 ,1,21.
,1,21. 2.
3.
4.
5.
Analysis of normal tissues adjacent to the tumour-specific expressed genes in breast cancer
Qichao Yu1,2, Bin Song1,2, Xuanxuan Zou1,2,3, Ling Wang4, Dequan Liu5, Bo Li1,2, Kun Ma,1,21. 2.
3.
4.
5.
通讯作者:
第一联系人:
编委: 周钢桥
收稿日期:2019-04-9修回日期:2019-05-15网络出版日期:2019-07-20
| 基金资助: |
Received:2019-04-9Revised:2019-05-15Online:2019-07-20
| Fund supported: |
作者简介 About authors
禹奇超,硕士,专业方向:肿瘤基因组学与生物信息学E-mail:yuqichao@genomics.cn。
宋彬,硕士,专业方向:肿瘤基因组学与生物信息学E-mail:songbin@genomics.cn。

摘要
关键词:
Abstract
Keywords:
PDF (1251KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
禹奇超, 宋彬, 邹轩轩, 王岭, 刘德权, 李波, 马昆. 乳腺癌癌旁组织特异性表达基因分析[J]. 遗传, 2019, 41(7): 625-633 doi:10.16288/j.yczz.19-099
Qichao Yu, Bin Song, Xuanxuan Zou, Ling Wang, Dequan Liu, Bo Li, Kun Ma.
已有研究表明,距离肿瘤组织(tumour tissues, T)1 cm以上的癌旁组织(normal tissues adjacent to the tumour, NAT)与肿瘤组织在pH[1]、转录图谱和表观遗传修饰[2]等方面都存在显著的差异。由于NAT可以在肿瘤切除手术中获得,样本采集相对容易,长期以来,绝大多数实体瘤研究都是以NAT作为对照,如癌症基因组图谱计划(The Cancer Genome Atlas, TCGA)[3]和国际癌症基因组联盟(International Cancer Genome Consortium, ICGC)[4]。然而,针对NAT的转录组研究却十分有限,NAT作为对照能否获得真实可靠的肿瘤组织差异表达基因(differentially expressed gene, DEG)仍存在疑问[5]。
Slaughter等[6]认为癌症的形成是一个变异逐渐积累的过程,将NAT视为一种中间状态,即肿瘤形成前形态学正常但分子水平已经发生变化的一群细胞。根据这个理论,NAT在分子生物学水平并不是真正的“正常”。因此,用NAT作为对照可能难以获得准确的分子水平上的差异。
人类的多种器官,包括肺、肾脏、乳房等都是成对存在的,但是在原发肿瘤患者中左右两侧同时产生肿瘤的比例却非常低。由此推测,无肿瘤的对侧正常组织(contralateral breast normal tissues, CBN)在分子生物学水平更接近真实的“正常”,用作癌症研究的对照应更为合适。基于此,本研究收集了14例在身体一侧患有乳腺癌的患者,对CBN、NAT和T 3种组织进行RNA测序[7],并通过对每个患者的CBN、NAT和T的全转录组各基因进行差异表达分析,结合基因富集和蛋白-蛋白互作(protein-protein interaction, PPI)分析,检测到NAT相比CBN有102个差异表达基因。进一步分析发现,这些差异基因主要富集到肿瘤坏死因子(tumour necrosis factor, TNF)和上皮间质转化(epithelial-mesenchymal transition, EMT)基因集中,其中有23个基因是癌旁特异激活基因,仅显著富集到TNF基因集中。此外,本研究还发现一些基因在NAT中的表达水平与肿瘤组织无显著差异,但与CBN相比有显著差异,而这些基因往往在以NAT为对照的研究中被遗漏。
