 ,1,3, 邢清和2,3, 贺林,1,3
,1,3, 邢清和2,3, 贺林,1,3Retrospect and prospect of the genetic research on birth defects
in China
Liya Sunin China
,1,3, Qinghe Xing2,3, Lin He,1,3通讯作者:
编委: 史庆华
收稿日期:2018-07-2修回日期:2018-09-4网络出版日期:2018-10-20
Received:2018-07-2Revised:2018-09-4Online:2018-10-20
作者简介 About authors
孙丽雅,博士,研究方向:精准医学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (4479KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孙丽雅, 邢清和, 贺林. 中国出生缺陷遗传学研究的回顾与展望[J]. 遗传, 2018, 40(10): 800-813 doi:10.16288/j.yczz.18-181
Liya Sun, Qinghe Xing, Lin He.
in China
目前,我国正处在为“健康中国”乃至“强大中国”打基础的新时代,其中降低出生缺陷率在提高人口健康水平方面起到根本性的作用。同时,根据“健康与疾病的发育起源(DOHaD)”学说[1],出生缺陷是人类各类疾病的“源头杀手”,因此研究出生缺陷可以为研究其他疾病提供不可或缺的参考信息,抓住了“出生缺陷”才有可能抓住其他疾病。
2012年9月,原国家卫生部发布的《中国出生缺陷防治报告》将“出生缺陷”定义为对婴儿出生前发生的身体结构、功能或代谢等方面异常的一种统称,通常包括先天畸形、染色体异常、遗传代谢性疾病以及功能异常如盲、聋和智力障碍等。目前已知的出生缺陷类疾病至少有8000~10000种,据在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man, OMIM)统计(截至2018年8月30日),其中有5208种孟德尔单基因疾病(或特征)已经发现了明确的致病基因(涉及3575个基因)。截至2012年,我国出生缺陷总发生率约为5.6%,处于中等收入国家水平,但由于人口基数大,每年新增出生缺陷病例高达约90万例,相当于每30秒就有1名出生缺陷患儿出生[2]。《中国出生缺陷防治报告》中显示,2011年我国最常见的5类围产期出生缺陷疾病分别为先天性心脏病、多指(趾)、总唇裂、脑积水和马蹄内翻。2015年10月,党的十八届五中全会正式宣布“全面放开二胎”政策,随着社会发展而伴发的婚龄孕龄的逐渐推迟导致高龄产妇逐年增加,进一步增加了潜在的出生缺陷发生率。出生缺陷儿童的死亡率极高,约30%会在5岁之前死亡[3]。2012年中国城市和农村小于1岁的居民疾病死因构成比中,“先天畸形、变形和染色体异常”类的因素占近1/4[4]。约40%的出生缺陷儿童会发展为终身残疾,其中肢体残疾、听力残疾和智力残疾是主要类型,严重影响儿童的生命和生活质量,也给家庭和社会带来沉重的精神和经济负担[3]。此外,在对2000~ 2015年间全球各国小于5岁儿童的死亡率分析中发现,死亡率越低的国家(通常是发达国家),先天畸形等出生缺陷所致儿童死亡的比例越高,可见减少出生缺陷是世界各国共同面临的挑战[5]。
出生缺陷的病因多种多样,其作用时间窗口位于出生前的受精卵形成及胎儿发育的时期。这些病因大致可分为遗传因素和环境因素。据估计,由遗传因素为主所致的出生缺陷占比约20%~30%,由环境因素(如母体疾病、营养不足、宫内病原体感染或环境有害化学物质、药物或射线等因素)所致的出生缺陷占比约10%,而剩余的60%~70%的出生缺陷多是由遗传和环境因素共同作用的结果[6]。因此,遗传因素直接或间接地导致了超过80%的出生缺陷,遗传学病因研究是出生缺陷防治工作的关键基础。相较于成人疾病,开展出生缺陷类的遗传学病因研究有其自身明显的优势:(1) 绝大部分出生缺陷在婴儿刚出生或儿童时期即会表现出相关的临床症状,进而被鉴别和诊断,因此出生缺陷的发生受后天环境影响少;(2) 个体层面的致病因素(无论是环境因素还是遗传因素)往往与发育密切相关,且具有较强的致病效应。近半个世纪以来,国内外的遗传学研究者已经成功鉴定了几千种出生缺陷类疾病的遗传学病因,为出生缺陷防控提供了有力的技术支撑。出生缺陷可以说是临床医学遗传学的前沿阵地和人类遗传学知识的资源宝库。本文首先回顾了我国出生缺陷遗传学的研究历史,继而介绍当前国内外出生缺陷遗传学研究的现状和热点,最后对未来的研究方向及相关的临床应用趋势进行展望和讨论,为读者提供一个相对全局性的视角来了解我国出生缺陷的遗传学研究概况。
1 我国出生缺陷遗传学研究历史回顾
我国出生缺陷遗传学研究的正式起航可以追溯至20世纪60年代,主要分为两个阶段:(1) 学习跟踪阶段。1962年,中国****吴旻从苏联学成回国后,发表了中国人类染色体的组型,并率先把对染色体组型的观察用于人类疾病研究,开创了国内的临床细胞遗传学研究领域[7]。1963年,上海儿童医院苏祖斐报告了出生缺陷中唐氏综合征患儿的染色体研究结果,发现21号染色体三体是该病的致病病因。同年,中国医学科学院詹宝光和吴旻在对羊水细胞中性染色质的检查中确认了一例XXY性染色体异常患者。从此,全国各地陆续开展了大量的出生缺陷相关的染色体异常检查和临床应用。1972年,中南大学夏家辉等向国内引进了国际上1971年建立的人类染色体G显带技术;1979年进一步引进了国际上1977年建立的人类染色体高分辨技术,并于1981年运用染色体高分辨技术将睾丸决定基因TDF定位在Y染色体的p11.32区域,为先天性性器官发育缺陷提供了重要的遗传学依据。继1983年的工作,他们于1990年报道了运用各种染色体技术在产前诊断中发现的约1200种染色体异常,引起了国内外的广泛关注。1984年,中国医学科学院宿远和吴旻提出了人类高分辨显带核型模式图,使我国的染色体显带研究进入微细胞遗传学领域。1986年,夏家辉等提出将1985年美国发现的PCR与染色体显微切割技术相结合,建立定点克隆基因的技术。这一尝试将我国出生缺陷遗传学研究从染色体水平进一步精细化到基因水平,并将主要的研究样本从遗传突变细胞拓展到家系样本[8]。此外,在临床转化方面,上海交通大学曾溢滔自1978年起发展了一整套遗传病分子诊断技术,先后攻克了地中海贫血、苯丙酮尿症、杜氏肌萎缩症、血友病和亨廷顿舞蹈症等国内主要遗传病的临床诊断和产前诊断,有力推动了我国遗传诊断学科的发展;
(2) 引领国际前沿发展。突破的契机往往在于对最新资源的及时把握和有效运用。1987年,美国能源部与国家健康研究院(National Institutes of Health, NIH)启动了“人类基因组计划”,无疑对出生缺陷的解决注入一剂兴奋剂。中国也承担了其中1%的任务[9]。1998年,夏家辉团队在中国的出生缺陷遗传家系中率先发现了具有新功能的神经性耳聋的致病基因GJB3,这一工作发表在具有国际重要影响力的专业杂志Nature Genetics[10]上。
中华民族包含相对独立通婚的56个民族,其中55个为少数民族,加上中国人民安土重迁的文化性格,使得这片土地孕育生养着很多大家族,因此我国保留有丰富的遗传家系资源。2000年前后,国内的遗传学研究者充分运用国内独有的样本资源,同时利用“人类基因组计划”的研究成果,再结合国际先进的分析方法,为中国出生缺陷的遗传学研究迎来了百花齐放的春天(图1)[12]。1996年,上海交通大学贺林团队首先把精力放在揭示A-1型短指(趾)症的研究上。A-1型短指(趾)症是1903年报道的世界上第一例孟德尔常染色体显性遗传病,被遗传学和生物学教科书广为引用,倍受世人关注,但近百年来也未鉴定其致病基因。贺林团队在中国家系样本中运用连锁分析方法,率先完成了致病基因IHH的精确定位、克隆与突变检测,还开展了后续的功能研究,相关成果分别于2000年、2001年和2009年发表在American Journal of Human Genetics[13]、Nature Genetics[14]和Nature[15]上,这是我国出生缺陷遗传学研究里程碑式的成果,体现了我国在该领域的引领地位。另外,贺林团队还对出生缺陷“贺-赵缺陷症”做出了积极的贡献。“贺-赵缺陷症”又称“家族性恒齿缺失”,由陕西省中学教师赵双民与内科医生赵万里于1985年首先发现[16]。2001年,贺林团队成功对该病的致病基因进行了精确定位,使该病成为国际上首次以中国人姓氏命名的遗传疾病[17]。中国医学科学院沈岩团队也于2001年在世界上首次定位并克隆了DSPP基因,发现其为遗传性乳光牙本质的致病基因[18];同年,中国科学院上海生命科学研究院孔祥银团队在遗传性牙本质发育不全Ⅰ型的疾病家系中也克隆了DSPP基因,发现该基因的部分突变还会引起进行性高频耳聋,建立了牙齿发育和内耳发育之间的联系,表明同一致病基因可以引发多种疾病表型[19]。这两项研究都相继发表在当年的Nature Genetics上。2002年,孔祥银团队再次在该杂志报道了成功定位并克隆遗传性儿童白内障的致病基因HSF4的工作,发现遗传突变会影响HSF4蛋白的DNA结合区功能,在白内障发病过程中具有重要作用[20]。2003年,同济大学医学遗传研究所陈义汉和国家人类基因组南方研究中心的徐世杰合作,他们在一个家族性房颤大家系中定位并克隆了致病基因KCNQ1,并在后续的功能学研究中发现该基因的致病突变(S140G)是通过改变动作电位时长引发房颤,这一研究成果发表在Science上[21]。在国内遗传学家的共同努力下,以上述优秀工作为代表的中国出生缺陷遗传学研究在21世纪伊始便走在了世界先进行列[11]。此后,复旦大学邢清和[22]、王红艳[23]和王磊[24],中国医学科学院张学[25],南方医科大学徐湘民[26],解放军总医院王秋菊[27]和袁慧军[28]等一批****亦为此领域分别做了重要贡献。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12000年前后中国出生缺陷遗传学研究迎来百花齐发的春天
