 ,石河子大学生命科学学院,石河子 832000
,石河子大学生命科学学院,石河子 832000A teaching design to introduce chromosomal aberration in genetics using case studies of chimeric genes
Lei Ma, Tingting Zhang,College of Life Science, Shihezi University, Shihezi 832000, China通讯作者:
编委: 史庆华
收稿日期:2018-06-6修回日期:2018-08-15网络出版日期:2018-12-20
| 基金资助: |
Received:2018-06-6Revised:2018-08-15Online:2018-12-20
| Fund supported: |
作者简介 About authors
马磊,博士,副教授,研究方向:生物信息学与分子遗传学E-mail:malei1979@hotmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (593KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马磊, 张婷婷. 应用嵌合基因实例拓展遗传学染色体畸变的教学[J]. 遗传, 2018, 40(12): 1129-1135 doi:10.16288/j.yczz.18-158
Lei Ma, Tingting Zhang.
染色体结构的稳定,是细胞有规律分裂及增殖的基础[1,2]。然而,稳定是相对的,变异是绝对的。染色体结构变异既可自发产生,也可人为诱变而生,会扰乱机体历经百万年进化而孕育的平衡系统,从而影响生物的生殖和发育[3,4,5]。在本科遗传学教学中,如何进一步诠释导致平衡系统紊乱的机理,是教师常面临的教学难题。
染色体相互易位,能导致位于不同染色体,或同一染色体不同区域的基因发生融合,产生嵌合基因(chimeric gene)[6,7]。嵌合基因可连接不同来源基因的功能域,组合出新功能,改变原有分子特性,引起功能失常[8],可能是致使固有平衡系统紊乱的原因之一。然而,因学时数、通识性和书籍容量等因素,鲜见在课堂或教科书中讲述与嵌合基因相关的内容,制约了学生对染色体结构变异的深度探究。为了满足学生的探索欲,本团队结合近年对嵌合基因的研究经验及遗传学教学实践,选取了一些嵌合基因实例,设计了相应教学环节,予以拓展染色体畸变的教学,以期在染色体结构变异教学过程中,使学生对基因的变异及功能有更深入的理解,为遗传学教学提供参考。
1 以一个司法案例创设情境,提升教与学的趣味性
美国一对父母曾将一个产前遗传诊断公司告上法庭,起诉理由是因该公司疏忽大意,导致原告竭力避免的遗传缺陷不幸地出现在了他们的新生儿体中。最终,相关公司被判赔偿原告及新生儿上千万美元。事情的经过大致如下:Hook (化名)的表妹患有严重癫痫病,基因检测诊断出患有22号和9号染色体非平衡易位。知情后,Hook也做了检测,未见异常,但有平衡染色体易位的问题。Hook夫妇第一胎生育正常,但随后多次怀孕,每次孕检胎儿均有染色体易位问题,只能流产。直到这一次,检验公司告之一切正常,结果却产下了一个患有染色体出生缺陷的新生儿。因此,该父母将检验公司告上法庭。引发不幸的原因,是BCR-ABL1嵌合基因的功能异常所致[9]。人类的22号染色体长臂区段易位至9号染色体长臂上,形成了一个较小的新22号染色体。此染色体因首先在美国费城一例慢性粒细胞白血病患者中发现,而被命名为费城染色体。易位致使22号染色体的断裂点簇区域(breakpoint cluster region, BCR)和9号染色体的ABL1融合,形成嵌合基因。正常的ABL1蛋白的酪氨酸激酶的活性受严格调控,而BCR-ABL1却有连续的、自发的和显著增强的酪氨酸激酶活性。90%的慢性粒细胞白血病由BCR-ABL1酪氨酸激酶的异常活性引起。
在本科生遗传学教学中,引入这个司法案例,既创设了趣味情境,又有利于引发好奇心,从而产生“课伊始趣亦生”的教学效果,激发学生对拓展知识的学习兴趣。
2 引入嵌合基因,补充理论知识
嵌合基因可转录为嵌合mRNA,翻译成嵌合蛋白。融合前的原基因一般称为亲本基因。嵌合蛋白通过连接不同基因的功能域,可改变亲本基因的功能,增加转录组和蛋白质组的多样性和复杂性,甚至阻碍正常的信号通路,起始或激活癌细胞生长。例如,多数前列腺癌携带的嵌合基因TMPRSS2- ERG,由TMPRSS2启动子与ERG的编码区融合而成,会驱动一种独特的转录程序,诱导DNA损伤、癌细胞侵袭和转移[10]。亲本基因可位于不同染色体上,也可位于同一染色体的不同DNA链上,亦可在同一条DNA链上,但嵌合基因与亲本基因的外显子排列顺序不同。嵌合基因概念的提出对传统经典基因定义提出了疑问和挑战,一个基因是否仅对应于染色体上的某一特定区段,而非来源于染色体不同的区段[7]?
