 ,, 吴强,上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240
,, 吴强,上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240Chromatin architectural protein CTCF regulates gene expression of the UGT1 cluster
Xiaofei Zheng, Haiyan Huang,, Qiang Wu,Center for Comparative Biomedicine, Key laboratory of Systems Biomedicine (Ministry of Education), Institute of Systems Biomedicine, Shanghai Jiao Tong University, Shanghai 200240, China通讯作者:
责任编辑: 方向东
收稿日期:2019-03-15修回日期:2019-04-8网络出版日期:2019-06-20
| 基金资助: |
Received:2019-03-15Revised:2019-04-8Online:2019-06-20
| Fund supported: |
作者简介 About authors
郑晓飞,硕士研究生,专业方向:遗传学E-mail:1159171993@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (1277KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郑晓飞, 黄海燕, 吴强. 染色质架构蛋白CTCF调控UGT1基因簇的表达[J]. 遗传, 2019, 41(6): 509-523 doi:10.16288/j.yczz.19-072
Xiaofei Zheng, Haiyan Huang, Qiang Wu.
脊椎动物基因组包含一系列能够编码多种蛋白质的特殊基因簇,它们由可变区和恒定区组成[1,2],这些基因簇包括免疫球蛋白(Ig)、T细胞受体(Tcr)、原钙粘蛋白(Pcdh)和尿苷二磷酸葡糖醛酸转移酶1 (UGT1)等[1,3]。其中,UGT1基因簇编码的UGT (UDP- glucuronosyltransferase)蛋白质家族是一类Ⅱ相药物代谢酶,这类酶能够将葡萄糖醛酸供体基团转移到各种内外源疏水小分子化合物受体底物上,将它们转化为水溶性物质而排出体外,或者影响小分子药物的药代动力学和药物最终的生物学效应[4]。内源性底物包括许多在机体内起重要作用的小分子化合物,如胆红素、胆汁酸、脂酸、类固醇、甲状腺激素和脂溶性维生素等;外源性底物则包括小分子药物、环境污染物和致癌化合物等。它们先经过Ⅰ相代谢酶P450氧化成极性化合物,再被UGT等Ⅱ相代谢酶进一步极化后排出体外。
哺乳动物UGT分为两大类:UGT1和UGT2[3,5],分别被UGT1和UGT2两个基因簇编码。其中UGT2又可分为两个亚家族:UGT2A和UGT2B。UGT2A分为可变区和恒定区,可变区内有2个外显子,恒定区内有5个外显子,而UGT2B有7个基因,每个基因都由一串外显子单独构成[6,7]。UGT1基因簇结构与UGT2基因簇明显不同,但却类似于原钙粘蛋白基因簇[8],由可变区和恒定区组成,可变区包含9个成串排列的外显子,恒定区包含4个外显子,每一个可变区外显子都有其自身的启动子,启动子激活后转录出的信使RNA前体能够通过可变剪接形成9种不同的转录本[1,5]。每个可变区外显子编码信号肽和N端糖苷受体结合域,4个恒定区外显子编码高度保守的UDPGA供体结合域和C端内质网锚定跨膜段。因此,UGT1基因簇编码的葡醛酸转移酶家族可以催化大量亲脂内外源化合物与UDP-葡萄糖醛酸的接合反应,将它们转化为亲水的葡萄糖醛酸化合物[9]。尽管UGT1基因簇结构类似于原钙粘蛋白基因簇,但其架构蛋白CTCF (CCCTC-结合因子)结合位点的分布特征与原钙粘蛋白基因簇截然不同,解析UGT1复杂基因簇的三维转录调控机制将会进一步拓宽人们对拓扑结构域(topological domain)内这一类复杂基因簇的转录调控的认知。
基因的表达调控和三维基因组中染色质高级结构密切相关,特别是远端增强子和目标启动子之间的特异性远程相互作用对于启动子激活至关重要。CTCF是一种高度保守和广泛表达的转录因子,是组织发育必需的一种染色质架构蛋白[10],在染色质折叠中起关键作用[11],依靠11个锌指蛋白域(ZFs)方向性结合到人类基因组约4万个特定序列元件(CTCF binding sites, CBSs)上[12,13],其中60%以上的结合都没有组织特异性(tissue-invariant)[14,15,16]。早期认为CTCF起到转录抑制作用,后来发现它也具有转录激活功能,近年来发现其在三维基因组架构中起到关键作用。总之,CTCF蛋白具有多种复杂甚至相反的功能,包括转录激活和抑制、基因印迹、RNA聚合酶暂停、选择性剪接、DNA复制和修复、染色体缩合或凝结(chromosome condensation)和易位(translocation)、X染色体失活、肿瘤发生、免疫系统V(D)J重组和神经系统启动子选择等[12,13]。CTCF蛋白可能通过自二聚化[17],或与cohesin蛋白质复合体相互作用参与形成染色质环[18,19,20,21,22],一般成环的一对CTCF结合位点的方向是相向的[13,23~25]。染色质架构蛋白CTCF通过方向性结合决定着染色质的环化方向[25]。在与UGT1基因簇结构高度相似的原钙粘蛋白基因簇中,CTCF通过方向性识别各增强子和启动子,形成长距离染色质环化结构,从而实现了对原钙粘蛋白基因时空表达的精密调控[23]。
