 ,武汉科技大学生命科学与健康学院,武汉 430065
,武汉科技大学生命科学与健康学院,武汉 430065The role of actin cytoskeleton in regulating the deployment process of mouse cardiac second heart field progenitor cells
Zhongying Liu, Xia Huang, Ziyi Li, Zihao Yang, Baiyin Yuan,College of Life Science and Health, Wuhan University of Science and Technology, Wuhan 430065, China通讯作者:
编委: 杨中州
收稿日期:2018-10-29修回日期:2019-01-18网络出版日期:2019-02-25
| 基金资助: |
Editorial board:
Received:2018-10-29Revised:2019-01-18Online:2019-02-25
| Fund supported: |
作者简介 About authors
刘钟颖,硕士研究生,专业方向:遗传及细胞生物学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (524KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘钟颖, 黄霞, 李紫怡, 杨子豪, 袁白银. 肌动蛋白细胞骨架在小鼠第二生心区祖细胞部署发育中的作用[J]. 遗传, 2019, 41(2): 125-136 doi:10.16288/j.yczz.18-293
Zhongying Liu, Xia Huang, Ziyi Li, Zihao Yang, Baiyin Yuan.
先天性心脏病是目前人类最常见的出生缺陷,并且一直是引起婴幼儿死亡的首要原因,其中约30%的先天性心脏缺陷为流出道(outflow tract, OFT)发育缺陷[1,2]。OFT和右心室(right ventricle, RV)是由第二生心区(second heart field, SHF)祖细胞发育形成,SHF祖细胞发育缺陷导致OFT和RV发育异常。因此,阐明SHF的发育过程和深入探讨其调控机制对于破译OFT发育缺陷的起源、控制和降低先天性心脏病的发病率、寻找临床相关致病基因对其诊断、防治以及试管婴儿的筛选具有重要意义。
哺乳动物的心脏是由第一生心区(first heart field, FHF)和SHF两种主要的心脏祖细胞发育而来。位于前部侧板中胚层的FHF祖细胞贡献于早期分化的心肌细胞,形成原始心管,将来发育成左心室(left ventricle, LV)、房室通道、极小部分的右心室和心房心肌细胞[3,4,5]。位于咽中胚层(pharyngeal mesoderm, PM)和内脏中胚层(splanchnic mesoderm, SpM)的SHF祖细胞添加晚分化的心肌细胞于伸展的心管,贡献于OFT、RV及心房心肌细胞[4,5,6]。SHF祖细胞向心管贡献、部署的缺陷使OFT的长度不足以在心脏间隔形成期间正确的旋转,升主动脉和肺动脉干与左心室和右心室对齐,导致心室分隔和心室/动脉排列缺陷,引起双出口右室(double outlet right ventricle, DORV)、大动脉转位(transposition of the great arteries, TGA)、永存动脉干(persistent truncus arteriosus, PTA)等畸形[6,7]。
研究发现,SHF细胞构成非经典顶端-基底极性上皮,并具有顶部单纤毛、动态肌动蛋白(actin)富集基底端的丝状伪足等特点,这表明actin细胞骨架在SHF发育中具有重要作用[8,9]。本文对哺乳动物SHF的发现及特性、actin细胞骨架的特性、actin与细胞运动、actin与横纹肌肌纤维以及actin与哺乳动物SHF发育等方面进行了阐述,以期为科研人员更深入地解析actin细胞骨架与哺乳动物SHF发育之间的联系提供参考,为阐明和理解SHF细胞迁移、部署的细胞生物学行为及调控网络提供思路和方向,为寻找临床相关致病基因并对先天性心脏病的诊断、防治提供理论基础。
1 小鼠心脏第二生心区
1.1 第一生心区与第二生心区
心脏发育和形成是一个复杂的过程,涉及不同空间、不同细胞群体的整合,并被复杂的级联调控网络调控。心脏是由两种主要的心脏祖细胞发育而来,分别是FHF和SHF祖细胞[5]。研究表明,FHF和SHF祖细胞在心脏新月(cardiac crescent, CC)期之前与共同的心脏祖细胞分离;FHF首先与心脏共同的祖细胞分离出来,并贡献于心脏新月期和早期心管心肌细胞;而SHF是另外一个祖细胞群,它随后将新分化的心肌细胞贡献于生长的心管[4,6,10]。FHF来源的原始心管将来发育为左心室、小部分的右心室、房室管和大部分的心房[3,5]。SHF祖细胞通过心脏动脉极贡献于OFT和右心室,并通过静脉极贡献于心房心肌细胞[4,5,6]。在小鼠(Mus musculus)胚胎发育的E8.5~E10.5时期,位于心管后部咽中胚层和内脏中胚层的SHF祖细胞向原始心管添加分化的心肌细胞,使心管延伸并向右环化[11]。SHF祖细胞对心管的贡献使心管得以足够的伸展和生长,这是心脏形态发生所必 需的。SHF祖细胞向心管贡献、部署失败导致OFT缩短,使OFT的长度不足以在心脏间隔形成期间 正确的旋转以使升主动脉和肺动脉干与左心室和 右心室对齐,导致心室分隔和心室/动脉排列缺 陷,如双出口右室、大动脉转位、永存动脉干及其他异常[6,12,13]。
1.2 第二生心区的发现及性质
20世纪70年代,de la Cruz及其同事利用体内氧化铁颗粒标记法揭示鸡(Gallus gallus)心脏远端OFT和RV实际是由后来添加的细胞形成,并且心脏的生长是通过添加位于早期心脏之外的细胞而实现[14]。另一项在小鼠中的实验也暗示,除了心脏新月以外,还有另外一种细胞群贡献于发育的心脏[15]。然而,这些早期心脏之外细胞的来源直到2001年才被确定。2001年发表的3项研究确定了在咽中胚层中的祖细胞群贡献于胚胎期延伸的OFT。Mjaatvedt等[16]确定了由de la Cruz等[14]发现的前部心脏形成区,该前部心脏形成区是由围绕在紧邻现有心管的主动脉囊周围的中胚层组成。此外,Waldo等[17]鉴定出在贡献于OFT期间,位于心包背壁(SpM)中表达Nkx2.5和Gata4以及Hnk1的祖细胞。同时,Kelly等[18]构建了受成纤维细胞生长因子10 (Fgf10)基因调控元件驱动的nlacZ转基因小鼠,发现在胚胎心脏的右心室和OFT以及相邻的PM和SpM中具有β-半乳糖苷酶活性;在Fgf10-nlacZ转基因小鼠中使用DiI标记法确定RV和OFT中的心肌细胞是从咽弓核心和SpM中添加。这3项研究揭示了位于PM和SpM中的另外一个祖细胞群,它们在原始心管形成以后贡献于心脏。目前认为心脏SHF是由位于PM和SpM中的祖细胞群共同组成[4,6],研究SHF细胞成为心脏发育领域的焦点。
SHF又可分为两个亚细胞群:前部-SHF (A-SHF)和后部SHF (P-SHF),A-SHF细胞通过动脉极贡献于OFT和右心室,P-SHF在静脉极贡献于心房及心房的分隔[4,6,10]。到目前为止,尚未确定背侧心包壁中A-SHF祖细胞和P-SHF祖细胞之间的界限。利用染料-标记和谱系-追踪实验表明,P-SHF中存在能够同时贡献于OFT和心房心肌细胞的祖细胞群[19,20]。持续增殖和分化延迟是SHF祖细胞区别于FHF祖细胞的两个限定特性,这两个特性使SHF祖细胞能够被逐渐添加到伸展的心管中,并受动态咽信号环境和PM转录程序的调节[21]。FGF10是由Kelly团队鉴定得到的鼠科动物SHF细胞的第一个分子标记物,随后的研究发现SHF祖细胞具有表达Fgf8、Isl1、Tbx1、Prdm1和Slx1等基因的特征[22,23,24,25,26]。
2 肌动蛋白细胞骨架
2.1 Actin细胞骨架及其解聚和加聚动态
Actin是真核生物中表达量最丰富的骨架蛋白之一[27]。动物细胞在不同环境中具有改变细胞形态的能力,这种能力主要是依赖actin细胞骨架系统[28]。细胞中actin具有两种存在形式,即游离的单体肌动蛋白(globular actin, G-actin)和丝状肌动蛋白(filamentous actin, F-actin)。G-actin自身能够聚合形成F-actin。F-actin是肌纤维细肌丝系统的核心组分,并被组织成各种不同的结构以参与行使不同的生物学过程,如细胞周质、应力纤维、丝状伪足和板状伪足等[27,28]。Actin细胞骨架是高度动态的骨架蛋白,在生理条件下F-actin能够持续在一端(正极端)聚合,在另一端(负极端)解聚。在细胞内,被组装成不同结构的F-actin不断进行动态且强烈地重组,使细胞适应环境。因此,actin细胞骨架的动态聚合和解聚在各种生理过程中发挥重要的调控作用[27],如细胞运动[29,30,31,32]、细胞分裂和胞质分裂[33,34]、细胞形状调节[35]及转录调控[36]等。在没有调节蛋白存在的生理离子条件下,actin自身能够经历自发的聚合、解聚和成核过程,但是其更新远比在生物体内缓慢的多。因此,为了适应体内F-actin快速更新的需求,需要大量的actin调节蛋白来调控actin成核、断裂、聚合和解聚等过程[37]。细胞内存在60多类G-actin和F-actin结合蛋白,协同调节actin的组装和拆卸[38,39]。例如,作为成核因子的Arp2/3复合体,具有促进分支状F-actin成核和交联F-actin的功能[40,41];WASP 家族(其中最有名的是WASP蛋白(Wiskott-Aldrich syndrome protein)和SCAR/WAVE (suppressor of cyclic AMP receptor/WASP-family verprolin-homologous protein))作为Arp2/3复合体的主要活化因子,能够结合并激活Arp2/3复合体,促进新的F-actin成核[42,43];Formins是另外一类actin成核因子,主要通过稳定actin二聚体,促进非分支F-actin成核和G-actin聚合[44,45];Profilin促进formins介导的actin聚合作用[41,44];ADF/cofilin是actin解聚的主要调控因子,通过促进F-actin切割和负极端G-actin解聚,增强actin的周转[46,47,48];WDR1/AIP1是ADF/cofilin的主要辅助因子,它能促进与ADF/cofilin结合的F-actin的解聚作用[49,50]。
2.2 Actin细胞骨架在细胞迁移、横纹肌中的调控作用
2.2.1 Actin细胞骨架与细胞迁移细胞运动作为动物细胞的重要特征,是与器官发育、宿主防御功能以及疾病发展密切相关的基本生命过程[51]。细胞迁移在许多生理和病理过程中发挥重要作用,如在后生动物胚胎发育过程中,原肠胚呈现出广泛的细胞迁移[52],且在器官和腺体形成期间,存在细胞群的协调迁移[53];在正常生理条件下,成纤维细胞、血管内皮细胞等细胞迁移是伤口愈合所必需的[54,55];免疫细胞通过迁移穿过血管和淋巴管并寻找入侵物质[56,57]等。
尽管在不同细胞、组织及研究系统中,细胞迁移会有不同的类型和机制,但它们可被有效的概念化为一个循环过程:首先细胞发生极化,随后在其前部形成突起,建立、稳定细胞-基质粘附作为细胞迁移的牵引位点,最后在其后部细胞脱离和缩回。细胞迁移过程是actin细胞骨架发生重大重塑的极端情况,而actin细胞骨架重塑是调控细胞迁移过程的关键和必需事件[29,58,59]。在细胞向前运动过程中,actin在细胞前端聚合以形成突起,同时这种聚合与后端板状伪足的解聚相匹配,为细胞随后的actin聚合反应提供所需要的再循环G-actin[28,37];另外,肌球蛋白马达拉动F-actin在迁移细胞尾部产生的收缩活性,提供细胞向前移动的动力[29,60]。F-actin作为肌球蛋白Ⅱ马达的支架,是肌动球蛋白收缩活动的先决条件[61]。目前,已有大量研究确定actin细胞骨架的动态特性是细胞运动所必需,使用actin解聚或稳定F-actin的药物处理运动细胞,将会终止细胞运动[62,63]。
2.2.2 横纹肌中的actin细胞骨架
脊椎动物的骨骼肌和心肌是典型的横纹肌,秀丽隐杆线虫(Caenorhabditis elegans)的体壁肌是斜横纹肌[64]。在横纹肌纤维中,基于actin的细肌丝、基于肌球蛋白的粗肌丝和其他相关蛋白被组织成收缩装置的最小重复单元—肌小节(sarcomere),以产生肌肉组织的收缩力[65,66]。肌小节是位于两个相邻Z盘之间的区域,粗肌丝位于肌小节的中部,并具有双极方向的肌球蛋白头部;被原肌球蛋白-肌钙蛋白复合物包裹的细肌丝在每个肌小节单元的末端以相反的方向取向;actin细肌丝的正极端被α-actinin蛋白锚定并交联在Z带上,并被加帽蛋白CapZ所加帽,而被加帽蛋白Tropomodulin加帽的细肌丝负极端则未被锚定在特定的结构上[67,68]。
虽然在迁移的成纤维细胞和肿瘤细胞中,F-actin形成的调控机制已经被广泛研究,但在发育的肌细胞中,肌小节细肌丝的形成及调控过程目前仍不清楚。针对肌纤维的组装,Sanger等提出了前肌原纤维模型[69,70]。在肌肉发育过程中,肌原纤维组装是actin细胞骨架发生的主要形态改变,此外G-actin和F-actin的比率也发生剧烈的变化[71]。在成熟的横纹肌肌小节中,整个actin细肌丝经历了不同速率的周转[72,73,74,75,76,77,78]。有趣的是,虽然成熟肌小节细肌丝经历actin解聚和加聚动态,但细肌丝却具有相似的长度[79,80]。横纹肌肌纤维的形成、成熟肌小节细肌丝的结构及动态等特点预示在肌小节组装和功能维持过程中,必然存在一套精确的调控系统来调节actin细肌丝的组装和分解(表1)。大量的研究已经表明,细肌丝正极端和负极端的加帽蛋白CapZ[81,82] 和Tropomodulin[76,83~86]能够调节肌小节细肌丝的组织和actin解聚和加聚动态;其他actin相关蛋白如Nubulin[87,88,89,90]、Tropomyosin[91,92]、ADF/cofilin[78,93,94]、WDR1[95,96]、Leiomodin[86,97~99]和FHOD3[100]等也参与了肌小节细肌丝的组织和actin动态的调控。作为actin解聚因子ADF/cofilin的辅助因子,WDR1能够促进与ADF/cofilin结合的F-actin的解聚。本课题组的研究表明,分别在胚胎期、幼年期和成年期心肌细胞中敲除Wdr1,均会导致F-actin堆积、肌纤维的组装和功能维持受损等表型[101,102]。
Table 1
表1
表1 横纹肌中重要的肌动蛋白动态调控因子
Table 1
| 调控因子 | 功能 | 参考文献 |
|---|---|---|
| CapZ | 调节细肌丝的组装及结构 | [81,82] |
| Tropomodulin | 抑制负极端actin dynamics,细肌丝组装及长度调节等 | [76,83~86] |
| Nubulin | 稳定F-actin,调节细肌丝长度及肌小节结构等 | [87~90] |
| Tropomyosin | 稳定F-actin,调控肌小节细肌丝组装 | [91,92] |
| ADF/Cofilin | 促进actin dynamics,调控细肌丝的正确组装 | [78,93,94] |
| WDR1 | 促进actin dynamics,调节细肌丝的正确组装 | [95,96,101,102] |
| Leiomodin | 促进F-actin聚合,调节细肌丝组装及长度 | [86,97~99] |
| FHOD3 | 促进actin聚合,调节肌小节组装 | [100] |
新窗口打开|下载CSV
在遗传和/或环境因素等影响的病理条件下,肌小节细肌丝会发生改变。先天性肌病是一种遗传性的肌肉病变,其特征是骨骼肌无力,并存在含有actin或其他肌纤维蛋白的棒状物或聚集体[103]。在杆状体肌病(nemaline myopathy)、肌动蛋白肌病和细胞核内杆状体肌病这3种主要的先天性肌病中,均涉及编码ACTIN及相关蛋白的基因突变[68,103],包括cofilin (CFL2)[104,105]、α-actin (ACTA1)[106,107,108,109,110]、α-和β-原肌球蛋白(TPM3和TPM2)[111,112]、肌钙蛋白T (TNNT1)[113,114,115]和nubulin (NEB)[116,117,118]等。
3 Actin细胞骨架在第二生心区祖细胞部署发育中的调控作用
近年来,一些调控SHF祖细胞部署发育的转录因子和信号通路相继被报道,这些转录因子及信号通路缺失或改变导致OFT缩短、心脏环化受损等SHF发育的缺陷,并伴随SHF细胞内actin细胞骨架的破坏。这些结果揭示了actin细胞骨架在SHF细胞发育特别是SHF祖细胞向OFT部署过程的重要性(表2)。Table 2
表2
表2 影响SHF祖细胞actin细胞骨架的调控因子及信号通路
Table 2
| 调控因子/信号通路 | 突变体SHF表型 | 突变体SHF细胞是否存在actin异常 | 参考文献 |
|---|---|---|---|
| Wnt5a-Dvl-PCP通路 | OFT缩短,心脏环化缺陷,OFT错误排列等 | 是 | [7,8,119] |
| TBX1 | OFT缩短,OFT环化缺陷 | 是 | [9] |
| Arid3b | OFT和流入道缩短 | 是 | [120] |
| WDR1 | 近端OFT和RV缩小,肌纤维组装受损 | 是 | [102] |
新窗口打开|下载CSV
3.1 Wnt5a-Dvl-PCP通路与SHF祖细胞actin细胞骨架