嵌合基因可作为细胞癌变的分子标记和药物靶标。例如,在人乳腺癌、卵巢癌、前列腺癌、非白血性白血病、急性白血病和非小细胞肺癌等病变组织中,发现了许多可作为分子诊断标记的嵌合基因,RNA干扰试验显示一些嵌合基因可促癌细胞生长,体外实验显示一些嵌合基因可致瘤[11,12,13,14]。鉴于嵌合基因与细胞癌变的特殊联系,一些研究以嵌合基因为治疗靶标,开发了许多抑制嵌合基因功能的抗癌药物。例如,抗癌药Gleevec,能与ATP竞争性结合BCR-ABL1酪氨酸激酶催化域上的ATP结合位点,从而抑制BCR-ABL1激酶上的磷酸基团转移,致使酪氨酸激酶信号传导通路中断,癌细胞停止分裂增殖而死亡[15]。因而,嵌合基因在临床上具有良好的应用前景和重要意义,可作为前诊断筛查的分子标记,也可作为潜在的药物靶标进行临床治疗。
可见,嵌合基因不仅与癌症相关,而且还可用于开发抑癌药物。在遗传学课程中,讲解嵌合基因与癌症的联系,不仅可提升学生对嵌合基因的关注度,还有利于知识点的扩展。
3 分析嵌合基因实例,延伸学习兴趣
基因融合所导致的功能异常,是嵌合基因致癌的潜在机制之一,常表现为激酶活性异常、细胞定位异常和靶基因互作异常等。下面以BCR-ABL1、PAX5-JAK2和RUNX1-ETO为例,说明嵌合蛋白的功能异常特点;此外,引入嵌合基因相关数据库,扩充和延伸知识点。3.1 BCR-ABL1与激酶活性异常有关
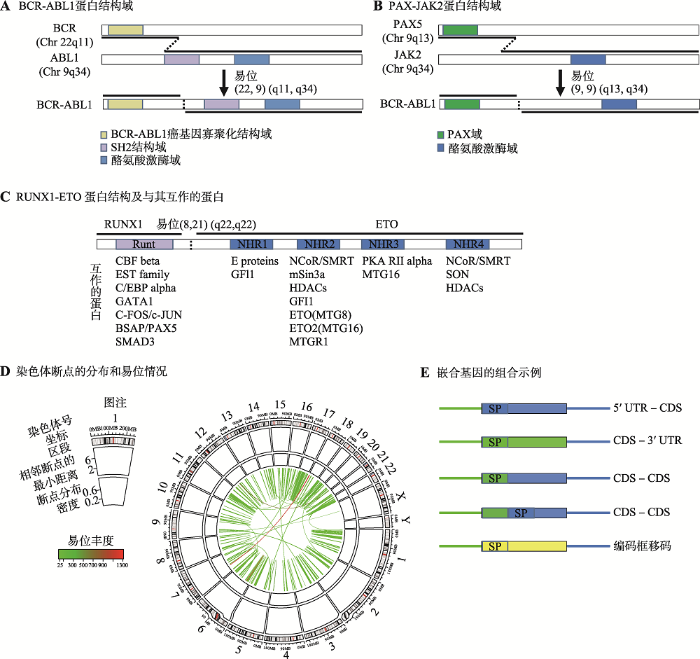
前面提及的费城染色体,即是染色体交互易位产生嵌合癌基因的例子。那么,为什么BCR-ABL1嵌合基因会致癌呢?在正常细胞内,ABL1所编码蛋白N端含有一个抑制激酶活性的区域,激酶活性受控。然而,BCR与ABL1融合后(图1 A),ABL1的激酶抑制区失活,分子构象改变,激酶活性异常增高,活化了许多调控细胞周期的蛋白和酶,细胞分裂加速,进而致癌。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1嵌合蛋白的结构域及染色体易位的分布
A:BCR-ABL1 蛋白结构域。B:PAX-JAK2蛋白结构域。C:RUNX1-ETO 蛋白结构域及与其互作靶基因(修改自文献[17]),互作基因标注于相应的结构域下方。D:染色体易位断点的分布及染色体间易位的丰度,自外而内显示:(1)染色体号、坐标和染色体区段;(2)相邻的易位断点的最小距离(log10转换);(3)易位断点在染色体上的分布密度;(4)以连线示意染色体间的易位情况,连线的颜色示意某一位点的易位发生数量。所有圈横坐标为染色体坐标。E:嵌合基因的结构域组合形式(修改自文献[8]),直线为非编码区(UTR),方框为编码区(CDS),绿色和红色分别示意5°上游和3°下游亲本的序列,黄色方框为移码框突变形成的新编码区,SP:信号肽序列。
Fig. 1Chimeric protein domains and chromosome translocation distribution
3.2 PAX5-JAK2与细胞定位异常有关
PAX5位于人类9号染色体的负链,具有保守的结合DNA的结构域,是一种调控B细胞早期发育的转录因子;JAK2位于9号染色体正链,具有酪氨酸激酶结构域,可调控许多细胞因子的信号转导(图1 B)[16]。二者嵌合之前受严格调控,蛋白质分别定位于细胞核和细胞质,而嵌合后PAX5-JAK2却兼备了DNA结合域和酪氨酸激酶活性,成为了一种细胞核内的活性激酶,致使原PAX5和JAK2的部分靶基因的表达和下游信号通路受干扰,从而诱导B细胞的肿瘤性转化[16]。3.3 RUNX1-ETO竞争结合底物
嵌合蛋白可与亲本蛋白竞争底物,从而引发功能紊乱致癌。例如,在急性髓系白血病中发现的RUNX1-ETO融合蛋白[17,18]。RUNX1是造血干细胞分化的关键转录因子,其上含有结合DNA的Runt同源结构域。ETO是转录阻遏因子。RUNX1-ETO嵌合蛋白,保留了RUNX1的DNA结合域,继承了其结合靶基因启动子调控区的能力;然而,同时含有转录阻遏因子ETO的大部分结构域(图1 C)。该嵌合蛋白会与亲本基因竞争性结合靶基因,抑制转录、干扰正常功能、阻断分化进而引发白血病[17]。3.4 引入嵌合基因数据库,提高科研检索乐趣
随着DNA测序技术的进步和人类基因组的研究深入,越来越多的嵌合基因被鉴定出来,相关结果汇集于公共数据库中。Chimer DB (http://203.255.191.229:8080/chimerdbv31/mindex.cdb)是一个关于人类基因组染色体重排和嵌合基因的数据库,收集了大量嵌合转录本,包含染色体间易位、缺失、重复和倒位等结构变异数据,是确定癌症标志物和药物靶点的一个有价值的工具[19]。
ProteinPaint (https://pecan.stjude.org/)是St. Jude儿童研究医院开发的一个研究基因变异的强大交互式工具[20]。在ProteinPaint的交互式信息图中,可显示基因上的突变,涵盖癌症亚型、突变类型、突变频率和突变位置等一系列信息。这些信息可以用于探究突变在癌症发生、发展和复发中的潜在机制。
Mitelman染色体畸变和癌症嵌合基因数据库(http://cgap.nci.nih.gov/Chromosomes/Mitelman),储存了大量与染色体畸变、肿瘤有关的数据[21]。
ChiTaRS (http://chitars.bioinfo.cnio.es)数据收录了人类、小鼠、果蝇、斑马鱼、奶牛、大鼠、猪和酵母的嵌合转录本,以及人类致癌的染色体断点数据[22]。
dbCRID (http://dbCRID.biolead.org)是一个存储人类染色体重排数据的库,包括了大量染色体重排数据,以及所致的疾病及临床征兆等数据,并介绍了重排染色体的断点、基因的位置、连接序列等[23]。
