 ,中国药科大学多肽药物创制工程中心,南京 211198
,中国药科大学多肽药物创制工程中心,南京 211198Common cancer genetic analysis methods and application study based on TCGA database
Xin Li, Mengwei Li, Yinan Zhang, Hanmei Xu,Engineering Research Center of Peptide Drug Discovery and Development, China Pharmaceutical University, Nanjing 211198, China通讯作者:
编委: 方向东
收稿日期:2018-11-20修回日期:2019-01-27网络出版日期:2019-02-25
| 基金资助: |
Received:2018-11-20Revised:2019-01-27Online:2019-02-25
| Fund supported: |
作者简介 About authors
李鑫,硕士研究生,专业方向:海洋药学E-mail:cpu_lixin@163.com。

摘要
关键词:
Abstract
Keywords:
PDF (411KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李鑫, 李梦玮, 张依楠, 徐寒梅. 常用肿瘤基因分析方法及基于TCGA数据库的分析应用[J]. 遗传, 2019, 41(3): 234-242 doi:10.16288/j.yczz.18-279
Xin Li, Mengwei Li, Yinan Zhang, Hanmei Xu.
近年来,随着高性能计算机集群技术支持的新一代测序机和自动化分析的高通量测序平台不断问世、基因组测序分析成本大幅降低、基因组数据共享平台层出不穷,以及大量的基因组数据被上传至互联网,为研究人员开展大规模的基因组学研究创造了便利条件,同时肿瘤基因组学的研究也越来越深入。由此,整合多种癌症基因组数据的The Cancer Genome Atlas (TCGA)数据库应运而生,为研究人 员快速、准确地获取肿瘤基因组数据提供了很好的途径。
数据挖掘是一门随着计算机科学发展而快速发展的学科,其在生命科学领域的作用随着大量测序数据的累计而逐渐显现。现阶段,国内大部分实验室对基因组数据挖掘和处理还处于起步阶段,不仅缺乏相应的数据处理平台,更缺乏具有相应知识背景的科研人员,而在国际上基因组数据研究已经是一个迅猛发展的领域。本文重点介绍了常见基因数据分析方法、TCGA数据库以及近年来围绕TCGA数据库所得到的研究成果,期望为相关科研人员提供一些利用数据库资源研究肿瘤基因组学的新思路。
1 常见基因数据分析方法
1.1 生存分析
生存分析是一类用于计算在一个集合内对于给定的时间段中影响因素与给定结果或时间事件之间关联的统计学方法,该方法的特点是可以对时间事件进行分析,其中Kaplan-Meier生存分析和Cox回归分析是两种最常用的时间事件标准化统计学方法。Kaplan-Meier生存分析可以基于一个影响因素对事件进行分析,每个独立个体的时间范围由记录点开始一直延续至事件发生点。Cox回归分析是一种多参数回归模型,该模型以生存结局和生存时间为因变量,可同时分析多种因素对生存期的影响[1]。在随机对照临床试验中,Kaplan-Meier生存分析是首选的数据分析方法[2]。对于多影响因素事件,可选用Cox回归分析。基于这两种分析方法的特点,在基因数据分析中,Kaplan-Meier多用于分析基因表达与生存周期的关系,而Cox回归多用于分析预后影响因素与生存周期的关系[3]。1.2 差异表达分析和聚类分析
差异表达是指同一基因在两个条件中的检测结果在排除系统误差、人为误差等因素后具有较为明显的差异,通常用P值来表示。这种差异可以通过外显子测序、芯片筛选等方法检测。比较同一基因在不同条件下的表达量差异是筛选潜在功能基因的第一步,通常由统计学工具辅助完成。常用的算法包括倍数法、t检验法、方差分析、SAM法、贝叶斯法和信息熵法等[4],这些统计学方法各有其优势和不足(表1)。Table 1
表1
表1 基因差异表达分析方法优缺点
Table 1
| 分析方法 | 优点 | 缺点 |
|---|---|---|
| 倍数法 | 计算量小,一般用于大规模初筛 | 具体阈值较难确定 |
| t检验法 | 能充分利用样本信息,检验效率高 | 在数据量较小时,对总体方差的估计不准确 |
| 方差分析 | 不受比较组数的限制,且可以同时分析多个因素的作用 | 多重假设检验可能带来放大的假阳性率 |
| SAM法 | 假阳性率低 | 诊断能力较差,ROC指数相对偏低 |
| 贝叶斯法 | 样本量小时也可得到较好的分析结果 | 对卡方分布和指数分布的数据不敏感 |
| 信息熵法 | 无需样本的类别信息即可进行筛选 | 不能得到差异表达的基因 |
新窗口打开|下载CSV
聚类分析在基因表达数据研究中被大量应用且在不断优化,它可以在模式分类数不确定的情况下对基因数据进行分组,其数学意义是将研究对象分为相对同质的群组。从生物学的角度,这种方法就是将具有潜在相同作用的基因分为同一组,如对于一组肿瘤组织高表达基因可以假定其存在促肿瘤生长活性,对于一组低表达基因则可假定其存在抗肿瘤活性,或认为同一组基因可能受同一转录因子的调控等。
两个影响聚类分析结果的重要指标是评价研究对象相似性程度的距离尺度和将研究对象分组的聚类算法,其中距离尺度可以根据不同的筛选目的分为几何距离、线性相关系数和非线性相关系数3种,分别对应的是衡量样本间的相似性、衡量样本间是否具有相同变化趋势和衡量样本间在同一时间节点的波动趋势是否相似。而常用的聚类算法主要包括简单聚类、层次聚类、模糊聚类、k均值聚类、双向聚类和自组织映射神经网络聚类等。对于聚类结果,一般选择对其进行可视化处理,使其更易于接受和直观的分析,常用的有热图(heatmap)、点线图和冰柱图等[5]。
1.3 受试者工作特征曲线分析
受试者工作特征曲线分析(receiver operating characteristic, ROC)最早起源于第二次世界大战时期,最初用来降低雷达兵们的误报率和漏报率,现多用于临床疾病诊断临界点寻找、不同检测方法对同一疾病的识别能力的比较、单一生物标志物对疾病的诊断准确度和筛选对疾病发生发展有显著影响的潜在基因。ROC曲线是一条通过二分类方式拟合的非线性曲线,其纵坐标为敏感度,横坐标为(1-特异性),评价指标为曲线下面积(area under the curve, AUC)。与生存分析最大的不同点在于ROC曲线分析不考虑时间因素,且不需要将试验结果分为两类,因此一般不用于分析预后等时间相关事件。ROC曲线分析的优点是直观、简单,可用肉眼看出结果。而缺点是对临界点的寻找没有明确的限定,可能一定程度上影响数据分析结果。在许多生物信息学分类分析时,ROC分析经常出现正相关显著低于负相关的现象,因此研究人员对其进行了改进,加入了精确率与反馈率曲线 (precision-recall, PR),这一优化使正负分类结果相对平衡,已经在R语言中实现了应用。对于不同条件间ROC比较,则需要分别对其AUC进行处理,消除抽样误差带来的影响,常用的处理方法有Delong法和Hanley法[6,7]。1.4 Meta分析