1 材料与方法
1.1 样本采集
用于本研究的组织材料来自云南省肿瘤医院和第四军医大学西京医院,从14例单体侧乳腺癌患者(表1)获得肿瘤组织、癌旁组织和对侧乳房正常乳腺组织,这些患者的另一个乳房有非肿瘤良性病变,为防止恶化,良性病变组织被切除。本研究取得了患者的知情同意,并通过了上述医院和华大基因生命伦理生物安全与遗传资源管理委员会(BGI- IRB)的伦理审查。医院采集了这14例患者的肿瘤组织(T),癌旁组织(NAT,距离肿瘤组织5 cm以上)和对侧正常乳腺组织(CBN),共计42个组织样本。所有样本的RNA提取、逆转录、扩增、建库等实验操作由深圳国家基因库完成。cDNA文库由BGISEQ-500测序仪完成单端50 bp (SE50)和双端100 bp (PE100)测序。Table 1
表1
表1 患者临床信息
Table 1
| 患者编号 | 年龄 | 病理类型 | 患病部位 | TNM |
|---|---|---|---|---|
| P01 | 46 | 乳腺浸润癌 | 左侧 | T2N1aM0 |
| P02 | 58 | 乳腺浸润性导管癌 | 左侧 | T1cN1M0 |
| P03 | 53 | 乳腺浸润性导管癌 | 左侧 | T2N1aM0 |
| P04 | 46 | 乳腺浸润性导管癌 | 左侧 | T1cN0M0 |
| P05 | 50 | 乳腺浸润性小叶癌 | 左侧 | T2N0M0 |
| P06 | 43 | 乳腺浸润癌 | 左侧 | T2N1aM0 |
| P07 | 44 | 乳腺浸润性导管癌 | 左侧 | T1N0M0 |
| P08 | 44 | 乳腺浸润性导管癌 | 左侧 | T2N0M0 |
| P09 | 49 | 乳腺浸润性导管癌 | 左侧 | T2N3M0 |
| P10 | 44 | 乳腺浸润性导管癌 | 右侧 | T1N0M0 |
| P11 | 60 | 乳腺浸润性导管癌 | 右侧 | T1cN0M0 |
| P12 | 50 | 乳腺浸润性导管癌 | 左侧 | T1cN0M0 |
| P13 | 60 | 乳腺浸润性导管癌 | 左侧 | T1cN1aM0 |
| P14 | 32 | 乳腺浸润癌 | 左侧 | T1bN2aM0 |
新窗口打开|下载CSV
1.2 数据获取
本研究产生的测序数据存放于国家基因库数据库(CNGBdb)的核酸序列归档系统(CNSA, https:// db.cngb.org/cnsa/)中,检索码(accession code)为CNP0000385。1.3 测序数据处理
首先用SOAPaligner将原始序列比对到rRNA上,映射到rRNA的序列被去除,剩下的序列用Bowtie2[8]比对到人类RefSeq参考基因上(hg19)。本研究使用RSEM[9]获得样本的基因读段(read)数目及表达水平的结果。1.4 基因表达谱分析