在教学中,讲解嵌合基因的实例,有助于提高学生对知识的综合分析能力,但限于时间,课堂讲解仅能举少量例子。为此,引入相关数据库,让学生自己查询,这样既不占用教学时间,又让学生体验了科研检索的乐趣,为课堂教学营造科研气氛。
4 融汇嵌合基因的共性,贯通知识点
尽管嵌合基因的实例较多,稍显庞杂,但亦有规律可循。为了深入了解嵌合基因的结构域特征,本文作者曾对人和猪的嵌合RNA及亲本基因[6,7,8]进行了结构域和组合模式的分析,下面将从染色体断点特征、嵌合基因的翻译、细胞定位和转录调控等方面,总结嵌合基因的通性,以提升学生的对知识的融汇贯通能力和培养学生的思维梳理能力。4.1 染色体断点具有非随机性和复发性的特点
在癌症中,产生嵌合基因的染色体断点位置,具有非随机性和复发性的特点,易受染色体的空间位置和DNA序列特征的影响,如碱基序列的重复、脆性位点和酶识别位点等[8,24]。为总结染色体易位的特性,本文作者利用Chimer DB[19]数据库中与人类嵌合基因相关的染色体易位数据(含46 492个嵌合基因),分析了染色体易位的分布和丰度(图1 D)。易位断点在染色体上分布不均匀,断点趋向聚集。例如,在图1 D第二圈(自外向内)的散点图中,横坐标为染色体坐标,纵坐标为邻近断点之间的最小距离,越向内圈距离越小。图中,散点示意染色体的易位断点。整体上散点位于内圈的底端,说明断点之间的距离较近,倾向聚集分布。第三圈的密度图也证实了这一点,显示在一些染色体区段上,断点的发生频率较高,说明复发性较高。
染色体之间相互易位的频率也不同。例如,在图1 D最内圈,用线条显示了不同染色体之间或不同区域之间的易位情况,线的颜色代表了易位发生的频率,低发频率的易位偏多,高发频率的易位偏少。整体上易位呈现非随机性和复发性。
4.2 嵌合基因的翻译特点
一些嵌合基因会保留亲本基因的阅读框(reading frame)[25],编码原亲本的结构域或新蛋白[26]。本文作者分析人和猪嵌合转录本时[7,8],发现亲本基因融合之后,在嵌合分子中会出现以下情况(图1 E): (1)上游亲本基因不编码,仅下游亲本基因编码蛋白,形成5° UTR–CDS形式;(2)上游编码,下游不编码,形成CDS–3° UTR;(3)二者都编码,形成CDS–CDS形式;(4)嵌合基因的阅读框与亲本基因的阅读框错位,编码新蛋白。4.3 嵌合基因的细胞定位
嵌合基因的结构域,可在两亲本结构域的基础上,形成新的结构域组合,甚至是新的结构域,增加转录组和蛋白质组的多样性。下面以信号肽和跨膜结构域为例,说明结构域组合对嵌合蛋白的细胞定位的影响。4.3.1 信号肽对嵌合蛋白细胞定位的影响
信号肽是在起始密码子后一段疏水性肽段,可引导新合成的蛋白质向分泌通路转移,将其定位到细胞不同膜结构内。嵌合基因的细胞定位,可因所融合的信号肽而改变(图1 E):(1)在嵌合分子中,上游亲本的信号肽,会改变下游亲本蛋白的细胞定位;(2)嵌合分子融合后,下游亲本的信号肽位于编码区中部,可能会失去分子引导功能,而改变细胞的定位;(3)原一对亲本基因都没有信号肽,而嵌合蛋白却含信号肽,可能来自移码突变。此外,可能会因移码框突变,或者原亲本基因成为非编码序列,嵌合分子失去原亲本基因的信号肽,而改变定位。
4.3.2 跨膜结构域对嵌合蛋白细胞定位的影响
跨膜区域是指蛋白质序列中跨越细胞膜的区域。嵌合蛋白的跨膜结构域也有几种来源:(1)上游亲本基因有跨膜结构域,下游亲本没有:此时,嵌合蛋白中的下游亲本结构域,有可能因上游亲本的跨膜结构域而改变细胞定位;(2)下游亲本有跨膜结构域,而上游亲本没有跨膜结构域:可能会改变上游亲本的细胞定位;(3)一对亲本都有跨膜结构域:嵌合跨膜结构域即可能来自上游亲本,也有可能来自下游亲本;(4)一对亲本都没有跨膜结构域,但嵌合蛋白含有跨膜结构域:其可能来自阅读框移位;(5)嵌合基因发生了阅读框移位或结构域异常,也会致使原亲本的跨膜结构域丢失。
4.4 嵌合蛋白的表达调控
4.4.1 嵌合蛋白与亲本蛋白的竞争嵌合转录的产物可与亲本蛋白竞争底物,对抗正常蛋白,从而在癌细胞中出现显著负效应。当融合涉及转录激活因子或抑制因子时,与亲本蛋白的竞争倾向性更强,如前述RUNX1-ETO融合蛋白 (图 1 C)。
4.4.2 亲本基因对融合蛋白表达调控的影响
有时,嵌合基因的上游亲本基因含有强启动子,可促进嵌合蛋白中的下游亲本的结构域表达上调;或者下游亲本有稳定的3°端UTR,保持表达稳定。例如,嵌合基因致癌方式,常表现为:(1)原癌基因与强启动子基因融合,激活肿瘤转化功能;(2)异源基因的结构域融合,编码致癌功能蛋白;(3)融合导致肿瘤抑制基因失活。
4.4.3 正常组织中融合蛋白的表达调控
嵌合蛋白的表达不仅局限于癌组织,也存在于正常细胞[27,28]。例如,JAZF1-JJAZ1融合蛋白在正常组织中表达水平很低,但当表达水平升高时,会与子宫内膜间质肉瘤有关[29]。同样,在前列腺癌和良性前列腺组织,都检测到了SLC45A3-ELK4融合转录本[30]。融合蛋白在正常组织与癌变组织中表达水平的区别与联系,仍未知。
5 教学拓展的延伸
案例教学是激发学习兴趣,培养科研素质的有效方法之一[31,32]。为了拓展染色体结构变异的课堂教学,分以下几步设计了教学环节:从一个司法案例创设情境,提升教与学的趣味性,引出嵌合基因知识点;逐步介绍嵌合基因,补充理论知识;列举嵌合基因实例,分析染色体结构变异的相关问题;结合教师对嵌合基因的研究,总结其通性,提升知识的层面,提高学生融汇贯通的能力;力求开拓学生的视野,提升教学水平。教学设计的效果分析如下:(1)有利于拓展学生视野,激发科研兴趣
由于教科书的容量限制以及通识性要求,一般很少见到深入解析染色体结构变异的具体分子机理。本文关于嵌合基因的知识拓展,增加了课本以外的内容,并且以受人关注的官司作为引入,逐步深入,激发了学生的学习兴趣,培养了好奇心;此外,通过讲解教师自身对嵌合基因研究的感悟,使学生学到了科研技能,增强了科研自信心,培养了创新意识。
(2)有利于促进教师成长,创造教研相长的氛围