2005年,Kirby研究团队发现鸡(Gallus gallus)中与远端OFT相邻的SpM SHF细胞具有假复层柱状上皮细胞层的特点[121]。直到2012年,这种紧密结合、上皮样细胞层的SHF细胞形态才在小鼠SpM中被发现,而靠近尾部SpM处的SHF细胞具有板状伪足状突起的细胞形态。因此有研究者提出一种假说:位于尾部SpM中松散堆积的间充质样SHF祖细胞经历间充质细胞到上皮细胞的转化过程,以形成SpM中上皮样SHF细胞层,通过细胞插入(cell intercalation)的方式促进SHF祖细胞对OFT的贡献;此外,在PCP活性被抑制的突变体中,尾部SpM处SHF祖细胞中actin聚合和板状伪足的活性被破坏;该研究推测Wnt5a-PCP信号可能通过调节尾部SpM处SHF祖细胞内actin聚合作用促进SHF祖细胞的迁移部署,影响OFT延伸和心脏环化过程[8]。2014年,王建波研究团队的结果表明:位于小鼠SpM中的SHF祖细胞被组织成上皮样细胞层,在细胞层中的单个细胞显示出多边形形态,并且在细胞顶端皮质周围富集F-actin 的细胞被紧密挤在一起;SHF祖细胞在SpM某处经历间充质到上皮样细胞的转变,被组织形成上皮样细胞层,并以紧密结合、上皮样细胞层的形式而非单细胞的方式向OFT迁移[7]。该研究与Sinha等[8]在Wnt5a缺失突变体小鼠中的研究结果一致,即抑制Wnt5a导致SpM处SHF祖细胞表现出减少且紊乱的actin聚合、细胞的形态和细胞排列方向被破坏等表型,进一步揭示了SpM处SHF祖细胞中actin细胞骨架、细胞形态及排列方向在SHF祖细胞向OFT部署中的重要性。
2016年,王建波研究团队报道了利用Wnt5a突变体小鼠研究SpM中SHF祖细胞向OFT部署的另一项研究成果,进一步揭示Wnt5a-PCP信号通路在SpM SHF祖细胞向OFT部署过程中的时空调控作用[119]。在Wnt5a缺失的突变体小鼠中,通过Islet1- Cre驱动SHF细胞过表达Wnt5a,能够挽救SpM处SHF细胞中actin聚合、细胞极性的缺陷。在Islet1- Cre驱动SHF细胞过表达Wnt5a的突变体小鼠中,与远端OFT相邻的SpM处上皮样SHF细胞的细胞粘附连接被破坏,SHF细胞滞留在该处,形成突起而不向OFT部署。虽然该研究并未通过实验证明Wnt5a过表达影响SpM处SHF细胞粘附连接的具体机制,然而研究表明上皮细胞极性和细胞连接相互影响,两者的建立和维持需要actin细胞骨架的协同调控作用[35,122]。该研究进一步揭示actin细胞骨架及细胞排列和极性在SHF祖细胞部署中的必要作用。
3.2 TBX1与SHF祖细胞actin细胞骨架
2014年,Francou等[9]研究证明:位于SpM及远端OFT处的SHF细胞组成非经典顶端-基底极性的上皮,并具有顶部单纤毛、动态actin富集基底端的丝状伪足等特点;该研究还发现,除了调节SHF祖细胞的增殖和分化外,Tbx1还通过参与调节SHF细胞上皮细胞极性、SHF祖细胞基底端actin动态和丝状伪足活性,影响SHF祖细胞向OFT的部署。该研究推测,在心管伸展过程中,SHF祖细胞基底端动态actin重塑驱动的丝状伪足运动可能在维持SpM处SHF祖细胞状态所需的信号通路中发挥作用。3.3 Arid3b与SHF祖细胞actin细胞骨架
2014年,Uribe等[120]研究发现,通过基因捕获(gene trap)方法构建的Arid3b突变体胚胎具有OFT和流入道(inflow tract, IFT)长度缩短、房室垫发育受损等一系列心脏发育缺陷的表型;在E9.5的突变体胚胎中观察到Islet1阳性的SHF祖细胞在OFT区域积累,且SpM处SHF祖细胞中actin细胞骨架和细胞结构均发生改变;Dil标记、追踪实验证明在突变体胚胎中,SHF细胞向心管的贡献受损。该研究推测Arid3b可能通过actin细胞骨架调节SHF祖细胞向心管的运动,表明actin细胞骨架在SHF祖细胞迁移部署中的重要作用。3.4 WDR1与SHF祖细胞actin细胞骨架
本课题组最近的研究揭示actin解聚调控因子WDR1在哺乳动物SHF发育中的必要作用[102]。通过Mef2c-SHF-Cre构建SHF细胞特异性敲除Wdr1小鼠,该突变体小鼠从E11.5开始死亡,且在E10.5表现出近端OFT和RV缩小的表型;敲除Wdr1并不影响SHF细胞的数量以及SHF祖细胞由SpM向远端OFT的部署过程,但近端OFT和右心室细胞的空间排列和心肌细胞肌纤维的组装过程被严重破坏(图1)。我们推测:WDR1介导的actin解聚和加聚动态可能通过调节上皮样SHF祖细胞向心肌细胞分化过程中心肌细胞空间排列的重塑过程,进一步调控OFT和RV大小。关于Mef2c-SHF-Cre敲除Wdr1并不影响SHF祖细胞由SpM向远端OFT的部署过程,推测可能有两方面原因:首先,基因不完全删除导致WDR1的少量存留;其次,前人的研究表明SpM和远端OFT处的SHF细胞是伸展、极化的上皮细胞,且在这种上皮样细胞层中可能存在一种推动力以驱动SHF细胞向远端OFT部署[7,119],暗示SHF祖细胞向远端OFT部署过程中可能对actin解聚和加聚动态的需求较少。图1
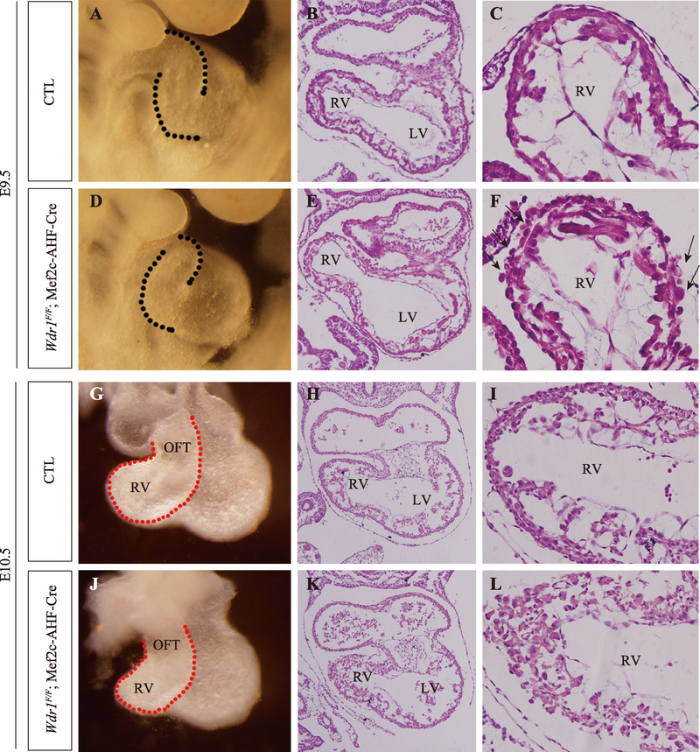 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1SHF祖细胞特异性敲除Wdr1小鼠的表型
E9.5 (A~F)、E10.5 (G~L)对照组(CTL)和SHF祖细胞敲除Wdr1组(Wdr1F/F; Mef2c-AHF-Cre)胚胎的显微和组织学分析。与对照组胚胎相比,Wdr1敲除组胚胎具有RV缩小、近端OFT和RV心肌细胞排列紊乱(黑箭头)的表型。
Fig. 1The phenotypes of SHF progenitors-specific Wdr1 deletion mice
4 结语与展望
SHF祖细胞整合、部署的精确、具体细胞机制尚不完全清楚,如是否通过主动细胞迁移、细胞凝聚及细胞张力、细胞插入还是有方向的细胞分裂等机制,仍需要进行深入的研究来阐明。然而,进一步研究SHF祖细胞部署的细胞生物学过程及生物力学等特性,需要开发高分辨率动态成像技术。SHF细胞具有非典型上皮样的顶端-基底细胞极性、细胞连接、排列和动态actin在基底端细胞膜富集等特点,这些研究揭示actin细胞骨架在SHF祖细胞部署、发育中发挥关键的调控作用。因此,利用actin细胞骨架作为切入点,通过分子细胞生物学手段构建SHF祖细胞标记、追踪体系,结合活细胞成像和高穿透性电子显微成像系统,将会为深入阐明、理解SHF祖细胞迁移、部署的细胞生物学特征以及生物力学提供新的研究思路和方向。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:18288184 [本文引用: 1]

Congenital heart disease is the leading cause of infant morbidity in the Western world, but only in the past ten years has its aetiology been understood. Recent studies have uncovered the genetic basis for some common forms of the disease and provide new insight into how the heart develops and how dysregulation of heart development leads to disease.
URLPMID:24821700Magsci [本文引用: 1]
Abstract Congenital heart defects affect at least 0.8% of newborn children and are a major cause of lethality prior to birth. Malformations of the arterial pole are particularly frequent. The myocardium at the base of the pulmonary trunk and aorta and the arterial tree associated with these great arteries are derived from splanchnic mesoderm of the second heart field (SHF), an important source of cardiac progenitor cells. These cells are controlled by a gene regulatory network that includes Fgf8, Fgf10 and Tbx1. Prdm1 encodes a transcriptional repressor that we show is also expressed in the SHF. In mouse embryos, mutation of Prdm1 affects branchial arch development and leads to persistent truncus arteriosus (PTA), indicative of neural crest dysfunction. Using conditional mutants, we show that this is not due to a direct function of Prdm1 in neural crest cells. Mutation of Prdm1 in the SHF does not result in PTA, but leads to arterial pole defects, characterized by mis-alignment or reduction of the aorta and pulmonary trunk, and abnormalities in the arterial tree, defects that are preceded by a reduction in outflow tract size and loss of caudal pharyngeal arch arteries. These defects are associated with a reduction in proliferation of progenitor cells in the SHF. We have investigated genetic interactions with Fgf8 and Tbx1, and show that on a Tbx1 heterozygote background, conditional Prdm1 mutants have more pronounced arterial pole defects, now including PTA. Our results identify PRDM1 as a potential modifier of phenotypic severity in TBX1 haploinsufficient DiGeorge syndrome patients. The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
[本文引用: 2]
[本文引用: 6]
[本文引用: 5]
URLPMID:2794420 [本文引用: 7]
Although de la Cruz and colleagues showed as early as 1977 that the outflow tract was added after the heart tube formed, the source of these secondarily added cells was not identified for nearly 25years. In 2001, three pivotal publications described a secondary or anterior heart field that contributed to the developing outflow tract. This review details the history of the heart field, the discovery and continuing elucidation of the secondarily adding myocardial cells, and how the different populations identified in 2001 are related to the more recent lineage tracing studies that defined the first and second myocardial heart fields/lineages. Much recent work has focused on secondary heart field progenitors that give rise to the myocardium and smooth muscle at the definitive arterial pole. These progenitors are the last to be added to the arterial pole and are particularly susceptible to abnormal development, leading to conotruncal malformations in children. The major signaling pathways (Wnt, BMP, FGF8, Notch, and Shh) that control various aspects of secondary heart field progenitor behavior are discussed.
URLPMID:4381755 [本文引用: 3]
Abstract Outflow tract (OFT) malformation accounts for 30% of human congenital heart defects and manifests frequently in TBX1 haplo-insufficiency associated DiGeorge (22q11.2 deletion) syndrome. OFT myocardium originates from second heart field (SHF) progenitors in the pharyngeal and splanchnic mesoderm (SpM), but how these progenitors are deployed to the OFT is unclear. We find that SHF progenitors in the SpM gradually gain epithelial character and are deployed to the OFT as a cohesive sheet. Wnt5a, a non-canonical Wnt, is expressed specifically in the caudal SpM and may regulate oriented cell intercalation to incorporate SHF progenitors into an epithelial-like sheet, thereby generating the pushing force to deploy SHF cells rostrally into the OFT. Using enhancer trap and Cre transgenes, our lineage tracing experiments show that in Wnt5a null mice, SHF progenitors are trapped in the SpM and fail to be deployed to the OFT efficiently, resulting in a reduction in the inferior OFT myocardial wall and its derivative, subpulmonary myocardium. Concomitantly, the superior OFT and subaortic myocardium are expanded. Finally, in chick embryos, blocking the Wnt5a function in the caudal SpM perturbs polarized elongation of SHF progenitors, and compromises their deployment to the OFT. Collectively, our results highlight a critical role for Wnt5a in deploying SHF progenitors from the SpM to the OFT. Given that Wnt5a is a putative transcriptional target of Tbx1, and the similar reduction of subpulmonary myocardium in Tbx1 mutant mice, our results suggest that perturbing Wnt5a-mediated SHF deployment may be an important pathogenic mechanism contributing to OFT malformations in DiGeorge syndrome. The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
URLPMID:22841628Magsci [本文引用: 3]
78 Dvl-mediated PCP signaling is required in the SHF lineage for OFT development. 78 Dvl, Vangl2 and Wnt5a play key roles in OFT lengthening during cardiac looping. 78 Wnt5a and Dvl2 co-express in SHF progenitors in the caudal SpM. 78 PCP mutant SpM show actin polymerization, filopodia formation and cell packing defects. 78 PCP-induced protrusive behavior in SpM cells may promote SHF deployment to the OFT.