利用Ⅱ型成簇规律间隔短回文重复系统CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nucelase 9)发展起来的基因组编辑技术为探索染色质环化机制和研究染色质高级结构提供了良好的技术平台[26,27]。CRISPR基因组编辑依赖于sgRNA (single-guide RNA)将Cas9酶定位到靶基因并进行切割,然后通过非同源末端连接(nonhomologous end-joining, NHEJ)、同源重组(homologous recombination, HR)、微同源介导的末端连接(microhomology- mediated end joining, MMEJ)或单链退火(single-strand annealing, SSA)方式修复切割末端[13,28,29]。采用双位点的DNA片段编辑(DNA fragment editing)方法可以删除、反转或重复所研究的目标片段[13,30]。本研究通过敲低细胞中CTCF或SMC3(cohesin亚基)的转录水平并利用CRISPR介导的DNA片段编辑技术对人类UGT1基因簇中保守的CBS元件进行编辑,旨在探索UGT1基因簇中CTCF结合位点对UGT1基因簇转录调控的影响,对研究UGT1基因表达调控网络的结构基础具有重要意义。
1 材料与方法
1.1 实验材料
人肺癌细胞系A549由上海交通大学电信学院仪器系付华林老师提供;人胚肾细胞系293T购买于中国科学院细胞库;胎牛血清、青霉素/链霉素双抗和DMEM购自美国Gibico公司;psPax2和pMD2.G质粒购自美国Addgene公司;pLKO.1质粒购自美国Sigma公司;CTCF、SMC3抗体购自英国Abcam公司;Cas9质粒由北京大学席建忠教授馈赠;pGL3- U6-sgRNA-puro质粒由南京大学黄行许教授馈赠;逆转录试剂购自南京诺唯赞生物科技有限公司;荧光定量PCR试剂购自瑞士Roche公司;Annealing buffer、T4 DNA ligase以及RNA-seq试剂盒购自美国NEB公司;Lipo3000购自美国Invitrogen公司;rTaq酶购自日本TaKaRa公司;质粒中抽试剂盒购自德国Qiagen公司;Trizol购自美国Ambion公司;所有引物均由上海生物工程股份有限公司合成。1.2 CTCF方向性及结合基序分析
ChIP-seq数据来自NCBI,抽取富集CTCF的DNA序列。所分析的人源细胞系数据包括人肺癌细胞系A549 (GSM822289)、人肝癌细胞系HepG2 (GSM733645)、人慢性髓系白血病细胞系K562 (GSM749733)、人胚肾细CBS胞系HEK293 (GSM749668)、正常人表皮角质形成细胞NHEK (GSM749707)、人乳腺癌细胞系MCF7 (GSM1022658)和人子宫内膜腺癌细胞系HEC-1-B (来自于本课题组)。所分析的小鼠细胞系包括B细胞淋巴瘤CH12 (GSM923568)、GATA 1-红系祖细胞G1E (GSM923570)和第0天的胚胎干细胞ES-B4 E0 (GSM918748);小鼠组织包括8周龄心脏(GSM918756)、肾脏(GSM918731)、肝脏(GSM918715)和肺(GSM918722)。利用在线软件CTCF BSDB2.0预测每个CTCF结合的motif,并判别其方向[31] (http://insulatordb. uthsc.edu/storm_new.php)。利用软件Clustal X和在线软件BoxShade对CTCF结合基序进行序列分析比对(https://embnet.vital-it.ch/software/BOX_form.html)。
1.3 cohesin与增强子标志分析
ChIP-seq数据来自NCBI,分析cohesin亚基SMC3及增强子标志p300和H3K27ac在不同人源细胞系的分布。人肺癌细胞系A549的SMC3、p300和H3K27ac GEO序列号分别是GSM3106366、GSM1010827和GSM2421872;人肝癌细胞系HepG2的SMC3、p300和H3K27ac GEO序列号分别是GSM935542、GSM935545和GSM733743;人宫颈癌细胞系HeLa-S3的CTCF、SMC3、p300和H3K27ac GEO序列号分别是GSM822285、GSM935384、GSM935500和GSM733684。1.4 细胞培养
A549细胞和293T细胞的培养基配方为DMEM中加入10%的胎牛血清和1%的青霉素/链霉素双抗。培养条件为37℃、5% CO2。1.5 shRNA慢病毒包装和感染细胞
shRNA (short hairpin RNA)引物序列采用文献[23]中的序列(表1)。寡核苷酸的两条链经过变性、退火后连接到pLKO.1载体,通过转化感受态细菌和质粒抽提得到shRNA的重组质粒。293T细胞用无抗培养基传代到10 cm培养皿,密度为8×105 cells/mL。第2天用Lipo3000转染质粒DNA (5.4 μg psPax2、0.6 μg pMD2.G、6 μg载体质粒),24 h后换为正常培养基,分两次回收病毒悬液,过滤得到病毒初始液,加入5×PEG母液,混匀、4℃放置过夜后4℃、4000×g离心30 min,得到shRNA病毒沉淀,加入适量DMEM溶解沉淀并分装存于-80℃。
