out of 130 genome sequences were grouped into 12 different clusters; 30 were characterized as singletons, which do not have close relationships with other phage genomes. For 5/12 clusters, constituent phage members originated from two or more different Pseudomonas host species, suggesting that phage in these clusters can traverse bacterial species boundaries. An analysis of CRISPR spacers in Pseudomonas bacterial genome sequences supported this finding. Substantial diversity was revealed in analyses of phage gene families; out of 4,462 total families, the largest had only 39 members and there were 2,992 families with only one member. An evolutionary analysis of 72 phage gene families, based on patterns of nucleotide diversity at non-synonymous and synonymous sites, revealed strong and consistent signals for purifying selection. Our study revealed highly diverse and dynamic Pseudomonas phage genomes, and evidence for a dominant role of purifying selection in shaping the evolution of genes encoded in them.
DOI:10.1186/1743-422X-11-2URL [本文引用: 1]
DOI:10.1186/1743-422X-8-142URLPMID:21439081 [本文引用: 1]
BACKGROUND: Phage varphiIBB-PF7A is a T7-like bacteriophage capable of infecting several Pseudomonas fluorescens dairy isolates and is extremely efficient in lysing this bacterium even when growing in biofilms attached to surfaces. This work describes the complete genome sequence of this phage. RESULTS: The genome consists of a linear double-stranded DNA of 40,973 bp, with 985 bp long direct terminal repeats and a GC content of approximately 56%. There are 52 open reading frames which occupy 94.6% of the genome ranging from 137 to 3995 nucleotides. Twenty eight (46.7%) of the proteins encoded by this virus exhibit sequence similarity to coliphage T7 proteins while 34 (81.0%) are similar to proteins of Pseudomonas phage gh-1. CONCLUSIONS: That this phage is closely related to Pseudomonas putida phage gh-1 and coliphage T7 places it in the
DOI:10.1371/journal.pone.0062933URLPMID:23675441 [本文引用: 2]
We isolated and characterized a new Pseudomonas aeruginosa myovirus named PaP1. The morphology of this phage was visualized by electron microscopy and its genome sequence and ends were determined. Finally, genomic and proteomic analyses were performed. PaP1 has an icosahedral head with an apex diameter of 68-70 nm and a contractile tail with a length of 138-140 nm. The PaP1 genome is a linear dsDNA molecule containing 91,715 base pairs (bp) with a G+C content of 49.36% and 12 tRNA genes. A strategy to identify the genome ends of PaP1 was designed. The genome has a 1190 bp terminal redundancy. PaP1 has 157 open reading frames (ORFs). Of these, 143 proteins are homologs of known proteins, but only 38 could be functionally identified. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and high-performance liquid chromatography-mass spectrometry allowed identification of 12 ORFs as structural protein coding genes within the PaP1 genome. Comparative genomic analysis indicated that the Pseudomonas aeruginosa phage PaP1, JG004, PAK_P1 and vB_PaeM_C2-10_Ab1 share great similarity. Besides their similar biological characteristics, the phages contain 123 core genes and have very close phylogenetic relationships, which distinguish them from other known phage genera. We therefore propose that these four phages be classified as PaP1-like phages, a new phage genus of Myoviridae that infects Pseudomonas aeruginosa.
DOI:10.1128/JB.188.3.1184-1187.2006URLPMID:16428425 [本文引用: 2]
A genomic analysis of 18 P. aeruginosa phages, including nine newly sequenced DNA genomes, indicates a tremendous reservoir of proteome diversity, with 55% of open reading frames (ORFs) being novel. Comparative sequence analysis and ORF map organization revealed that most of the phages analyzed displayed little relationship to each other.
DOI:10.3390/v10060331URL [本文引用: 1]
DOI:10.1007/s00705-018-3979-3URL [本文引用: 1]
DOI:10.1186/1471-2164-15-803URL [本文引用: 1]
DOI:10.1002/biot.v14.1URL [本文引用: 2]
DOI:10.1007/s00705-018-3866-yURL [本文引用: 1]
Siphoviridae Phages.