Meta分析是一种对同类研究结果进行整合定量分析的统计学方法,其目的是通过整合多个已有的研究数据来增大样本含量,从而减少由随机误差所导致的数据差异,进而增大检验学效能。在临床研究中常用于病因学、诊断性试验、发病机制、病人费用和效益、流行病学、干预措施评价、随访和预后测评等方面的分析。一般的分析流程为提出研究问题、文献与资料收集、数据构建、Meta分析和实验验证。其中文献与资料收集是影响Meta分析结果的关键步骤,涉及到文献搜索策略和数据纳入排除标准的建立[8]。一般来说,同一领域不同研究组之间的操作和研究方法会存在一定区别,进而带来一些人为误差。这种差异被称为异质性,一般分为方法异质性、临床异质性和统计学异质性。异质性检验是验证所构建标准是否良好的常用方法。对于基因表达常用的芯片Meta分析,一般选用同一测序平台来源的数据以避免测序方法对分析结果的干扰。Meta分析根据实际要求不同可以分为多种类型包括单组率Meta分析、网状Meta分析和诊断性Meta分析等,其具体分类依据在许多文章中都有报道过,因此不再叙述[9]。
2 TCGA数据库
2.1 数据库简介
肿瘤被认为是人类最复杂疾病之一,目前为止人类已经发现了超过200种肿瘤亚型。肿瘤病人基因中发生的变化如体细胞突变、拷贝数变异、基因表达量差异和表观修饰变化与其特定的肿瘤亚型是相对应的。因此,为了更好地发现、诊断和治疗肿瘤,对其基因变化进行深入研究和建立相应数据库是目前所急需的[10]。2006年,美国国立癌症研 究院(National Cancer Institute, NCI)和美国国立人 类基因组研究院(National Human Genome Research Institute, NHGRI)合作开展了The Cancer Genome Atlas (TCGA)数据库计划,该计划旨在通过大规模基因测序和综合性、多维度的分析手段来寻找由肿瘤发生发展造成的基因变化,构建肿瘤基因相关的全方位“地图集”[11]。TCGA计划分为两个部分:第一部分从2006~ 2008年选择了具有严重不良预后且危害公共健康的3种常见肿瘤(脑癌、肺癌和卵巢癌)进行数据采集和分析,从而对其数据库整体框架的构建进行基本测试;从2009年开始进入第二阶段,扩大肿瘤类型至33种并扩大样本量进行6种数据类型的记录和分析(图1,A和B),这一过程虽然耗资巨大但成果显著。近年来科研人员已经依据TCGA数据库在多种肿瘤中发现了潜在的临床标志物和治疗靶点[12,13,14,15]。
图1
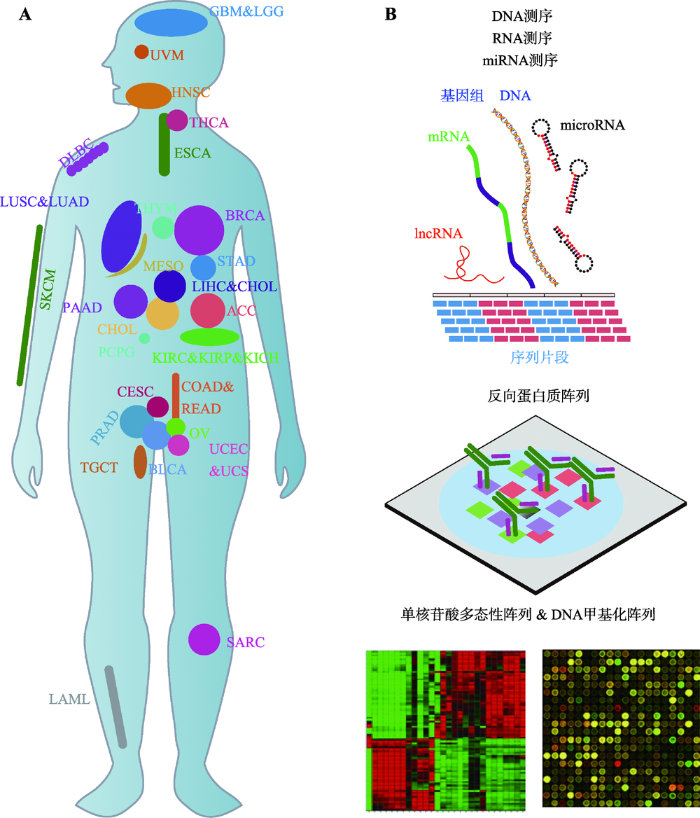 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1TCGA数据库收录的肿瘤类型和数据类型
Fig. 1Tumor types and numeric types of TCGA database
2.2 TCGA数据类型
TCGA使用基于芯片技术的高通量测序方法和二代测序技术来精确记录肿瘤基因组的全方位信息,除此之外,TCGA还记录并追踪了病人的临床信息包括性别、年龄、肿瘤分期、复发和预后情况等,从而有利于对其开展多因素综合性的分析。以下为TCGA数据库中较为常见的数据类型。2.2.1 RNA测序数据
RNA测序(RNA-seq)是一种针对转录组进行测序的高通量技术,其特点是可以在大量样本中快速识别和量化不同表达水平的转录组,检测异构体变化、找到新的转录组、筛选融合基因和非编码RNA (ncRNA)。TCGA数据库中提供了RNA序列、基因表达量、外显子序列和突变点等信息的记录,这一数据库为肿瘤转录组研究人员提供了大量数据和样本信息支持[16,17]。
2.2.2 MicroRNA测序数据
MicroRNA是一种长度约20nt的非编码小RNA分子,通过与mRNA相互作用影响目标mRNA的稳定性及转录翻译等过程,最终调控基因表达、诱导靶基因沉默、影响细胞生长、发育等生物过程[18],近年来也有研究以miRNA作为靶点的抗肿瘤药物[19]。TCGA数据库提供了肿瘤样本的miRNA表达、异构体情况,可以用于分析肿瘤相关基因的互作网络关系和探索未被发现的miRNA[20,21]。
2.2.3 DNA测序数据
DNA测序(DNA-seq)是一种高通量手段来测定DNA序列从而找到DNA的变化如插入、缺失、点突变、多态性、拷贝数改变、突变频率和病毒基因组侵入。TCGA数据库以Sanger测序技术为基础构建了DNA测序数据集,构建该数据集是为了探究在不同肿瘤类型中基因组的多样性,从而进一步找到具有诊断和治疗意义的新靶点[22,23]。
2.2.4 单核苷酸多态性检测数据
单核苷酸多态性检测(single nucleotide polymorphisms, SNPs)是指由单一核苷酸的改变所引起的序列多态性,TCGA选择了Illumina平台的分子量阵列技术来检测多种肿瘤基因组中SNP水平的变化,此外还能记录拷贝数变异(copy number variation, CNV)和杂合性缺失(loss of heterozygosity, LOH)[24]。
2.2.5 DNA甲基化测序数据
DNA甲基化测序可以检测全基因组的表观遗传学改变,在CpG位点上的甲基化和去甲基化修饰是最早和最常见的肿瘤相关表观遗传变异,这些表观遗传变异具有成为特异性肿瘤标志物的可能。TCGA数据库中的甲基化数据是基于lllumina测序平台获得的,保证了单碱基对的分辨率,高测量精度和低样品DNA需要量,不仅记录了信号强度、探查可信度还收载了用于进一步确定DNA甲基化水平的的计算β值等[25,26,27]。
2.2.6 反向蛋白质阵列表达数据
反向蛋白质阵列(reverse-phase protein array, RPPA)A: TCGA收录的33种肿瘤类型的体内分布示意图。ACC:肾上腺皮质癌;BLCA:膀胱癌;BRCA:乳腺癌;CESC:宫颈鳞状细胞癌;CHOL:胆癌;COAD:结肠腺癌;DLBC:弥漫性大B细胞淋巴瘤;ESCA:食管癌;GBM:多形性胶质母细胞瘤;HNSC:头颈部鳞癌;KICH:肾嫌色细胞癌;KIRC:肾透明细胞癌;KIRP:乳头状肾细胞癌;LAML:骨髓癌;LBB:低分化脑胶质细胞瘤;LIHC:肝癌;LUAD:肺腺癌;LUSC:肺鳞状细胞癌;MESO:间皮瘤;OV:卵巢癌;PAAD:胰腺癌;PCPG:肾上腺癌;PRAD:前列腺癌;READ:直肠癌;SARC:肉瘤;SKCM:皮肤黑色素瘤;STAD:胃癌;TGCT:睾丸癌;THCA:甲状腺癌;THYM:胸腺癌;UCEC:子宫内膜癌;UCS:子宫癌;UVM:葡萄膜黑色素瘤。B:TCGA记录的6种测序数据类型。是一种高通量、高灵敏度、可重复的蛋白检测技术,可同时用500个抗体对超过1000个样本进行检测,可以用于分子标志物筛选、分子靶标识别、肿瘤细胞亚型分析和药效学评价。TCGA数据库收录了RPPA分析的原始图片,原始信号强度,相对蛋白表达量以及标准化后的蛋白信号[28]。