使用上四分位数(upper quartile)标准化的方法校正了基因的读段数目,然后使用edgeR包[10]基于标准化后的读段数据进行差异表达基因分析。对每个患者的3种组织两两之间都进行了差异表达基因分析。在2个或2个以下样本中检测到少于10条读段的基因被去除。使用错误发现率(false discovery rate, FDR)、倍数变化(fold change, FC)和基因覆盖的读段数目对差异表达基因进行过滤。基因覆盖深度使用CPM (counts per million),即每100万条读段中映射到某基因上的读段数进行量化。同时满足FDR<0.05、log2(FC)的绝对值大于1且log2(CPM)>3的基因被视作差异表达基因。校正后的CPM数据用于Rtsne降维分析。1.5 蛋白-蛋白相互作用分析
将得到的差异表达基因集输入在线工具STRING (https://string-db.org/)[11],筛选出差异表达基因间的相互作用关系,获得蛋白-蛋白相互作用网络。同时,该工具还可对得到的PPI网络进行图形可视化展示。1.6 差异表达基因的富集分析
使用50个Hallmark基因集在基因富集分析(gene set enrichment analysis, GSEA)[12]网站(http:// software.broadinstitute.org/gsea/msigdb/annotate.jsp)对各组差异表达基因进行富集分析。通过计算了富集分数(enrichment score, ES),即-log10(P value),对不同的富集结果进行排序。当P值为0.05时,对应的富集分数为1.3。富集分数越大,说明富集的显著性越高。1.7 统计方法
统计分析由R程序实现。双侧t检验P<0.05被视为具有统计学显著性。2 结果与分析
2.1 差异表达基因分析
14例患者CBN、NAT和T 3种组织样本平均测序深度分别为168.4×、207.5×和186.4×,平均测序深度达186.4× (hg19);平均检测基因数分别为10 998.7、10 769.1和10 580.8,平均每个组织能检测到10 782.9个基因。平均每个患者CBN与NAT的差异表达基因的有967.6个,在55%以上患者共享的DEG有102个。NAT特异性表达基因(即与CBN和T相比,在NAT中都上调或下调)有2045个,平均每个患者携带246个。
CBN、NAT和T 3种组织t-SNE降维分析的结果显示T形成了一个独立的簇,说明肿瘤组织与CBN和NAT的表达谱有明显差异(图1A)。CBN与NAT形成一个簇,说明二者的表达谱有一定的相似性。通过比较3个组织两两之间检测到的DEG数目,发现NAT与T的差异表达基因数目(平均值2477.6),显著小于CBN与T的DEG数目(平均值3069.2,P<0.05,图1B)。从DEG数目上看,与CBN相比,NAT与肿瘤组织的基因表达谱一致性更高,即癌旁组织的基因表达谱是处于对侧正常组织与肿瘤组织中间的一种状态。
图1
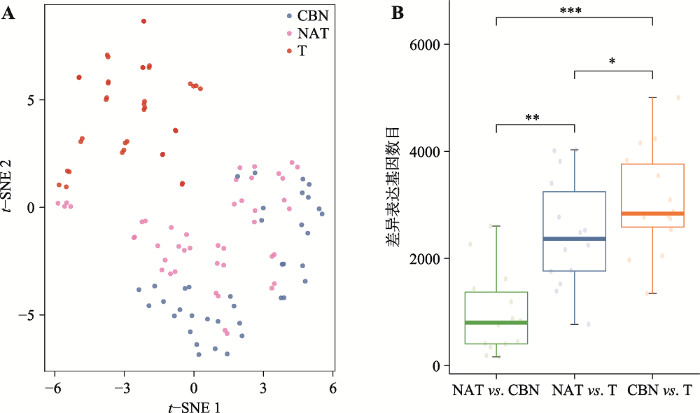 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CBN、NAT和T的转录组特征
A:3种组织的t-SNE图;B:3种组织两两比较DEG的数目。*:P<0.05, **:P<0.01, ***:P<0.001。NAT:癌旁组织,CBN:对侧正常乳腺组织,T:肿瘤组织。
Fig. 1Transcriptomic features of CBN, NAT and T
2.2 NAT相比CBN的差异表达基因分析
对14例患者CBN相比NAT的DEG分别进行Hallmark基因富集分析,发现“响应TNF受NF-κB调控的基因”(HALLMARK_TNFA_SIGNALING_ VIA_NFKB,以下简称TNF基因集)和EMT基因集是富集最为显著的两个基因集(图2A)。图2
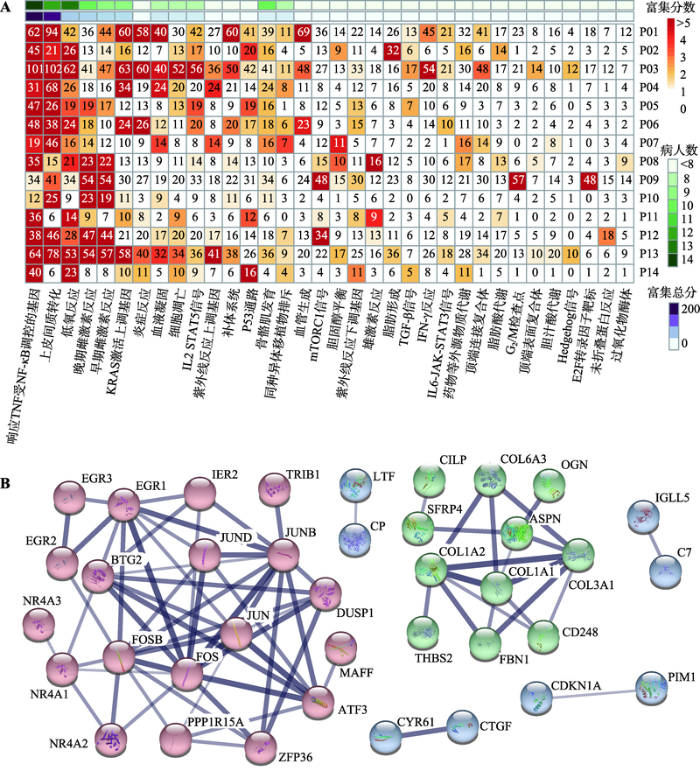 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2NAT相比CBN的差异表达基因
