 ,上海交通大学系统生物医学研究院,系统生物医学教育部重点实验室,上海 200240
,上海交通大学系统生物医学研究院,系统生物医学教育部重点实验室,上海 200240Identification and single-cell sequencing analysis of rare tumor cells in malignant pleural effusion of lung cancer patients
Baojun Wu, Zhuo Wang, Yu Dong, Yuliang Deng, Qihui Shi,Key Laboratory of Systems Biomedicine (Ministry of Education), Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai, 200240, China通讯作者:
编委: 周钢桥
收稿日期:2018-10-31修回日期:2018-12-27网络出版日期:2019-02-25
| 基金资助: |
Editorial board:
Received:2018-10-31Revised:2018-12-27Online:2019-02-25
| Fund supported: |
作者简介 About authors
吴保军,硕士研究生,专业方向:循环肿瘤细胞单细胞测序E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (642KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吴保军, 王卓, 董宇, 邓宇亮, 施奇惠. 肺癌恶性胸腔积液中稀有肿瘤细胞的鉴定与单细胞测序分析[J]. 遗传, 2019, 41(2): 175-184 doi:10.16288/j.yczz.18-230
Baojun Wu, Zhuo Wang, Yu Dong, Yuliang Deng, Qihui Shi.
肺癌是威胁人类健康的主要疾病,其发病率及致死率在我国稳居各项恶性肿瘤之首[1]。对于同一种癌症,不同患者间的肿瘤具有很大的异质性[2,3];即使对于同一患者而言,由于基因组的不稳定,其恶性肿瘤在增殖过程中会产生不同的子代细胞亚群,它们在基因组[4]或表型[5]层面具有不同的分子与功能特征,不同的亚群其细胞增殖、侵袭转移以及耐药潜能方面都存在差异,尤其是存在一部分具有转移能力的肿瘤细胞能脱离原位组织并在远处器官形成转移灶[6],因此,肿瘤细胞的异质性为其发生转移和耐药提供了可能。但传统的肿瘤异质性研究主要基于不同位置原位组织样本中的群体细胞。虽然近年来迅速发展的高通量测序技术提高了肿瘤基因组解析的速度,但针对群体肿瘤细胞的基因组高通量测序不可避免的会引入大量非肿瘤细胞的遗传信息。有研究表明,从实体瘤组织内提取的肿瘤细胞,其中真正肿瘤细胞的DNA占总DNA的量不到50%,这样整体平均化的测序结果使得很多频率较低但非常重要的肿瘤基因组信息被湮没[7]。单细胞测序技术从根本上克服了这样的局限,它能够从单个细胞水平探究不同肿瘤细胞的基因组和转录组特征。Xu等[8]利用单细胞测序技术揭示了肾癌细胞单核苷酸突变特性;Hou 等[9]同样利用单细胞测序技术探究了骨髓增殖性肿瘤的发生发展机制,为肿瘤异质性研究提供了全新的思路。而Navin等[10]利用单细胞测序技术绘制了200多个肿瘤细胞的进化图谱,可见单细胞测序技术在肿瘤生物学方面有着极大的应用前景。
过去肿瘤异质性研究主要聚焦于肿瘤组织的异质性,比如对肿瘤组织的不同位置取样进行异质性分析,缺乏对具有转移能力的肿瘤细胞进行深入的分析。众所周知,转移是肿瘤致死的最重要因素,因此非常有必要对转移性的肿瘤细胞进行单细胞尺度上的分子分析,解析其异质性[11,12]。由于肺癌患者常伴有恶性胸腔积液的产生,胸腔积液中所含有的肿瘤细胞是肺癌胸膜转移的标志[13]。因此,肺癌患者恶性胸腔积液中的肿瘤细胞代表了从肿瘤组织脱落、具有转移能力的肿瘤细胞群体[14]。本研究以肺癌患者恶性胸腔积液中单个肿瘤细胞为研究对象,通过单细胞测序检测其基因组特征并分析这些肿瘤细胞在基因组层面上的异质性。由于胸腔积液中细胞成分复杂,包含多种类型的细胞,因此需首先使用本实验室前期开发的高代谢活性恶性肿瘤细胞的鉴定方法,并以多重置换扩增的单细胞基因组扩增技术和高通量测序技术为主要技术路线,建立一整套转移性肿瘤细胞的单细胞分析方案,从而进一步解析肿瘤细胞在分子层面的异质性,为更深入理解肿瘤转移机制提供更多信息。
1 材料与方法
1.1 样本收集
肺癌患者的胸腔积液样本收集自上海市胸科医院,5名患者均为非小细胞肺癌患者,临床分期均为Ⅳ期,其中男性患者3名,女性患者2名,样本收集及后续研究均通过伦理委员会审核,且在患者充分知情的情况下进行。1.2 微孔阵列芯片制作及表面修饰
参照本实验室前期开发的微孔阵列芯片制作方法制作实验所需微孔阵列芯片[15]。将聚二甲基硅氧烷(polydimethylsiloxane, PDMS)的两种组分按照1∶10质量比充分搅拌混匀,真空抽去气泡,均匀倒于放置硅片模具的10 cm培养皿中。将皿置于80℃电热烘箱中烘烤2 h。将PDMS从模具上剥离,按照设计的边沿切割成具有400个小型数字编号区域,共计11万个直径为30 μm微孔的长方形芯片,然后与经等离子体表面处理的玻片紧密贴合,最后使用2%的Matrigel对PDMS芯片的微孔区域进行修饰。1.3 高代谢活性肿瘤细胞的鉴定
临床收集的肺癌患者胸腔积液样本置于4℃条件下送至实验室。首先经100 μm孔径的细胞筛去除细胞聚团和杂质,然后在4℃条件下通过300 g离心5 min收集细胞沉淀,再加入一定量红细胞裂解液重悬细胞沉淀并室温避光裂解15 min。裂解结束后于4℃条件200 g离心5 min,用含0.1% BSA的HBSS重悬细胞,按照每1×107细胞加入20 μL CD45- APC抗体染色,室温翻转孵育,然后将细胞均匀铺至事先处理好的PDMS微孔芯片上并用无糖Dulbeccos Moddified Eagle Medium(DMEM)培养基洗去CD45-APC。在芯片上加入使用无糖DMEM培养基配制2-NBDG染色液,使其浓度达到0.4 mmol/L,37℃避光条件下孵育10 min,再用预冷的HBSS溶液快速清洗芯片,在低温下使用高内涵显微成像系统(ImageXpress. XLS. Molecular Devices公司, 美国)对细胞进行荧光成像。根据本实验室建立的方法鉴定芯片上的高代谢活性肿瘤细胞。1.4 肿瘤单细胞回收与单细胞全基因组扩增
借助显微操作平台准确回收高代谢活性肿瘤单细胞,将肿瘤单细胞移入含有4 μL PBS的PCR管中。使用REPLI-g single cell kit (Qiagen, 德国)对单细胞全基因组进行扩增,具体实验操作参照试剂盒说明书。首先按照11∶1混合DLB与DTT配制单细胞裂解液,向肿瘤单细胞中加入3 μL细胞裂解液,65℃裂解10 min,加入3 μLStop solution终止裂解。按照表1配制单细胞基因组扩增反应液,向每个肿瘤单细胞样本中加入40 μL单细胞基因组扩增反应液,具体成分为29 μL REPLI-g sc Reaction Buffer、9 μL H2O、2 μL scPhi29 DNA polymerase。PCR仪中30℃孵育5 h;65℃,3 min使酶灭活。Table 1
表1
表1 单细胞基因组扩增反应液成分
Table 1
| 试剂 | 体积(μL) |
|---|---|
| REPLI-g sc Reaction Buffer | 29 |
| H2O | 9 |
| scPhi29 DNA polymerase | 2 |
| Total | 40 |
新窗口打开|下载CSV
1.5 单细胞全基因组扩增产物基因突变检测
