 ,1, 徐讯,1,2,4, 魏晓锋,1
,1, 徐讯,1,2,4, 魏晓锋,1CNGBdb: China National GeneBank DataBase
Fengzhen Chen1, Lijin You1, Fan Yang1, Lina Wang1, Xueqin Guo1, Fei Gao1, Cong Hua1, Cong Tan1, Lin Fang2, Riqiang Shan3, Wenjun Zeng1, Bo Wang1, Ren Wang,1, Xun Xu,1,2,4, Xiaofeng Wei,1通讯作者: 徐讯,博士,研究员,研究方向:基因组学、生物信息学等。E-mail:xuxun@genomics.cn王韧,博士,研究员,研究方向:农学。E-mail:wangren@cngb.org魏晓锋,本科,研究方向:生物大数据。E-mail:weixiaofeng@cngb.org
编委: 胡松年
收稿日期:2020-03-23修回日期:2020-05-23网络出版日期:2020-08-20
| 基金资助: |
Received:2020-03-23Revised:2020-05-23Online:2020-08-20
| Fund supported: |
作者简介 About authors
陈凤珍,本科,研究方向:生物大数据。E-mail:

摘要
国家基因库生命大数据平台(China National GeneBank DataBase, CNGBdb)是一个致力于生命科学多组学数据归档和开放共享的数据库平台,是深圳国家基因库的核心功能“三库两平台”中生物信息数据库的对外服务平台,拥有深圳国家基因库丰富的样本资源、数据资源、合作项目资源和强大的数据计算和分析能力等优势。生命科学研究已经进入到了一个以高通量多组学数据为基础的大数据时代,迫切需要加强国际合作和信息共享。随着中国经济的发展和在生命科学研究领域的研究项目投入力度的加大,需要建立相关的生命大数据归档和共享的平台, 来促进我国生命科学研究项目中生成的基因组学数据的系统管理、开放共享与合理利用。目前,CNGBdb主要提供生命科学研究相关的数据归档、知识搜索、数据管理、数据计算和数据服务等服务。其归档和共享的数据类型,主要包括项目、样本、实验、测序、组装、变异、序列等。截止2020年5月22号, CNGBdb已接受了全球生命科学科研工作者提交的研究项目达2176个,归档的基因组学数据量超过2221 TB。未来,CNGBdb将继续推动生命科学研究多组学数据的开放共享和产业应用,完善基因组学数据的归档和共享功能,提升其服务生命科学数据开放共享的能力。CNGBdb的网址是:https://db.cngb.org/。
关键词:
Abstract
China National GeneBank DataBase (CNGBdb) is a data platform aiming to systematically archiving and sharing of multi-omics data in life science. As the service portal of Bio-informatics Data Center of the core structure, namely, "Three Banks and Two Platforms" of China National GeneBank (CNGB), CNGBdb has the advantages of rich sample resources, data resources, cooperation projects, powerful data computation and analysis capabilities. With the advent of high throughput sequencing technologies, research in life science has entered the big data era, which is in the need of closer international cooperation and data sharing. With the development of China's economy and the increase of investment in life science research, we need to establish a national public platform for data archiving and sharing in life science to promote the systematic management, application and industrial utilization. Currently, CNGBdb can provide genomic data archiving, information search engines, data management and data analysis services. The data schema of CNGBdb has covered projects, samples, experiments, runs, assemblies, variations and sequences. Until May 22, 2020, CNGBdb has archived 2176 research projects and more than 2221 TB sequencing data submitted by researchers globally. In the future, CNGBdb will continue to be dedicated to promoting data sharing in life science research and improving the service capability. CNGBdb website is: https://db.cngb.org/.
Keywords:
PDF (573KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈凤珍, 游丽金, 杨帆, 王丽娜, 郭学芹, 高飞, 华聪, 谈聪, 方林, 单日强, 曾文君, 王博, 王韧, 徐讯, 魏晓锋. CNGBdb:国家基因库生命大数据平台. 遗传[J], 2020, 42(8): 799-809 doi:10.16288/j.yczz.20-080
Fengzhen Chen.
国家基因库生命大数据平台(China National Gene Bank DataBase, CNGBdb),是深圳国家基因库(China National GeneBank, CNGB)[1](以下简称“国家基因库”)核心功能“三库两平台”中生物信息数据库的对外服务平台。CNGB是以公益性、开放性、支撑性、引领性为宗旨,服务于国家战略的国家级创新科研及产业基础设施建设项目。其中,生物信息数据库致力于存储人类健康及生物多样性相关的数字化遗传资源,构建生物数据库及数据分析平台,实现数据存储、分析的贯穿,为后续科研及产业提供大数据源头保障。
随着基因组测序技术的飞速发展和测序成本的大幅下降,生命科学研究已经进入到了以高通量多组学技术为基础的大数据时代。为了解决人类生存面临的诸多问题,在过去的20多年里,世界各国相继实施了一些大规模的包括人类、动植物和微生物在内基因组测序项目,如千人基因组项目[2]、国际癌症基因组项目[3]、水稻参考基因组项目[4,5]、全球3000份水稻(Oryza sativa L.)种质资源测序项目[6]、全球超过2万份大麦种质资源测序项目[7]等。这些项目的实施促进了生命科学相关领域研究的快速发展,特别是人类遗传疾病致病机制发现和动植物分子设计育种应用等领域。迄今,世界范围有多达11,508种真核生物,245,875种原核生物和35,746种病毒样本经完成测序(依据2020年4月17日的NCBI已测序物种统计)。同时,还有大量的正在进行或即将开始的大型基因组测序项目,将导致基因组数据的爆炸式增长。
为了实现这些数据的安全保存和开放共享,全球生命科学研究组织相继建立了3个国际生物数据库,分别依托于美国国家生物信息中心(National Centre of Biotechnology, NCBI)的相关数据库[8],欧洲分子生物实验室(European Molecular Biology Laboratory)的欧洲生物信息研究所(European Bioinformatics Institute, EBI)系列数据库[9]和日本国家遗传研究所的DNA数据库(the DNA Database of Japan, DDBJ)[10]。这3个数据库的主要功能包括:(1)接收生物学领域研究人员提交在研究项目过程中生成的基因组测序数据,如测序仪下机数据,以及后续的生物信息分析结果数据,如组装的基因组序列和基因注释结果等;(2)维护覆盖人类、动植物及微生物的物种的参考基因组及基因注释信息,方便生物研究人员交流和使用。另外,还有大量由生物信息领域研究人员维护,同时由分子生物学领域研究人员逐一审核的高质量生物大分子知识数据库[11],如依托于瑞士生物信息研究所(Swiss Institute of Bioinformatics, SIB)的系列生物数据库[12]和由日本京都大学和东京大学联合开发的代谢途径/通路相关数据库(Kyoto Encyclopedia of Genes and Genomes, KEGG)[13]。其中,NCBI、EMBL-EBI和DDBJ的核酸数据库组成了国际核酸序列数据库联盟(International Nucleotide Sequence Database Collaboration, INSDC)[14],这3个核酸数据库之间,每日进行数据交换,在促进国际生物学数据的共享和利用方面发挥了重要作用。 但是国外这3个核酸数据库的目的,主要还是促进其本国生物研究机构之间生命大数据的共享和合作。当其他国家人员使用这些数据库时,还是存在诸多不方便的地方,如网络基础设施、国家与国家之间合作态度的倾向,以及数据库维护人员与科研人员在沟通语言和方式等方面的限制。
