2.
3.
Research progress of super enhancer in cancer
Zhiqiang Wu1,2, Zeyun Mi31. 2.
3.
编委: 方向东
收稿日期:2018-07-13修回日期:2018-09-21网络出版日期:2019-01-20
| 基金资助: |
Editorial board:
Received:2018-07-13Revised:2018-09-21Online:2019-01-20
| Fund supported: |
作者简介 About authors
吴志强,博士,讲师,研究方向:放射生物学,肿瘤分子生物学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (652KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吴志强, 米泽云. 超级增强子在肿瘤研究中的进展[J]. 遗传, 2019, 41(1): 41-51 doi:10.16288/j.yczz.18-152
Zhiqiang Wu, Zeyun Mi.
20世纪80年代,研究发现SV40病毒的一段DNA序列对于家兔(Oryctolagus cuniculus)的β-珠蛋白(β-globin,一种能够通过铁卟啉环可逆性结合氧的呼吸性蛋白质)的转录具有增强作用,因此将这一段DNA称为增强子(enhancer)[1]。随后的研究发现在哺乳动物细胞内也存在类似特性的DNA序列,可以远距离、无方向性的增强基因转录[2,3,4]。近30年研究证明增强子具有以下特征(图1)[2,3,4,5]:(1) 增强子DNA序列处于染色体疏松的区域,与核小体中组蛋白的修饰,转录因子的结合有关;(2) 增强子活性与其DNA序列结合的组蛋白H3的第4位赖氨酸单甲基化(H3K4me1)和第27位赖氨酸乙酰化(H3K27ac)修饰程度成正相关[6];(3) 增强子发挥功能需要增强子区域和启动子的区域的直接相互作用,形成三维环状结构(3D-loop)。增强子和启动子的相互作用由多种蛋白介导,如Mediator复合体、Cohesin等[6,7]。
图1
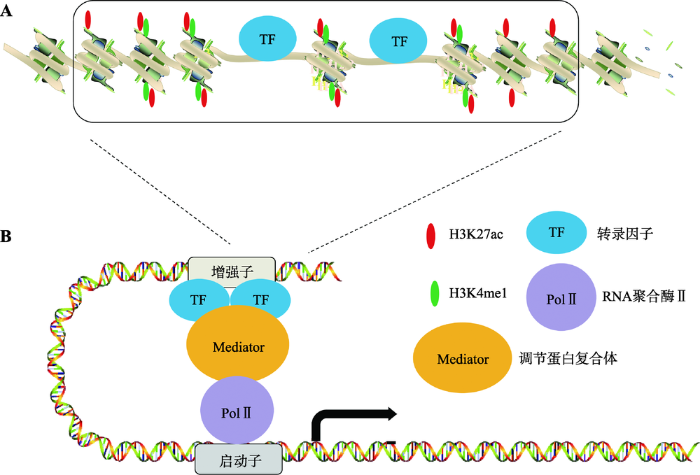 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1增强子的结构特征和功能
A:增强子的结构特征和表观修饰。增强子区域染色体比较疏松,部分DNA暴露并且富集转录因子;增强子区域H3组蛋白具有H3K4me1和H3K27ac修饰。B:转录因子结合到增强子区域之后进一步招募Mediator复合体,其主要介导增强子与PolⅡ相互作用,这样增强子与启动子之间形成三维环状结构,从而增强基因的转录水平。
Fig. 1The structure and function of enhancer
随着DNA测序技术的发展,人类对基因有了进一步的认识,对于增强子的研究也越来越深入。2013年,美国Young R.A.教授(Whitehead Institute for Biomedical Research)基于当时增强子的研究首次提出超级增强子(super enhancers,SEs)这一概念。他们发现胚胎干细胞(embryonic stem cells,ESC)的主要转录因子结合在一些特殊的增强子上,这些特殊的增强子对于维持胚胎干细胞的干性至关重要,并将这些特殊的增强子定义为超级增强子[8]。超级增强子简单来说就是由多个增强子组成的一个大簇,富集高密度的转录因子、辅因子和增强子表观修饰。它和普通增强子在序列大小、转录因子的结合密度、激活转录的能力以及对转录因子抑制剂的敏感性均不同[8]。随后的研究不仅发现超级增强子存在于多种细胞类型中,也进一步明确了超级增强子区别于普通增强子的功能特性(图2)[6]:(1) 超级增强子具有高密度的H3K27ac和H3K4me1修饰,以及Mediator复合体和Bromodomain containing 4蛋白(BRD4,与组蛋白乙酰化修饰位点结合)的结合;(2) 超级增强子结合的转录因子以及与转录活性相关的染色体的标记比普通增强子高很多;(3) 超级增强子调控的基因比普通增强子调控的基因表达水平高很多;(4) 组成超级增强子的单个增强子也可以像普通增强子一样激活基因转录;(5) 超级增强子可以结合组织中特异的转录因子;(6) 与普通增强子相比,超级增强子活性对于转录因子的阻断更敏感[9,10]。这些现象支持一个假说:超级增强子发挥功能需要结合到超级增强子上的转录因子的合作协同,具有大量转录因子结合的增强子对于基因转录的调控会对转录因子浓度的改变更敏感[11](图2)。有趣的是,富集在超级增强子上的主要的转录因子也受超级增强子的调控转录,这就意味着超级增强子调控基因转录存在正反馈协同作用,也就形成了细胞中的核心转录调控环路(core transcription regulatory circuitry, CRC)[12,13]。正是由于超级增强子调控基因表达的特性和其敏感性,因而才能够协调细胞在生长、发育、分化和疾病等各种状态的过渡[9,14~16]。本文主要从超级增强子与肿瘤细胞的关系、在肿瘤细胞中的调控以及该靶点药物在肿瘤治疗中的现状这3个方面阐述超级增强子在肿瘤细胞中的作用。
图2
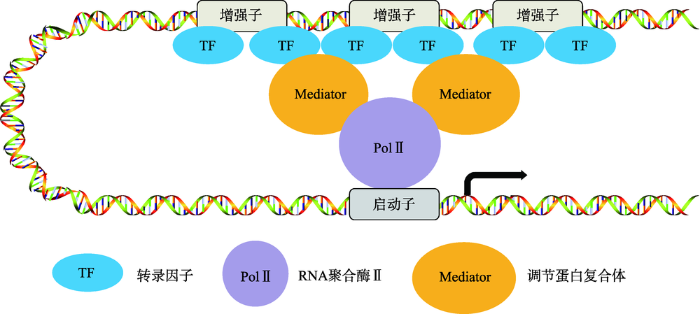 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2超级增强子的结构
超级增强子是由多个增强子组成的一个大簇。组成超级增强子的单个增强子基因组的距离比较近,它们均可以独立的结合转录因子、Mediator复合体等辅因子,共同调控同一启动子的转录活性。
Fig. 2The structure of super enhancer
1 超级增强子与肿瘤的关系
2013年,Young R.A.教授发现在多发性骨髓瘤细胞中超级增强子的区域募集了高浓度的Mediator复合体和BRD4[17],这意味着在多发性骨髓瘤细胞中超级增强子处于活化状态。功能分析实验表明,超级增强子调控的基因(MYC、IRF4、PRDM1、XBP1)对于多发性骨髓瘤的发生和发展起到关键的促进作用[17]。后续研究发现,超级增强子在多种肿瘤中均有报道,如弥漫性大B细胞淋巴瘤[18]、T细胞急性淋巴细胞白血病[19,20]、默克尔细胞癌[21]、急性髓性白血病[22]、小细胞肺癌[10]、卵巢癌[23]、上皮癌[24]、鳞状细胞癌[25]、黑色素瘤[15]、乳腺癌[26]、食管鳞状细胞癌[27]和结肠癌[28]等。在肿瘤细胞中超级增强子调控的关键癌基因在正常细胞中是不表达的,这就提示超级增强子通过调控这些基因而对肿瘤生成和肿瘤特性维持起到关键作用[9,29,30]。由于细胞在癌变过程中大多数的超级增强子是重新形成具有功能性的元件,因此超级增强子的活化可以作为细胞癌变的一种标志[9,29,30]。综上所述目前研究均表明超级 增强子的激活可以促使正常细胞向肿瘤细胞的恶性转化。超级增强子不但对蛋白编码基因具有转录激活作用,对非编码基因,如microRNA (miRNA,一种长度约22nt的小RNA)的转录及成熟也具有调控功能。美国麻省理工学院生物系Phillip A. S.教授研究组利用CRISPR/Cas9基因组编辑方法发现超级增强子不仅促进miRNA的转录,也可以通过招募Drosha/DGCR8蛋白复合体促进前体miRNA (pri- miRNA)的成熟,以此来调控细胞种类特异性miRNA的生成[31]。对18种肿瘤细胞分析发现,在有些肿瘤细胞中超级增强子活性上调,而有些肿瘤细胞中超级增强子活性下降。进一步分析表明在细胞癌变过程中激活的超级增强子往往与促癌miRNA相关,而失活的超级增强子主要调控抑癌miRNA的生成[31]。以上研究提示,调控miRNA的超级增强子活性与肿瘤发生发展密切相关。因此,超级增强子联合多个miRNA (SE-miRNA)将有潜力成为细胞癌变的生物标志物[31],对于肿瘤的早期诊断以及治疗具有重要的临床意义。除此之外,超级增强子还可以调控长链非编码RNA (long non-coding RNA, lncRNA)的转录[32]。在鳞状细胞癌组织中发现受超级增强子调控的lncRNA LINC01503明显上调。进一步研究发现,LINC01503的表达水平与鳞状细胞癌病人预后呈负相关:LINC01503高表达的病人生存率低。研究表明激活的增强子或超级增强子区域也可以被转录产生RNA,称为enhancer RNA (eRNA),eRNA可以协同超级增强子激活转录[33,34]。超级增强子发挥功能不仅依赖于和启动子之间的3D-loop的形成,也依赖于超级增强子转录的eRNA的生成。因此,在临床上可以结合lncRNA以及eRNA的水平对病人进行精准治疗。
肿瘤的异质性很大一方面是由于一个肿瘤内的细胞通常可能来源于多个不同的细胞克隆,而这些不同克隆来源的肿瘤细胞其超级增强子的激活也存在差异,这就为区分肿瘤亚型或肿瘤细胞亚群提供了一种新的鉴定方法。例如,通过以往的方法对成神经管细胞瘤的生物化学和遗传学分析把其分成4个亚型。但是通过对这4个亚型的增强子图谱分析发现了一种新的亚型,这种新型的成神经管细胞瘤细胞中都具有与肿瘤异质性相关的超级增强子群[35]。更为重要的是通过分析在这类肿瘤细胞的超级增强子调控的转录因子可以明确细胞特异性的核心转录调控环路(CRC)。通过对于CRC分析确定了LIM homeobox transcription factor 1 alpha (LMX1A,一种转录因子)在第4类亚型的成神经管细胞瘤是一个主要转录因子(master transcription factor)[36]。同样,在其他基因异质性癌中也发现类似情况,如三阴性乳腺癌依靠超级增强子调控的特异性的基因群来维持细胞生长和增殖[26]。可见通过对于不同肿瘤细胞的增强子的图谱分析可以独立预测肿瘤亚型,发现之前治疗的不足以及新的潜在治疗靶点,为肿瘤治疗提供新思路、新方向[6]。
综上所述,在多种肿瘤细胞中均发现超级增强子处于异常激活状态,其对于靶基因的调控呈多样化:促进mRNA的生成、促进miRNA的转录以及成熟、促进lncRNA的转录生成以及超级增强子自身转录生成的eRNA对于其活性也起到协同作用。除此之外,通过绘制肿瘤细胞的增强子图谱可以预测肿瘤亚型,为基因异质性肿瘤提供统一的治疗平台。
2 肿瘤细胞中超级增强子的调控
在肿瘤细胞中超级增强子的调控是如何实现的呢?早期对于小鼠胚胎干细胞发育的研究提出一个模型:组成超级增强子的每一个增强子都具有活性,而超级增强子的功能类似于一个平台,这个平台汇集了与发育相关的信号通路传递过来的信号,这些信号协同调控超级增强子活性启动基因转录(图3)[11]。同样,与癌基因相关的超级增强子也富集了肿瘤细胞依赖的信号通路的转录因子。在Wnt信号通路异常引起的结肠癌细胞中,相关的超级增强子区域富集了很多由Wnt信号通路终端的转录因子4 (transcription factor 4, TCF4),通过激活或者抑制Wnt信号通路,可以控制超级增强子调控的基因转录[11,37]。在雌激素受体(estrogen receptor, ER)阳性的乳腺癌细胞中,相关的超级增强子区域聚集了大量的ERα;而在三阴性乳腺癌细胞中缺少类固醇激素的表达,与其相关的超级增强子区域富集了完全不同的转录因子[26,37]。图3
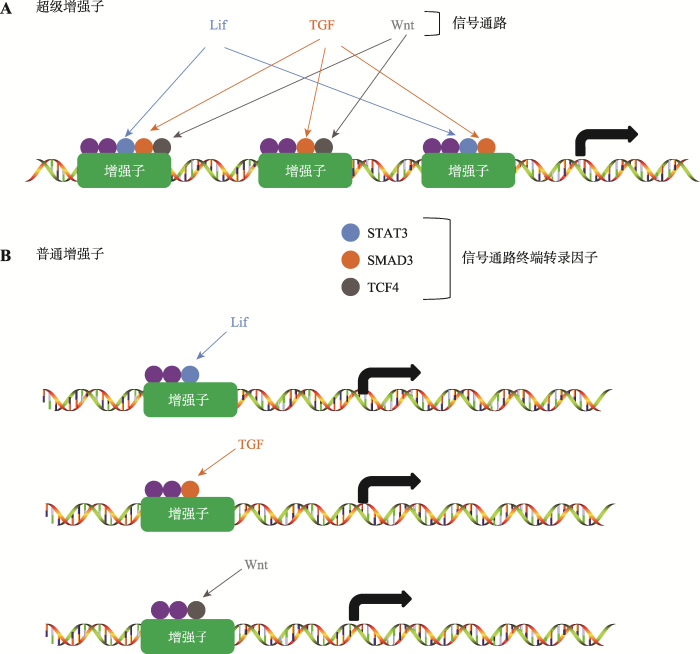 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3信号通路对于超级增强子与普通增强子的调控
A:组成超级增强子的每一个增强子都具有活性,每个增强子富集了各种信号通路终端转录因子,这些转录因子协同调控基因转录;B:普通增强子只富集一种信号通路的终端转录因子。
Fig. 3The regulation of super enhancers and typical enhancers by signal pathway
在肿瘤细胞中,信号通路从多方面对超级增强子的活性进行调控。2015年美国西北大学Licht J.D.教授团队研究发现Ras-Erk活性与超级增强子的活性密切相关:抑制Ras蛋白的活性会导致超级增强子相关的特征(如H3K27ac)消失、活性下降、降低相关基因转录;激活Ras可以增强调控癌基因的超级增强子活性[38]。另一方面,促癌信号通路可以通过操纵转录机器调节超级增强子的活性。转录暂停是激活的RNA聚合酶Ⅱ(RNA polymerase Ⅱ,Pol Ⅱ)在启动子附近停止转录的一种状态[39]。在正常的肝细胞中,Hippo信号通路可以通过限制暂停的Pol Ⅱ释放,因而抑制了增强子或超级增强子调控的基因转录[40]。然而,在肝癌细胞中Hippo信号通路的缺失导致YAP(Yes associated protein)入核,YAP蛋白结合到超级增强子上,招募Mediator复合体和细胞周期素依赖性激酶9(Cyclin-dependent kinase 9,CDK9),使暂停的Pol Ⅱ进入到延伸状态,促进癌基因转录[41]。因此,在肝癌中YAP通过激活超级增强子促进癌基因的转录。