URLPMID:253713661Magsci [本文引用: 2]
Abstract Elongation of the vertebrate heart occurs by progressive addition of second heart field (SHF) cardiac progenitor cells from pharyngeal mesoderm to the poles of the heart tube. The importance of these cells in the etiology of congenital heart defects has led to extensive research into the regulation of SHF deployment by signaling pathways and transcription factors. However, the basic cellular features of these progenitor cells, including epithelial polarity, cell shape and cell dynamics, remain poorly characterized. Here, using immunofluorescence, live imaging and embryo culture, we demonstrate that SHF cells constitute an atypical, apicobasally polarized epithelium in the dorsal pericardial wall, characterized by apical monocilia and dynamic actin-rich basal filopodia. We identify the 22q11.2 deletion syndrome gene Tbx1, required in the SHF for outflow tract development, as a regulator of the epithelial properties of SHF cells. Cell shape changes in mutant embryos include increased circularity, a reduced basolateral membrane domain and impaired filopodial activity, and are associated with elevated aPKC0209 levels. Activation of aPKC0209 in embryo culture similarly impairs filopodia activity and phenocopies proliferative defects and ectopic differentiation observed in the SHF of Tbx1 null embryos. Our results reveal that epithelial and progenitor cell status are coupled in the SHF, identifying control of cell shape as a regulatory step in heart tube elongation and outflow tract morphogenesis. 0008 2014. Published by The Company of Biologists Ltd.
[本文引用: 2]
[本文引用: 1]
URLPMID:3006184 [本文引用: 1]
Conotruncal and ventricular septal congenital heart anomalies result from defects in formation and division of the embryonic outflow tract. Cardiac remodeling during outflow tract and ventricular septation converts the tubular embryonic heart into a parallel circulatory system with an independent left ventricular outlet and right ventricular inlet. Tbx3 encodes a T-box–containing transcription factor expressed in the developing conduction system of the heart. Mutations in TBX3 cause ulnar–mammary syndrome. Here we show that mice lacking Tbx3 develop severe outflow tract defects, including connection of both the aorta and pulmonary trunk with the right ventricle, in addition to aortic arch artery anomalies and abnormal communication between the right atrium and left ventricle. Alignment defects are preceded by a delay in caudal displacement of the arterial pole of the heart during aortic arch artery formation. Embryonic anterior–posterior patterning and cardiac chamber development are unaffected in Tbx3 mutant embryos. However, the contribution of second heart field derived progenitor cells to the arterial pole of the heart is impaired. Tbx3 is expressed in pharyngeal epithelia and neural crest cells in the pharyngeal region, suggesting an indirect role in second heart field deployment. Loss of Tbx3 affects multiple signaling pathways regulating second heart field proliferation and outflow tract morphogenesis, including fibroblast growth factor signaling, leading to a failure of normal heart tube extension and consequent atrioventricular and ventriculoarterial alignment defects.
URLPMID:29301846 [本文引用: 1]
Abstract The vertebrate heart tube forms from epithelial progenitor cells in the early embryo and subsequently elongates by progressive addition of second heart field (SHF) progenitor cells from adjacent splanchnic mesoderm. Failure to maximally elongate the heart results in a spectrum of morphological defects affecting the cardiac poles, including outflow tract alignment and atrioventricular septal defects, among the most common congenital birth anomalies. SHF cells constitute an atypical apicobasally polarized epithelium with dynamic basal filopodia, located in the dorsal wall of the pericardial cavity. Recent studies have highlighted the importance of epithelial architecture and cell adhesion in the SHF, particularly for signaling events that control the progenitor cell niche during heart tube elongation. The 22q11.2 deletion syndrome gene Tbx1 regulates progenitor cell status through modulating cell shape and filopodial activity and is required for SHF contributions to both cardiac poles. Noncanonical Wnt signaling and planar cell polarity pathway genes control epithelial polarity in the dorsal pericardial wall, as progenitor cells differentiate in a transition zone at the arterial pole. Defects in these pathways lead to outflow tract shortening. Moreover, new biomechanical models of heart tube elongation have been proposed based on analysis of tissue-wide forces driving epithelial morphogenesis in the SHF, including regional cell intercalation, cell cohesion, and epithelial tension. Regulation of the epithelial properties of SHF cells is thus emerging as a key step during heart tube elongation, adding a new facet to our understanding of the mechanisms underlying both heart morphogenesis and congenital heart defects.
URLPMID:885781 [本文引用: 2]
Abstract The development of the truncus and the conus was studied in the chick embryo by in vivo labelling techniques. The earliest labels were placed at the stage of fusion of the myocardial troughs (stage 9-) and they were traced until the mature heart stage (stage 35). Microdissections and light microscopic studies were also carried out. The results are discussed in relation to the human heart. Our experiments permit the following conclusions: (1) At stage 9- fusion of the myocardial troughs takes place at the level of the primordium of the trabeculated portion of the right ventricle, when neither the conus nor the truncus are present. (2) At stage 12 (loop stage) there appears the caudal portion of the conus, which constitutes the cephalic end of the cardiac tube. (3) The truncus appears between stages 13 and 22. (4) At stage 22 angular junction between the conus and the truncus is the area where the semilunar valve cusps of the great arteries will develop and that, at this same stage, the junction between the conus and the trabeculated portion of the right ventricle seen from the right surface corresponds to the inferior edge of the crista supraventricularis. (5) It was confirmed that the pulmonary semilunar valve cusps originate from the walls of the truncus. (6) The development of the conus and truncus are similar in chick and man. (7) Histologically, in the chick, the wall of the truncus and the conus contain cardiac muscle as late as stage 28, but from then on the walls of the truncus are transformed into connective tissue and plain muscle.
URLPMID:4684039 [本文引用: 1]
The origin and differentiation of heart muscle and epicardial cells was studied in 8- to 11-day mouse embryos. The truncoventricular and atrial portions of the primitive tubular heart, as well as the dorsolateral wall of the sinus venosus, were seen to originate from the epithelial layer of the splanchnic mesoderm. The transformation of mesenchymal cells into heart muscle is marked by the appearance of thick myosin-type filaments, although thin filamentous material, attached in part to the intercellular junctions, was observed in these cells before the appearance of myosin filaments. Evidence was obtained suggesting that the primitive terminal bars of the epithelial cells develop into the embryonic intercalated disks. The ventrocaudal wall of the sinus venosus differentiates by a progressive condensation of the loose mesenchymal tissue of the septum transversum. Some indications were obtained suggesting that a few cells of the mesenchyme which invade the cardiac jelly may also transform into muscle cells in the truncus arteriosus. After the establishment of the ventricular loop, where the development of the muscle cells was found to be the most advanced, transformation of the mesenchyme proceeds not only at the venosus end but also at the arterial end of the heart. The epicardial cells migrate to the heart surface from the septum transversum. When the epicardial investment has been established on the total ventricular surface, the blood vessels begin to grow craniocaudally into the subepicardial layer.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:25190705Magsci [本文引用: 1]
Rationale: Cardiac progenitor cells from the second heart field (SHF) contribute to rapid growth of the embryonic heart, giving rise to right ventricular and outflow tract (OFT) myocardium at the arterial pole of the heart, and atrial myocardium at the venous pole. Recent clonal analysis and cell-tracing experiments indicate that a common progenitor pool in the posterior region of the SHF gives rise to both OFT and atrial myocytes. The mechanisms regulating deployment of this progenitor pool remain unknown. Objective: To evaluate the role of TBX1, the major gene implicated in congenital heart defects in 22q11.2 deletion syndrome patients, in posterior SHF development. Methods and Results: Using transcriptome analysis, genetic tracing, and fluorescent dye-labeling experiments, we show that Tbx1-dependent OFT myocardium originates in Hox-expressing cells in the posterior SHF. In Tbx1 null embryos, OFT progenitor cells fail to segregate from this progenitor cell pool, leading to failure to expand the dorsal pericardial wall and altered positioning of the cardiac poles. Unexpectedly, addition of SHF cells to the venous pole of the heart is also impaired, resulting in abnormal development of the dorsal mesenchymal protrusion, and partially penetrant atrioventricular septal defects, including ostium primum defects. Conclusions: Tbx1 is required for inflow as well as OFT morphogenesis by regulating the segregation and deployment of progenitor cells in the posterior SHF. Our results provide new insights into the pathogenesis of congenital heart defects and 22q11.2 deletion syndrome phenotypes.
URLPMID:22955731Magsci [本文引用: 1]
The second heart field (SHF) contains progenitors of all heart chambers, excluding the left ventricle. The SHF is patterned, and the anterior region is known to be destined to form the outflow tract and right ventricle.The aim of this study was to map the fate of the posterior SHF (pSHF).We examined the contribution of pSHF cells, labeled by lipophilic dye at the 4- to 6-somite stage, to regions of the heart at 20 to 25 somites, using mouse embryo culture. Cells more cranial in the pSHF contribute to the atrioventricular canal (AVC) and atria, whereas those more caudal generate the sinus venosus, but there is intermixing of fate throughout the pSHF. Caudal pSHF contributes symmetrically to the sinus venosus, but the fate of cranial pSHF is left/right asymmetrical. Left pSHF moves to dorsal left atrium and superior AVC, whereas right pSHF contributes to right atrium, ventral left atrium, and inferior AVC. Retrospective clonal analysis shows the relationships between AVC and atria to be clonal and that right and left progenitors diverge before first and second heart lineage separation. Cranial pSHF cells also contribute to the outflow tract: proximal and distal at 4 somites, and distal only at 6 somites. All outflow tract-destined cells are intermingled with those that will contribute to inflow and AVC.These observations show asymmetric fate of the pSHF, resulting in unexpected left/right contributions to both poles of the heart and can be integrated into a model of the morphogenetic movement of cells during cardiac looping.
[本文引用: 1]
URLPMID:14667410 [本文引用: 1]
Hearts of mice lacking Isl1, a LIM homeodomain transcription factor, are completely missing the outflow tract, right ventricle, and much of the atria. isl1 expression and lineage tracing of isl1-expressing progenitors demonstrate that Isl1 is a marker for a distinct population of undifferentiated cardiac progenitors that give rise to the cardiac segments missing in isl1 mutants. Isl1 function is required for these progenitors to contribute to the heart. In isl1 mutants, isl1-expressing progenitors are progressively reduced in number, and FGF and BMP growth factors are downregulated. Our studies define two sets of cardiogenic precursors, one of which expresses and requires Isl1 and the other of which does not. Our results have implications for the development of specific cardiac lineages, left-right asymmetry, cardiac evolution, and isolation of cardiac progenitor cells.
URLPMID:19745164 [本文引用: 1]
RATIONALE: TBX1 encodes a T-box transcription factor implicated in DiGeorge syndrome, which affects the development of many organs, including the heart. Loss of Tbx1 results into hypoplasia of heart regions derived from the second heart field, a population of cardiac progenitors cells (CPCs). Thus, we hypothesized that Tbx1 is an important player in the biology of CPCs. OBJECTIVE: We asked whether Tbx1 is expressed in multipotent CPCs and, if so, what role it may play in them. METHODS AND RESULTS: We used clonal analysis of Tbx1-expressing cells and loss and gain of function models, in vivo and in vitro, to define the role of Tbx1 in CPCs. We found that Tbx1 is expressed in multipotent heart progenitors that, in clonal assays, can give rise to 3 heart lineages expressing endothelial, smooth muscle and cardiomyocyte markers. In multipotent cells, Tbx1 stimulates proliferation, explaining why Tbx1(-/-) embryos have reduced proliferation in the second heart field. In this population, Tbx1 is expressed while cells are undifferentiated and it disappears with the onset of muscle markers. Loss of Tbx1 results in premature differentiation, whereas gain results in reduced differentiation in vivo. We found that Tbx1 binds serum response factor, a master regulator of muscle differentiation, and negatively regulates its level. CONCLUSIONS: The Tbx1 protein marks CPCs, supports their proliferation, and inhibits their differentiation. We propose that Tbx1 is a key regulator of CPC homeostasis as it modulates positively their proliferation and negatively their differentiation.
URLPMID:21364285Magsci [本文引用: 1]
Shared molecular programs govern the formation of heart and head during mammalian embryogenesis. Development of both structures is disrupted in human chromosomal microdeletion of 22q11.2 (del22q11), which causes DiGeorge syndrome (DGS) and velo-cardio-facial syndrome (VCFS). Here, we have identified a genetic pathway involving the Six1/Eya1 transcription complex that regulates cardiovascular and craniofacial development. We demonstrate that murine mutation of both Six1 and Eya1 recapitulated most features of human del22q11 syndromes, including craniofacial, cardiac outflow tract, and aortic arch malformations. The mutant phenotypes were attributable in part to a reduction of fibroblast growth factor 8 (Fgf8), which was shown to be a direct downstream effector of Six1 and Eya1. Furthermore, we showed that Six1 and Eya1 genetically interacted with Fgf8 and the critical del22q11 gene T-box transcription factor 1 (Tbx1) in mice. Together, these findings reveal a Tbx1-Six1/Eya1-Fgf8 genetic pathway that is crucial for mammalian cardiocraniofacial morphogenesis and provide insights into the pathogenesis of human del22q11 syndromes.
URLPMID:16720880 [本文引用: 1]
In the mouse embryo, the splanchnic mesodermal cells of the anterior heart field (AHF) migrate from the pharynx to contribute to the early myocardium of the outflow tract (OT) and right ventricle (RV). Recent studies have attempted to distinguish the AHF from other precardiac populations, and to determine the genetic and molecular mechanisms that regulate its development. Here, we have used an Fgf8lacZ allele to demonstrate that Fgf8 is expressed within the developing AHF. In addition, we use both a hypomorphic Fgf8 allele (Fgf8neo) and Cre-mediated gene ablation to show that Fgf8 is essential for the survival and proliferation of the AHF. Nkx2.5Cre is expressed in the AHF, primary heart tube and pharyngeal endoderm, while TnT-Cre is expressed only within the specified heart tube myocardium. Deletion of Fgf8 by Nkx2.5Cre results in a significant loss of the Nkx2.5Cre lineage and severe OT and RV truncations by E9.5, while the remaining heart chambers (left ventricle and atria) are grossly normal. These defects result from significant decreases in cell proliferation and aberrant cell death in both the pharyngeal endoderm and splanchnic mesoderm. By contrast, ablation of Fgf8 in the TnT-Cre domain does not result in OT or RV defects, providing strong evidence that Fgf8 expression is crucial in the pharyngeal endoderm and/or overlying splanchnic mesoderm of the AHF at a stage prior to heart tube elongation. Analysis of downstream signaling components, such as phosphorylated-Erk and Pea3, identifies the AHF splanchnic mesoderm itself as a target for Fgf8 signaling.