随着中国经济的快速发展,中国政府正在加大科学研究的资助力度,特别是生物医学和现代农业领域。在过去的20年里,中国也相继实施了一些重大的基因组学研究项目,如炎黄基因组项目[15]和大熊猫基因组项目[16]等,生成了海量的基因组测序数据和大量珍贵的项目研究成果。目前,由中国不同研究机构分别承担的基因组学项目生成的生命科学相关数据和结果,面临着“数据孤岛”、“数据主权”等实际问题。为了更好地服务于中国的科研人员,管理好中国在基因组学领域重大项目实施过程中生成的数据,中国政府相关部门和生命科学研究共同体近几年已经开始布局并着手建设国家级的生命大数据平台或大数据中心,以解决中国生命科学大数据产出面临的实际问题,促进基因组学数据的开放共享。建设属于中国自己的大型基因组数据库的基础设施,不仅可以更好地服务中国的科研人员,还可以在符合国家的利益和法律的前提下,促进与国际同行的信息数据合作与共享。目前,国内已经建成一定规模的生命科学数据中心主要有:依托于北京基因组研究所的国家基因组数据中心(National Genomics Data Center, NGDC)[17,18]、依托于中科院微生物研究所的国家微生物科学数据中心(National Microbiology Data Center, NMDC)和依托于深圳国家基因库CNGBdb等。NGDC平台(https://bigd. big.ac.cn/),除了支持组学原始数据归档,参考基因组及基因注释信息存储和查询,还建立了甲基化数据库,单核苷酸多态性数据库等多组学数据库系统以及以表观组关联分析为代表的综合数据系统[19,20,21]。NMDC平台(http://nmdc.cn/),主要致力于微生物资源信息和微生物基因组数据的保存和共享,其整合的数据资源总量超过1 PB,数据记录数超过40亿条。由NMDC平台维护的具有代表性的数据库资源主要有:微生物宏基因组数据库[22],全球微生物菌种目录数据库[23]和全球流感病毒数据库。
依托于国家基因库[1]的生命大数据中心有以下优势:(1)国家基因库多年来开展的重大基因组项目,如万种鸟类基因组项目[24]、万种鱼类基因组项目[25]、千种植物转录组项目[26]等,积累了海量珍贵数据资源;(2)国家基因库多年来已建成了世界级基因组高通量测序平台和高性能计算平台;(3)国家基因库与国内各省及其他国家相继合作开展的生物样本资源库及其数字化项目,如海洋生物样本资源库及数字化、云南药用植物资源样本资源库及数字化等项目;(4)国家基因库在长期大量基因组学项目中积累的生物信息分析能力和多组学数据深度整合的能力。国家基因库多年来积累的海量基因组学数据和强大的多组学数据计算分析和整合能力,将为CNGBdb提供丰富的生物数据资源和强有力的维护支撑能力。
本文将主要从数据归档、知识搜索、数据管理、数据计算和数据服务等方面介绍CNGBdb的相关功能模块和数据服务。目前,CNGBdb不仅归类存档了CNGB内部项目及与国内国际大量合作项目实施中产生的海量生物学数据,而且还支持研究人员在线提交包括项目、样本、实验、测序、组装和变异数据信息。另外,CNGBdb还积极与NGDC、NMDC、SRA、ENA和DDBJ等平台的依托单位开展合作交流,促进与各大数据库平台之间数据交流与共享,进而推动全球生命大数据资源的利用。
2 数据共享服务
2.1 数据归档
为提供便捷的测序数据归档和数据管理服务,CNGBdb已构建了国家基因库序列归档系统(CNSA, https://db.cngb.org/cnsa)。CNSA可以接受全球用户在线提交的生物研究项目、样本、实验、测序数据及后期项目研究结果等信息。CNSA数据归档系统主要遵循了在全球生命科学领域广泛达成共识的INSDC和DataCite等数据库标准。CNSA是一个测序数据归档和分享系统,还提供早期数据的共享等服务,方便科研文章在投稿过程中杂志编辑检查投稿文章中的数据是否已经全部成功上传。CNSA系统采用了项目(project)、样本(sample)、实验(experiment)和测序(run) 4个元数据结构进行原始测序数据的组织和归档。除原始数据归档外,CNSA还支持组装数据、变异数据的在线批量归档。为了提高数据的通用性,CNSA支持各种常用格式的数据文件的递交,例如,原始数据格式包括FASTQ、BAM、SFF和PacBio_HDF5,组装数据格式包含FASTA,变异数据格式包含VCF等。为了确保归档数据的完整性和提高其后续的可用性,CNSA对用户递交的数据进行校验和质控。在CNSA归档的数据,递交者可以根据项目的保密级别以及研究进度,自由决定归档数据的开放权限和开放时间等。CNSA自2018年10月上线以来,其归档数据量快速增长。截至2020年5月22日,在该平台归档的项目有2176个,提交的数据量达到2221 TB (图1),支撑文章发表115篇。为便于研究人员查找和利用数据,CNSA为每个归档的项目分配DOI,索引项目。通过DOI为CNSA归档的数据能够在互联网环境下的访问建立便利的途径,以增加人们对研究数据的认可,将其作为对科学记录合法的、可引用的成果支持数据存档,并允许这些数据在未来的研究中被验证以及被重新利用[27]。
图1
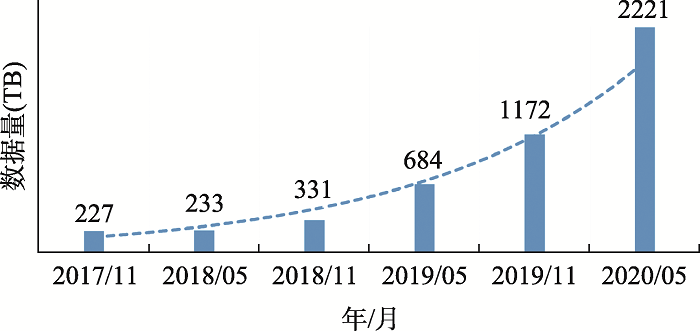 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CNSA归档数据量统计图
Fig. 1Data statistics of CNSA
为实现活体资源、样本资源和数据资源的贯穿,使得生命数据在全生命周期可追溯,除归档核酸数据,CNGBdb还构建了国家基因库样本信息共享平台(E-BioBank, EBB,
2.2 知识搜索
除了国家基因库“三库两平台”的生命科学大数据资源,CNGBdb还整合很多外部数据库的优秀数据资源,如科研文献、基因、变异、蛋白质和序列等知识数据。为了使用户能够快速准确的检索到其需要的数据和信息,CNGBdb平台中搭建了生命大数据搜索引擎。CNGBdb知识搜索的数据类型主要包括文献、项目、样本、实验、测序、组装、变异、基因、蛋白质、序列等。目前CNGBdb中可检索的知识条目数超过30亿条(表1),其中可被检索的文献数量超过2947万条记录,基因序列超过2274万条记录,蛋白质序列超过22.7亿条记录。CNGBdb中的科研文献信息,来源于对多个文献数据库系统的数据的整合,包括GigaScience、PubMed和Europe PMC等。CNGBdb知识检索服务,可通过平台首页(
Table 1
表1
表1知识搜索服务数据
Table 1
| 数据类型 | 索引量(万) | 主要外源数据库 | 主要信息 |
|---|---|---|---|
| 文献 | 2947.19 | GigaScience、PubMed和Europe PMC | 文献标题、摘要、医学关键词、引用和参考文献和文献相关数据等 |
| 基因 | 2274.41 | NCBI Gene | 基因名称、染色体位置、基因产物和它的属性、基因所在的基因组、基因序列和基因变异等 |
| 变异 | 76323.01 | dbSNP[28]、dbVar和ClinVar[29] | 变异名称(HGVS名称)、基因组位置、相关物种、人群频率以及变异数据与疾病、表型和文献等 |
| 蛋白 | 13406.59 | Uniprot[30] | 蛋白名称、蛋白长度、物种和编码蛋白的基因等 |
| 序列 | 213665.12 | NCBI Refseq[31]和GenBank[8] | 序列名称、序列长度、物种和fastq序列文件等 |
| 项目 | 35.63 | NCBI BioProject[32] | 项目的名称、描述和数据类型等 |
| 样本 | 1007.36 | NCBI BioSample[8] | 样本的名称、物种、样本类型和描述等 |
| 实验 | 5515.46 | NCBI SRA[33] | 实验的题目、测序平台、文库构建策略、文库来源和文库选项等 |
| 组装 | 0.24 | NCBI Assembly[34] | 组装的名称、分子类型、测序技术和组装方法等 |
新窗口打开|下载CSV
CNGBdb的知识搜索服务,基于Elasticsearch搜索引擎,支持全文检索功能[2,35],检索速度快。搜索引擎可对检索的结果进行综合评分排序,将最匹配的最符合用户检索目的数据排在前列,通过数据编号索引可以查看检索出的每一条数据的详细信息。CNGBdb搜索引擎还实现了分布式的实时文件存储,每个字段都被索引并可被搜索,可以扩展到上百台服务器,处理PB级结构化或非结构化数据,提供更加深层次的数据、信息和知识的关联关系。
在Elasticsearch的基础上,CNGBdb还拓展了基于生物数据特征的辅助搜索功能,如文献推荐功能、同义词转换功能、高级检索功能和过滤检索功能。文献推荐功能,根据文献的发表年份、杂志影响因子、作者、医学主题词等构建算法模型,综合打分,进行文献推荐,帮助用户查找到与正在查阅的文献最相关的文献,有助于其进行深入阅读和研究。为更深入地理解用户的检索意图,CNGBdb搜索配置了物种同义词(同义词表主要来源于NCBI物种分类数据库[3,36])及医学主题词(同义词表主要来源于NCBI医学主题词库[37]),在检索某个关键词的时候,该关键词的同义词也能检索到,例如Oryza sativa,其学名为Oryza sativa L.,常用名为 rice,Inherited blast name为monocots。您在检索Oryza sativa时,它的同义词Oryza sativa L.、rice和monocots也能被检索到。高级检索功能,可以帮助用户实现对指定字段进行检索,如指定文献的标题、作者、期刊等字段进行检索。过滤检索功能,可以根据用户设置的过滤条件实现对检索结果快速准确的过滤,如根据文献是否免费进行免费全文检索,根据文献发表年限,对不同年限的数据进行过滤检索,根据物种类型,对不同类型物种数据进行过滤检索,使得CNGBdb检索更加准确。