2.3 TCGA数据库资源获取方法
TCGA数据库提供的数据量较大,一般需要专业的工具下载和处理,研究人员可以直接访问TCGA数据库网站(https://portal.gdc.cancer.gov/),使用其自带的GDC-Client进行下载。也可以利用编程语言R中的多种包如TCGA2STAT、RTCGA等进行下载。此外,还可以使用一些研究人员制作的第三方工具如TCGA-Assemble等进行数据下载和初始化处理。3 基于TCGA数据库分析的应用实例
3.1 针对单一类型数据的研究
三阴性乳腺癌(triple negative breast cancer, TNBC)是一种高异质性和侵略性的疾病,且目前为止没有明确有效的治疗靶点,在依据肿瘤亚型为基准的个体化医疗时代,TNBC相比于其他类型的乳腺癌有更高的死亡率。但在临床中发现,约有1/3的病人通过常规化疗手段使病情得到完全缓解。因此,Jiang等[29]以对化疗敏感为条件在TCGA、METAVRIC等数据库中选择了约400例样本的肿瘤组织和正常组织外显子序列进行研究。在分析中他们发现以BRCA1分子为核心的AR-和FOXA-调节网络的突变与化疗敏感性有较高的相关度。进一步分析发现以BRCA1/2低表达为表型的BRCA基因缺陷型TNBC病人有更高的化疗敏感性和更长的化疗后生存周期。除此之外,通过体外实验发现BRCA缺陷型TNBC病人体内不仅有相对更高的突变率且体内表达了一种可以增强免疫细胞活性的新抗原。因此,BRCA缺陷可以作为一个潜在的三阴性乳腺癌分类标签。IsomiRNA是一类序列或长度发生变化的异构体miRNA,这类RNA的靶点和功能会较原有的标准miRNA有所变化。在肿瘤发生过程中,这类miRNA被认为对其有潜在的调控作用。Omar等[30]通过对TCGA-miRNA数据集中乳腺癌的数据进行分析,发现has-miR-140-3p和5°isomiR-140-3p在乳腺癌中均高表达。他们对这两种miRNA进行功能分析发现,两者均能通过作用于增殖和迁移相关的基因从而对肿瘤细胞的生存和转移有显著的调控作用,且二者之间存在协同作用关系。
3.2 针对多组学数据的研究
由于胰管腺癌病患的异质性高导致现阶段的治疗效果不理想,Gibori等[31]尝试利用RNAi技术进行多靶点给药,从而解决这一问题。他们首先通过对TCGA数据库中胰管腺癌的蛋白质阵列数据和microRNA测序数据进行分析,结合病人的生存情况找出与生存时间显著正相关的microRNA和显著负相关的蛋白质,分别为miR-34a和PLK1。他们还利用两亲性谷氨酰胺聚合物作为纳米载体,将miR-34a的类似物(miR-34a mimic)和抑制PLK1蛋白表达的siRNA共同偶联至载体表面进行体内外给药实验。小鼠移植瘤模型研究发现这种双靶点纳米制剂可以有效靶向至胰管腺癌的发病部位并抑制肿瘤生长,这为胰管腺癌的治疗提供了新思路。TCGA数据库提供了30余种肿瘤类型的相关数据,这使得泛肿瘤研究的进展大大提升,Thorsso等[32]对TCGA中33种肿瘤类型的超过10000例样本的全部6种数据进行免疫基因组分析,使用160个免疫表达特征进行打分,通过聚类分析将这10000余个样本进行分类,最终基于不同的免疫表达特征分为6类,包括IFN-γ主导型、炎症型、淋巴细胞耗尽型、免疫沉默型和TGF-β主导型等。基于这6种分类,研究人员对不同类别中的肿瘤免疫浸润构成、免疫反应与体细胞多样性的相关性、免疫反应与预后的相关性、不同免疫亚型与预后的相关性、免疫原性的变化、免疫调节剂的表达差异等进行了进一步的关联分析,从而证明了这种分类的准确性。这一分类几乎包括了人类所有的恶性肿瘤类型,这为从免疫基因组学角度预测疾病走向和病人预后提供了帮助。
Berger等[33]通过对TCGA数据库中包括乳腺癌在内的5种妇科肿瘤类型的2579例样本进行综合的多平台分析并与其余肿瘤类型样本数据进行对比,发现了这5种肿瘤病人样本中特有的基因组和表观基因组特征,包括3个体细胞拷贝数变异、46个显著突变基因以及与之前报道相同的多种miRNA和lncRNA异常表达,研究人员通过多种聚类分析将这5种具有共性的妇科肿瘤类型基于16个特异性分子指标分为了5个亚型,进一步验证发现这5种亚型病人的生存时间存在显著差异,最终研究人员在保证分类精确度的基础上,使用二分决策树将16个特异性分子优化至6个,这为未来妇科肿瘤的分类和诊断提供了帮助。
精准肿瘤学是一门分析个体差异从而指导肿瘤治疗的学科。近年来研究发现,多组学特征可以用来预测肿瘤患者的临床特征,但多组学数据计算量大,分析难度高且大部分医生没有学习过相关的生物信息学知识,因此Yu等[34]建立了Omics Analysis System for PRecision Oncology (OASISPRO)系统,用于挖掘和量化TCGA数据库中的多组学数据。该系统可以将临床样本数据可视化,并基于机器学习相关算法找出与临床分期相关的基因,以及预测患者生存时间,这对精准治疗和个体化用药提供了指导。
Omics Pipe是一个模块化的云计算平台,该平台可以根据用户要求自动获取TCGA数据库中的相关数据集,并进行多组学整合分析,此外还可以自定义组学分析和在平台框架基础上加入自己的计算模块,自由度更高。该平台是用python代码构建而来,所有的计算与分析工作都是依托亚马逊云服务器完成,平台构建的目的是为广大生物学家提供一个模块化的高通量数据分析框架,使数据分析变得更简单和高效[35]。
4 结语与展望
二代测序技术作为21世纪的重大科学技术进步之一,为肿瘤基因组学研究提供了极大的帮助,随着肿瘤基因组数据库和患者样本信息的不断丰富,科研人员对肿瘤基因的分析日趋深入,而对分析方法和工具的选择要求也不断提高。目前对肿瘤基因组的分析仍然处于起步阶段,虽然TCGA构建了立体化的多元素肿瘤基因组数据库,但多组学的基因数据很少作为一个整体进行立体化的分析,大多数研究都只局限于某一特定的数据类型如SNP、miRNA和表观修饰等。这也从侧面体现了现阶段统计学算法的局限性。计算机性能的不断提升使数据量不再是限制科研人员的主要因素,而如何将多组学数据整合到一起才更为关键。现阶段的多组学分析还比较简单,大多数研究都围绕聚类分析展开,将多组学数据依照临床样本信息进行分类,筛选出潜在的肿瘤标志物。而这种分析对肿瘤的多组学发病机理研究帮助较小,无法系统的阐明不同组学水平之间的关联性。但由于机器学习等人工智能算法的出现,科研人员将从更宏观的角度来分析肿瘤基因组数据,TCGA数据库也已经与多个高校及科研机构合作,尝试进行高通量多组学的肿瘤基因数据分析,但其分析结果的准确性还有待进一步的验证,同时,分析结果的具体临床应用也有待开发。随着算法的不断发展,多组学分析将为肿瘤学研究提供强有力的支持,并从宏观的角度阐述不同分子水平对肿瘤的调控作用以及之间的联系。相信未来会出现基于多组学基因数据的整合分析方法,更全面的阐述肿瘤的发生和发展过程,为肿瘤诊断和治疗提供帮助。