URLPMID:18039967 [本文引用: 1]
The zinc-finger transcriptional repressor Blimp1 (Prdm1) controls geneexpression patterns during differentiation of B lymphocytes and regulatesepigenetic changes required for specification of primordial germ cells.Blimp1 is dynamically expressed at diverse tissue sites in thedeveloping mouse embryo, but its functional role remains unknown becauseBlimp1 mutant embryos arrest at E10.5 due to placental insufficiency.To explore Blimp1 activities at later stages in the embryo proper,here we used a conditional inactivation strategy. A Blimp1-Cretransgenic strain was also exploited to generate a fate map ofBlimp1-expressing cells. Blimp1 plays essential roles inmultipotent progenitor cell populations in the posterior forelimb, caudalpharyngeal arches, secondary heart field and sensory vibrissae and maintainskey signalling centres at these diverse tissues sites. Interestingly, embryoscarrying a hypomorphic Blimp1gfp reporter allele surviveto late gestation and exhibit similar, but less severe developmentalabnormalities, whereas transheterozygous Blimp1gfp/-embryos with further reduced expression levels, display exacerbated defects.Collectively, the present experiments demonstrate that Blimp1requirements in diverse cell types are exquisitely dose dependent.
URLPMID:17909522 [本文引用: 3]
Actin filaments are major components of at least 15 distinct structures in metazoan cells. These filaments assemble from a common pool of actin monomers, but do so at different times and places, and in response to different stimuli. All of these structures require actin-filament assembly factors. To date, many assembly factors have been identified, including Arp2/3 complex, multiple formin isoforms and spire. Now, a major task is to figure out which factors assemble which actin-based structures. Here, we focus on structures at the plasma membrane, including both sheet-like protrusive structures (such as lamellipodia and ruffles) and finger-like protrusions (such as filopodia and microvilli). Insights gained from studies of adherens junctions and the immunological synapse are also considered.
Magsci [本文引用: 3]
Tight coupling between biochemical and mechanical properties of the actin cytoskeleton drives a large range of cellular processes including polarity establishment, morphogenesis, and motility. This is possible because actin filaments are semi-flexible polymers that, in conjunction with the molecular motor myosin, can act as biological active springs or "dashpots" (in laymen's terms, shock absorbers or fluidizers) able to exert or resist against force in a cellular environment. To modulate their mechanical properties, actin filaments can organize into a variety of architectures generating a diversity of cellular organizations including branched or crosslinked networks in the lamellipodium, parallel bundles in filopodia, and antiparallel structures in contractile fibers. In this review we describe the feedback loop between biochemical and mechanical properties of actin organization at the molecular level in vitro, then we integrate this knowledge into our current understanding of cellular actin organization and its physiological roles.
URLPMID:29295889 [本文引用: 3]
Abstract SUMMARYThe actin cytoskeleton-a collection of actin filaments with their accessory and regulatory proteins-is the primary force-generating machinery in the cell. It can produce pushing (protrusive) forces through coordinated polymerization of multiple actin filaments or pulling (contractile) forces through sliding actin filaments along bipolar filaments of myosin II. Both force types are particularly important for whole-cell migration, but they also define and change the cell shape and mechanical properties of the cell surface, drive the intracellular motility and morphogenesis of membrane organelles, and allow cells to form adhesions with each other and with the extracellular matrix.
URLPMID:26827283 [本文引用: 1]
Eukaryotic cell movement is characterized by very diverse migration modes. Recent studies show that cells can adapt to environmental cues, such as adhesion and geometric confinement, thereby readily switching their mode of migration. Among this diversity of motile behavior, actin flows have emerged as a highly conserved feature of both mesenchymal and amoeboid migration, and have also been identified as key regulators of cell polarity. This suggests that the various observed migration modes are continuous variations of elementary locomotion mechanisms, based on a very robust physical property of the actin/myosin system - its ability to sustain flows at the cell scale. This central role of actin/myosin flows is shown to affect the large scale properties of cell trajectories.
URLPMID:28283221 [本文引用: 1]
Actin filaments and associated proteins undergo wave-like movement in various types of cells. The prevailing models to explain the wave-like dynamics of actin filaments involve an activator-nhibitor mechanism, in which a combination of autocatalytic positive feedback and slower negative feedback confers excitability on cells for generation and propagation of actin waves. Axonal actin waves migrate through directional assembly and disassembly of membrane-anchored actin filaments and serve as a new type of intracellular protein transport system that supplies actin and associated proteins toward the growth cone. Actin waves provide a potential framework for understanding how actin and associated proteins are spatiotemporally organized into the molecular machinery for cell protrusion, polarization, and migration.
URLPMID:22985541Magsci [本文引用: 1]
Much of current understanding of cell motility arose from studying steady treadmilling of actin arrays. Recently, there have been a growing number of observations of a more complex, non-steady, actin behavior, including self-organized waves. It is becoming clear that these waves result from activation and inhibition feedbacks in actin dynamics acting on different scales, but the exact molecular nature of these feedbacks and the respective roles of biomechanics and biochemistry are still unclear. Here, we review recent advances achieved in experimental and theoretical studies of actin waves and discuss mechanisms and physiological significance of wavy protrusions.
URLPMID:23764118Magsci [本文引用: 1]
In contrast to symmetric division in mitosis, mammalian oocyte maturation is characterized by asymmetric cell division that produces a large egg and a small polar body. The asymmetry results from oocyte polarization, which includes spindle positioning, migration, and cortical reorganization, and this process is critical for fertilization and the retention of maternal components for early embryo development. Although actin dynamics are involved in this process, the molecular mechanism underlying this remained unclear until the use of confocal microscopy and live cell imaging became widespread in recent years. Information obtained through a PubMed database search of all articles published in English between 2000 and 2012 that included the phrases “oocyte, actin, spindle migration,” “oocyte, actin, polar body,” or “oocyte, actin, asymmetric division” was reviewed. The actin nucleation factor actin-related protein 2/3 complex and its nucleation-promoting factors, formins and Spire, and regulators such as small GTPases, partitioning-defective/protein kinase C, Fyn, microRNAs, cis-Golgi apparatus components, myosin/myosin light-chain kinase, spindle stability regulators, and spindle assembly checkpoint regulators, play critical roles in asymmetric cell division in oocytes. This review summarizes recent findings on these actin-related regulators in mammalian oocyte asymmetric division and outlines a complete signaling pathway.
URLPMID:25208553 [本文引用: 1]
Abstract Maternal effect genes play critical roles in early embryogenesis of model organisms where they have been intensively investigated. However, their molecular function in mammals remains largely unknown. Recently, we identified a subcortical maternal complex (SCMC) that contains four proteins encoded by maternal effect genes (Mater, Filia, Floped and Tle6). Here we report that TLE6, similar to FLOPED and MATER, stabilizes the SCMC and is necessary for cleavage beyond the two-cell stage of development. We document that the SCMC is required for formation of the cytoplasmic F-actin meshwork that controls the central position of the spindle and ensures symmetric division of mouse zygotes. We further demonstrate that the SCMC controls formation of the actin cytoskeleton specifically via Cofilin, a key regulator of F-actin assembly. Our results provide molecular insight into the physiological function of TLE6, its interaction with the SCMC and their roles in the symmetric division of the zygote in early mouse development.
URLPMID:24859003 [本文引用: 2]
Abstract Mechanical stress is increasingly being shown to be a potent modulator of cell-cell junctional morphologies in developmental and homeostatic processes. Intercellular force sensing is thus expected to be an important regulator of cell signalling and tissue integrity. In particular, the interplay between myosin contractility, actin dynamics and E-cadherin recruitment largely remains to be uncovered. We devised a suspended cell doublet assay to quantitatively assess the correlation between myosin II activity and local E-cadherin recruitment. The single junction of the doublet exhibited a stereotypical morphology, with E-cadherin accumulating into clusters of varied concentrations at the rim of the circular contact. This local recruitment into clusters derived from the sequestration of E-cadherin through a myosin-II-driven modulation of actin turnover. We exemplify how the regulation of actin dynamics provides a mechanism for the mechanosensitive response of cell contacts.
URLPMID:20414257 [本文引用: 1]
Abstract Numerous physiological and pathological stimuli promote the rearrangement of the actin cytoskeleton, thereby modulating cellular motile functions. Although it seems intuitively obvious that cell motility requires coordinated protein biosynthesis, until recently the linkage between cytoskeletal actin dynamics and correlated gene activities remained unknown. This knowledge gap was filled in part by the discovery that globular actin polymerization liberates myocardin-related transcription factor (MRTF) cofactors, thereby inducing the nuclear transcription factor serum response factor (SRF) to modulate the expression of genes encoding structural and regulatory effectors of actin dynamics. This insight stimulated research to better understand the actin-MRTF-SRF circuit and to identify alternative mechanisms that link cytoskeletal dynamics and genome activity.
URLPMID:12600310 [本文引用: 2]
Motile cells extend a leading edge by assembling a branched network of actin filaments that produces physical force as the polymers grow beneath the plasma membrane. A core set of proteins including actin, Arp2/3 complex, profilin, capping protein, and ADF/cofilin can reconstitute the process in vitro, and mathematical models of the constituent reactions predict the rate of motion. Signaling pathways converging on WASp/Scar proteins regulate the activity of Arp2/3 complex, which mediates the initiation of new filaments as branches on preexisting filaments. After a brief spurt of growth, capping protein terminates the elongation of the filaments. After filaments have aged by hydrolysis of their bound ATP and dissociation of the 纬 phosphate, ADF/cofilin proteins promote debranching and depolymerization. Profilin catalyzes the exchange of ADP for ATP, refilling the pool of ATP-actin monomers bound to profilin, ready for elongation.
URLPMID:10940259 [本文引用: 1]
Abstract We review how motile cells regulate actin filament assembly at their leading edge. Activation of cell surface receptors generates signals (including activated Rho family GTPases) that converge on integrating proteins of the WASp family (WASp, N-WASP, and Scar/WAVE). WASP family proteins stimulate Arp2/3 complex to nucleate actin filaments, which grow at a fixed 70 degrees angle from the side of pre-existing actin filaments. These filaments push the membrane forward as they grow at their barbed ends. Arp2/3 complex is incorporated into the network, and new filaments are capped rapidly, so that activated Arp2/3 complex must be supplied continuously to keep the network growing. Hydrolysis of ATP bound to polymerized actin followed by phosphate dissociation marks older filaments for depolymerization by ADF/cofilins. Profilin catalyzes exchange of ADP for ATP, recycling actin back to a pool of unpolymerized monomers bound to profilin and thymosin-beta 4 that is poised for rapid elongation of new barbed ends.
URLPMID:15246432Magsci [本文引用: 1]
The actin cytoskeleton is a vital component of several key cellular and developmental processes in eukaryotes. Many proteins that interact with filamentous and/or monomeric actin regulate the structure and dynamics of the actin cytoskeleton. Actin-filament-binding proteins control the nucleation, assembly, disassembly and crosslinking of actin filaments, whereas actin-monomer-binding proteins regulate the size, localization and dynamics of the large pool of unpolymerized actin in cells. In this article, we focus on recent advances in understanding how the six evolutionarily conserved actin-monomer-binding proteins – profilin, ADF/cofilin, twinfilin, Srv2/CAP, WASP/WAVE and verprolin/WIP – interact with actin monomers and regulate their incorporation into filament ends. We also present a model of how, together, these ubiquitous actin-monomer-binding proteins contribute to cytoskeletal dynamics and actin-dependent cellular processes.
URLPMID:12504682 [本文引用: 1]
The Arp2/3 complex, a 220 kDa macromolecular assembly comprising two actin-related proteins and five other subunits, plays a key role in cellular motility by initiating the polymerization of new actin filaments. Crystal and cryo-electron microscopic structures of the Arp2/3 complex and branch junctions have clarified extensive biochemical data on the mechanism of this process.
URLPMID:19758556 [本文引用: 2]
Cell migration is an essential feature of eukaryotic life, required for processes ranging from feeding and phagoctyosis to development, healing, and immunity. Migration requires the actin cytoskeleton, specifically the localized polymerization of actin filaments underneath the plasma membrane. Here we summarize recent developments in actin biology that particularly affect structures at the leading edge of the cell, including the structure of actin branches, the multiple pathways that lead to cytoskeleton assembly and disassembly, and the role of blebs. Future progress depends on connecting these processes and components to the dynamic behavior of the whole cell in three dimensions.
URLPMID:14561714 [本文引用: 1]
Rapid reorganization of the actin cytoskeleton underlies morphological changes and motility of cells. WASP family proteins have received a great deal of attention as the signal-regulated molecular switches that initiate actin polymerization. The first member, WASP, was identified as the product of a gene of which dysfunction causes the human hereditary disease Wiskott-Aldrich syndrome. There are now five members in this protein family, namely WASP, N-WASP, WAVE/Scar1, 2, and 3. WASP and N-WASP have functional and physical associations with Cdc42, a Rho family small GTPase involved in filopodium formation. In contrast, there is evidence that links the WAVE/Scar proteins with another Rho family protein, Rac, which is a regulator of membrane ruffling. All WASP family members have a VCA domain at the C-terminus through which Arp2/3 complex is activated to nucleate actin polymerization. Analyses of model organisms have just begun to reveal unexpected functions of WASP family proteins in multicellular organisms.
Magsci [本文引用: 1]
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">Signal-dependent regulation of actin dynamics is essential for a variety of cellular processes, including formation of the membrane protrusions required for cell locomotion. Wiskott–Aldrich syndrome protein (WASP), neural (N)-WASP and WASP family verprolin-homologous (WAVE, also named Scar) proteins are thought to play a role in these processes by relaying activation signals from small GTPases, such as Cdc42 and Rac, to the actin-nucleating complex Arp2/3. Although much biochemical and structural biological work has defined the paradigms through which WASP and N-WASP regulation is achieved, only recently have the mechanisms that control WAVE proteins begun to be clarified. WAVE proteins assemble into macromolecular complexes, which are essential for the regulation of WAVE nucleation-promoting activity, dynamic localization and stability. In this article, we discuss recent studies that highlight novel modalities through which WAVE proteins are regulated and, in turn, mediate the site-directed actin polymerization required for membrane protrusion, thus enabling cell motility.</p>
URLPMID:12134165 [本文引用: 2]
Abstract Formins are required for cell polarization and cytokinesis, but do not have a defined biochemical activity. In Saccharomyces cerevisiae, formins and the actin-monomer-binding protein profilin are specifically required to assemble linear actin structures called 'actin cables'. These structures seem to be assembled independently of the Arp2/3 complex, the only well characterized cellular mediator of actin nucleation. Here, an activated yeast formin was purified and found to promote the nucleation of actin filaments in vitro. Formin-dependent actin nucleation was stimulated by profilin. Thus, formin and profilin mediate actin nucleation by an Arp2/3-independent mechanism. These findings suggest that distinct actin nucleation mechanisms may underlie the assembly of different actin cytoskeletal structures.