在六孔板准备A549细胞,密度为1×105 cells/mL。第2天用慢病毒感染细胞,培养24 h后换培养基,48 h后收集细胞。
1.6 Western blot鉴定
收集细胞样品中加入100 μL RIPA裂解液,冰上放置30 min,4℃、13000×g离心15 min,收集上清并分装,液氮速冻后放-80℃冰箱待用。配制10%分离胶:4 mL MilliQ水、2.4 mL 1.5 mol/L Tris-HCl (pH 8.8)、3.4 mL 30% Arc-Bis、100 μL 10% SDS、100 μL 10%过硫酸铵、10 μL四甲基乙二胺。4%积层胶包括:3 mL MilliQ水、500 μL 1 mol/L Tris-HCl (pH 6.8)、522 μL 30% Arc-Bis、40 μL 10% SDS、40 μL 10%过硫酸铵、6 μL四甲基乙二胺。取10 μL蛋白样品于PCR管中,补MilliQ水至16 μL,加入4 μL 5× SDS Loading buffer混匀后,95℃ 5 min。点样并电泳,首先80 V电泳30 min,然后120 V电泳2 h,剪切所需蛋白胶,按照滤纸-硝酸纤维素滤膜-凝胶-滤纸的顺序叠加,在电泳槽中0.16 A电泳2 h。然后将印记后的膜转入5%的脱脂牛奶中,室温避光摇晃1 h后,按照目的条带大小剪切膜,分别加入一抗稀释液,4℃摇晃过夜。加入PBST,室温摇晃5 min,重复3次。加入1 mL二抗稀释液,室温摇晃1 h,再用PBST洗膜3次。最后用Odyssey双色红外激光成像系统扫膜成像。Table 1
表1
表1 本研究使用的引物序列
Table 1
| 类型 | 引物 | 序列(5′→3′) |
|---|---|---|
| qPCR | hCTCF F | TCTGGACGACAATGAGGATGAG |
| hCTCF R | GCACCTGTATTCTGGTCTTCAAC | |
| 1A6F1 | CAACTGTAAGAAGAGGAAAGAC | |
| 1A7F1 | CCCCTATTTTTTCAAAAATGTCTT | |
| 1A9F1 | GAACATTTATTATGCCACCG | |
| 1AR | ATTGATCCCAAAGAGAAAACCAC | |
| hSMC3 F | AATGATGTGATGAACCTCCTTG | |
| hSMC3 R | TGAGAATCTGGTGCTGTTGC | |
| PCR | C1F | GGCTGCTCCTCCTATTTCCTG |
| C1F2 | GACCAGGACAAGGAGGCGT | |
| C1F5 | TCTCCCCAGTGCTCCCATGT | |
| C1F8 | AGGTTTGGGTGGAAAGAGGT | |
| C1R | GCAAGTCTGCTTAGAGACTGAAGTG | |
| C1R2 | ACCTGGGAGGGTGCTCTCA | |
| C1R6 | CCTTAAAACTGTATGCATCCTAAGC | |
| C1R9 | GTGAGGTCACCCCAGAAAAA | |
| sgRNA | sgC1fF | ACCGAATCATTCAGATCAGGCTAT |
| sgC1fR | AAACATAGCCTGATCTGAATGATT | |
| sgC1uF | ACCGGGTCTAGGAGTCCTAGACGT | |
| sgC1uR | AAACACGTCTAGGACTCCTAGACC | |
| sgC1dF | ACCGTTAAACGTGTGCCTTTCTAA | |
| sgC1dR | AAACTTAGAAAGGCACACGTTTAA | |
| shRNA | pLKO.1-hCTCF-1F | CCGGGAGGAGCCTGCCGTAGAAATTCTCGAGAATTTCTACGGCAGGCTCCTCTTTTTG |
| pLKO.1-hCTCF-1R | AATTCAAAAAGAGGAGCCTGCCGTAGAAATTCTCGAGAATTTCTACGGCAGGCTCCTC | |
| pLKO.1-hSMC3-1F | CCGGGCAGTGCAACACAGAATTAAACTCGAGTTTAATTCTGTGTTGCACTGCTTTTTG | |
| pLKO.1-hSMC3-1R | AATTCAAAAAGGCAGTGCAACACAGAATTAAACTCGAGTTTAATTCTGTGTTGCACTGC | |
| P7-index | P7-index-wyh14 | CAAGCAGAAGACGGCATACGAGATGCTCCGTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| P7-index-wyh15 | CAAGCAGAAGACGGCATACGAGATCATGAGAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-wyh18 | CAAGCAGAAGACGGCATACGAGATCTGAATGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-wyh20 | CAAGCAGAAGACGGCATACGAGATTCGCATGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-wyh21 | CAAGCAGAAGACGGCATACGAGATAATAGCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-wyh22 | CAAGCAGAAGACGGCATACGAGATGTCGCGTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