此外,现有数据库主要针对白种人构建,而亚洲人种数据库还尚处于起步阶段,存在数据量少、数据类型单一、临床信息不全面等缺陷,但近年来也有一些成果出现,如中国科学院的生命与健康大数据中心等[36],相信随着政府部门的重视和国内测序产业的发展,黄种人多组学数据库也将逐步完善,成为肿瘤基因组学研究的新支柱。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:4111957 [本文引用: 1]

Time-to-event outcomes are common in medical research as they offer more information than simply whether or not an event occurred. To handle these outcomes, as well as censored observations where the event was not observed during follow-up, survival analysis methods should be used. Kaplan-Meier estimation can be used to create graphs of the observed survival curves, while the log-rank test can be used to compare curves from different groups. If it is desired to test continuous predictors or to test multiple covariates at once, survival regression models such as the Cox model or the accelerated failure time model (AFT) should be used. The choice of model should depend on whether or not the assumption of the model (proportional hazards for the Cox model, a parametric distribution of the event times for the AFT model) is met. The goal of this paper is to review basic concepts of survival analysis. Discussions relating the Cox model and the AFT model will be provided. The use and interpretation of the survival methods model are illustrated using an artificially simulated dataset.
URL [本文引用: 1]
URLPMID:29717943 [本文引用: 1]
Abstract We consider the situation of estimating Cox regression in which some covariates are subject to missing, and there exists additional information (including observed event time, censoring indicator and fully observed covariates) which may be predictive of the missing covariates. We propose to use two working regression models: one for predicting the missing covariates and the other for predicting the missing probabilities. For each missing covariate observation, these two working models are used to define a nearest neighbor imputing set. This set is then used to non-parametrically impute covariate values for the missing observation. Upon the completion of imputation, Cox regression is performed on the multiply imputed datasets to estimate the regression coefficients. In a simulation study, we compare the nonparametric multiple imputation approach with the augmented inverse probability weighted (AIPW) method, which directly incorporates the two working models into estimation of Cox regression, and the predictive mean matching imputation (PMM) method. We show that all approaches can reduce bias due to non-ignorable missing mechanism. The proposed nonparametric imputation method is robust to mis-specification of either one of the two working models and robust to mis-specification of the link function of the two working models. In contrast, the PMM method is sensitive to misspecification of the covariates included in imputation. The AIPW method is sensitive to the selection probability. We apply the approaches to a breast cancer dataset from Surveillance, Epidemiology and End Results (SEER) Program.