[本文引用: 1]
URLPMID:10611961 [本文引用: 1]
Ubiquitous among eukaryotes, the ADF/cofilins are essential proteins responsible for the high turnover rates of actin filaments in vivo. In vertebrates, ADF and cofilin are products of different genes. Both bind to F-actin cooperatively and induce a twist in the actin filament that results in the loss of the phalloidin-binding site. This conformational change may be responsible for the enhancement of the off rate of subunits at the minus end of ADF/cofilin-decorated filaments and for the weak filament-severing activity. Binding of ADF/cofilin is competitive with tropomyosin. Other regulatory mechanisms in animal cells include binding of phosphoinositides, phosphorylation by LIM kinases on a single serine, and changes in pH. Although vertebrate ADF/cofilins contain a nuclear localization sequence, they are usually concentrated in regions containing dynamic actin pools, such as the leading edge of migrating cells and neuronal growth cones. ADF/cofilins are essential for cytokinesis, phagocytosis, fluid phase endocytosis, and other cellular processes dependent upon actin dynamics.
URLPMID:17018289 [本文引用: 1]
ADF/cofilins are key regulators of actin dynamics during cellular motility, yet their precise role and mechanism of action are shrouded in ambiguity. Direct observation of actin filaments by evanescent wave microscopy showed that cofilins from fission yeast and human do not increase the rate that pointed ends of actin filaments shorten beyond the rate for ADP-actin subunits, but both cofilins inhibit elongation and subunit dissociation at barbed ends. Direct observation also showed that cofilins from fission yeast, Acanthamoeba, and human sever actin filaments optimally at low-cofilin binding densities well below their K ds, but not at high binding densities. High concentrations of cofilin nucleate actin assembly. Thus, the action of cofilins in cells will depend on the local concentration of active cofilins: low concentrations favor severing, whereas high concentrations favor nucleation. These results establish a clear paradigm for actin turnover by coflin in cells.
URLPMID:2849908Magsci [本文引用: 1]
Recent findings have significantly expanded our understanding of the regulation of actin-depolymerizing factor (ADF)/cofilin proteins and the profound multifaceted impact that these well-established regulators of actin dynamics have on cell biology. In this review we discuss new aspects of previously documented regulation, such as phosphorylation, but also cover novel recently established modes of regulation and functions of ADF (also known as destrin)/cofilin. We now understand that their activity responds to a vast array of inputs far greater than previously appreciated and that these proteins not only feed back to the crucially important dynamics of actin, but also to apoptosis cascades, phospholipid metabolism, and gene expression. We argue that this ability to respond to physiological changes by modulating those same changes makes the ADF/cofilin protein family a homeostatic regulator or ‘functional node’ in cell biology.
URLPMID:14621980 [本文引用: 1]
Actin depolymerizing factor (ADF)/cofilin enhances turnover of actin filaments by severing and depolymerizing filaments. A number of proteins functionally interact with ADF/cofilin to modulate the dynamics of actin filaments. Actin-interacting protein 1 (AIP1) has emerged as a conserved WD-repeat protein that specifically enhances ADF/cofilin-induced actin dynamics. Interaction of AIP1 with actin was originally characterized by a yeast two-hybrid system. However, biochemical studies revealed its unique activity on ADF/cofilin-bound actin filaments. AIP1 alone has negligible effects on actin filament dynamics, whereas in the presence of ADF/cofilin, AIP1 enhances filament fragmentation by capping ends of severed filaments. Studies in model organisms demonstrated that AIP1 genetically interacts with ADF/cofilin and participates in several actin-dependent cellular events. The crystal structure of AIP1 revealed its unique structure with two seven-bladed beta-propeller domains. Thus, AIP1 is a new class of actin regulatory proteins that selectively enhances ADF/cofilin-dependent actin filament dynamics.
URLPMID:25448002Magsci [本文引用: 1]
Nadkarni and Brieher show that Aip1 is a cofilin-dependent actin disassembly factor that destabilizes filaments even in the presence of stabilizing amounts of cofilin. Filaments depolymerize by increased severing and depolymerization from ends. They also show that Aip1 is not a high-affinity barbed-end-capping protein as was previously described.
URLPMID:21807492Magsci [本文引用: 1]
Highlights? Essential factors driving distinct actin structures in migration are defined. ? Functions and regulation of lamellipodium versus lamella are compared. ? Ultrastructures of lamellipodia and focal adhesions are discussed. ? Role and crosstalk of different Rho-GTPases are summarized. ? The signalling network driving contractility is uncovered.
[本文引用: 1]
[本文引用: 1]
URLPMID:27085790 [本文引用: 1]
A skin wound requires several cell lineages to exhibit considerable plasticity as they migrate towards and over the site of damage to contribute to repair. The keratinocytes that re-epithelialize the tissue, the dermal fibroblasts and potentially other mesenchymal stem cell populations that repopulate damaged connective tissue, the immune cells that counter infections, and endothelial cells that re-establish blood supply and facilitate the immune response – all of these cells are ‘dynamic’ in that they are activated by immediate wound cues, they reprogram to adopt cell behaviours essential for repair including migration, and finally they must resolve. In adult tissues, repair is unique in its requirement for dramatic cell changes and movements otherwise associated only with development and disease.
URLPMID:19726630 [本文引用: 1]
This article is part of a Minifocus on collective cell migration. For further reading, please see related articles: `Mechanisms of collective cell migration at a glance' by Olga Ilina and Peter Friedl ( J. Cell Sci. 122 , [3203-3208][1]) and `Collective cell migration in development' by Cornelis
URLPMID:25517612Magsci [本文引用: 1]
A critical step for leukocyte access to sites of inflammation is the transmigration of cells across vascular endothelium for access to the tissue space. Nourshargh and Alon discuss our current knowledge on the molecular mechanisms used by different effector leukocytes, in particular myeloid cells, to breach inflamed blood vessels.
URLPMID:24603165 [本文引用: 1]
Leukocyte migration through interstitial tissues is essential for mounting a successful immune response. Interstitial motility is governed by a vast array of cell-intrinsic and cell-extrinsic factors that together ensure the proper positioning of immune cells in the context of specific microenvironments. Recent advances in imaging modalities, in particular intravital confocal and multi-photon microscopy, have helped to expand our understanding of the cellular and molecular mechanisms that underlie leukocyte navigation in the extravascular space. In this Review, we discuss the key factors that regulate leukocyte motility within three-dimensional environments, with a focus on neutrophils and T cells in non-lymphoid organs.
URLPMID:8608589 [本文引用: 1]
Cell. 1996 Feb 9;84(3):359-69. Research Support, U.S. Gov't, P.H.S.; Review
URLPMID:8608590 [本文引用: 1]
Cell. 1996 Feb 9;84(3):371-9. Research Support, Non-U.S. Gov't; Research Support, U.S. Gov't, P.H.S.; Review
URL [本文引用: 1]
ABSTRACTActomyosin contractility is a highly regulated process that affects many fundamental biological processes in each and every cell in our body. In this Cell Science at a Glance article and the accompanying poster, we mined the literature and databases to map the contractome of non-muscle cells. Actomyosin contractility is involved in at least 49 distinct cellular functions that range from providing cell architecture to signal transduction and nuclear activity. Containing over 100 scaffolding and regulatory proteins, the contractome forms a highly complex network with more than 230 direct interactions between its components, 86 of them involving phosphorylation. Mapping these interactions, we identify the key regulatory pathways involved in the assembly of actomyosin structures and in activating myosin to produce contractile forces within non-muscle cells at the exact time and place necessary for cellular function.
URLPMID:28715714 [本文引用: 1]
Collective cell migration is essential during physiological processes such as development or wound healing and in pathological conditions such as cancer dissemination. Cells migrating within multicellular tissues experiment different forces which play an intricate role during tissue formation, development and maintenance. How cells are able to respond to these forces depends largely on how they interact with the extracellular matrix. In this review, we focus on mechanics and mechanotransduction in collective migration. In particular, we discuss current knowledge on how cells integrate mechanical signals during collective migration and we highlight actomyosin contractility as a central hub coordinating mechanosensing and mechanotransduction responses.
URLPMID:11379633 [本文引用: 1]
Spatially controlled polymerization of actin is at the origin of cell motility and is responsible for the formation of cellular protrusions like lamellipodia. The pathogens Listeria monocytogenes and Shigella flexneri, which undergo actin-based propulsion, are acknowledged models of the leading edge of lamellipodia. Actin-based motility of the bacteria or of functionalized microspheres can be reconstituted in vitro from only five pure proteins. Movement results from the regulated site-directed treadmilling of actin filaments, consistent with observations of actin dynamics in living motile cells and with the biochemical properties of the components of the synthetic motility medium.
[本文引用: 1]
[本文引用: 1]
URLPMID:9094325 [本文引用: 1]
Striated muscle sarcomeres in vertebrates comprise ordered arrays of actin and myosin filaments, organized by an elaborate protein scaffold. Recent innovative work in a number of laboratories has greatly improved our knowledge of these structures, their organization and their interactions. Structural details have been reported on myosin filaments, actin filaments, Z-bands, M-bands, titin, and nebulin. Time-resolved X-ray diffraction and electron microscopy are revealing the molecular movements involved in force production and regulation.
URLPMID:12142273 [本文引用: 1]
Striated muscle is an intricate, efficient, and precise machine that contains complex interconnected cytoskeletal networks critical for its contractile activity. The individual units of the sarcomere, the basic contractile unit of myofibrils, include the thin, thick, titin, and nebulin filaments. These filament systems have been investigated intensely for some time, but the details of their functions, as well as how they are connected to other cytoskeletal elements, are just beginning to be elucidated. These investigations have advanced significantly in recent years through the identification of novel sarcomeric and sarcomeric-associated proteins and their subsequent functional analyses in model systems. Mutations in these cytoskeletal components account for a large percentage of human myopathies, and thus insight into the normal functions of these proteins has provided a much needed mechanistic understanding of these disorders. In this review, we highlight the components of striated muscle cytoarchitecture with respect to their interactions, dynamics, links to signaling pathways, and functions. The exciting conclusion is that the striated muscle cytoskeleton, an exquisitely tuned, dynamic molecular machine, is capable of responding to subtle changes in cellular physiology.
[本文引用: 1]
URLPMID:2963174 [本文引用: 2]
In striated muscle, the actin cytoskeleton is differentiated into myofibrils. Actin and myosin filaments are organized in sarcomeres and specialized for producing contractile forces. Regular arrangement of actin filaments with uniform length and polarity is critical for the contractile function. However, the mechanisms of assembly and maintenance of sarcomeric actin filaments in striated muscle are not completely understood. Live imaging of actin in striated muscle has revealed that actin subunits within sarcomeric actin filaments are dynamically exchanged without altering overall sarcomeric structures. A number of regulators for actin dynamics have been identified, and malfunction of these regulators often result in disorganization of myofibril structures or muscle diseases. Therefore, proper regulation of actin dynamics in striated muscle is critical for assembly and maintenance of functional myofibrils. Recent studies have suggested that both enhancers of actin dynamics and stabilizers of actin filaments are important for sarcomeric actin organization. Further investigation of the regulatory mechanism of actin dynamics in striated muscle should be a key to understanding how myofibrils develop and operate. 2010 Wiley-Liss, Inc.
URLPMID:16465476 [本文引用: 1]
Building a myofibril from its component proteins requires the interactions of many different proteins in a process whose details are not understood. Several models have been proposed to provide a framework for understanding the increasing data on new myofibrillar proteins and their localizations during muscle development. In this article we discuss four current models that seek to explain how the assembly occurs in vertebrate cross-striated muscles. The models hypothesize: (a) stress fiber-like structures as templates for the assembly of myofibrils, (b) assembly in which the actin filaments and Z-bands form subunits independently from A-band subunits, with the two subsequently joined together to form a myofibril, (c) premyofibrils as precursors of myofibrils, or (d) assembly occurring without any intermediary structures. The premyofibril model, proposed by the authors, is discussed in more detail as it could explain myofibrillogenesis under a variety of different conditions: in ovo , in explants, and in tissue culture studies on cardiac and skeletal muscles.
URLPMID:20625425 [本文引用: 1]
We review some of the problems in determining how myofibrils may be assembled and just as importantly how this contractile structure may be renewed by sarcomeric proteins moving between the sarcomere and the cytoplasm. We also address in this personal review the recent evidence that indicates that the assembly and dynamics of myofibrils are conserved whether the cells are analyzed in situ or in tissue culture conditions. We suggest that myofibrillogenesis is a fundamentally conserved process, comparable to protein synthesis, mitosis, or cytokinesis, whether examined in situ or in vitro.
URLPMID:3519593 [本文引用: 1]
The actin concentration and monomer-polymer ratio in developing chicken skeletal muscle were determined by means of a DNase I inhibition assay. The concentration of G-actin in embryonic muscle was much higher than the critical concentration for polymerization of purified actin. As muscle development progressed, the amount of total actin remarkably increased, whereas the concentration of G-actin markedly decreased, and finally in adults reached the critical concentration for polymerization of purified actin. When the monomeric actin in the soluble fraction of embryonic muscle was purified, the critical concentration for polymerization of the embryonic actin decreased to the same value as that of adult skeletal muscle actin. On the other hand, there was no difference between the crude and purified actin in the type of actin. They consisted of alpha-, beta-, and gamma-actins; their amounts were in the order, beta greater than gamma greater than alpha. Furthermore, polymerization of the monomeric actin in the soluble fraction of embryonic muscle was induced by the addition of myosin or HMM. The large amount of monomeric actin in the embryonic skeletal muscle may be due to the presence of some factor(s) which inhibits actin polymerization and also to an insufficiency of myosin.
URLPMID:2453294 [本文引用: 1]
The two major proteins in the I-bands of skeletal muscle, actin and tropomyosin, were each labeled with fluorescent dyes and microinjected into cultured cardiac myocytes and skeletal muscle myotubes. Actin was incorporated along the entire length of the I-band in both types of muscle cells. In the myotubes, the incorporation was uniform, whereas in cardiac myocytes twice as much actin was incorporated in the Z-bands as in any other area of the I-band. Labeled tropomyosin that had been prepared from skeletal or smooth muscle was incorporated in a doublet in the I-band with an absence of incorporation in the Z-band. Tropomyosin prepared from brain was incorporated in a similar pattern in the I-bands of cardiac myocytes but was not incorporated in myotubes. These results in living muscle cells contrast with the patterns obtained when labeled actin and tropomyosin are added to isolated myofibrils. Labeled tropomyosins do not bind to any region of the isolated myofibrils, and labeled actin binds to A-bands. Thus, only living skeletal and cardiac muscle cells incorporate exogenous actin and tropomyosin in patterns expected from their known myofibrillar localization. These experiments demonstrate that in contrast to the isolated myofibrils, myofibrils in living cells are dynamic structures that are able to exchange actin and tropomyosin molecules for corresponding labeled molecules. The known overlap of actin filaments in cardiac Z-bands but not in skeletal muscle Z-bands accounts for the different patterns of actin incorporation in these cells. The ability of cardiac myocytes and non-muscle cells but not skeletal myotubes to incorporate brain tropomyosin may reflect differences in the relative actin-binding affinities of non-muscle tropomyosin and the respective native tropomyosins. The implications of these results for myofibrillogenesis are presented.