以上研究表明超级增强子可以作为连接癌基因信号通路和维持肿瘤细胞特性的基因转录表达的渠道。然而进一步研究发现信号通路对于超级增强子的调控与转录因子在超级增强子区动态结合有关。例如,在NOTCH1异常导致的T细胞白血病(T-ALL)细胞中,NOTCH1在基因组上具有普遍的结合,但是只有不到10%的基因对于NOTCH1信号通路的改变有应答,而这些应答基因的NOTCH1结合在对应的超级增强子上。如果这些位点丢失NOTCH1的结合就会导致超级增强子的特征消失[42]。
3 针对超级增强子在肿瘤治疗中的作用
在肿瘤细胞中癌基因被转录激活,进而介导细胞的增殖和永生化[29,30]。因此,抑制癌基因的转录是一个潜在的治疗靶点。但是针对这一靶点面临着巨大的挑战:转录是细胞最基本的功能,对癌基因的转录抑制可能会引起细胞基因转录的广谱抑制[43]。因此,临床上用到的转录抑制剂应该特异性的抑制癌基因,对于正常细胞的转录影响不大[6,44~46]。转录起始、暂停、延伸等过程的有序转变都是通过转录因子调控。目前研究发现超级增强子调控的转录依赖于BRD4、Mediator复合体、包含细胞周期素依赖性激酶7 (cyclin-dependent kinase 7, CDK7)的TF ⅡH复合体和包含CDK9的转录延伸复合体(P-TEFb)[39]。CDK7对Pol Ⅱ C末端区域(C terminal domain,CTD)第5位丝氨酸磷酸化起始转录[47];CDK9主要对Pol Ⅱ CTD 第2位丝氨酸磷酸化促进转录暂停的Pol Ⅱ进入转录延伸阶段,也称作Pol Ⅱ的释放[39]。另外,BRD4通过招募Mediator复合体促进超级增强子的装配进而促进暂停状态的Pol II的释放[48]。CDK12/13可以加速Pol Ⅱ的转录延伸[49,50]。因此,目前普遍认为超级增强子调控转录的关键调节点Mediator复合体、BRD4和关键的CDK有利于开发成治疗肿瘤的新靶点[6]。基于上述的转录抑制的关键节点,目前对于超级增强子这一靶点的药物主要有以下几类:(1) 针对BRD家族蛋白的抑制剂或者降解剂;(2) CDK7抑制剂;(3) 其他类型抑制剂(表1)。
Table 1
表1
表1 在肿瘤治疗中以超级增强子为靶点的小分子抑制剂研究现状
Table 1
| 小分子抑制剂 | 靶点 | 疾病 | 临床研究 | 文献 |
|---|---|---|---|---|
| JQ1 | BRD4 | 多发性骨髓瘤 | - | [17] |
| Merkel细胞癌 | - | [21] | ||
| iBET151 | BRD4 | 白血病 | - | [51] |
| iBET762 | BRD2. BRD3. BRD4 | NUT中线癌 | Ⅰ期临床(NCT01587703) | [51] |
| BETd-246 | BRD2. BRD3. BRD4 | 三阴性乳腺癌 | - | [52] |
| OTX015 | BRD2. BRD3. BRD4 | 成神经细胞瘤 | 临床前研究 | [52] |
| 弥漫性大B细胞淋巴瘤 | Ⅰ期临床(NCT01713582) | [52] | ||
| 急性淋巴细胞白血病 | Ⅰ期临床(NCT01713582) | [53,54] | ||
| NUT中线癌 | Ⅰ期临床 (NCT02259114) | [51] | ||
| 多行性成胶质细胞瘤 | Ⅱ期临床(NCT02296476) | [51] | ||
| CPI0610 | BRD4 | 多发性骨髓瘤 | Ⅰ期临床(NCT02157636) | [51] |
| 淋巴瘤 | Ⅰ期临床(NCT01949883) | [51] | ||
| THZ1 | CDK7 | 食管鳞状细胞癌 | - | [27] |
| 成神经细胞瘤 | - | [55] | ||
| 成人T细胞白血病 | - | [56] | ||
| 小细胞肺癌 | - | [10] | ||
| SY-1365 | CDK7 | 成人晚期实体瘤 | Ⅰ期临床(NCT03134638) | Syros公司 |
| 乳腺癌(与氟维司群联合) | 临床前研究 | Syros公司 | ||
| SY-1425 | RARα | 急性髓样细胞样白血病 | Ⅱ期临床(NCT02807558) | Syros公司 |
| 骨髓增生异常综合征 | Ⅱ期临床(NCT02807558) | Syros公司 | ||
| THZ531 | CDK12/13 | 急性T细胞淋巴瘤 | - | [57] |
| Lee011 | CDK4/6 | 尤因肉瘤 | - | [58] |
新窗口打开|下载CSV
JQ1通过与BRD4的bromodomain结构域结 合而抑制BRD4与发生乙酰化修饰的蛋白相互作用[59,60],也就限制了BRD4和超级增强子的H3K27ac位点结合,抑制超级增强子和启动子的相互作用,进而影响癌基因的转录[17]。由于超级增强子所调控的转录对于转录因子的浓度变化特别敏感,JQ1处理可以优先阻止BRD4和超级增强子上的乙酰化修饰位点结合,进而特异性的抑制超级增强子介导的转录激活[9,17]。除此之外,还发现BRD抑制剂iBET762、OTX015、CPI0610和iBET151等,前3者已经进入临床实验阶段[22,51,61]。dBET系列化合物是基于JQ1的化学结构研发的特异性更高的BRD4抑制剂,其可以特异性的介导BRD家族蛋白的降解[62],从而阻止BRD家族蛋白识别超级增强子的乙酰化位点,影响超级增强子活性,抑制转录[63]。研究表明,BETd- 246可以靶向特异的降解BRD家族蛋白,相对于iBET- 211在三阴性乳腺癌中也表现出更好的治疗效果[52]。
THZ1是CDK7特异的抑制剂,依赖于超级增强子介导的肿瘤细胞对于THZ1高度敏感[64]。THZ1可以和CDK7的第312位半胱氨酸共价结合,抑制CDK7的激酶活性,从而抑制CDK7对于Pol Ⅱ CTD的第五位丝氨酸磷酸化,抑制转录起始,进一步阻止Pol Ⅱ在启动子近端暂停。超级增强子主要调控暂停的Pol Ⅱ释放,THZ1处理之后在启动子近端的Pol Ⅱ减少,也减少在增强子处的Pol Ⅱ结合,最终抑制转录[55,64]。THZ1处理之后超级增强子活性下降,导致多种癌基因转录抑制,从而抑制多种肿瘤细胞的生长和增殖[64]。SY-1365是Syros公司研发的CDK7的特异性抑制剂,可以选择性抑制多种实体瘤(乳腺癌、卵巢癌和小细胞肺癌等)和血癌(急性髓细胞样白血病和急性淋巴细胞白血病),目前对于晚期实体瘤的实验处于一期临床阶段。该公司通过对肿瘤细胞的基因检测分析,发现急性髓细胞白血病病人和骨髓增生异常综合征病人具有受超级增强子调控的RARA和IRF8基因的高表达,并且发现SY- 1425可以作用于维甲酸受体α(RARα),在临床上对上述两类病人具有较好的治疗效果。
CDK12是调节转录延伸的一个激酶,在T细胞白血病中THZ531可以特异性抑制CDK12/13,有效抑制超级增强子介导的基因表达[57]。在急性髓性白血病中抑制Mediator激酶(CDK8/19)活性可以上调肿瘤抑制因子相关的超级增强子活性,激活肿瘤抑制基因的表达,最终达到抗白血病的活性[22]。类似的,CDK4/6抑制剂LEE011选择性抑制CDK4,下调cyclin D1相关的超级增强子活性,有效的促进尤因肉瘤细胞的凋亡[58]。
4 结语与展望
目前关于超级增强子的研究,发现超级增强子在多种肿瘤细胞中均有激活,而激活的这些超级增强子往往促进癌基因的产生,维持癌细胞特性。通过抑制CDK、BRD4和Mediator复合体均可以干扰超级增强子的活性。超级增强子的先驱者Young R.A.与JQ1/iBET研发者Bradner J.E.这两位科学家曾预言超级增强子具有广阔的研发前景和价值,必将成为下一个药物研发的黄金靶点,因为针对这一靶点有望开发一种精确影响基因调控元件的药物,为此这两位科学家联手成立Syros公司专门研发针对超级增强子这一靶点的抗癌药。但是到目前为止对于超级增强子各个组分的研究还有欠缺:在癌细胞中组成超级增强子的每个增强子的活性是否和正常细胞中对应的增强子活性一致?信号通路活性的改变是怎样影响单个增强子组装成超级增强子的?从治疗角度考虑,也需要探索清楚超级增强子各个组分之间是怎么发挥作用的,药物是怎样抑制超级增强子各组分的活性。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:6277502 [本文引用: 1]

We have studied the transient expression of a cloned rabbit hemoglobin β1 gene after its introduction into HeLa cells. Two and one-half days after transfection using the calcium phosphate technique, we extracted RNA from the entire cell population and analyzed it by the S1 nuclease hybridization assay. Transcripts were barely detectable when β-globin gene-plasmid recombinants were used. However, 200 times more β-globin gene transcripts were found when the β-globin gene recombinants also contained SV40 DNA, and 90% of these transcripts (about 1000 per cell) had the same 5′ end as authentic rabbit globin mRNA. In the latter case, abundant production of β-globin protein was readily detected in a fraction of transfected cells by immunofluorescent staining. Enhancement of globin gene expression was dependent on SV40 sequences acting in cis, but independent of the viral origin of DNA replication. The enhancing activity was associated with the 72 bp repeated sequence element located at the beginning of the viral late gene region. Viral DNA fragments containing the transcriptional enhancer element could act in either orientation at many positions, including 1400 bp upstream or 3300 bp downstream from the transcription initiation site of the rabbit β-globin gene. These studies define a class of DNA elements with a mode of action that has not been heretofore described. The activation of genes by specific enhancer elements seems to be a widespread mechanism that may be used for the regulation of gene expression.
URLPMID:9679020 [本文引用: 2]
In eukaryotes, transcription of genes by RNA polymerase II yields messenger RNA intermediates from which protein products are synthesized. Transcriptional enhancers are discrete DNA elements that contain specific sequence motifs with which DNA-binding proteins interact and transmit molecular signals to genes. Here, current models regarding the role of enhancers in the regulation of transcription by RNA polymerase II are presented.
URLPMID:3742076 [本文引用: 2]
