 ,1,2
,1,2Controlling the spatiotemporal expression of germ line specific genes by PRC1.6 complex
Xiaowei Sun1, Hongyang Li1, Jian Wang1, Bo Cheng,1,2通讯作者:
第一联系人:
编委: 苗龙
收稿日期:2019-01-27修回日期:2019-03-14网络出版日期:2019-04-20
| 基金资助: |
Received:2019-01-27Revised:2019-03-14Online:2019-04-20
| Fund supported: |
作者简介 About authors
孙晓伟,在读博士研究生,专业方向:细胞生物学E-mail:
李宏阳,在读硕士研究生,专业方向:细胞生物学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (555KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孙晓伟, 李宏阳, 王健, 程博. PRC1.6复合体表观遗传调控生殖谱系特异性基因的时空表达 [J]. 遗传, 2019, 41(4): 271-284 doi:10.16288/j.yczz.18-332
Xiaowei Sun, Hongyang Li, Jian Wang, Bo Cheng.
表观遗传调控是实现真核生物基因选择性表达的主要途径。多梳蛋白家族(polycomb groups,PcGs)是一类重要的表观遗传调控因子,与多种干细胞的干性维持、细胞分化、细胞周期的调控、细胞衰老、X染色体失活等一系列细胞生理活动密切相关[1,2]。它主要通过两类蛋白复合体发挥功能——多梳抑制复合体Ⅰ和Ⅱ (polycomb repressive complex 1/2, PRC1/2)。近年来在高等动物细胞中不断鉴定出组分不同的PRC1亚型,且它们的生物学功能在靶基因群的选择及调控基因表达的作用机制中有所差异。PRC1.6复合体属于其中一类PRC1亚型,主要包含RING1B、PCGF6 (polycomb group ring finger protein 6)、MAX (Myc-associated factor X)、MGA (MAX gene associated)、E2F6和L3MBTL2 (lethal (3) malignant brain tumour like 2)等组分。近年来通过对PRC1.6复合体不同核心组分在生物化学、分子生物学、细胞生物学及发育生物学等方面的研究,发现该复合体对于哺乳动物发育过程中多种相关细胞谱系的建成及维持是必需的。本文在介绍该复合体的发现、核心组分的分子生物学功能的基础上,对该复合体在高等动物胚胎发育、性腺发育、精子发生、胚胎干细胞(embryonic stem cells, ESCs)及生殖干细胞(germ line stem cells, GSCs)维持等过程中发挥的生物学功能展开了系统论述。
1 多梳蛋白家族PcGs
PcGs是一大类通过催化和识别组蛋白表观遗传修饰来改变染色质构象、对靶基因进行转录调控的蛋白。第一个多梳蛋白编码基因Polycomb (Pc)是Pamela Lewis于1947年在黑腹果蝇(Drosophila melanogaster)中发现的,1978年Edward Lewis发现Pc突变导致果蝇发生同源异型转化(如雄性果蝇产生多对性梳的异常发育表型),进一步研究发现多梳蛋白是通过抑制同源异型基因的表达来调控果蝇的体节发育[1,3,4],其功能与发挥基因激活作用的三胸蛋白家族TrxGs(trithorax groups)相拮抗[5,6]。但是近几年也有关于PcGs蛋白激活基因表达的报道[7,8,9],表明PcGs蛋白在执行转录调控方面的机制可能是非常复杂且多效的。PcGs蛋白主要通过PRC1和PRC2两类蛋白复合体形式发挥作用。其中,EZH2是PRC2中具有催化活性的核心组分,可以催化组蛋白H3第27位赖氨酸的三甲基化(H3K27me3)[10]。其他的PRC2组分参与复合体组装及活性维持[11]。PRC1具有E3泛素连接酶活性,该活性由RING1A/ RING1B及PCGF的环指结构域(ring finger domain)的部分亲水性表面构成[12,13],催化组蛋白H2A第119位赖氨酸的单泛素化(H2AK119ub1)[14]。PRC1和PRC2 调控的靶基因群存在部分重叠,在多数情况下二 者存在相互招募的关系,互助实现对靶基因的转录 调控[15]。高等动物中PRC1的组分较果蝇要复杂的多,除了对应果蝇PRC1的每个组分都衍生出多个同源蛋白之外,还增添了很多其他组分,且组分之间的组合方式非常复杂多样[16]。目前在哺乳动物中发现PRC1亚型有6类,称为PRC1.1~PRC1.6,其中PRC1.2和PRC1.4因包含有经典的Pc蛋白同系物CBX蛋白而被称作经典PRC1 (canonical PRC1),其余几种亚型因缺乏CBX蛋白而被列为非典型PRC1 (non-canonical PRC1)。本文着重讨论了PRC1.6亚型,关于其他亚型的组成及功能均有较多报道,请读者参考相关综述[17,18]。
2 PRC1.6复合体的发现、核心组分及其分子功能
2.1 PRC1.6复合体的发现
PRC1.6复合体先后被不同的研究团队发现,并被赋予了不同的名称(表1)。2002年,Ogawa等[19]在HeLa及成纤维细胞中利用免疫沉降技术、甘油密度梯度离心并结合质谱检测发现了与E2F6-标签融合蛋白结合的蛋白复合体—E2F6复合物(E2F6.com)。该复合体中除了包含转录因子E2F6/DP-1,还有另一对转录因子异二聚体MAX/MGA;同时发现该复合体在甘油密度梯度分离中与组蛋白甲基转移酶G9a、Eu-HMTase1 (GLP)部分重叠,具有H3K9的甲基转移酶活性[19]。2011年,美国纽约大学医学院Danny Reinberg课题组的Trojer等[20]研究L3MBTL2的功能时再次发现了该复合体,将其命名为PRC1L4 (PRC1-like4)复合体;2012年,同课题组的Gao等[21]通过免疫沉降/质谱实验发现了人源细胞中包含不同PCGF因子(PCGF1~PCGF6)的PRC1复合体,并根据包含PCGF因子的编号将PRC1复合体细分为PRC1.1~PRC1.6复合体亚型,各亚型除了共同组分是PRC1复合体的酶学核心组分RING1B (RING2)外其余组分差异较大。其中PRC1.6组分包括PCGF6、RING1B、RYBP、MAX、MGA、L3MBTL2、E2F6、DP-1/2、HP1γ和HDAC1/2等。2017年,Endoh等[22] 在小鼠ESCs中利用带标签的PCGF6蛋白也分离到了类似的复合体并将其称为PCGF6-PRC1。以上4种复合体虽然名称各异,但是核心组分都含有PCGF6、E2F6、RING1B、L3MBTL2和HP1γ等因子(表1),本质上属于同一复合体或十分相近的同类复合体。为了叙述简洁,本文在后续介绍中将其统称为PRC1.6复合体。Table 1
表1
表1 PRC1.6复合体的发现及组分
Table 1
| 复合体命名 | 组分 | 细胞系 | 文献 |
|---|---|---|---|
| E2F6.com | E2F6、PCGF6、RING1B、MAX、MGA、L3MBTL2、HP1γ、DP-1/2、YAF2、G9a和Eu-HMTase1(GLP) | HeLa | [19] |
| PRC1-like 4 (PRC1L4) | E2F6、PCGF6、RING1B/RING1A、L3MBTL2、HP1γ和MBLR | HEK293T | [20] |
| PRC1.6 | E2F6、PCGF6、RING1B、MAX、MGA、L3MBTL2、HP1γ、DP-1/2、HDAC1/2、RYBP/YAF2和WDR5 | 293TREx | [21] |
| PCGF6-PRC1 | PCGF6、RING1B/RING1A、MGA、L3MBTL2、HP1γ、HP1β、DP-1和RYBP/YAF2 | mESCs | [22] |
新窗口打开|下载CSV
2.2 PRC1.6复合体各组分的分子功能
如上所述,PRC1.6复合体是一个多组分蛋白复合体,其组分大致可以分成与催化组蛋白修饰相关的因子和与复合体装配或靶向定位相关的因子。同其他PRC1亚型类似,PRC1.6复合体最主要的酶学活性是由RING1B提供的E3泛素连接酶活性,催化产生H2AK119ub1;而RYBP、PCGF6等因子对于RING1B的活性发挥起到重要的促进作用[14,21,23]。在该复合体中包含异染色质结合蛋白HP1γ,说明其可能介导PRC1.6与甲基化的H3K9之间存在功能互作;除此之外,Gao等[21]还在PRC1.6复合体中检测到了组蛋白去乙酰化酶HDAC1/2,说明该复合体在某些情况下可能与组蛋白去乙酰化酶协同作用抑制靶基因的转录。PRC1.6蛋白复合体各组分的已知分子功能见表2。在ESCs中通过免疫共沉淀实验发现PCGF6与L3MBTL2、MAX、RING1B和RYBP具有相互作用,敲除Pcgf6时阻断了L3MBTL2与复合体中RING1B、RYBP和MAX等组分的互作,这说明整个复合体的完整性在一定程度上依赖于PCGF6[24]。另有研究表明PCGF6可通过招募RING1B到靶基因上催化H2AK119位点的泛素化并抑制其表达[22]。MGA对于PRC1.6复合体的装配和稳定性发挥着更加关键的作用,在HEK293T细胞中敲除Mga发现不仅该复合体各特异性组分在靶基因上的富集程度均受到明显抑制,且其中几个核心特异组分的蛋白总量明显下降,如PCGF6、L3MBTL2和E2F6[25]。从L3MBTL2的RNA干扰实验可知,部分靶基因的上调与其基因上富集的H2K119ub1水平的明显下降相伴发生,说明该因子对于PRC1.6复合体在靶基因区域组蛋白H2AK119位点的泛素化是必需的。同时,体外实验还发现L3MBTL2能够诱发染色质凝缩[20]。
Table 2
表2
表2 PRC1.6复合体中各组分的主要生化及分子功能
Table 2
| 组分名称 | 主要生化及分子生物学功能 | 文献 |
|---|---|---|
| RING1B/ RYBP | RING1B具有E3泛素连接酶活性,催化H2AK119泛素化从而抑制基因转录;RYBP与RING1B直接互作,辅助增强RING1B酶活性,其泛素结合结构域可结合H2A上的泛素化修饰 | [21,23,30] |
| HDAC1/2 | 去乙酰化酶,降低组蛋白特定位点的乙酰化水平,抑制靶基因转录 | [21,31] |
| HP1γ | 与H3K9甲基化转移酶互作,参与染色质凝缩和异染色质形成与维持,并在转录延伸和RNA形成中发挥作用 | [21,32] |
| PCGF6 | 招募RING1B到PRC1靶基因形成H2AK119单泛素化及维持H3K27me3水平来抑制基因表达;在复合体组装中发挥作用 | [21,22,24, 33,34] |
| MAX/MGA | 是识别特定DNA序列结构域的转录因子异二聚体,帮助招募复合体到特定靶基因;主要抑制靶基因表达;MGA对于该复合体装配及其他组分的蛋白稳定性十分重要 | [21,25,35] |
| E2F6/DP-1 | 是识别特定DNA序列结构域的转录因子异二聚体,帮助招募复合体到特定靶基因。E2F6在细胞G0期结合至E2F应答基因启动子并沉默其表达;DP-1调控DNA复制及细胞周期相关基因 | [19,36,37] |
| L3MBTL2 | 具有组蛋白结合能力,参与染色质凝缩和转录抑制;其MBT结构域参与复合体装配及复合体靶向定位 | [20,21,25,35] |
新窗口打开|下载CSV
目前,关于PRC1.6复合体的靶向定位的研究结果显示该复合体的定位机制较为复杂,由多个组分协同执行,包括MAX/MGA和E2F6/DP-1/2这两对转录因子形成的异二聚体以及L3MBTL2[25] (表2)。MAX和MGA通过各自的bHLHZip (basic Helix- Loop-Helix-Zipper)结构域结合形成异二聚体,并特异性识别E-boxes序列CACGTG;此外MGA的氮端还包含另一个DNA结合结构域,可识别T-box序列AGGC/TGC/TGA,该异二聚体在PRC1.6复合体的组装、稳定性及靶基因识别上都发挥着重要作用[25,26]。另一对转录因子是E2F6和DP-1或DP-2,二者结合共同识别E2F家族识别序列GCGGGA[27]。与E2F家族的其他转录激活因子不同,E2F6通常对其结合的序列调控的靶基因产生转录抑制效应。L3MBTL2包含4个MBT结构域,结合组蛋白H3和H4,但该结合是否依赖于组蛋白的甲基化状态目前还有争议[28,29]。
3 PRC1.6复合体在胚胎发育中抑制生殖谱系相关基因的表达
近年来不断积累的针对PRC1.6组分的RNA干扰或基因敲除实验已经充分证实PRC1.6复合体在维持ESCs的自我更新及调控其分化能力方面,尤其在抑制生殖谱系相关基因表达中发挥重要作用。在小鼠中,该复合体组分的敲除往往造成胚层及/或胚外谱系不同程度的发育异常,多数情况下产生胚胎致死表型(表3),由此可见,PRC 1.6复合体在ESCs以及胚胎发育过程中发挥至关重要的调控功能。Table 3
表3
表3 PRC1.6组分下调对ESCs及小鼠胚胎发育或性腺发育的影响
Table 3
| 组分 | 敲低/敲除表型(ESCs) | 敲低/敲除表型(小鼠胚胎发育) |
|---|---|---|
| RING1B | 敲除后干细胞分化相关基因(含印记基因)转录去抑制,ESCs异常分化[47,48,49];与Ring1a双敲除导致H2A K119泛素化标记消失,同时释放对polⅡ的转录延伸抑制[50] | 敲除后胚胎及胚外组织发育迟缓,不能正常进入原肠胚阶段,E8.5左右出现胚胎致死[51];半合子体轴和胸骨发育异常[52];在生殖细胞特异性敲除的雄鼠可存活但睾丸明显变小且不 育[53];对原始生殖细胞的性别分化至关重要[54] |
| RYBP | 缺失后ESCs不能形成收缩的心肌细胞[55];对ESCs自我更新维持非必需,但敲除导致ESCs不能形成囊胚,生殖谱系相关基因及内源性反转录病毒表达去抑制[38] | 敲除后在受精卵着床后早期胚胎致死(E5.5~6.0),对于胚胎存活及胚外组织的结构建成是必需的;敲除Rybp的杂合子CNS系统发育异常[56] |
| HDAC1/2 | 同时条件性敲除Hdac1和2导致细胞活力丧失,基因失活从而导致有丝分裂纺锤丝异常增加和染色体分离缺陷,多能性核心因子Oct4,Nanog等表达下调[57] | 敲除Hdac1会导致胚胎E10.5致死[58];在E8.5时诱导敲除Hdac1和2导致从E12.5开始产生突变的肺上皮细胞,并且出生后全部死于呼吸窘迫[59] |
| HP1γ | 敲除导致ESCs趋于分化且内胚层和神经发育缺陷,细胞增殖能力下降[60] | 纯合敲除的小鼠只有约1%活到成年,且雄性表现出性腺功能低下,精子发生缺陷,转座子活性升高[61];对于减数分裂中组蛋白H3K9甲基化修饰的识别至关重要[62] |