新窗口打开|下载CSV
1.7 各细胞系RNA的提取
对12孔板中长满的细胞进行胰酶消化后,3000×g离心10 min,收集沉淀加入500 μL Trizol溶液,室温放置5 min,再加入100 μL氯仿,剧烈摇15 s,室温放置3 min后,4℃、12000×g离心15 min,取上层透明相于干净1.5 mL的EP管中。在该管中加入250 μL异丙醇,充分混匀,室温放置10 min后,4℃、12000×g离心10 min,去上清收集沉淀,用75%的乙醇洗沉淀,4℃、7500×g离心5 min,收集沉淀,倒置10 min,最后加入无核酸酶的水30 μL溶解沉淀得到RNA溶液。用NanoDrop2000测RNA浓度,并放-80℃冰箱待用。1.8 荧光定量PCR检测表达量
RNA逆转录为cDNA的方法为:在无核酸酶的PCR管加入4 μL gDNA wiper混合液、0.5 μg RNA模板,补无核酸酶水至总体积16 μL,轻轻吹打混匀,42℃ 2 min。继续加入4 μL 5×HiScript II qRT SuperMix II,轻轻吹打混匀,50℃ 15 min;85℃ 5 s。产物即可用于qPCR反应或存于-20℃冰箱待用。qPCR反应体系:5 μL 2×SYBR Green Master混合液、0.3 μL正向引物(10 μmol/L)、0.3 μL反向引物(10 μmol/L)、2 μL模板、2.4 μL MilliQ水。所用到正、反向引物序列见表1。qPCR扩增条件:95℃ 10 min;95℃ 15 s,60℃ 33 s,40个循环;95℃ 15 s,60℃ 1 min,95℃ 15 s。分析其ΔΔCt值及RQ (relative quantity)值,RQ=2-ΔΔCt。显著性差异分析使用t-test。
1.9 CBS元件的基因片段编辑
根据CRISPR/Cas9基因编辑原理设计sgRNA并合成(表1)。每一对sgRNA的两条链经过变性、退火后连接到pGL3-U6-sgRNA-puro载体上。通过转化感受态细菌和质粒抽提得到sgRNA的重组质粒。hCBS1反转和删除的单克隆细胞株的获得采用了两组sgRNA。在24孔板准备细胞,密度为8× 105 cells/mL。第2天用Lipo3000转染sgRNAs (均为169.9 ng)和Cas9 (940.392 ng)。转染18 h后换培养基,24 h后加嘌呤霉素(1 mg/mL),每孔1 μL,连续4天,将存活下来的细胞继续培养。待细胞足够多时,取一部分做模板。设计PCR引物,进行细胞鉴定。鉴定到目标基因型后,将该混合细胞逐渐放大培养。按照30 cells/mL的浓度接种到96孔板进行单克隆化。两周后对单克隆细胞进行鉴定。在此过程,要保证这些单克隆细胞生长状态良好。
1.10 CRISPR单克隆细胞的鉴定
采用细胞裂解法和PCR方法鉴定单克隆细胞。细胞DNA模板的制备方法:取部分细胞于PCR管,3000×g离心10 min,留下细胞沉淀;加入20 μL裂解液,沸水煮5 min;再加入20 μL中和液,混匀即可。可存于-20℃。裂解液配方为25 mmol/L NaOH、0.2 mmol/L EDTA;中和液配方为40 mmol/L Tris- HCl。PCR反应体系:6.3 μL MilliQ水、1 μL 10 × Buffer (含Mg2+)、1 μL dNTP混合液(各2.5 mmol/L)、0.3 μL正向引物(10 μmol/L)、0.3 μL反向引物(10 μmol/L)、0.1 μL rTaq酶(5 U/μL)和1 μL DNA模板。所用到正、反向引物序列见表1。PCR 扩增条件:94℃ 3 min;94℃ 15 s,60℃ 30 s,72℃ 1min,38个循环;72℃ 7 min (退火温度根据引物Tm适当调整,延伸时间根据产物长度调整)。PCR扩增产物用2%琼脂糖凝胶进行电泳分析鉴定。
1.11 RNA-seq建库和高通量测序
待检测细胞系RNA的抽提方法和上面方法相同。poly(A) mRNA的分离、cDNA合成和文库构建根据NEB RNA-sequencing试剂盒说明书操作,所用P7-index序列见表1。文库采用Invitrogen公司的Qubit 3 Fluorometer仪器进行定量,并送至苏州金唯智生物科技有限公司进行质检和高通量测序,测序仪器为Illumina HiSeq平台,测序长度双端150 bp。RNA-seq数据用TopHat和Cufflinks进行分析[32],显著性差异分析使用t-test。2 结果与分析
2.1 CTCF和cohesin调控UGT1基因簇表达
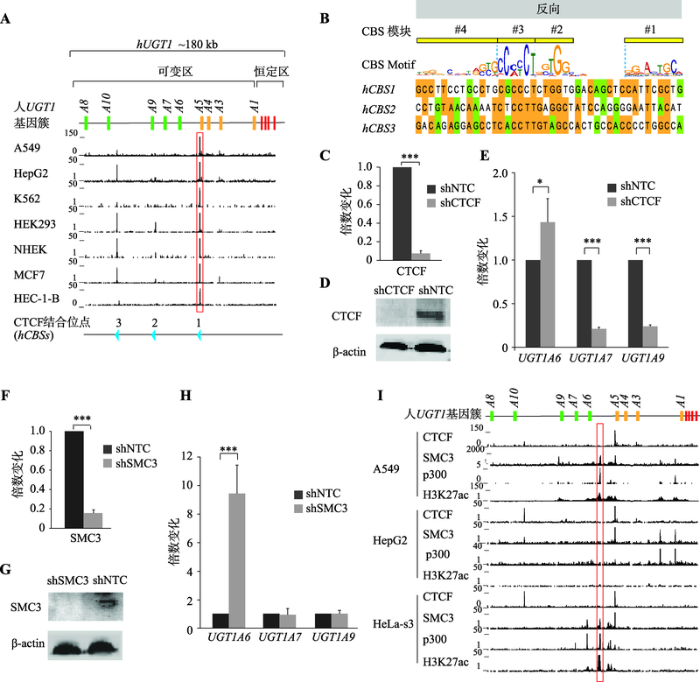