A:上海交通大学贺林团队攻克A-1型短指(趾)症的百年遗传谜题。图为所使用的中国家系信息;B:2001年,Nature杂志报道我国参与“人类基因组计划”后,在遗传学研究和产业领域表现出了巨大进步[11]。
Fig. 1Around 2000, China’s genetic research on birth defects experienced a rapid development
2 当前国内外出生缺陷遗传学研究的现状和热点
“人类基因组计划”的实施促进了遗传学检测技术,特别是DNA测序技术的飞速发展,这一发展也给医学遗传学研究模式和效率带来了日新月异的变化[29]。DNA测序技术的发展始于1977年,Sanger等开创的链终止法测序技术标志着人类第一代DNA测序技术的诞生[30]。Sanger测序法准确率高,但是测序通量低,不适用于大规模的筛查,因此早期的出生缺陷遗传学研究多是先采用PCR和电泳技术,对遗传标记进行分型和分析来间接定位候选基因,然后再采用Sanger测序法寻找较小范围内的致病基因及其突变。“人类基因组计划”结束后不久的2005年,美国罗氏公司推出了首款高通量的基因组测序系统—454基因组测序仪,代表了第二代DNA测序技术的诞生[31]。随后市场上相继出现多款高通量测序系统,性能愈加优化,测序成本快速下降。最初个人全基因组的测序费用高达1亿美金,而时至今日同样数据量的人类全基因组只需要1000美金左右。测序技术成本的“亲民化”使得遗传学研究者在面对小规模的疾病样本集,特别是家系样本时,可直接采用全基因组测序(whole genome sequencing, WGS)或全外显子组测序(whole exome sequencing, WES)[32]的策略从基因组水平筛检致病基因及其突变[33]。2.1 单基因突变导致的出生缺陷的研究
阵发性运动源性运动障碍(paroxysmal kinesigenit dyskinesia, PKD)是一种多发于儿童和青少年时期的出生缺陷疾病。2011年,复旦大学吴志英和福建医科大学王柠合作,利用WES技术结合Sanger测序验证的策略,在具有PKD病史的8个汉族家系中鉴别出了PRRT2基因的3个截短突变为该病的致病突变[34]。2015年,德国Maass等[35]利用WGS技术在一个兼具短趾和高血压遗传特征的土耳其大家系中鉴定出了PDE3A基因上的致病突变,并在5个其他民族的独立家系中得到验证。播散性浅表性光线性汗孔角化症(disseminated superfical actinic porokeratosis, DSAP)一般在20~40岁之间发病,是一种常染色体显性遗传病。2012年,安徽医科大学张学军团队利用WES技术并结合功能学研究分别在家系和散发样本中鉴定出MVK基因为DSAP疾病的致病基因之一[36]。2016年,复旦大学的王磊团队利用WES平台在一个由于卵子成熟障碍导致多名女性成员不孕的大家系中发现,TUBB8基因的突变可能是这个家族不孕的遗传学病因。这一发现在其他23个患病家庭中得到验证,并在功能学实验中发现TUBB8基因突变会通过影响微管结构的组装,阻遏卵母细胞的减数分裂,从而导致不孕[24]。可以看出,与基于遗传标记的家系连锁分析策略不同,目前基于新一代测序技术的出生缺陷家系研究,一般是先评估疾病遗传模式(如外显率为100%的常染色体显性遗传),再挑选先证者家系中少数几名诊断明确的患者和正常人开展二代测序(大多为WES),筛选出与表型共分离的功能性罕见遗传突变及其所在基因;然后用Sanger测序等方法靶向鉴定候选基因在其他家系或同种族正常人群中发生突变的情况。那些在验证人群中仍然严格表现为与表型共分离的基因,便是值得进一步开展病理功能学研究的候选致病基因。这个策略对单基因外显子区域的单碱基变异(single nucleotide variant, SNV)或插入缺失突变(insertion and deletion variation, InDel)引发的遗传因素高度外显的家族性出生缺陷疾病尤其有效,而且目前临床上基于WES技术已经能够对相当一部分儿科遗传病做出基因诊断[37](图2)。不过,对于由基因组结构变异(structural variation, SV)或基因拷贝数变异(copy number variations, CNV)[38]引发的出生缺陷疾病或存在外显不全、拟表型、多基因遗传等复杂情况的疾病,则需要基于WGS平台或家族更多成员的连锁分析来进行更为深入的探索[39]。图2
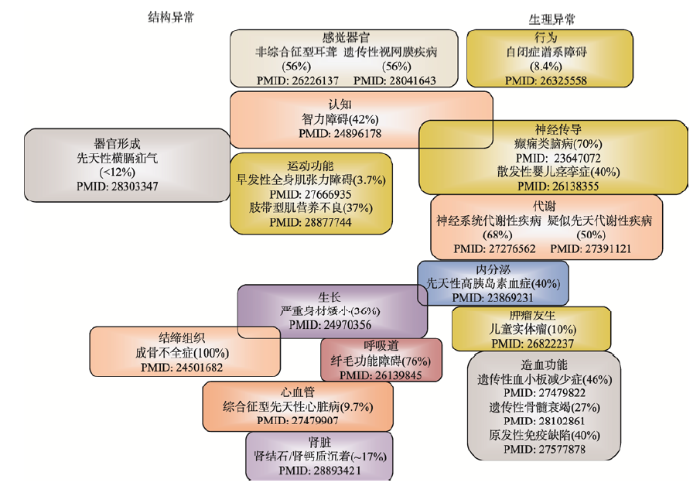 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2全外显子组基因检测在部分儿童遗传病中的诊断率
Fig. 2Available diagnostic rates based on whole-exome sequencing in classes of paediatric genomic diseases
括号中的百分比数据为诊断率;PMID为参考文献在PubMed数据库中的索引号;临床诊断主要基于已知的或较大概率致病的基因突变;方框的大小近似反映了临床相应疾病的患病率。根据参考文献[37]修改绘制。
2.2 多基因复杂出生缺陷的研究
出生缺陷除了由单基因或单位点的遗传突变引起,还有相当一部分是多基因和环境因素综合作用的结果,如高发的非综合征型先天性心脏病和总唇裂。单基因或单位点遗传疾病的主要研究样本类型为染色体变异细胞系或疾病家系,一般符合“rare diseases rare variants”的致病假设,而对于复杂疾病,国际上曾提出“common disease common variants”的致病假设[40],即疾病的发生与多个相对微效的致病基因或变异有关,由于这些变异的致病性较弱,它们可以在疾病或正常人群中以较高的频率存在,只有当这类变异在个体中累积到一定数量时才会导致疾病发生。群体遗传学中的关联分析是研究这类疾病的常用策略。群体遗传学是计算遗传变异在人群中分布频率的统计性学科,因此为达到足够的统计效力,往往需要对成百上千甚至上万的散发性样本进行遗传变异检测,和测序技术一同发展起来的基因芯片技术在该领域最先得到应用。不同于DNA测序,基因芯片是针对已知位点的靶向性检测。当它与标记数目多、覆盖密度大的第三代遗传多态性标记—单核苷酸多态性(single nucleotide polymerphisms, SNP)结合应用时,可以实现全基因组高分辨率的关联分析(genome wide association study, GWAS)[41]。上海交通大学Bio-X研究院师咏勇课题组将GWAS应用于精神分裂症,发现了一系列中国汉族人群精神分裂症高风险位点,为多基因复杂疾病的研究做出了重要贡献[42, 43]。在出生缺陷疾病方面,2013年南京医科大学沈洪兵团队在4225名先天性心脏畸形患儿和5112名对照的样本集中,采用覆盖90万个SNP位点的基因芯片,检测和定位到染色体1p12位置的SNP rs2474937 (TBX15基因附近)和4q31.1位置的SNP rs1531070 (位于MAML3基因中)与先天性心脏畸形密切相关,表明这两个位点本身或与之相连锁的某些基因变异具有一定的致病性[44]。2015年,南京医科大学的胡志斌团队在6053个先天性心脏畸形病例和7410名对照人群中开展了多阶段的GWAS分析,发现了4个新的全基因组范围内的显著关联位点,其中染色体20q12上PTPRT基因中的rs490514位点在欧洲人群中也表现出一致的关联性[45]。但总的来说,基于高频SNP标记的GWAS研究在出生缺陷类疾病中并不普及,究其原由:一是因为大多数出生缺陷疾病相对发病率低,研究者往往无法收集到满足研究需求的大量样本;二是如前所述,导致出生缺陷的遗传变异的致病效应往往较其他有更多后天因素参与的疾病相关位点的致病效应更为显著,而这类具有较强致病效应的遗传变异由于自然选择,较少以“common variants”的形式在人群中存在,因此在某种程度上可以说,基于基因多态性的GWAS研究对于出生缺陷遗传病因的发现效率有限。