[本文引用: 1]
DOI:10.3389/fmicb.2018.02561URLPMID:30410478 [本文引用: 1]
Phages have attracted a renewed interest as alternative to chemical antibiotics. Although the number of phages is 10-fold higher than that of bacteria, the number of genomically characterized phages is far less than that of bacteria. In this study, phage TC6, a novel lytic virus of Pseudomonas aeruginosa, was isolated and characterized. TC6 consists of an icosahedral head with a diameter of approximately 54 nm and a short tail with a length of about 17 nm, which are characteristics of the family Podoviridae. TC6 can lyse 86 out of 233 clinically isolated P. aeruginosa strains, thus showing application potentials for phage therapy. The linear double-stranded genomic DNA of TC6 consisted of 49796 base pairs and was predicted to contain 71 protein-coding genes. A total of 11 TC6 structural proteins were identified by mass spectrometry. Comparative analysis revealed that the P. aeruginosa phages TC6, O4, PA11, and IME180 shared high similarity at DNA sequence and proteome levels, among which PA11 was the first phage discovered and published. Meanwhile, these phages contain 54 core genes and have very close phylogenetic relationships, which distinguish them from other known phage genera. We therefore proposed that these four phages can be classified as Pa11virus, comprising a new phage genus of Podoviridae that infects Pseudomonas spp. The results of this work promoted our understanding of phage biology, classification, and diversity.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s12223-016-0471-xURL [本文引用: 1]
DOI:10.1186/s12864-017-3842-zURLPMID:28606056 [本文引用: 1]
BACKGROUND: Pseudomonas aeruginosa ATCC 27853 was isolated from a hospital blood specimen in 1971 and has been widely used as a model strain to survey antibiotics susceptibilities, biofilm development, and metabolic activities of Pseudomonas spp.. Although four draft genomes of P. aeruginosa ATCC 27853 have been sequenced, the complete genome of this strain is still lacking, hindering a comprehensive understanding of its physiology and functional genome. RESULTS: Here we sequenced and assembled the complete genome of P. aeruginosa ATCC 27853 using the Pacific Biosciences SMRT (PacBio) technology and Illumina sequencing platform. We found that accessory genes of ATCC 27853 including prophages and genomic islands (GIs) mainly contribute to the difference between P. aeruginosa ATCC 27853 and other P. aeruginosa strains. Seven prophages were identified within the genome of P. aeruginosa ATCC 27853. Of the predicted 25 GIs, three contain genes that encode monoxoygenases, dioxygenases and hydrolases that could be involved in the metabolism of aromatic compounds. Surveying virulence-related genes revealed that a series of genes that encode the B-band O-antigen of LPS are lacking in ATCC 27853. Distinctive SNPs in genes of cellular adhesion proteins such as type IV pili and flagella biosynthesis were also observed in this strain. Colony morphology analysis confirmed an enhanced biofilm formation capability of ATCC 27853 on solid agar surface compared to Pseudomonas aeruginosa PAO1. We then performed transcriptome analysis of ATCC 27853 and PAO1 using RNA-seq and compared the expression of orthologous genes to understand the functional genome and the genomic details underlying the distinctive colony morphogenesis. These analyses revealed an increased expression of genes involved in cellular adhesion and biofilm maturation such as type IV pili, exopolysaccharide and electron transport chain components in ATCC 27853 compared with PAO1. In addition, distinctive expression profiles of the virulence genes lecA, lasB, quorum sensing regulators LasI/R, and the type I, III and VI secretion systems were observed in the two strains. CONCLUSIONS: The complete genome sequence of P. aeruginosa ATCC 27853 reveals the comprehensive genetic background of the strain, and provides genetic basis for several interesting findings about the functions of surface associated proteins, prophages, and genomic islands. Comparative transcriptome analysis of P. aeruginosa ATCC 27853 and PAO1 revealed several classes of differentially expressed genes in the two strains, underlying the genetic and molecular details of several known and yet to be explored morphological and physiological potentials of P. aeruginosa ATCC 27853.
DOI:10.1038/nrmicro.2017.61URLPMID:28649138 [本文引用: 1]
Species in the genus Pseudomonas thrive in a diverse set of ecological niches and include crucial pathogens, such as the human pathogen Pseudomonas aeruginosa and the plant pathogen Pseudomonas syringae. The bacteriophages that infect Pseudomonas spp. mirror the widespread and diverse nature of their hosts. Therefore, Pseudomonas spp. and their phages are an ideal system to study the molecular mechanisms that govern virus-host interactions. Furthermore, phages are principal catalysts of host evolution and diversity, which directly affects the ecological roles of environmental and pathogenic Pseudomonas spp. Understanding these interactions not only provides novel insights into phage biology but also advances the development of phage therapy, phage-derived antimicrobial strategies and innovative biotechnological tools that may be derived from phage-bacteria interactions.