除此之外,CNGBdb搜索引擎还结合了人工智能的智能语义识别和知识图谱技术,使得搜索更加智能。在智能语义识别方面,CNGBdb搜索系统可以实现自动补全功和文本纠错功能。自动补全功能是能根据用户的输入的检索词自动识别用户的检索意图,进行自动补全。文本纠错功能,可对用户输入的错别词进行自动纠错,使用正确的检索词进行检索,如输入“Oryza sativa”,系统将识别为“Oryza sativa”后进行检索。在知识图谱技术方面,CNGBdb构建了文献-作者-研究领域知识图谱,通过文献引用与被引用的关系,文献、作者和医学主题词(Medical Subject Headings,MeSH)关联关系,构建文献-作者-研究领域知识图谱。文献知识图谱,旨在帮助用户快速锁定某个研究方向的重要文献和同领域内具有重要影响力研究人员,建立某个研究领域的发展脉络。以“千人基因组计划”(1,000 Genomes Project, 1KGP) 2010年发表于Nature上的“A map of human genome variation from population-scale sequencing”为例,在CNGBdb的文献库中搜索这篇文章,在文献详情页面,通过知识图谱技术,可视化地展示了该篇文献的文献-作者-研究领域关联图谱(图2)。CNGBdb平台中的知识图谱算法,主要是基于文章和作者的权重。权重越大,图谱中的圆点越大,文献在该领域的影响力越大,作者在该领域的影响力越高,通过图谱的方式,可比较快速锁定领域内的重要文献和重要研究人员。
图2
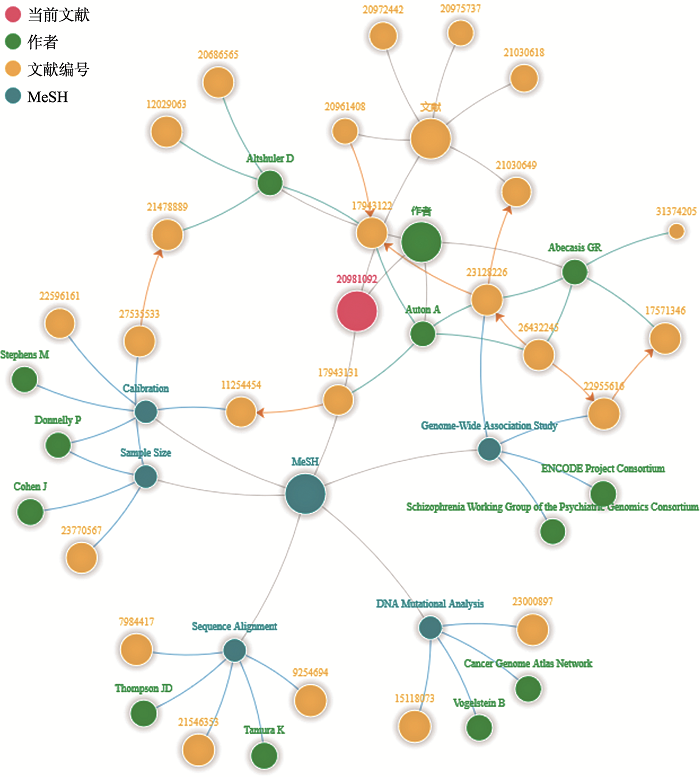 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图21KGP文献推荐知识图谱
当前文献(红色圆点)是整个图谱的中心和起点,与其相连的绿、黄、蓝3个节点分别代表这篇文献的作者、相关推荐文献和MeSH(医学主题词)。这4个大的主节点构成了图谱的主干。
Fig. 21KGP literature recommendation knowledge map
2.3 数据管理
2.3.1 用户管理CNGBdb基于独立的用户统一登录系统(UMS)进行用户登陆注册和管理。UMS具备单点登录、用户管理和权限管理3大核心功能,其中单点登录实现了在同一个集群里面,用户只需登录一次即可访问已授权的系统。用户在UMS系统注册后,可以使用同一个ID和密码访问CNGBdb所有的独立数据库或服务,无需重复注册。UMS给每个用户都赋予唯一识别编码,作为各系统数据贯穿的核心索引,用于打通CNGBdb的各数据库数据。UMS系统还可以对用户在各个数据库的数据权限进行统一的授权和管理。为了最大化地提高平台的利用率,UMS系统提供了各种丰富的API接口供各数据库使用,主要有注册API、登录验证API、用户信息修改API和密码修改API等。
2.3.2 数据分类分级管理
CNGBdb制定了数据资源分类和数据访问形式分类机制,进行数据分类分级保护和统一管理。
在数据资源分类方面,CNGBdb数据的资源类型分为去身份识别的人类遗传资源、生物多样性资源,以及人源微生物资源,定义如下:(1)人类遗传资源数据:是指利用人类遗传资源材料产生的数据等信息资料,是未经过深层处理,未过滤掉人体基因组信息的数据;(2)生物多样性资源数据:是指动物、植物以及微生物等物种资源的数据;(3)人源微生物资源数据:人源微生物是指微生物研究(包括培养以及宏基因组测序研究)的样本来源是人,其本质是微生物。人源微生物资源数据又分为已过滤掉人体基因组的人源微生物资源数据和未过滤掉人体基因组的人源微生物资源数据。
在数据访问形式方面,CNGBdb数据的访问形式包括公开、受控管理形式。(1)公开:数据公开是指元数据和数据文件都公开。数据递交者需要设置一个公开日期后,元数据和数据文件都将在该公开日期公开,公开数据将展示在CNGBdb,且面向全球开放,用户可在CNGBdb自由访问或使用。(2)受控:即项目关联的元数据公开和数据文件受控。数据递交者需要设置一个元数据的公开日期,元数据都将在该公开日期公开,数据文件受控。受控数据仅在CNGBdb上展示元数据,数据文件受控管理,具有数据访问权限的用户可使用受控的数据。
2.4 数据计算
CNGBdb数据计算服务是基于CNGBdb清洗和归档的数据部署的BLAST序列比对服务(Table 2
表2
表2BLAST工具数据资源
Table 2
| 数据源 | 数据库名称 | 数据库编号 | 数据库格式版本 |
|---|---|---|---|
| CNGB | The 1000 Plants Project | onekp | v5 |
| Microbiome DataBase | microbiome | v5 | |
| Pan Immune Repertoire Database | pird | v5 | |
| The Transcriptomes of 1,000 Fishes Project | fisht1k | v5 | |
| The Bird 10,000 Genomes Project | b10k | v5 | |
| NCBI | Nucleotide collection | nt | v5 |
| Reference proteins | refseq_protein | v4 | |
| 16S ribosomal RNA sequences | 16smicrobial | v4 | |
| Human genomic | human_genomic | v4 | |
| RefSeq Representative genomes | refseq_representative_genomes | v4 | |
| Non-human organisms genomic | other_genomic | v4 | |
| Human RefSeqGene sequences | refseqgene | v4 | |
| Genomic survey sequences | gss | v4 | |
| Reference genomic sequences | refseq_genomic | v4 | |
| High throughput genomic sequences | htgs | v4 | |
| Transcriptome Shotgun Assembly | tsa | v4 | |
| Expressed sequence tags | est | v4 | |
| Patent sequences | pat | v4 | |
| Sequence tagged sites | sts | v4 | |
| Protein Data Bank | pdb | v4 | |
| Metagenomic sequences | env | v4 |
新窗口打开|下载CSV
2.5 数据应用
CNGBdb基于底层数据结构和数据,构建了包括动物、植物、微生物等不同专题数据库及分析数据库系统。目前CNGBdb已上线的上层应用数据库包括:千种植物数据库(OneKP,为便捷和及时地共享科研数据,在CNGBdb数据库平台,除CNGBdb已经构建的不同研究领域的数据库,还允许用户自定义创建数据集并共享发布。相比于传统的数据库共享,用户不需要开发数据库、运营和维护数据库。在CNGBdb仅需上传数据、创建数据集和分享数据集3步,即可将科研数据分享给科研领域的研究人员。CNGBdb用户已创建的部分数据集见表3。
Table 3
表3
表3CNGBdb用户已创建的部分数据集
Table 3
| 分类 | 数据集名称 | 简要介绍 | 网址 |
|---|---|---|---|
| 植物 | 10,000 Plant Genomes Project | 万种植物基因组项目数据集 | https://db.cngb.org/datamart/plant/DATApla1/ |
| The 3000 Rice Genomes Project | 3000水稻项目数据集 | https://db.cngb.org/datamart/plant/DATApla2/ | |
| 1000 Plant Transcriptomes | 千种植物转录组项目数据集 | https://db.cngb.org/datamart/plant/DATApla4/ | |
| Data of Ruili Botanical Garden | 瑞丽珍稀植物园689种植物 基因组测序数据 | https://db.cngb.org/datamart/plant/DATApla5/ | |
| 动物 | The B10K Genomes Project | 万种鸟基因组项目数据集 | https://db.cngb.org/datamart/animal/DATAani1/ |
| 1K Insect Transcriptome | 千种昆虫转录组数据集 | https://db.cngb.org/datamart/animal/DATAani3/ | |
| Transcriptomes of 1000 Fishes | 千种鱼转录组数据集 | https://db.cngb.org/datamart/animal/DATAani2/ | |