Biological differences among metazoans and between cell types in a given organism arise in large part due to differences in gene expression patterns. Gene-distal enhancers are key contributors to these expression patterns, exhibiting both sequence diversity and cell type specificity. Studies of long-range interactions indicate that enhancers are often important determinants of nuclear organization, contributing to a general model for enhancer function that involves direct enhancer-promoter contact. However, mechanisms for enhancer function are emerging that do not fit solely within such a model, suggesting that enhancers as a class of DNA regulatory element may be functionally and mechanistically diverse.
URL [本文引用: 2]
超级增强子是具有转录活性增强子的一个大簇,驱动控制细胞身份基因的表达,在发育和肿瘤等疾病发生过程中起到重要作用。和普通增强子相比,许多肿瘤细胞关键致癌基因是由超级增强子驱动,常见疾病如阿尔茨海默病等相关的变异显著富集于超级增强子。超级增强子在关键致癌基因的鉴定、疾病关联变异位点的发现等领域显示了巨大的应用潜力。本文首先概述了如何在全基因组水平进行增强子的鉴定,随后引入超级增强子的概念和鉴定方法,最后阐述了超级增强子的主要结构和功能特征,并对其在研究中的应用做了展望。
URL [本文引用: 2]
超级增强子是具有转录活性增强子的一个大簇,驱动控制细胞身份基因的表达,在发育和肿瘤等疾病发生过程中起到重要作用。和普通增强子相比,许多肿瘤细胞关键致癌基因是由超级增强子驱动,常见疾病如阿尔茨海默病等相关的变异显著富集于超级增强子。超级增强子在关键致癌基因的鉴定、疾病关联变异位点的发现等领域显示了巨大的应用潜力。本文首先概述了如何在全基因组水平进行增强子的鉴定,随后引入超级增强子的概念和鉴定方法,最后阐述了超级增强子的主要结构和功能特征,并对其在研究中的应用做了展望。
URLPMID:20833320 [本文引用: 1]
Regulatory DNAs serve as templates to bring weakly interacting transcription factors into close proximity so they can work synergistically to switch genes on and off in time and space. Most of these regulatory DNAs are enhancers that can work over long distances — a million base pairs or more in mammals — to control gene expression. Critical enhancers are sometimes even found within the introns of neighboring genes. This review summarizes well-defined examples of enhancers controlling key processes in animal development. Potential mechanisms of transcriptional synergy are discussed with regard to enhancer structure and contemporary ChIP-sequencing assays, whereby just a small fraction of the observed binding sites represent bona fide regulatory DNAs. Finally, there is a discussion of how enhancer evolution can produce novelty in animal morphology and of the prospects for reconstructing transitions in animal evolution by introducing derived enhancers in basal ancestors.
URLPMID:28718439 [本文引用: 6]
Transcriptional deregulation is one of the core tenets of cancer biology and is underpinned by alterations in both protein-coding genes and noncoding regulatory elements. Large regulatory elements, so-called super-enhancers (SEs), are central to the maintenance of cancer cell identity and promote oncogenic transcription to which cancer cells become highly addicted. Such dependence on SE-driven transcription for proliferation and survival offers an Achilles heel for the therapeutic targeting of cancer cells. Indeed, inhibition of the cellular machinery required for the assembly and maintenance of SEs dampens oncogenic transcription and inhibits tumor growth. In this article, we review the organization, function, and regulation of oncogenic SEs and their contribution to the cancer cell state.
URLPMID:25677180 [本文引用: 1]
In addition to mediating sister chromatid cohesion during the cell cycle, the cohesin complex associates with CTCF and with active gene regulatory elements to form long-range interactions between its binding sites. Genome-wide chromosome conformation capture had shown that cohesin's main role in interphase genome organization is in mediating interactions within architectural chromosome compartments, rather than specifying compartments per se. However, it remains unclear how cohesin-mediated interactions contribute to the regulation of gene expression. We have found that the binding of CTCF and cohesin is highly enriched at enhancers and in particular at enhancer arrays or "super-enhancers" in mouse thymocytes. Using local and global chromosome conformation capture, we demonstrate that enhancer elements associate not just in linear sequence, but also in 3D, and that spatial enhancer clustering is facilitated by cohesin. The conditional deletion of cohesin from noncycling thymocytes preserved enhancer position, H3K27ac, H4K4me1, and enhancer transcription, but weakened interactions between enhancers. Interestingly, 50% of deregulated genes reside in the vicinity of enhancer elements, suggesting that cohesin regulates gene expression through spatial clustering of enhancer elements. We propose a model for cohesin-dependent gene regulation in which spatial clustering of enhancer elements acts as a unified mechanism for both enhancer-promoter "connections" and "insulation."
URLPMID:23582322 [本文引用: 2]
Master transcription factors Oct4, Sox2, and Nanog bind enhancer elements and recruit Mediator to activate much of the gene expression program of pluripotent embryonic stem cells (ESCs). We report here that the ESC master transcription factors form unusual enhancer domains at most genes that control the pluripotent state. These domains, which we call super-enhancers, consist of clusters of enhancers that are densely occupied by the master regulators and Mediator. Super-enhancers differ from typical enhancers in size, transcription factor density and content, ability to activate transcription, and sensitivity to perturbation. Reduced levels of Oct4 or Mediator cause preferential loss of expression of super-enhancer-associated genes relative to other genes, suggesting how changes in gene expression programs might be accomplished during development. In other more differentiated cells, super-enhancers containing cell-type-specific master transcription factors are also found at genes that define cell identity. Super-enhancers thus play key roles in the control of mammalian cell identity.