URLPMID:4402510 [本文引用: 1]
Abstract limma is an R/Bioconductor software package that provides an integrated solution for analysing data from gene expression experiments. It contains rich features for handling complex experimental designs and for information borrowing to overcome the problem of small sample sizes. Over the past decade, limma has been a popular choice for gene discovery through differential expression analyses of microarray and high-throughput PCR data. The package contains particularly strong facilities for reading, normalizing and exploring such data. Recently, the capabilities of limma have been significantly expanded in two important directions. First, the package can now perform both differential expression and differential splicing analyses of RNA sequencing (RNA-seq) data. All the downstream analysis tools previously restricted to microarray data are now available for RNA-seq as well. These capabilities allow users to analyse both RNA-seq and microarray data with very similar pipelines. Second, the package is now able to go past the traditional gene-wise expression analyses in a variety of ways, analysing expression profiles in terms of co-regulated sets of genes or in terms of higher-order expression signatures. This provides enhanced possibilities for biological interpretation of gene expression differences. This article reviews the philosophy and design of the limma package, summarizing both new and historical features, with an emphasis on recent enhancements and features that have not been previously described. The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
URLPMID:10568750 [本文引用: 1]
Analysis procedures are needed to extract useful information from the large amount of gene expression data that is becoming available. This work describes a set of analytical tools and their application to yeast cell cycle data. The components of our approach are (1) a similarity measure that reduces the number of false positives, (2) a new clustering algorithm designed specifically for grouping gene expression patterns, and (3) an interactive graphical cluster analysis tool that allows user feedback and validation. We use the clusters generated by our algorithm to summarize genome-wide expression and to initiate supervised clustering of genes into biologically meaningful groups.
URL [本文引用: 1]
URLPMID:4514923 [本文引用: 1]
Precision-recall (PR) and receiver operating characteristic (ROC) curves are valuable measures of classifier performance. Here, we present the R-package PRROC, which allows for computing and visualizing both PR and ROC curves. In contrast to available R-packages, PRROC allows for computing PR and ROC curves and areas under these curves for soft-labeled data using a continuous interpolation between the points of PR curves. In addition, PRROC provides a generic plot function for generating publication-quality graphics of PR and ROC curves.
URLPMID:27153689 [本文引用: 1]
Motivation: A dominant approach to genetic association studies is to perform univariate tests between genotype-phenotype pairs. However, analyzing related traits together increases statistical power, and certain complex associations become detectable only when several variants are tested jointly. Currently, modest sample sizes of individual cohorts, and restricted availability of individual-level genotype-phenotype data across the cohorts limit conducting multivariate tests. Results: We introducemetaCCA, a computational framework for summary statistics-based analysis of a single or multiple studies that allows multivariate representation of both genotype and phenotype. It extends the statistical technique of canonical correlation analysis to the setting where original individual-level records are not available, and employs a covariance shrinkage algorithm to achieve robustness. Multivariate meta-analysis of two Finnish studies of nuclear magnetic resonance metabolomics bymetaCCA, using standard univariate output from the program SNPTEST, shows an excellent agreement with the pooled individual-level analysis of original data. Motivated by strong multivariate signals in the lipid genes tested, we envision that multivariate association testing usingmetaCCAhas a great potential to provide novel insights from already published summary statistics from high-throughput phenotyping technologies. Availability and implementation: Code is available athttps://github.com/aalto-ics-kepaco Contacts:anna.cichonska@helsinki.fiormatti.pirinen@helsinki.fi Supplementary information:Supplementary dataare available atBioinformaticsonline.
URLPMID:28108451 [本文引用: 1]
Motivation: In the context of genome-wide association studies (GWAS), there is a variety of statistical techniques in order to conduct the analysis, but, in most cases, the underlying genetic model ...
URLPMID:21383744 [本文引用: 1]
Recent advances in genome technologies and the ensuing outpouring of genomic information related to cancer have accelerated the convergence of discovery science and clinical medicine. Successful examples of translating cancer genomics into therapeutics and diagnostics reinforce its potential to make possible personalized cancer medicine. However, the bottlenecks along the path of converting a genome discovery into a tangible clinical endpoint are numerous and formidable. In this Perspective, we emphasize the importance of establishing the biological relevance of a cancer genomic discovery in realizing its clinical potential and discuss some of the major obstacles to moving from the bench to the bedside.
URLPMID:4322527 [本文引用: 1]
The Cancer Genome Atlas (TCGA) is a public funded project that aims to catalogue and discover major cancer-causing genomic alterations to create a comprehensive "atlas" of cancer genomic profiles. So far, TCGA researchers have analysed large cohorts of over 30 human tumours through large-scale genome sequencing and integrated multi-dimensional analyses. Studies of individual cancer types, as well as comprehensive pan-cancer analyses have extended current knowledge of tumorigenesis. A major goal of the project was to provide publicly available datasets to help improve diagnostic methods, treatment standards, and finally to prevent cancer. This review discusses the current status of TCGA Research Network structure, purpose, and achievements.
URL [本文引用: 1]
URLPMID:26851671 [本文引用: 1]
Translational bioinformatics and clinical research (biomedical) informatics are the primary domains related to informatics activities that support translational research. Translational bioinformatics focuses on computational techniques in genetics, molecular biology, and systems biology. Clinical research (biomedical) informatics involves the use of informatics in discovery and management of new knowledge relating to health and disease. This article details 3 projects that are hybrid applications of translational bioinformatics and clinical research (biomedical) informatics: The Cancer Genome Atlas, the cBioPortal for Cancer Genomics, and the Memorial Sloan Kettering Cancer Center clinical variants and results database, all designed to facilitate insights into cancer biology and clinical/therapeutic correlations.
[本文引用: 1]
URL [本文引用: 1]
URLPMID:26996076 [本文引用: 1]
With the emergence of RNA sequencing (RNA-seq) technologies, RNA-based biomolecules hold expanded promise for their diagnostic, prognostic and therapeutic applicability in various diseases, including cancers and infectious diseases. Detection of gene fusions and differential expression of known disease-causing transcripts by RNA-seq represent some of the most immediate opportunities. However, it is the diversity of RNA species detected through RNA-seq that holds new promise for the multi-faceted clinical applicability of RNA-based measures, including the potential of extracellular RNAs as non-invasive diagnostic indicators of disease. Ongoing efforts towards the establishment of benchmark standards, assay optimization for clinical conditions and demonstration of assay reproducibility are required to expand the clinical utility of RNA-seq.