本文作者在讲授遗传学课程期间,正对嵌合基因进行相关研究,通过不断的教研结合,慢慢体会到如何把嵌合基因的相关研究进展与遗传教学中染色体结构变异教学联系起来,以研促教;另外,通过备课阅览众多遗传学教材和讲解经典理论,对遗传学中的科学大师们所做的贡献更加崇敬,促进了科研立志,培养了科学精神。
事实证明科研与教学并不孤立,因为,随着教龄和研龄的增长,逐渐会发现许多环节都有共鸣之处,正所谓:“科研是教学的源头活水,教学是科研的隐形动力”[31]。
总之,本文作者期望能将在嵌合基因方面的科研与教学相结合的经验,与众多从事遗传学课程教学的前辈和同行共勉。经验总结与行文,难免错漏,结合模式也非独有,但希望能为染色体结构变异教学提供拓展素材,能为教研相长提供参考经验。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
10th ed.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:3531304 [本文引用: 2]

pAbstract/p pBackground/p pGene fusion is ubiquitous over the course of evolution. It is expected to increase the diversity and complexity of transcriptomes and proteomes through chimeric sequence segments or altered regulation. However, chimeric mRNAs in pigs remain unclear. Here we identified some chimeric mRNAs in pigs and analyzed the expression of them across individuals and breeds using RNA-sequencing data./p pResults/p pThe present study identified 669 putative chimeric mRNAs in pigs, of which 251 chimeric candidates were detected in a set of RNA-sequencing data. The 618 candidates had clear trans-splicing sites, 537 of which obeyed the canonical GU-AG splice rule. Only two putative pig chimera variants whose fusion junction was overlapped with that of a known human chimeric mRNA were found. A set of unique chimeric events were considered middle variances in the expression across individuals and breeds, and revealed non-significant variance between sexes. Furthermore, the genomic region of the 5sup /sup partner gene shares a similar DNA sequence with that of the 3sup /sup partner gene for 458 putative chimeric mRNAs. The 81 of those shared DNA sequences significantly matched the known DNA-binding motifs in the JASPAR CORE database. Four DNA motifs shared in parental genomic regions had significant similarity with known human CTCF binding sites./p pConclusions/p pThe present study provided detailed information on some pig chimeric mRNAs. We proposed a model that trans-acting factors, such as CTCF, induced the spatial organisation of parental genes to the same transcriptional factory so that parental genes were coordinatively transcribed to give birth to chimeric mRNAs./p
Beijing: Jiuzhou Press,
[本文引用: 4]
北京: 九州出版社,
[本文引用: 4]
URL [本文引用: 5]
天然嵌合基因(natural chimeric gene)是由两个或两个以上的独立基因天然融合而成的新基因,该类型基 因的发现,突破了"一个基因对应一个染色体座位"的经典认知,扩展了基因的概念.在人类癌症研究过程中, 诸多的嵌合基因可导致肿瘤相关疾病,并作为癌症分子的诊断标志而受到人们的广泛关注.本文基于嵌合基因 生物信息学方面的相关研究,以癌基因为切入点,从天然嵌合基因的融合特点,转录,调控,以及融合蛋白的 结构域组合形式和功能等方面,结合本研究组前期的相关工作,综述了嵌合基因融合结构和功能的研究进展, 探讨了当前研究工作的困难与挑战,并对嵌合规律在新基因设计的应用作了展望.