事实上,在其他复杂疾病(如精神分裂症[43])的研究中,研究者也发现GWAS研究找到的显著关联的多态性位点往往只能解释疾病遗传因素中的一小部分,相关基因只能称作疾病易感基因,而不是致病基因。因此近年来,越来越多的研究者开始考虑复杂疾病的“common disease, rare variants”的致病研究假设[46],即某些多基因复杂疾病的遗传病因也可能是由几个主效致病基因的罕见突变配合其他微效基因的易感突变共同构成。在这种假设下,能够覆盖各种突变类型的新一代测序技术成为了研究多基因复杂疾病的重要手段[47]。智障是一种在新生儿中发病率约为0.5%的出生缺陷疾病,遗传因素在疾病病因中占据主要地位。2014年,荷兰Joris A. Veltman实验室对50个智障3口之家(子女为智障患者,父母为正常人)的DNA样本进行了全基因组测序,以寻找致病性的遗传突变。最终他们在这些样本中发现了84个新发的(de novo)SNV和8个de novo的CNV[48]。这些变异显著富集在基因编码区域以及已发现的智障相关基因区域。根据测序结果,其中20位患者被诊断为携带显性遗传的de novo致病突变,1位患者被诊断为携带复合杂合致病突变,诊断率达到42%。从这个例子中可以看出,不同于家族聚集性遗传的出生缺陷,散发的复杂性出生缺陷疾病的遗传异质性非常高,体现出“多基因”的特征:对个体患者来说,疾病可能只是由一个或少数几个基因的主效性罕见突变导致,但在群体水平,很多基因上的主效性罕见突变或某个主效基因上的多种突变都可以导致类似的临床表型。迄今为止,研究者在智障患者中已发现超过700个以显性或隐性模式致病的主效基因[37]。美国Michael Wigler实验室于2014年报道了他们在自闭症儿童中的研究,表明高致病性的多基因罕见de novo突变在出生缺陷遗传学中扮演重要作用[49]。他们对2500多个核心家系(患者及其父母)或受累同胞对(患者及其正常的兄弟姐妹)的样本进行WES测序,发现13%的de novo错义突变和43%的de novo潜在基因破坏性突变(likely gene-disrupting mutations, LGM,包括无义、移码和剪切突变),解释了近21%的患儿的遗传性病因,其中约400个基因上的LGM对低智力的自闭症儿童亚群贡献更大。从样本的角度来看,以上两个研究利用的都是核心家系或受累同胞对样本,这类样本能够高效率地发现致病突变,特别是de novo类型的突变。近年来,随着各国加大对测序类遗传研究的投入,在大型散发样本中开展的基于新一代测序技术的疾病-对照研究也发现了一些重要的罕见致病突变[50]。2017年,美国Evan E Eichler实验室在大于11 730例的儿童期神经发育障碍(包括自闭症、智障/精神发育迟滞、注意力缺失/多动症,运动发育障碍和语言交流障碍)患者和大于2867例对照人群中运用二代测序技术靶向检测了208个神经发育相关的候选基因的外显子序列,发现91个基因上的罕见突变与精神发育障碍显著相关,其中25个基因上的罕见突变更倾向于发生在自闭症中。这些突变基因在神经发育障碍疾病中发挥的具体作用还需要功能学实验来做进一步的研究[51]。
我国基于新一代测序技术的出生缺陷遗传学研究也在紧锣密鼓地开展之中:2015年起,贺林领导的中国遗传学会遗传咨询分会开始联合有关医疗机构和科研单位组织开展“人类单靶标基因组计划”,旨在利用新一代测序技术分别解决单个生命体现象或医学疾病问题,特别是出生缺陷相关的遗传学病因问题。2015年11月8日,由中国遗传学会遗传咨询分会与中国医疗保健国际交流促进会耳内科学分会领衔的“中国聋病基因组计划”正式启动。计划拟在5年内完成10~20万例的遗传性聋病患者的基因组检测,通过对收集的数据进行汇总统计和分析,建立中国聋病人群的致病基因与表型数据库,并依此制定临床聋病的分子筛查与诊治指南。2016年8月7日,中国遗传学会遗传咨询分会联合复旦大学附属儿科医院发起了中国新生儿基因组计划。该计划将在未来5年开展10万例样本的新生儿基因检测,同样会构建起一个中国新生儿的基因组数据库,并依此建立新生儿各类遗传病的基因检测和临床诊断标准。同年10月28日,“人类单靶标基因组计划”中首个针对中国儿童出生缺陷发病率最高的先天性心脏病基因组研究计划正式在上海交通大学医学院附属上海儿童医学中心启动。该研究计划将对约2000例包括法洛四联症在内的先心病患者及2000例正常对照进行全基因组基因测序,通过父母基因组序列的过滤及与正常基因的对比,发现导致先天性心脏病的罕见致病突变。同时,该计划还将采集孕期相关危险环境因素,采用统计遗传学的方法对基因-基因、基因-环境交互作用和基因通路进行分析,以期发现更多的先心病致病/易感基因以及相关的流行病学依据。此外,自2016年起,国家科技部启动了“生殖健康及重大出生缺陷防控研究”重点专项,以每年设立一定数量的项目的形式,支持“建立和完善中国人群育龄人口队列和出生人口队列,开展生殖健康相关疾病临床防治研究”、“生殖健康与出生缺陷相关疾病发病机制研究”和“出生缺陷、不孕不育和避孕节育防治技术及产品研发”等多个方向的重点任务。上述研究计划目前都在实施过程中,有望为我国出生缺陷的遗传学研究增添浓重的一笔,为复杂出生缺陷疾病的临床遗传学诊疗奠定关键性的理论基础。
3 未来我国出生缺陷遗传学研究和应用展望
自20世纪60年代以来,我国出生缺陷遗传学研究发展迅速,不断取得国际一流水平的工作成果。随着研究者对各类出生缺陷疾病更深入地了解以及对基因检测技术更灵活地运用,目前出生缺陷类疾病的遗传学研究已经可以通过对各研究要素的合理选择以及优化组合来有效地设计思路,挖掘相关病因(表1)。未来我国出生缺陷的遗传学研究和应用工作还有以下几个方面会有进一步的发展和完善:Table 1
表1
表1 出生缺陷遗传学研究要素
Table 1
| 合理的病因假设 (突变类型和遗传模式假设) | 有效的研究 样本 | 正确的疾病 诊断和全面 的表型描述 | 灵敏准确的检测技术 | 完善的遗传数据库和生物信息学支持 | 充分的验证 实验 |
|---|---|---|---|---|---|
| 1. 遗传因素 (1) 染色体异常 染色体数目异常 染色体结构变异(large SVs) 拷贝数变异(CNVs) (2) 单基因突变(SNVs, small InDels) 常染色体显性突变 常染色体隐性突变 X染色体显性突变 X染色体隐性突变 Y染色体显性突变 (3) 非经典孟德尔遗传 线粒体突变 印迹基因突变 其他:嵌合体、单亲二倍体、杂合性缺失等 2. 遗传和环境因素共同作用 (1) 主效基因+微效基因+环境因素模式 (2) 多微效基因+环境因素模式 3. 环境因素 | 1. 较为完整的大家系 2. 数量充足的核心家系 3. 数量充足的同胞对 4. 数量充足的疾病散发 样本 | 1. 严格按照诊断标准进行诊断 2. 结构化的表型描述(HPO表型组计划) | 1. 遗传标记法(间接) (1) 第一代遗传标记(RFLP) (2) 第二代遗传标记(STR) (3) 第三代遗传标记(SNP) 2. 变异扫描法(直接) (1) 核型分析平台 G显带分析 FISH技术 (2) 芯片平台 CGH芯片 SNP芯片 (3) 测序平台 靶向基因测序 全外显子测序(WES) 全基因组学测序(WGS) | 1. DNA序列数据库 同源序列分析 2. 遗传突变数据库 (1) 人群突变频率分析 (2) 突变的生物学效应分析 (3) 突变和疾病表型共分离分析 | 1. 独立样本验证(正常人或疾病样本) 2. 基因表达组织验证 3. 基因突变的病理功能研究 |
新窗口打开|下载CSV
(1) 研究样本的分层处理。以先天性心脏病为 例[52],染色体核型异常、基因拷贝数变异、单基因突变以及多基因缺陷均会导致先天性心脏病,先天性心脏疾病作为综合征的疾病表型之一,也可以以单病的形式存在。因此大规模的先天性心脏病散发样本(或独立家系样本)往往具有很高的遗传异质性,这种异质性会增加数据噪声,降低对特定遗传病因的发现效力。若能根据表型谱的相似度或特异性来对疾病样本进行分层纯化处理,则能更有效地发现样本之间共享的遗传变异[37]。
(2) 疾病表型数据的收集和整理。测序技术大大提高了研究者对基因组信息的掌握水平,但在疾病基因组和表型的关联研究中,如此高分辨率的遗传信息需要明确、周全、清晰的疾病表型信息与之相辅相成,才能推进更为客观和精确的发现[53]。为此,国际上的Human Phenotype Ontology (HPO)项目(https://hpo.jax.org)对来自医学文献的表型信息进行结构化归纳,并对表型相关词汇及其语义相互关系进行仔细定义,建立分层关系。截止至2018年3月,HPO数据库已包含了各类遗传疾病的13 000个词条和156 000对“疾病实体-症状词条”的关联注释。2015年至2016年间,华大基因杨焕明和HPO创始人Peter Robinson联合推动成立了中文的人类表型标准用语联盟(The Chinese Human Phenotype Ontology Consortium, CHPO),现已为5271个OMIM词条加入了中文译名(http://www.chinahpo.org)。