URLPMID:24119843 [本文引用: 5]
Super-enhancers in 86 human cell types have been cataloged. Disease-associated sequence variation is enriched in super-enhancers, and cancer cells generate super-enhancers at key tumor pathogenesis genes.
URLPMID:4261156 [本文引用: 2]
Small cell lung cancer (SCLC) is an aggressive disease for which effective therapies are needed. Christensen et al. discover that SCLC is sensitive to THZ1, which is a covalent inhibitor of the transcriptional regulator CDK7 and downregulates the expression of key transcription factors that drive the disease.
URLPMID:4402134 [本文引用: 3]
How large clusters of enhancers, super-enhancers (SEs), drive key cell identity genes is unclear. Hnisz et al. find that SEs contain constituent enhancers that respond to multiple signaling pathways and enhance responsiveness and sensitivity of pluripotency genes in ESCs or tumor-promoting genes in cancer cells to changes in these pathways.
URLPMID:26843070 [本文引用: 1]
Abstract A small set of core transcription factors (TFs) dominates control of the gene expression program in embryonic stem cells and other well-studied cellular models. These core TFs collectively regulate their own gene expression, thus forming an interconnected auto-regulatory loop that can be considered the core transcriptional regulatory circuitry (CRC) for that cell type. There is limited knowledge of core TFs, and thus models of core regulatory circuitry, for most cell types. We recently discovered that genes encoding known core TFs forming CRCs are driven by super-enhancers, which provides an opportunity to systematically predict CRCs in poorly studied cell types through super-enhancer mapping. Here, we use super-enhancer maps to generate CRC models for 75 human cell and tissue types. These core circuitry models should prove valuable for further investigating cell-type-specific transcriptional regulation in healthy and diseased cells. 0008 2016 Saint-Andr0108 et al.; Published by Cold Spring Harbor Laboratory Press.
URLPMID:26829750 [本文引用: 1]
Translocation events are frequent in cancer and may create chimeric fusions or ‘regulatory rearrangements’ that drive oncogene overexpression. Here we identify super-enhancer translocations that drive overexpression of the oncogenic transcription factorMYBas a recurrent theme in adenoid cystic carcinoma (ACC). Whole-genome sequencing data and chromatin maps reveal distinct chromosomal rearrangements that juxtapose super-enhancers to theMYBlocus. Chromosome conformation capture confirms that the translocated enhancers interact with theMYBpromoter. Remarkably, MYB protein binds to the translocated enhancers, creating a positive feedback loop that sustains its expression. MYB also binds enhancers that drive different regulatory programs in alternate cell lineages in ACC, cooperating with TP63 in myoepithelial cells and a Notch program in luminal epithelial cells. Bromodomain inhibitors slow tumor growth in ACC primagraft modelsin vivo. Thus, our study identifies super-enhancer translocations that driveMYBexpression and provides insight into downstream MYB functions in the alternate ACC lineages.
URLPMID:26689127 [本文引用: 1]
Adult stem cells coordinate niche signals and chromatin states to choose appropriate fates. Upon changes in the local niche environment, stem cells remodel chromatin to survive in transitional states, before undergoing fate selection. Epigenetic repressors put a brake on precocious lineage commitment. DNA methylation and Polycomb-silencing complexes cooperate to ensure that stem cells are robustly maintained during homeostasis. Cell identity depends on combinatorial transcription factor complexes on lineage-specific enhancers. Pioneer factors select unique enhancer repertoires by making condensed chromatin accessible for robust gene activation. Epigenetic memory is achieved by coupling pioneer factors with super-enhancers, allowing stem cells to retain their unique identities in different microenvironments.
URLPMID:4868069 [本文引用: 1]
The "cancerized field" concept posits that cancer-prone cells in a given tissue share an oncogenic mutation, but only discreet clones within the field initiate tumors. Most benign nevi carry oncogenic BRAF(V600E) mutations but rarely become melanoma. The zebrafish crestin gene is expressed embryonically in neural crest progenitors (NCPs) and specifically reexpressed in melanoma. Live imaging of transgenic zebrafish crestin reporters shows that within a cancerized field (BRAF(V600E)-mutant; p53-deficient), a single melanocyte reactivates the NCP state, revealing a fate change at melanoma initiation in this model. NCP transcription factors, including sox10, regulate crestin expression. Forced sox10 overexpression in melanocytes accelerated melanoma formation, which is consistent with activation of NCP genes and super-enhancers leading to melanoma. Our work highlights NCP state reemergence as a key event in melanoma initiation.
URLPMID:4636788 [本文引用: 1]
Transcriptional enhancers are frequently bound by a set of transcription factors that collaborate to activate lineage-specific gene expression. Recently, it was appreciated that a subset of enhancers comprise extended clusters dubbed stretch- or super-enhancers (SEs). These SEs are located near key cell identity genes, and enriched for non-coding genetic variations associated with disease. Previously, SEs have been defined as having the highest density of Med1, Brd4 or H3K27ac by ChIP-seq. The histone acetyltransferase P300 has been used as a marker of enhancers, but little is known about its binding to SEs. We establish that P300 marks a similar SE repertoire in embryonic stem cells as previously reported using Med1 and H3K27ac. We also exemplify a role for SEs in mouse T helper cell fate decision. Similarly, upon activation of macrophages by bacterial endotoxin, we found that many SE-associated genes encode inflammatory proteins that are strongly up-regulated. These SEs arise from small, low-density enhancers in unstimulated macrophages. We also identified expression quantitative trait loci (eQTL) in human monocytes that lie within such SEs. In macrophages and Th17 cells, inflammatory SEs can be perturbed either genetically or pharmacologically thus revealing new avenues to target inflammation. Our findings support the notion that P300-marked SEs can help identify key nodes of transcriptional control during cell fate decisions. The SE landscape changes drastically during cell differentiation and cell activation. As these processes are crucial in immune responses, SEs may be useful in revealing novel targets for treating inflammatory diseases. The online version of this article (doi:10.1186/s12864-015-1905-6) contains supplementary material, which is available to authorized users.
URLPMID:23582323 [本文引用: 4]
A small set of super-enhancers associated with oncogenes such as MYC was co-occupied by BRD4 and mediator in multiple myeloma. Inhibition of BRD4 leads to selective repression of these genes.
URLPMID:4018722 [本文引用: 1]
Diffuse large B cell lymphoma (DLBCL) is a biologically heterogeneous and clinically aggressive disease. Here, we explore the role of bromodomain and extra-terminal domain (BET) proteins in DLBCL, using integrative chemical genetics and functional epigenomics. We observe highly asymmetric loading of bromodomain 4 (BRD4) at enhancers, with approximately 33% of all BRD4 localizing to enhancers at 1.6% of occupied genes. These super-enhancers prove particularly sensitive to bromodomain inhibition, explaining the selective effect of BET inhibitors on oncogenic and lineage-specific transcriptional circuits. Functional study of genes marked by super-enhancers identifies DLBCLs dependent on OCA-B and suggests a strategy for discovering unrecognized cancer dependencies. Translational studies performed on a comprehensive panel of DLBCLs establish a therapeutic rationale for evaluating BET inhibitors in this disease.
URLPMID:25194570 [本文引用: 1]
Efforts to identify and annotate cancer driver genetic lesions have been focused primarily on the analysis of protein-coding genes; however, most genetic abnormalities found in human cancer are located in intergenic regions. Here we identify a new long range-acting MYC enhancer controlled by NOTCH1 that is targeted by recurrent chromosomal duplications in human T cell acute lymphoblastic leukemia (T-ALL). This highly conserved regulatory element, hereby named N-Me for NOTCH MYC enhancer, is located within a broad super-enhancer region +1.47 Mb from the MYC transcription initiating site, interacts with the MYC proximal promoter and induces orientation-independent MYC expression in reporter assays. Moreover, analysis of N-Me knockout mice demonstrates a selective and essential role of this regulatory element during thymocyte development and in NOTCH1-induced T-ALL. Together these results identify N-Me as a long-range oncogenic enhancer implicated directly in the pathogenesis of human leukemia and highlight the importance of the NOTCH1-MYC regulatory axis in T cell transformation and as a therapeutic target in T-ALL.
URLPMID:4720521 [本文引用: 1]
In certain human cancers, the expression of critical oncogenes is driven from large regulatory elements, called super-enhancers, that recruit much of the cell's transcriptional apparatus and are defined by extensive acetylation of histone H3 lysine 27 (H3K27ac). In a subset of T-cell acute lymphoblastic leukemia (T-ALL) cases, we found that heterozygous somatic mutations are acquired that introduce binding motifs for the MYB transcription factor in a precise noncoding site, which creates a super-enhancer upstream of the TAL1 oncogene. MYB binds to this new site and recruits its H3K27 acetylase-binding partner CBP, as well as core components of a major leukemogenic transcriptional complex that contains RUNX1, GATA-3, and TAL1 itself. Additionally, most endogenous super-enhancers found in T-ALL cells are occupied by MYB and CBP, which suggests a general role for MYB in super-enhancer initiation. Thus, this study identifies a genetic mechanism responsible for the generation of oncogenic super-enhancers in malignant cells.
URLPMID:25941994 [本文引用: 1]
Pathologic c-Myc expression is frequently detected in human cancers, including Merkel cell carcinoma (MCC), an aggressive skin cancer with no cure for metastatic disease. Bromodomain protein 4 (BRD4) regulates gene transcription by binding to acetylated histone H3 lysine 27 (H3K27Ac) on the chromatin. Super-enhancers of transcription are identified by enrichment of H3K27Ac. BET inhibitor JQ1 disrupts BRD4 association with super-enhancers, downregulates proto-oncogenes, such as c-Myc, and displays antitumor activity in preclinical animal models of human cancers. Here we show that an enhancer proximal to the c-Myc promoter is enriched in H3K27Ac and associated with high occupancy of BRD4, and coincides with a putative c-Myc super-enhancer in MCC cells. This observation is mirrored in tumors from MCC patients. Importantly, depleted BRD4 occupancy at the putative c-Myc super-enhancer region by JQ1 correlates with decreased c-Myc expression. Thus, our study provides initial evidence that super-enhancers regulate c-Myc expression in MCC.