URL [本文引用: 5]
天然嵌合基因(natural chimeric gene)是由两个或两个以上的独立基因天然融合而成的新基因,该类型基 因的发现,突破了"一个基因对应一个染色体座位"的经典认知,扩展了基因的概念.在人类癌症研究过程中, 诸多的嵌合基因可导致肿瘤相关疾病,并作为癌症分子的诊断标志而受到人们的广泛关注.本文基于嵌合基因 生物信息学方面的相关研究,以癌基因为切入点,从天然嵌合基因的融合特点,转录,调控,以及融合蛋白的 结构域组合形式和功能等方面,结合本研究组前期的相关工作,综述了嵌合基因融合结构和功能的研究进展, 探讨了当前研究工作的困难与挑战,并对嵌合规律在新基因设计的应用作了展望.
URLPMID:12476311 [本文引用: 1]
Abstract The Philadelphia chromosome (Ph), a minute chromosome that derives from the balanced translocation between chromosomes 9 and 22, was first described in 1960 and was for a long time the only genetic lesion consistently associated with human cancer. This chromosomal translocation results in the fusion between the 5' part of BCR gene, normally located on chromosome 22, and the 3' part of the ABL gene on chromosome 9 giving origin to a BCR/ABL fusion gene which is transcribed and then translated into a hybrid protein. Three main variants of the BCR/ABL gene have been described, that, depending on the length of the sequence of the BCR gene included, encode for the p190(BCR/ABL), P210(BCR/ABL), and P230(BCR/ABL) proteins. These three main variants are associated with distinct clinical types of human leukemias. Herein we review the data on the correlations between the type of BCR/ABL gene and the corresponding leukemic clinical features. Lastly, drawing on experimental data, we provide insight into the different transforming power of the three hybrid BCR/ABL proteins.
URLPMID:16254181 [本文引用: 1]
Recurrent chromosomal rearrangements have not been well characterized in common carcinomas. We used a bioinformatics approach to discover candidate oncogenic chromosomal aberrations on the basis of outlier gene expression. Two ETS transcription factors, ERG and ETV1, were identified as outliers in prostate cancer. We identified recurrent gene fusions of the 5' untranslated region of TMPRSS2 to ERG or ETV1 in prostate cancer tissues with outlier expression. By using fluorescence in situ hybridization, we demonstrated that 23 of 29 prostate cancer samples harbor rearrangements in ERG or ETV1. Cell line experiments suggest that the androgen-responsive promoter elements of TMPRSS2 mediate the overexpression of ETS family members in prostate cancer. These results have implications in the development of carcinomas and the molecular diagnosis and treatment of prostate cancer.
URLPMID:10448117 [本文引用: 1]
Abstract Tumor development in different cell types and tissue locations involves many pathways, distinct genes and exogenous factors. Tumor type-specific chromosome rearrangements resulting in fusion genes or promoter swapping are believed to be involved in the early development of many tumor types. They are present in almost all cases of a particular tumor type and cases have been described that carry only tumor type-specific translocations without any signs of other cytogenetic changes. The mechanisms behind chromosome rearrangements in solid tumors are largely unknown. Radiation is an important factor in thyroid carcinomas but no com-$bmon sequence motifs are made out in the break points of solid tumors. The fusion genes found in sarcomas are dominated by the transcription factor type of genes with the TLS/FUS and EWS series of fusion genes as the largest group. More than 50% of papillary thyroid carcinomas carry fusion proteins with tyrosine kinase activity. Rearrangements involving HMGIC, HMGIY, and PLAG1 are common in benign mesenchymal tumors and salivary gland adenomas. Many recurrent tumor translocations show a strict specificity for tumor type. This specificity can most likely be explained by the specific sets of target genes that are deregulated by the fusion gene products. Identification of the downstream target genes is currently the object of intense research and may provide us with information that will help design better diagnostic tools and eventually find a cure for these diseases. Copyright 1999 Academic Press.
URLPMID:3845546 [本文引用: 1]
Studies over the past decades have uncovered fusion genes, a class of oncogenes that provide immense diagnostic and therapeutic advantages because of their tumor-specific expression. Originally associated with hemotologic cancers, fusion genes have recently been discovered in a wide array of solid tumors, including sarcomas, carcinomas, and tumors of the central nervous system. Fusion genes are attractive as both therapeutic targets and diagnostic tools due to their inherent expression in tumor tissue alone. Therefore, the discovery and elucidation of fusion genes in various cancer types may provide more effective therapies in the future for cancer patients.