(3) 测序技术的发展和优化。目前测序市场产出的多是短读长(如150 bp)的原始数据,这类数据适用于检出SNV和小型InDel,在阅读基因组的高度重复区域和确定长链结构方面还存在缺陷,整合长读长的检测平台进行综合分析是未来的发展趋势。另外,单细胞测序技术的发展也在降低对临床测序DNA样本量的要求[54],它在辅助生殖中胚胎筛选方面的应用也令人振奋[55, 56]。此外,对于常见复杂疾病到底是基于“common variants”还是“rare variants”的假设之争,目前仍未尘埃落定,虽然目前应用于出生缺陷遗传学研究的多为全外显子组测序和目标区域测序[57],随着测序成本的下降,未来基于大样本的深度足够的全基因组测序(能全面检测到各类变异)将得到更多应用。
(4) 多组学检测和精准医学理念的普及。60%~ 70%的出生缺陷是遗传和环境因素相互作用的结 果。因此,遗传变异和疾病表型之间往往不存在简单的对应关系,环境因素在其中扮演重要的调节作用。这种作用在生物体中可以表现为表观基因组的改变、mRNA或蛋白质表达的改变、细胞信号通路或物质代谢通路等多个方面的改变。后基因组时代发展起来的表观基因组学、转录组学、蛋白质组学和代谢组学等技术可以高通量地观察机体在这些层面的改变,从而评估环境和遗传因素在生物体内综合作用的结果,以更好地理解表型产生的机理[58]。精准医学即提倡利用现代高通量的检测技术,更精确地认识疾病成因及其个体化差异,从而设计有针对性的方案实现有效的治疗[59]。此外,上述多个层面之间的数据还可以相互印证和补充,提高临床诊断率。比如运用转录组学,可以在二代测序显示为阴性的患者中额外诊断出21%存在基因转录水平异常的病例[37]。
(5) 疾病相关信息化系统的建立。“人类基因组计划”的实施及其效果已经向世人展示了现代大数据信息资源的重要性。随着各类大型出生缺陷研究项目和临床基因检测服务的开展,巨量的基因组和临床信息数据在日复一日地产出和累积,如何合理地组织、整理、保存、利用以及共享这些宝贵的数据资源是出生缺陷遗传学研究和应用工作的一大挑战:需要不同领域的专家和工作人员之间通力合作,在政府或有关机构的协调下,搭建出整合型的信息学平台,从而使这些公共资源汇集后能够发挥出乘和效应,更好地为我国临床医学事业服务。
(6) 遗传咨询和检测在出生缺陷遗传学研究成果临床转化中发挥核心作用。对子代的遗传物质进行评估并判断其致病性,能够在婚前筛查、辅助生殖以及产前诊断等环节中及早发现潜在患儿。临床上若能借助遗传咨询发挥“桥梁”作用,将现代医学遗传知识有效地传递给普通民众,促成理性的互动,帮助育龄夫妻或患儿父母做出合理选择,则能够有效降低实际出生缺陷率,提高我国人口健康水平。作为一门新兴学科,我国遗传咨询体系尚不成熟,亟待完善。2015年2月9日中国遗传学会遗传咨询分会在上海正式成立,贺林任主任委员。此后中国遗传学会遗传咨询分会在贺林院士的领导下(图3)开始陆续在全国各主要城市开办遗传咨询师培训班,目前已开办13期初级班,3期中级班[60]和1期高级班,总共培训相关人员近4000人,有效地促进了我国遗传咨询和基因检测的健康发展。不过目前许多医疗机构包括第三方遗传检测机构仍存在较为严重的过度诊断或诊断不足等无序状况,因此除了建设相关人才队伍还需要制定相应的行业规范或指南。2016年10月,国家卫生和计划生育委员会发布了《规范有序开展孕妇外周血胎儿游离DNA产前筛查与诊断工作的通知》;2017年2月,中国遗传学会遗传咨询分会联合多位遗传咨询专家形成了《中国遗传咨询标准专家共识指南》和《遗传变异分类标准与指南》[61]。这些文件都为临床遗传咨询工作起到了及时而有效的指导。2017年6月,上海市妇幼保健中心正式挂牌成立“上海市‘健康孩’协同创新中心”,旨在联合贺林院士团队,以开展遗传咨询、遗传检测、加强三级预防管理为抓手,全方位整链条地打造出生缺陷防控示范服务体系,尤其是遗传咨询服务的示范和出生缺陷检测技术的引领,以期在机构层面带动出生缺陷防控事业的有序发展。在临床新技术方面,当前的无创产前筛查(non-invasive prenatal screening, NIPS)还只能检出明显的染色体变异,未来可进一步拓展到对单基因疾病的无创性检出(如利用胎儿有核红细胞)。此外还有染色体芯片(chromosomal microarray, CMA)在产前诊断中的普及应用[62]。CMA在CNV和SNP类型的突变检出中具有明显的优势[63]。在遗传咨询的临床实践中,临床专家还发现目前检测中会出现大量无法进行临床注释的突变(variants of unknown significance, VUS),这些突变为致病性的理论概率低至10%,也可以高至90%,存在较大的不确定性,未来如果能够建立起大规模的中国人群遗传参考数据库(类似千人基因组计划,可以同时包含正常人群以及疾病人群),这一类型的突变可以得到更好的解释和判断。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图32015年2月9日中国遗传学会遗传咨询分会在上海正式成立(贺林任主任委员)
Fig. 3Chinese Board of Genetic Counseling was founded in Shanghai on Feb 9, 2015 (Professor Lin He was elected as the chairman of the board)
(7) 基因治疗技术的发展。出生缺陷遗传学的研究对主要病因的揭示可以指导临床上的靶向基因治疗。脊髓性肌萎缩症(spinal muscular atrophy, SMA)是一种严重的出生缺陷疾病,其中I型的患儿活不过2岁,SMN基因的失活是其首要病因,一直以来被认为无药可医。2017年,New England Journal of Medicine杂志报道了美国一项针对I型SMA患儿的临床I期实验,实验采用腺相关病毒(adeno-associated viruses, AAV)载体向大脑细胞传递正常的SMN基因,结果发现参与这项临床试验的15名患儿全部都活过了两岁,而正常情况下能活到20个月的SMA患儿只有8%[64]。目前,我国将最新的CRISPR技术应用于临床基因治疗方面走在世界前列[65]。遗传病因的准确发现配合日益成熟的基因治疗技术,将为成千上万的出生缺陷患儿带来福音[66]。基因治疗还可以应用在胚胎阶段,即在患儿出生前就纠正其致病突变,从而避免可预测的出生缺陷的发生[67]。中山大学生命科学学院黄军就团队分别于2015年[68]和2017年[69]在Protein & Cell杂志上报道了对人类胚胎进行精准基因修复的工作,引起世界范围内的广泛关注和伦理学的探讨。目前普遍认为,以消除疾病为目的的胚胎基因编辑研究及治疗性应用是值得鼓励的[70]。
总而言之,我国出生缺陷遗传学研究自20世纪60年代以来经历了飞跃式的发展,相关成果为我国医学遗传学及其临床应用提供了坚实的理论基础。同时,以遗传咨询和检测为核心的临床转化近年来也在不断地完善和规范之中,是我国“健康中国”战略不可或缺的一部分。随着出生缺陷遗传学研究以及临床应用工作的深入开展,相信未来将如每一位参与其中的****、医生和有关工作人员所期待的,我国的出生缺陷发生率将显著降低,人口健康水平得到稳步上升,更多的父母能够告别出生缺陷,拥抱“健康孩”。
(责任编委: 史庆华)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:28870276 [本文引用: 1]

Abstract Since its debut in a ground-breaking report by Barker and Osmond in 1986, the concept of the Developmental Origins of Health and Disease (DOHaD) has been further developed in several aspects. Its methodology and conclusions relating to proposed origins and outcomes of early life events have been developing and spreading internationally. Indeed, the DOHaD concept now seems to have influenced many fields of research. This article aims to briefly review why the DOHaD concept is important in biomedical science, how it has developed, is currently developing, and how it should develop in future.