| Vertebrate Genomes 10K | 万种脊椎动物数据集 | https://db.cngb.org/datamart/animal/DATAani5/ | |
| Life Periodic Plan (LPP) | 生命周期表项目数据集 | https://db.cngb.org/datamart/animal/DATAani6/ | |
| 微生物 | 1520 reference genomes | 覆盖人体肠道中所有主要细菌门和 属的1520个基因组数据集 | https://db.cngb.org/datamart/microbe/DATAmic1/ |
| Earth Microbiome Project | 地球微生物组项目数据集 | https://db.cngb.org/datamart/microbe/DATAmic4/ | |
| 1000 Fungal Genomes Project | 千种真菌基因组项目数据集 | https://db.cngb.org/datamart/microbe/DATAmic7/ | |
| MetaHIT (metagenomics of humanintestinal tract) | 欧盟的肠道微生物组计划数据集 | https://db.cngb.org/datamart/microbe/DATAmic5/ | |
| Human Microbiome Project | 人类微生物组项目数据集 | https://db.cngb.org/datamart/microbe/DATAmic3/ | |
| 人群 | WGS of 175 Mongolians | 175个蒙古人基因组数据集 | https://db.cngb.org/datamart/other/DATAoth1/ |
| 1000 Genomes Project (human) | 千人基因组项目数据集 | https://db.cngb.org/datamart/other/DATAoth2/ |
新窗口打开|下载CSV
3 结语与展望
CNGBdb是一个自由开放的生命科学大数据 共享平台,致力于促进生命科学研究项目中生成的测序数据及研究项目所取得的成果的开发共享和合作利用。目前,CNGBdb提供生物大数据归档、管理、搜索、计算、分析及应用一体化的生命大数据服务。随着对生命科学大数据共享的需求的不断变化,CNGBdb将在以下几个方面做出改进和提升。在数据归档上,除归档项目、样本、实验/测序数据、组装、变异数据和样本实体信息,在实现样本实体到组学数据的贯穿基础上,CNGBdb还将扩展序列、蛋白、代谢、表达、临床和影像等多组学数据,实现数据的组学贯穿。在知识搜索上,除提供给用户主动搜索,CNGBdb还将提供数据及知识推荐搜索,实现主被动搜索联动,提升搜索的准确度和搜索体验。在数据管理上,CNGBdb将依据现有的伦理规范、现行法律、法规、条例、国际条约等,制定更加完善的数据共享和应用政策。同时,CNGBdb还将逐步建立数据可信计算环境和工具,使得数据在可用而不可见的环境下进行安全计算,并依托区块链技术,对数据生命周期进行记账和监控,实现生命科学数据的安全管理和应用。在数据应用上,CNGBdb将在现有数据集的功能基础上,提供更个性化、便捷化的多维度的统计分析工具,数据比对工具,数据可视化工具等,实现数据的分享到数据应用的个性化、自动化。
CNGBdb的建设和发展,将促进我国生物遗传数据与生命科学数据的规范管理和利用,为生物医药、生物农业和海洋生物等诸多生物产业的科学研究提供数据共享平台,推动我国生命科学向更深入、更为广阔和更多创新的领域发展。CNGBdb作为国家基因库的对外数据共享平台,不仅促进扩大国内、国际交流与合作的范围,还促进国内外生命科学数据的汇集、交流和互通。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
DOI:10.1093/nar/gkw829URLPMID:27638885 [本文引用: 2]

The International Genome Sample Resource (IGSR; http://www.internationalgenome.org) expands in data type and population diversity the resources from the 1000 Genomes Project. IGSR represents the largest open collection of human variation data and provides easy access to these resources. IGSR was established in 2015 to maintain and extend the 1000 Genomes Project data, which has been widely used as a reference set of human variation and by researchers developing analysis methods. IGSR has mapped all of the 1000 Genomes sequence to the newest human reference (GRCh38), and will release updated variant calls to ensure maximal usefulness of the existing data. IGSR is collecting new structural variation data on the 1000 Genomes samples from long read sequencing and other technologies, and will collect relevant functional data into a single comprehensive resource. IGSR is extending coverage with new populations sequenced by collaborating groups. Here, we present the new data and analysis that IGSR has made available. We have also introduced a new data portal that increases discoverability of our data-previously only browseable through our FTP site-by focusing on particular samples, populations or data sets of interest.
DOI:10.1038/nature08987URLPMID:20393554 [本文引用: 2]
The International Cancer Genome Consortium (ICGC) was launched to coordinate large-scale cancer genome studies in tumours from 50 different cancer types and/or subtypes that are of clinical and societal importance across the globe. Systematic studies of more than 25,000 cancer genomes at the genomic, epigenomic and transcriptomic levels will reveal the repertoire of oncogenic mutations, uncover traces of the mutagenic influences, define clinically relevant subtypes for prognosis and therapeutic management, and enable the development of new cancer therapies.
DOI:10.1126/science.1068037URLPMID:11935017 [本文引用: 1]
We have produced a draft sequence of the rice genome for the most widely cultivated subspecies in China, Oryza sativa L. ssp. indica, by whole-genome shotgun sequencing. The genome was 466 megabases in size, with an estimated 46,022 to 55,615 genes. Functional coverage in the assembled sequences was 92.0%. About 42.2% of the genome was in exact 20-nucleotide oligomer repeats, and most of the transposons were in the intergenic regions between genes. Although 80.6% of predicted Arabidopsis thaliana genes had a homolog in rice, only 49.4% of predicted rice genes had a homolog in A. thaliana. The large proportion of rice genes with no recognizable homologs is due to a gradient in the GC content of rice coding sequences.