针对表皮生长因子受体基因(epidermal growth factor receptor, EGFR)的18、19、20和21外显子分别设计对应特异性扩增引物用于基因突变检测[16] (引物见表2)。使用2×Ex-Taq Premix扩增目标片段,配制反应体系。具体PCR反应程序:94℃,3 min;94℃,15 s,60℃,20 s,72℃,30 s,进行30个循环,72℃,5 min。扩增产物经过凝胶电泳检验,有明显条带则证明扩增成功,剩余扩增产物进行Sanger测序。Table 2
表2
表2 非小细胞肺癌EGFR基因检测扩增引物
Table 2
| 引物名称 | 序列(5'→3') | 片段大小(bp) |
|---|---|---|
| EGFR18-F | TGGAGAAGCTCCCAACCAA | 231 |
| EGFR18-R | TTCCCAAACACTCAGTGAAACA | |
| EGFR19-F | GTGGCACCATCTCACAATT | 451 |
| EGFR19-R | ATGCTCCAGGCTCACCAAG | |
| EGFR20-F | CTTTATCCAATGTGCTCCTC | 486 |
| EGFR20-R | TCTCCCTTCCCTGATTACCT | |
| EGFR21-F | TTCGCCAGCCATAAGTCCT | 500 |
| EGFR21-R | TCATTCACTGTCCCAGCAAG |
新窗口打开|下载CSV
1.6 单细胞全基因组测序
使用实验室设计的针对22条染色体特定片段的扩增引物对单细胞扩增产物进行质控,通过质检的单细胞进行后续建库测序。单细胞全基因组测序文库构建使用NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB,美国)。首先使用3 μL NEBNest dsDNA fragmentase在片段化缓冲液中37℃反应4 min,立即加入5 μL EDTA终止片段化;再使用0.65×恢复至室温的Agencourt AMPure XP beads吸附>500 bp的DNA片段并弃去,使用0.1×Agencourt AMPure XP beads吸附300~500 bp大小的片段,经现配的80%酒精洗涤两次后,用无核酸酶水洗脱建库DNA片段;使用Qubit dsDNA HS Assay Kit (Thermo, 美国)测定样本浓度;末端修复阶段使用样本量为60~100 ng,并在End Prep Enzyme Mix作用下,20℃孵育30 min,65℃孵育30 min;修复后的平末端加A及接头阶段,使用NEB Next Adaptor在Blunt/TA Ligase Master Mix及Ligation Enhancer的作用下,DNA平末端加上腺嘌呤及连接有胸腺嘧啶的特异性接头;为保证后续反应,需将U型环状特异性接头剪开形成双链,加入3 μL USER Enzyme在37℃条件下孵育15 min完成酶切;为纯化连上特异性接头的DNA片段,使用0.8×恢复至室温的Agencourt AMPure XP beads吸附目的DNA,使用现配的80%酒精洗去多余的接头,并用16 μL无核酸酶水室温孵育beads 2 min,置于磁力架吸附3 min,将上清移入0.2 mL PCR管中;全基因组测序文库预扩增阶段使用Q5 DNA Polymerase PreMix体系,针对每一个不同的单细胞全基因组文库引入不同的Index进行标记;文库扩增产物经Agencourt AMPure XP beads两步法纯化,使用现配的80%酒精洗涤beads两次,再用20 μL无核酸酶水洗脱文库DNA;最后使用Qubit dsDNA HS Assay Kit (Thermo, 美国)测定文库浓度,Agilent 2100 Bioanalyzer (Agilent, 美国)检测DNA片段分布,按照等摩尔质量混合不同样本文库,高通量测序使用HiSeq X Ten平台,按照PE150测序方案完成。1.7 数据质控与分析
测序数据质控采用MAPD值法。基于Multiple absolute pairwise difference算法:MAPD=Median (log2nCNRi+1-log2nCNRi),其中i代表单个窗口,MAPD值越大,噪音越大,数据质量越差。本研究采用0.45为阈值,去除MAPD值大于0.45的样本[17]。以通过质控的单细胞下一代测序(Next generation sequencing, NGS)数据为分析单元,使用FASTQC工具评估数据质量,使用Trimmomatic-0.36程序[18]去除质量不好的碱基和多余的接头,采用处理PE测序模式数据的命令得到clean reads和数据处理日志。Clean reads获取原则为切除首端碱基质量小于3和末端碱基质量小于3的碱基,Windows的size为4个碱基,切除平均碱基质量小于15,舍弃小于36 bp的reads。针对每一个去除接头和低质量reads的单细胞测序数据,使用BWA比对软件[19] (Burrows- Wheeler Alignment tool)将clean reads数据比对到UCSC hg19的参考基因组上并且输出SAM文件。为了后续计算,用Samtools工具[20]将SAM文件压缩成BAM文件,并进行染色体排序。为了提高计算速度,用Samtools对其建立索引,然后使用Picard (Picard-tools-1.119)将测序文库构建过程中由PCR预扩增引入的重复进行标记和删除,排除PCR过程造成的干扰。处理完成的每一个单细胞NGS数据,我们采用500 kb长度为窗口将其分割,记录每一个窗口内的reads数目,并对其进行GC含量的标准化。最后,采用R语言中DNAcopy package [21]环状二元分割法来计算染色体拷贝数,用于染色体拷贝数变异分析,最后用R语言画图可视化。
2 结果与分析
2.1 微孔阵列芯片设计
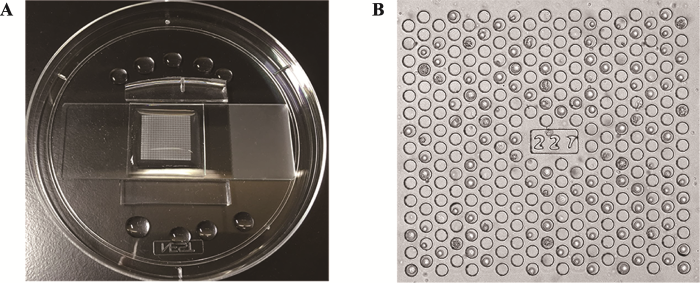
本研究采用本实验室前期设计的微孔阵列芯片,该芯片包含11万个直径为30 μm,深度为30 μm的微孔,分布于400个数字编号的方形区域中(图1,A和B)。微孔区域经过等离子修饰亲水,并滴加上400 μL PBS保持亲水性。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1微孔阵列芯片实物图
A:芯片与干净的玻片粘合,置于培养皿中固定;B:编号为“227”的方形区域,细胞较均匀铺满微孔。
Fig. 1The photos of the microwell chip
2.2 分选高代谢活性肿瘤单细胞
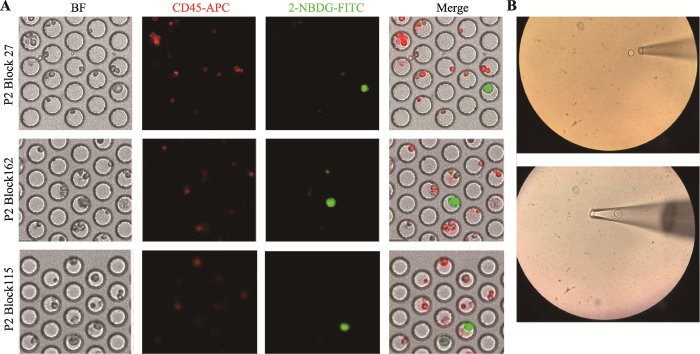
基于本实验室之前的研究,可通过荧光标记的葡萄糖类似物2-NBDG表征细胞的葡萄糖摄取能力,并将白细胞标志物CD45表达阴性,2-NBDG高摄取的细胞鉴定为高代谢活性的疑似肿瘤细胞,其恶性可通过单细胞测序进一步确认。图2A所示为2号患者胸腔积液中细胞成像结果,FITC通道强绿色荧光而APC通道无红色荧光(CD45-,NBDG+)即为CD45阴性的高代谢活性疑似肿瘤细胞,APC通道有红色荧光(CD45+)即表征白细胞,在5名患者胸腔积液中均发现此类疑似肿瘤细胞。基于芯片设计的可寻址功能,使用显微操作仪将目的细胞取出置于PBS中进行后续实验(图2B)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2胸腔积液中高代谢活性肿瘤细胞的鉴定及显微操作挑取单细胞示意图
A:2号患者3个典型的高代谢活性肿瘤细胞。BF表示明场下的成像图,CD45-APC表示在APC通道的成像图,2-NBDG-FITC表示在FITC通道的成像图,Merge表示将明场以及各荧光通道叠加之后的成像效果图;B:使用毛细玻璃管吸取和吹出目的细胞的示意图。