| PCGF6 | 对于维持ESCs干性是必需的[24],抑制小鼠ESCs过早分化[22];敲除导致干性相关基因表达下调,中胚层及精子发生特异性基因上调,在iPS形成实验中可有效取代Sox2[33,34] | 敲除小鼠可以存活并且可育,但是存在部分胚胎致死现象,出现骨骼同源异型转化及胎盘发育异常,约有1/3的Pcgf6-/-胚胎在E10.5体现出明显的发育迟缓[22] |
| E2F6 | 结合在生殖相关基因的启动子区抑制其表达[63];可以与G0期相关基因的靶启动子区结合,沉默E2F-和Myc-应答基因[19] | E2f6敲除小鼠可存活,但是轴向骨骼出现同源异型转化;2~3个月龄的雄鼠睾丸发育异常,支持细胞数目异常增加,精母细胞及成熟的精子数目减少,但未达到不孕程度[64];E2f6与Bmi1双敲小鼠生长迟缓,且严重贫血[65];体细胞广谱表达生殖细胞特异性基因[42] |
| DP-1 | DP-1的敲除不影响ESCs中细胞周期相关基因的表达[66] | 敲除小鼠胚胎外谱系发育及DNA复制异常,于E12.5胚胎致死;但对胚胎本身发育不是必需的[66,67] |
| MAX | 敲除后诱发ESCs进入减数分裂[39,40] | 影响胚胎及胚外组织发育,在受精卵着床后早期发育停滞并于E5.5~E6.5胚胎致死[44] |
| MGA | 敲低导致ESCs明显分化甚至死亡,生殖谱系发育及减数分裂相关基因去抑制[45,68] | 敲除导致小鼠胚胎中多能性的内细胞团细胞死亡(可能与调控腐胺合成酶Odc1有关),于E4.5~E5.5胚胎致死[45] |
| L3MBTL2 | 敲除后ESCs的增殖能力下降,细胞周期改变,干细胞分化异常;生殖谱系发育及减数分裂及其他发育相关基因去抑制[41] | 缺失导致内细胞团不能形成正常的上胚层,胚胎于E7.5~9.5致死[41];在生殖细胞中特异性敲除后,雄鼠可育但精子数目减少,睾丸小且呈现生育能力下降[69] |
新窗口打开|下载CSV
3.1 PRC1.6复合体参与ESCs干性维持及调控ESCs正常分化
PRC1.6复合体的组分RING1B、MAX、MGA、HP1γ、PCGF6和L3MBTL2等都已被报道对于ESCs的干性维持是必需的,这些组分在被敲除后均会引起ESCs分化异常(表3)。另有一些因子如RYBP虽然对于ESCs的自我更新是非必需的,但对于ESCs的正常分化却是不可缺少的[38]。2013年,Maeda等[39]在小鼠ESCs中通过siRNA文库从864个候选基因中筛选能够抑制生殖细胞分化路径的重要基因,筛选到多个PRC1.6复合体组分,包括Max、Mga和L3mbtl2,其中Max敲低效应最为显著,可诱发ESCs进入类似减数分裂的状态。Suzuki等[40]在ESCs中诱导性敲除Max后也发现表达上调的基因主要包括减数分裂和精子发生过程中的相关基因,如Hormad1、Dazl和Slc25a31等。通过细胞免疫荧光实验发现在Dox诱导Max敲除10天后,ESCs发生类似减数分裂细胞的形态学变化,出现减数分裂前期(细线期和偶线期)相关蛋白SYCP3的表达,因此PRC1.6复合体功能异常导致减数分裂相关基因的异常高表达,使ESCs越过原始生殖细胞(primordial germ cells, PGCs)直接形成类似减数分裂前期的细胞,说明MAX对于调控减数分裂的起始至关重要[40]。在小鼠ESCs中PRC1.6复合体不同组分的ChIP-Seq数据显示,该复合体在全基因组中靶向上万个位点(由PCGF6、L3MBTL2和MGA共同靶向);当逐个敲除不同组分时,这些PRC1.6复合体靶基因中转录本水平发生上调的基因数目一般为数百个,如敲除Pcgf6导致882个基因上调[25,26];敲除L3mbtl2导致421个[25]或167个[41]基因上调。这类对PRC1.6复合体调控最敏感的基因中包含不同类型的发育相关基因(如神经发育基因),但最显著的是特异性地包含生殖谱系及减数分裂关键基因[24,25,40,41],这是其区分于其他PRC1复合体亚型所独具的功能。在敲除Pcgf6的ESCs中表达显著上调(>10倍)的基因中有49个都是在精子发生过程中必不可少的基因[22]。在ESCs中敲除Mga后发现与减数分裂相 关的基因Taf7l、Slc25a31、Stra8和Sycp3表达量上调[25]。同样,敲除Rybp的ESCs也呈现出生殖谱系相关基因特异性上调的现象[38]。另有研究表明,PRC1.6复合体同时抑制体细胞中生殖谱系相关基因的表达,如敲除E2f6基因的小鼠呈现出生殖谱系相关基因(SMC1β和STAG3等)在体细胞中的异常表达[42,43]。这些实验证据都支持PRC1.6复合体在ESCs等其他非生殖细胞类型中发挥稳定抑制生殖谱系特异性基因表达的作用,维持这些细胞类型的身份,限制其向生殖细胞谱系异常分化。
3.2 PRC1.6复合体对于正常胚胎发育必不可少
PRC1.6复合体对于哺乳动物胚胎发育的正常进行是必需的,但每个组分被敲除后小鼠所呈现的胚胎发育异常状况却有所区别,这一定程度上反映出每个组分在该复合体功能发挥中的权重不同以及部分组分可能因为参与多种复合体而具备更为多样的发育调控功能(表3)。有研究表明,Max敲除小鼠在E5.5~6.5时表现出胚胎及胚外组织发育阻滞,且不具备明显的胚胎特征或胚胎与胚外组织分界不清晰[44]。Mga的表达在小鼠E3.5时的内细胞团中以及E4.5~E6.5时具全能性的上胚层(epiblast)中均可检测到,当使用MgaGt/MgaGt纯合敲除Mga,内细胞团在E4.5后随即发育停滞,细胞增殖无明显变化但细胞凋亡增加,上胚层不能正常形成[45]。由MgaGt衍生出来的MgaInv等位基因呈现出Mga低表达表型,约一半MgaInv/MgaInv纯合小鼠在出生及哺乳期死亡,其余能存活并可育[45]。早期通过原位杂交实验发现Mga在胚胎发育E9.5~10.5间广谱表达,在肢芽、腮弓及尾部区域高表达[26]。为进一步澄清其在E4.5之后的发育调控功能,近期研究发现表达低剂量Mga的MgaGt/MgaInv杂合转基因小鼠在E7.5~E12.5之间死亡,主要由于E7.5之前上胚层全能性细胞发育停滞导致[46],所以Mga对于上胚层的正常分化是必不可少的。L3mbtl2缺失会导致多能性ESCs增殖及分化异常,在E7.5左右胚胎因不能正常形成原肠胚而致死,致死原因尚不明确[41]。4 PRC1.6复合体调控雄性性腺发育及精子生成过程中生殖谱系基因的顺次激活
PRC1.6复合体不仅可以影响胚胎发育中生殖谱系相关基因的表达,还可以影响精子生成和雄性性腺发育。从表3中可知,当分别敲除E2f6、Hp1γ、L3mbtl2和Ring1b时,小鼠都可存活,但会表现出不同程度的睾丸发育不良及精子生成障碍。特异性敲除生殖细胞中Ring1b的小鼠呈现出睾丸发育异常和不育表型,GSCs数目明显下降并且异常分化[53]。最新研究表明,L3MBTL2在小鼠精原细胞减数分裂前期高表达,特异性敲除生殖细胞中L3mbtl2的雄鼠虽然可育但表现出睾丸重量减轻,附睾中的精子数目减少及异常精子增多,并出现睾丸早衰现象[69]。进一步研究发现这些雄鼠的精原细胞在减数分裂前期出现染色体异常联会,且影响延长型精子中组蛋白乙酰化水平和精子成熟过程中组蛋白向鱼精蛋白的转变,说明L3mbtl2参与精子生成过程进而影响性腺发育。这些研究结果证明了PRC1.6复合体确实在精子发生及性腺发育过程中具有重要的作用。近期关于PRC1.6复合体对生殖细胞分化过程中生殖谱系基因的表达调控也有相应的报道。Maezawa等[53]构建了生殖细胞特异性敲除Ring1b的小鼠,并从出生7天的雄鼠中分离出未分化的精原细胞(Thy1+标记)和已初步分化的精原细胞(c-Kit+标记)。RNA-Seq检测结果揭示,与野生型相比Ring1b-/-小鼠的Thy1+细胞中分别有116个基因表达下调和69个基因表达上调,c-Kit+细胞中有1381基因表达下调和269个基因表达上调。GO分析发现大多数上调基因的功能与精子发生没有直接的关联,而下调基因中富含调控精子分化的重要功能基因。Thy1+和c-Kit+的细胞中因Ring1b敲除表达下调的基因中有78个重叠,包含许多精子发生过程中必要的调控基因(如Sall4、Plzf、Lin28a、Tdrd9和Piwil2),其中下调最显著的是Sall4,后续ChIP实验证实Sall4是RING1B的直接靶基因。c-Kit+细胞中因Ring1b敲除特异性下调的基因主要用于调节精原细胞分化(如Dmrt、Nxf2和Soh1h1d),减数分裂的开启(如Dazl和Stra8)和piRNA的调控(如Tdrkh、Mael和Tdrd1);在正常精子发生中这些基因会在Thy1+精原细胞向c-Kit+精原细胞分化的过程中被高度激活,且进一步研究证实RING1B与SALL4可以共同靶向并上调这群基因的表达。该研究表明RING1B可与SALL4协同调控生殖谱系基因的顺次激活,这对生殖谱系细胞的正常分化及进入减数分裂等过程至关重要。
5 PRC1.6复合体是精子发生表观遗传调控网络的重要组成部分
精子发生是一个受到多因素调控的复杂发育过程。表4简要概括了精子发生过程中已知的表观遗传调控机制的大致类型。参与甲基化的各类DNA甲基转移酶(DNMTs)的表达及活性在生殖细胞发育过程中呈现明显的动态变化,这些DNMTs的异常往往直接导致生殖细胞发育异常以及个体雄性不育[70]。胚胎发育及生殖细胞发育过程中呈现动态变化的组蛋白甲基化、乙酰化等修饰对于建立细胞谱系的特异性身份至关重要[71,72]。其他组蛋白修饰,如泛素化、磷酸化以及近几年新发现的组蛋白巴豆酰化等对精子发育也十分重要[73,74]。在精子发育过程中,组装核小体的经典组蛋白在特定发育阶段被不同睾丸特异性组蛋白变体所替换,逐渐减弱DNA和组蛋白之间的作用,为大多数组蛋白在单倍体精子成熟的过程中被鱼精蛋白所替代做准备[75,76]。非编码RNA如piRNA介导的 Piwi基因沉默机制是精子发育及其他情况下抑制可转座元件活性的重要途径,对于精子的正常发育必不可少[77];同时生殖细胞特异性表达的lncRNAs或miRNAs等在精子发育过程中也都起到重要的调控作用[78,79,80,81]。另外,近几年发现RNA的修饰尤其是甲基化修饰可参与RNA的转录后加工、核输出、翻译及RNA稳定性调节等,在造血干细胞分化、精子发生、神经发育等发育过程中发挥重要作用[82,83]。Table 4
表4
表4 精子发育过程中的表观遗传调控方式
Table 4
| 调控类型 | 分类 | 作用靶点及分子功能举例 | 文献 |
|---|---|---|---|
| DNA甲基化 | 与原始生殖细胞的增殖状态以及染色体联会等有关 | [84,85] | |
| 组蛋白修饰 | 甲基化 | H3K4甲基化水平在精原干细胞(SSCs)阶段最高;H3K9和H3K27甲基化水平在SSCs阶段中较低,在减数分裂过程中增加 | [86] |
| 乙酰化 | 精子形成过程中,组蛋白H4高度乙酰化,有助于置换组蛋白变体 | [87] | |
| 磷酸化 | 小鼠精子组蛋白磷酸化影响染色质解凝集、减数分裂后染色质折叠和压缩、双线期精原细胞减数分裂等过程,是组蛋白变体和/或鱼精蛋白替换的前提 | [88] | |
| 泛素化 | E3泛素连接酶RNF8异常导致精子细胞发育后期缺陷 | [89,90] | |
| 巴豆酰化 | Cdyl敲除小鼠组蛋白巴豆酰化失调和雄性生育能力降低,附睾精子数和精子运动能力降低 | [74] | |
| 组蛋白变体 | 睾丸H1变体H1t2影响精子形态和生育力 | [91] | |
| 非编码RNA | piRNA | 在生殖细胞中抑制可转座元件的表达并指导该位置的新生甲基化;MILI和MIWI2在影响piRNA生物合成及指导在不同类型转座元件上DNA新生甲基化方面功能有所区别 | [92, 93] |
| miRNA | 调控SSCs自我更新相关转录因子的表达;维持SSCs总数并调控SSCs动态平衡 | [94, 95] | |
| lncRNA | 小鼠精原细胞特异性表达的lncRNA033862调控Gfra1基因表达,对于SSCs生存所必需 | [96] | |
| RNA甲基化 | mRNA的N6-腺苷甲基化修饰(m6A)确保在小鼠精子发生的不同阶段协调翻译 | [97] |
新窗口打开|下载CSV
如表4所示,生殖谱系基因的表达受到非常复杂精密的表观遗传调控网络的调控,PRC1.6复合体作为该表观遗传调控网络的一部分,与该调控网络的其他“版块”之间存在着紧密的交互作用。目前已知PRC1.6复合体与DNA甲基化、组蛋白H3K9me3和H3K27me3等重要的表观遗传修饰存在协同作用,且这些协同互作呈现出明显的动态变化(图1)。例如,在胚胎发育早期E2F6对于减数分裂相关基因的转录抑制主要通过与PRC2协同完成,但不依赖于DNA甲基转移酶DNMT3B[98];然而,在小鼠体细胞中,E2F6则更多地通过招募DNMT3B对靶基因启动子进行甲基化来实现对这些生殖谱系特异性基因表达的有效抑制[99]。有趣的是,虽然小鼠ESCs中E2F6与DNMT3B之间没有协同关系,但MAX与DNA甲基转移酶DNMT1,DNMT3A,DNMT3B之间存在明显的协同作用并抑制生殖谱系相关基因的转录[100]。除了PRC1.6复合体自身包含HP1γ,可实现对转录抑制标记H3K9me3的结合之外,最近有研究揭示MAX与H3K9甲基转移酶SETDB1 可协同介导生殖谱系相关基因的转录抑制,但该机制并不依赖于PRC1.6复合体[100]。另外,在PCGF6 敲除的小鼠ESCs中,piRNA互作蛋白PIWIL1和PIWIL2的表达量均明显上升,意味着PRC1.6复合体通过抑制piRNA调控途径中重要RNA结合蛋白的表达水平来控制其活性[24]。除此之外,PRC1.6复合体组分自身的表达水平受到严密调控,目前这方面的报道相对较少,已知RYBP[101]和E2F6[102]等均可以受到miRNA的调控。
图1
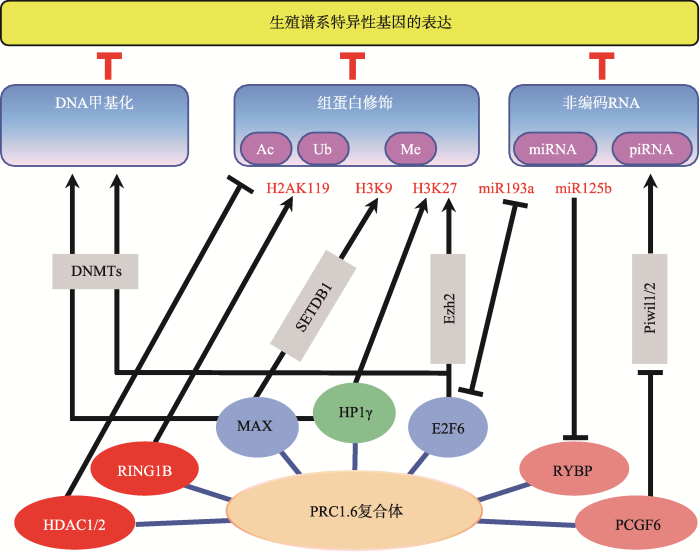 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1PRC1.6复合体与其他表观遗传调控机制的相互作用
Fig. 1The crosstalk between PRC1.6 and other epigenetic regulators
6 结语与展望
本文总结了多梳蛋白PRC1.6复合体主要组分的分子功能及其在胚胎发育和精子发生中所扮演的重要角色(图2)。胚胎发育过程中PRC1.6复合体通过抑制生殖谱系相关基因及其他发育调控基因的表达确保了胚胎的正常分化。PRC1.6复合体也参与精子发生的表观遗传调控,顺次激活其不同阶段生殖谱系相关基因的表达,是正常精子发生及性腺发育的必要条件。尽管在过去的几年中有关PRC1.6复合体的组成及作用机制等方面取得了可喜的进展,但是对于该复合体具体的工作机制包括组分之间及整个复合体与其他表观调控途径之间的协调关系等的研究还有待深入。如PRC1.6各组分在调节减数分裂及精子形成过程中的具体分工还不够明确,以及该复合体如何实现对体细胞及不同分化阶段的生殖细胞中相关特异性基因的差异调控等。随着基因编辑技术以及现代分子生物学的进一步深入发展,PRC1.6各组分的具体功能以及它们之间的相互作用机制一定会研究得更加清晰,并将为与PRC1.6复合体组分突变或功能缺陷相关的男性不育的诊断与治疗带来新的思路。图2