URLPMID:29625054 [本文引用: 1]
Abstract The role of enhancers, a key class of non-coding regulatory DNA elements, in cancer development has increasingly been appreciated. Here, we present the detection and characterization of a large number of expressed enhancers in a genome-wide analysis of 8928 tumor samples across 33 cancer types using02TCGA RNA-seq data. Compared with matched normal tissues, global enhancer activation was observed in most cancers. Across cancer types, global enhancer activity was positively associated with aneuploidy, but not mutation load, suggesting02a02hypothesis centered on "chromatin-state" to explain their interplay. Integrating eQTL, mRNA co-expression, and Hi-C data analysis, we developed a02computational method to infer causal enhancer-gene interactions, revealing enhancers of clinically actionable genes. Having identified an enhancer 65140 kb downstream of PD-L1, a major immunotherapy target, we validated it experimentally. This study provides a systematic view of enhancer activity in diverse tumor contexts and suggests the clinical implications of enhancers.
URLPMID:29230018 [本文引用: 1]
Abstract CD8 + memory T (Tm) cells are fundamental for protective immunity against infections and cancers 1-5 . Metabolic activities are crucial in controlling memory T-cell homeostasis, but mechanisms linking metabolic signals to memory formation and survival remain elusive. Here we show that CD8 + Tm cells markedly upregulate cytosolic phosphoenolpyruvate carboxykinase (Pck1), the hub molecule regulating glycolysis, tricarboxylic acid cycle and gluconeogenesis, to increase glycogenesis via gluconeogenesis. The resultant glycogen is then channelled to glycogenolysis to generate glucose-6-phosphate and the subsequent pentose phosphate pathway (PPP) that generates abundant NADPH, ensuring high levels of reduced glutathione in Tm cells. Abrogation of Pck1-glycogen-PPP decreases GSH/GSSG ratios and increases levels of reactive oxygen species (ROS), leading to impairment of CD8 + Tm formation and maintenance. Importantly, this metabolic regulatory mechanism could be readily translated into more efficient T-cell immunotherapy in mouse tumour models.
URLPMID:28209991 [本文引用: 1]
In just over two decades since the discovery of the first microRNA (miRNA), the field of miRNA biology has expanded considerably. Insights into the roles of miRNAs in development and disease, particularly in cancer, have made miRNAs attractive tools and targets for novel therapeutic approaches. Functional studies have confirmed that miRNA dysregulation is causal in many cases of cancer, with miRNAs acting as tumour suppressors or oncogenes (oncomiRs), and miRNA mimics and molecules targeted at miRNAs (antimiRs) have shown promise in preclinical development. Several miRNA-targeted therapeutics have reached clinical development, including a mimic of the tumour suppressor miRNA miR-34, which reached phase I clinical trials for treating cancer, and antimiRs targeted at miR-122, which reached phase II trials for treating hepatitis. In this article, we describe recent advances in our understanding of miRNAs in cancer and in other diseases and provide an overview of current miRNA therapeutics in the clinic. We also discuss the challenge of identifying the most efficacious therapeutic candidates and provide a perspective on achieving safe and targeted delivery of miRNA therapeutics.
URL [本文引用: 1]
URLPMID:4862407 [本文引用: 1]
Although clinical studies have shown promise for targeting PD1/PDL1 signaling in non–small cell lung cancer (NSCLC), the regulation of PDL1 expression is poorly understood. Here, we show that PDL1 is regulated by p53 via miR-34. p53 wild-type and p53-deficient cell lines (p53–/–and p53+/+HCT116, p53-inducible H1299, and p53-knockdown H460) were used to determine if p53 regulates PDL1 via miR-34. PDL1 and miR-34a expression were analyzed in samples from patients with NSCLC and mutated p53 vs wild-type p53 tumors from The Cancer Genome Atlas for Lung Adenocarcinoma (TCGA LUAD). We confirmed that PDL1 is a direct target of miR-34 with western blotting and luciferase assays and used a p53R172HΔg/+K-rasLA1/+syngeneic mouse model (n = 12) to deliver miR-34a–loaded liposomes (MRX34) plus radiotherapy (XRT) and assessed PDL1 expression and tumor-infiltrating lymphocytes (TILs). A two-sidedttest was applied to compare the mean between different treatments. We found that p53 regulates PDL1 via miR-34, which directly binds to thePDL13’ untranslated region in models of NSCLC (fold-change luciferase activity to control group, mean for miR-34a = 0.50, SD = 0.2,P< .001; mean for miR-34b = 0.52, SD = 0.2,P= .006; and mean for miR-34c = 0.59, SD = 0.14, andP= .006). Therapeutic delivery of MRX34, currently the subject of a phase I clinical trial, promoted TILs (mean of CD8 expression percentage of control group = 22.5%, SD = 1.9%; mean of CD8 expression percentage of MRX34 = 30.1%, SD = 3.7%,P= .016, n = 4) and reduced CD8+PD1+cells in vivo (mean of CD8/PD1 expression percentage of control group = 40.2%, SD = 6.2%; mean of CD8/PD1 expression percentage of MRX34 = 20.3%, SD = 5.1%,P= .001, n = 4). Further, MRX34 plus XRT increased CD8+cell numbers more than either therapy alone (mean of CD8 expression percentage of MRX34 plus XRT to control group = 44.2%, SD = 8.7%,P= .004, n = 4). Finally, miR-34a delivery reduced the numbers of radiation-induced macrophages (mean of F4-80 expression percentage of control group = 52.4%, SD = 1.7%; mean of F4-80 expression percentage of MRX34 = 40.1%, SD = 3.5%,P= .008, n = 4) and T-regulatory cells. We identified a novel mechanism by which tumor immune evasion is regulated by p53/miR-34/PDL1 axis. Our results suggest that delivery of miRNAs with standard therapies, such as XRT, may represent a novel therapeutic approach for lung cancer.
URLPMID:23121054 [本文引用: 1]
Advances in DNA sequencing technology have allowed comprehensive investigation of the genetics of human beings and human diseases. Insights from sequencing the genomes, exomes, or transcriptomes of healthy and diseased cells in patients are already enabling improved diagnostic classification, prognostication, and therapy selection for many diseases. Understanding the data obtained using new high-throughput DNA sequencing methods, choices made in sequencing strategies, and common challenges in data analysis and genotype-phenotype correlation is essential if pathologists, geneticists, and clinicians are to interpret the growing scientific literature in this area. This review highlights some of the major results and discoveries stemming from high-throughput DNA sequencing research in our understanding of Mendelian genetic disorders, hematologic cancer biology, infectious diseases, the immune system, transplant biology, and prenatal diagnostics. Transition of new DNA sequencing methodologies to the clinical laboratory is under way and is likely to have a major impact on all areas of medicine.