URL [本文引用: 1]
URLPMID:23821214 [本文引用: 1]
AbstractSalivary gland tumors constitute a heterogeneous group of uncommon diseases that pose significant diagnostic and therapeutic challenges. However, the recent discovery of a translocation-generated gene fusion network in salivary gland carcinomas as well in benign salivary gland tumors opens up new avenues for improved diagnosis, prognostication, and development of specific targeted therapies. The gene fusions encode novel fusion oncoproteins or ectopically expressed normal or truncated oncoproteins. The major targets of the translocations are transcriptional coactivators, tyrosine kinase receptors, and transcription factors involved in growth factor signaling and cell cycle regulation. Notably, several of these targets or pathways activated by these targets are druggable. Examples of clinically significant gene fusions in salivary gland cancers are the
URLPMID:12957454 [本文引用: 1]
The aim of this study was to assess the antitumour response and time to progression (TTP) of patients treated with imatinib mesylate (Glivec , Gleevec , formerly STI-571) who had advanced and/or metastatic gastrointestinal stroma tumours (GIST) or other soft tissue sarcomas (STS). Patients with measurable lesions and adequate organ function were entered. They were treated with imatinib mesylate at the dose of 400 mg twice daily (bid). All tumours were subject to a stringent pathological review by an expert panel. Immunohistochemical expression of KIT expression was evaluated. A total of 51 patients (27 GIST, 24 other STS), median age 53 years, median World Health Organization (WHO) performance score 1, were entered. 71% of the patients had received prior chemotherapy. The most frequent side-effects were anaemia (92%), periorbital oedema (84%), skin rash (69%), fatigue (76%), nausea (57%), granulocytopenia (47%) and diarrhoea (47%). Most of these side-effects were mild to moderate and no patient was taken off study due to side-effects. Skin rash and periorbital oedema frequently seem to be self limiting, despite continued treatment. In GIST patients, the current response rates (RRs) are 4% complete remission (CR), 67% partial remission (PR), 18% stable disease (SD) and 11% progression (PD). 73% of GIST patients are free from progression at 1 year. In the other STS group, there were no objective responses. The median time to progression in this subgroup was only 58 days. Imatinib mesylate is well tolerated at a dose of 400 mg bid. This dose is active in patients with KIT-positive GIST, but patients with other STS subtypes unselected for a molecular target are unlikely to benefit.
URLPMID:25515960 [本文引用: 2]
PAX5-JAK2 has recently been identified as a novel recurrent fusion gene in B-cell precursor acute lymphoblastic leukemia, but the function of the encoded chimeric protein has not yet been characterized in detail. Herein we show that the PAX5-JAK2 chimera, which consists of the DNA-binding paired domain of PAX5 and the active kinase domain of JAK2, is a nuclear protein that has the ability to bind to wild-type PAX5 target loci. Moreover, our data provide compelling evidence that PAX5-JAK2 functions as a nuclear catalytically active kinase that autophosphorylates and in turn phosphorylates and activates downstream signal transducers and activators of transcription (STATs) in an apparently noncanonical mode. The chimeric protein also enables cytokine-independent growth of Ba/F3 cells and therefore possesses transforming potential. Importantly, the kinase activity of PAX5-JAK2 can be efficiently blocked by JAK2 inhibitors, rendering it a potential target for therapeutic intervention. Together, our data show that PAX5-JAK2 simultaneously deregulates the PAX5 downstream transcriptional program and activates the Janus kinase-STAT signaling cascade and thus, by interfering with these two important pathways, may promote leukemogenesis.
URLPMID:22201794 [本文引用: 3]
RUNX1 is a transcription factor that regulates critical processes in many aspects of hematopoiesis. RUNX1 is also integral in defining the definitive hematopoietic stem cell. In addition, many hematological diseases like myelodysplastic syndrome and myeloproliferative neoplasms have been associated with mutations in RUNX1. Located on chromosomal 21, the RUNX1 gene is involved in many forms of chromosomal translocations in leukemia. t(8;21) is one of the most common chromosomal translocations found in acute myeloid leukemia (AML), where it results in a fusion protein between RUNX1 and ETO. The RUNX1-ETO fusion protein is found in approximately 12% of all AML patients. In this review, we detail the structural features, functions, and models used to study both RUNX1 and RUNX1-ETO in hematopoiesis over the past two decades.
URL [本文引用: 1]
URLPMID:27899563 [本文引用: 2]
Fusion gene is an important class of therapeutic targets and prognostic markers in cancer. ChimerDB is a comprehensive database of fusion genes encompassing analysis of deep sequencing data and manual curations. In this update, the database coverage was enhanced considerably by adding two new modules of The Cancer Genome Atlas (TCGA) RNA-Seq analysis and PubMed abstract mining. ChimerDB 3.0 is composed of three modules of ChimerKB, ChimerPub and ChimerSeq. ChimerKB represents a knowledgebase including 1066 fusion genes with manual curation that were compiled from public resources of fusion genes with experimental evidences. ChimerPub includes 2767 fusion genes obtained from text mining of PubMed abstracts. ChimerSeq module is designed to archive the fusion candidates from deep sequencing data. Importantly, we have analyzed RNA-Seq data of the TCGA project covering 4569 patients in 23 cancer types using two reliable programs of FusionScan and TopHat-Fusion. The new user interface supports diverse search options and graphic representation of fusion gene structure. ChimerDB 3.0 is available athttp://ercsb.ewha.ac.kr/fusiongene/.