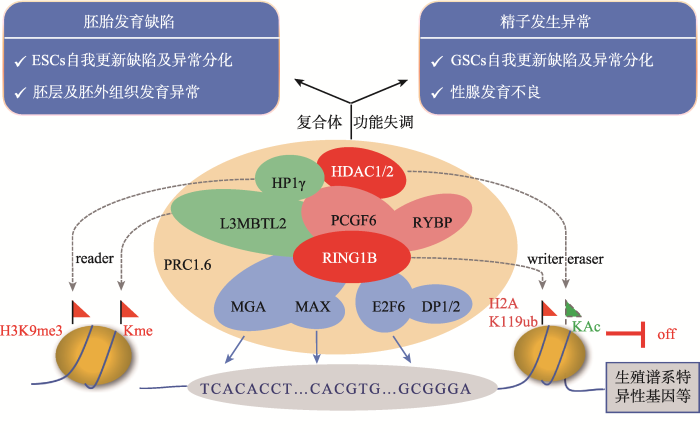 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2PRC1.6复合体的组成、分子功能及活性异常产生的发育缺陷
标注红色的组分为具有酶活性或辅助酶活性的因子;标注蓝色的组分为具有DNA结合能力的转录因子;标注绿色的组分为具有染色质结合能力的因子。
Fig. 2The composition, molecular functions of PRC1.6 complex and the developmental defects originated from its dysregulation
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URLPMID:7254358 [本文引用: 1]

Insects are thought to have evolved from millipede-like ancestors composed largely of a series of identical, leg-bearing segments. This view of insect evolution is supported by the existence of homoeotic mutations which transform particular abdominal and head segments into thoracic segments 1 3 . Such transformations are described as atavistic 3 , because they return specialized segments to a more primitive condition. In Drosophila , several dominant mutations of the homoeotic locus Antennapedia ( Antp ) lead to a transformation of the antenna to the second leg 4 8 . Here, I describe the isolation and characterization of apparent null alleles of the Antp locus. These mutations lead to a homoeotic phenotype which is the reverse of the dominant Antennapedia phenotype, namely, they result in the transformation of the second leg into an antenna but do not alter the development of the normal antenna itself. This result indicates that (1) the product of the wild-type Antp gene is normally active in the mesothorax where it promotes a mesothoracic pathway of development instead of an antennal pathway, (2) the Antp + gene product is normally absent or inactive in the antenna and (3) dominant mutations of the locus result in the inappropriate activity of the wild-type gene product in the antenna and hence in the transformation of antenna to leg. Thus, unlike most other homoeotic gene products, the product of the Antp + gene seems to promote, not to repress or modify, an atavistic condition.
URLPMID:26329598 [本文引用: 1]
Intricate layers of regulation determine the unique gene expression profiles of a given cell and, therefore, underlie the immense phenotypic diversity observed among cell types. Understanding the mechanisms that govern which genes are expressed and which genes are silenced is a fundamental focus in biology. The Polycomb and Trithorax group chromatin proteins play important roles promoting the stable and heritable repression and activation of gene expression, respectively. These proteins, which are conserved across metazoans, modulate post-translational modifications on histone tails and regulate nucleosomal structures. Here, we review recent advances that have shed light on the mechanisms by which these two classes of proteins act to maintain epigenetic memory and allow dynamic switches in gene expression during development.
URLPMID:24937863 [本文引用: 1]
Each body segment of Drosophila follows a unique developmental pathway, controlled by the selective expression of homoeotic genes such as Sex combs reduced ( Scr ) 1,2 and the bithorax complex ( BX-C ) 3,4 . Little is known about the regulation of these genes, though several potential activators or repressers have been described 5鈥8 . For instance, absence of the extra sex combs ( esc ) gene product apparently causes adventitious expression of all the BX-C genes in most or all larval body segments 5 . Absence of the trithorax ( trx ) gene appears to prevent Scr and BX-C expression 7,9,10 but only in adult cells; differentiation of the larval segments is only slightly affected 11 . I show here that the correct segmental differentiation of the larva does not require maternally deposited trx + product, but that the esc mutant phenotype is suppressed by the removal of the trx gene, which implies that the BX-C can be differentially expressed in the absence of both the trx gene and the esc gene product.
URLPMID:42633981004851 [本文引用: 1]
Polycomb proteins play an essential role in maintaining the repression of developmental genes in self-renewing embryonic stem cells. The exact mechanism allowing the derepression of polycomb target genes during cell differentiation remains unclear. Our project aimed to identify Cbx8 binding sites in differentiating mouse embryonic stem cells. Therefore, we used a genome-wide chromatin immunoprecipitation of endogenous Cbx8 coupled to direct massive parallel sequencing (ChIP-Seq). Our analysis identified 171 high confidence peaks. By crossing our data with previously published microarray analysis, we show that several differentiation genes transiently recruit Cbx8 during their early activation. Depletion of Cbx8 partially impairs the transcriptional activation of these genes. Both interaction analysis, as well as chromatin immunoprecipitation experiments support the idea that activating Cbx8 acts in the context of an intact PRC1 complex. Prolonged gene activation results in eviction of PRC1 despite persisting H3K27me3 and H2A ubiquitination. The composition of PRC1 is highly modular and changes when embryonic stem cells commit to differentiation. We further demonstrate that the exchange of Cbx7 for Cbx8 is required for the effective activation of differentiation genes. Taken together, our results establish a function for a Cbx8-containing complex in facilitating the transition from a Polycomb-repressed chromatin state to an active state. As this affects several key regulatory differentiation genes this mechanism is likely to contribute to the robust execution of differentiation programs.
URLPMID:25519132 [本文引用: 1]
Abstract Naturally occurring variations of Polycomb repressive complex 1 (PRC1) comprise a core assembly of Polycomb group proteins and additional factors that include, surprisingly, autism susceptibility candidate 2 (AUTS2). Although AUTS2 is often disrupted in patients with neuronal disorders, the mechanism underlying the pathogenesis is unclear. We investigated the role of AUTS2 as part of a previously identified PRC1 complex (PRC1-AUTS2), and in the context of neurodevelopment. In contrast to the canonical role of PRC1 in gene repression, PRC1-AUTS2 activates transcription. Biochemical studies demonstrate that the CK2 component of PRC1-AUTS2 neutralizes PRC1 repressive activity, whereas AUTS2-mediated recruitment of P300 leads to gene activation. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) demonstrated that AUTS2 regulates neuronal gene expression through promoter association. Conditional targeting of Auts2 in the mouse central nervous system (CNS) leads to various developmental defects. These findings reveal a natural means of subverting PRC1 activity, linking key epigenetic modulators with neuronal functions and diseases.