CTCF在细胞中的多样性功能与其在三维基因组中长距离染色质环化作用相关。本研究分析了多类人源细胞系的CTCF ChIP-seq (chromatin immunoprecipitation and massive parallel sequencing)数据,发现人UGT1基因簇中存在3个CTCF可结合位点:hCBS1~3,其中hCBS1位点在所分析的人肺癌细胞系(A549)、人肝癌细胞系(HepG2)、人慢性髓系白血病细胞系(K562)、人胚肾细胞系(HEK293)、正常人表皮角质形成细胞系(NHEK)、人乳腺癌细胞系(MCF7)和人子宫内膜腺癌细胞系(HEC-1-B)中均高度富集CTCF蛋白(图1A)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CTCF和cohesin参与人UGT1基因簇的转录调控
A:人UGT1基因簇内CBS分布。人UGT1位于2号染色体,类似于原钙粘蛋白基因簇,由可变区和恒定区组成,可变区内串联排列着9个高度相似的可变区外显子,下游恒定区包含4个恒定区外显子。垂直框代表UGT1基因簇外显子并用不同颜色标记,绿色表示苯酚组可变外显子,橘黄色表示胆红素组可变外显子,红色表示恒定外显子;人肺癌细胞系A549、人肝癌细胞系HepG2、人慢性髓系白血病细胞K562、人胚肾细胞HEK293、正常人表皮角质形成细胞NHEK、人乳腺癌细胞MCF7和人子宫内膜腺癌细胞HEC-1-B的CTCF ChIP-seq数据显示人UGT1基因簇内3个CTCF可结合位点:hCBS1~3,每个CBS方向由下方蓝色三角形指示;B:人UGT1基因簇3个CBS序列及方向性预测;Module#4~1的CBS定义为反向CBS;C:荧光定量PCR技术检测到A549细胞中敲低CTCF后CTCF的转录水平显著降低,shNTC是对照组;;D:蛋白免疫印迹实验证明CTCF蛋白被敲低;E:荧光定量PCR技术检测敲低CTCF对A549细胞中高度表达的UGT1A6、UGT1A7和UGT1A9三个基因的转录的影响;F:荧光定量PCR技术检测A549细胞中敲低SMC3后SMC3的转录水平显著降低;G:蛋白免疫印迹实验证明SMC3蛋白被敲低;H:荧光定量PCR技术检测敲低SMC3对A549细胞中高表达基因UGT1A6、UGT1A7和UGT1A9的转录影响;I:人肺癌细胞系A549、肝癌细胞系HepG2和宫颈癌细胞系HeLa-S3的cohesin蛋白亚基SMC3以及增强子标志p300和H3K27ac在人UGT1基因簇的分布。*:P<0.05 表示有统计学差异,**:P<0.01表示有显著的统计学差异,***:P<0.001 表示有非常显著的统计学差异。
Fig. 1CTCF and cohesin regulate the transcription of human UGT1 gene cluster
CBS基序通常被分为4个模块(Module#1~4)[25,33],核心序列为Module#2~4,其中Module#2~3由CTCF的ZF4~7特异识别,Module#4由ZF3识别。核心序列上有一段保守的11 bp回文序列“CCACCAGGTGG”,位于Module#2~3内,中心的核苷酸碱基“A”被CTCF蛋白ZF6上的Gln418残基特异性识别,对鉴别CBS的方向至关重要。约15%的CBS在核心序列上游存在Module#1,CTCF的ZF9~11缠绕在Module#1的DNA双链上并将它们的α-螺旋插入Module#1的大沟里,对CTCF方向性结合也至关重要[34]。根据上述CBS的结合特征,结合BSDB2.0分析软件,预测人UGT1基因簇中CBS1~3的方向为反向(图1B),它们在核心序列Module#2~3处保守度较高(图1B)。
A549细胞系中UGT1基因簇各基因的表达差异较大,UGT1A6、UGT1A7和UGT1A9高度表达,UGT1A1和UGT1A3低度表达,而UGT1A2、UGT1A4、UGT1A5、UGT1A8和UGT1A10则处于沉默状态。为研究CTCF是否参与调控UGT1基因簇的转录表达,以人肺癌细胞系A549为模型,通过向细胞感染表达shRNA的慢病毒来敲低CTCF。CTCF靶向的shRNA病毒感染A549细胞后,CTCF的转录水平降低了93% (图1C),CTCF的蛋白表达水平显著降低(图1D)。进一步采用荧光定量PCR技术检测了UGT1的表达,发现UGT1A6转录水平升高了43%,UGT1A7和UGT1A9分别降低了79%和76% (图1E)。表明CTCF显著影响UGT1A6、UGT1A7和UGT1A9的表达,参与调控UGT1基因簇的转录表达。
CTCF在染色质高级架构中的绝缘作用依赖于cohesin蛋白复合体,它们往往共定位于染色质上的CBS处,本研究进一步利用慢病毒介导的shRNA敲低了SMC3的表达,SMC3的转录水平降低86.7% (图1F)、蛋白量显著降低(图1G)。SMC3敲低后,UGT1A6转录水平升高8.25倍,而UGT1A7和UGT1A9转录水平几乎没有变化(图1H),这和CTCF敲低时的情况不一样,可能与cohesin在组织特异性转录中的CTCF非依赖性功能相关[20,35,36]。于是,进一步分析了人肺癌细胞系A549、肝癌细胞系HepG2和宫颈癌细胞系HeLa-S3的cohesin亚基SMC3蛋白以及增强子标志p300和H3K27ac在UGT1基因簇的结合分布,结果发现SMC3除了在CTCF位点富集,还在p300和H3K27ac位点处富集(图1I)。
2.2 CRISPR删除hCBS1片段对人细胞系A549中UGT1基因簇转录的影响
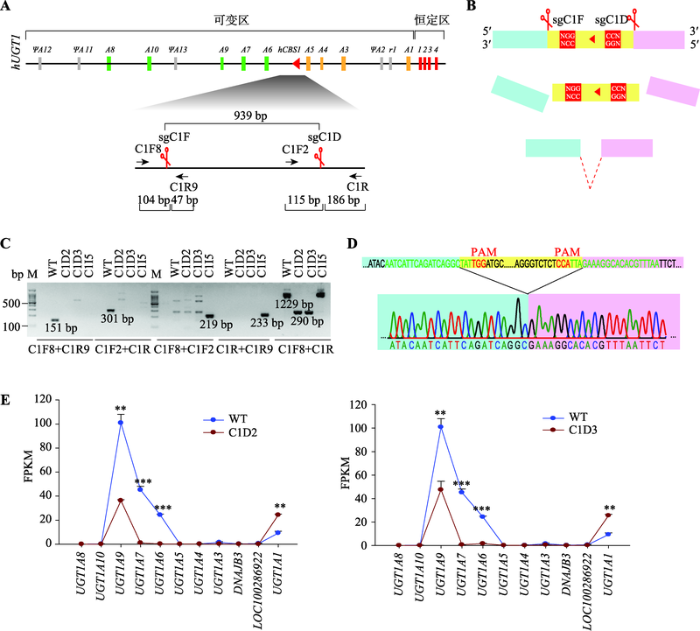