URLPMID:26416749 [本文引用: 3]
Super-enhancers (SEs), which are composed of large clusters of enhancers densely loaded with the Mediator complex, transcription factors and chromatin regulators, drive high expression of genes implicated in cell identity and disease, such as lineage-controlling transcription factors and oncogenes(1,2). BRD4 and CDK7 are positive regulators of SE-mediated transcription(3-5). By contrast, negative regulators of SE-associated genes have not been well described. Here we show that the Mediator-associated kinases cyclin-dependent kinase 8 (CDK8) and CDK19 restrain increased activation of key SE-associated genes in acute myeloid leukaemia (AML) cells. We report that the natural product cortistatin A (CA) selectively inhibits Mediator kinases, has anti-leukaemic activity in vitro and in vivo, and disproportionately induces upregulation of SE-associated genes in CA-sensitiveAML cell lines but not in CA-insensitive cell lines. In AML cells, CA upregulated SE-associated genes with tumour suppressor and lineage-controlling functions, including the transcription factors CEBPA, IRF8, IRF1 and ETV6 (refs 6-8). The BRD4 inhibitor I-BET151 downregulated these SE-associated genes, yet also has anti-leukaemic activity. Individually increasing or decreasing the expression of these transcription factors suppressed AML cell growth, providing evidence that leukaemia cells are sensitive to the dosage of SE-associated genes. Our results demonstrate that Mediator kinases can negatively regulate SE-associated gene expression in specific cell types, and can be pharmacologically targeted as a therapeutic approach to AML.
URLPMID:26152742 [本文引用: 1]
PURPOSE: Chemotherapy resistance remains a major challenge in the treatment of ovarian cancer. We hypothesize that germline polymorphisms might be associated with clinical outcome. EXPERIMENTAL DESIGN: We analyzed approximately 2.8 million genotyped and imputed SNPs from the iCOGS experiment for progression-free survival (PFS) and overall survival (OS) in 2,901 European epithelial ovarian cancer (EOC) patients who underwent first-line treatment of cytoreductive surgery and chemotherapy regardless of regimen, and in a subset of 1,098 patients treated with ≥ 4 cycles of paclitaxel and carboplatin at standard doses. We evaluated the top SNPs in 4,434 EOC patients, including patients from The Cancer Genome Atlas. In addition, we conducted pathway analysis of all intragenic SNPs and tested their association with PFS and OS using gene set enrichment analysis. RESULTS: Five SNPs were significantly associated (P ≤ 1.0 × 10(-5)) with poorer outcomes in at least one of the four analyses, three of which, rs4910232 (11p15.3), rs2549714 (16q23), and rs6674079 (1q22), were located in long noncoding RNAs (lncRNAs) RP11-179A10.1, RP11-314O13.1, and RP11-284F21.8, respectively (P ≤ 7.1 × 10(-6)). ENCODE ChIP-seq data at 1q22 for normal ovary show evidence of histone modification around RP11-284F21.8, and rs6674079 is perfectly correlated with another SNP within the super-enhancer MEF2D, expression levels of which were reportedly associated with prognosis in another solid tumor. YAP1- and WWTR1 (TAZ)-stimulated gene expression and high-density lipoprotein (HDL)-mediated lipid transport pathways were associated with PFS and OS, respectively, in the cohort who had standard chemotherapy (pGSEA ≤ 6 × 10(-3)). CONCLUSIONS: We have identified SNPs in three lncRNAs that might be important targets for novel EOC therapies.
URLPMID:4857881 [本文引用: 1]
Whole-genome analysis approaches are identifying recurrent cancer-associated somatic alterations in noncoding DNA regions. We combined somatic copy number analysis of 12 tumor types with tissue-specific epigenetic profiling to identify significant regions of focal amplification harboring super-enhancers. Copy number gains of noncoding regions harboring super-enhancers near KLF5, USP12, PARD6B and MYC are associated with overexpression of these cancer-related genes. We show that two distinct focal amplifications of super-enhancers 3' to MYC in lung adenocarcinoma (MYC-LASE) and endometrial carcinoma (MYC-ECSE) are physically associated with the MYC promoter and correlate with MYC overexpression. CRISPR/Cas9-mediated repression or deletion of a constituent enhancer within the MYC-LASE region led to significant reductions in the expression of MYC and its target genes and to the impairment of anchorage-independent and clonogenic growth, consistent with an oncogenic function. Our results suggest that genomic amplification of super-enhancers represents a common mechanism to activate cancer driver genes in multiple cancer types.
URLPMID:4739765 [本文引用: 1]
Tumor-initiating stem cells (SCs) exhibit distinct patterns of transcription factors and gene expression compared to healthy counterparts. Here, we show that dramatic shifts in large open-chromatin domain (super-enhancer) landscapes underlie these differences and reflect tumor microenvironment. By in vivo super-enhancer and transcriptional profiling, we uncover a dynamic cancer-specific epigenetic network selectively enriched for binding motifs of a transcription factor cohort expressed in squamous cell carcinoma SCs (SCC-SCs). Many of their genes, includingEts2andElk3,are themselves regulated by SCC-SC super-enhancers suggesting a cooperative feed-forward loop. Malignant progression requires these genes, whose knockdown severely impairs tumor growth and prohibits progression from benign papillomas to SCCs. ETS2-deficiency disrupts the SCC-SC super-enhancer landscape and downstream cancer genes while ETS2-overactivation in epidermal-SCs induces hyperproliferation and SCC super-enhancer-associated genesFos, JunbandKlf5. Together, our findings unearth an essential regulatory network required for the SCC-SC chromatin landscape and unveil its importance in malignant progression. DOI:http://dx.doi.org/10.7554/eLife.10870.001 Many cancers contain a mixture of different types of cells. Of these, cells known as cancer stem cells can form new tumours and drive the growth and spread of the cancer around the body. A central question is how cancer stem cells differ from healthy adult stem cells. Recent evidence suggests that, in addition to having genetic mutations, cancer stem cells live in a very different environment to other cells within the tumour. This 'microenvironment'also has a major impact on how these cells behave compared to normal stem cells. Together, the genetic and environmental differences profoundly change the way genes are expressed in the cancer cells. In 2013, a group of researchers identified regions of DNA called super-enhancers. These regions are long stretches of DNA that proteins called transcription factors can interact with to coordinate the expression of nearby genes to alter the production of certain proteins. Super-enhancers contain several transcription factor-binding sites that are close to each other with the different sites being associated with transcription factors that are only active in specific types of cells. Furthermore, super-enhancers are often self-regulatory, meaning that the binding of transcription factors to a super-enhancer can lead to an increase in the expression of the genes that encode the same transcription factors. Yang, Schramek et al. have now identified the super-enhancers in a skin cancer called squamous cell carcinoma and showed that they differ dramatically from the super-enhancers of normal skin stem cells. Their experiments show that the active super-enhancers in cancer stem cells are associated with a very different set of genes that are highly and often specifically expressed in cancer stem cells. In the cancer stem cells, a transcription factor called ETS2 binds to the super-enhancers and reprograms the expression of genes to promote the development of cancer. Yang, Schramek et al. also show that over-active ETS2 is a major driver of squamous cell carcinoma. Furthermore, ETS2 also increases the expression of genes that cause inflammation and promote the growth of cancers. Yang, Schramek et al. 檚 findings reveal a new regulatory network that governs the expression of genes involved in cancer. Furthermore, the experiments show that high levels of ETS2 are linked with poor outcomes for patients with head and neck squamous cell carcinoma, which is one of the most life-threatening cancers world-wide. In the future, these findings might lead to the development of new therapies to treat these cancers. DOI:http://dx.doi.org/10.7554/eLife.10870.002
URLPMID:26406377 [本文引用: 3]
A potential therapy for triple-negative breast cancer is suggested by its strong dependence on the transcriptional kinase CDK7 and the cluster of genes the kinase regulates.
URLPMID:27196599 [本文引用: 1]
Abstract OBJECTIVES: Oesophageal squamous cell carcinoma (OSCC) is an aggressive malignancy and the major histological subtype of oesophageal cancer. Although recent large-scale genomic analysis has improved the description of the genetic abnormalities of OSCC, few targetable genomic lesions have been identified, and no molecular therapy is available. This study aims to identify druggable candidates in this tumour. DESIGN: High-throughput small-molecule inhibitor screening was performed to identify potent anti-OSCC compounds. Whole-transcriptome sequencing (RNA-Seq) and chromatin immunoprecipitation sequencing (ChIP-Seq) were conducted to decipher the mechanisms of action of CDK7 inhibition in OSCC. A variety of in vitro and in vivo cellular assays were performed to determine the effects of candidate genes on OSCC malignant phenotypes. RESULTS: The unbiased high-throughput small-molecule inhibitor screening led us to discover a highly potent anti-OSCC compound, THZ1, a specific CDK7 inhibitor. RNA-Seq revealed that low-dose THZ1 treatment caused selective inhibition of a number of oncogenic transcripts. Notably, further characterisation of the genomic features of these THZ1-sensitive transcripts demonstrated that they were frequently associated with super-enhancer (SE). Moreover, SE analysis alone uncovered many OSCC lineage-specific master regulators. Finally, integrative analysis of both THZ1-sensitive and SE-associated transcripts identified a number of novel OSCC oncogenes, including PAK4, RUNX1, DNAJB1, SREBF2 and YAP1, with PAK4 being a potential druggable kinase. CONCLUSIONS: Our integrative approaches led to a catalogue of SE-associated master regulators and oncogenic transcripts, which may significantly promote both the understanding of OSCC biology and the development of more innovative therapies. Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
URLPMID:26983878 [本文引用: 1]
Abstract Inhibitors of the bromodomain and extraterminal domain (BET) protein family attenuate the proliferation of several tumor cell lines. These effects are mediated, at least in part, through repression of c-MYC. In colorectal cancer, overexpression of c-MYC due to hyperactive WNT/0205-catenin/TCF signaling is a key driver of tumor progression; however, effective strategies to target this oncogene remain elusive. Here, we investigated the effect of BET inhibitors (BETi) on colorectal cancer cell proliferation and c-MYC expression. Treatment of 20 colorectal cancer cell lines with the BETi JQ1 identified a subset of highly sensitive lines. JQ1 sensitivity was higher in cell lines with microsatellite instability but was not associated with the CpG island methylator phenotype, c-MYC expression or amplification status, BET protein expression, or mutation status of TP53, KRAS/BRAF, or PIK3CA/PTEN Conversely, JQ1 sensitivity correlated significantly with the magnitude of c-MYC mRNA and protein repression. JQ1-mediated c-MYC repression was not due to generalized attenuation of 0205-catenin/TCF-mediated transcription, as JQ1 had minimal effects on other 0205-catenin/TCF target genes or 0205-catenin/TCF reporter activity. BETi preferentially target super-enhancer-regulated genes, and a super-enhancer in c-MYC was recently identified in HCT116 cells to which BRD4 and effector transcription factors of the WNT/0205-catenin/TCF and MEK/ERK pathways are recruited. Combined targeting of c-MYC with JQ1 and inhibitors of these pathways additively repressed c-MYC and proliferation of HCT116 cells. These findings demonstrate that BETi downregulate c-MYC expression and inhibit colorectal cancer cell proliferation and identify strategies for enhancing the effects of BETi on c-MYC repression by combinatorial targeting the c-MYC super-enhancer. Mol Cancer Ther; 15(6); 1217-26. 00082016 AACR. 00082016 American Association for Cancer Research.