DOI:10.1038/nature03895URLPMID:16100779 [本文引用: 1]
Rice, one of the world's most important food plants, has important syntenic relationships with the other cereal species and is a model plant for the grasses. Here we present a map-based, finished quality sequence that covers 95% of the 389 Mb genome, including virtually all of the euchromatin and two complete centromeres. A total of 37,544 non-transposable-element-related protein-coding genes were identified, of which 71% had a putative homologue in Arabidopsis. In a reciprocal analysis, 90% of the Arabidopsis proteins had a putative homologue in the predicted rice proteome. Twenty-nine per cent of the 37,544 predicted genes appear in clustered gene families. The number and classes of transposable elements found in the rice genome are consistent with the expansion of syntenic regions in the maize and sorghum genomes. We find evidence for widespread and recurrent gene transfer from the organelles to the nuclear chromosomes. The map-based sequence has proven useful for the identification of genes underlying agronomic traits. The additional single-nucleotide polymorphisms and simple sequence repeats identified in our study should accelerate improvements in rice production.
DOI:10.1186/2047-217X-3-7URLPMID:24872877 [本文引用: 1]
BACKGROUND: Rice, Oryza sativa L., is the staple food for half the world's population. By 2030, the production of rice must increase by at least 25% in order to keep up with global population growth and demand. Accelerated genetic gains in rice improvement are needed to mitigate the effects of climate change and loss of arable land, as well as to ensure a stable global food supply. FINDINGS: We resequenced a core collection of 3,000 rice accessions from 89 countries. All 3,000 genomes had an average sequencing depth of 14x, with average genome coverages and mapping rates of 94.0% and 92.5%, respectively. From our sequencing efforts, approximately 18.9 million single nucleotide polymorphisms (SNPs) in rice were discovered when aligned to the reference genome of the temperate japonica variety, Nipponbare. Phylogenetic analyses based on SNP data confirmed differentiation of the O. sativa gene pool into 5 varietal groups - indica, aus/boro, basmati/sadri, tropical japonica and temperate japonica. CONCLUSIONS: Here, we report an international resequencing effort of 3,000 rice genomes. This data serves as a foundation for large-scale discovery of novel alleles for important rice phenotypes using various bioinformatics and/or genetic approaches. It also serves to understand the genomic diversity within O. sativa at a higher level of detail. With the release of the sequencing data, the project calls for the global rice community to take advantage of this data as a foundation for establishing a global, public rice genetic/genomic database and information platform for advancing rice breeding technology for future rice improvement.
DOI:10.1038/s41588-018-0266-xURLPMID:30420647 [本文引用: 1]
Genebanks hold comprehensive collections of cultivars, landraces and crop wild relatives of all major food crops, but their detailed characterization has so far been limited to sparse core sets. The analysis of genome-wide genotyping-by-sequencing data for almost all barley accessions of the German ex situ genebank provides insights into the global population structure of domesticated barley and points out redundancies and coverage gaps in one of the world's major genebanks. Our large sample size and dense marker data afford great power for genome-wide association scans. We detect known and novel loci underlying morphological traits differentiating barley genepools, find evidence for convergent selection for barbless awns in barley and rice and show that a major-effect resistance locus conferring resistance to bymovirus infection has been favored by traditional farmers. This study outlines future directions for genomics-assisted genebank management and the utilization of germplasm collections for linking natural variation to human selection during crop evolution.