为进一步研究人UGT1基因簇内保守的CTCF结合元件hCBS1对UGT1基因簇的转录调控的影响,以人肺癌细胞系A549为模型,利用CRISPR/Cas9片段编辑技术原位删除包含hCBS1的DNA片段。本研究采用使用最广泛的酿脓链球菌(Streptococcus pyogenes)来源的SpCas9酶。该酶被sgRNA带入靶标DNA处,其两个核酸酶结构域HNH和RuvC在5′ PAM (protospacer adjacent motif)序列(NGG)上游3 bp处分别对互补链和非互补链进行切割,产生具有平头末端的DSBs (double strand breaks)[37,38,39,40]。最新的研究发现,SpCas9切割位点不局限于PAM上游3 bp处,非互补链上的切割可能发生在更上游位置,进而产生突出末端[27]。在A549细胞中转入SpCas9和sgRNAs (sgC1F和sgC1D,图2A)表达质粒后,在转染细胞群中检测到hCBS1删除的基因型,进一步对转染细胞群进行单克隆化,获得89个单克隆细胞株,对这些单克隆细胞进行PCR鉴定,发现C1D2和C1D3是hCBS1删除纯合型单克隆细胞株。hCBS1片段删除示意图如图2B所示,SpCas9在sgCIF和sgC1D引导下,特异性识别hCBS1片段两侧靶标位置并切割产生两个切口,两个切口连接到一起形成hCBS1片段删除的编辑细胞。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2CRISPR删除hCBS1片段对人细胞系A549中UGT1基因簇转录的影响
A:CRISPR删除hCBS1片段所使用的成对sgRNA (sgCIF和sgC1D)以及各鉴定引物的位置示意图;B:hCBS1片段删除示意图。Cas9在sgCIF和sgC1D引导下,特异性识别hCBS1片段两侧靶标位置并切割产生两个切口,两个切口连接到一起形成hCBS1片段删除的编辑细胞;C:hCBS1片段删除的单克隆A549细胞株C1D2和C1D3的基因型鉴定结果。野生型A549细胞(WT)和hCBS1片段原位反转型A549细胞(C1I5)被作为PCR鉴定对照;用C1F8/C1R9或C1F2/C1R引物对可在WT中扩增出切点处151 bp或301 bp片段,用C1F8/C1F2或C1R/C1R9可在C1I5中扩增出反转后剪接处的219 bp或233 bp片段,C1D2和CID3中未检测到以上4个片段;用hCBS1片段两侧引物C1F8/C1R可在WT和C1I5中扩增出1229 bp条带,C1D2和CID3中扩增条带缩短为290 bp;D:C1F8/C1R在C1D2和C1D3细胞中的扩增产物的测序结果示意图;E:RNA-seq数据分析比较WT和C1D2或C1D3细胞中UGT1基因簇的转录水平。
Fig. 2Effect of CRISPR deletion of hCBS1 fragment on the transcription of human UGT1 gene cluster in A549 cell line
C1D2和C1D3的基因型鉴定结果如图2C所示,切口处原野生型片段(上游C1F8/C1R9引物对PCR产物151 bp、下游C1F2/C1R引物对PCR产物301 bp)全部消失,反转检测条带(C1F8/C1F2引物对PCR产物219 bp、C1R/C1R9引物对PCR产物233 bp)呈阴性,hCBS1片段两侧引物C1F8/C1R扩增产物从野生型细胞的1229 bp缩短为290 bp,表明编辑片段被删除。对hCBS1片段两侧引物C1F8/C1R扩增的290 bp产物进一步测序分析,发现C1D2和C1D3单克隆细胞株中删除片段上下游的切割位点均恰好在PAM上游3 bp处(图2D),这可能因为即使SpCas9在非互补链造成的切割位点超过PAM上游3 bp,细胞内修复体系也会使其3°端补齐,然后再连接到一起。
采用RNA-seq分析方法检测比较C1D2和C1D3单克隆与野生型A549细胞的转录水平(图2E),发现在C1D2单克隆细胞中,UGT1A9表达降低64%,UGT1A6表达降低99%,UGT1A7表达降低97.6%,但是UGT1A1表达升高1.62倍,UGT1基因簇其他基因没有变化。在C1D3单克隆细胞中,UGT1A9表达降低52%,UGT1A6表达降低93%,UGT1A7表达降低99%,但是UGT1A1表达升高1.75倍,UGT1基因簇其他基因没有变化,这与C1D2克隆结果一致。总之,hCBS1删除显著影响UGT1基因簇的基因转录。
2.3 CRISPR反转hCBS1片段对人细胞系A549中UGT1基因簇转录的影响
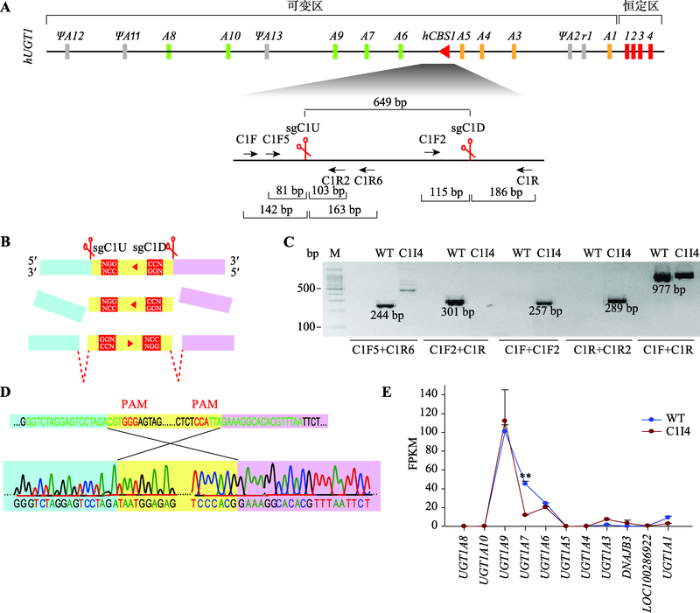
CTCF在DNA上的结合具有方向性,参与形成染色质环化,在三维基因组复杂的空间结构的形成和维持中起关键作用,上述研究证明删除hCBS1显著影响UGT1基因簇的转录,表明hCBS1是UGT1基因簇的一个重要的转录调控元件。hCBS1是反向的,那么如果把hCBS1反转,变为正向,UGT1基因簇局部的基因表达会有怎样的变化呢?本研究在A549细胞中转入SpCas9和sgRNAs (sgC1U和sgC1D,图3A)表达质粒后,在转染细胞群中检测到hCBS1反转的基因型,进一步对转染细胞群进行单克隆化,获得90个单克隆细胞株,对这些单克隆细胞进行基因型鉴定,发现C1I4单克隆细胞株为hCBS1反转纯合型。hCBS1片段反转示意图如图3B所示,Cas9在sgCIU和sgC1D引导下,特异性识别hCBS1片段两侧靶标位置并切割产生两个切口,hCBS1反转后两个切口分别连接到染色体断点处形成hCBS1片段反转的编辑细胞。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3CRISPR反转hCBS1片段对人细胞系A549中UGT1基因簇转录的影响