URLPMID:26711108 [本文引用: 1]
Genomic alterations frequently occur in many cancer patients and play important mechanistic roles in the pathogenesis of cancer. Furthermore, they can modify the expression level of genes due to altered copy number in the corresponding region of the chromosome. An accumulating body of evidence supports the possibility that strong genome-wide correlation exists between DNA content and gene expression. Therefore, more comprehensive analysis is needed to quantify the relationship between genomic alteration and gene expression. A well-designed bioinformatics tool is essential to perform this kind of integrative analysis. A few programs have already been introduced for integrative analysis. However, there are many limitations in their performance of comprehensive integrated analysis using published software because of limitations in implemented algorithms and visualization modules.To address this issue, we have implemented the Java-based program CHESS to allow integrative analysis of two experimental data sets: genomic alteration and genome-wide expression profile. CHESS is composed of a genomic alteration analysis module and an integrative analysis module. The genomic alteration analysis module detects genomic alteration by applying a threshold based method or SW-ARRAY algorithm and investigates whether the detected alteration is phenotype specific or not. On the other hand, the integrative analysis module measures the genomic alteration's influence on gene expression. It is divided into two separate parts. The first part calculates overall correlation between comparative genomic hybridization ratio and gene expression level by applying following three statistical methods: simple linear regression, Spearman rank correlation and Pearson's correlation. In the second part, CHESS detects the genes that are differentially expressed according to the genomic alteration pattern with three alternative statistical approaches: Student's t-test, Fisher's exact test and Chi square test. By successive operations of two modules, users can clarify how gene expression levels are affected by the phenotype specific genomic alterations. As CHESS was developed in both Java application and web environments, it can be run on a web browser or a local machine. It also supports all experimental platforms if a properly formatted text file is provided to include the chromosomal position of probes and their gene identifiers.CHESS is a user-friendly tool for investigating disease specific genomic alterations and quantitative relationships between those genomic alterations and genome-wide gene expression profiling.
.
URL [本文引用: 1]
URLPMID:4383979 [本文引用: 1]
Chimeric RNAs that comprise two or more different transcripts have been identified in many cancers and among the Expressed Sequence Tags (ESTs) isolated from different organisms; they might represent functional proteins and produce different disease phenotypes. The ChiTaRS 2.1 database of chimeric transcripts and RNA-Seq data (http://chitars.bioinfo.cnio.es/) is the second version of the ChiTaRS database and includes improvements in content and functionality. Chimeras from eight organisms have been collated including novel sense ntisense (SAS) chimeras resulting from the slippage of the sense and anti-sense intragenic regions. The new database version collects more than 29 000 chimeric transcripts and indicates the expression and tissue specificity for 333 entries confirmed by RNA-seq reads mapping the chimeric junction sites. User interface allows for rapid and easy analysis of evolutionary conservation of fusions, literature references and experimental data supporting fusions in different organisms. More than 1428 cancer breakpoints have been automatically collected from public databases and manually verified to identify their correct cross-references, genomic sequences and junction sites. As a result, the ChiTaRS 2.1 collection of chimeras from eight organisms and human cancer breakpoints extends our understanding of the evolution of chimeric transcripts in eukaryotes as well as their functional role in carcinogenic processes.
URLPMID:3013658 [本文引用: 1]
Chromosomal rearrangement (CR) events result from abnormal breaking and rejoining of the DNA molecules, or from crossing-over between repetitive DNA sequences, and they are involved in many tumor and non-tumor diseases. Investigations of disease-associated CR events can not only lead to important discoveries about DNA breakage and repair mechanisms, but also offer important clues about the pathologic causes and the diagnostic/therapeutic targets of these diseases. We have developed a database of Chromosomal Rearrangements In Diseases (dbCRID, http://dbCRID.biolead.org), a comprehensive database of human CR events and their associated diseases. For each reported CR event, dbCRID documents the type of the event, the disease or symptoms associated, and--when possible--detailed information about the CR event including precise breakpoint positions, junction sequences, genes and gene regions disrupted and experimental techniques applied to discover/analyze the CR event. With 2643 records of disease-associated CR events curated from 1172 original studies, dbCRID is a comprehensive and dynamic resource useful for studying DNA breakage and repair mechanisms, and for analyzing the genetic basis of human tumor and non-tumor diseases.
URLPMID:24691255 [本文引用: 1]
Abstract Chromosome translocations are catastrophic genomic events and often play key roles in tumorigenesis. Yet the biogenesis of chromosome translocations is remarkably poorly understood. Recent work has delineated several distinct mechanistic steps in the formation of translocations, and it has become apparent that non-random spatial genome organization, DNA repair pathways and chromatin features, including histone marks and the dynamic motion of broken chromatin, are critical for determining translocation frequency and partner selection.
URL [本文引用: 1]
URLPMID:3371848 [本文引用: 1]
Motivation: Chimeric RNA transcripts are generated by different mechanisms including pre-mRNA trans-splicing, chromosomal translocations and/or gene fusions. It was shown recently that at least some of chimeric transcripts can be translated into functional chimeric proteins. Results: To gain a better understanding of the design principles underlying chimeric proteins, we have analyzed 7,424 chimeric RNAs from humans. We focused on the specific domains present in these proteins, comparing their permutations with those of known human proteins. Our method uses genomic alignments of the chimeras, identification of the gene ene junction sites and prediction of the protein domains. We found that chimeras contain complete protein domains significantly more often than in random data sets. Specifically, we show that eight different types of domains are over-represented among all chimeras as well as in those chimeras confirmed by RNA-seq experiments. Moreover, we discovered that some chimeras potentially encode proteins with novel and unique domain combinations. Given the observed prevalence of entire protein domains in chimeras, we predict that certain putative chimeras that lack activation domains may actively compete with their parental proteins, thereby exerting dominant negative effects. More generally, the production of chimeric transcripts enables a combinatorial increase in the number of protein products available, which may disturb the function of parental genes and influence their protein rotein interaction network. Availability: our scripts are available upon request. Contact: avalencia@cnio.es Supplementary information: Supplementary data are available at Bioinformatics online.