URLPMID:26340528 [本文引用: 1]
Morey et02al. reveal that Mel18, a Polycomb-complex-associated protein, is an essential epigenetic regulator of cardiac differentiation. During directed differentiation of embryonic stem cells, Morey et02al. find that Mel18-PRC1 complexes exchange subunits in a stage-specific manner and instruct sequential gene activation and repression programs to specify mesoderm fate, prevent alternate lineage commitment, and promote cardiac differentiation.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:16714294 [本文引用: 1]
Polycomb group proteins Bmi-1 and Ring1B are core subunits of the PRC1 complex, which plays important roles in the regulation of Hox gene expression, X-chromosome inactivation, tumorigenesis, and stem cell self-renewal. The RING finger protein Ring1B is an E3 ligase that participates in the ubiquitination of lysine 119 of histone H2A, and the binding of Bmi-1 stimulates the E3 ligase activity. We have mapped the regions of Bmi-1 and Ring1B required for efficient ubiquitin transfer and determined a 2.5-A structure of the Bmi-1-Ring1B core domain complex. The structure reveals that Ring1B "hugs" Bmi-1 through extensive RING domain contacts and its N-terminal tail wraps around Bmi-1. The two regions of interaction have a synergistic effect on the E3 ligase activity. Our analyses suggest a model where the Bmi-1-Ring1B complex stabilizes the interaction between the E2 enzyme and the nucleosomal substrate to allow efficient ubiquitin transfer.
URLPMID:15525528 [本文引用: 2]
In many higher organisms, 5% 15% of histone H2A is ubiquitylated at lysine 119 (uH2A). The function of this modification and the factors involved in its establishment, however, are unknown. Here we demonstrate that uH2A occurs on the inactive X chromosome in female mammals and that this correlates with recruitment of Polycomb group (PcG) proteins belonging to Polycomb repressor complex 1 (PRC1). Based on our observations, we tested the role of the PRC1 protein Ring1B and its closely related homolog Ring1A in H2A ubiquitylation. Analysis of Ring1B null embryonic stem (ES) cells revealed extensive depletion of global uH2A levels. On the inactive X chromosome, uH2A was maintained in Ring1A or Ring1B null cells, but not in double knockout cells, demonstrating an overlapping function for these proteins in development. These observations link H2A ubiquitylation, X inactivation, and PRC1 PcG function, suggesting an unanticipated and novel mechanism for chromatin-mediated heritable gene silencing.
URLPMID:24857660 [本文引用: 1]
Polycomb group proteins are important repressors of developmentally regulated genes, but how these complexes are recruited to their target genes is still largely unknown. In this study, Cooper et al. show that Polycomb group protein recruitment is a combinatorial readout of unmethylated CpG density and antagonism by specific histone tail modifications. Unexpectedly, they also show that monoubiquitylated histone H2A, the modification produced by Polycomb repressor complex 1 (PRC1), is sufficient to recruit PRC2.
URLPMID:4076598 [本文引用: 1]
Polycomb group (PcG) complexes are epigenetic regulatory complexes that conduct transcriptional repression of target genes via modifying the chromatin. The two best characterized forms of PcG complexes, polycomb repressive complexes 1 and 2 (PRC1 and PRC2), are required for maintaining the stemness of embryonic stem cells and many types of adult stem cells. The spectra of target genes for PRCs are dynamically changing with cell differentiation, which is essential for proper decisions on cell fate during developmental processes. Chromobox (CBX) family proteins are canonical components in PRC1, responsible for targeting PRC1 to the chromatin. Recent studies highlight the function specifications among CBX family members in undifferentiated and differentiated stem cells, which reveal the interplay between compositional diversity and functional specificity of PRC1. In this review, we summarize the current knowledge about targeting and functional mechanisms of PRCs, emphasizing the recent breakthroughs related to CBX proteins under a number of physiological and pathological conditions.
URLPMID:25065329 [本文引用: 1]
Polycomb group proteins (PcGs) are essential epigenetic regulators that play key roles in development, pluripotency, senescence, and cancer. Recent reports have found that the composition of mammalian Polycomb repressive complex 1 (PRC1) is far more varied than previously thought. PRC1 diversity largely depends on the presence of CBX proteins, dividing them into canonical and non-canonical, the existence of redundant subunits, and different binding affinities and/or regulation. However, there is no clear insight into how many functional PRC1 complexes exist and what the biological relevance is for such diversification. In this review we focus on mammalian PRC1 and discuss the mechanisms by which canonical and non-canonical PRC1 are recruited to chromatin, their role in normal development and disease, and emerging evidence for PRC1 as a transcriptional activator.
URLPMID:28007606 [本文引用: 1]
The compositional complexity of Polycomb Repressive Complex 1 (PRC1) increased dramatically during vertebrate evolution. What is considered the “canonical” PRC1 complex consists of four subunits originally identified as regulators of body segmentation in Drosophila . In mammals, each of these four canonical subunits consists of two to six paralogs that associate in a combinatorial manner to produce over a hundred possible distinct PRC1 complexes with unknown function. Genetic studies have begun to define the phenotypic roles for different PRC1 paralogs; however, relating these phenotypes to unique biochemical and transcriptional function for the different paralogs has been challenging. In this review, we attempt to address how the compositional diversity of canonical PRC1 complexes relates to unique roles for individual PRC1 paralogs in transcriptional regulation. This review focuses primarily on PRC1 complex composition, genome targeting, and biochemical function.
URL [本文引用: 3]
URLPMID:3142354 [本文引用: 2]
We have identified human MBT domain-containing protein L3MBTL2 as an integral component of a protein complex that we termed Polycomb repressive complex 1 (PRC1)-like 4 (PRC1L4), given the copresence of PcG proteins RING1, RING2, and PCGF6/MBLR. PRC1L4 also contained E2F6 and CBX3/HP1 , known to function in transcriptional repression. PRC1L4-mediated repression necessitated L3MBTL2 that compacted chromatin in a histone modification-independent manner. Genome-wide location analyses identified several hundred genes simultaneously bound by L3MBTL2 and E2F6, preferentially around transcriptional start sites that exhibited little overlap with those targeted by other E2Fs or by L3MBTL1, another MBT domain-containing protein that interacts with RB1. L3MBTL2-specific RNAi resulted in increased expression of target genes that exhibited a significant reduction in H2A lysine 119 monoubiquitination. Our findings highlight a PcG/MBT collaboration that attains repressive chromatin without entailing histone lysine methylation marks.Highlights? L3MBTL2 is a component of a PcG protein complex with H2AK119ub1 E3-ligase activity ? L3MBTL2 is preferentially bound to TSSs and functions as transcriptional repressor ? L3MBTL2 and E2F6 cooperate on a genome scale to regulate transcription ? L3MBTL2 compacts chromatin independent of histone lysine methylation marks
URL [本文引用: 3]
URL [本文引用: 5]
URLPMID:17070805 [本文引用: 1]
The Rybp protein has been promoted as a Polycomb group (PcG)-associated protein, but its molecular function has remained elusive. Here we show that Rybp is a novel ubiquitin binding protein and is itself ubiquitinated. The Rybp interacting PcG protein Ring1B, a known ubiquitin E3 ligase, promotes Rybp ubiquitination. Moreover, one target of Rybp’s ubiquitin binding domain appears to be ubiquitinated histone H2A; this histone is a substrate for Ring1B’s E3 ligase activity in association with gene silencing processes. These findings on Rybp provide a further link between the ubiquitination system and PcG transcriptional repressors.
URLPMID:28049731 [本文引用: 4]
The Polycomb group (PcG) proteins have an important role in controlling the expression of key genes implicated in embryonic development, differentiation, and decision of cell fates. Emerging evidence suggests that Polycomb repressive complexes 1 (PRC1) is defined by the six Polycomb group RING finger protein (Pcgf) paralogs, and Pcgf proteins can assemble into noncanonical PRC1 complexes. However, little is known about the precise mechanisms of differently composed noncanonical PRC1 in the maintenance of the pluripotent cell state. Here we disrupt the Pcgf genes in mouse embryonic stem cells by CRISPR-Cas9 and find Pcgf6 null embryonic stem cells display severe defects in self-renewal and differentiation. Furthermore, Pcgf6 regulates genes mostly involved in differentiation and spermatogenesis by assembling a noncanonical PRC1 complex PRC1.6. Notably, Pcgf6 deletion causes a dramatic decrease in PRC1.6 binding to target genes and no loss of H2AK119ub1. Thus, Pcgf6 is essential for recruitment of PRC1.6 to chromatin. Our results reveal a previously uncharacterized, H2AK119ub1-independent chromatin assembly associated with PRC1.6 complex.
URL [本文引用: 7]
URLPMID:10601024 [本文引用: 3]
The basic-helix-loop-helix-leucine zipper (bHLHZip) proteins Myc, Mad and Mnt are part of a transcription activation/repression system involved in the regulation of cell proliferation. The function of these proteins as transcription factors is mediated by heterodimerization with the small bHLHZip protein Max, which is required for their specific DNA binding to E-box sequences. We have identified a novel Max-interacting protein, Mga, which contains a Myc-like bHLHZip motif, but otherwise shows no relationship with Myc or other Max-interacting proteins. Like Myc, Mad and Mnt proteins, Mga requires heterodimerization with Max for binding to the preferred Myc090009Max-binding site CACGTG. In addition to the bHLHZip domain, Mga contains a second DNA-binding domain: the T-box or T-domain. The T-domain is a highly conserved DNA-binding motif originally defined in Brachyury and characteristic of the Tbx family of transcription factors. Mga binds the preferred Brachyury-binding sequence and represses transcription of reporter genes containing promoter-proximal Brachyury-binding sites. Surprisingly, Mga is converted to a transcription activator of both Myc090009Max and Brachyury site-containing reporters in a Max-dependent manner. Our results suggest that Mga functions as a dual-specificity transcription factor that regulates the expression of both Max-network and T-box family target genes.
[本文引用: 1]
URLPMID:3950706 [本文引用: 1]
Lethal(3) malignant brain tumour like 2 (L3MBTL2) is an integral component of the polycomb repressive complex 1.6 (PRC1.6) and has been implicated in transcriptional repression and chromatin compaction. Here, we show that L3MBTL2 is modified by SUMO2/3 at lysine residues 675 and 700 close to the C-terminus. SUMOylation of L3MBTL2 neither affected its repressive activity in reporter gene assays nor it binding to histone tails in vitro. In order to analyse whether SUMOylation affects binding of L3MBTL2 to chromatin, we performed ChIP-Seq analysis with chromatin of wild-type HEK293 cells and with chromatin of HEK293 cells stably expressing either FLAG-tagged SUMOylation-competent or SUMOylation-defective L3MBTL2. Wild-type FLAG-L3MBTL2 and the SUMOylation-defective FLAG-L3MBTL2 K675/700R mutant essentially occupied the same sites as endogenous L3MBTL2 suggesting that SUMOylation of L3MBTL2 does not affect chromatin binding. However, a subset of L3MBTL2-target genes, particularly those with low L3MBTL2 occupancy including pro-inflammatory genes, was de-repressed in cells expressing the FLAG-L3MBTL2 K675/700R mutant. Finally, we provide evidence that SUMOylation of L3MBTL2 facilitates repression of these PRC1.6-target genes by balancing the local H2Aub1 levels established by the ubiquitinating enzyme RING2 and the de-ubiquitinating PR UB complex.
URLPMID:19233876 [本文引用: 1]
The MBT repeat has been recently identified as a key domain capable of methyl-lysine histone recognition. Functional work has pointed to a role for MBT domain-containing proteins in transcriptional repression of developmental control genes such as Hox genes. In this study, L3MBTL2, a human homolog of Drosophila Sfmbt critical for Hox gene silencing, is demonstrated to preferentially recognize lower methylation states of several histone-derived peptides through its fourth MBT repeat. High-resolution crystallographic analysis of the four MBT repeats of this protein reveals its unique asymmetric rhomboid architecture, as well as binding mechanism, which preclude the interaction of the first three MBT repeats with methylated peptides. Structural elucidation of an L3MBTL2-H4K20me1 complex and comparison with other MBT-histone peptide complexes also suggests that an absence of distinct surface contours surrounding the methyl-lysine-binding pocket may underlie the lack of sequence specificity observed for members of this protein family.