A:CRISPR反转hCBS1片段所使用的成对sgRNA (sgCIU和sgC1D)以及各鉴定引物的位置示意图;B:hCBS1片段反转示意图。Cas9在sgCIU和sgC1D引导下,特异性识别hCBS1片段两侧靶标位置并切割产生两个切口,hCBS1反转后两个切口分别连接到染色体断点处形成hCBS1片段反转的编辑细胞;C:hCBS1片段反转的单克隆A549细胞株C1I4的基因型鉴定结果。野生型A549细胞(WT)被作为PCR鉴定对照;用C1F5/C1R6或C1F2/C1R引物对可在WT中扩增出切点处244 bp或301 bp的野生型条带,C1I4中检测结果均为阴性;用CBS1反转检测引物对C1F/C1F2或C1R/C1R2可在C1I4中扩增出257 bp或289 bp片段;用hCBS1片段两侧引物C1F8/C1R可在WT、C1I4中扩增出约977 bp条带;D:C1F/C1R在C1I4细胞中的扩增产物的测序结果示意图;E:RNA-seq数据分析比较WT和C1I4细胞中UGT1基因簇的转录水平。
Fig. 3Effect of CRISPR inversion of hCBS1 fragment on the transcription of human UGT1 gene cluster in A549 cell line
C1I4的基因型鉴定结果如图3C所示,切口处原野生型片段(上游C1F5/C1R6引物对PCR产物244 bp、下游C1F2/C1R引物对PCR产物301 bp)检测为阴性,表明野生基因型不存在;用hCBS1反转检测引物对C1F/C1F2或C1R/C1R2可在C1I4中扩增出257 bp或289 bp大小的反转条带,表明hCBS1发生反转;用hCBS1片段两侧引物C1F/C1R可扩增出和野生型条带大小相当约977 bp的条带。对hCBS1片段两侧引物C1F/C1R扩增的产物进一步测序分析,发现C1I4单克隆细胞株中编辑片段上下游的切割位点均恰好在PAM上游3 bp处(图3D)。
采用RNA-seq分析方法检测比较C1I4单克隆与野生型A549细胞中UGT1基因簇的转录水平(图3E),发现当hCBS1反转后,C1I4单克隆中UGT1A6和UGT1A9表达没有明显变化,但是UGT1A7表达降低74%,低表达的UGT1A1和UGT1A3变化幅度并不显著,UGT1其余基因(UGT1A4、UGT1A5、UGT1A8、UGT1A10)依然处于沉默状态,几乎不转录。总之,hCBS1反转主要影响UGT1A7基因的转录。
2.4 人和小鼠UGT1基因簇中CBS分布比较
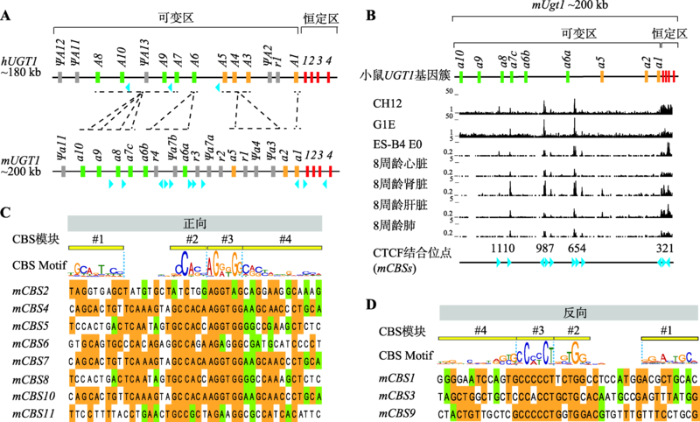
人和小鼠UGT1基因簇分别长180 kb和200 kb,基因结构相似,由高度同源且串联排列的9个可变外显子组成的可变区和4个恒定外显子组成的恒定区组成(图4A),可变区任意外显子可被选择性剪接到下游全套恒定外显子上,共可形成9种转录本,表达9种UGT1同工酶。根据催化底物特异性,UGT1同工酶被分为胆红素组和苯酚组,其编码基因的可变区分别对应图4A中的橘黄色和绿色外显子。胆红素组中,人UGT1A1和小鼠Ugt1a1基因为直系同源关系,蛋白氨基酸序列相似度为66%。人UGT1A3-5与小鼠Ugt1a2、1a5为直系同源关系,它们的平均氨基酸序列同源性约61%。苯酚组也有两个分支,人UGT1A6与小鼠Ugt1a6a-b为直系同源关系,它们之间相似度为70%,小鼠的Ugt1a6a和1a6b基因是在两个物种分开后复制衍生形成的,两者编码的氨基酸相似度为95%。人UGT1A7-10与小鼠Ugt1a7-10基因直系同源,平均氨基酸序列相似度约67%[1,41]。除了9个功能性可变外显子外,人和小鼠UGT1基因簇可变区内还有多片分散排列的UGT1假基因和残骸(relic)序列。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4小鼠Ugt1基因簇内CTCF结合位点分布和方向性预测
A:人和小鼠UGT1基因簇内各同工酶基因的线性进化关系示意图。人UGT1位于2号染色体,小鼠Ugt1位于1号染色体,它们都类似于原钙粘蛋白基因簇,由可变区和恒定区组成,可变区内串联排列着9个高度相似的可变区外显子,下游恒定区包含4个恒定区外显子。人UGT1基因簇可变区还有4个假基因和1个残骸(relic)序列,而小鼠Ugt1可变区含有5个假基因和4个残骸序列。垂直框代表UGT1基因簇外显子并用不同颜色标记,绿色表示苯酚组可变外显子,橘黄色表示胆红素组可变外显子,红色表示恒定外显子,灰色表示假基因或残骸序列,分别用Ψ和r表示,直系同源基因之间由黑色虚线相连,蓝色三角形表示CBS方向;B:小鼠Ugt1基因簇内CBS分布。小鼠B细胞淋巴瘤CH12、GATA 1-红系祖细胞G1E和第0天的胚胎干细胞ES-B4 E0,以及小鼠(C57BL/6)8周龄心脏、肾脏、肝脏和肺的CTCF ChIP-seq数据显示小鼠Ugt1基因簇内至少有11个CTCF可结合位点:mCBS1~11,每个CBS方向由下方蓝色三角形指示;C:小鼠Ugt1基因簇内正向CBS序列及方向性预测。Module#1~4的CBS定义为正向CBS;D:小鼠Ugt1基因簇内反向CBS序列及方向性预测。Module#4~1的CBS定义为反向CBS。