DOI:10.1093/nar/gky989URLPMID:30365038 [本文引用: 3]
GenBank(R) (www.ncbi.nlm.nih.gov/genbank/) is a comprehensive database that contains publicly available nucleotide sequences for 420 000 formally described species. Most GenBank submissions are made using BankIt, the NCBI Submission Portal, or the tool tbl2asn, and are obtained from individual laboratories and batch submissions from large-scale sequencing projects, including whole genome shotgun (WGS) and environmental sampling projects. Daily data exchange with the European Nucleotide Archive (ENA) and the DNA Data Bank of Japan (DDBJ) ensures worldwide coverage. GenBank is accessible through the NCBI Nucleotide database, which links to related information such as taxonomy, genomes, protein sequences and structures, and biomedical journal literature in PubMed. BLAST provides sequence similarity searches of GenBank and other sequence databases. Complete bimonthly releases and daily updates of the GenBank database are available by FTP. Recent updates include an expansion of sequence identifier formats to accommodate expected database growth, submission wizards for ribosomal RNA, and the transfer of Expressed Sequence Tag (EST) and Genome Survey Sequence (GSS) data into the Nucleotide database.
DOI:10.1093/nar/gkz268URLPMID:30976793 [本文引用: 1]
The EMBL-EBI provides free access to popular bioinformatics sequence analysis applications as well as to a full-featured text search engine with powerful cross-referencing and data retrieval capabilities. Access to these services is provided via user-friendly web interfaces and via established RESTful and SOAP Web Services APIs (https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/EMBL-EBI+Web+Services+APIs+-+Data+Retrieval). Both systems have been developed with the same core principles that allow them to integrate an ever-increasing volume of biological data, making them an integral part of many popular data resources provided at the EMBL-EBI. Here, we describe the latest improvements made to the frameworks which enhance the interconnectivity between public EMBL-EBI resources and ultimately enhance biological data discoverability, accessibility, interoperability and reusability.
DOI:10.1093/nar/gky1002URLPMID:30357349 [本文引用: 1]
The Genomic Expression Archive (GEA) for functional genomics data from microarray and high-throughput sequencing experiments has been established at the DNA Data Bank of Japan (DDBJ) Center (https://www.ddbj.nig.ac.jp), which is a member of the International Nucleotide Sequence Database Collaboration (INSDC) with the US National Center for Biotechnology Information and the European Bioinformatics Institute. The DDBJ Center collects nucleotide sequence data and associated biological information from researchers and also services the Japanese Genotype-phenotype Archive (JGA) with the National Bioscience Database Center for collecting human data. To automate the submission process, we have implemented the DDBJ BioSample validator which checks submitted records, auto-corrects their format, and issues error messages and warnings if necessary. The DDBJ Center also operates the NIG supercomputer, prepared for analyzing large-scale genome sequences. We now offer a secure platform specifically to handle personal human genomes. This report describes database activities for INSDC and JGA over the past year, the newly launched GEA, submission, retrieval, and analysis services available in our supercomputer system and their recent developments.
DOI:10.1093/nar/gkx1235URLPMID:29316735 [本文引用: 1]
The 2018 Nucleic Acids Research Database Issue contains 181 papers spanning molecular biology. Among them, 82 are new and 84 are updates describing resources that appeared in the Issue previously. The remaining 15 cover databases most recently published elsewhere. Databases in the area of nucleic acids include 3DIV for visualisation of data on genome 3D structure and RNArchitecture, a hierarchical classification of RNA families. Protein databases include the established SMART, ELM and MEROPS while GPCRdb and the newcomer STCRDab cover families of biomedical interest. In the area of metabolism, HMDB and Reactome both report new features while PULDB appears in NAR for the first time. This issue also contains reports on genomics resources including Ensembl, the UCSC Genome Browser and ENCODE. Update papers from the IUPHAR/BPS Guide to Pharmacology and DrugBank are highlights of the drug and drug target section while a number of proteomics databases including proteomicsDB are also covered. The entire Database Issue is freely available online on the Nucleic Acids Research website (https://academic.oup.com/nar). The NAR online Molecular Biology Database Collection has been updated, reviewing 138 entries, adding 88 new resources and eliminating 47 discontinued URLs, bringing the current total to 1737 databases. It is available at http://www.oxfordjournals.org/nar/database/c/.
DOI:10.1093/nar/gkv1310URLPMID:26615188 [本文引用: 1]
The SIB Swiss Institute of Bioinformatics (www.isb-sib.ch) provides world-class bioinformatics databases, software tools, services and training to the international life science community in academia and industry. These solutions allow life scientists to turn the exponentially growing amount of data into knowledge. Here, we provide an overview of SIB's resources and competence areas, with a strong focus on curated databases and SIB's most popular and widely used resources. In particular, SIB's Bioinformatics resource portal ExPASy features over 150 resources, including UniProtKB/Swiss-Prot, ENZYME, PROSITE, neXtProt, STRING, UniCarbKB, SugarBindDB, SwissRegulon, EPD, arrayMap, Bgee, SWISS-MODEL Repository, OMA, OrthoDB and other databases, which are briefly described in this article.
DOI:10.1093/nar/gkw1092URLPMID:27899662 [本文引用: 1]
KEGG (http://www.kegg.jp/ or http://www.genome.jp/kegg/) is an encyclopedia of genes and genomes. Assigning functional meanings to genes and genomes both at the molecular and higher levels is the primary objective of the KEGG database project. Molecular-level functions are stored in the KO (KEGG Orthology) database, where each KO is defined as a functional ortholog of genes and proteins. Higher-level functions are represented by networks of molecular interactions, reactions and relations in the forms of KEGG pathway maps, BRITE hierarchies and KEGG modules. In the past the KO database was developed for the purpose of defining nodes of molecular networks, but now the content has been expanded and the quality improved irrespective of whether or not the KOs appear in the three molecular network databases. The newly introduced addendum category of the GENES database is a collection of individual proteins whose functions are experimentally characterized and from which an increasing number of KOs are defined. Furthermore, the DISEASE and DRUG databases have been improved by systematic analysis of drug labels for better integration of diseases and drugs with the KEGG molecular networks. KEGG is moving towards becoming a comprehensive knowledge base for both functional interpretation and practical application of genomic information.
DOI:10.1093/nar/gkx1097URLPMID:29190397 [本文引用: 1]
For more than 30 years, the International Nucleotide Sequence Database Collaboration (INSDC; http://www.insdc.org/) has been committed to capturing, preserving and providing access to comprehensive public domain nucleotide sequence and associated metadata which enables discovery in biomedicine, biodiversity and biological sciences. Since 1987, the DNA Data Bank of Japan (DDBJ) at the National Institute for Genetics in Mishima, Japan; the European Nucleotide Archive (ENA) at the European Molecular Biology Laboratory's European Bioinformatics Institute (EMBL-EBI) in Hinxton, UK; and GenBank at National Center for Biotechnology Information (NCBI), National Library of Medicine, National Institutes of Health in Bethesda, Maryland, USA have worked collaboratively to enable access to nucleotide sequence data in standardized formats for the worldwide scientific community. In this article, we reiterate the principles of the INSDC collaboration and briefly summarize the trends of the archival content.
DOI:10.1038/nature07484URLPMID:18987735 [本文引用: 1]
Here we present the first diploid genome sequence of an Asian individual. The genome was sequenced to 36-fold average coverage using massively parallel sequencing technology. We aligned the short reads onto the NCBI human reference genome to 99.97% coverage, and guided by the reference genome, we used uniquely mapped reads to assemble a high-quality consensus sequence for 92% of the Asian individual's genome. We identified approximately 3 million single-nucleotide polymorphisms (SNPs) inside this region, of which 13.6% were not in the dbSNP database. Genotyping analysis showed that SNP identification had high accuracy and consistency, indicating the high sequence quality of this assembly. We also carried out heterozygote phasing and haplotype prediction against HapMap CHB and JPT haplotypes (Chinese and Japanese, respectively), sequence comparison with the two available individual genomes (J. D. Watson and J. C. Venter), and structural variation identification. These variations were considered for their potential biological impact. Our sequence data and analyses demonstrate the potential usefulness of next-generation sequencing technologies for personal genomics.