URLPMID:5065315
10.7554/eLife.18591.001Polycomb group (PcG) proteins function as chromatin-based transcriptional repressors that are essential for normal gene regulation during development. However, how these systems function to achieve transcriptional regulation remains very poorly understood. Here, we discover that the histone H2AK119 E3 ubiquitin ligase activity of Polycomb repressive complex 1 (PRC1) is defined by the composition of its catalytic subunits and is highly regulated by RYBP/YAF2-dependent stimulation. In mouse embryonic stem cells, RYBP plays a central role in shaping H2AK119 mono-ubiquitylation at PcG targets and underpins an activity-based communication between PRC1 and Polycomb repressive complex 2 (PRC2) which is required for normal histone H3 lysine 27 trimethylation (H3K27me3). Without normal histone modification-dependent communication between PRC1 and PRC2, repressive Polycomb chromatin domains can erode, rendering target genes susceptible to inappropriate gene expression signals. This suggests that activity-based communication and histone modification-dependent thresholds create a localized form of epigenetic memory required for normal PcG chromatin domain function in gene regulation.DOI: http://dx.doi.org/10.7554/eLife.18591.001
URLPMID:25648995
Abstract Histone deacetylases (HDACs) are posttranslational modifiers that deacetylate proteins. Despite their crucial role in numerous biological processes, the use of broad-range HDAC inhibitors (HDACi), has shown clinical efficacy. However, undesired side effects highlight the necessity to better understand the biology of different HDACs and target the relevant HDACs. Using a novel mouse model, in which HDAC1 and HDAC2 can be simultaneously deleted in the intestine of adult mice, we show that the simultaneous deletion of HDAC1 and HDAC2 leads to a rapid loss of intestinal homeostasis. Importantly, this deletion cannot be sustained, and 8 days after initial ablation, stem cells that have escaped HDAC1 or HDAC2 deletion swiftly repopulate the intestinal lining. In vitro ablation of HDAC1 and HDAC2 using intestinal organoid cultures resulted in a down-regulation of multiple intestinal stem cell markers and functional loss of clonogenic capacity. Importantly, treatment of wild-type organoids with class I-specific HDACi MS-275 also induced a similar loss of stemness, providing a possible rationale for the gastrointestinal side effects often observed in HDACi-treated patients. In conclusion, these data show that HDAC1 and HDAC2 have a redundant function and are essential to maintain intestinal homeostasis.
URLPMID:16061184
Methylation of histones modulates chromatin structure and function. Whereas methylation of histone H3 on lysines 4, 36, and 79 has been linked with gene activation, methylation of H3 on lysines 9 and 27 and histone H4 on lysine 20 is associated with heterochromatin and some repressed genes within euchromatin. Here, we show that H3K9 di- and trimethylation occur in the transcribed region of active genes in mammalian chromatin. This modification is dynamic, as it increases during activation of transcription and is rapidly removed upon gene repression. Heterochromatin Protein 1γ (HP1γ), a protein containing a chromo-domain that recognizes H3K9 methylation, is also present in the transcribed region of all active genes examined. Both the presence of HP1γ and H3K9 methylation are dependent upon elongation by RNA polymerase II. These findings demonstrate novel roles for H3K9 methylation and HP1γ in transcription activation.
URLPMID:27247273 [本文引用: 1]
The polycomb repressive complex 1 (PRC1) is a multi-subunit complex that plays critical roles in the epigenetic modulation of gene expression. Here, we show that the PRC1 component polycomb group ring finger 6 (Pcgf6) is required to maintain embryonic stem cell (ESC) identity. In contrast to canonical PRC1, Pcgf6 acts as a positive regulator of transcription and binds predominantly to promoters bearing active chromatin marks.Pcgf6is expressed at high levels in ESCs, and knockdown reduces the expression of the core ESC regulatorsOct4,Sox2, andNanog. Conversely,Pcgf6overexpression prevents downregulation of these factors and impairs differentiation. In addition, Pcgf6 enhanced reprogramming in both mouse and human somatic cells. The genomic binding profile of Pcgf6 is highly similar to that of trithorax group proteins, but not of PRC1 or PRC2 complexes, suggesting that Pcgf6 functions atypically in ESCs. Our data reveal novel roles for Pcgf6 in directly regulatingOct4,Nanog,Sox2, andLin28expression to maintain ESC identity.
URLPMID:25187489 [本文引用: 1]
Abstract Polycomb group (PcG) proteins comprise evolutionary conserved factors with essential functions for embryonic development and adult stem cells. PcG proteins constitute two main multiprotein polycomb repressive complexes (PRC1 and PRC2) that operate in a hierarchical manner to silence gene transcription. Functionally distinct PRC1 complexes are defined by Polycomb group RING finger protein (Pcgf) paralogs. So far, six Pcgf paralogs (Pcgf1 6) have been identified as defining components of different PCR1-type complexes. Paralog-specific functions are not well understood. Here, we show that Pcgf6 is the only Pcgf paralog with high expression in undifferentiated embryonic stem cells (ESCs). Upon differentiation Pcgf6 expression declines. Following Pcgf6 kockdown (KD) in ESCs, the expression of pluripotency genes decreased, while mesodermal- and spermatogenesis-specific genes were derepressed. Concomitantly with the elevated expression of mesodermal lineage markers, Pcgf6 KD ESCs showed increased hemangioblastic and hematopoietic activities upon differentiation suggesting a function of Pcgf6 in repressing mesodermal-specific lineage genes. Consistant with a role in pluripotency, Pcgf6 replaced Sox2 in the generation of germline-competent induced pluripotent stem (iPS) cells. Furthermore, Pcgf6 KD in mouse embryonic fibroblasts reduced the formation of ESC-like colonies in OSKM-driven reprogramming. Together, these analyses indicate that Pcgf6 is nonredundantly involved in maintaining the pluripotent nature of ESCs and it functions in iPS reprogramming. S tem C ells 2014;32:3112 3125
URL
Though genetic data suggest that Polycomb group proteins (PcGs) are central chromatin modifiers and repressors that have been implicated in control of embryonic stem cell (ESC) pluripotency, the precise mechanism of PcG complex recruitment remains elusive, especially in mammals. We now report that the first and second MBT repeats of L3mbtl2 are important structural and functional features that arenecessary and sufficient for L3mbtl2-mediated recruitment of PRC1.6 complex to target promoters. Interestingly, this region of L3mbtl2 harbors the evolutionarily conserved Pho-binding pocketalso present in Drosophila Sfmbt, and mutation of the critical residues within this pocket completely abolishes its interaction with target promoters. Additionally, decreased PRC1.6 chromatin occupancy was observed following loss of individual components (L3mbtl2, Pcgf6, and Max) of the complex. Our findings suggest that the recruitment of noncanonical PRC1.6 complex in ESCs might be the result of L3mbtl2 interaction with multiple components of the complex.
URLPMID:8223441
Abstract It is widely believed that the cellular transcription factor DRTF1/E2F integrates cell cycle events with the transcription apparatus because during cell cycle progression in mammalian cells it interacts with molecules that are important regulators of cellular proliferation, such as the retinoblastoma tumour suppressor gene product (pRb), p107, cyclins and cyclin-dependent kinases. Thus, pRb, which negatively regulates early cell cycle progression and is frequently mutated in tumour cells, and the Rb-related protein p107, bind to and repress the transcriptional activity of DRTF1/E2F. Viral oncoproteins, such as adenovirus E1a and SV40 large T antigen, overcome such repression by sequestering pRb and p107 and in so doing are likely to activate genes regulated by DRTF1/E2F, such as cdc2, c-myc and DHFR. Two sequence-specific DNA binding proteins, E2F-1 and DP-1, which bind to the E2F site, contain a small region of similarity. The functional relationship between them has, however, been unclear. We report here that DP-1 and E2F-1 exist in a DNA binding complex in vivo and that they bind efficiently and preferentially as a heterodimer to the E2F site. Moreover, studies in yeast and Drosophila cells indicate that DP-1 and E2F-1 interact synergistically in E2F site-dependent transcriptional activation.
URLPMID:17129771
Activation of E2F transcription factors is thought to drive the expression of genes essential for the transition of cells from G1 to S phase and for the initiation of DNA replication. However, this textbook view of E2Fs is increasingly under challenge. Here we discuss an alternative model for how E2Fs may work.
URLPMID:3295013 [本文引用: 3]
Abstract Polycomb repressive complexes (PRCs) are important chromatin regulators of embryonic stem (ES) cell function. RYBP binds Polycomb H2A monoubiquitin ligases Ring1A and Ring1B and has been suggested to assist PRC localization to their targets. Moreover, constitutive inactivation of RYBP precludes ES cell formation. Using ES cells conditionally deficient in RYBP, we found that RYBP is not required for maintenance of the ES cell state, although mutant cells differentiate abnormally. Genome-wide chromatin association studies showed RYBP binding to promoters of Polycomb targets, although its presence is dispensable for gene repression. We discovered, using Eed-knockout (KO) ES cells, that RYBP binding to promoters was independent of H3K27me3. However, recruiting of PRC1 subunits Ring1B and Mel18 to their targets was not altered in the absence of RYBP. In contrast, we have found that RYBP efficiently represses endogenous retroviruses (murine endogenous retrovirus [MuERV] class) and preimplantation (including zygotic genome activation stage)- and germ line-specific genes. These observations support a selective repressor activity for RYBP that is dispensable for Polycomb function in the ES cell state. Also, they suggest a role for RYBP in epigenetic resetting during preimplantation development through repression of germ line genes and PcG targets before formation of pluripotent epiblast cells.
URLPMID:23612295 [本文引用: 2]
Embryonic stem cells and primordial germ cells (PGCs) express many pluripotency-associated genes, but embryonic stem cells do not normally undergo conversion into primordial germ cells. Thus, we predicted that there is a mechanism that represses primordial germ cell-related gene expression in embryonic stem cells. Here we identify genes involved in this putative mechanism, by using an embryonic stem cell line with a Vasa reporter in an RNA interference screen of transcription factor genes expressed in embryonic stem cells. We identify five genes that result in the expression of Vasa when silenced. Of these, Max is the most striking. Transcriptome analysis reveals that Max knockdown in embryonic stem cells results in selective, global derepression of germ cell-specific genes. Max interacts with histone H3K9 methyltransferases and associates with the germ cell-specific genes in embryonic stem cells. In addition, Max knockdown results in a decrease in histone H3K9 dimethylation at their promoter regions. We propose that Max is part of protein complex that acts as a repressor of germ cell-related genes in embryonic stem cells.
URLPMID:27025988 [本文引用: 4]
Meiosis is a unique process that allows the generation of reproductive cells. It remains largely unknown how meiosis is initiated in germ cells and why non-germline cells do not undergo meiosis. We previously demonstrated that knockdown ofMaxexpression, a gene encoding a partner of MYC family proteins, strongly activates expression of germ cell-related genes in ESCs. Here we find that complete ablation ofMaxexpression in ESCs results in profound cytological changes reminiscent of cells undergoing meiotic cell division. Furthermore, our analyses uncovers thatMaxexpression is transiently attenuated in germ cells undergoing meiosisin vivoand its forced reduction induces meiosis-like cytological changes in cultured germline stem cells. Mechanistically,Maxdepletion alterations are, in part, due to impairment of the function of an atypical PRC1 complex (PRC1.6), in which MAX is one of the components. Our data highlight MAX as a new regulator of meiotic onset. The mechanisms that trigger meiosis in germ cells and halt this process in non-germline cells are unclear. Here, the authors show that knockout ofMaxin embryonic stem cells results in meiotic onset in a mechanism that involves the PRC1 complex.
URLPMID:3647456 [本文引用: 5]
The chromatin protein L3mblt2 plays an essential role in early differentiation in02vitro and in02vivo through an atypical PRC1 complex and repressive histone modifications.
URL [本文引用: 2]
URL [本文引用: 1]
[本文引用: 2]
URLPMID:25516968 [本文引用: 4]
Abstract The maintenance and control of pluripotency is of great interest in stem cell biology. The dual specificity T-box/basic-helix-loop-helix-zipper transcription factor Mga is expressed in the pluripotent cells of the inner cell mass (ICM) and epiblast of the peri-implantation mouse embryo, but its function has not been investigated previously. Here, we use a loss-of-function allele and RNA knockdown to demonstrate that Mga depletion leads to the death of proliferating pluripotent ICM cells in vivo and in vitro, and the death of embryonic stem cells (ESCs) in vitro. Additionally, quiescent pluripotent cells lacking Mga are lost during embryonic diapause. Expression of Odc1, the rate-limiting enzyme in the conversion of ornithine into putrescine in the synthesis of polyamines, is reduced in Mga mutant cells, and the survival of mutant ICM cells as well as ESCs is rescued in culture by the addition of exogenous putrescine. These results suggest a mechanism whereby Mga influences pluripotent cell survival through regulation of the polyamine pool in pluripotent cells of the embryo, whether they are in a proliferative or quiescent state. 2015. Published by The Company of Biologists Ltd.