Fig. 4Distributions and orientations of the CTCF binding sites(CBSs)in mouse Ugt1 gene cluster
为解析UGT1基因簇内分布的CBS在人和模式动物小鼠间是否高度保守,本研究分析和比较了小鼠不同细胞和组织中Ugt1基因簇的CTCF ChIP-seq数据(图4B),包括B细胞淋巴瘤细胞系(CH12)、GATA 1-红系祖细胞系(G1E)、第0天的胚胎干细胞(ES-B4 E0)和8周龄的心脏、肾脏、肝脏、肺,发现小鼠Ugt1基因簇含有至少11个CTCF可结合位点:mCBS1~11,其中8个为正向、3个为反向,在核心序列Module#2~3处保守性较高(图4,C和D)。比较人和小鼠UGT1基因簇中的CBS分布和方向性,发现小鼠Ugt1基因簇含有CBS数目比人UGT1基因簇更多。
3 讨论
脊椎动物UGT1表达具有组织特异性,同一个体不同器官的UGT1表达水平有很大的差别[1,42],这和组织特异性转录因子及其配体激活密切相关[7]。UGT葡醛酸转移酶的多样性和其在各种组织中的表达差异与特定疾病以及治疗药物的疗效或毒性有关,新生儿黄疸、克里格勒-纳贾尔(Crigler-Najjar)综合征(Ⅰ型和Ⅱ型)和吉尔伯氏(Gilbert)综合征等遗传性高胆红素疾病[43,44]均是由UGT1多态性或基因突变造成的胆红素代谢功能降低或缺失引起,其中大部分变异位点位于非编码区。非编码区在UGT1基因簇的转录调控中发挥重要作用[45]。为了解析这些非编码区如何在复杂的染色质三维空间结构上调控UGT1基因簇的表达以及在染色体拓扑结构域内各UGT1启动子与远端DNA调控元件之间的特异性成环机制,本研究首次聚焦UGT1基因簇中染色质架构蛋白CTCF及其结合元件CBS,探索其对UGT1基因簇表达的影响。本研究分析比较了人和模式动物小鼠的不同细胞或组织中CTCF在UGT1基因簇的富集情况,发现人和小鼠的UGT1基因簇中CBS分布差异较大。通过对人和小鼠UGT1基因簇内的CBS方向以及保守性的初步分析,发现这些CBS基序在Module#2-3处保守度较大。然后以人肺癌细胞系A549为模型,通过CTCF和cohesin蛋白靶向shRNA病毒感染细胞以敲低细胞中的CTCF和cohesin蛋白,发现CTCF敲低后,A549细胞中高度表达的UGT1A7和UGT1A9的转录水平显著降低,而UGT1A6升高。cohesin蛋白敲低后,UGT1A6表达显著升高,而UGT17和UGT1A9没有变化。该现象表明CTCF和cohesin蛋白参与调控UGT1基因簇的转录表达,可能通过影响UGT1局部基因簇的染色质构象而改变各启动子和增强子之间的相互作用。最后,本研究发现人和小鼠的所有UGT1基因启动子附近没有CBS位点,这与基因结构类似的原钙粘蛋白基因簇[8,46,47]存在明显不同:原钙粘蛋白基因簇中几乎每个启动子附近都有CBS位点,且CBS位点的位置和方向在人和小鼠原钙粘蛋白基因簇中高度保守[48]。
进一步通过CRISPR介导的DNA片段编辑技术研究了人UGT1基因簇内高度保守的CTCF结合位点hCBS1对UGT1转录的影响。同样以人肺癌细胞系A549为研究模型,利用CRISPR/Cas9系统将hCBS1片段删除。从RNA-seq数据可以看出,hCBS1删除明显降低了UGT1A6、UGT1A7和UGT1A9的表达。当把hCBS1方向反转为正向时,UGT1A7转录水平明显降低。这可能是因为删除或反转hCBS1会改变染色质空间构象,影响增强子和启动子之间远距离相互作用,例如,hCBS1删除使得调控UGT1A6、UGT1A7和UGT1A9启动子的增强子空间上远离这些启动子,使它们的转录活性降低。A549细胞中主要分布有2个CBS:hCBS1和hCBS3,两者方向均向左(图1A),hCBS1反转后仅UGT1A7表达下调,可能是因为hCBS1反转后与hCBS3方向相背,造成hCBS1与下游CBS成环、hCBS3与上游CBS成环,使得hCBS1和hCBS3之间的3个基因UGT1A6、UGT1A7和UGT1A9不再处于同一个调控拓扑域内,它们具体受哪些增强子调控有待进一步研究。尽管如此,该现象表明hCBS1参与调节UGT1A6、UGT1A7和UGT1A9的转录,是UGT1基因簇的潜在转录调控元件。
综上所述,本研究证明了CTCF和cohesin蛋白参与调控人UGT1基因簇的转录,删除或反转该基因簇内高度保守的CTCF结合位点hCBS1显著改变该基因簇活性基因的转录,这可能源于CTCF带来的染色质构象改变。本研究工作将为后续对UGT1的三维基因转录调控机制研究奠定基础,为临床合理用药和人类疾病的预防提供科学依据。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 5]
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 2]
URL [本文引用: 5]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 3]
URL
URL [本文引用: 3]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}