Fig. 2Identification of tumor cells in pleural effusion samples and capture of single cells by micromanipulation
2.3 单细胞全基因组扩增及Sanger测序
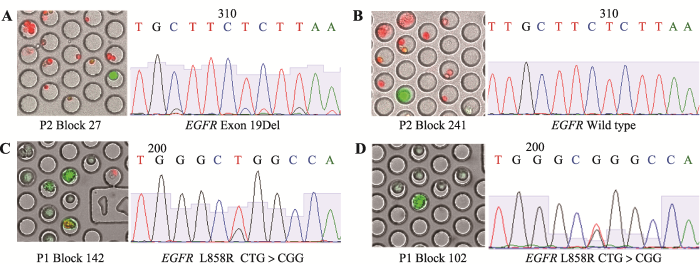
通过显微操作挑取的肿瘤单细胞进行全基因组扩增,扩增产物经过凝胶电泳确认单细胞扩增成功。由于非小细胞肺癌中负责信号转导的表皮生长因子受体EGFR发生突变导致其表达量升高,促进肿瘤的进展和转移,而针对EGFR突变的患者使用酪氨酸激酶抑制剂药物有着很好的效果,因此对其进行针对EGFR基因的Sanger测序可以判定其靶向治疗有效性,有着重大的临床意义[22,23,24]。结果显示:患者P2单细胞测序为EGFR-19Del (图3A),存在一个EGFR野生型(图3B),患者P1的肿瘤单细胞测序结果为EGFR-L858R突变(图3,C和D),患者P3和P5为EGFR-19Del,患者P4为EGFR-L858R突变,5名患者恶性胸腔积液中高代谢活性肿瘤单细胞Sanger测序结果与临床医生提供的患者突变信息吻合。同时,在同一个患者的单细胞样本中,用CD45-/2- NBDG+为标准筛选的高代谢活性肿瘤细胞,有一定比例的野生型,2号患者有1个单细胞是EGFR野生型,提示利用高代谢活性的标准筛选出的肿瘤细胞可能有着不存在驱动基因突变的亚群;P1、P3、,P4和P5号患者所有高代谢活性肿瘤单细胞均带有EGFR点突变。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3肺癌患者恶性胸腔积液中高代谢活性肿瘤细胞Sanger测序结果
A:2号患者2-NBDG高摄取的单细胞,Sanger测序为EGFR基因19Del;B:2号患者2-NBDG高摄取的单细胞,Sanger测序为EGFR野生型;C,D:为1号患者2-NBDG高摄取的两个不同单细胞,Sanger测序均为EGFR基因21号外显子L858R点突变。
Fig. 3Sanger sequencing of tumor cells from NSCLC MPE
2.4 高代谢活性肿瘤单细胞基因组拷贝数变异分析
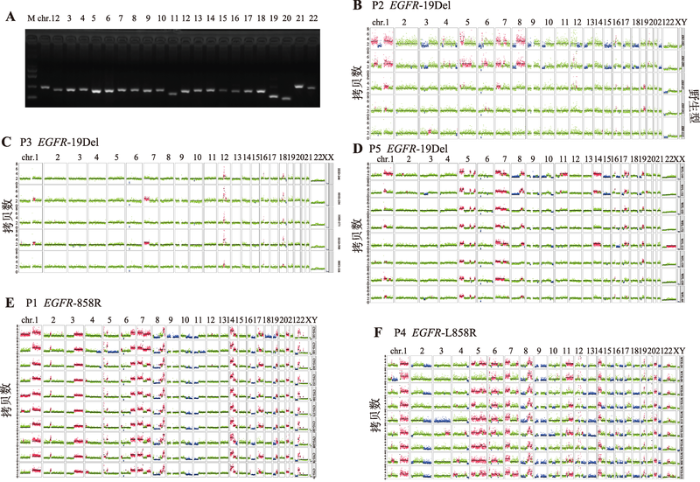
对于通过22条染色体检测的单细胞样本(图4A),认为其全基因组扩增产物覆盖度较好适合进行后续拷贝数变异分析。肿瘤细胞的基因组极不稳定,容易发生基因组重排[25],且重排基因组的大小都在1 kb以上,亚显微结构上体现出扩增[26]或者缺失[27]的现象。基于实验室对单细胞建立的染色体拷贝数变异(copy number variants, CNV)分析路线,对每个患者的所有高代谢活性的肿瘤细胞都进行了CNV分析。结果表明,P2、P3和P5患者都是EGFR-19Del突变类型,但3者的CNV变异情况存在很大差异(图4,B~D)。P3患者的5个肿瘤细胞均存在驱动基因突变但是在基因组水平上没有大范围的变异情况可能属于早期转移的细胞,P2患者肿瘤细胞可细分为2个亚群,可能代表了转移能力不同的两个群体,P5患者存在较好的一致性,但其基因组变异谱与P2和P3均不同;P1患者的10个单细胞基因组拷贝数变异一致性好,大部分染色体都发生了扩增或者缺失,认为此类肿瘤细胞的突变比较多,恶性程度比较高(图4E);P4患者在驱动基因突变检测上有很好的一致性,但是从CNV水平看每个单细胞的拷贝数变异都有较大的异质性(图4F)。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4单细胞全基因组质控及染色体拷贝数变异分析
A:单细胞全基因组扩增产物质量评估。泳道1:DNA marker;泳道2~23:分别表示22条染色体上特定基因片段的扩增产物;B:2号患者CNV结果,其EGFR突变类型为19Del;C:3号患者CNV结果,其EGFR突变类型为19Del;D:5号患者CNV结果,其EGFR突变类型为19Del;E:1号患者CNV结果,其EGFR突变类型为L858R;F:4号患者CNV结果,其EGFR突变类型为L858R。每一横排为一个单细胞的22对常染色体及一对性染色体拷贝数变异情况,每条染色体的绿色标注为正常的2倍体,红色标注为染色体拷贝数发生扩增,蓝色标注为染色体拷贝数发生缺失。
Fig. 4Quality control and CNV analysis for single-cell WGA products
3 讨论
肿瘤异质性是目前肿瘤治疗面临的最大难题,同一癌种在不同患者之间存在基因或表型的差异,且在一个患者肿瘤不同区域也存在分子层面的显著差异[28]。有研究表明,患者体内实体瘤本身就是由含有不同基因或表型特征的亚克隆细胞群体组成[29],它们在增殖速度、侵袭转移能力等方面都具有显著差异,当所处微环境[30,31]发生改变时,一部分不能适应的肿瘤细胞失巢凋亡[32],而另一部分则存活下来,更有一部分潜在转移性肿瘤细胞在外界不利因子的压力下,突破屏障进入到循环系统进而实现远端转移。自肿瘤异质性首次被提出之后,越来越多的临床医生开始关注肿瘤异质性与患者的治疗方案之间的关系。临床上对EGFR突变的晚期肺癌患者使用靶向药物可以杀死突变肿瘤细胞,但是通常患者会在几个月内表现出药物抵抗性,需不断更换治疗方案以延长其生存期,药物等外界环境刺激还可能会诱导体内部分转移潜能的肿瘤细胞从原位脱落实现远端转移。因此深入解析肿瘤异质性是解释肿瘤耐药、侵袭和转移机制的关键。葡萄糖高摄取的肿瘤细胞代表了恶性胸腔积液中高代谢活性的细胞亚群[33],这部分细胞通过大量糖酵解方式获取能量[34,35]。本实验室前期研究认为它们是具有高转移潜能的细胞亚群,因此本研究以肺癌患者恶性胸腔积液中的高代谢活性细胞为研究对象对深入解析肿瘤异质性及转移机制有着重要意义。结果发现:突变类型均为EGFR-L858R的患者(P1和P4),同一个患者多个高代谢活性肿瘤细胞的拷贝数变异存在高度一致性,但是患者之间基因组水平上的拷贝数变异存在极大的差异,P1患者在染色体拷贝数扩增现象多于缺失,而P4患者的扩增和缺失现象都非常明显;同样突变类型均为EGFR-19Del的患者(P2、P3和P5),同一个患者的多个高代谢活性肿瘤细胞的拷贝数变异存在差异,可细分为若干亚群,患者之间的CNV变异类型也完全不一样,由此推测这样的差异可能是导致患者出现的不同转移和耐药情况的原因。针对转移性肿瘤细胞之前的研究主要有两种观点,其一是这种差异来自于原位灶本来就不同的细胞亚克隆[36],另一种观点认为是具有高度干性的肿瘤细胞亚群转移之后再形成的不同分子特点[37,38]。从同一个患者(P2和P5)的多个肿瘤细胞CNV结果看,可细分成不同的细胞亚群,虽然其突变类型一致,但是其基因组上明显的分群现象可能与肿瘤后期进展相关。恶性胸腔积液中高代谢活性肿瘤细胞应该同时具备这两种情况。本研究认为一些早期转移的患者(P3)其基因组水平变异是比较平的,显著区别于P1和P4高度变异的情况,这对于临床医生针对性治疗和最大程度使患者获益有极大的帮助。
综上所述,本研究建立了一整套针对肺癌患者恶性胸腔积液中转移性肿瘤细胞的单细胞分析方案。利用代谢标志物分选高代谢活性肿瘤细胞避免了胸腔积液中其他杂细胞的遗传信息干扰,从单细胞角度解析了同一患者多个转移性肿瘤细胞在基因组水平上的一致性,而相同驱动基因的患者之间存在显著差异,认为其差异可能导致患者在转移和耐药方面存在差异;而同一患者不同的细胞亚群可能是早期转移患者仍然存在潜在过渡态细胞群体。本文对于肿瘤异质性的研究尤其是对转移性肿瘤细胞的异质性研究将有望对肺癌患者临床治疗,耐药监测和复发转移提供更精确的生物学信息。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:24639115Magsci [本文引用: 1]