URL [本文引用: 1]
出生缺陷是影响我国人口素质的主要原因,是引起新生儿死亡和婴儿死亡的重要因素。现今先天性心脏病已成为发生率最高的出生缺陷,其后依次为多指、唇裂、脑积水、马蹄内反足、尿道下裂、并指、肢体短缩和小耳等。出生缺陷的产前筛查及诊断主要包括血清学筛查、影像学筛查、介入性产前诊断、分子生物学筛查及诊断等。出生缺陷严重影响儿童健康和生活质量,故应加强三级预防制度,增进产前筛查和诊断准确率,全面推动出生缺陷的诊治工作,提高我国的出生人口素质。
URL [本文引用: 2]
2012年下半年,前卫生部发布了《出生缺陷防治报告》,具体由前卫生部妇幼监测中心根据全国的数据编撰而成。1. 本文分中、英文两部分。2. 每一部分涵盖三章:一、出生缺陷发生现状;二、工作进展与成效;三、出生缺陷防治策略。
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
正出生缺陷是指由于遗传因素、环境因素或两者共同作用于孕前或孕期,引起胚胎或胎儿在发育过程中发生的解剖学结构和/或功能上的异常。据报道,全世界每年生育缺陷儿约790万,确定
[本文引用: 1]
URL [本文引用: 1]
介绍了中国医学遗传学国家重点实验室在遗传病家系收集、疾病基因定位、疾病基因克隆和疾病基因功能研究方面的研究工作.用细胞遗传学G显带技术于1975年发现了一条与鼻咽癌相关的标记染色体t(1;3)(q44;p11);1981年将睾丸决定基因(TDF)定位于Yp11.32带;1991年以来收集遗传病家系345种共590个;1996年用显微切割、PCR、微克隆技术克隆了EXT2基因;1998年用基因家族-候选疾病基因克隆方法克隆了遗传性神经性耳聋基因GJB3;1999年用连锁分析和全基因组扫描将一种遗传性弥漫性浅表性光敏性汗孔角化症定位于12q23.2带,并在基因功能研究中发现了一个新的细胞内转运蛋白.
URL [本文引用: 1]
正随着2004年10月人类基因组精细图在Nature杂志上发表,标志着人类基因组DNA全序列测定的目标已基本达到:最近,绘制人类基因组SNP单体型图谱(HapMap)的国际合作第一阶段的任务也基本完成,识别人体内所有基因的结构与功能,解读包括非编码序列在内的人类遗传物质的全部信息,已是人类基因组研究的主要任务。在老一辈科学家的创导和积极推动下,我国的人类基因组研究于1994年由国家自然科学基金的资助下正式启动。基于当时有限的资助,起步工作主要是建立基本的方法与技术,收集和保存人体遗
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
Brachydactyly type A-1 (BDA1) was, in 1903, the first recorded example of a human anomaly with Mendelian autosomal dominant inheritance. Two large families, the affected members of which were radiographed, were recruited in the study we describe here. Two-point linkage analysis for pedigree 1 (maximum LOD score [ Z max] 6.59 at recombination fraction [胃] 0.00) and for pedigree 2 ( Z max=5.53 at 胃=0.00) mapped the locus for BDA1 in the two families to chromosome 2q. Haplotype analysis of pedigree 1 confined the locus for family 1 within an interval of <8.1 cM flanked by markers D2S2248 and D2S360, which was mapped to chromosome 2q35-q36 on the cytogenetic map. Haplotype analysis of pedigree 2 confined the locus for family 2 within an interval of <28.8 cM flanked by markers GATA30E06 and D2S427, which was localized to chromosome 2q35-q37. The two families had no identical haplotype within the defined region, which suggests that the two families were not related.
URLPMID:11455389 [本文引用: 1]
Abstract Brachydactyly type A-1 (BDA-1; MIM 112500) is characterized by shortening or missing of the middle phalanges (Fig. 1a). It was first identified by Farabee in 1903 (ref. 2), is the first recorded example of a human anomaly with Mendelian autosomal-dominant inheritance and, as such, is cited in most genetic and biological textbooks. Here we show that mutations in IHH, which encodes Indian hedgehog, cause BDA-1. We have identified three heterozygous missense mutations in the region encoding the amino-terminal signaling domain in all affected members of three large, unrelated families. The three mutant amino acids, which are conserved across all vertebrates and invertebrates studied so far, are predicted to be adjacent on the surface of IHH.
URLPMID:19252479 [本文引用: 1]
Brachydactyly type A1 (BDA1) was the first recorded disorder of the autosomal dominant Mendelian trait in humans, characterized by shortened or absent middle phalanges in digits. It is associated with heterozygous missense mutations in indian hedgehog (IHH). Hedgehog proteins are important morphogens for a wide range of developmental processes. The capacity and range of signalling is thought to be regulated by its interaction with the receptor PTCH1 and antagonist HIP1. Here we show that a BDA1 mutation (E95K) in Ihh impairs the interaction of IHH with PTCH1 and HIP1. This is consistent with a recent paper showing that BDA1 mutations cluster in a calcium-binding site essential for the interaction with its receptor and cell-surface partners. Furthermore, we show that in a mouse model that recapitulates the E95K mutation, there is a change in the potency and range of signalling. The mice have digit abnormalities consistent with the human disorder. 2009 Macmillan Publishers Limited.