A:55%以上患者共有的102个DEG的Hallmark基因富集热图。富集分数ES=-log10(P value),富集总分为各样本富集分数之和,热图中的数值表明患者富集到该基因集的DEG数目。B:102个DEG的蛋白-蛋白互作网络。未展示64个不在PPI网络中的DEG。包含大于等于3个基因的簇(cluster)有2个,其中A簇(红色):TNF基因集;B簇(绿色):EMT基因集。
Fig. 2DEGs of NAT vs. CBN
筛选出在55%以上患者中复现的102个DEG进行蛋白-蛋白互作分析,在互作关系的筛选过程中去掉了可靠性较低的(来源于文本挖掘和数据库)蛋白-蛋白互作关系对,得到更可靠的蛋白互作网络(图2B)。这些DEG中,有38个基因出现在PPI网络中(75条边,P<1.0e-16),形成2个相互独立的簇(cluster)。通过对每个簇中的基因进行富集分析发现,A簇(红色)显著富集于TNF基因集(包含17个基因,P=6.6e-40),B簇(绿色)显著富集于EMT基因集(包含6个基因,P=5.2e-13)。其中A簇富集的基因集与以往的研究结果一致[5]。
2.3 对侧正常乳腺组织、癌旁组织和肿瘤组织的基因表达模式分析
对CBN、NAT与T进行差异表达分析,根据基因从CBN到NAT再到T的表达水平变化,可将基因的表达模式划分成4种类型,即NAT特异性表达型(NAT-specific,A型),梯度型(gradient,G类),类肿瘤型(tumour-like,T型)和类正常型(normal-like,N型)[5]。同时用“1”和“2”分别表示基因上调和下调,可以将基因的表达模式细分为8个小类。对每个病人的DEG划分类型后统计每种类型的平均基因数目和高频复现基因数目(定义为55%以上患者共有的基因),并分别做了富集分析,计算了平均富集分数(表2)。Table 2
表2
表2 基因在对侧正常乳腺组织、癌旁组织和肿瘤组织中的表达模式
Table 2
| 类型 | 模式 | MGC | RGC | 富集结果1(MS) | 富集结果2(MS) |
|---|---|---|---|---|---|
| A | UD/DU | 246 | 23 | 响应TNF受NF-κB调控的基因(14.17) | 低氧反应(3.43) |
| G | UU/DD | 91 | 0 | 上皮间质转化(6.39) | - |
| T | US/DS | 463 | 1 | 上皮间质转化(5.70) | 响应TNF受NF-κB调控的基因(2.69) |
| N | SU/SD | 1672 | 335 | 上皮间质转化(10.19) | IFN-γ反应(5.38) |
| A1 | UD | 190 | 23 | 响应TNF受NF-κB调控的基因(15.32) | 低氧反应(3.72) |
| A2 | DU | 56 | 0 | - | - |
| G1 | UU | 37 | 0 | 上皮间质转化(7.34) | - |
| G2 | DD | 54 | 0 | - | - |
| T1 | US | 154 | 0 | 上皮间质转化(7.52) | 响应TNF受NF-κB调控的基因(4.24) |
| T2 | DS | 309 | 1 | - | - |
| N1 | SU | 828 | 111 | 上皮间质转化(10.29) | IFN-γ反应(10.12) |
| N2 | SD | 844 | 224 | 骨骼肌发育(4.26) | 早期雌激素反应(3.88) |
新窗口打开|下载CSV