URL [本文引用: 1]
URLPMID:16855402 [本文引用: 1]
Stem cells are characterised by a capacity to self renew and generate progenycapable of differentiating along several defined lineage paths. Embryonic Stem (ES) cellsare derived from the inner cell mass (ICM) of early-stage embryos and can contribute toall tissues of the developing embryo. Discovering how ES cell pluripotency and lineageinduction is achieved is important for understanding normal development and forsuccessfully applying stem cell-based therapies. A series of recent studies have shownthat the chromatin profile of ES cells is unusual and have revealed a critical role for thePolycomb Repressive Complexes (PRCs) in maintaining pluripotency. In human andmouse ES cells many genes that encode transcription factors that are required for lineagespecification bind PRC2 and carry bivalent (or opposing) histone signatures, beingenriched for conventional indicators of active chromatin such as acetylated H3K9 andmethylated H3K4, while lying within domains of repressive trimethylated H3K27.Mutant ES cells that lack H3K27 methylation inappropriately expressed these genesshowing that PRC2 represses lineage-specific gene programs in ES cells. Here wediscuss the implications of these new discoveries and explore the interdependence ofPRC1 and PRC2 in regulating lineage-specific gene expression in ES cells.
URLPMID:18848501 [本文引用: 1]
Genomic imprinting regulates parental-specific expression of particular genes and is required for normal mammalian development. How imprinting is established during development is, however, largely unknown. To address this question, we studied the mouse Kcnq1 imprinted cluster at which paternal-specific silencing depends on expression of the noncoding RNA Kcnq1ot1. We show that Kcnq1ot1 is expressed from the zygote stage onward and rapidly associates with chromatin marked by Polycomb group (PcG) proteins and repressive histone modifications, forming a discrete repressive nuclear compartment devoid of RNA polymerase II, a configuration also observed at the Igf2r imprinted cluster. In this compartment, the paternal Kcnq1 cluster exists in a three-dimensionally contracted state. In vivo the PcG proteins Ezh2 and Rnf2 are independently required for genomic contraction and imprinted silencing. We propose that the formation of a parental-specific higher-order chromatin organization renders imprint clusters competent for monoallelic silencing and assign a central role to PcG proteins in this process.
URLPMID:18339675 [本文引用: 1]
The Polycomb group (PcG) proteins mediate heritable silencing ofdevelopmental regulators in metazoans, participating in one of two distinctmultimeric protein complexes, the Polycomb repressive complexes 1 (PRC1) and 2(PRC2). Although PRC2 has been shown to share target genes with the coretranscription network, including Oct3/4, to maintain embryonic stem (ES)cells, it is still unclear whether PcG proteins and the core transcriptionnetwork are functionally linked. Here, we identify an essential role for thecore PRC1 components Ring1A/B in repressing developmental regulators in mouseES cells and, thereby, in maintaining ES cell identity. A significantproportion of the PRC1 target genes are also repressed by Oct3/4. Wedemonstrate that engagement of PRC1 at target genes is Oct3/4-dependent,whereas engagement of Oct3/4 is PRC1-independent. Moreover, upondifferentiation induced by Gata6 expression, most of the Ring1A/B target genesare derepressed and the binding of Ring1A/B to their target loci is alsodecreased. Collectively, these results indicate that Ring1A/B-mediatedPolycomb silencing functions downstream of the core transcriptional regulatorycircuitry to maintain ES cell identity.
URL [本文引用: 1]
URLPMID:12589020 [本文引用: 1]
The highly homologous Rnf2 (Ring1b) and Ring1 (Ring1a) proteins were identified as in vivo interactors of the Polycomb Group (PcG) protein Bmi1. Functional ablation of Rnf2 results in gastrulation arrest, in contrast to relatively mild phenotypes in most other PcG gene null mutants belonging to the same functional group, among which is Ring1. Developmental defects occur in both embryonic and extraembryonic tissues during gastrulation. The early lethal phenotype is reminiscent of that of the PcG-gene knockouts Eed and Ezh2, which belong to a separate functional PcG group and PcG protein complex. This finding indicates that these biochemically distinct PcG complexes are both required during early mouse development. In contrast to the strong skeletal transformation in Ring1 hemizygous mice, hemizygocity for Rnf2 does not affect vertebral identity. However, it does aggravate the cerebellar phenotype in a Bmi1 null-mutant background. Together, these results suggest that Rnf2 or Ring1-containing PcG complexes have minimal functional redundancy in specific tissues, despite overlap in expression patterns. We show that the early developmental arrest in Rnf2-null embryos is partially bypassed by genetic inactivation of the Cdkn2a (Ink4a/ARF) locus. Importantly, this finding implicates Polycomb-mediated repression of the Cdkn2a locus in early murine development.
URLPMID:12183370 [本文引用: 1]
The products of the Polycomb group of genes form complexes that maintain the state of transcriptional repression of several genes with relevance to development and in cell proliferation. We have identified Ring1B, the product of the Ring1B gene (Rnf2 Mouse Genome Informatics), by means of its interaction with the Polycomb group protein Mel18. We describe biochemical and genetic studies directed to understand the biological role of Ring1B. Immunoprecipitation studies indicate that Ring1B form part of protein complexes containing the products of other Polycomb group genes, such as Rae28/Mph1 and M33, and that this complexes associate to chromosomal DNA. We have generated a mouse line bearing a hypomorphic Ring1B allele, which shows posterior homeotic transformations of the axial skeleton and a mild derepression of some Hox genes (Hoxb4, Hoxb6 and Hoxb8) in cells anterior to their normal boundaries of expression in the mesodermal compartment. By contrast, the overexpression of Ring1B in chick embryos results in the repression of Hoxb9 expression in the neural tube. These results, together with the genetic interactions observed in compound Ring1B/Mel18 mutant mice, are consistent with a role for Ring1B in the regulation of Hox gene expression by Polycomb group complexes.
URLPMID:28924034 [本文引用: 3]
A biweekly scientific journal publishing high-quality research in molecular biology and genetics, cancer biology, biochemistry, and related fields
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:1190233 [本文引用: 1]
Abstract The Rybp/DEDAF protein has been implicated in both transcriptional regulation and apoptotic signaling, but its precise molecular function is unclear. To determine the physiological role of Rybp, we analyzed its expression during mouse development and generated mice carrying a targeted deletion of Rybp using homologous recombination in embryonic stem cells. Rybp was found to be broadly expressed during embryogenesis and was particularly abundant in extraembryonic tissues, including trophoblast giant cells. Consistent with this result, rybp homozygous null embryos exhibited lethality at the early postimplantation stage. At this time, Rybp was essential for survival of the embryo, for the establishment of functional extraembryonic structures, and for the execution of full decidualization. Through the use of a chimeric approach, the embryonic lethal phenotype was circumvented and a role for Rybp in central nervous system development was uncovered. Specifically, the presence of Rybp-deficient cells resulted in marked forebrain overgrowth and in localized regions of disrupted neural tube closure. Functions for Rybp in the brain also were supported by the finding of exencephaly in about 15% of rybp heterozygous mutant embryos, and by Rybp's distinct neural expression pattern. Together, these findings support critical roles for Rybp at multiple stages of mouse embryogenesis.
URL [本文引用: 1]
Histone deacetylases 1 and 2 (HDAC1/2) form the core catalytic components of corepressor complexes that modulate gene expression. In most cell types, deletion of both Hdac1 and Hdac2 is required to generate a discernible phenotype, suggesting their activity is largely redundant. We have therefore generated an ES cell line in which Hdac1 and Hdac2 can be inactivated simultaneously. Loss of HDAC1/2 resulted in a 60% reduction in total HDAC activity and a loss of cell viability. Cell death is dependent upon cell cycle progression, because differentiated, nonproliferating cells retain their viability. Furthermore, we observe increased mitotic defects, chromatin bridges, and micronuclei, suggesting HDAC1/2 are necessary for accurate chromosome segregation. Consistent with a critical role in the regulation of gene expression, microarray analysis of Hdac1/2-deleted cells reveals 1,708 differentially expressed genes. Significantly for the maintenance of stem cell self-renewal, we detected a reduction in the expression of the pluripotent transcription factors, Oct4, Nanog, Esrrb, and Rex1. HDAC1/2 activity is regulated through binding of an inositol tetraphosphate molecule (IP4) sandwiched between the HDAC and its cognate corepressor. This raises the important question of whether IP4 regulates the activity of the complex in cells. By rescuing the viability of double-knockout cells, we demonstrate for the first time (to our knowledge) that mutations that abolish IP4 binding reduce the activity of HDAC1/2 in vivo. Our data indicate that HDAC1/2 have essential and pleiotropic roles in cellular proliferation and regulate stem cell self-renewal by maintaining expression of key pluripotent transcription factors.
URLPMID:12032080 [本文引用: 1]
Histone deacetylases (HDACs) modulate chromatin structure and transcription, but little is known about their function in mammalian development. HDAC1 was implicated previously in the repression of genes required for cell proliferation and differentiation. Here we show that targeted disruption of both HDAC1 alleles results in embryonic lethality before E10.5 due to severe proliferation defects and retardation in development. HDAC1-deficient embryonic stem cells show reduced proliferation rates, which correlate with decreased cyclin-associated kinase activities and elevated levels of the cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p27KIP1. Similarly, expression of p21 and p27 is up-regulated in HDAC1-null embryos. In addition, loss of HDAC1 leads to significantly reduced overall deacetylase activity, hyperacetylation of a subset of histones H3 and H4 and concomitant changes in other histone modifications. The expression of HDAC2 and HDAC3 is induced in HDAC1-deficient cells, but cannot compensate for loss of the enzyme, suggesting a unique function for HDAC1. Our study provides the first evidence that a histone deacetylase is essential for unrestricted cell proliferation by repressing the expression of selective cell cycle inhibitors.
URLPMID:3586315 [本文引用: 1]
Wang et al. show that development and regeneration of Sox2+ endoderm progenitors in the lung require both histone deacetylases 1 and 2 but via regulation of target genes distinct between developing and postnatal tissue. Context-specific Hdac activity output may underlie the regulation of proper Sox2+ cell regeneration in multiple tissues.
URL [本文引用: 1]
The unique properties of embryonic stem cells (ESC) rely on long-lasting self-renewal and their ability to switch in all adult cell type programs. Recent advances have shown that regulations at the chromatin level sustain both ESC properties along with transcription factors. We have focused our interest on the epigenetic modulator HP1γ (Heterochromatin Protein 1, isoform γ) that binds histones H3 methylated at lysine 9 (meH3K9) and is highly plastic in its distribution and association with the transcriptional regulation of specific genes during cell fate transitions. These characteristics of HP1γ make it a good candidate to sustain the ESC flexibility required for rapid program changes during differentiation. Using RNA interference, we describe the functional role of HP1γ in mouse ESC. The analysis of HP1γ deprived cells in proliferative and in various differentiating conditions was performed combining functional assays with molecular approaches (RT-qPCR, microarray). We show that HP1γ deprivation slows down the cell cycle of ESC and decreases their resistance to differentiating conditions, rendering the cells poised to differentiate. In addition, HP1γ depletion hampers the differentiation to the endoderm as compared with the differentiation to the neurectoderm or the mesoderm. Altogether, our results reveal the role of HP1γ in ESC self-renewal and in the balance between the pluripotent and the differentiation programs.
URL [本文引用: 1]
URLPMID:21896631 [本文引用: 1]
During meiosis, specific histone modifications at pericentric heterochromatin (PCH), especially histone H3 tri- and dimethylation at lysine 9 (H3K9me3 and H3K9me2, respectively), are required for proper chromosome interactions. However, the molecular mechanism by which H3K9 methylation mediates the synapsis is not yet understood. We have generated a Cbx3-deficient mouse line and performed comparative analysis on Suv39h1/h2-, G9a- and Cbx3-deficient spermatocytes. This study revealed that H3K9me2 at PCH depended on Suv39h1/h2-mediated H3K9me3 and its recognition by the Cbx3 gene product HP1 . We further found that centromere clustering and synapsis were commonly affected in G9a- and Cbx3-deficient spermatocytes. These genetic observations suggest that HP1纬/G9a-dependent PCH-mediated centromere clustering is an axis for proper chromosome interactions during meiotic prophase. We propose that the role of the HP1 /G9a axis is to retain centromeric regions of unpaired homologous chromosomes in close alignment and facilitate progression of their pairing in early meiotic prophase. This study also reveals considerable plasticity in the interplay between different histone modifications and suggests that such stepwise and dynamic epigenetic modifications may play a pivotal role in meiosis.
URL [本文引用: 1]
URLPMID:12101104 [本文引用: 1]
Abstract Top of page Abstract Introduction Results Discussion Methods Acknowledgements References E2F transcription factors play an important role in regulating mammalian cell proliferation. E2F6, the most recently identified E2F family member, is a transcriptional repressor. In an effort to ascertain the in vivo biological function of E2F6, we have generated an E2f6 mutant mouse strain. Mice lacking E2F6 are viable and healthy. Surprisingly, E2f6 61/61 embryonic fibroblasts proliferate normally. However, E2f6 61/61 animals display overt homeotic transformations of the axial skeleton that are strikingly similar to the skeletal transformations observed in polycomb mutant mice. This observation is compatible with the recent finding that endogenous E2F6 and one or more mammalian polycomb proteins are components of the same multiprotein complex. The accumulated evidence suggests that, during development, E2F6 participates in the recruitment of polycomb proteins to specific target promoters.