[本文引用: 3]
URL [本文引用: 3]
URLPMID:28283057 [本文引用: 3]
Abstract Super-enhancers are an emerging subclass of regulatory regions controlling cell identity and disease genes. However, their biological function and impact on miRNA networks are unclear. Here, we report that super-enhancers drive the biogenesis of master miRNAs crucial for cell identity by enhancing both transcription and Drosha/DGCR8-mediated primary miRNA (pri-miRNA) processing. Super-enhancers, together with broad H3K4me3 domains, shape a tissue-specific and evolutionarily conserved atlas of miRNA expression and function. CRISPR/Cas9 genomics revealed that super-enhancer constituents act cooperatively and facilitate Drosha/DGCR8 recruitment and pri-miRNA processing to boost cell-specific miRNA production. The BET-bromodomain inhibitor JQ1 preferentially inhibits super-enhancer-directed cotranscriptional pri-miRNA processing. Furthermore, super-enhancers are characterized by pervasive interaction with DGCR8/Drosha and DGCR8/Drosha-regulated mRNA stability control, suggesting unique RNA regulation at super-enhancers. Finally, super-enhancers mark multiple miRNAs associated with cancer hallmarks. This study presents principles underlying miRNA biology in health and disease and an unrecognized higher-order property of super-enhancers in RNA processing beyond transcription. Copyright 2017 Elsevier Inc. All rights reserved.
[本文引用: 1]
URLPMID:29511351 [本文引用: 1]
Previous studies have indicated that as the only mammalian endo- -D-glucuronidase, heparanase (HPSE) is up-regulated and associated with poor prognosis in gastric cancer, while the underlying mechanisms still remain to be determined. Herein, through integrative analysis of public datasets, we found microRNA-558 (miR-558) and SMAD family member 4 (Smad4) as the crucial transcription regulators... [Show full abstract]
URL [本文引用: 1]
增强子是真核生物基因表达调控的主要顺式作用元件,能有效促进基因表达。活化的增强子可以转录生成增强子RNA (enhancer RNAs, eRNAs),其合成受到信号系统和信号转录因子的约束。eRNAs与其他转录本(如lncRNAs和mRNAs)相比,其长度更短、稳定性更差、组织特异性更强。此外,eRNAs对增强子与启动子之间的染色质环(looping)的形成和稳定有一定的作用,并能促进靶基因的表达。目前,越来越多的研究发现eRNAs在发育和疾病发生等生物学过程中扮演着重要角色,但是其功能研究一直进展缓慢,调控机制尚不清楚。本文概述了eRNAs的特征、研究方法和功能特性,探讨了eRNAs作为潜在治疗靶标的可能性,以期为eRNAs的后续研究提供参考。
URL [本文引用: 1]
增强子是真核生物基因表达调控的主要顺式作用元件,能有效促进基因表达。活化的增强子可以转录生成增强子RNA (enhancer RNAs, eRNAs),其合成受到信号系统和信号转录因子的约束。eRNAs与其他转录本(如lncRNAs和mRNAs)相比,其长度更短、稳定性更差、组织特异性更强。此外,eRNAs对增强子与启动子之间的染色质环(looping)的形成和稳定有一定的作用,并能促进靶基因的表达。目前,越来越多的研究发现eRNAs在发育和疾病发生等生物学过程中扮演着重要角色,但是其功能研究一直进展缓慢,调控机制尚不清楚。本文概述了eRNAs的特征、研究方法和功能特性,探讨了eRNAs作为潜在治疗靶标的可能性,以期为eRNAs的后续研究提供参考。
URLPMID:26814967 [本文引用: 1]
Medulloblastoma is a highly malignant paediatric brain tumour, often inflicting devastating consequences on the developing child. Genomic studies have revealed four distinct molecular subgroups with divergent biology and clinical behaviour. An understanding of the regulatory circuitry governing the transcriptional landscapes of medulloblastoma subgroups, and how this relates to their respective developmental origins, is lacking. Here, using H3K27ac and BRD4 chromatin immunoprecipitation followed by sequencing (ChIP-seq) coupled with tissue-matched DNA methylation and transcriptome data, we describe the active cis-regulatory landscape across 28 primary medulloblastoma specimens. Analysis of differentially regulated enhancers and super-enhancers reinforced inter-subgroup heterogeneity and revealed novel, clinically relevant insights into medulloblastoma biology. Computational reconstruction of core regulatory circuitry identified a master set of transcription factors, validated by ChIP-seq, that is responsible for subgroup divergence, and implicates candidate cells of origin for Group 4. Our integrated analysis of enhancer elements in a large series of primary tumour samples reveals insights into cis-regulatory architecture, unrecognized dependencies, and cellular origins.
URLPMID:26560027 [本文引用: 1]
Neuroblastoma is a paediatric malignancy that typically arises in early childhood, and is derived from the developing sympathetic nervous system. Clinical phenotypes range from localized tumours with excellent outcomes to widely metastatic disease in which long-term survival is approximately 40% despite intensive therapy. A previous genome-wide association study identified common polymorphisms at the LMO1 gene locus that are highly associated with neuroblastoma susceptibility and oncogenic addiction to LMO1 in the tumour cells. Here we investigate the causal DNA variant at this locus and the mechanism by which it leads to neuroblastoma tumorigenesis. We first imputed all possible genotypes across the LMO1 locus and then mapped highly associated single nucleotide polymorphism (SNPs) to areas of chromatin accessibility, evolutionary conservation and transcription factor binding sites. We show that SNP rs2168101 G>T is the most highly associated variant (combined P65=657.4765×6510[superscript 6129], odds ratio 0.65, 95% confidence interval 0.60–0.70), and resides in a super-enhancer defined by extensive acetylation of histone H3 lysine 27 within the first intron of LMO1. The ancestral G allele that is associated with tumour formation resides in a conserved GATA transcription factor binding motif. We show that the newly evolved protective TATA allele is associated with decreased total LMO1 expression (P65=650.028) in neuroblastoma primary tumours, and ablates GATA3 binding (P65<650.0001). We demonstrate allelic imbalance favouring the G-containing strand in tumours heterozygous for this SNP, as demonstrated both by RNA sequencing (P65<650.0001) and reporter assays (P65=650.002). These findings indicate that a recently evolved polymorphism within a super-enhancer element in the first intron of LMO1 influences neuroblastoma susceptibility through differential GATA transcription factor binding and direct modulation of LMO1 expression in cis, and this leads to an oncogenic dependency in tumour cells.
URLPMID:25813350 [本文引用: 2]
Abstract Super-enhancers increase responsiveness to developmental and oncogenic signaling pathways. 082015 American Association for Cancer Research.
URLPMID:26279576 [本文引用: 1]
Aberrant receptor tyrosine kinase signaling mediated by oncogenic Ras or loss of Sprouty promotes tumorigenesis. Nabet et al. find that unrestrained receptor tyrosine signaling driven by these lesions alters distinct super-enhancers, transcription factors, and target genes. Gata4 and Prkcb are identified as mediators of the oncogenic program upon Ras transformation.
URL [本文引用: 3]
URLPMID:26439301 [本文引用: 1]
The transcriptional coactivators YAP and TAZ are critical regulators of stem cell activity and tumorigenesis. Galli et al. show that YAP/TAZ binding is restricted to a relatively small number of the most potent enhancers in the genome. They show that YAP/TAZ regulate transcriptional elongation from these elements by recruiting the Mediator complex.
URL [本文引用: 1]
URLPMID:24374627 [本文引用: 1]
The main oncogenic driver in T-lymphoblastic leukemia is NOTCH1, which activates genes by forming chromatin-associated Notch transcription complexes. Gamma-secretase-inhibitor treatment prevents NOTCH1 nuclear localization, but most genes with NOTCH1-binding sites are insensitive to gamma-secretase inhibitors. Here, we demonstrate that fewer than 10% of NOTCH1-binding sites show dynamic changes in NOTCH1 occupancy when T-lymphoblastic leukemia cells are toggled between the Notch-on and -off states with gamma-secretase inhibiters. Dynamic NOTCH1 sites are functional, being highly associated with Notch target genes, are located mainly in distal enhancers, and frequently overlap with RUNX1 binding. In line with the latter association, we show that expression of IL7R, a gene with key roles in normal T-cell development and in T-lymphoblastic leukemia, is coordinately regulated by Runx factors and dynamic NOTCH1 binding to distal enhancers. Like IL7R, most Notch target genes and associated dynamic NOTCH1-binding sites cooccupy chromatin domains defined by constitutive binding of CCCTC binding factor, which appears to restrict the regulatory potential of dynamic NOTCH1 sites. More remarkably, the majority of dynamic NOTCH1 sites lie in superenhancers, distal elements with exceptionally broad and high levels of H3K27ac. Changes in Notch occupancy produces dynamic alterations in H3K27ac levels across the entire breadth of superenhancers and in the promoters of Notch target genes. These findings link regulation of superenhancer function to NOTCH1, a master regulatory factor and potent oncoprotein in the context of immature T cells, and delineate a generally applicable roadmap for identifying functional Notch sites in cellular genomes.
URLPMID:27364481 [本文引用: 1]
Enhancer elements function as the logic gates of the genetic regulatory circuitry. One of their most important functions is the integration of extracellular signals with intracellular cell fate information to generate cell type-specific transcriptional responses. Mutations occurring in cancer often misregulate enhancers that normally control the signal-dependent expression of growth-related genes. This misregulation can result from trans-acting mechanisms, such as activation of the transcription factors or epigenetic regulators that control enhancer activity, or can be caused in cis by direct mutations that alter the activity of the enhancer or its target gene specificity. These processes can generate tumour type-specific super-enhancers and establish a 'locked' gene regulatory state that drives the uncontrolled proliferation of cancer cells. Here, we review the role of enhancers in cancer, and their potential as therapeutic targets.
URL [本文引用: 1]
URLPMID:2302121520
Myc-induced transcriptional amplification, rather than the switching on of “Myc target genes” is important for tumorigenesis, suggesting that therapies targeting the apparatus involved in transcriptional amplification may be useful in the treatment of cancer.
