 ,2,4,5, 徐湘民,11.
,2,4,5, 徐湘民,11. 2.
3.
4.
5.
A comprehensive repository of mutation data and a clinical assistant decision system for hemoglobinopathy in the Chinese population
Qianqian Zhang1, Li Zhang1, Yaohua Tang2, Xiarong Li3, Xiaopeng Xu3, Ming Qi,2,4,5, Xiangmin Xu,11. 2.
3.
4.
5.
通讯作者:
编委: 杨昭庆
收稿日期:2019-05-13修回日期:2019-07-24网络出版日期:2019-08-20
| 基金资助: |
Received:2019-05-13Revised:2019-07-24Online:2019-08-20
| Fund supported: |
作者简介 About authors
张倩倩,博士研究生,专业方向:遗传学E-mail:zqq.smu@foxmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (547KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张倩倩, 张立, 唐耀华, 李厦戎, 徐晓鹏, 祁鸣, 徐湘民. 中国人血红蛋白病突变数据集和临床辅助决策管理系统 [J]. 遗传, 2019, 41(8): 746-753 doi:10.16288/j.yczz.19-136
Qianqian Zhang, Li Zhang, Yaohua Tang, Xiarong Li, Xiaopeng Xu, Ming Qi, Xiangmin Xu.
随着高通量测序技术的日益发展,个体基因组信息逐步被应用于遗传病的临床诊断与分子筛查,为满足对基因组信息的全面解读,建立系统综合的疾病表型关联变异注释库势在必行。目前常用的人类基因突变注释库有人类孟德尔遗传病在线数据库(Online Mendelian Inheritance in Man, OMIM),OMIM收录已报道的单基因遗传病约6400种,致病基因约4000个,但疾病表型关联的变异信息较少[1]。ClinVar是美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)对外免费开放的人类基因组致病变异数据库[2],近期Shah 等[3]深度分析大样本病例队列全基因组测序数据,以美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南评估ClinVar中收录的变异条目,发现有11.5%的基因变异被错误释义,亟待校正,因此建议ClinVar管理者对上传的信息进行严格的审核和过滤。
为充分利用目前的高通量数据资源,需系统、高效收集疾病表型关联的致病变异及非致病变异数据,接轨基础科学研究与临床应用,充分解读变异信息,以期将其转化应用于临床疾病精准基因分型和个性化诊治方案。人类变异组计划(Human Variome Project, HVP)倡导以国家为计划节点,以特定疾病为单位创建基因变异数据库[4]。中国遗传资源十分丰富且自身科研力量不断发展壮大,许多科学家在国内外科学杂志上发表了大量的科研成果,亟需整合这些资源建立具有中国人种族特异性的疾病变异谱。因此在2008年,浙江大学遗传与基因组医学中心领衔建立了莱顿开放变异-中国数据库(Leiden Open Variation Database-China, LOVD-China),作为HVP的重要组成部分,现已发展为包含乳腺癌、结肠癌、长QT综合征等多种重要疾病的变异谱和表型组信息的大型综合数据库[5,6]。目前在LOVD基因变异数据管理系统的基础上,本项目团队新建立了中国人群血红蛋白病变异谱及表型谱数据库,一同整合进LOVD- China (http://www.genomed.zju.edu.cn/LOVD3/genes)。