NAT相比CBN和T都上调的基因(A1型)被称为癌旁特异激活基因(tumour-adjacent speci?c activation, TASA)。本研究共计检测到1403个TASA基因,平均每个患者有190个。高频复现的TASA基因有23个,其中包括8个已报道的基因:ATF3、CSRNP1、EGR1、EGR2、EGR3、FOS、FOSB和NR4A3,15个新的TASA基因:ADAMTS1、APOLD1、CYR61、DUSP1、JCHAIN、JUN、KLF2、KLF4、MAFF、NR4A3、PPP1R15A、RASD1、TNXB、VEGFD和ZFP36。对上述23个TASA基因进行PPI分析,得到一个包含19个节点、27条边的蛋白互作网络(P<1.0e-16)。富集分析发现,这些基因显著富集在TNF基因集中(包含16个基因,P=1.8e-34,图3C),与以往的研究结果相似[5]。值得注意的是,新发现的基因中,DUSP1、JUN、KLF2、KLF4、MAFF、NR4A1、PPP1R15A和ZFP36也包含在TNF基因集中,其中KLF4和JUN是COSMIC数据库(https://cancer.sanger.ac.uk/census)收录的癌基因(图3,A和B)。NAT相比CBN和T都下调的基因(A2型)基因有642个,平均每个患者56个,没有发现高频复现基因和显著富集结果。此外,A2型表达模式的基因数目显著低于A1型(P<0.05)。
图3
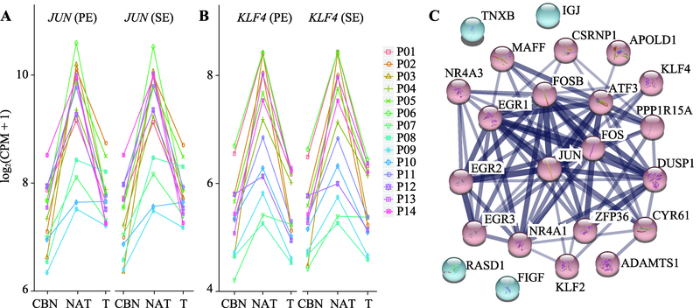 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3多个患者中复现的癌旁特异激活基因
A:JUN基因在3种组织中的表达水平;B:KLF4基因在3种组织中的表达水平;C:高频复现的TASA基因PPI网络。SE:单端测序数据;PE:双端测序数据。
Fig. 3TASA genes in multiple patients
平均每个患者检测到37个G1型(从CBN、NAT到T持续上调)基因和54个G2型(从CBN、NAT到T持续下调)基因,未发现高频复现基因,G1类型基因同样显著富集到了EMT基因集。平均每个患者检测到154个T1型基因和309个T2型基因,而这些基因在常规的肿瘤-癌旁成对转录组研究的DEG分析中可能会被遗漏。其中T1型显著富集到EMT和TNF等基因集中,T2型没有显著富集结果,仅有一个高频复现基因MIRLET7BHG。这些基因中有85个的复现率大于35%,包含COSMIC数据库中4个已知的癌基因:BRIP1、CDKN1A、COL3A1和POU5F1。平均每个患者检测到828个N1型基因和844个N2型基因,高频复现基因数目分别为111和224个。N1型基因显著富集到EMT基因集和IFN-γ反应等基因集中,N2型基因显著富集到骨骼肌发育和早期雌激素反应等基因集中(表2)。
A2、G2和T2型基因几乎没有高频复现基因,且没有显著富集结果。放宽过滤条件发现它们都能富集到早期雌激素反应基因集中,与N2型基因的富集结果类似,但富集分数远低于A1、G1和T1型基因,说明在肿瘤组织中下调的基因的共表达模式与上调基因相比随机性升高。此外,本研究还分别对A、G、T、N 4种模式的基因进行富集分析,富集结果与A1、G1、T1、N1高度一致(表2)。
3 讨论