URLPMID:8299146 [本文引用: 1]
The purpose of this study was to determine how quickly contractile proteins are incorporated into the myofibrils of freshly isolated cardiomyocytes and to determine whether there are regions of the cells that are more dynamic than others in their ability to incorporate the proteins. Paired cardiomyocytes joined at intercalated discs and single cells were isolated from adult rats, and microinjected 3 hours later with fluorescently labeied actin, alpha-actinin, myosin light chains, and vinculin. The cells were fixed and permeabilized at various period, 5 seconds and longer, after microinjection. Actin became incorporated throughout the I-Bands in as short a time as 5 seconds. The free edges of the cells, which were formerly intercalated discs, exhibited concentrations of actin greater than that incorporated in the I-Bands. This extra concentration of actin was not detected, however, at intact intercalated discs connecting paired cells. Alpha-actinin was incorporated immediately into Z-Bands and intercalated discs. Vinculin, also, was localized at the Z-Bands and at intercalated discs, but in contrast to alpha-actinin, there was a higher concentration of vinculin in the region of the intact intercalated discs. Both alpha-actinin and vinculin were concentrated at the free ends of the cells that were formerly parts of intercalated discs. Myosin light chains were observed to incorporate into the A-Bands in periods as short as 5 seconds. These results suggest that the myofibrils of adult cardiomyocytes may be capable of rapid isoform transitions along the length of the myofibrils. The rapid accumulation of fluorescent actin, alpha-actinin, and vinculin in membrane sites that were previously parts of intercalated discs, may reflect the response to locomotory activity that is initiated in these areas as cells spread in culture. A similar response after an injury in the intact heart could allow repair to occur. ? 1993 Wiley-Liss, Inc.
URLPMID:9113391 [本文引用: 1]
The exchangeability of in and fibroblast was investigated using fluorescent analogue cytochemistry in combination with fluorescence recovery (FR) after photobleaching. Living embryonic cardiac myocytes and fibroblasts were microinjected with rhodamine (rh)-labeled muscle and nonmuscle . After incorporation of the fluorescent analogue into cellular structures, small areas of labeled structures were photobleached with a laser pulse. In , FR in their proximal striated portions occurred at a slower rate than that in their proximal nonstriated and distal terminal portions with each rh-isoactin injected. Thus, nascent at different developmental stages display different exchangeabilities. Further, in all portions of , FR of rh-muscle was faster than that of rh-nonmuscle . This indicates that molecules in cannot be readily exchanged by heterotypic nonmuscle . In fibroblasts, photobleaching of yielded similar results in both their proximal mid-points and distal terminal portions, and the FR rate was consistently faster than that observed in any part of the . This result seems to be related to the dynamic properties of filaments in at all portions. Further, the fact that possessed a similar exchange rate with muscle and nonmuscle appears to be related to a more primitive nature of than .
URLPMID:9661299 [本文引用: 1]
Rhodamine (Rho)-labeled muscle and non-muscle actins were microinjected into cultured embryonic chicken cardiac myocytes and fibroblasts. After incorporation of the fluorescent actin analog into cellular structures, small areas of labeled structures were photobleached with a laser pulse, and fluorescence recovery (FR) was measured to determine the exchangeability of isoactins in these structures. With both Rho-muscle and Rho-non-muscle actins, the FR rate in any part of stress fibers was consistently faster than that observed in any part of myofibrils. Thus, although non-striated (proximal and terminal) portions of nascent myofibrils are similar in appearance and composition to stress fibers, our data clearly revealed differences in actin stability between these two structures. Further, although cardiomyocytes were incapable of discriminating between the incorporation of muscle and non-muscle actin isoforms into myofibrils, FR after photobleaching of Rho-muscle actin was faster than that of Rho-non-muscle actin in immature non-striated portions. This indicates that actin molecules in cardiac myofibrils cannot be readily exchanged by heterotypic non-muscle actin. Fluorescently labeled actin incorporated into non-striated (proximal and terminal) portions of myofibrils and terminal portions of stress fibers was found to be more stable than alpha-actinin. The relative stability of actin could facilitate the formation of nascent Z-bands of myofibrils and the reorganization of stress fibers at these portions.
URLPMID:11389438 [本文引用: 2]
Regulation of dynamics at filament ends determines the organization and turnover of cytoskeletal structures. In striated muscle, it is believed that tight capping of the fast-growing (barbed) ends by CapZ and of the slow-growing (pointed) ends by () stabilizes the uniform lengths of (thin) filaments in . Here we demonstrate for the first time that both CapZ and are dynamic on the basis of the rapid incorporation of microinjected rhodamine-labelled (-) at both barbed and pointed ends and from the photobleaching of ()-labelled . Unexpectedly, the inhibition of dynamics at pointed ends by -overexpression results in shorter thin filaments, whereas the inhibition of dynamics at barbed ends by D has no effect on length. These data demonstrate that the filaments in are relatively dynamic despite the presence of capping proteins, and that regulated assembly at pointed ends determines the length of thin filaments.
URLPMID:15810059 [本文引用: 1]
During myofibril formation, Z-bodies, small complexes of alpha-actinin and associated proteins, grow in size, fuse and align to produce Z-bands. To determine if there were changes in protein dynamics during the assembly process, Fluorescence Recovery after Photobleaching was used to measure the exchange of Z-body and Z-band proteins with cytoplasmic pools in cultures of quail myotubes. Myotubes were transfected with plasmids encoding Yellow, Green, or Cyan Fluorescent Protein linked to the Z-band proteins: actin, alpha-actinin, cypher, FATZ, myotilin, and telethonin. Each Z-band protein showed a characteristic recovery rate and mobility. All except telethonin were localized in both Z-bodies and Z-bands. Proteins that were present both early in development in Z-bodies and later in Z-bands had faster exchange rates in Z-bodies. These results suggest that during myofibrillogenesis, molecular interactions develop between the Z-band proteins that decrease their mobility and increase the stability of the Z-bands. A truncated construct of alpha-actinin, which localized in Z-bands in myotubes and exhibited a very low rate of exchange, led to disruption of myofibrils, suggesting the importance of dynamic, intact alpha-actinin molecules for the formation and maintenance of Z-bands. Our experiments reveal the Z-band to be a much more dynamic structure than its appearance in electron micrographs of cross-striated muscle cells might suggest. Cell Motil. Cytoskeleton 61:34-48, 2005. ? 2005 Wiley-Liss, Inc.
URLPMID:19470580 [本文引用: 2]
In contrast to the highly dynamic actin cytoskeleton in non-muscle cells, actin filaments in muscle sarcomeres are thought to be relatively stable and undergo dynamics only at their ends. However, many proteins that promote rapid actin dynamics are also expressed in striated muscles. We show that a subset of actin filaments in cardiomyocyte sarcomeres displays rapid turnover. Importantly, we found that turnover of these filaments depends on contractility of the cardiomyocytes. Studies using an actin-polymerization inhibitor suggest that the pool of dynamic actin filaments is composed of filaments that do not contribute to contractility. Furthermore, we provide evidence that ADF/cofilins, together with myosin-induced contractility, are required to disassemble non-productive filaments in developing cardiomyocytes. These data indicate that an excess of actin filaments is produced during sarcomere assembly, and that contractility is applied to recognize non-productive filaments that are subsequently destined for depolymerization. Consequently, contractility-induced actin dynamics plays an important role in sarcomere maturation.
[本文引用: 1]
URLPMID:18793739Magsci [本文引用: 1]
The actin (thin) filaments in striated muscle are highly regulated and precisely specified in length to optimally overlap with the myosin (thick) filaments for efficient myofibril contraction. Here, we review and critically discuss recent evidence for how thin filament lengths are controlled in vertebrate skeletal, vertebrate cardiac, and invertebrate (arthropod) sarcomeres. Regulation of actin polymerization dynamics at the slow-growing (pointed) ends by the capping protein tropomodulin provides a unified explanation for how thin filament lengths are physiologically optimized in all three muscle types. Nebulin, a large protein thought to specify thin filament lengths in vertebrate skeletal muscle through a ruler mechanism, may not control pointed-end actin dynamics directly, but instead may stabilize a large core region of the thin filament. We suggest that this stabilizing function for nebulin modifies the lengths primarily specified by pointed-end actin dynamics to generate uniform filament lengths in vertebrate skeletal muscle. We suggest that nebulette, a small homolog of nebulin, may stabilize a correspondingly shorter core region and allow individual thin filament lengths to vary according to working sarcomere lengths in vertebrate cardiac muscle. We present a unified model for thin filament length regulation where these two mechanisms cooperate to tailor thin filament lengths for specific contractile environments in diverse muscles.
URLPMID:2120327 [本文引用: 1]
The actin filaments of myofibrils are highly organized; they are of a uniform length and polarity and are situated in the sarcomere in an aligned array. We hypothesized that the barbed-end actin-binding protein, CapZ, directs the process of actin filament assembly during myofibrillogenesis. We tested this hypothesis by inhibiting the actin- binding activity of CapZ in developing myotubes in culture using two different methods. First, injection of a monoclonal antibody that prevents the interaction of CapZ and actin disrupts the non-striated bundles of actin filaments formed during the early stages of myofibril formation in skeletal myotubes in culture. The antibody, when injected at concentrations lower than that required for disrupting the actin filaments, binds at nascent Z-disks. Since the interaction of CapZ and the monoclonal antibody are mutually exclusive, this result indicates that CapZ binds nascent Z-disks independent of an interaction with actin filaments. In a second approach, expression in myotubes of a mutant form of CapZ that does not bind actin results in a delay in the appearance of actin in a striated pattern in myofibrils. The organization of alpha-actinin at Z-disks also is delayed, but the organization of titin and myosin in sarcomeres is not significantly altered. We conclude that the interaction of CapZ and actin is important for the organization of actin filaments of the sarcomere.
URLPMID:10601341 [本文引用: 1]
Actin capping protein (CP) binds barbed ends of actin filaments to regulate actin assembly. CP is an α/β heterodimer. Vertebrates have conserved isoforms of each subunit. Muscle cells contain two β isoforms. β1 is at the Z-line; β2 is at the intercalated disc and cell periphery in general. To investigate the functions of the isoforms, we replaced one isoform with another using expression in hearts of transgenic mice. Mice expressing β2 had a severe phenotype with juvenile lethality. Myofibril architecture was severely disrupted. The β2 did not localize to the Z-line. Therefore, β1 has a distinct function that includes interactions at the Z-line. Mice expressing β1 showed altered morphology of the intercalated disc, without the lethality or myofibril disruption of the β2-expressing mice. The in vivo function of CP is presumed to involve binding barbed ends of actin filaments. To test this hypothesis, we expressed a β1 mutant that poorly binds actin. These mice showed both myofibril disruption and intercalated disc remodeling, as predicted. Therefore, CPβ1 and CPβ2 each have a distinct function that cannot be provided by the other isoform. CPβ1 attaches actin filaments to the Z-line, and CPβ2 organizes the actin at the intercalated discs.
URLPMID:7544875 [本文引用: 1]
CONTROL of actin filament length and dynamics is important for cell motility and architecture and is regulated in part by capping proteins that block elongation and depolymerization at both the fast-growing (barbed) and slow-growing (pointed) ends 1-4 . Tropomodulin is a capping protein for the pointed end of the actin filament 5,6 ; it is associated with the free, pointed ends of the thin filaments in striated muscle, where it is thought to bind to both tropomyosin and actin 7,8 . In embryonic chick cardiac myocytes, tropomodulin assembles after the thin, as well as the thick, filaments have become organized into periodic I and A bands 8 , suggesting that tropomodulin might be involved in maintaining actin filament length. Here we show that microinjection of an antibody that inhibits tropomodulin's pointed-end-capping activity in vitro results in a marked elongation of actin filaments from their pointed ends and a >80% reduction in the percentage of beating cells. This demonstrates that pointed-end capping by tropomodulin is required to maintain actin filament length in vivo and that this is essential for contractile function in embryonic chick cardiac myocytes.
URLPMID:9440708
Abstract Tropomodulin is a tropomyosin-binding protein that terminates "pointed-end" actin filament polymerization. To test the hypothesis that regulation of tropomodulin:actin filament stoichiometry is critical for maintenance of actin filament length, tropomodulin levels were altered in cells by infection with recombinant adenoviral expression vectors, which produce either sense or antisense tropomodulin mRNA. Neonatal rat cardiomyocytes were infected, and sarcomeric actin filament organization was examined. Confocal microscopy indicated that overexpression of tropomodulin protein shortened actin filaments and caused myofibril degeneration. In contrast, decreased tropomodulin content resulted in the formation of abnormally long actin filament bundles. Despite changes in myofibril structure caused by altered tropomodulin expression, total protein turnover of the cardiomyocytes was unaffected. Biochemical analyses of infected cardiomyocytes indicated that changes in actin distribution, rather than altered actin content, accounted for myofibril reorganization. Ultrastructural analysis showed thin-filament disarray and revealed the presence of leptomeres after tropomodulin overexpression. Tropomodulin-mediated effects constitute a novel mechanism to control actin filaments, and our findings demonstrate that regulated tropomodulin expression is necessary to maintain stabilized actin filament structures in cardiac muscle cells.
URLPMID:26586224
The sarcomeric tropomodulin (Tmod) isoforms Tmod1 and Tmod4 cap thin filament pointed ends and functionally interact with the leiomodin (Lmod) isoforms Lmod2 and Lmod3 to control myofibril organization, thin filament lengths, and actomyosin crossbridge formation in skeletal muscle fibers. Here, we show that Tmod4 is more abundant than Tmod1 at both the transcript and protein level in a variety of muscle types, but the relative abundances of sarcomeric Tmods are muscle specific. We then generate Tmod4(-/-) mice, which exhibit normal thin filament lengths, myofibril organization, and skeletal muscle contractile function owing to compensatory upregulation of Tmod1, together with an Lmod isoform switch wherein Lmod3 is downregulated and Lmod2 is upregulated. However, RNAi depletion of Tmod1 from either wild-type or Tmod4(-/-) muscle fibers leads to thin filament elongation by 15%. Thus, Tmod1 per se, rather than total sarcomeric Tmod levels, controls thin filament lengths in mouse skeletal muscle, whereas Tmod4 appears to be dispensable for thin filament length regulation. These findings identify Tmod1 as the key direct regulator of thin filament length in skeletal muscle, in both adult muscle homeostasis and in developmentally compensated contexts.