URLPMID:29695137 [本文引用: 1]
Aberrant activation of hedgehog (Hh) signaling has been observed in a wide variety of tumors and accounts for more than 25% of human cancer deaths. Inhibitors targeting the Hh signal transducer Smoothened (SMO) are widely used and display a good initial efficacy in patients suffering from basal cell carcinoma (BCC); however, a large number of patients relapse. Though SMO mutations may explain acquired therapy resistance, a growing body of evidence suggests that the non-canonical, SMO-independent activation of the Hh pathway in BCC patients can also account for this adverse effect. In this review, we highlight the importance of glioma-associated oncogene (GLI) transcription factors (the main downstream effectors of the canonical and the non-canonical Hh cascade) and their putative role in the regulation of multiple oncogenic signaling pathways. Moreover, we discuss the contribution of the Hh signaling to malignant transformation and propose GLIs as central hubs in tumor signaling networks and thus attractive molecular targets in anti-cancer therapies.
URLPMID:26257281 [本文引用: 1]
Nilson et al. use THZ1, a Cdk7 inhibitor, to uncover defects in Pol II phosphorylation, co-transcriptional capping, pausing, and productive elongation. THZ1 disrupts an ordered exchange of factors after initiation, blocking capping and pausing. These results provide mechanistic insights into the anti-proliferative and super-enhancer-selective effects of THZ1 seen by others.
URLPMID:4317728 [本文引用: 1]
Di Micco et al. now dissect the mechanisms by which BRD4 regulates embryonic stem cell (ESC) identity by binding to super-enhancers of core pluripotency genes and recruiting active transcription complexes. BRD4 inhibition results in defective elongation of super-enhancer-associated gene transcripts, loss of ESC self-renewal/pluripotency, and commitment to the neuroectodermal lineage.
URLPMID:20952539 [本文引用: 1]
Abstract Drosophila contains one (dCDK12) and humans contain two (hCDK12 and hCDK13) proteins that are the closest evolutionary relatives of yeast Ctk1, the catalytic subunit of the major elongation-phase C-terminal repeat domain (CTD) kinase in Saccharomyces cerevisiae, CTDK-I. However, until now, neither CDK12 nor CDK13 has been demonstrated to be a bona fide CTD kinase. Using Drosophila, we demonstrate that dCDK12 (CG7597) is a transcription-associated CTD kinase, the ortholog of yCtk1. Fluorescence microscopy reveals that the distribution of dCDK12 on formaldehyde-fixed polytene chromosomes is virtually identical to that of hyperphosphorylated RNA polymerase II (RNAPII), but is distinct from that of P-TEFb (dCDK9 + dCyclin T). Chromatin immunoprecipitation (ChIP) experiments confirm that dCDK12 is present on the transcribed regions of active Drosophila genes. Compared with P-TEFb, dCDK12 amounts are lower at the 5' end and higher in the middle and at the 3' end of genes (both normalized to RNAPII). Appropriately, Drosophila dCDK12 purified from nuclear extracts manifests CTD kinase activity in vitro. Intriguingly, we find that cyclin K is associated with purified dCDK12, implicating it as the cyclin subunit of this CTD kinase. Most importantly, we demonstrate that RNAi knockdown of dCDK12 in S2 cells alters the phosphorylation state of the CTD, reducing its Ser2 phosphorylation levels. Similarly, in human HeLa cells, we show that hCDK13 purified from nuclear extracts displays CTD kinase activity in vitro, as anticipated. Also, we find that chimeric (yeast/human) versions of Ctk1 containing the kinase homology domains of hCDK12/13 (or hCDK9) are functional in yeast cells (and also in vitro); using this system, we show that a bur1(ts) mutant is rescued more efficiently by a hCDK9 chimera than by a hCDK13 chimera, suggesting the following orthology relationships: Bur1 芒聠聰 CDK9 and Ctk1 CDK12/13. Finally, we show that siRNA knockdown of hCDK12 in HeLa cells results in alterations in the CTD phosphorylation state. Our findings demonstrate that metazoan CDK12 and CDK13 are CTD kinases, and that CDK12 is orthologous to yeast Ctk1.
URLPMID:4333096 [本文引用: 1]
Cyclin-dependent kinase 9 (CDK9) and CDK12 have each been demonstrated to phosphorylate the RNA polymerase II C-terminal domain (CTD) at serine 2 of the heptad repeat, both in vitro and in vivo. CDK9, as part of P-TEFb and the super elongation complex (SEC), is by far the best characterized of CDK9, CDK12, and CDK13. We employed both in vitro and in vivo assays to further investigate the molecular properties of CDK12 and its paralog CDK13. We isolated Flag-tagged CDK12 and CDK13 and found that they associate with numerous RNA processing factors. Although knockdown of CDK12, CDK13, or their cyclin partner CCNK did not affect the bulk CTD phosphorylation levels in HCT116 cells, transcriptome sequencing (RNA-seq) analysis revealed that CDK12 and CDK13 losses in HCT116 cells preferentially affect expression of DNA damage response and snoRNA genes, respectively. CDK12 and CDK13 depletion also leads to a loss of expression of RNA processing factors and to defects in RNA processing. These findings suggest that in addition to implementing CTD phosphorylation, CDK12 and CDK13 may affect RNA processing through direct physical interactions with RNA processing factors and by regulating their expression.
URLPMID:29754476 [本文引用: 1]
The transcriptional regulation of genes determines the fate of animal cell differentiation and subsequent organ development. With the recent progress in genome-wide technologies, the genomic landscapes of enhancers have been broadly explored in mammalian genomes, which led to the discovery of novel specific subsets of enhancers, termed super-enhancers. Super-enhancers are large clusters of enhancers covering the long region of regulatory DNA and are densely occupied by transcription factors, active histone marks, and co-activators. Accumulating evidence points to the critical role that super-enhancers play in cell type-specific development and differentiation, as well as in the development of various diseases. Here, I provide a comprehensive description of the optimal approach for identifying functional units of super-enhancers and their unique chromatin features in normal development and in diseases, including cancers. I also review the recent updated knowledge on novel approaches of targeting super-enhancers for the treatment of specific diseases, such as small-molecule inhibitors and potential gene therapy. This review will provide perspectives on using super-enhancers as biomarkers to develop novel disease diagnostic tools and establish new directions in clinical therapeutic strategies.
URLPMID:28209615 [本文引用: 1]
Abstract Triple-negative breast cancers (TNBC) remain clinically challenging with a lack of options for targeted therapy. In this study, we report the development of a second-generation BET protein degrader, BETd-246, which exhibits superior selectivity, potency, and antitumor activity. In human TNBC cells, BETd-246 induced degradation of BET proteins at low nanomolar concentrations within 1 hour of exposure, resulting in robust growth inhibition and apoptosis. BETd-246 was more potent and effective in TNBC cells than its parental BET inhibitor compound BETi-211. RNA-seq analysis revealed predominant downregulation of a large number of genes involved in proliferation and apoptosis in cells treated with BETd-246, as compared with BETi-211 treatment that upregulated and downregulated a similar number of genes. Functional investigations identified the MCL1 gene as a critical downstream effector for BET degraders, which synergized with small-molecule inhibitors of BCL-xL in triggering apoptosis. In multiple murine xenograft models of human breast cancer, BETd-246 and a further optimized analogue BETd-260 effectively depleted BET proteins in tumors and exhibited strong antitumor activities at well-tolerated dosing schedules. Overall, our findings show that targeting BET proteins for degradation represents an effective therapeutic strategy for TNBC treatment. Cancer Res; 77(9); 2476-87. 2017 AACR . 2017 American Association for Cancer Research.
URLPMID:26341814
Abstract BACKGROUND AND OBJECTIVES: OTX015 (MK-8628) is a novel inhibitor of the bromodomain and extraterminal (BET)-bromodomain (BRD) protein family, binding specifically to bromodomains BRD2/3/4 and impacting the epigenetic regulation of several oncogenes. We characterized the pharmacokinetics of this first-in-class BET-BRD inhibitor administered as a single agent, including population pharmacokinetic modelling. METHODS: A dose-escalation, phase Ib study was performed with oral OTX015 in patients with haematologic malignancies, at doses starting from 100002mg once daily (QD) with continuous or discontinuous schedules. Five or eight blood samples were collected per patient for pharmacokinetic analysis. OTX015 plasma concentrations were determined using validated ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) and analysed using a nonlinear mixed-effects modelling software program. A population pharmacokinetic model was fitted to the data, and patient demographics and clinical chemistry parameters were tested as predictive covariates on the model parameters. RESULTS: Blood samples were analysed from 81 patients treated with OTX015 at doses ranging from 10 to 1600002mg QD or 400002mg twice daily (BID), and 633 time-plasma concentrations were available for analysis. A one-compartment open model with linear elimination adequately described OTX015 pharmacokinetics. The most significant covariate was lean body mass (LBM), which decreased the between-subject variability in apparent total body clearance (CL) and the volume of distribution (V). The estimated pharmacokinetic parameters were the absorption rate constant (k a)0002=00020.7310002h(-1), V0002=000271.40002L and CL0002=00028.470002L00·h(-1). CONCLUSION: The pharmacokinetics of oral OTX015 in patients with haematologic malignancies can be described with a one-compartment model. Population pharmacokinetic modelling of OTX015 plasma concentrations showed that LBM influences V and CL. These findings do not suggest the need for dose adjustment.
URLPMID:27063977
Bromodomain and extraterminal (BET) proteins are chromatin readers that preferentially affect the transcription of genes with super-enhancers, including oncogenes. BET proteins bind acetylated histone tails via their bromodomain, bringing the elongation complex to the promoter region. OTX015 (MK-8628) specifically binds to BRD2, BRD3, and BRD4, preventing BET proteins from binding to the chromatin, thus inhibiting gene transcription. OTX015 inhibits proliferation in many haematological malignancy cell lines and patient cells, in vitro and in vivo. We aimed to establish the recommended dose of OTX015 in patients with haematological malignancies. We report the results of patients with acute leukaemia (leukaemia cohort). In this dose-escalation, phase 1 study we recruited patients from seven university hospital centres (in France [five], UK [one], and Canada [one]). Adults with acute leukaemia who had failed or had a contraindication to standard therapies were eligible to participate. OTX015 was given orally at increasing doses from 10 mg/day to 160 mg/day (14 of 21 days), using a conventional 364+64364design. In this open-label trial, OTX015 was initially administered once a day, with allowance for exploration of other schedules. The primary endpoint was dose-limiting toxicity (DLT), assessed during the first treatment cycle (21 days). The study is ongoing and is registered withClinicalTrials.gov,NCT01713582. Between Jan 18, 2013, and Sept 9, 2014, 41 patients, 36 with acute myeloid leukaemia, a median age of 70 years (IQR 60–75) and two lines of previous therapy, were recruited and treated across six dose levels of OTX015. No DLT was recorded until 160 mg/day, when one patient had grade 3 diarrhoea and another had grade 3 fatigue. However, concomitant grade 1–2 non-DLT toxic effects (ie, gastrointestinal, fatigue, or cutaneous) from 120 mg doses hampered patient compliance and 80 mg once a day was judged the recommended dose with a 14 days on, 7 days off schedule. Common toxic effects for all OTX015 doses were fatigue (including grade 3 in three patients) and bilirubin concentration increases (including grade 3–4 in two patients). OTX015 plasma exposure increased proportionally up to 120 mg/day with trough concentrations in the in-vitro active range from 80 mg/day (274 nmol/L). Three patients (receiving 40 mg/day, 80 mg/day, and 160 mg/day) achieved complete remission or complete remission with incomplete recovery of platelets lasting 2–5 months, and two additional patients had partial blast clearance. No predictive biomarkers for response have been identified so far. The once-daily recommended dose for oral, single agent oral OTX015 use in patients with acute leukaemia for further phase 2 studies is 80 mg on a 14 days on, 7 days off schedule. Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme Corp.