通过对14例乳腺癌患者3种组织(CBN、NAT和T)高深度RNA测序数据分析,本研究在比较NAT和CBN的转录图谱分析中得到了102个在大于55%患者中复现的DEG,表明NAT与CBN的基因表达谱存在明显差异。通过对DEG进行Hallmark基因富集分析,发现这些DEG显著富集到TNF基因集和EMT基因集中。蛋白-蛋白互作分析将这些DEG分为2个显著的簇,而这两个簇分别对应了TNF基因集(A簇)和EMT基因集(B簇)。富集到TNF基因集的基因包括JUN、FOS和FOSB等,富集到EMT基因集的基因包括COL1A1、COL3A1和COL1A2等(图2B)。Aran等[5]根据GTEx (the Genotype-Tissue Expression)[13]的非肿瘤组织样本和TCGA的多种癌症组织和癌旁组织样本的基因表达谱发现了262个NAT特异性表达基因,通过PPI分析将蛋白互作网络分成4个簇,包括细胞分裂(Ⅰ簇)、免疫反应(Ⅱ簇)、细胞刺激(Ⅲ簇)和ATP(Ⅳ簇)。本研究发现A簇中的基因与Aran等[5]报道的Ⅲ簇基因基本一致,这些基因能够显著富集到TNF基因集。其中FOS和JUN被认为是促炎性即刻早期基因(immediate-early gene, IEG),可能参与免疫系统的早期反应。此外,本研究的结果与Aran等[5]有所不同:(1)免疫反应(Ⅱ簇)在本研究的结果中不是富集最显著的基因集,其富集总分(32.9)远小于TNF(208.8)和EMT(191.9)基因集;(2) ATP (Ⅳ簇)没有出现在本研究的显著富集基因集中。
本研究还发现了23个高频复现的TASA基因显著富集到TNF基因集中。与已报导的18个TASA基因相比[5],发现了15个新基因,其中8个包含在TNF基因集中。TNF信号通路能够影响细胞增殖、分化和免疫反应,同时又可激活促进凋亡或抑制凋亡通路:TNF-α活化caspase-8和caspase-10诱导凋亡,又能通过NF-κB抑制凋亡[14]。本研究发现的TASA基因富集在“响应TNF受NF-κB调控的基因”集合,说明在癌旁组织中这些TASA基因可能激活了抑制凋亡通路,癌旁组织很可能受到了癌细胞的影响或者部分癌旁组织的细胞已经开始癌变。此外,FOS、ATF3、JUN、PPP1R15A和KFL4同时富集在TP53通路中,表明TP53通路在癌旁组织中可能被激活。除此之外,新发现的DUSP1、NR4A1和VEGFD都包含在MAPK信号通路中。以往研究表明,异常活化的MAPK信号通路在细胞恶性转化及演进中发挥重要作用,并且与乳腺癌、卵巢癌、胃癌和肝癌等癌症的发生与进展密切相关[15,16]。新发现的KLF4和JUN都是COSMIC数据库收录的重要的癌基因。KLF4在细胞中扮演了双重角色,它既能够促进细胞存活,在一定条件下又能够促进细胞死亡。有研究报道在原发乳腺导管癌中,KLF4的活化起到促进癌症进展的作用[17]。然而,也有研究报道在乳腺癌SK-BR-3细胞系中激活KLF4能够促进细胞凋亡,从而抑制肿瘤发生过程[18]。在乳腺癌MCF-7细胞系中,JUN基因的过表达显著增加了癌细胞的致瘤性和侵入性[19],在乳腺癌肝转移过程中也发挥了关键作用[20]。本研究基于14例散发乳腺癌患者,证实了TASA基因在乳腺癌癌旁组织中普遍存在,并且与免疫系统的TNF信号有关。
Aran等[5]报导的TASA基因有10个在本研究的数据集中复现率较低,除了JUND基因的复现率(50%)接近阈值之外,其他基因复现率都小于40%。据此推测,造成这些差异的原因可能有以下3点:(1)本研究的每个患者都包含CBN、NAT和T3种组织,来源统一,而Aran等[5]的研究显然使用了不同患者;(2)本研究的样本都是来源于同一种癌症患者,而Aran等[5]使用的对照来自GTEx,而这些样本不是肿瘤患者;(3)本研究的数据来源于相同的测序平台(BGISEQ-500),而Aran等[5]使用的数据来源于GTEx和TCGA,产出数据的平台不同。来源于同一个患者、同一类癌症、同一种测序平台的数据则更为可靠。