According to the World Cancer Research Foundation, the newly diagnosed annual lung cancer cases all over the world are alarmingly high at 12.5 %. It also shows the highest mortality rate among all the cancer types. Nearly 225,000 new lung cancer patients are reported annually in the USA. The lung cancer cells also have very fast growth rates. As a result of this rapid proliferation rate, the lung cancer cells are sensitive to the available therapeutics like the radiation, surgical, or chemo therapy. Notwithstanding all the advances in the field of tumor biology, the mortality rate with lung cancer has remained significantly high. Precise and early diagnosis of the disease can be an important step in the proper and successful setting up of the treatment modalities. There are no comprehensive reviews available that discusses all the basic and updated aspects of lung cancer. This review focuses on the basic aspects of lung cancer like the etiology, risk factors, and clonal evolution. Exposure to smoking comes up as a single major environmental cause of the disease. The classification of lung cancer has also been discussed in detail based on immunohistochemistry. The existing therapeutic approaches as well as the upcoming modern day interventions have been discussed with their pros and cons. Recent techniques like molecular profiling can prove to be highly beneficial if properly standardized. With such advancements in therapy in conjunction with the updated diagnostics, there is a real hope in the treatment of lung cancer.
URLPMID:25315013 [本文引用: 1]
The term heterogeneity covers many aspects of the variability in tumor phenotypes, which are a characteristic of human malignancies. Morphologists of the late 19 th century first described the multiple cell types composing tumors and began to recognize cancers of different types. Over the past half century the molecular underpinnings of the variability in human cancers has been gradually revealed but within the last 5 years there has been an explosion in our ability to determine and learn from cancer heterogeneity, through the use of next-generation sequencing and related methods. The complexity and variation in the structure of cancers can seem daunting, but important lessons in cancer biology and the approaches to therapy can be learned from studying how much of the complexity is subject to change and how much is a consequence of stochastic rather than deterministic processes. The evolution of clones, individual variation in response to therapy, distinct biological subtypes of cancer ...
URLPMID:23002210 [本文引用: 1]
Recent technologic advances have permitted higher resolution and more rapid analysis of individual cancer genomes at the single-nucleotide level. Such advances have shown bewildering intertumor heterogeneity with limited somatic alterations shared between tumors of the same histopathologic subtype. Exacerbating such complexity, increasing evidence of intratumor genetic heterogeneity (ITH) is emerging, both within individual tumor biopsies and spatially separated between biopsies of the same tumor. Sequential analysis of tumors has also revealed evidence that ITH temporally evolves during the disease course. ITH has implications for predictive or prognostic biomarker strategies, where the tumor subclone that may ultimately