URLPMID:10678655 [本文引用: 1]
We report on rare, heritable, permanent tooth agenesis in a large Chinese kindred. The congenital absence of permanent teeth except the first and second accessory teeth was observed in 52 individuals through six successive generations in the kindred comprising 328 members. Clinical assessments were carried out, and inheritance mode and spousal influence of the anomaly on their offspring were analyzed. Consequently, the anomaly was transmitted in an autosomal dominant fashion with incomplete penetrance (P = 0.88), and no significant clinical manifestations other than the oligodontia were found. A geographical or environmental effect on the affected individuals was obviously eliminated, because any who are related to the kindred but live under the same conditions are fully healthy. The disorder we describe, therefore, differs from any previously reported oligodontia/anodontia syndromes. The oligodontia ranged from a few teeth to the whole set of teeth, and usually occurred at a period from age 7 or 8 years, the time when primary teeth are normally replaced by permanent teeth, to the forties. Roentgenography of the affected persons indicated that only the first and/or second accessory teeth with tooth buds developed as permanent teeth. In fact, the diphyodontic germination sometimes occurred in the oral cavity of the affected individuals. Am. J. Med. Genet. 90:193-198, 2000. ? 2000 Wiley-Liss, Inc.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:20558380 [本文引用: 1]
Correspondence from The New England Journal of Medicine — VANGL2 Mutations in Human Cranial Neural-Tube Defects
URLPMID:26789871 [本文引用: 2]
Abstract Background Human reproduction depends on the fusion of a mature oocyte with a sperm cell to form a fertilized egg. The genetic events that lead to the arrest of human oocyte maturation are unknown. Methods We sequenced the exomes of five members of a four-generation family, three of whom had infertility due to oocyte meiosis I arrest. We performed Sanger sequencing of a candidate gene, TUBB8, in DNA samples from these members, additional family members, and members of 23 other affected families. The expression of TUBB8 and all other β-tubulin isotypes was assessed in human oocytes, early embryos, sperm cells, and several somatic tissues by means of a quantitative reverse-transcriptase-polymerase-chain-reaction assay. We evaluated the effect of the TUBB8 mutations on the assembly of the heterodimer consisting of one α-tubulin polypeptide and one β-tubulin polypeptide (α/β-tubulin heterodimer) in vitro, on microtubule architecture in HeLa cells, on microtubule dynamics in yeast cells, and on spindle assembly in mouse and human oocytes. Results We identified seven mutations in the primate-specific gene TUBB8 that were responsible for oocyte meiosis I arrest in 7 of the 24 families. TUBB8 expression is unique to oocytes and the early embryo, in which this gene accounts for almost all the expressed β-tubulin. The mutations affect chaperone-dependent folding and assembly of the α/β-tubulin heterodimer, disrupt microtubule behavior on expression in cultured cells, alter microtubule dynamics in vivo, and cause catastrophic spindle-assembly defects and maturation arrest on expression in mouse and human oocytes. Conclusions TUBB8 mutations have dominant-negative effects that disrupt microtubule behavior and oocyte meiotic spindle assembly and maturation, causing female infertility. (Funded by the National Basic Research Program of China and others.).
URLPMID:20929727 [本文引用: 1]
Abstract Acne inversa (AI), also known as hidradenitis suppurativa, is a chronic, recurrent, inflammatory disease of hair follicles that often runs in families. We studied six Chinese families with features of AI as well as additional skin lesions on back, face, nape, and waist and found independent loss-of-function mutations in PSENEN, PSEN1, or NCSTN, the genes encoding essential components of the 0206-secretase multiprotein complex. Our results identify the 0206-secretase component genes as the culprits for a subset of familial AI, implicate the 0206-secretase-Notch pathway in the molecular pathogenesis of AI, and demonstrate that familial AI can be an allelic disorder of early-onset familial Alzheimer's disease.
URL [本文引用: 1]
URLMagsci [本文引用: 1]
<br><br>在进行中国人群的遗传性耳聋发病情况的调查中,发现了一个5代隔代遗传的聋哑家系(L021家系)。研究中调查家系成员64人,对其中的31人进行了系统的听力学检查,发现聋哑男性8位,听力表型为全聋及极重度聋,获得家系成员的血样31人份。家系图谱分析显示该家系为X连锁隐性遗传性耳聋家系,为先天性聋哑疾病分子病理机制的研究提供了模板。<br><br>Abstract: In studying genetic factors in hearing loss among Chinese hearing-impaired population, a Chinese family with deaf-mute that had been reversion inherited through five generations was found (named pedigree L021). X linked recessive inheritance was hypothesized to be the transmission in this family. A total of 64 members in this family were investigated. Of these, audiometric evaluation was performed on 31 members, including 8 males with deaf-mute. Most affected individuals showed deafness or profound sensorineural hearing loss. Blood samples were obtained from 31 consented individuals in this family. Pedigree analysis indicates a X-linked recessive inheritance pattern in pedigree L021. The pedigree described herein provides an excellent model for further study on the molecular mechanism of congenital deaf-mute.<br>
URLMagsci [本文引用: 1]
<br><br>在进行中国人群的遗传性耳聋发病情况的调查中,发现了一个5代隔代遗传的聋哑家系(L021家系)。研究中调查家系成员64人,对其中的31人进行了系统的听力学检查,发现聋哑男性8位,听力表型为全聋及极重度聋,获得家系成员的血样31人份。家系图谱分析显示该家系为X连锁隐性遗传性耳聋家系,为先天性聋哑疾病分子病理机制的研究提供了模板。<br><br>Abstract: In studying genetic factors in hearing loss among Chinese hearing-impaired population, a Chinese family with deaf-mute that had been reversion inherited through five generations was found (named pedigree L021). X linked recessive inheritance was hypothesized to be the transmission in this family. A total of 64 members in this family were investigated. Of these, audiometric evaluation was performed on 31 members, including 8 males with deaf-mute. Most affected individuals showed deafness or profound sensorineural hearing loss. Blood samples were obtained from 31 consented individuals in this family. Pedigree analysis indicates a X-linked recessive inheritance pattern in pedigree L021. The pedigree described herein provides an excellent model for further study on the molecular mechanism of congenital deaf-mute.<br>
URLMagsci [本文引用: 1]
<P>耳聋具有高度的遗传异质性, 迄今已定位了51个常染色体显性遗传非综合征型耳聋(autosomal dominant non-syndromic sensorineural hearing loss, DFNA)基因位点, 20个DFNA相关基因被克隆。文章收集了一个DFNA巨大家系, 家系中有血缘关系的家族成员共170人, 对73名家族成员进行了详细的病史调查、全身检查和耳科学检查, 提示39人有不同程度的迟发性感音神经性听力下降, 未见前庭及其他系统的异常。应用ABI公司382个常染色体微卫星多态标记进行全基因组扫描连锁分析, 将该家系致聋基因定位于14q12-13处D14S1021-D14S70之间约7.6 cM (3.18 Mb)的区域, 最大LOD值为6.69 (D14S1040), 与已知DFNA9位点有4.7 cM (2.57 Mb)的重叠区, DFNA9致病基因<EM>COCH</EM>位于重叠区域内。下一步拟进行<EM>COCH</EM>基因的突变筛查, 以揭示该家系耳聋的分子致病机制。</P>
URLMagsci [本文引用: 1]
<P>耳聋具有高度的遗传异质性, 迄今已定位了51个常染色体显性遗传非综合征型耳聋(autosomal dominant non-syndromic sensorineural hearing loss, DFNA)基因位点, 20个DFNA相关基因被克隆。文章收集了一个DFNA巨大家系, 家系中有血缘关系的家族成员共170人, 对73名家族成员进行了详细的病史调查、全身检查和耳科学检查, 提示39人有不同程度的迟发性感音神经性听力下降, 未见前庭及其他系统的异常。应用ABI公司382个常染色体微卫星多态标记进行全基因组扫描连锁分析, 将该家系致聋基因定位于14q12-13处D14S1021-D14S70之间约7.6 cM (3.18 Mb)的区域, 最大LOD值为6.69 (D14S1040), 与已知DFNA9位点有4.7 cM (2.57 Mb)的重叠区, DFNA9致病基因<EM>COCH</EM>位于重叠区域内。下一步拟进行<EM>COCH</EM>基因的突变筛查, 以揭示该家系耳聋的分子致病机制。</P>
URLPMID:29019985 [本文引用: 1]
This review commemorates the 40th anniversary of DNA sequencing, a period in which we have already witnessed multiple technological revolutions and a growth in scale from a few kilobases to the first human genome, and now to millions of human and a myriad of other genomes. DNA sequencing has been extensively and creatively repurposed, including as a ‘counter’ for a vast range of molecular phenomena. We predict that in the long view of history, the impact of DNA sequencing will be on a par with that of the microscope.
URL [本文引用: 1]
URLPMID:1464427 [本文引用: 1]
Nature is the international weekly journal of science: a magazine style journal that publishes full-length research papers in all disciplines of science, as well as News and Views, reviews, news, features, commentaries, web focuses and more, covering all branches of science and how science impacts upon all aspects of society and life.