URLPMID:18366140 [本文引用: 1]
Bmi1 is a Polycomb Group protein that functions as a component of Polycomb Repressive Complex 1 (PRC1) to control axial skeleton development through Hox gene repression. Bmi1 also represses transcription of the Ink4a-Arf locus and is consequently required to maintain the proliferative and self-renewal properties of hematopoietic and neural stem cells. Previously, one E2F family member, E2F6, has been shown to interact with Bmi1 and other known PRC1 components. However, the biological relevance of this interaction is unknown. In this study, we use mouse models to investigate the interplay between E2F6 and Bmi1. This analysis shows that E2f6 and Bmi1 cooperate in the regulation of Hox genes, and consequently axial skeleton development, but not in the repression of the Ink4a-Arf locus. These findings underscore the significance of the E2F6-Bmi1 interaction in vivo and suggest that the Hox and Ink4a-Arf loci are regulated by somewhat different mechanisms. Developmental Dynamics 237:1232-1242, 2008. 2008 Wiley-Liss, Inc.
URLPMID:479717 [本文引用: 2]
E2F/DP complexes activate or repress the transcription of E2F target genes, depending on the association of a pRB family member, thereby regulating cell cycle progression. Whereas the E2F family consists of seven members, the DP family contains only two (Dp1 and Dp2), Dp1 being the more highly expressed member. In contrast to the inactivation of individual E2F family members, we have recently demonstrated that loss of Dp1 results in embryonic lethality by embryonic day 12.5 (E12.5) due to the failure of extraembryonic lineages to develop and replicate DNA properly. To bypass this placental requirement and search for roles of Dp1 in the embryo proper, we generated Dp1-deficient embryonic stem (ES) cells that carry the ROSA26-LacZ marker and injected them into wild-type blastocysts to construct Dp1-deficient chimeras. Surprisingly, we recovered mid- to late gestational embryos (E12.5 to E17.5), in which the Dp1-deficient ES cells contributed strongly to most chimeric tissues as judged by X-Gal (5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside) staining and Western blotting. Importantly, the abundance of DP2 protein does not increase and the expression of an array of cell cycle genes is virtually unchanged in Dp1-deficient ES cells or chimeric E15.5 tissues with the absence of Dp1. Thus, Dp1 is largely dispensable for embryonic development, despite the absolute extraembryonic requirement for Dp1, which is highly reminiscent of the restricted roles for Rb and cyclins E1/E2 in vivo.
URL [本文引用: 1]
URLPMID:19339689 [本文引用: 1]
We performed a genome-wide siRNA screen in mouse embryonic stem (ES) cells to identify genes essential for self-renewal, and found 148 genes whose down-regulation caused differentiation. Many of the identified genes function in gene regulation and/or development, and are highly expressed in ES cells and embryonic tissues. We further identified target genes of two transcription regulators Cnot3 and Trim28. We discovered that Cnot3 and Trim28 co-occupy many putative gene promoters with c-Myc and Zfx, but not other pluripotency-associated transcription factors. They form a unique module in the self-renewal transcription network, separate from the core module formed by Nanog, Oct4, and Sox2. The transcriptional targets of this module are enriched for genes involved in cell cycle, cell death, and cancer. This supports the idea that regulatory networks controlling self-renewal in stem cells may also be active in certain cancers and may represent novel anti-cancer targets. Our screen has implicated over 100 new genes in ES cell self-renewal, and illustrates the power of RNAi and forward genetics for the systematic study of self-renewal.
[本文引用: 2]
URLPMID:27687053 [本文引用: 1]
DNA methylation is one of the epigenetic marks and plays critically important functions during spermatogenesis in mammals. DNA methylation is catalysed by DNA methyltransferase (DNMT) enzymes, which are responsible for the addition of a methyl group to the fifth carbon atom of the cytosine residues within cytosine-phosphate-guanine (CpG) and non-CpG dinucleotide sites. Structurally and functionally five different DNMT enzymes have been identified in mammals, including DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. These enzymes mainly play roles in two DNA methylation processes: maintenance andde novo. While DNMT1 is primarily responsible for maintenance methylation via transferring methyl groups to the hemi-methylated DNA strands following DNA replication, both DNMT3A and DNMT3B are capable of methylating unmodified cytosine residues, known asde novomethylation. However, DNMT3L indirectly participates inde novomethylation, and DNMT2 carries out methylation of the cytosine 38 in the anticodon loop of aspartic acid transfer RNA. To date, many studies have been performed to determine spatial and temporal expression levels and functional features of the DNMT in the male germ cells. This review article comprehensively discusses dynamic expression of the DNMT during spermatogenesis and their relationship with male infertility development in the light of existing investigations.
URLPMID:5581151 [本文引用: 1]
Mapping epigenetic modifications and identifying their roles in the regulation of spermatogenesis and embryogenesis are essential for gaining fundamental medical understandings and for clinical applications. More and more evidence has shown that specific epigenetic modifications are established during spermatogenesis, which will be transferred into oocyteviafertilisation, and play an important role in the early embryo development. Defects in epigenetic patterns may increase the risk of abnormal spermatogenesis, fertilisation failure, early embryogenesis abnormality and several other complications during pregnancy. This review mainly discusses the relationship between altered epigenetic profiles and reproductive diseases, highlighting how epigenetic defects affect the quality of sperm and embryo.
URLPMID:3115959 [本文引用: 1]
Multiglycosides of Tripterygium wilfordii Hook f (GTW), a Chinese herb-derived medicine used as a remedy for rheumatoid arthritis, are considered to be a reversible anti-fertility drug affecting the mammalian spermatids. However, the mechanism behind this effect is still unknown. To study the possible mechanism behind the impact of GTW on spermatogenesis, we administered 4 groups of 4-week-old male mice with different doses of GTW. We found a dose-dependent decrease in the number of germ cells after 40 days of GTW treatment, and an increase in apoptotic cells from the low-dose to the high-dose group. During this same period the dimethylated level of histone H3 lysine 9 (H3K9me2) in GTW-treated testes germ cells declined. Additionally, spermatogonial stem cells (SSCs) from 6-day-old mice were isolated to evaluate the possible effect of GTW or triptolide on development of SSCs. We found a significantly higher incidence of apoptosis and lower dimethylation level of H3K9me2 in the SSCs of GTW or triptolide treatment than in controls. Thus, these data suggest that the GTW-induced apoptosis might be responsible for the fertility impairment in mice. This damage could be traced back to the early stages of spermatogenesis. GTW also affected the epigenetic modification of H3K9 in spermatogenesis. Molecular dynamics simulation suggested that triptolide and dimethylated or trimethylated H3K9 might have similar interaction mechanisms with EED (embryonic ectoderm development). These candidate activation mechanisms provide the first glimpse into the pathway of GTW-induced gonad toxicity, which is crucial for further research and clinical application.
URLPMID:4052122 [本文引用: 1]
Protein ubiquitin-proteasome (ubiquitin-proteasome) system is the major mechanism responsible for protein degradation in eukaryotic cell. During spermatogenesis, the replacement of histone by protamine is vital for normal sperm formation, which is involved in ubiquitination enzymes expressed in testis. Recently, histone ubiquitin ligases have been shown to play critical roles in several aspects of spermatogenesis, such as meiotic sex chromosome inactivation (MSCI), DNA damage response, and spermiogenesis. In this review, we highlight recent progress in the discovery of several histone ubiquitin ligases and elaborate mechanisms of how these enzymes are involved in these processes through knockout mouse model. Using Huwe1, UBR2, and RNF8 as examples, we emphasized the diverse functions for each enzyme and the broad involvement of these enzymes in every stage, from spermatogonia differentiation and meiotic division to spermiogenesis; thus histone ubiquitin ligases represent a class of enzymes, which play important roles in spermatogenesis through targeting histone for ubiquitination and therefore are involved in transcription regulation, epigenetic modification, and other processes essential for normal gametes formation.
URLPMID:28803779 [本文引用: 1]
Abstract Lysine crotonylation (Kcr) is a newly identified histone modification that is associated with active transcription in mammalian cells. Here we report that the chromodomain Y-like transcription corepressor CDYL negatively regulates histone Kcr by acting as a crotonyl-CoA hydratase to convert crotonyl-CoA to -hydroxybutyryl-CoA. We showed that the negative regulation of histone Kcr by CDYL is intrinsically linked to its transcription repression activity and functionally implemented in the reactivation of sex chromosome-linked genes in round spermatids and genome-wide histone replacement in elongating spermatids. Significantly, Cdyl transgenic mice manifest dysregulation of histone Kcr and reduction of male fertility with a decreased epididymal sperm count and sperm cell motility. Our study uncovers a biochemical pathway in the regulation of histone Kcr and implicates CDYL-regulated histone Kcr in spermatogenesis, adding to the understanding of the physiology of male reproduction and the mechanism of the spermatogenic failure in AZFc (Azoospermia Factor c)-deleted infertile men. Copyright 2017 Elsevier Inc. All rights reserved.
URLPMID:24614311 [本文引用: 1]
Abstract Despite a conserved role for histones as general DNA packaging agents, it is now clear that another key function of these proteins is to confer variations in chromatin structure to ensure dynamic patterns of transcriptional regulation in eukaryotes. The incorporation of histone variants is particularly important to this process. Recent knockdown and knockout studies in various cellular systems, as well as direct mutational evidence from human cancers, now suggest a crucial role for histone variant regulation in processes as diverse as differentiation and proliferation, meiosis and nuclear reprogramming. In this Review, we provide an overview of histone variants in the context of their unique functions during mammalian germ cell and embryonic development, and examine the consequences of aberrant histone variant regulation in human disease.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
Spermatogonial stem cells (SSCs), a unique population of male germ cells with self-renewal ability, are the foundation for maintenance of spermatogenesis throughout the life of the male. Although many regulatory molecules essential for SSC self-renewal have been identified, the fundamental mechanism underlying how SSCs acquire and maintain their self-renewal activity remains largely to be elucidated. In recent years, many types of noncoding RNAs (ncRNAs) have been suggested to regulate the SSC self-renewal through multiple ways, indicating ncRNAs paly crucial roles in SSC self-renewal. In this paper, we mainly focus on four types of ncRNAs including microRNA (miRNA), long noncoding RNA (lncRNA), piwi-interacting RNA (piRNA), as well as circular RNAs (circRNAs), and reviewed their potential roles in SSC self-renewal that discovered recently to help us gain a better understanding of molecular mechanisms by which ncRNAs perform their function in regulating SSC self-renewal. This article is protected by copyright. All rights reserved
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:29125941 [本文引用: 1]
Abstract Cellular RNAs can be modified post-transcriptionally with dynamic and reversible chemical modifications. These modifications can alter the structure and metabolism of mRNA, but only recent methodological and conceptual advances allowed systematic mapping and functional analysis to unfold the role they play in mRNA biology. Mapping the most common internal mRNA modification, N 6 -methyladenosine (m 6 A), paved the way for the deciphering of other types of mRNA modifications, such as N 1 -methyladenosine (m 1 A). RNA methylation provides dynamic regulation to the processing, export, translation and stability of mRNA molecules, thereby influencing fundamental biological and pathological processes such as differentiation, cellular response to stress and tumorigenesis. This review summarizes the key methods and the recent discoveries in the field of epitranscriptomics through the prism of post-transcriptional mRNA methylation in eukaryotes. Copyright 2017. Published by Elsevier Ltd.
URLPMID:17190809
In the mammalian lifecycle, the two periods of genome-wide epigenetic reprogramming are in the early embryo, when somatic patterns are set, and during germ cell development. Although some differences between the reprogrammed states of somatic and germ cells have been reported, overall patterns of genomic methylation are considered to be similar. Using restriction landmark genomic scanning to examine 鈮2,600 loci distributed randomly throughout the genome, we find that the methylation status of testicular DNA is highly distinct, displaying eightfold the number of hypomethylated loci relative to somatic tissues. Identification and analysis of >300 loci show that these regions are generally located within nonrepetitive sequences that are away from CpG islands and 5鈥 regions of genes. We show that a contributing factor for these differences is that the methylation state of non-CpG-island DNA is correlated with the regional level of GC content within chromosomes and that this relationship is inverted between the testis and somatic tissues. We also show that in Dnmt3L-deficient mice, which exhibit infertility associated with abnormal chromosomal structures in germ cells, this unique testicular DNA methylation pattern is not established. These special properties of testicular DNA point to a broad, distinct epigenetic state that may be involved in maintaining a unique chromosomal structure in male germ cells.
URLPMID:16211598
The DNA methyltransferase-like protein Dnmt3L is necessary for the establishment of genomic imprints in oogenesis and for normal spermatogenesis (Bourc'his et al., [ 2001 ]; Hata et al., [ 2002 ]). Also, a paternally imprinted gene, H19 , loses DNA methylation in Dnmt3L -/- spermatogonia (Bourc'his and Bestor, [ 2004 ]; Kaneda et al., [ 2004 ]). To determine the reason for the impaired spermatogenesis in the Dnmt3L -/- testes, we have carried out a series of histological and molecular studies. We show here that Dnmt3L -/- germ cells were arrested and died around the early meiotic stage. A microarray-based gene expression-profiling analysis revealed that various gonad-specific and/or sex-chromosome-linked genes were downregulated in the Dnmt3L -/- testes. In contrast, expression of retrovirus-like intracisternal A-particle (IAP) sequences was upregulated; consistent with this observation, a specific IAP copy showed complete loss of DNA methylation. These findings indicate that Dnmt3L regulates germ cell-specific gene expression and IAP suppression, which are critical for male germ cell proliferation and meiosis. Mol. Reprod. Dev. 2005 Wiley-Liss, Inc.