influence therapeutic outcome may evade detection because of its absence or presence at low frequency at diagnosis or because of its regional separation from the tumor biopsy site. In this review, the implications of "trunk and branch" tumor evolution for drug discovery approaches and emerging evidence that low-frequency somatic events may drive tumor growth through paracrine signaling fostering a tumor ecologic niche are discussed. The concept of an "actionable mutation" is considered within a model of clonal dominance and heterogeneous tumor cell dependencies. Evidence that cancer therapeutics may augment ITH and the need to track the tumor subclonal architecture through treatment are defined as key research areas. Finally, if combination therapeutic approaches to limit the consequences of ITH prove challenging, identification of drivers or suppressors of ITH may provide attractive therapeutic targets to limit tumor evolutionary rates and adaptation.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:29215634 [本文引用: 1]
Abstract The development of personalized cancer therapy depends on a robust system to monitor the patient's individual response to anticancer treatment. Anticancer drug efficacy has been tested on circulating tumor cells (CTCs) derived from patient blood samples after ex vivo expansion into CTC clusters. Current attempts to culture these primary cancer cells focus on long-term maintenance under growth factor supplements into cell lines, which usually takes >6 months and results in a CTC expansion efficiency of <20%. We recently developed a simple but unique microfluidics-based culture approach that requires minimal preprocessing (6530 min) and does not require prior enrichment of CTCs or depend on the use of growth factor supplements. The approach capitalizes on co-culture of immune cells from the same patient blood sample within specially designed microwells that promote CTC cluster formation within 2 weeks, with an overall cluster formation success rate of 6550%. Drug screening is facilitated by the incorporation of a gradient generator for parallel exposure to two or more drugs at various concentrations. Owing to the cost-effectiveness and less-invasive nature of this procedure, routine monitoring of disease progression can be achieved. The described microfluidics system can be operated with a single syringe pump to introduce drug compounds (which takes 656 min), followed by incubation of the CTC clusters for 48 h before analysis. In addition to its applications in biomedical research, the rapid readout of our platform will enable clinicians to assess or predict a patient's response to various therapeutic strategies, so as to enable personalized or precision therapy.
URLPMID:4878653 [本文引用: 1]
Original Article from The New England Journal of Medicine — Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing
URLPMID:22385958Magsci [本文引用: 1]
Whole-exome sequencing of 25 tumor and somatic cells from a single patient with clear cell renal cell carcinoma (ccRCC) reveals the heterogeneity of cancer cells in a tumor that does not exhibit driver mutations in the two most commonly mutated genes in ccRCC. This analysis points to the importance of understanding the mutation landscape in individual patients to make informed treatment decisions and may pave the way to discovery of new driver mutations.