DOI:10.1038/nature08696URLPMID:20010809 [本文引用: 1]
Using next-generation sequencing technology alone, we have successfully generated and assembled a draft sequence of the giant panda genome. The assembled contigs (2.25 gigabases (Gb)) cover approximately 94% of the whole genome, and the remaining gaps (0.05 Gb) seem to contain carnivore-specific repeats and tandem repeats. Comparisons with the dog and human showed that the panda genome has a lower divergence rate. The assessment of panda genes potentially underlying some of its unique traits indicated that its bamboo diet might be more dependent on its gut microbiome than its own genetic composition. We also identified more than 2.7 million heterozygous single nucleotide polymorphisms in the diploid genome. Our data and analyses provide a foundation for promoting mammalian genetic research, and demonstrate the feasibility for using next-generation sequencing technologies for accurate, cost-effective and rapid de novo assembly of large eukaryotic genomes.
DOI:10.1093/nar/gkz913URLPMID:31702008 [本文引用: 1]
The National Genomics Data Center (NGDC) provides a suite of database resources to support worldwide research activities in both academia and industry. With the rapid advancements in higher-throughput and lower-cost sequencing technologies and accordingly the huge volume of multi-omics data generated at exponential scales and rates, NGDC is continually expanding, updating and enriching its core database resources through big data integration and value-added curation. In the past year, efforts for update have been mainly devoted to BioProject, BioSample, GSA, GWH, GVM, NONCODE, LncBook, EWAS Atlas and IC4R. Newly released resources include three human genome databases (PGG.SNV, PGG.Han and CGVD), eLMSG, EWAS Data Hub, GWAS Atlas, iSheep and PADS Arsenal. In addition, four web services, namely, eGPS Cloud, BIG Search, BIG Submission and BIG SSO, have been significantly improved and enhanced. All of these resources along with their services are publicly accessible at https://bigd.big.ac.cn.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.gpb.2017.01.001URLPMID:28387199 [本文引用: 1]
With the rapid development of sequencing technologies towards higher throughput and lower cost, sequence data are generated at an unprecedentedly explosive rate. To provide an efficient and easy-to-use platform for managing huge sequence data, here we present Genome Sequence Archive (GSA; http://bigd.big.ac.cn/gsa or http://gsa.big.ac.cn), a data repository for archiving raw sequence data. In compliance with data standards and structures of the International Nucleotide Sequence Database Collaboration (INSDC), GSA adopts four data objects (BioProject, BioSample, Experiment, and Run) for data organization, accepts raw sequence reads produced by a variety of sequencing platforms, stores both sequence reads and metadata submitted from all over the world, and makes all these data publicly available to worldwide scientific communities. In the era of big data, GSA is not only an important complement to existing INSDC members by alleviating the increasing burdens of handling sequence data deluge, but also takes the significant responsibility for global big data archive and provides free unrestricted access to all publicly available data in support of research activities throughout the world.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gky1008URLPMID:30365027 [本文引用: 1]
Meta-omics approaches have been increasingly used to study the structure and function of the microbial communities. A variety of large-scale collaborative projects are being conducted to encompass samples from diverse environments and habitats. This change has resulted in enormous demands for long-term data maintenance and capacity for data analysis. The Global Catalogue of Metagenomics (gcMeta) is a part of the 'Chinese Academy of Sciences Initiative of Microbiome (CAS-CMI)', which focuses on studying the human and environmental microbiome, establishing depositories of samples, strains and data, as well as promoting international collaboration. To accommodate and rationally organize massive datasets derived from several thousands of human and environmental microbiome samples, gcMeta features a database management system for archiving and publishing data in a standardized way. Another main feature is the integration of more than ninety web-based data analysis tools and workflows through a Docker platform which enables data analysis by using various operating systems. This platform has been rapidly expanding, and now hosts data from the CAS-CMI and a number of other ongoing research projects. In conclusion, this platform presents a powerful and user-friendly service to support worldwide collaborative efforts in the field of meta-omics research. This platform is freely accessible at https://gcmeta.wdcm.org/.
DOI:10.1186/1471-2164-14-933URLPMID:24377417 [本文引用: 1]
BACKGROUND: Throughout the long history of industrial and academic research, many microbes have been isolated, characterized and preserved (whenever possible) in culture collections. With the steady accumulation in observational data of biodiversity as well as microbial sequencing data, bio-resource centers have to function as data and information repositories to serve academia, industry, and regulators on behalf of and for the general public. Hence, the World Data Centre for Microorganisms (WDCM) started to take its responsibility for constructing an effective information environment that would promote and sustain microbial research data activities, and bridge the gaps currently present within and outside the microbiology communities. DESCRIPTION: Strain catalogue information was collected from collections by online submission. We developed tools for automatic extraction of strain numbers and species names from various sources, including Genbank, Pubmed, and SwissProt. These new tools connect strain catalogue information with the corresponding nucleotide and protein sequences, as well as to genome sequence and references citing a particular strain. All information has been processed and compiled in order to create a comprehensive database of microbial resources, and was named Global Catalogue of Microorganisms (GCM). The current version of GCM contains information of over 273,933 strains, which includes 43,436 bacterial, fungal and archaea species from 52 collections in 25 countries and regions.A number of online analysis and statistical tools have been integrated, together with advanced search functions, which should greatly facilitate the exploration of the content of GCM. CONCLUSION: A comprehensive dynamic database of microbial resources has been created, which unveils the resources preserved in culture collections especially for those whose informatics infrastructures are still under development, which should foster cumulative research, facilitating the activities of microbiologists world-wide, who work in both public and industrial research centres. This database is available from http://gcm.wfcc.info.
DOI:10.1038/522034dURLPMID:26040883 [本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41586-019-1693-2URLPMID:31645766 [本文引用: 1]
Green plants (Viridiplantae) include around 450,000-500,000 species(1,2) of great diversity and have important roles in terrestrial and aquatic ecosystems. Here, as part of the One Thousand Plant Transcriptomes Initiative, we sequenced the vegetative transcriptomes of 1,124 species that span the diversity of plants in a broad sense (Archaeplastida), including green plants (Viridiplantae), glaucophytes (Glaucophyta) and red algae (Rhodophyta). Our analysis provides a robust phylogenomic framework for examining the evolution of green plants. Most inferred species relationships are well supported across multiple species tree and supermatrix analyses, but discordance among plastid and nuclear gene trees at a few important nodes highlights the complexity of plant genome evolution, including polyploidy, periods of rapid speciation, and extinction. Incomplete sorting of ancestral variation, polyploidization and massive expansions of gene families punctuate the evolutionary history of green plants. Notably, we find that large expansions of gene families preceded the origins of green plants, land plants and vascular plants, whereas whole-genome duplications are inferred to have occurred repeatedly throughout the evolution of flowering plants and ferns. The increasing availability of high-quality plant genome sequences and advances in functional genomics are enabling research on genome evolution across the green tree of life.