通过分析A、G、T、N 4种表达模式的基因,本研究发现A1、G1、T1、N1和N2型的基因能够显著富集在与癌症相关的基因集中,如TNF、EMT和IFN-γ反应,而A2、G2、T2型的基因未能富集到类似的基因集中,推测这3种类型的基因可能与癌症发生和进展的关系较小。
由于NAT相对容易获取,包括TCGA和ICGC在内的绝大多数实体肿瘤转录组研究都使用NAT作对照。但是,类肿瘤型(T1和T2型)表达模式的基因在癌旁组织和肿瘤组织中的表达水平无显著差异,而与对侧正常组织相比是有显著差异的。在这种情况下,癌旁-肿瘤配对的转录组分析几乎不可避免地会遗漏这种类型的DEG。另一方面,癌旁特异性表达基因(A1和A2型)的存在会导致对DEG结果的误判,即一些在正常组织和肿瘤组织中表达水平没有显著差异的基因会被错误的认为是DEG。这会对分析结果造成很大的影响,进而干扰研究的结论。
Ma[21]提出“两侧对称动物胚胎发育左-右分隔原理”:人的左、右体侧是分别由胚胎8-细胞期左侧四个细胞衍生的后代和右侧4个细胞所衍生的后代构成的。分布在人体左、右体侧的成对器官在发育过程中由于细胞分裂DNA复制而产生的体细胞变异是独立的和随机的。如果这些变异会导致某些基因表达的变化,那么建造左、右体侧成对器官细胞的转录组是可能有明显差异的。然而,除非不同的人在同种成对器官上患同类疾病,在构成他们左、右体侧同种成对器官的细胞间复现共同的转录组差异的概率是很小的。根据该原理,建议在癌生物学研究,尤其是成对器官一侧患癌,另一侧未患癌的癌症研究中,尽量获得对侧正常组织样本作为对照,以减少癌旁组织特异性表达基因带来的干扰,提高组学数据分析结果的准确性。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 12]
URL [本文引用: 1]
Magsci [本文引用: 1]

转录组是特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的集合。转录组研究能够从整体水平研究基因功能以及基因结构, 揭示特定生物学过程以及疾病发生过程中的分子机理。RNA-Seq作为一种新的高效、快捷的转录组研究手段正在改变着人们对转录组的认识。RNA-Seq利用高通量测序技术对组织或细胞中所有RNA反转录而成的cDNA文库进行测序, 通过统计相关读段(reads)数计算出不同RNA的表达量, 发现新的转录本; 如果有基因组参考序列, 可以把转录本映射回基因组, 确定转录本位置、剪切情况等更为全面的遗传信息, 已广泛应用于生物学研究、医学研究、临床研究和药物研发等。文章主要介绍了RNA-Seq原理、技术特点及其应用, 并就RNA-Seq技术面临的挑战和未来发展前景进行了讨论, 为今后该技术的研究与应用提供参考。
Magsci [本文引用: 1]
转录组是特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的集合。转录组研究能够从整体水平研究基因功能以及基因结构, 揭示特定生物学过程以及疾病发生过程中的分子机理。RNA-Seq作为一种新的高效、快捷的转录组研究手段正在改变着人们对转录组的认识。RNA-Seq利用高通量测序技术对组织或细胞中所有RNA反转录而成的cDNA文库进行测序, 通过统计相关读段(reads)数计算出不同RNA的表达量, 发现新的转录本; 如果有基因组参考序列, 可以把转录本映射回基因组, 确定转录本位置、剪切情况等更为全面的遗传信息, 已广泛应用于生物学研究、医学研究、临床研究和药物研发等。文章主要介绍了RNA-Seq原理、技术特点及其应用, 并就RNA-Seq技术面临的挑战和未来发展前景进行了讨论, 为今后该技术的研究与应用提供参考。
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}