[本文引用: 1]
[本文引用: 1]
DOI:10.1128/mBio.00896-14URLPMID:24736222 [本文引用: 1]
CRISPR-Cas systems are one of the most widespread phage resistance mechanisms in prokaryotes. Our lab recently identified the first examples of phage-borne anti-CRISPR genes that encode protein inhibitors of the type I-F CRISPR-Cas system of Pseudomonas aeruginosa. A key question arising from this work was whether there are other types of anti-CRISPR genes. In the current work, we address this question by demonstrating that some of the same phages carrying type I-F anti-CRISPR genes also possess genes that mediate inhibition of the type I-E CRISPR-Cas system of P. aeruginosa. We have discovered four distinct families of these type I-E anti-CRISPR genes. These genes do not inhibit the type I-F CRISPR-Cas system of P. aeruginosa or the type I-E system of Escherichia coli. Type I-E and I-F anti-CRISPR genes are located at the same position in the genomes of a large group of related P. aeruginosa phages, yet they are found in a variety of combinations and arrangements. We have also identified functional anti-CRISPR genes within nonprophage Pseudomonas genomic regions that are likely mobile genetic elements. This work emphasizes the potential importance of anti-CRISPR genes in phage evolution and lateral gene transfer and supports the hypothesis that more undiscovered families of anti-CRISPR genes exist. Finally, we provide the first demonstration that the type I-E CRISPR-Cas system of P. aeruginosa is naturally active without genetic manipulation, which contrasts with E. coli and other previously characterized I-E systems. IMPORTANCE The CRISPR-Cas system is an adaptive immune system possessed by the majority of prokaryotic organisms to combat potentially harmful foreign genetic elements. This study reports the discovery of bacteriophage-encoded anti-CRISPR genes that mediate inhibition of a well-studied subtype of CRISPR-Cas system. The four families of anti-CRISPR genes described here, which comprise only the second group of anti-CRISPR genes to be identified, encode small proteins that bear no sequence similarity to previously studied phage or bacterial proteins. Anti-CRISPR genes represent a newly discovered and intriguing facet of the ongoing evolutionary competition between phages and their bacterial hosts.
DOI:10.1111/mmi.14231URLPMID:30811056 [本文引用: 1]
Bacteriophages (phages) are the most abundant entities in nature, yet little is known about their capacity to acquire new hosts and invade new niches. By exploiting the Gram-positive soil bacterium Bacillus subtilis (B. subtilis) and its lytic phage SPO1 as a model, we followed the coevolution of bacteria and phages. After infection, phage-resistant bacteria were readily isolated. These bacteria were defective in production of glycosylated wall teichoic acid (WTA) polymers that served as SPO1 receptor. Subsequently, a SPO1 mutant phage that could infect the resistant bacteria evolved. The emerging phage contained mutations in two genes, encoding the baseplate and fibers required for host attachment. Remarkably, the mutant phage gained the capacity to infect non-host Bacillus species that are not infected by the wild-type phage. We provide evidence that the evolved phage lost its dependency on the species-specific glycosylation pattern of WTA polymers. Instead, the mutant phage gained the capacity to directly adhere to the WTA backbone, conserved among different species, thereby crossing the species barrier.
DOI:10.3390/v11020188URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.molcel.2016.01.030URLPMID:26949040 [本文引用: 1]
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute an adaptive immune system in prokaryotes. The system preserves memories of prior infections by integrating short segments of foreign DNA, termed spacers, into the CRISPR array in a process termed adaptation. During the past 3 years, significant progress has been made on the genetic requirements and molecular mechanisms of adaptation. Here we review these recent advances, with a focus on the experimental approaches that have been developed, the insights they generated, and a proposed mechanism for self- versus non-self-discrimination during the process of spacer selection. We further describe the regulation of adaptation and the protein players involved in this fascinating process that allows bacteria and archaea to harbor adaptive immunity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}