URLPMID:25431137 [本文引用: 2]
Precise regulation of thin filament length is essential for optimal force generation during muscle contraction. The thin filament capping protein tropomodulin (Tmod) contributes to thin filament length uniformity by regulating elongation and depolymerization at thin filament ends. The leiomodins (Lmod1-3) are structurally related to Tmod1-4 and also localize to actin filament pointed ends, but in vitro biochemical studies indicate that Lmods act instead as robust nucleators. Here, we examined the roles of Tmod4 and Lmod3 during Xenopus skeletal myofibrillogenesis. Loss of Tmod4 or Lmod3 resulted in severe disruption of sarcomere assembly and impaired embryonic movement. Remarkably, when Tmod4-deficient embryos were supplemented with additional Lmod3, and Lmod3-deficient embryos were supplemented with additional Tmod4, sarcomere assembly was rescued and embryonic locomotion improved. These results demonstrate for the first time that appropriate levels of both Tmod4 and Lmod3 are required for embryonic myofibrillogenesis and, unexpectedly, both proteins can function redundantly during in vivo skeletal muscle thin filament assembly. Furthermore, these studies demonstrate the value of Xenopus for the analysis of contractile protein function during de novo myofibril assembly.
[本文引用: 1]
[本文引用: 1]
URLPMID:16902413 [本文引用: 1]
The precise assembly of the highly organized filament systems found in muscle is critically important for its function. It has been hypothesized that nebulin, a giant filamentous protein extending along the entire length of the thin filament, provides a blueprint for muscle thin filament assembly. To test this hypothesis, we generated a KO mouse model to investigate nebulin functions in vivo. Nebulin KO mice assemble thin filaments of reduced lengths and 15% of their Z-disks are abnormally wide. Our data demonstrate that nebulin functions in vivo as a molecular ruler by specifying pointed- and barbed-end thin filament capping. Consistent with the shorter thin filament length of nebulin deficient mice, maximal active tension was significantly reduced in KO animals. Phenotypically, the murine model recapitulates human nemaline myopathy (NM), that is, the formation of nemaline rods combined with severe skeletal muscle weakness. The myopathic changes in the nebulin KO model include depressed contractility, loss of myopalladin from the Z-disk, and dysregulation of genes involved in calcium homeostasis and glycogen metabolism; features potentially relevant for understanding human NM.
URLPMID:20498015 [本文引用: 1]
Efficient muscle contraction requires regulation of actin filament lengths. In one highly cited model, the giant protein nebulin has been proposed to function as a molecular ruler specifying filament lengths. We directly challenged this hypothesis by constructing a unique, small version of nebulin (mini-nebulin). When endogenous nebulin was replaced with mini-nebulin in skeletal myocytes, thin filaments extended beyond the end of mini-nebulin, an observation which is inconsistent with a strict ruler function. However, under conditions that promote actin filament depolymerization, filaments associated with mini-nebulin were remarkably maintained at lengths either matching or longer than mini-nebulin. This indicates that mini-nebulin is able to stabilize portions of the filament it has no contact with. Knockdown of nebulin also resulted in more dynamic populations of thin filament components, whereas expression of mini-nebulin decreased the dynamics at both filament ends (i.e., recovered loss of endogenous nebulin). Thus, nebulin regulates thin filament architecture by a mechanism that includes stabilizing the filaments and preventing actin depolymerization.
URLPMID:2173459 [本文引用: 1]
Tropomyosin binds to actin filaments and is implicated in stabilization of actin cytoskeleton. We examined biochemical and cell biological properties of Caenorhabditis elegans tropomyosin (CeTM) and obtained evidence that CeTM is antagonistic to ADF/cofilin-dependent actin filament dynamics. We purified CeTM, actin, and UNC-60B (a muscle-specific ADF/cofilin isoform), all of which are derived from C. elegans, and showed that CeTM and UNC-60B bound to F-actin in a mutually exclusive manner. CeTM inhibited UNC-60B-induced actin depolymerization and enhancement of actin polymerization. Within isolated native thin filaments, actin and CeTM were detected as major components, whereas UNC-60B was present at a trace amount. Purified UNC-60B was unable to interact with the native thin filaments unless CeTM and other associated proteins were removed by high-salt extraction. Purified CeTM was sufficient to restore the resistance of the salt-extracted filaments from UNC-60B. In muscle cells, CeTM and UNC-60B were localized in different patterns. Suppression of CeTM by RNA interference resulted in disorganized actin filaments and paralyzed worms in wild-type background. However, in an ADF/cofilin mutant background, suppression of CeTM did not worsen actin organization and worm motility. These results suggest that tropomyosin is a physiological inhibitor of ADF/cofilin-dependent actin dynamics.
URLPMID:1705952 [本文引用: 1]
Tropomyosin is a well-characterized regulator of muscle contraction. It also stabilizes actin filaments in a variety of muscle and non-muscle cells. Although these two functions of tropomyosin could have different impacts on actin cytoskeletal organization, their functional relationship has not been studied in the same experimental system. Here, we investigated how tropomyosin stabilizes actin filaments and how this function is influenced by muscle contraction in Caenorhabditis elegans body wall muscle. We confirmed the antagonistic role of tropomyosin against UNC-60B, a muscle-specific ADF/cofilin isoform, in actin filament organization using multiple UNC-60B mutant alleles. Tropomyosin was also antagonistic to UNC-78 (AIP1) in vivo and protected actin filaments from disassembly by UNC-60B and UNC-78 in vitro, suggesting that tropomyosin protects actin filaments from the ADF/cofilin-AIP1 actin disassembly system in muscle cells. A mutation in the myosin heavy chain caused greater reduction in contractility than tropomyosin depletion. However, the myosin mutation showed much weaker suppression of the phenotypes of ADF/cofilin or AIP1 mutants than tropomyosin depletion. These results suggest that muscle contraction has only minor influence on the tropomyosin's protective role against ADF/cofilin and AIP1, and that the two functions of tropomyosin in actin stability and muscle contraction are independent of each other. Cell Motil. Cytoskeleton 2006. 2006 Wiley-Liss, Inc.
URLPMID:4223631 [本文引用: 1]
Generation of contractile force in skeletal muscles relies on careful regulation of actin dynamics. Kremneva etal. identify unique features of cofilin-2, a muscle-specific isoform of the actin binding protein, that allow it to efficiently disassemble both ATP- and ADP-Pi-actin filaments to regulate their length in cardiac muscle sarcomeres.
URLPMID:29878125 [本文引用: 1]
URLPMID:11257131 [本文引用: 1]
Assembly and maintenance of myofibrils require dynamic regulation of the actin cytoskeleton. In Caenorhabditis elegans, UNC-60B, a muscle-specific actin depolymerizing factor (ADF)/cofilin isoform, is required for proper actin filament assembly in body wall muscle. Here, I show that UNC-78 is a homologue of actin-interacting protein 1 (AIP1) and functions as a novel regulator of actin organization in myofibrils. In unc-78 mutants, the striated organization of actin filaments is disrupted, and large actin aggregates are formed in the body wall muscle cells, resulting in defects in their motility. Point mutations in unc-78 alleles change conserved residues within different WD repeats of the UNC-78 protein and cause less severe phenotypes than a deletion allele, suggesting that these mutations partially impair the function of UNC-78. UNC-60B is normally localized in the diffuse cytoplasm and to the myofibrils in wild type but mislocalized to the actin aggregates in unc-78 mutants. Similar Unc-78 phenotypes are observed in both embryonic and adult muscles. Thus, AIP1 is an important regulator of actin filament organization and localization of ADF/cofilin during development of myofibrils.
URLPMID:1446098 [本文引用: 1]
Abstract Regulated disassembly of actin filaments is involved in several cellular processes that require dynamic rearrangement of the actin cytoskeleton. Actin-interacting protein (AIP) 1 specifically enhances disassembly of actin-depolymerizing factor (ADF)/cofilin-bound actin filaments. In vitro, AIP1 actively disassembles filaments, caps barbed ends, and binds to the side of filaments. However, how AIP1 functions in the cellular actin cytoskeletal dynamics is not understood. We compared biochemical and in vivo activities of mutant UNC-78 proteins and found that impaired activity of mutant UNC-78 proteins to enhance disassembly of ADF/cofilin-bound actin filaments is associated with inability to regulate striated organization of actin filaments in muscle cells. Six functionally important residues are present in the N-terminal beta-propeller, whereas one residue is located in the C-terminal beta-propeller, suggesting the presence of two separate sites for interaction with ADF/cofilin and actin. In vitro, these mutant UNC-78 proteins exhibited variable alterations in actin disassembly and/or barbed end-capping activities, suggesting that both activities are important for its in vivo function. These results indicate that the actin-regulating activity of AIP1 in cooperation with ADF/cofilin is essential for its in vivo function to regulate actin filament organization in muscle cells.
URLPMID:4382254Magsci [本文引用: 1]
Nemaline myopathy (NM) is a genetic muscle disorder characterized by muscle dysfunction and electron-dense protein accumulations (nemaline bodies) in myofibers. Pathogenic mutations have been described in 9 genes to date, but the genetic basis remains unknown in many cases. Here, using an approach that combined whole-exome sequencing (WES) and Sanger sequencing, we identified homozygous or compound heterozygous variants in LMOD3 in 21 patients from 14 families with severe, usually lethal, NM. LMOD3 encodes leiomodin-3 (LMOD3), a 65-kDa protein expressed in skeletal and cardiac muscle. LMOD3 was expressed from early stages of muscle differentiation; localized to actin thin filaments, with enrichment near the pointed ends; and had strong actin filament-nucleating activity. Loss of LMOD3 in patient muscle resulted in shortening and disorganization of thin filaments. Knockdown of lmod3 in zebrafish replicated NM-associated functional and pathological phenotypes. Together, these findings indicate that mutations in the gene encoding LMOD3 underlie congenital myopathy and demonstrate that LMOD3 is essential for the organization of sarcomeric thin filaments in skeletal muscle.
URLPMID:20736303 [本文引用: 1]
http://jcs.biologists.org/cgi/doi/10.1242/jcs.071837
URLPMID:2785617 [本文引用: 1]
Actin filament assembly in nonmuscle cells is regulated by the actin polymerization machinery, including the Arp2/3 complex and formins. However, little is known about the regulation of actin assembly in muscle cells, where straight actin filaments are organized into the contractile unit sarcomere. Here, we show that Fhod3, a myocardial formin that localizes to thin actin filaments in a striated pattern, regulates sarcomere organization in cardiomyocytes. RNA interference-mediated depletion of Fhod3 results in a marked reduction in filamentous actin and disruption of the sarcomeric structure. These defects are rescued by expression of wild-type Fhod3 but not by that of mutant proteins carrying amino acid substitution for conserved residues for actin assembly. These findings suggest that actin dynamics regulated by Fhod3 are critical for sarcomere organization in striated muscle cells.
URLPMID:24840128 [本文引用: 1]
Actin dynamics are critical for muscle development and function, and mutations leading to deregulation of actin dynamics cause various forms of heritable muscle diseases. AIP1 is a major cofactor of the actin depolymerizing factor/cofilin in eukaryotes, promoting actin depolymerizing factor/cofilin-mediated actin disassembly. Its function in vertebrate muscle has been unknown. To investigate functional roles of AIP1 in myocardium, we generated conditional knockout (cKO) mice with cardiomyocyte-specific deletion of Wdr1, the mammalian homolog of yeast AIP1. Wdr1 cKO mice began to die at postnatal day 13 (P13), and none survived past P24. At P12, cKO mice exhibited cardiac hypertrophy and impaired contraction of the left ventricle. Electrocardiography revealed reduced heart rate, abnormal P wave, and abnormal T wave at P10 and prolonged QT interval at P12. Actin filament (F-actin) accumulations began at P10 and became prominent at P12 in the myocardium of cKO mice. Within regions of F-actin accumulation in myofibrils, the sarcomeric components 伪-actinin and tropomodulin-1 exhibited disrupted patterns, indicating that F-actin accumulations caused by Wdr1 deletion result in disruption of sarcomeric structure. Ectopic cofilin colocalized with F-actin aggregates. In adult mice, Wdr1 deletion resulted in similar but much milder phenotypes of heart hypertrophy, F-actin accumulations within myofibrils, and lethality. Taken together, these results demonstrate that AIP1-regulated actin dynamics play essential roles in heart function in mice.
URLPMID:29654745 [本文引用: 2]
Outflow tract (OFT) anomalies account for about 30% of human congenital heart defects detected at birth. The second heart field (SHF) progenitors contribute to OFT and right ventricle (RV) development, but the process largely remains unknown. WDR1 (WD-repeat domain 1) is a major co-factor of actin depolymerizing factor (ADF)/cofilin that actively disassembles ADF/cofilin-bound actin filaments. Its function in embryonic heart development has been unknown. Using Wdr1 floxed mice and Nkx2.5-Cre, we deleted Wdr1 in embryonic heart ( Wdr1 F/F ;Nkx2.5-Cre) and found that these mice exhibited embryonic lethality, and hypoplasia of OFT and RV. To investigate the role of WDR1 in OFT and RV development, we generated SHF progenitors-specific Wdr1 deletion mice (shfKO). shfKO mice began to die at embryonic day 11.5 (E11.5), and displayed decreased size of the proximal OFT and RV at E10.5. In shfKO embryos, neither the number of SHF cells deployment to OFT nor cell proliferation and the cell number were changed, whereas the cellular organization and myofibrillar assembly of cardiomyocytes were severely disrupted. In the proximal OFT and RV of both shfKO and Wdr1 F/F ;Nkx2.5-Cre embryos, cardiomyocytes were dissociated from the outer compact myocardial layer and loosely and disorderly arranged into multilayered myocardium. Our results demonstrate that WDR1 is indispensable for normal OFT and RV development, and suggest that WDR1-mediated actin dynamics functions in controlling the size of OFT and RV, which might through regulating the spatial arrangement of cardiomyocytes.
URLPMID:15495263 [本文引用: 2]
Abstract Congenital myopathies are clinical and genetic heterogeneous disorders characterized by skeletal muscle weakness ranging in severity. Three major forms have been identified: actin myopathy, intranuclear rod myopathy, and nemaline myopathy. Nemaline myopathy is the most common of these myopathies and is further subdivided into seven groups according to severity, progressiveness, and age of onset. At present, five genes have been linked to congenital myopathies. These include alpha-actin (ACTA1), alpha- and beta-tropomyosin (TPM3 and TPM2), troponin T (TNNT1), and nebulin (NEB). Their protein products are all components of the thin filament of the sarcomere. The mutations identified within these genes have varying impacts on protein structure and give rise to different forms of congenital myopathies. Greater understanding of muscle formation and cause of disease can be established through the study of the effect of mutations on the functional proteins. However, a major limitation in the understanding of congenital myopathies is the lack of correlation between the degree of sarcomeric disruption and disease severity. Consequently, great difficulty may be encountered when diagnosing patients and predicting the progression of the disorders. There are no existing cures for congenital myopathies, although improvements can be made to both the standard of living and the life expectancy of the patient through various therapies. Copyright (c) 2004 Pathological Society of Great Britain and Ireland.
URLPMID:17160903Magsci [本文引用: 1]
Nemaline myopathy (NM) is a congenital myopathy characterized by muscle weakness and nemaline bodies in affected myofibers. Five NM genes, all encoding components of the sarcomeric thin filament, are known. We report identification of a sixth gene, CFL2, encoding the actin-binding protein muscle cofilin-2, which is mutated in two siblings with congenital myopathy. The proband’s muscle contained characteristic nemaline bodies, as well as occasional fibers with minicores, concentric laminated bodies, and areas of F-actin accumulation. Her affected sister’s muscle was reported to exhibit nonspecific myopathic changes. Cofilin-2 levels were significantly lower in the proband’s muscle, and the mutant protein was less soluble when expressed in Escherichia coli, suggesting that deficiency of cofilin-2 may result in reduced depolymerization of actin filaments, causing their accumulation in nemaline bodies, minicores, and, possibly, concentric laminated bodies.