URLPMID:25416950 [本文引用: 1]
CDK7 inhibition induces cytotoxic effects in neuroblastoma cells through massive downregulation of MYC-induced transcriptional amplification and preferential disruption of super-enhancer-associated oncogenic drivers.
URLPMID:28978570
Abstract A number of studies have recently demonstrated that "super-enhancers", which are large cluster of enhancers typically marked by a high level of acetylation of histone H3 lysine 27 and mediator bindings, are frequently associated with genes that control and define cell identity during normal development. Super-enhancers are also often enriched at cancer genes in various malignancies. Identification of such enhancers would pinpoint critical factors that directly contribute to pathogenesis. Here, we performed enhancer profiling using primary leukemia samples from adult T-cell leukemia/lymphoma (ATL), which is a genetically heterogeneous intractable cancer. Super-enhancers were enriched at genes involved in the T-cell activation pathway including IL2RA/CD25 , CD30 and FYN , in both ATL and normal mature T-cells, which reflected the origin of the leukemic cells. Super-enhancers were found at several known cancer gene loci, including CCR4 , PIK3R1 and TP73 , in multiple ATL samples but not in normal mature T-cells, which implicated those genes in ATL pathogenesis. A small-molecule CDK7 inhibitor THZ1 efficiently inhibited cell growth, induced apoptosis and downregulated the expression of super-enhancer-associated genes in ATL cells. Furthermore, enhancer profiling combined with gene expression analysis identified a previously uncharacterized gene, TIAM2 that was associated with super-enhancers in all ATL samples but not in normal T-cells. Knockdown of TIAM2 induced apoptosis in ATL cell lines, whereas overexpression of this gene promoted cell growth. Our study provides a novel strategy for identifying critical cancer genes. Copyright 2017 American Society of Hematology.
URLPMID:5033074 [本文引用: 1]
Editorial summaryA small molecule inhibits CDK12 and CDK13 activity through covalent modification of Cys residues and reveals a role of the two kinases in regulating Pol II processivity and super-enhancer-driven transcription factor and DNA damage response gene expression.
URLPMID:26337082 [本文引用: 1]
Ewing sarcoma is an aggressive bone and soft tissue tumor in children and adolescents, with treatment remaining a clinical challenge. This disease is mediated by somatic chromosomal translocations of the EWS gene and a gene encoding an ETS transcription factor, most commonly, FLI1. While direct targeting of aberrant transcription factors remains a pharmacological challenge, identification of dependencies incurred by EWS/FLI1 expression would offer a new therapeutic avenue. We used a combination of super-enhancer profiling, near-whole genome shRNA-based and small-molecule screening to identify cyclin D1 and CDK4 as Ewing sarcoma-selective dependencies. We revealed that super-enhancers mark Ewing sarcoma specific expression signatures and EWS/FLI1 target genes in human Ewing sarcoma cell lines. Particularly, a super-enhancer regulates cyclin D1 and promotes its expression in Ewing sarcoma. We demonstrated that Ewing sarcoma cells require CDK4 and cyclin D1 for survival and anchorage-independent growth. Additionally, pharmacologic inhibition of CDK4 with selective CDK4/6 inhibitors led to cytostasis and cell death of Ewing sarcoma cell lines in vitro and growth delay in an in vivo Ewing sarcoma xenograft model. These results demonstrated a dependency in Ewing sarcoma on CDK4 and cyclin D1 and support exploration of CDK4/6 inhibitors as a therapeutic approach for patients with this disease.
URLPMID:21889194 [本文引用: 1]
Small-molecule inhibition of chromatin-reading bromodomoain proteins leads to transcriptional downregulation of the oncogene c-Myc, an intervention that is efficacious in mouse models of multiple myeloma.
URL [本文引用: 1]
URLPMID:27063978 [本文引用: 1]
The first-in-class small molecule inhibitor OTX015 (MK-8628) specifically binds to bromodomain motifs BRD2, BRD3, and BRD4 of bromodomain and extraterminal (BET) proteins, inhibiting them from binding to acetylated histones, which occurs preferentially at super-enhancer regions that control oncogene expression. OTX015 is active in haematological preclinical entities including leukaemia, lymphoma, and myeloma. We aimed to establish the recommended dose of OTX015 in patients with haematological malignancies. We report the results from a cohort of patients with lymphoma or multiple myeloma (non-leukaemia cohort). In this dose-escalation, open-label, phase 1 study, we recruited patients from seven university hospital centres (in France [four], Switzerland [one], UK [one], and Italy [one]). Adult patients with non-leukaemia haematological malignancies who had disease progression on standard therapies were eligible to participate. Patients were treated with oral OTX015 once a day continuously over five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg), using a conventional 3 + 3 design, with allowance for evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity (DLT) in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered withClinicalTrials.gov, numberNCT01713582. Between Feb 4, 2013, and Sept 5, 2014, 45 patients (33 with lymphoma and 12 with myeloma), with a median age of 66 years (IQR 55-72) and a median of four lines of prior therapy (IQR 3 5), were enrolled and treated. No DLTs were observed in the doses up to and including 80 mg once a day (first three patients). We then explored a schedule of 40 mg twice a day (21 of 21 days). DLTs were reported in five of six patients receiving OTX015 at this dose and schedule (all five patients had grade 4 thrombocytopenia). We explored various schedules at 120 mg once a day but none was tolerable, with DLTs of thrombocytopenia, gastrointestinal events (diarrhoea, vomiting, dysgeusia, mucositis), fatigue, and hyponatraemia in 11 of 18 evaluable patients. At this point, the Safety Monitoring Committee decided to establish the feasibility of 80 mg once a day on a continuous basis, and four additional patients were enrolled at this dose. DLTs (grade 4 thrombocytopenia) was noted in two of the patients. In light of these DLTs and other toxicities noted at 120 mg, the dose of 80 mg once a day was selected, although on a schedule of 14 days on, 7 days off. Common toxic effects reported in the study were thrombocytopenia (43 [96%] patients), anaemia (41 [91%]), neutropenia (23 [51%]), diarrhoea (21 [47%]), fatigue (12 [27%]), and nausea (11 [24%]). Grade 3 4 adverse events were infrequent other than thrombocytopenia (26 [58%]). OTX015 plasma peak concentrations and areas under the concentration versus time curve increased proportionally with dose. Trough concentrations increased less than proportionally at lower doses, but reached or exceeded the in-vitro active range at 40 mg twice a day and 120 mg once a day. Three patients with diffuse large B-cell lymphoma achieved durable objective responses (two complete responses at 120 mg once a day, and one partial response at 80 mg once a day), and six additional patients (two with diffuse large B-cell lymphoma, four with indolent lymphomas) had evidence of clinical activity, albeit not meeting objective response criteria. The once-daily recommended dose for oral, single agent oral OTX015 in patients with lymphoma is 80 mg on a 14 days on, 7 days off schedule, for phase 2 studies. OTX015 is under evaluation in expansion cohorts using this intermittent administration (14 days every 3 weeks) to allow for recovery from toxic effects. Oncoethix GmbH (a wholly owned subsidiary of Merck Sharp & Dohme Corp).
URL [本文引用: 1]
URLPMID:27099234 [本文引用: 1]
Abstract Oncogene-induced senescence is a potent barrier to tumorigenesis that limits cellular expansion following certain oncogenic events. Senescent cells display a repressive chromatin configuration thought to stably silence proliferation-promoting genes while simultaneously activating an unusual form of immune surveillance involving a secretory program referred to as the senescence-associated secretory phenotype (SASP). Here, we demonstrate that senescence also involves a global remodeling of the enhancer landscape with recruitment of the chromatin reader BRD4 to newly activated super-enhancers adjacent to key SASP genes. Transcriptional profiling and functional studies indicate that BRD4 is required for the SASP and downstream paracrine signaling. Consequently, BRD4 inhibition disrupts immune cell-mediated targeting and elimination of premalignant senescent cells in vitro and in vivo Our results identify a critical role for BRD4-bound super-enhancers in senescence immune surveillance and in the proper execution of a tumor-suppressive program. SIGNIFICANCE: This study reveals how cells undergoing oncogene-induced senescence acquire a distinctive enhancer landscape that includes formation of super-enhancers adjacent to immune-modulatory genes required for paracrine immune activation. This process links BRD4 and super-enhancers to a tumor-suppressive immune surveillance program that can be disrupted by small molecule inhibitors of the bromo and extra terminal domain family of proteins. Cancer Discov; 6(6); 612-29. 2016 AACR.See related commentary by Vizioli and Adams, p. 576This article is highlighted in the In This Issue feature, p. 561. 2016 American Association for Cancer Research.
URLPMID:4244910 [本文引用: 3]
Tumour oncogenes include transcription factors that co-opt the general transcriptional machinery to sustain the oncogenic state, but direct pharmacological inhibition of transcription factors has so far proven difficult. However, the transcriptional machinery contains various enzymatic cofactors that can be targeted for the development of new therapeutic candidates, including cyclin-dependent kinases (CDKs). Here we present the discovery and characterization of a covalent CDK7 inhibitor, THZ1, which has the unprecedented ability to target a remote cysteine residue located outside of the canonical kinase domain, providing an unanticipated means of achieving selectivity for CDK7. Cancer cell-line profiling indicates that a subset of cancer cell lines, including human T-cell acute lymphoblastic leukaemia (T-ALL), have exceptional sensitivity to THZ1. Genome-wide analysis in Jurkat T-ALL cells shows that THZ1 disproportionally affects transcription of RUNX1 and suggests that sensitivity to THZ1 may be due to vulnerability conferred by the RUNX1 super-enhancer and the key role of RUNX1 in the core transcriptional regulatory circuitry of these tumour cells. Pharmacological modulation of CDK7 kinase activity may thus provide an approach to identify and treat tumour types that are dependent on transcription for maintenance of the oncogenic state.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}