URLPMID:25091868 [本文引用: 1]
Abstract The vast majority of microbial species remain uncultivated and, until recently, about half of all known bacterial phyla were identified only from their 16S ribosomal RNA gene sequence. With the advent of single-cell sequencing, genomes of uncultivated species are rapidly filling in unsequenced branches of the microbial phylogenetic tree. The wealth of new insights gained from these previously inaccessible groups is providing a deeper understanding of their basic biology, taxonomy and evolution, as well as their diverse roles in environmental ecosystems and human health.
URLPMID:18776908 [本文引用: 1]
Abstract Dissecting the genetic basis of disease risk requires measuring all forms of genetic variation, including SNPs and copy number variants (CNVs), and is enabled by accurate maps of their locations, frequencies and population-genetic properties. We designed a hybrid genotyping array (Affymetrix SNP 6.0) to simultaneously measure 906,600 SNPs and copy number at 1.8 million genomic locations. By characterizing 270 HapMap samples, we developed a map of human CNV (at 2-kb breakpoint resolution) informed by integer genotypes for 1,320 copy number polymorphisms (CNPs) that segregate at an allele frequency >1%. More than 80% of the sequence in previously reported CNV regions fell outside our estimated CNV boundaries, indicating that large (>100 kb) CNVs affect much less of the genome than initially reported. Approximately 80% of observed copy number differences between pairs of individuals were due to common CNPs with an allele frequency >5%, and more than 99% derived from inheritance rather than new mutation. Most common, diallelic CNPs were in strong linkage disequilibrium with SNPs, and most low-frequency CNVs segregated on specific SNP haplotypes.
URLPMID:28729483 [本文引用: 1]
Abstract Chromatin and associated epigenetic mechanisms stabilize gene expression and cellular states while also facilitating appropriate responses to developmental or environmental cues. Genetic, environmental, or metabolic insults can induce overly restrictive or overly permissive epigenetic landscapes that contribute to pathogenesis of cancer and other diseases. Restrictive chromatin states may prevent appropriate induction of tumor suppressor programs or block differentiation. By contrast, permissive or "plastic" states may allow stochastic oncogene activation or nonphysiologic cell fate transitions. Whereas many stochastic events will be inconsequential "passengers," some will confer a fitness advantage to a cell and be selected as "drivers." We review the broad roles played by epigenetic aberrations in tumor initiation and evolution and their potential to give rise to all classic hallmarks of cancer. Copyright 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
URLPMID:26216839 [本文引用: 1]
Colorectal cancer (CRC) is a leading cause of cancer deaths worldwide. One of the fundamental processes driving the initiation and progression of CRC is the accumulation of a variety of genetic and epigenetic changes in colon epithelial cells. Over the past decade, major advances have been made in our understanding of cancer epigenetics, particularly regarding aberrant DNA methylation, microRNA (miRNA) and noncoding RNA deregulation, and alterations in histone modification states. Assessment of the colon cancer pigenome has revealed that virtually all CRCs have aberrantly methylated genes and altered miRNA expression. The average CRC methylome has hundreds to thousands of abnormally methylated genes and dozens of altered miRNAs. As with gene mutations in the cancer genome, a subset of these epigenetic alterations, called driver events, is presumed to have a functional role in CRC. In addition, the advances in our understanding of epigenetic alterations in CRC have led to these alterations being developed as clinical biomarkers for diagnostic, prognostic and therapeutic applications. Progress in this field suggests that these epigenetic alterations will be commonly used in the near future to direct the prevention and treatment of CRC.
URL [本文引用: 1]
URLPMID:27663479 [本文引用: 1]
The majority of the targeted therapeutic agents in clinical use target proteins and protein function. Although DNA and RNA analyses have been used extensively to identify novel targets and patients likely to benefit from targeted therapies, these are indirect measures of the levels and functions of most therapeutic targets. More importantly, DNA and RNA analysis is ill-suited for determining the pharmacodynamic effects of target inhibition. Assessing changes in protein levels and function is the most efficient way to evaluate the mechanisms underlying sensitivity and resistance to targeted agents. Understanding these mechanisms is necessary to identify patients likely to benefit from treatment and to develop rational drug combinations to prevent or bypass therapeutic resistance. There is an urgent need for a robust approach to assess protein levels and protein function in model systems and across patient samples. While hot gun mass spectrometry can provide in-depth analysis of proteins across a limited number of samples, and emerging approaches such as multiple reaction monitoring have the potential to analyze candidate markers, mass spectrometry has not entered into general use because of the high cost, requirement of extensive analysis and support, and relatively large amount of material needed for analysis. Rather, antibody-based technologies, including immunohistochemistry, radio immunoassays, ELISAs and more recently protein arrays, remain the most common approaches for multiplexed protein analysis. Reverse-phase protein array (RPPA) technology has emerged as a robust, sensitive, cost-effective approach to the analysis of large numbers of samples for quantitative assessment of key members of functional pathways that are affected by tumor-targeting therapeutics. The RPPA platform is a powerful approach for identifying and validating targets, classifying tumor subsets, assessing pharmacodynamics, and identifying prognostic and predictive markers, adaptive responses and rational drug combinations in model systems and patient samples. Its greatest utility has been realized through integration with other analytic platforms such as DNA sequencing, transcriptional profiling, epigenomics, mass spectrometry, and metabolomics. The power of the technology is becoming apparent through its use in pathology laboratories and integration into trial design and implementation.