血红蛋白病是一种广泛流行于世界范围内的遗传性血液病,具有致死、致残率高的特点,是全球重点关注的出生缺陷之一。根据其基因型及临床表现分为3种:异常血红蛋白、地中海贫血(地贫)和遗传性持续性胎儿血红蛋白增高症(hereditary persistence of fetal hemoglobin, HPFH)[7],现已报道有超过2000种致病变异和600种疾病修饰变异,血红蛋白病的变异谱有地理和种族特异性[8]。在美洲和非洲大陆,广泛流行的是由于β-肽链第6位氨基酸谷氨酸被缬氨酸代替生成血红蛋白S (hemoglobin S, Hb S)所致的镰状细胞贫血,该病危害极大,纯合子患者一般较难活过30岁,但此病在亚洲较罕见[9]。除Hb S外,全球已报道上千种异常血红蛋白(abnormal hemoglobin)变异,但90%以上的异常血红蛋白是良性的,对人体无影响,极少数异常血红蛋白可致贫血表型,如东南亚最常见的血红蛋白E (hemoglobin E, Hb E)等。在我国高发且危害巨大的血红蛋白病为地贫,长江以南区域致病基因携带率高达20%,严重影响我国出生人口质量。地贫是由于珠蛋白链合成不平衡造成的溶血性贫血,根据致病基因分为α-地中海贫血(α-地贫)和β-地中海贫血(β-地贫),中国已报道α-地贫变异80种,其中以3种缺失型变异(- -SEA/、-α3.7/、-α4.2/)和3种非缺失型变异(αWSα/、αCSα/、αQSα/)最为常见,当α-珠蛋白基因发生严重缺陷,即4个α-基因中有3个受损(- -/-α或- -/αTα),α-链合成量显著减少,多余β-链形成不稳定四聚体结构,继而再分解沉积形成H包涵体,损伤红细胞,造成中度溶血性贫血,称为血红蛋白H病(hemoglobin H disease, Hb H)。已报道β-地贫变异123种,最新流行病学调查显示我国最常见β-地贫变异为CD41-42 (-CTTT)、CD17 (AAG>TAG)、IVS-Ⅱ-654 (C>T)、-28(A>G)、CD 26 (GAG>AAG)和-100 (G>A),此6种变异携带率占总变异的94%[10,11]。β-地贫变异根据珠蛋白基因残留活性程度分为β+和β0,一般来说,患者临床表型从重到轻为:β0/β0>β0/β+>β+/β+。但是β-地贫具有较强的遗传异质性,除了致病基因HBB外,尚有大量可影响β-地贫表型的遗传修饰因素,部分基因如BCL11A、HBS1L-MYB和KLF1变异可以通过调控胎儿血红蛋白(fetal hemoglobin, Hb F)重开放表达,缓解患者贫血症状,这些基因被称作修饰基因。经本项目团队研究发现,KLF1锌指结构非同义变异对中国β-地贫患者表型修饰作用最显著,但是当同一个体携带该类变异纯合子或双重杂合子时,个体表现为非典型地中海贫血症状(atypical thalassemia)[12]。HPFH是一种良性表型的血红蛋白病,对应个体Hb F异常高表达,研究发现个体复合HPFH和β-地中海贫血变异临床贫血程度较轻,在少数病例中甚至无症状,HPFH是研究β-地贫遗传修饰机制的重要模型[13]。随着高通量测序技术的发展,越来越多的修饰变异被鉴定,部分修饰变异已经被应用于β-地贫和镰状细胞贫血的治疗方案研发中[14]。现在临床实验室广泛应用的跨越断裂点聚合酶链式反应(gap-polymerase chain reaction, Gap-PCR)、反向点杂交(reverse dot blotting, RDB)等传统检测疾病变异的技术存在筛查漏诊、不能给出精准风险预测及诊断结果等弊端,未来临床和实验室的分子筛查与疾病诊断建议采用二代测序等先进的高通量方法,以适应当前和未来快速、精准、全面的地贫筛查和临床精确诊断的需求[10]。但是,要将这类依赖大量基因测序数据的诊断方法应用于临床,医生及其团队需能够对大规模检测结果数据进行分析和解读,并关联众多临床数据库和知识库进行验证辨别,从而得到诊断结论,这对其能力提出了较高的要求。如果能推出一个全面综合的系统,可以快速注释血红蛋白病变异,做出精准的基因分型和综合诊断结果,将会大大有助于提高我国该病防治水平。因此,本项目团队开创了世界上首个血红蛋白病在线辅助精确诊断及风险评估系统DASH (diagnosis and at-risk assessment system of hemoglobinopath),辅助临床医生做出快速、精准的疾病诊断和遗传咨询,同时可为该病的预防、筛查和风险评估提供高效的分析手段。
1 中国人群血红蛋白病变异谱LOVD数据库
1.1 变异信息收集及整合