Magsci [本文引用: 1]
近年来, 众多研究小组开展了大量的全基因组关联研究(Genome-wide association studies, GWAS), 发现并鉴定了许多与复杂疾病/性状相关联的遗传变异, 为复杂疾病发病机制的研究提供了重要线索。由于GWAS的结果存在假阳性、假阴性、检测到的单核苷酸多态性很少位于功能区以及对稀有变异和结构变异不敏感等问题, 导致了其应用的局限性。而新一代测序技术的进步, 促进了全基因组测序和全基因组外显子测序的快速发展, 为解决上述问题提供了契机。全基因组外显子测序是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。由于其具有对常见和罕见变异高灵敏度, 能发现外显子区绝大部分疾病相关变异以及仅需要对约1%的基因组进行测序等优点, 促使全基因组外显子测序成为鉴定孟德尔疾病的致病基因最有效的策略, 也被运用于复杂疾病易感基因的研究和临床诊断中。
Magsci [本文引用: 1]
近年来, 众多研究小组开展了大量的全基因组关联研究(Genome-wide association studies, GWAS), 发现并鉴定了许多与复杂疾病/性状相关联的遗传变异, 为复杂疾病发病机制的研究提供了重要线索。由于GWAS的结果存在假阳性、假阴性、检测到的单核苷酸多态性很少位于功能区以及对稀有变异和结构变异不敏感等问题, 导致了其应用的局限性。而新一代测序技术的进步, 促进了全基因组测序和全基因组外显子测序的快速发展, 为解决上述问题提供了契机。全基因组外显子测序是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。由于其具有对常见和罕见变异高灵敏度, 能发现外显子区绝大部分疾病相关变异以及仅需要对约1%的基因组进行测序等优点, 促使全基因组外显子测序成为鉴定孟德尔疾病的致病基因最有效的策略, 也被运用于复杂疾病易感基因的研究和临床诊断中。
URL [本文引用: 1]
基因突变是当今生命科学研究的热点之一,其检测方法和诊断技术发展迅猛。测序技术在基因病的确诊、分型等方面发挥着不可或缺的作用。文章重点围绕第一~三代测序技术的研究进展、优缺点及其在基因诊断中的应用进行评述,并对未来的发展趋势进行了预测和展望。
URL [本文引用: 1]
基因突变是当今生命科学研究的热点之一,其检测方法和诊断技术发展迅猛。测序技术在基因病的确诊、分型等方面发挥着不可或缺的作用。文章重点围绕第一~三代测序技术的研究进展、优缺点及其在基因诊断中的应用进行评述,并对未来的发展趋势进行了预测和展望。
URL [本文引用: 1]
URLPMID:25961942 [本文引用: 1]
Cardiovascular disease is the most common cause of death worldwide, and hypertension is the major risk factor. Mendelian hypertension elucidates mechanisms of blood pressure regulation. Here we report six missense mutations in PDE3A (encoding phosphodiesterase 3A) in six unrelated families with mendelian hypertension and brachydactyly type E (HTNB). The syndrome features brachydactyly type E (BDE), severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, altered baroreflex blood pressure regulation and death from stroke before age 50 years when untreated. In vitro analyses of mesenchymal stem cell-derived vascular smooth muscle cells (VSMCs) and chondrocytes provided insights into molecular pathogenesis. The mutations increased protein kinase A-mediated PDE3A phosphorylation and resulted in gain of function, with increased cAMP-hydrolytic activity and enhanced cell proliferation. Levels of phosphorylated VASP were diminished, and PTHrP levels were dysregulated. We suggest that the identified PDE3A mutations cause the syndrome. VSMC-expressed PDE3A deserves scrutiny as a therapeutic target for the treatment of hypertension.
URLPMID:22983302 [本文引用: 1]
Disseminated superficial actinic porokeratosis (DSAP) is an autosomal dominantly inherited epidermal keratinization disorder whose etiology remains unclear. We performed exome sequencing in one unaffected and two affected individuals from a DSAP family. The mevalonate kinase gene (MVK) emerged as the only candidate gene located in previously defined linkage regions after filtering against existing SNP databases, eight HapMap exomes and 1000 Genomes Project data and taking into consideration the functional implications of the mutations. Sanger sequencing in 57 individuals with familial DSAP and 25 individuals with sporadic DSAP identified MVK mutations in 33% and 16% of these individuals (cases), respectively. All 14 MVK mutations identified in our study were absent in 676 individuals without DSAP. Our functional studies in cultured primary keratinocytes suggest that MVK has a role in regulating calcium-induced keratinocyte differentiation and could protect keratinocytes from apoptosis induced by type A ultraviolet radiation. Our results should help advance the understanding of DSAP pathogenesis.
URLPMID:329398702 [本文引用: 5]
Abstract The majority of rare diseases affect children, most of whom have an underlying genetic cause for their condition. However, making a molecular diagnosis with current technologies and knowledge is often still a challenge. Paediatric genomics is an immature but rapidly evolving field that tackles this issue by incorporating next-generation sequencing technologies, especially whole-exome sequencing and whole-genome sequencing, into research and clinical workflows. This complex multidisciplinary approach, coupled with the increasing availability of population genetic variation data, has already resulted in an increased discovery rate of causative genes and in improved diagnosis of rare paediatric disease. Importantly, for affected families, a better understanding of the genetic basis of rare disease translates to more accurate prognosis, management, surveillance and genetic advice; stimulates research into new therapies; and enables provision of better support.
URL [本文引用: 1]
基因诊断依赖于有效的遗传标记,但多数遗传病的发病机制尚不清楚,也缺乏有效的分子标记,本文介绍了拷贝数变异有可能成为寻找遗传病致病基因的新途径,为基因诊断和产前诊断提供有力的支持。
URLPMID:25824869 [本文引用: 1]
For many years, linkage analysis was the primary tool used for the genetic mapping of Mendelian and complex traits with familial aggregation. Linkage analysis was largely supplanted by the wide adoption of genome-wide association studies (GWASs). However, with the recent increased use of whole-genome sequencing (WGS), linkage analysis is again emerging as an important and powerful analysis method for the identification of genes involved in disease aetiology, often in conjunction with WGS filtering approaches. Here, we review the principles of linkage analysis and provide practical guidelines for carrying out linkage studies using WGS data.
URLPMID:2758134 [本文引用: 1]
Abstract The identification of complex disease susceptibility loci has been accelerated considerably by advances in high-throughput genotyping technologies, improved insight into correlation patterns of common variants and the availability of large-scale sample sets. Linkage scans and small-scale candidate gene studies have now given way to genome-wide association scans. In this review, we summarize insights gained from the past, highlight practical issues relating to the design and analysis of current state-of-the-art GWA studies and look into future trends in the field of human complex trait genetics.
[本文引用: 1]
[本文引用: 1]
URLPMID:20 [本文引用: 1]
Schizophrenia is a severe mental disorder affecting 651% of the world population, with heritability of up to 80%. To identify new common genetic risk factors, we performed a genome-wide association study (GWAS) in the Han Chinese population. The discovery sample set consisted of 3,750 individuals with schizophrenia and 6,468 healthy controls (1,578 cases and 1,592 controls from northern Han Chinese, 1,238 cases and 2,856 controls from central Han Chinese, and 934 cases and 2,020 controls from the southern Han Chinese). We further analyzed the strongest association signals in an additional independent cohort of 4,383 cases and 4,539 controls from the Han Chinese population. Meta-analysis identified common SNPs that associated with schizophrenia with genome-wide significance on 8p12 (rs16887244, P = 1.27 × 10(-10)) and 1q24.2 (rs10489202, P = 9.50 × 10(-9)). Our findings provide new insights into the pathogenesis of schizophrenia.
URLPMID:28991256 [本文引用: 2]
Abstract We conducted a genome-wide association study (GWAS) with replication in 36,180 Chinese individuals and performed further transancestry meta-analyses with data from the Psychiatry Genomics Consortium (PGC2). Approximately 95% of the genome-wide significant (GWS) index alleles (or their proxies) from the PGC2 study were overrepresented in Chinese schizophrenia cases, including 09080450% that achieved nominal significance and 09080475% that continued to be GWS in the transancestry analysis. The Chinese-only analysis identified seven GWS loci; three of these also were GWS in the transancestry analyses, which identified 109 GWS loci, thus yielding a total of 113 GWS loci (30 novel) in at least one of these analyses. We observed improvements in the fine-mapping resolution at many susceptibility loci. Our results provide several lines of evidence supporting candidate genes at many loci and highlight some pathways for further research. Together, our findings provide novel insight into the genetic architecture and biological etiology of schizophrenia.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:28449691 [本文引用: 1]
Abstract Despite thousands of genetic loci identified to date, a large proportion of genetic variation predisposing to complex disease and traits remains unaccounted for. Advances in sequencing technology enable focused explorations on the contribution of low-frequency and rare variants to human traits. Here we review experimental approaches and current knowledge on the contribution of these genetic variants in complex disease and discuss challenges and opportunities for personalised medicine.
URLPMID:20479773 [本文引用: 1]
Although genome-wide association (GWA) studies for common variants have thus far succeeded in explaining only a modest fraction of the genetic components of human common diseases, recent advances in next-generation sequencing technologies could rapidly facilitate substantial progress. This outcome is expected if much of the missing genetic control is due to gene variants that are too rare to be picked up by GWA studies and have relatively large effects on risk. Here, we evaluate the evidence for an important role of rare gene variants of major effect in common diseases and outline discovery strategies for their identification.