[本文引用: 1]
URLPMID:21399628 [本文引用: 1]
Abstract Genomic analysis provides insights into the role of copy number variation in disease, but most methods are not designed to resolve mixed populations of cells. In tumours, where genetic heterogeneity is common, very important information may be lost that would be useful for reconstructing evolutionary history. Here we show that with flow-sorted nuclei, whole genome amplification and next generation sequencing we can accurately quantify genomic copy number within an individual nucleus. We apply single-nucleus sequencing to investigate tumour population structure and evolution in two human breast cancer cases. Analysis of 100 single cells from a polygenomic tumour revealed three distinct clonal subpopulations that probably represent sequential clonal expansions. Additional analysis of 100 single cells from a monogenomic primary tumour and its liver metastasis indicated that a single clonal expansion formed the primary tumour and seeded the metastasis. In both primary tumours, we also identified an unexpectedly abundant subpopulation of genetically diverse 'pseudodiploid' cells that do not travel to the metastatic site. In contrast to gradual models of tumour progression, our data indicate that tumours grow by punctuated clonal expansions with few persistent intermediates. 2011 Macmillan Publishers Limited. All rights reserved
URLPMID:22000009Magsci [本文引用: 1]
Metastases represent the end products of a multistep cell-biological process termed the invasion-metastasis cascade, which involves dissemination of cancer cells to anatomically distant organ sites and their subsequent adaptation to foreign tissue microenvironments. Each of these events is driven by the acquisition of genetic and/or epigenetic alterations within tumor cells and the co-option of nonneoplastic stromal cells, which together endow incipient metastatic cells with traits needed to generate macroscopic metastases. Recent advances provide provocative insights into these cell-biological and molecular changes, which have implications regarding the steps of the invasion-metastasis cascade that appear amenable to therapeutic targeting.
[本文引用: 1]
[本文引用: 1]
URLPMID:7793501 [本文引用: 1]
Abstract: Malignant pleural effusions (MPEs) complicate the clinical course of patients with a broad array of malignancies, which are most often due to lymphomas or carcinomas of the breast, lung, gastrointestinal tract or ovaries. Patients may present with a MPE as the initial manifestation of a cancer or develop an effusion during the advanced phases of a known malignancy. In either circumstance, the median survival after presentation with a MPE is 4 months. Effusions may result from direct pleural invasion (MPE) or indirect effects (paraneoplastic effusions), such as impairment of fluid efflux from the pleural space by lymphatic obstruction or pleural effects of cancer radiation or drug therapy. Because only 50% of patients with cancer who develop a pleural effusion during their clinical course have a MPE, careful evaluation of the effusion to establish its aetiology is required to direct therapy. Management is palliative with interventions directed towards decreasing the volume of intrapleural fluid and the severity of associated symptoms.
[本文引用: 1]
URLPMID:28223509 [本文引用: 1]
Abstract Malignant pleural effusion (MPE), the presence of malignant cells in pleural fluid, is often the first sign of many cancers and occurs in patients with metastatic malignancies. Accurate detection of tumor cells in pleural fluid is crucial because the presence of MPE denotes an advanced stage of disease and directs a switch in clinical managements. Cytology, as a traditional diagnostic tool, has limited sensitivity especially when tumor cells are not abundant, and may be confounded by reactive mesothelial cells in the pleural fluid. We describe a highly sensitive approach for rapid detection of metabolically active tumor cells in MPE via exploiting the altered glucose metabolism of tumor cells relative to benign cells. Metabolically active tumor cells with high glucose uptake, as evaluated by a fluorescent glucose analog (2-NBDG), are identified by high-throughput fluorescence screening within a chip containing 200,000 addressable microwells and collected for malignancy confirmation via single-cell sequencing. We demonstrate the utility of this approach through analyzing MPE from a cohort of lung cancer patients. Most candidate tumor cells identified are confirmed to harbor the same driver oncogenes as their primary lesions. In some patients, emergence of secondary mutations that mediate acquired resistance to ongoing targeted therapies is also detected before resistance is manifested in the clinical imaging. The detection scheme can be extended to analyze peripheral blood samples. Our approach may serve as a valuable complement to cytology in MPE diagnosis, helping identify the driver oncogenes and resistance-leading mutations for targeted therapies.
[本文引用: 1]
[本文引用: 1]
URLPMID:4272008 [本文引用: 1]
De novo copy-number variants (CNVs) can cause neuropsychiatric disease, but the degree to which they occur somatically, and during development, is unknown. Single-cell whole-genome sequencing (WGS) in >200 single cells, including >160 neurons from three normal and two pathological human brains, sensitively identified germline trisomy of chromosome 18 but found most (>= 95%) neurons in normal brain tissue to be euploid. Analysis of a patient with hemimegalencephaly (HMG) due to a somatic CNV of chromosome 1q found unexpected tetrasomy 1q in similar to 20% of neurons, suggesting that CNVs in a minority of cells can cause widespread brain dysfunction. Single-cell analysis identified large (>1 Mb) clonal CNVs in lymphoblasts and in single neurons from normal human brain tissue, suggesting that some CNVs occur during neurogenesis. Many neurons contained one or more large candidate private CNVs, including one at chromosome 15q13.2-13.3, a site of duplication in neuropsychiatric conditions. Large private and clonal somatic CNVs occur in normal and diseased human brains.
Magsci [本文引用: 1]
Motivation: Although many next-generation sequencing (NGS) read preprocessing tools already existed, we could not find any tool or combination of tools that met our requirements in terms of flexibility, correct handling of paired-end data and high performance. We have developed Trimmomatic as a more flexible and efficient preprocessing tool, which could correctly handle paired-end data.<br/>Results: The value of NGS read preprocessing is demonstrated for both reference-based and reference-free tasks. Trimmomatic is shown to produce output that is at least competitive with, and in many cases superior to, that produced by other tools, in all scenarios tested.
[本文引用: 1]
URL [本文引用: 1]
PubMed comprises more than 23 million citations for biomedical literature from MEDLINE, life science journals, and online books. Citations may include links to full-text content from PubMed Central and publisher web sites.