URLPMID:3507207
During the last phase of spermatogenesis, spermiogenesis, haploid round spermatids metamorphose towards spermatozoa. Extensive cytoplasmic reduction and chromatin remodelling together allow a dramatic decrease of cellular, notably nuclear volume. DNA packing by a nucleosome based chromatin structure is largely replaced by a protamine based one. At the cytoplasmic level among others the acrosome and perinuclear theca (PNT) are formed. In this study we describe the onset of chromatin remodelling to occur concomitantly with acrosome and PNT development. In spread human round spermatid nuclei, we show development of a DAPI-intense doughnut-like structure co-localizing with the acrosomal sac and sub acrosomal PNT. At this structure we observe the first gradual decrease of nucleosomes and several histones. Histone post-translational modifications linked to chromatin remodelling such as H4K8ac and H4K16ac also delineate the doughnut, that is furthermore marked by H3K9me2. During the capping phase of acrosome development, the size of the doughnut-like chromatin domain increases, and this area often is marked by uniform nucleosome loss and the first appearance of transition protein 2 and protamine 1. In the acrosome phase at nuclear elongation, chromatin remodelling follows the downward movement of the marginal ring of the acrosome. Our results indicate that acrosome development and chromatin remodelling are interacting processes. In the discussion we relate chromatin remodelling to the available data on the nuclear envelope and the linker of nucleoskeleton and cytoskeleton (LINC) complex of spermatids, suggesting a signalling route for triggering chromatin remodelling.
URLPMID:11152286
Here we report a detailed analysis of waves of histone acetylation that occurs throughout spermatogenesis in mouse. Our data showed that spermatogonia and preleptotene spermatocytes contained acetylated core histones H2A, H2B and H4, whereas no acetylated histones were observed throughout meiosis in leptotene or pachytene spermatocytes. Histones remained unacetylated in most round spermatids. Acetylated forms of H2A and H2B, H3 and H4 reappeared in step 9 to 11 elongating spermatids, and disappeared later in condensing spermatids. The spatial distribution pattern of acetylated H4 within the spermatids nuclei, analyzed in 3D by immunofluorescence combined with confocal microscopy, showed a spatial sequence of events tightly associated with chromatin condensation. In order to gain an insight into mechanisms controlling histone hyperacetylation during spermiogenesis, we treated spermatogenic cells with a histone deacetylase inhibitor, trichostatin A (TSA), which showed a spectacular increase of histone acetylation in round spermatids. This observation suggests that deacetylases are responsible for maintaining a deacetylated state of histones in these cells. TSA treatment could not induce histone acetylation in condensing spermatids, suggesting that acetylated core histones are replaced by transition proteins without being previously deacetylated. Moreover, our data showed a dramatic decrease in histone deacetylases in condensing spermatids. Therefore, the regulation of histone deacetylase activity/concentration appears to play a major role in controling histone hyperacetylation and probably histone replacement during spermiogenesis.
URLPMID:16222244
Histones are subject to numerous post-translational modifications(1). Some of these 'epigenetic' marks recruit proteins that modulate chromatin structure. For example, heterochromatin protein 1 (HP1) binds to histone H3 when its lysine 9 residue has been tri-methylated by the methyltransferase Suv39h ( refs 2 - 6). During mitosis, H3 is also phosphorylated by the kinase Aurora B-7. Although H3 phosphorylation is a hallmark of mitosis, its function remains mysterious. It has been proposed that histone phosphorylation controls the binding of proteins to chromatin(8), but any such mechanisms are unknown. Here we show that antibodies against mitotic chromosomal antigens that are associated with human autoimmune diseases(9) specifically recognize H3 molecules that are modified by both tri-methylation of lysine 9 and phosphorylation of serine 10 ( H3K9me3S10ph). The generation of H3K9me3S10ph depends on Suv39h and Aurora B, and occurs at pericentric heterochromatin during mitosis in different eukaryotes. Most HP1 typically dissociates from chromosomes during mitosis(10-12), but if phosphorylation of H3 serine 10 is inhibited, HP1 remains chromosome-bound throughout mitosis. H3 phosphorylation by Aurora B is therefore part of a 'methyl/ phos switch' mechanism(8) that displaces HP1 and perhaps other proteins from mitotic heterochromatin.
URLPMID:20153262
During spermatogenesis, global nucleosome removal occurs where histones are initially replaced by transition proteins and subsequently by protamines. This chromatin reorganization is thought to facilitate the compaction of the paternal genome into the sperm head and to protect the DNA from damaging agents. Histone ubiquitination has been suggested to be important for sex chromosome inactivation during meiotic prophase and nucleosome removal at postmeiotic stages. However, the mechanisms regulating these ubiquitin-mediated processes are unknown. In this study, we investigate the role of the ubiquitin ligase RNF8 during spermatogenesis and find that RNF8-deficient mice are proficient in meiotic sex chromosome inactivation (MSCI) but deficient in global nucleosome removal. Moreover, we show that RNF8-dependent histone ubiquitination induces H4K16 acetylation, which may be an initial step in nucleosome removal. Thus, our results show that RNF8 plays an important role during spermatogenesis through histone ubiquitination, resulting in Highlights? Histone ubiquitination is not required for meiotic sex chromosome inactivation ? RNF8 regulates histone removal and protamine deposition during spermiogenesis ? RNF8-dependent histone ubiquitination regulate H4K16 acetylation
URL
Sex chromosomes are uniquely subject to chromosome-wide silencing during male meiosis, and silencing persists into post-meiotic spermatids. Against this background, a select set of sex chromosome-linked genes escapes silencing and is activated in post-meiotic spermatids. Here, we identify a novel mechanism that regulates escape gene activation in an environment of chromosome-wide silencing in murine germ cells. We show that RNF8-dependent ubiquitination of histone H2A during meiosis establishes active epigenetic modifications, including dimethylation of H3K4 on the sex chromosomes. RNF8-dependent active epigenetic memory, defined by dimethylation of H3K4, persists throughout meiotic division. Various active epigenetic modifications are subsequently established on the sex chromosomes in post-meiotic spermatids. These RNF8-dependent modifications include trimethylation of H3K4, histone lysine crotonylation (Kcr), and incorporation of the histone variant H2AFZ. RNF8-dependent epigenetic programming regulates escape gene activation from inactive sex chromosomes in post-meiotic spermatids. Kcr accumulates at transcriptional start sites of sex-linked genes activated in an RNF8-dependent manner, and a chromatin conformational change is associated with RNF8-dependent epigenetic programming. Furthermore, we demonstrate that this RNF8-dependent pathway is distinct from that which recognizes DNA double-strand breaks. Our results establish a novel connection between a DNA damage response factor (RNF8) and epigenetic programming, specifically in establishing active epigenetic modifications and gene activation.
URL
URLPMID:16751777
Abstract Small RNAs bound to Argonaute proteins recognize partially or fully complementary nucleic acid targets in diverse gene-silencing processes. A subgroup of the Argonaute proteins--known as the 'Piwi family'--is required for germ- and stem-cell development in invertebrates, and two Piwi members--MILI and MIWI--are essential for spermatogenesis in mouse. Here we describe a new class of small RNAs that bind to MILI in mouse male germ cells, where they accumulate at the onset of meiosis. The sequences of the over 1,000 identified unique molecules share a strong preference for a 5' uridine, but otherwise cannot be readily classified into sequence families. Genomic mapping of these small RNAs reveals a limited number of clusters, suggesting that these RNAs are processed from long primary transcripts. The small RNAs are 26-31 nucleotides (nt) in length--clearly distinct from the 21-23 nt of microRNAs (miRNAs) or short interfering RNAs (siRNAs)--and we refer to them as 'Piwi-interacting RNAs' or piRNAs. Orthologous human chromosomal regions also give rise to small RNAs with the characteristics of piRNAs, but the cloned sequences are distinct. The identification of this new class of small RNAs provides an important starting point to determine the molecular function of Piwi proteins in mammalian spermatogenesis.
URLPMID:26279574
Two components in the piRNA pathway, MILI and MIWI2, were proposed to work in a linear hierarchy to generate piRNAs and methylate transposon sequences. Manakov et al. now profile transcription and methylation patterns in Mili and Miwi2 mutants and demonstrate that these proteins are in part functionally independent.
URLPMID:24184258
We elucidated the mechanism of spermatogonial stem cell disturbance of cryptorchidism and investigated the expression of miRNAs and their target genes in undescended testes. Using microarray analysis we compared total miRNA expression in unilateral undescended testes with that in contralateral descended and normal testes in a rat model of cryptorchidism. The model was derived by administering flutamide to pregnant Sprague Dawley rats. We identified mRNA targets of miRNAs by bioinformatic analysis, followed by in situ hybridization and immunohistochemistry to localize candidate miRNAs and mRNAs, respectively. We also investigated whether miRNAs could inhibit target protein expression in vitro. Microarray analysis and subsequent quantitative reverse transcriptase-polymerase chain reaction showed that only miR-135a was expressed at a lower level in undescended testes. We identified its target as FoxO1, which is essential for stem cell maintenance. miR-135a and FoxO1 localized to spermatogonial stem cells. FoxO1 localized to the spermatogonial stem cell nucleus less frequently in undescended testes, indicating that the activity of FoxO1, which acts as a transcription factor, is altered in undescended testes. Finally, miR-135a transfection into spermatogonia in vitro resulted in down-regulation of FoxO1 expression. In cryptorchid testes there is a decreased number of spermatogonial stem cells in which FoxO1 is activated, indicating that failure of spermatogonial stem cell maintenance results in spermatogenesis alteration. We also noted interaction between miR-135a and FoxO1, and propose that miR-135a contributes to spermatogonial stem cell maintenance through modulation of FoxO1 activity.
URL
URLPMID:4823932
Spermatogonial stem cells (SSCs) are unique male germline stem cells that support spermatogenesis and male fertility. Long non-coding RNAs (lncRNA) have been identified as key regulators of stem cell fate; however, their role in SSCs has not been explored. Here, we report that a novel spermatogonia-specific lncRNA (lncRNA033862) is essential for the survival of murine SSCs. LncRNA033862 is expressed in early spermatogonia including SSC and was among 805 lncRNAs identified by global expression profiling as responsive to glial cell-derived neurotrophic factor (GDNF), a growth factor required for SSC self-renewal and survival. LncRNA033862 is an antisense transcript of the GDNF receptor alpha1 (Gfra1) that lacks protein coding potential and regulatesGfra1expression levels by interacting withGfra1chromatin. Importantly, lncRNA033862 knockdown severely impairs SSC survival and their capacity to repopulate recipient testes in a transplantation assay. Collectively, our data provide the first evidence that long non-coding RNAs (lncRNAs) regulate SSC fate.
URLPMID:28914256
Cell death and differentiation is a monthly research journal focused on the exciting field of programmed cell death and apoptosis. It provides a single accessible source of information for both scientists and clinicians, keeping them up-to-date with advances in the field. It encompasses programmed cell death, cell death induced by toxic agents, differentiation and the interrelation of these with cell proliferation.
URL [本文引用: 1]
URL [本文引用: 1]
Methylation of cytosine residues within the CpG dinucleotide in mammalian cells is an important mediator of gene expression, genome stability, X-chromosome inactivation, genomic imprinting, chromatin structure, and embryonic development. The majority of CpG sites in mammalian cells is methylated in a nonrandom fashion, raising the question of how DNA methylation is distributed along the genome. Here, we focused on the functions of DNA methyltransferase-3b (Dnmt3b), of which deregulated activity is linked to several human pathologies. We generated Dnmt3b hypomorphic mutant mice with reduced catalytic activity, which first revealed a deregulation of Hox genes expression, consistent with the observed homeotic transformations of the posterior axis. In addition, analysis of deregulated expression programs in Dnmt3b mutant embryos, using DNA microarrays, highlighted illegitimate activation of several germ-line genes in somatic tissues that appeared to be linked directly to their hypomethylation in mutant embryos. We provide evidence that these genes are direct targets of Dnmt3b. Moreover, the recruitment of Dnmt3b to their proximal promoter is dependant on the binding of the E2F6 transcriptional repressor, which emerges as a common hallmark in the promoters of genes found to be up-regulated as a consequence of impaired Dnmt3b activity. Therefore, our results unraveled a coordinated regulation of genes involved in meiosis, through E2F6-dependant methylation and transcriptional silencing in somatic tissues.
URL [本文引用: 2]
URLPMID:28111200 [本文引用: 1]
DDX5 acts as a barrier to somatic cell reprogramming DDX5 loss of function upregulates RYBP through microRNA-125b Upregulated RYBP enhances H2AK119ub1 deposition at lineage-specific genes via PRC1 DDX5 silencing activates the OCT4-KDM2B network through RYBP independently of PRC1
URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}