URLPMID:29100211 [本文引用: 1]
react-text: 64 tumor origin /react-text react-text: 65 /react-text
URLPMID:28589684 [本文引用: 1]
Abstract Traditionally, chimeric RNAs were considered to be exclusive to cancer cells. When occasionally observed in normal samples, they were usually considered to be transcriptional 'noises,' or artifacts due to template switching during the reverse transcription and/or Polymerase chain reaction (PCR) steps of experimentation. However, with the advances being made in next generation sequencing technologies and software tools, as well as the accumulation of new experimental evidences, increasing numbers of chimeric transcripts are being identified in noncancerous tissues and cells. Recent studies have also demonstrated functional relevance, for at least a subset of chimeric RNAs in normal physiology. The advances have resulted in an influx of knowledge; this knowledge indicates that chimeric RNAs are a component of basic biology, and thus challenging traditional dogma. In addition to chromosomal rearrangement, chimeric RNAs can also be formed via different molecular mechanisms including cis-splicing of adjacent genes (cis-SAGe) and trans-splicing, as well as others. Little is known about the details of these noncanonical splicing processes. However, research in this new field promises to not only advance our basic understanding of the human genome and gene regulation, but also lead to improvements in clinical practice, especially in the areas of cancer diagnostics and treatment. For further resources related to this article, please visit the WIREs website.
URLPMID:19158498 [本文引用: 1]
Chimeric gene products, most often resulting from chromosome translocations, have been considered unique features of cancer, or at least of cells at high risk for becoming cancerous. Chimeric JAZF1-JJAZ1 mRNA transcribed from DNA spanning the site of recombination in the (7;17)(p15;q21) chromosomal translocation found in half of endometrial stromal sarcomas and most cases of benign stromal nodules is one such example. The recent finding that chimeric JAZF1-JJAZ1 mRNA can also be detected in normal endometrial stromal cells suggests that chimeric gene products are not limited to cancer or pre-cancerous cells. The JAZF1-JJAZ1 mRNA and the protein encoded by it appear to be identical to that synthesized from the gene fusion in neoplastic cells. In cultured cells, the chimeric protein has anti-apoptotic properties and is pro-proliferative when unrearranged JJAZ1 alleles are silenced, as they are in endometrial stromal sarcomas but not in the stromal nodules. These observations are consistent with the conclusion that chromosomal rearrangements and gene fusions in neoplastic cells may represent mechanisms for the deregulated expression of chimeric gene products that are generated at specific stages in cell development and have physiologic functions in normal cells. Furthermore, it may be possible that other means for abnormal production of chimeric gene products, such as hyperactive trans-splicing of RNA, may be another mechanism underlying the neoplastic properties of tumor cells.
URLPMID:28716526 [本文引用: 1]
Abstract Gene fusions in cancer typically lead to the expression of a fusion protein or disrupt the expression of one of the parental genes. Here we report a new phenomenon whereby a fusion transcript functions as a long non-coding chimeric RNA (lnccRNA). This fusion RNA, SLC45A3-ELK4, generated by cis-splicing between neighboring genes, was found in prostate cancer. The fusion RNA encodes the same protein as ELK4. Intriguingly, we found that the fusion RNA level is less than 1% of wild type ELK4, unlikely to perturb the general pool of ELK4 protein. Nonetheless, when the fusion RNA, but not ELK4 is silenced, cell proliferation is inhibited in both androgen-dependent and castration-resistant prostate cancer cells. This growth arrest can be rescued by exogenous expression of the fusion and a mutant designed to prevent translation of the ELK4 protein. In the same setting, the mutant could also suppress CDKN1A and several other targets of SLC45A3-ELK4. In addition, similar to many long non-coding RNAs, the fusion RNA is enriched in the nuclear fraction. Altogether, these results indicate that SLC45A3-ELK4 regulates cancer cell proliferation by its transcript, not translated protein. Copyright 2017 Elsevier B.V. All rights reserved.
URL [本文引用: 2]
课堂是本科教学的主要场地,充分利用课堂教学培养本科生的科研素质,是一个值得思考的教改问题。本文从系统架构课程内容体系、教学内容反映科研进展、知识点的讲解反映其科研活动、用PPT动画讲解科学原理和实验过程、用双语教学提高专业英语阅读能力、考试考察学生的科研分析能力6个方面介绍了作者多年来在遗传学课堂教学中培养学生科研素质的教改措施与实践经验。这些改革措施有利于激发本科生的学习积极性,培养本科生发现问题、分析问题和解决问题的科学思维能力,提高本科生的科技英语阅读能力和文献查阅能力,为本科生进入科研领域奠定了良好的基础。
URL [本文引用: 2]
课堂是本科教学的主要场地,充分利用课堂教学培养本科生的科研素质,是一个值得思考的教改问题。本文从系统架构课程内容体系、教学内容反映科研进展、知识点的讲解反映其科研活动、用PPT动画讲解科学原理和实验过程、用双语教学提高专业英语阅读能力、考试考察学生的科研分析能力6个方面介绍了作者多年来在遗传学课堂教学中培养学生科研素质的教改措施与实践经验。这些改革措施有利于激发本科生的学习积极性,培养本科生发现问题、分析问题和解决问题的科学思维能力,提高本科生的科技英语阅读能力和文献查阅能力,为本科生进入科研领域奠定了良好的基础。
URLMagsci [本文引用: 1]
<p>通过增设研究性较强或由学生自主设计的实验项目, 采用生动有趣的案例式教学和问题式教学, 用“多元性”的教学理念设计实验教学环节, 结合综合性的教学手段, 有效激发了学生对遗传学实验的学习兴趣。</p>
URLMagsci [本文引用: 1]
<p>通过增设研究性较强或由学生自主设计的实验项目, 采用生动有趣的案例式教学和问题式教学, 用“多元性”的教学理念设计实验教学环节, 结合综合性的教学手段, 有效激发了学生对遗传学实验的学习兴趣。</p>

{kind=link}
{kind=link}