数据库整合的中国人群血红蛋白病变异谱分为两个来源:一是已发表的数据,包括血红蛋白病国际权威数据库HbVar (http://globin.cse.psu.edu/)和IthaGenes (http://www.ithanet.eu/db/ithagenes/),另外通过PubMed (https://www.ncbi.nlm.nih.gov/pubmed/)和百度学术(http://xueshu.baidu.com/)文献资源库扫描挖掘国内外文献;二是南方医科大学遗传教研室地贫课题组经过近10年的积累,汇总2087例血红蛋白病患者和20222例中国地贫高发区域血红蛋白病筛查个体的表型数据和区域捕获测序所得的基因型数据(有部分数据未曾发表)。通过以上两个途径,本项目团队在珠蛋白基因(HBA1, MIM+ 141800; HBA2, MIM* 141850; HBB, MIM+ 141900; HBG1, MIM* 142200; HBG2, MIM* 142250; HBD, MIM* 142000)和修饰基因(BCL11A, MIM* 606557; KLF1, MIM* 600599; GATA1, MIM* 305371; HMIP, MIM% 142470)上总计收集了371个变异,其中265个致病变异,106个修饰变异,涵盖PLINK关联分析本地实验室β-地贫病例的基因型和表型数据所鉴定出的34个功能性修饰变异。该数据库将中国2087 例血红蛋白病患者的表型与基因型数据整合在一起,包括血液学指标:血红蛋白量(hemoglobin, HGB)、平均红细胞体积(erythrocyte mean corpuscular volume, MCV)和平均红细胞血红蛋白量(mean corpuscular hemoglobin, MCH)等,病史信息:发病年龄和输血频率,基因型:致病变异和修饰变异,以数据库条目的形式公开(http://www.genomed.zju.edu.cn/LOVD3/ individuals),这是世界上首次共享如此大队列血红蛋白病的病例信息。1.2 数据库结构
数据库总共分为8个子版块:基因、转录本、变异、个体、疾病、筛查、提交和帮助文档。通过基因列表的页面(http://www.genomed.zju.edu.cn/LOVD3/genes)可选择感兴趣的基因并点击查看该基因主页。以HBB为例(图1),在基因主页介绍了HBB的染色体位置、转录本号、相关联疾病、已记录变异数等信息;在变异版块下,既可选择查看所有基因的变异,也可选择查看某一特定基因的变异,每一个变异的主页里均记录了其基因组位置(hg19)、俗名、人类基因组变异协会(Human Genome Variation Society, HGVS)命名、致病性以及dbSNP、参考文献、OMIM和携带该变异个体的链接。个体的主页分为4部分:(1)基本信息:个体α-和β-地贫基因型、个体罹患疾病、性别和年龄;(2)表型:血液学指标和病史信息;(3)筛选:此个体进行了哪些基因的变异检测;(4)变异:所携带的变异和杂合度信息。在该数据库中,血红蛋白病被分为6种:异常血红蛋白、α-地贫、β-地贫、Hb H病、HPFH和非典型地中海贫血,在疾病版块中可以查看每种疾病的患者个体及表型与基因型信息。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1LOVD-China HBB基因主页
Fig. 1Homepage of the HBB gene from LOVD-China
1.3 数据提交与审核
收集共享疾病相关变异是一个艰巨而漫长的任务,需要基因组医学领域研究人员和医生共同努力才可完成。LOVD-China欢迎和鼓励业界同行上传新的变异信息,根据数据库要求,需请上传者先行注册及登录,上传者应尽可能详细填写变异数据和相应的表型信息,需同时上传支持变异临床意义的文献或证据,提交的所有变异需遵循HGVS命名规则[15],提交的变异相关信息及格式应符合LOVD系统的上传要求。该数据库变异及表型谱有望助力疾病基础科学研究和临床诊治,需保证所展示分享信息的可靠性和权威性。因此,LOVD-China具有一套较严格的人工数据审核流程,数据库后台管理者会手动审阅每个条目,提交至业内专家结合变异类型及临床表型校验突变功能,通过专家核查后,后台管理者会添加新条目、更新现有的变异和流调数据。有关更详细的数据库信息,请参阅系统文档(http:// www.genomed.zju.edu.cn/LOVD3/docs/)。2 血红蛋白病在线辅助诊断及风险评估系统