URLPMID:27959926 [本文引用: 1]
Triple negative breast cancer (TNBC) is a highly heterogeneous and aggressive disease, and although no effective targeted therapies are available to date, about one-third of patients with TNBC achieve pathologic complete response (pCR) from standard-of-care anthracycline/taxane (ACT) chemotherapy. The heterogeneity of these tumors, however, has hindered the discovery of effective biomarkers to identify such patients. We performed whole exome sequencing on 29 TNBC cases from the MD Anderson Cancer Center (MDACC) selected because they had either pCR (n= 18) or extensive residual disease (n= 11) after neoadjuvant chemotherapy, with cases from The Cancer Genome Atlas (TCGA;n= 144) and METABRIC (n= 278) cohorts serving as validation cohorts. Our analysis revealed that mutations in the AR- and FOXA1-regulated networks, in which BRCA1 plays a key role, are associated with significantly higher sensitivity to ACT chemotherapy in the MDACC cohort (pCR rate of 94.1% compared to 16.6% in tumors without mutations in AR/FOXA1 pathway, adjustedp= 0.02) and significantly better survival outcome in the TCGA TNBC cohort (log-rank test,p= 0.05). Combined analysis of DNA sequencing, DNA methylation, and RNA sequencing identified tumors of a distinct BRCA-deficient (BRCA-D) TNBC subtype characterized by low levels of wild-type BRCA1/2 expression. Patients with functionally BRCA-D tumors had significantly better survival with standard-of-care chemotherapy than patients whose tumors were not BRCA-D (log-rank test,p= 0.021), and they had significantly higher mutation burden (p <0.001) and presented clonal neoantigens that were associated with increased immune cell activity. A transcriptional signature of BRCA-D TNBC tumors was independently validated to be significantly associated with improved survival in the METABRIC dataset (log-rank test,p= 0.009). As a retrospective study, limitations include the small size and potential selection bias in the discovery cohort. The comprehensive molecular analysis presented in this study directly links BRCA deficiency with increased clonal mutation burden and significantly enhanced chemosensitivity in TNBC and suggests that functional RNA-based BRCA deficiency needs to be further examined in TNBC. Through molecular analyses of tissue from triple negative breast cancer patients, Christos Hatzis and colleagues report on a new tumor subtype with possible implications for treatment. Identifying chemosensitive triple negative breast cancers (TNBCs) could significantly impact the survival of patients with these difficult to treat cancers until novel targeted therapies become available. We hypothesized that genomic somatic aberrations may provide important molecular clues about chemosensitivity in TNBC. Our study used a carefully selected cohort of 29 uniformly treated TNBC patients who either achieved pathologic complete response (pCR) or had extensive residual disease after neoadjuvant anthracycline/taxane chemotherapy. We sequenced the coding genomic DNA of TNBC tumors and compared the somatic mutations found in the two groups at the two extremes of the chemosensitivity spectrum. Our analysis revealed that, although mutations in single genes were not individually predictive, TNBC tumors bearing mutations in genes involved in the androgen receptor (AR) and FOXA1 pathways were much more sensitive to chemotherapy. We also found that mutations that lowered the levels of functionalBRCA1orBRCA2RNA were associated with significantly better survival outcomes; we derived a BRCA deficiency signature to define this new, highly chemosensitive subtype of TNBC. BRCA-deficient TNBC tumors have a higher rate of clonal mutation burden, defined as more clonal tumors with a higher number of mutations per clone, and are also associated with a higher level of immune activation, which may explain their greater chemosensitivity. Mutations in the AR/FOXA1 pathway provide a novel marker for identifying chemosensitive TNBC patients who may benefit from current standard-of-care chemotherapy regimens. The newly defined RNA-based BRCA-deficient subtype includes up to 50% of the TNBC tumors that appear to be immune primed, and it would be of interest to investigate combinations of chemotherapy with immunotherapies, which could provide clinical benefit for these patients. Although our study showed concordant results in three different datasets, our key findings need to be further validated in a larger, prospectively designed study with archival samples.
URL [本文引用: 1]
URLPMID:29295989 [本文引用: 1]
Targeting pancreatic cancer with a polymeric nano carrier and RNAi combination
URLPMID:29628290 [本文引用: 1]
Abstract We performed an extensive immunogenomic analysis of more than 10,000 tumors comprising 33 diverse cancer types by utilizing data compiled by TCGA. Across cancer types, we identified six immune subtypes-wound healing, IFN-0206 dominant, inflammatory, lymphocyte depleted, immunologically quiet, and TGF-0205 dominant-characterized by differences in macrophage or lymphocyte signatures, Th1:Th2 cell ratio, extent of intratumoral heterogeneity, aneuploidy, extent of neoantigen load, overall cell proliferation, expression of immunomodulatory genes, and prognosis. Specific driver mutations correlated with lower (CTNNB1, NRAS, or IDH1) or higher (BRAF, TP53, or CASP8) leukocyte levels across all cancers. Multiple control modalities of the intracellular and extracellular networks (transcription, microRNAs, copy number, and epigenetic processes) were involved in tumor-immune cell interactions, both across and within immune subtypes. Our immunogenomics pipeline to characterize these heterogeneous tumors and the resulting data are intended to serve as a resource for future targeted studies to further advance the field.
URLPMID:29622464 [本文引用: 1]
Abstract We analyzed molecular data on 2,579 tumors from The Cancer Genome Atlas (TCGA) of four gynecological types plus breast. Our aims were to identify shared and unique molecular features, clinically significant subtypes, and potential therapeutic targets. We found 61 somatic copy-number alterations (SCNAs) and 46 significantly mutated genes (SMGs). Eleven SCNAs and 11 SMGs had not been identified in previous TCGA studies of the individual tumor types. We found functionally significant estrogen receptor-regulated long non-coding RNAs (lncRNAs) and gene/lncRNA interaction networks. Pathway analysis identified subtypes with high leukocyte infiltration, raising potential implications for immunotherapy. Using 16 key molecular features, we identified five prognostic subtypes and developed a decision tree that classified patients into the subtypes based on just six features that are assessable in clinical laboratories.
URLPMID:28968749 [本文引用: 1]
Abstract Precision oncology is an approach that accounts for individual differences to guide cancer management. Omics signatures have been shown to predict clinical traits for cancer patients. However, the vast amount of omics information poses an informatics challenge in systematically identifying patterns associated with health outcomes, and no general-purpose data-mining tool exists for physicians, medical researchers, and citizen scientists without significant training in programming and bioinformatics. To bridge this gap, we built the Omics AnalySIs System for PRecision Oncology (OASISPRO), a web-based system to mine the quantitative omics information from The Cancer Genome Atlas (TCGA). This system effectively visualizes patients' clinical profiles, executes machine-learning algorithms of choice on the omics data, and evaluates the prediction performance using held-out test sets. With this tool, we successfully identified genes strongly associated with tumor stage, and accurately predicted patients' survival outcomes in many cancer types, including mesothelioma and adrenocortical carcinoma. By identifying the links between omics and clinical phenotypes, this system will facilitate omics studies on precision cancer medicine and contribute to establishing personalized cancer treatment plans. Availability and implementation: This web-based tool is available at http://tinyurl.com/oasispro ;source codes are available at http://tinyurl.com/oasisproSourceCode . Contact: khyu@stanford.edu ; mpsnyder@stanford.edu.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}