[本文引用: 1]
DOI:10.1093/nar/28.1.352URLPMID:10592272 [本文引用: 1]
In response to a need for a general catalog of genome variation to address the large-scale sampling designs required by association studies, gene mapping and evolutionary biology, the National Cancer for Biotechnology Information (NCBI) has established the dbSNP database. Submissions to dbSNP will be integrated with other sources of information at NCBI such as GenBank, PubMed, LocusLink and the Human Genome Project data. The complete contents of dbSNP are available to the public at website: http://www.ncbi.nlm.nih.gov/SNP. Submitted SNPs can also be downloaded via anonymous FTP at ftp://ncbi.nlm.nih.gov/snp/
DOI:10.1093/nar/gkv1222URLPMID:26582918 [本文引用: 1]
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) at the National Center for Biotechnology Information (NCBI) is a freely available archive for interpretations of clinical significance of variants for reported conditions. The database includes germline and somatic variants of any size, type or genomic location. Interpretations are submitted by clinical testing laboratories, research laboratories, locus-specific databases, OMIM(R), GeneReviews, UniProt, expert panels and practice guidelines. In NCBI's Variation submission portal, submitters upload batch submissions or use the Submission Wizard for single submissions. Each submitted interpretation is assigned an accession number prefixed with SCV. ClinVar staff review validation reports with data types such as HGVS (Human Genome Variation Society) expressions; however, clinical significance is reported directly from submitters. Interpretations are aggregated by variant-condition combination and assigned an accession number prefixed with RCV. Clinical significance is calculated for the aggregate record, indicating consensus or conflict in the submitted interpretations. ClinVar uses data standards, such as HGVS nomenclature for variants and MedGen identifiers for conditions. The data are available on the web as variant-specific views; the entire data set can be downloaded via ftp. Programmatic access for ClinVar records is available through NCBI's E-utilities. Future development includes providing a variant-centric XML archive and a web page for details of SCV submissions.
DOI:10.1093/nar/gky1049URLPMID:30395287 [本文引用: 1]
The UniProt Knowledgebase is a collection of sequences and annotations for over 120 million proteins across all branches of life. Detailed annotations extracted from the literature by expert curators have been collected for over half a million of these proteins. These annotations are supplemented by annotations provided by rule based automated systems, and those imported from other resources. In this article we describe significant updates that we have made over the last 2 years to the resource. We have greatly expanded the number of Reference Proteomes that we provide and in particular we have focussed on improving the number of viral Reference Proteomes. The UniProt website has been augmented with new data visualizations for the subcellular localization of proteins as well as their structure and interactions. UniProt resources are available under a CC-BY (4.0) license via the web at https://www.uniprot.org/.
DOI:10.1093/nar/gkl842URLPMID:17130148 [本文引用: 1]
NCBI's reference sequence (RefSeq) database (http://www.ncbi.nlm.nih.gov/RefSeq/) is a curated non-redundant collection of sequences representing genomes, transcripts and proteins. The database includes 3774 organisms spanning prokaryotes, eukaryotes and viruses, and has records for 2,879,860 proteins (RefSeq release 19). RefSeq records integrate information from multiple sources, when additional data are available from those sources and therefore represent a current description of the sequence and its features. Annotations include coding regions, conserved domains, tRNAs, sequence tagged sites (STS), variation, references, gene and protein product names, and database cross-references. Sequence is reviewed and features are added using a combined approach of collaboration and other input from the scientific community, prediction, propagation from GenBank and curation by NCBI staff. The format of all RefSeq records is validated, and an increasing number of tests are being applied to evaluate the quality of sequence and annotation, especially in the context of complete genomic sequence.
DOI:10.1093/nar/gkr1163URL [本文引用: 1]
DOI:10.1093/nar/gkr854URLPMID:22009675 [本文引用: 1]
New generation sequencing platforms are producing data with significantly higher throughput and lower cost. A portion of this capacity is devoted to individual and community scientific projects. As these projects reach publication, raw sequencing datasets are submitted into the primary next-generation sequence data archive, the Sequence Read Archive (SRA). Archiving experimental data is the key to the progress of reproducible science. The SRA was established as a public repository for next-generation sequence data as a part of the International Nucleotide Sequence Database Collaboration (INSDC). INSDC is composed of the National Center for Biotechnology Information (NCBI), the European Bioinformatics Institute (EBI) and the DNA Data Bank of Japan (DDBJ). The SRA is accessible at www.ncbi.nlm.nih.gov/sra from NCBI, at www.ebi.ac.uk/ena from EBI and at trace.ddbj.nig.ac.jp from DDBJ. In this article, we present the content and structure of the SRA and report on updated metadata structures, submission file formats and supported sequencing platforms. We also briefly outline our various responses to the challenge of explosive data growth.
DOI:10.1093/nar/gkv1226URLPMID:26578580 [本文引用: 1]
The NCBI Assembly database (www.ncbi.nlm.nih.gov/assembly/) provides stable accessioning and data tracking for genome assembly data. The model underlying the database can accommodate a range of assembly structures, including sets of unordered contig or scaffold sequences, bacterial genomes consisting of a single complete chromosome, or complex structures such as a human genome with modeled allelic variation. The database provides an assembly accession and version to unambiguously identify the set of sequences that make up a particular version of an assembly, and tracks changes to updated genome assemblies. The Assembly database reports metadata such as assembly names, simple statistical reports of the assembly (number of contigs and scaffolds, contiguity metrics such as contig N50, total sequence length and total gap length) as well as the assembly update history. The Assembly database also tracks the relationship between an assembly submitted to the International Nucleotide Sequence Database Consortium (INSDC) and the assembly represented in the NCBI RefSeq project. Users can find assemblies of interest by querying the Assembly Resource directly or by browsing available assemblies for a particular organism. Links in the Assembly Resource allow users to easily download sequence and annotations for current versions of genome assemblies from the NCBI genomes FTP site.
[本文引用: 1]
DOI:10.1093/nar/gkr1178URLPMID:22139910 [本文引用: 1]
The NCBI Taxonomy database (http://www.ncbi.nlm.nih.gov/taxonomy) is the standard nomenclature and classification repository for the International Nucleotide Sequence Database Collaboration (INSDC), comprising the GenBank, ENA (EMBL) and DDBJ databases. It includes organism names and taxonomic lineages for each of the sequences represented in the INSDC's nucleotide and protein sequence databases. The taxonomy database is manually curated by a small group of scientists at the NCBI who use the current taxonomic literature to maintain a phylogenetic taxonomy for the source organisms represented in the sequence databases. The taxonomy database is a central organizing hub for many of the resources at the NCBI, and provides a means for clustering elements within other domains of NCBI web site, for internal linking between domains of the Entrez system and for linking out to taxon-specific external resources on the web. Our primary purpose is to index the domain of sequences as conveniently as possible for our user community.
URLPMID:25000040 [本文引用: 1]
The US federal government initiated the Open Government Directive where federal agencies are required to publish high value datasets so that they are available to the public. Data.gov and the community site Healthdata.gov were initiated to disperse such datasets. However, data searches and retrieval for these sites are keyword driven and severely limited in performance. The purpose of this paper is to address the issue of extracting relevant open-source data by proposing a method of adopting the MeSH framework for indexing and data retrieval. A pilot study was conducted to compare the performance of traditional keywords to MeSH terms for retrieving relevant open-source datasets related to

{kind=link}
{kind=link}
{kind=link}
{kind=link}