本项目整合了LOVD-China、22 309例个体表型-基因型数据和HbVar数据库,构建DASH后台血红蛋白病特异性注释的数据集。DASH提供云端标准化的血红蛋白病诊断知识库和辅助决策系统(http:// www.smuhemoglobinopathy.com/),参考临床分子筛查和诊断场景,建立了3个工作模块:血红蛋白病表型-基因型推导模块、血红蛋白病精确诊断模块和血红蛋白病风险评估模块。根据键入的个体血液学表型初步推断地中海贫血的类型;识别用户上传的CNV(拷贝数变异)和SNV(单核苷酸变异)信息并用特定的血红蛋白病注释库进行注释,综合分析评估致病基因变异和修饰基因变异,对血红蛋白病进行临床精确分子分型和诊断。此外,该系统可以对夫妇双方提交的突变进行风险评估,揭示后代罹患血红蛋白病的风险。2.1 血红蛋白病表型-基因型推导模块
此模块将临床和实验室诊断血红蛋白病的传统策略制成自动化的推导算法,个体年龄、性别和血液学指标(MCV、MCH、HGB、Hb F%和异常血红蛋白等)均需填写,根据国际通用标准可以基本判断出个体血红蛋白病特征。地贫个体血常规结果表现明显的小细胞低色素特征,MCH<28 pg和MCV< 80 fL是临床判断小细胞低色素的重要诊断标准[16],另外,需注明是否铁缺乏,因为缺铁无论是否合并地贫均可致小细胞低色素贫血表型,如果个体缺铁,建议在补铁后再做一次血液学检查[17]。血红蛋白检查可分析体内血红蛋白A、血红蛋白F、血红蛋白A2百分比含量,依此可初步区分α-地贫和β-地贫。依此模块,可辅助初步筛选出地贫同型高危夫妇,为后续基因诊断和遗传咨询打下基础。由于患有静止型地贫的个体常常具有正常或边缘的血液学指标,常常导致漏诊,我们在最终分析报告中强烈建议所有个体后续做分子遗传学检测,进一步精准基因分型。2.2 血红蛋白病精确诊断模块
该模块利用特定的血红蛋白病注释库解析上传的变异列表,实现个体的疾病精准诊断。系统精确诊断分析由两个进程构成,首先是血红蛋白病特异性注释输入的变异,用户上传与血红蛋白病相关的SNPs和CNVs,页面右侧提供了详细的输入规则及范例(http://www.smuhemoglobinopathy.com/clinical/),HGVS命名、GRCh37/hg19坐标和常用俗名均为可接受格式,键入框具有自动补全(模糊匹配)的功能。注释完成后,将致病变异与修饰变异进行疾病特异综合分析评估得到精准诊断结果,此部分分析涉及到一些血红蛋白病中的特殊组合规则,需要长期的临床和实验室诊断经验才可准确掌握,当临床上碰到此类案例,极易产生误诊或漏诊的情况。比如,一般β-地贫杂合子即携带者无贫血表型,当β-地贫杂合子个体合并α-珠蛋白基因多拷贝,个体会表现中间型地贫表型,需要医生制定出对应的临床处置方案[18];个体携带修饰基因KLF1锌指结构非同义突变纯合子或双重杂合子会表现出贫血表型[19]。建议在使用模块功能前详细阅读页面右侧的输入规则和系统右上角Q&A功能区输出格式及内容的释义(http://www.smuhemoglobinopathy.com/question/#tab=1)。2.3 血红蛋白病风险评估模块
风险评估模块是针对夫妇对设计的,个体及配偶的变异列表需要同时上传,变异上传格式等规则与精确诊断模块一致,详见模块页面右侧(http:// www.smuhemoglobinopathy.com/at_risk/)。上传变异后先会对每一个个体进行全面综合的血红蛋白病分析,得到精确诊断结果,首先判断夫妇是否携带/患有同型地贫,其次组合分析父母双方的所有变异,报告这对夫妇的后代是否有罹患血红蛋白病的风险,告知后代可能有疾病风险的基因型,以及可能影响患病后代表型严重程度的修饰变异,协助临床医生进行全面的遗传咨询和制定个性化优生优育的方案。比如,单个个体携带β-地贫变异,配偶β-基因正常,但携带α-基因多拷贝变异,系统会分析得出后代可能的基因型,并告知后代有罹患β-地贫的风险。3 结语与展望
基因测序技术的快速发展,对遗传疾病的基础研究与临床应用起到了巨大的推动作用,更多遗传病相关位点被发现,更有效精准的诊断方法被发明,疾病变异谱信息及临床表型系统的收集和管理工作是实现精准医学的基石[20,21]。本项目团队通过多渠道整合疾病数据库、变异数据库和文献,收录与中国人群血红蛋白病候选基因相对应的突变,并上传南方医科大学地贫课题组多年累积的临床表型和基因型数据,以此建立起 LOVD-中国血红蛋白病变异谱资源库。另外本项目开发了DASH提供云端标准化的血红蛋白病诊断知识库和辅助决策系统,可加速新的诊断方法应用于临床:由遗传专家持续更新数据分析方法,维护疾病知识库;临床医生则可基于互联网便捷地使用辅助决策系统,提升诊疗效率。通过DASH的构建也积累了遗传病辅助决策系统的普适方法论、工程实施技术和经验(图2):(1)从医学实践中获取诊疗场景;(2)采用循证医学、精准医学的方法,从医学文献、临床指南、公共数据库和自建数据库中获取知识;(3)通过分析数据、整合知识,建立标准化、规范化的疾病知识库;(4)使用云计算、大数据和人工智能等信息技术,实现疾病知识库的共享和持续更新。