URLPMID:17234643 [本文引用: 1]
Array CGH technologies enable the simultaneous measurement of DNA copy number for thousands of sites on a genome. We developed the circular binary segmentation (CBS) algorithm to divide the genome into regions of equal copy number. The algorithm tests for change-points using a maximal t-statistic with a permutation reference distribution to obtain the corresponding P-value. The number of computations required for the maximal test statistic is O(N2), where N is the number of markers. This makes the full permutation approach computationally prohibitive for the newer arrays that contain tens of thousands markers and highlights the need for a faster algorithm.We present a hybrid approach to obtain the P-value of the test statistic in linear time. We also introduce a rule for stopping early when there is strong evidence for the presence of a change. We show through simulations that the hybrid approach provides a substantial gain in speed with only a negligible loss in accuracy and that the stopping rule further increases speed. We also present the analyses of array CGH data from breast cancer cell lines to show the impact of the new approaches on the analysis of real data.An R version of the CBS algorithm has been implemented in the "DNAcopy" package of the Bioconductor project. The proposed hybrid method for the P-value is available in version 1.2.1 or higher and the stopping rule for declaring a change early is available in version 1.5.1 or higher.
[本文引用: 1]
URLPMID:15870435 [本文引用: 1]
Gefitinib is a selective inhibitor of the epidermal growth factor (EGFR) tyrosine kinase, which is overexpressed in many cancers, including non-small-cell lung cancer (NSCLC). We carried out a clinical study to compare the relationship between EGFR gene copy number, EGFR protein expression, EGFR mutations, and Akt activation status as predictive markers for gefitinib therapy in advanced NSCLC. Tumors from 102 NSCLC patients treated daily with 250 mg of gefitinib were evaluated for EGFR status by fluorescence in situ hybridization (FISH), DNA sequencing, and immunohistochemistry and for Akt activation status (phospho-Akt [P-Akt]) by immunohistochemistry. Time to progression, overall survival, and 95% confidence intervals (CIs) were calculated and evaluated by the Kaplan-Meier method; groups were compared using the log-rank test. Risk factors associated with survival were evaluated using Cox proportional hazards regression modeling and multivariable analysis. All statistical tests were two-sided. Amplification or high polysomy of the EGFR gene (seen in 33 of 102 patients) and high protein expression (seen in 58 of 98 patients) were statistically significantly associated with better response (36% versus 3%, mean difference = 34%, 95% CI = 16.6 to 50.3; P<.001), disease control rate (67% versus 26%, mean difference = 40.6%, 95% CI = 21.5 to 59.7; P<.001), time to progression (9.0 versus 2.5 months, mean difference = 6.5 months, 95% CI = 2.8 to 10.3; P<.001), and survival (18.7 versus 7.0 months, mean difference = 11.7 months, 95% CI = 2.1 to 21.4; P = .03). EGFR mutations (seen in 15 of 89 patients) were also statistically significantly related to response and time to progression, but the association with survival was not statistically significant, and 40% of the patients with mutation had progressive disease. In multivariable analysis, only high EGFR gene copy number remained statistically significantly associated with better survival (hazard ratio = 0.44, 95% CI = 0.23 to 0.82). Independent of EGFR assessment method, EGFR+/P-Akt+ patients had a statistically significantly better outcome than EGFR-, P-Akt-, or EGFR+/P-Akt- patients. High EGFR gene copy number identified by FISH may be an effective molecular predictor for gefitinib efficacy in advanced NSCLC.
[本文引用: 1]
URLPMID:11818139 [本文引用: 1]
An increasing number of human diseases are recognized to result from recurrent DNA rearrangements involving unstable genomic regions. These are termed genomic disorders, in which the clinical phenotype is a consequence of abnormal dosage of gene(s) located within the rearranged genomic fragments. Both inter- and intrachromosomal rearrangements are facilitated by the presence of region-specific low-copy repeats (LCRs) and result from nonallelic homologous recombination (NAHR) between paralogous genomic segments. LCRs usually span apprx10-400 kb of genomic DNA, share gtoreq97% sequence identity, and provide the substrates for homologous recombination, thus predisposing the region to rearrangements. Moreover, it has been suggested that higher order genomic architecture involving LCRs plays a significant role in karyotypic evolution accompanying primate speciation.
URLPMID:2563634 [本文引用: 1]
The human genome contains approximately 50,000 copies of an interspersed repeat with the sequence (dT-dG)n, where n = approximately 10-60. In humans, (TG)n repeats have been found in several sequenced regions. Since minisatellite regions with larger repeat elements often display extensive length polymorphisms, we suspected that (TG)n repeats ("microsatellites") might also be polymorphic. Using the polymerase chain reaction to amplify a (TG)n microsatellite in the human cardiac actin gene, we detected 12 different allelic fragments in 37 unrelated individuals, 32 of whom were heterozygous. Codominant Mendelian inheritance of fragments was observed in three families with a total of 24 children. Because of the widespread distribution of (TG)n microsatellites, polymorphisms of this type may be generally abundant and present in regions where minisatellites are rare, making such microsatellite loci very useful for linkage studies in humans.
[本文引用: 1]
URLPMID:28977762 [本文引用: 1]
Lung cancer heterogeneity plays an important role in the development of drug resistance. Comprehensive molecular characterizations of lung cancer can describe hereditary and somatic gene changes, mutation, and heterogeneity. We discuss heterogeneity specificity, characterization, and roles of PIK3CD, TP53, and KRAS, as well as target-driven therapies and strategies applied in clinical trials based on a proposed precise self-validation system. The system is a specifically selected strategy of treatment for patients with cancer gene mutations and heterogeneity based on gene sequencing, following validation of the strategies in the patient's own cancer cells or in patient-derived xenografts using their own cancer cells isolated during surgery or biopsies. These results will be more precise if the drugs used in the strategies are selected through protein structure-guided compound screening or a DNA-encoded chemical library before validation in the patient's own cancer cells. Thus, a deeper understanding of heterogeneity mechanisms and improved validation of the therapeutic strategy will result in more precise treatments for patients.