URLPMID:22560515 [本文引用: 1]
Nemaline myopathy and myofibrillar myopathy are heterogeneous myopathies that both comprise early-onset forms. We present two sisters from a consanguineous Iraqi Kurdish family with predominant axial and limb girdle weakness. Muscle biopsies showed features of both nemaline myopathy and myofibrillar myopathy. We performed homozygosity mapping in both siblings using an Affymetrix 250K Nspl SNP array. One of the overlapping homozygous regions harbored the gene CFL2. Because a mutation in CFL2 was identified in a family with nemaline myopathy, we performed sequence analysis of the gene and a novel homozygous missense mutation in exon 2 (c.19G>A, p.Val7Met) of CFL2 was identified in both siblings. CFL2 encodes the protein cofilin-2, which plays an important role in regulation of sarcomeric actin filaments. To our knowledge, this is the second family in which a mutation in CFL2 causes an autosomal recessive form of congenital myopathy with features of both nemaline and myofibrillar myopathy. Given the clinical variability and the multitude of histological features of congenital myopathies, CFL2 sequence analysis should be considered in patients presenting with an autosomal recessive form of congenital myopathy.
URLPMID:11333380 [本文引用: 1]
Nemaline myopathy (NM) is a clinically and genetically heterogeneous disorder characterized by muscle weakness and the presence of nemaline bodies (rods) in skeletal muscle. Disease-causing mutations have been reported in five genes, each encoding a protein component of the sarcomeric thin filament. Recently, we identified mutations in the muscle α-skeletal-actin gene ( ACTA1) in a subset of patients with NM. In the present study, we evaluated a new series of 35 patients with NM. We identified five novel missense mutations in ACTA1, which suggested that mutations in muscle α-skeletal actin account for the disease in 6515% of patients with NM. The mutations appeared de novo and represent new dominant mutations. One proband subsequently had two affected children, a result consistent with autosomal dominant transmission. The seven patients exhibited marked clinical variability, ranging from severe congenital-onset weakness, with death from respiratory failure during the 1st year of life, to a mild childhood-onset myopathy, with survival into adulthood. There was marked variation in both age at onset and clinical severity in the three affected members of one family. Common pathological features included abnormal fiber type differentiation, glycogen accumulation, myofibrillar disruption, and “whorling” of actin thin filaments. The percentage of fibers with rods did not correlate with clinical severity; however, the severe, lethal phenotype was associated with both severe, generalized disorganization of sarcomeric structure and abnormal localization of sarcomeric actin. The marked variability, in clinical phenotype, among patients with different mutations in ACTA1 suggests that both the site of the mutation and the nature of the amino acid change have differential effects on thin-filament formation and protein-protein interactions. The intrafamilial variability suggests that α-actin genotype is not the sole determinant of phenotype.
URLPMID:18976909 [本文引用: 1]
Mutations in the skeletal muscle actin gene, ACTA1 are responsible for up to 20% of congenital myopathies with a variety of pathologies that includes nemaline myopathy, intranuclear rod myopathy, actin myopathy and congenital fibre type disproportion. In their review of 2003, Sparrow et al. considered how these actin mutations might affect muscle function at the molecular level and thus cause the disease. Since then several laboratories have taken up the challenge of investigating genotype-henotype relationships experimentally. The objective of this review is to assess the current state of our understanding of the molecular mechanisms of skeletal myopathies and the prospects for future therapies based on this knowledge. Thirty congenital myopathy-causing ACTA1 mutations have been studied using a range of biochemical and in vitro approaches. They showed diverse molecular defects, and there is no obvious pattern seen in mutations resulting in the same histopathology.
[本文引用: 1]
URLPMID:24447884 [本文引用: 1]
Nemaline myopathy (NM) is a genetically and clinically heterogeneous disorder resulting from a disruption of the thin filament proteins of the striated muscle sarcomere. The disorder is typically characterized by muscle weakness including the face, neck, respiratory, and limb muscles and is clinically classified based on the age of onset and severity. Mutations in the ACTA1 gene contribute to a significant proportion of NM cases. The majority of ACTA1 gene mutations are missense mutations causing autosomal dominant NM by producing an abnormal protein. However, approximately 10% of ACTA1 gene mutations are associated with autosomal recessive NM; these mutations are associated with loss of protein function. We report the first case of a large deletion in the ACTA1 gene contributing to autosomal recessive NM. This case illustrates the importance of understanding disease mechanisms at the molecular level to accurately infer the inheritance pattern and potentially aid with clinical management.
URLPMID:22442437Magsci [本文引用: 1]
Abstract Nemaline myopathy, known to be caused by mutations in 7 genes, including skeletal muscle α-actin (ACTA1),(1) is characterized by muscle weakness, hypotonia, and nemaline rods in muscle biopsy. Here we report a patient with nemaline rods but the opposite phenotype of hypercontractility.
URLPMID:27726070 [本文引用: 1]
To date, only two splice-site mutations within the TPM2 gene have been shown to be causative for congenital myopathies. While the majority of TPM2 gene mutations are causative for nemaline myopathy, c
URLPMID:4200603Magsci [本文引用: 1]
ABSTRACT<p>Mutations affecting skeletal muscle isoforms of the tropomyosin genes may cause nemaline myopathy, cap myopathy, core-rod myopathy, congenital fiber-type disproportion, distal arthrogryposes, and Escobar syndrome. We correlate the clinical picture of these diseases with novel (19) and previously reported (31) mutations of the TPM2 and TPM3 genes. Included are altogether 93 families: 53 with TPM2 mutations and 40 with TPM3 mutations. Thirty distinct pathogenic variants of TPM2 and 20 of TPM3 have been published or listed in the Leiden Open Variant Database (
URLPMID:29931346 [本文引用: 1]
Abstract We describe the natural history of 'Amish' nemaline myopathy (ANM), an infantile-onset, lethal disease linked to a pathogenic c.505G>T nonsense mutation of TNNT1, which encodes the slow fiber isoform of troponin T (TNNT1; a.k.a TnT). The TNNT1 c.505G>T allele has a carrier frequency of 6.5% within Old Order Amish settlements of North America. We collected natural history data for 106 ANM patients born between 1923 and 2017. Over the last two decades, mean age of molecular diagnosis was 16-27 days. TNNT1 c.505G>T homozygotes were normal weight at birth but failed to thrive by age 9 months. Presenting neonatal signs were axial hypotonia, hip and shoulder stiffness, and tremors, followed by progressive muscle weakness, atrophy, and contractures. Affected children developed thoracic rigidity, pectus carinatum, and restrictive lung disease during infancy, and all succumbed to respiratory failure by 6 years of age (median survival 18 months, range 0.2-66 months). Muscle histology from two affected children showed marked fiber size variation due to both type 1 myofiber smallness (hypotrophy) and type 2 fiber hypertrophy, with evidence of nemaline rods, myofibrillar disarray, and vacuolar pathology in both fiber types. The truncated slow TNNT1 (TnT) fragment (p.Glu180Ter) was undetectable in ANM muscle, reflecting its rapid proteolysis and clearance from sarcoplasm. Similar functional and histological phenotypes were observed in other human cohorts and two transgenic murine models (Tnnt1-/- and Tnnt1 c.505G>T). These findings have implications for emerging molecular therapies, including the suitably of TNNT1 gene replacement for newborns with ANM or other TNNT1-associated myopathies.
URLPMID:5702563 [本文引用: 1]
Nemaline myopathy (NEM) is one of the three major forms of congenital myopathy and is characterized by diffuse muscle weakness, hypotonia, respiratory insufficiency, and the presence of nemaline rod structures on muscle biopsy. Mutations in troponin T1 (TNNT1) is 1 of 10 genes known to causeNEM. To date, only homozygous nonsense mutations or compound heterozygous truncating or internal deletion mutations inTNNT1gene have been identified inNEM. This extended family is of historical importance as some members were reported in the 1960s as initial evidence thatNEMis a hereditary disorder. Proband and extended family underwent Sanger sequencing forTNNT1. We performedRT-CRand immunoblot on muscle to assessTNNT1RNAexpression and protein levels in proband and father. We report a novel heterozygous missense mutation ofTNNT1c.311A>T (p.E104V) that segregated in an autosomal dominant fashion in a large family residing in the United States. Extensive sequencing of the other known genes forNEMfailed to identify any other mutant alleles. Muscle biopsies revealed a characteristic pattern of nemaline rods and severe myofiber hypotrophy that was almost entirely restricted to the type 1 fiber population. This novel mutation alters a residue that is highly conserved among vertebrates. This report highlights not only a family with autosomal dominant inheritance ofNEM, but that this novel mutation likely acts via a dominant negative mechanism.
URLPMID:26296490 [本文引用: 1]
Introduction : Nemaline myopathy is a rare disorder characterized by skeletal muscle weakness of varying severity and onset, with the presence of nemaline rods on muscle biopsy. Congenital nemaline body myopathy due to mutations in TNNT1 has hitherto only been described as a result of a single founder mutation in patients of Amish origin and in 2 other individuals with different recessive mutations. Methods : Autozygosity mapping and whole exome sequencing were applied after we identified 9 Palestinian patients from 7 unrelated families who have nemaline myopathy. Results : All patients were homozygous for a novel complex rearrangement of the TNNT1 gene (c.574_577delinsTAGTGCTGT | NM_003283) leading to C-terminal truncation of the protein (p.L203* | NP_003274.3). Their clinical course was remarkable for early respiratory failure and striking stiffness of the cervical spine. Conclusions : This report exemplifies the utility of combining autozygosity mapping and whole exome sequencing and expands the phenotype associated with TNNT1 mutations. Muscle Nerve 53: 564-569, 2016
URLPMID:27105866 [本文引用: 1]
Abstract Nemaline myopathy represents a group of clinically and genetically heterogeneous neuromuscular disorders. Different clinical-genetic entities have been characterized in the last few years, with implications for diagnostics and genetic counseling. Fifty percent of nemaline myopathy forms are due to NEB mutations, but genetic analysis of this large and complex gene by Sanger sequencing is time consuming and expensive. We selected 10 Italian patients with clinical and biopsy features suggestive for nemaline myopathy and negative for ACTA1, TPM2 and TPM3 mutations. We applied a targeted next-generation sequencing strategy designed to analyse NEB coding regions, the relative full introns and the promoter. We also evaluated copy number variations (by CGH array) and transcriptional changes by RNA Sanger sequencing, whenever possible. This combined strategy revealed 11 likely pathogenic variants in 8 of 10 patients. The molecular diagnosis was fully achieved in 3 of 8 patients, while only one heterozygous mutation was observed in 5 subjects. This approach revealed to be a fast and cost-effective way to analyse the large NEB gene in a small group of patients and might be promising for the detection of pathological variants of other genes featuring large coding regions and lacking mutational hotspots.
URLPMID:16917880 [本文引用: 1]
Nemaline myopathy (NM) is a clinically and genetically heterogeneous disorder of skeletal muscle caused by mutations in at least five different genes encoding thin filament proteins of the striated muscle sarcomere. We have previously described 18 different mutations in the last 42 exons of the nebulin gene ( NEB ) in 18 families with NM. Here we report 45 novel NEB mutations detected by denaturing high-performance liquid chromatography (dHPLC) and sequence analysis of all 183 NEB exons in NM patients from 44 families. Altogether we have identified, including the deletion of exon 55 identified in the Ashkenazi Jewish population, 64 different mutations in NEB segregating with autosomal recessive NM in 55 families. The majority (55%) of the mutations in NEB are frameshift or nonsense mutations predicted to cause premature truncation of nebulin. Point mutations (25%) or deletions (3%) affecting conserved splice signals are predicted in the majority of cases to cause in-frame exon skipping, possibly leading to impaired nebulin-tropomyosin interaction along the thin filament. Patients in 18 families had one of nine missense mutations (14%) affecting conserved amino acids at or in the vicinity of actin or tropomyosin binding sites. In addition, we found the exon 55 deletion in four families. The majority of the patients (in 49/55 families) were shown to be compound heterozygous for two different mutations. The mutations were found in both constitutively and alternatively expressed exons throughout the NEB gene, and there were no obvious mutational hotspots. Patients with more severe clinical pictures tended to have mutations predicted to be more disruptive than patients with milder forms. Hum Mutat 27(9), 946-956, 2006. Published 2006 Wiley-Liss, Inc.
URLPMID:28131200 [本文引用: 1]
react-text: 426 Background: Among several types of ischemic stroke (IS), branch atheromatous disease (BAD) is known to be the leading cause of disability. Methods: A total of 1919 patients with acute IS were retrospectively analyzed, and BAD patients were classified into anterior or posterior BAD, depending on the responsible vascular territories. These patients were further subcategorized with or without... /react-text react-text: 427 /react-text [Show full abstract]
[本文引用: 2]
URLPMID:25336743Magsci [本文引用: 1]
Arid3b, a member of the conserved ARID family of transcription factors, is essential for mouse embryonic development but its precise roles are poorly understood. Here, we show that Arid3b is expressed in the myocardium of the tubular heart and in second heart field progenitors. Arid3b-deficient embryos show cardiac abnormalities, including a notable shortening of the poles, absence of myocardial differentiation and altered patterning of the atrioventricular canal, which also lacks epithelial-to-mesenchymal transition. Proliferation and death of progenitors as well as early patterning of the heart appear normal. However, DiI labelling of second heart field progenitors revealed a defect in the addition of cells to the heart. RNA microarray analysis uncovered a set of differentially expressed genes in Arid3b-deficient tissues, including Bhlhb2, a regulator of cardiomyocyte differentiation, and Lims2, a gene involved in cell migration. Arid3b is thus required for heart development by regulating the motility and differentiation of heart progenitors. These findings identify Arid3b as a candidate gene involved in the aetiology of human congenital malformations.
[本文引用: 1]
URLPMID:27336173 [本文引用: 1]
Maintenance of epithelial cell polarity and epithelial barrier relies on the spatial organization of the actin cytoskeleton and proper positioning/assembly of intercellular junctions. However, how these processes are regulated is poorly understood. Here we reveal a key role for the multifunctional protein Alix in both processes. In a knockout mouse model of Alix, we identified overt structural changes in the epithelium of the choroid plexus and in the ependyma, such as asymmetrical cell shape and size, misplacement and abnormal beating of cilia, blebbing of the microvilli. These defects culminate in excessive cell extrusion, enlargement of the lateral ventricles and hydrocephalus. Mechanistically, we find that by interacting with F-actin, the Par complex and ZO-1, Alix ensures the formation and maintenance of the apically restricted actomyosin-ight junction complex. We propose that in this capacity Alix plays a role in the establishment of apical-asal polarity and in the maintenance of the epithelial barrier. Correct assembly of intercellular junctions is required to maintain epithelial polarity and barrier function. Here Camposet al. show that the scaffold protein Alix interacts with F-actin, the Par complex and ZO-1 to ensure the formation and maintenance of the actomyosin tight junction complex in choroid plexus epithelium.

{kind=link}
{kind=link}