图2
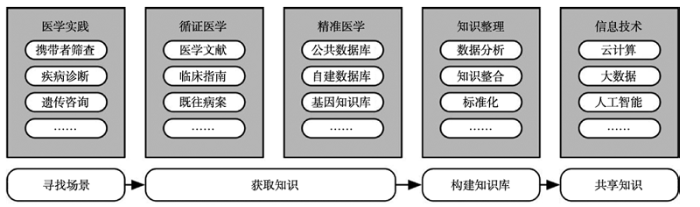 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2构建遗传病辅助决策系统的方法论
Fig. 2The methodology of constructing clinical decision support system for genetic diseases
2015年意大利研究团队开发了一套地中海贫血严重程度评分系统,通过890名β-地贫患者组成的测试集集中验证了5个修饰位点遗传变异特征与β-地贫首次输血年龄(临床严重程度)相关联,以此为基础构建了β-地贫表型严重程度打分矩阵,在http://tss.unica.it上提供了自动在线计算分数,以期作为筛查的预测评分和标准化严重程度量表,该系统强调了修饰基因在地贫诊断及筛查中的重要作用,并且将其修饰作用量化用于指导临床决策[22]。但该系统仍存在一定的局限,比如:需用户自主判断地贫类型,无法准确注释并解读地贫变异;系统只涵盖5个修饰位点,仍存在大量具有较强作用的修饰位点待补充更新。DASH是世界上首个全面整合和高效分析的血红蛋白病在线辅助精确诊断及风险评估系统,可服务于全世界基础科研人员和具有较高层次的医生群体,根据血液学指标对血红蛋白病类型进行判断和疾病特异变异综合分析评估,扩大我国在血液病和遗传病业界内的影响力,并希望获得国内外专家的评议及认可。DASH仍存在局限性和挑战,现在的页面及产出结果均为英文,较难满足国内基层医生和普通孕产妇用户的使用需求,在后续升级中将陆续推出中文版,适应不同层次用户群体的需求。此外,还计划推出一个病例分享版块,用于世界范围内的罕见病例分享,包括基因型以及临床表型,将不断吸纳扩容后台血红蛋白病诊断数据库,以此得出更为精准的诊断和遗传咨询。在数字健康应用发展迅速的今天,如何保护个体隐私是一项复杂的挑战,DASH目前不收集记录可能透漏个人身份的敏感信息,如姓名和联系方式等,在后续版块升级过程中,将应用规范的个体信息匿名化和知情同意等保护机制。
DASH是一个非营利项目,旨在为血红蛋白病的筛查、诊断和遗传咨询提供一站式的信息平台,在医学研究和产业应用中具有重要的价值。医学研究中面临数据标准化、分散病例收集和大规模数据汇集管理等诸多挑战。针对这些问题,DASH 相应实现了对血红蛋白病字段的标准化、增加电子数据采集系统和基因数据质控分析,以及采用可扩展的云端架构支持大规模数据存储和管理。在产业应用方面,DASH上的疾病数据,为诊疗技术的研发提供数据支持。DASH未来也将为多中心临床试验提供数据支持,在试验设计和患者入组等方面探索新的数据驱动解决方案。中华人民共和国国家卫生健康委员会等5部门于2018年5月22日联合发布了包含121种疾病的《第一批罕见病目录》,并在2019年2月12日发文建立全国罕见病诊疗协作网,第一批共有324家医院参与国家罕见病诊疗协作网。DASH符合国家罕见病诊疗协作网的建设思路,为单个罕见病网络化诊疗协作提供了可参考的模板(图3)。结合DASH的实践经验,可继续拓展更多的罕见疾病,鼓励更多专家参与建立公共的罕见病辅助决策系统,以云服务模式对区域和基层医疗机构赋能,以大数据和人工智能技术提升罕见病诊疗的效率,推进各个疾病领域的研究和临床进展,造福更广大的患者人群。
图3
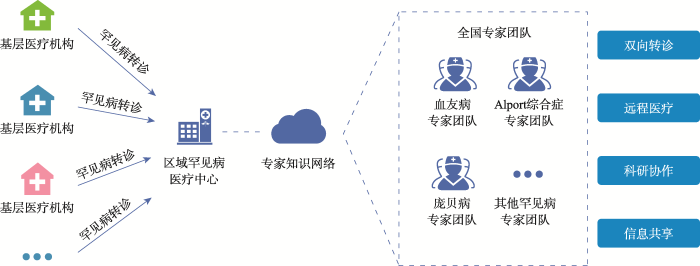 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3罕见病诊疗协作的新模式示意图
Fig. 3A new model of collaboration in the diagnosis and treatment of rare diseases
作者声明
本文作者、系统研发团队及机构之间无相关利益冲突。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}