Magsci [本文引用: 1]
Cancers evolve by a reiterative process of clonal expansion, genetic diversification and clonal selection within the adaptive landscapes of tissue ecosystems. The dynamics are complex, with highly variable patterns of genetic diversity and resulting clonal architecture. Therapeutic intervention may destroy cancer clones and erode their habitats, but it can also inadvertently provide a potent selective pressure for the expansion of resistant variants. The inherently Darwinian character of cancer is the primary reason for this therapeutic failure, but it may also hold the key to more effective control.
[本文引用: 1]
[本文引用: 1]
URLPMID:11357145 [本文引用: 1]
Abstract Throughout the entire process of cancer aetiology, progression and metastasis, the microenvironment of the local host tissue can be an active participant. Invasion occurs within a tumour-host microecology, where stroma and tumour cells exchange enzymes and cytokines that modify the local extracellular matrix, stimulate migration, and promote proliferation and survival. A new class of cancer therapies that targets this pathological communication interface between tumour cells and host cells is currently under development.
URLPMID:15329723 [本文引用: 1]
Metastasis is a major factor in the malignancy of cancers, and is often responsible for the failure of cancer treatment. Anoikis (apoptosis resulting from loss of cell-matrix interactions) has been suggested to act as a physiological barrier to metastasis; resistance to anoikis may allow survival of cancer cells during systemic circulation, thereby facilitating secondary tumour formation in distant organs. In an attempt to identify metastasis-associated oncogenes, we designed an unbiased, genome-wide functional screen solely on the basis of anoikis suppression. Here, we report the identification of TrkB, a neurotrophic tyrosine kinase receptor, as a potent and specific suppressor of caspase-associated anoikis of non-malignant epithelial cells. By activating the phosphatidylinositol-3-OH kinase/protein kinase B pathway, TrkB induced the formation of large cellular aggregates that survive and proliferate in suspension. In mice, these cells formed rapidly growing tumours that infiltrated lymphatics and blood vessels to colonize distant organs. Consistent with the ability of TrkB to suppress anoikis, metastases--whether small vessel infiltrates or large tumour nodules--contained very few apoptotic cells. These observations demonstrate the potent oncogenic effects of TrkB and uncover a specific pro-survival function that may contribute to its metastatic capacity, providing a possible explanation for the aggressive nature of human tumours that overexpress TrkB.
[本文引用: 1]
[本文引用: 1]
URLPMID:2509068 [本文引用: 1]
The effects of i.p. versus i.v. administration on laser Doppler flow (LDF) were studied in peripheral tissue areas of FSaII implanted s.c. in the hind foot dorsum and in normal skin of conscious C3Hf/Sed . LDF was monitored prior to and continuously for 90 min following the administration of , , or at doses of 5 or 10 mg/g. Results showed that i.p. administration of hyperosmolar solutions was followed by a substantial, dose-dependent flow reduction which was indistinguishable for the various agents at equal osmotic load, and similar in tissue and normal skin. Reductions in LDF are, therefore, primarily caused by hypovolemic hemoconcentration following i.p. administration of hyperosmolar sugar solutions. In contrast, i.v. administration of these solutions at 5 mg/g caused an initial flow increase (most probably due to a transient hypervolemic hemodilution), with a return to baseline readings within 5-10 min. At 10 mg/g i.v., a biphasic change in LDF occurred with an initial, temporary increase and a significant decline thereafter with no recovery within the observation period. This drop in LDF most probably is due to a decrease in cardiac output and an increase in viscous resistance to flow. Since comparable changes were observed with all agents and in both tissues investigated, it is concluded that the alterations in flow pattern following injection of hyperosmolar solutions are neither nor tissue specific. - or -specific effects, if present at all, must be of secondary importance in the animal model chosen.
URLPMID:19737509 [本文引用: 1]
The identification and characterization of cancer stem cells might lead to more effective treatments for some cancers by focusing therapy on the most malignant cells. To achieve this goal it will be necessary to determine which cancers follow a cancer stem cell model and which do not, to address technical issues related to tumorigenesis assays, and to test the extent to which cancer cell heterogeneity arises from genetic versus epigenetic differences.
URLPMID:14522905 [本文引用: 1]
Most current research on is focused on the molecular and cellular analysis of the bulk mass. However, there is overwhelming evidence in some that the clone is heterogeneous with respect to proliferation and differentiation. In , the clone is organized as a hierarchy that originates from rare leukemic stem cells that possess extensive proliferative and self-renewal potential, and are responsible for maintaining the clone. We report here the identification and purification of a stem cell from of different phenotypes that possesses a marked capacity for proliferation, self-renewal, and differentiation. The increased self-renewal capacity of the stem cell (BTSC) was highest from the most aggressive clinical samples of compared with low-grade . The BTSC was exclusively isolated with the expressing the neural stem marker CD133. These CD133+ cells could differentiate in culture into cells that phenotypically resembled the from the patient. The identification of a BTSC provides a powerful tool to investigate the tumorigenic process in the central nervous system and to develop therapies targeted to the BTSC.
URLPMID:17283135 [本文引用: 1]
Emerging evidence has suggested that the capability of a tumor to grow and propagate is dependent on a small subset of cells within a tumor, termed cancer stem cells. Although data have been provided to support this theory in human blood, brain, and breast cancers, the identity of pancreatic cancer stem cells has not been determined. Using a xenograft model in which primary human pancreatic adenocarcinomas were grown in immunocompromised mice, we identified a highly tumorigenic subpopulation of pancreatic cancer cells expressing the cell surface markers CD44, CD24, and epithelial-specific antigen (ESA). Pancreatic cancer cells with the CD44(+)CD24(+)ESA(+) phenotype (0.2-0.8% of pancreatic cancer cells) had a 100-fold increased tumorigenic potential compared with nontumorigenic cancer cells, with 50% of animals injected with as few as 100 CD44(+)CD24(+)ESA(+) cells forming tumors that were histologically indistinguishable from the human tumors from which they originated. The enhanced ability of CD44(+)CD24(+)ESA(+) pancreatic cancer cells to form tumors was confirmed in an orthotopic pancreatic tail injection model. The CD44(+)CD24(+)ESA(+) pancreatic cancer cells showed the stem cell properties of self-renewal, the ability to produce differentiated progeny, and increased expression of the developmental signaling molecule sonic hedgehog. Identification of pancreatic cancer stem cells and further elucidation of the signaling pathways that regulate their growth and survival may provide novel therapeutic approaches to treat pancreatic cancer, which is notoriously resistant to standard chemotherapy and radiation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}