URL [本文引用: 1]
URLPMID:25363768 [本文引用: 1]
Abstract Whole exome sequencing has proven to be a powerful tool for understanding the genetic architecture of human disease. Here we apply it to more than 2,500 simplex families, each having a child with an autistic spectrum disorder. By comparing affected to unaffected siblings, we show that 13% of de novo missense mutations and 43% of de novo likely gene-disrupting (LGD) mutations contribute to 12% and 9% of diagnoses, respectively. Including copy number variants, coding de novo mutations contribute to about 30% of all simplex and 45% of female diagnoses. Almost all LGD mutations occur opposite wild-type alleles. LGD targets in affected females significantly overlap the targets in males of lower intelligence quotient (IQ), but neither overlaps significantly with targets in males of higher IQ. We estimate that LGD mutation in about 400 genes can contribute to the joint class of affected females and males of lower IQ, with an overlapping and similar number of genes vulnerable to contributory missense mutation. LGD targets in the joint class overlap with published targets for intellectual disability and schizophrenia, and are enriched for chromatin modifiers, FMRP-associated genes and embryonically expressed genes. Most of the significance for the latter comes from affected females.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
先天性心脏病(CHD)是最常见的先天性出生缺陷,其发病率与病死率位居婴幼儿非感染性疾病的首位。CHD可分为非综合征性CHD和综合征性CHD。目前认为,CHD的发生主要与遗传因素、环境因素及多基因遗传相关,但CHD的发病机制复杂,并且仍未完全阐明。笔者拟就CHD发病机制中遗传因素的研究进展进行综述,旨在为CHD的预防和早期诊断提供理论基础。
URL [本文引用: 1]
先天性心脏病(CHD)是最常见的先天性出生缺陷,其发病率与病死率位居婴幼儿非感染性疾病的首位。CHD可分为非综合征性CHD和综合征性CHD。目前认为,CHD的发生主要与遗传因素、环境因素及多基因遗传相关,但CHD的发病机制复杂,并且仍未完全阐明。笔者拟就CHD发病机制中遗传因素的研究进展进行综述,旨在为CHD的预防和早期诊断提供理论基础。
URLPMID:25232173 [本文引用: 1]
Abstract In contemporary genomics research, phenotype-driven bioinformatics approaches hint at the potential of establishing a framework for quantifying phenotypic similarities among patients (Zemojtel et al., issue 252). Copyright 08 2014, American Association for the Advancement of Science.
URL [本文引用: 1]
URLPMID:29255258 [本文引用: 1]
Abstract DNA methylation is a crucial layer of epigenetic regulation during mammalian embryonic development 1-3 . Although the DNA methylome of early human embryos has been analyzed 4-6 , some of the key features have not been addressed thus far. Here we performed single-cell DNA methylome sequencing for human preimplantation embryos and found that tens of thousands of genomic loci exhibited de novo DNA methylation. This finding indicates that genome-wide DNA methylation reprogramming during preimplantation development is a dynamic balance between strong global demethylation and drastic focused remethylation. Furthermore, demethylation of the paternal genome is much faster and thorough than that of the maternal genome. From the two-cell to the postimplantation stage, methylation of the paternal genome is consistently lower than that of the maternal genome. We also show that the genetic lineage of early blastomeres can be traced by DNA methylation analysis. Our work paves the way for deciphering the secrets of DNA methylation reprogramming in early human embryos.
URLPMID:24360273 [本文引用: 1]
MALBAC genome amplification and high-throughput sequencing of the two polar bodies allowed inference of the health status of the oocyte, both in terms of aneuploidy and single-nucleotide variants associated with Mendelian diseases, demonstrating proof of principle for MALBAC-based preimplantation genomic screening in IVF.
URL [本文引用: 1]
耳聋是一种常见的严重出生缺陷,阐明遗传性耳聋的致病机理不仅能够在临床上辅助诊断,为遗传咨询及耳聋预防提供依据,而且能促进人们更深入地了解耳聋的致病机制,开发新的治疗方法。随着基因组研究技术不断创新,以全基因组测序、全外显子组测序、目标区域测序为代表的高通量测序技术在遗传性耳聋研究中已得到广泛应用。本文总结了近5年全外显子组测序和目标区域测序在遗传性耳聋致病基因研究及临床分子诊断中应用及研究进展,希望能够有助于我国临床耳聋基因诊断技术的发展及诊断水平的提升。
URL [本文引用: 1]
耳聋是一种常见的严重出生缺陷,阐明遗传性耳聋的致病机理不仅能够在临床上辅助诊断,为遗传咨询及耳聋预防提供依据,而且能促进人们更深入地了解耳聋的致病机制,开发新的治疗方法。随着基因组研究技术不断创新,以全基因组测序、全外显子组测序、目标区域测序为代表的高通量测序技术在遗传性耳聋研究中已得到广泛应用。本文总结了近5年全外显子组测序和目标区域测序在遗传性耳聋致病基因研究及临床分子诊断中应用及研究进展,希望能够有助于我国临床耳聋基因诊断技术的发展及诊断水平的提升。
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
2017年6月25日,中国遗传学会遗传咨询分会联合上海交通大学医学院附属上海儿童医学中心(国家儿童医学中心)、美洲华人遗传学会共同举行第二届遗传咨询师中级培训班。中国遗传学会遗传咨询分会主任委员、上海交通大学Bio-X研究院院长贺林院士,美国哈佛医学院医学遗传学培训课程主任、上海市****讲座教授、上海儿童医学中心医学遗传科主任沈亦平教授出席大会并作精彩演讲,100余位来自全国各地的临床医师、分子诊断实验室技术人员、遗传咨询行业从业者以及生命科学或医学相关研究人员共同参加。
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
Chromosomal microarray analysis (CMA) is performed either by array comparative genomic hybridization or by using a single nucleotide polymorphism array. In the prenatal setting, CMA is on par with traditional karyotyping for detection of major chromosomal imbalances such as aneuploidy and unbalanced rearrangements. CMA offers additional diagnostic benefits by revealing sub-microscopic imbalances or copy number variations that are too small to be seen on a standard G-banded chromosome preparation. These submicroscopic imbalances are also referred to as microdeletions and microduplications, particularly when they include specific genomic regions that are associated with clinical sequelae. Not all microdeletions/duplications are associated with adverse clinical phenotypes and in many cases, their presence is benign. In other cases, they are associated with a spectrum of clinical phenotypes that may range from benign to severe, while in some situations, the clinical significance may simply be unknown. These scenarios present a challenge for prenatal diagnosis, and genetic counseling prior to prenatal CMA greatly facilitates delivery of complex results. In prenatal diagnostic samples with a normal karyotype, chromosomal microarray will diagnose a clinically significant subchromosomal deletion or duplication in approximately 1% of structurally normal pregnancies and 6% with a structural anomaly. Pre-test counseling is also necessary to distinguish the primary differences between the benefits, limitations and diagnostic scope of CMA versus the powerful but limited screening nature of non-invasive prenatal diagnosis using cell-free fetal DNA.
URL [本文引用: 1]
URLPMID:28983027 [本文引用: 1]
CRISPR, the wildly popular genome-editing research tool, was invented in the West, but it is speeding toward potential human applications in China. Last week, the Chinese team that sparked a worldwide debate in 2015 when it reported the first use of CRISPR to edit a human embryo9s genome notched another first. In early embryos, they showed that a new CRISPR variant, which chemically modifies rather than cuts DNA, can correct the mutation causing a debilitating blood disease. But the most striking evidence of progress in China can be found on the clinicaltrials.gov database: Of the 10 listed trials of CRISPR in patients, nine are in China, where streamlined safety and ethical reviews have given researchers a head start. Three of the groups confirmed to Science that they are infusing cancer patients with their own immune cells modified using CRISPR.
URL [本文引用: 1]
基因组编辑技术的飞速发展,尤其是近年来CRISPR/Cas9基因组编辑体系的出现,使得研究人员能高效地在细胞系和动物模型中对基因组进行精确编辑。基于基因组编辑技术的各种实验研究平台被相继开发,包括通过在细胞系中引入疾病相关突变位点建立疾病模型,通过高通量筛选寻找导致肿瘤耐药性的突变基因,通过体内原位靶向致病基因并修改突变进行基因治疗等。这些基因组编辑技术研究平台极大推动了精准医学研究领域的发展。本文对基因组编辑技术在精准医学领域的基础研究、转化应用、目前存在的问题以及未来发展的方向进行了讨论。
URL [本文引用: 1]
基因组编辑技术的飞速发展,尤其是近年来CRISPR/Cas9基因组编辑体系的出现,使得研究人员能高效地在细胞系和动物模型中对基因组进行精确编辑。基于基因组编辑技术的各种实验研究平台被相继开发,包括通过在细胞系中引入疾病相关突变位点建立疾病模型,通过高通量筛选寻找导致肿瘤耐药性的突变基因,通过体内原位靶向致病基因并修改突变进行基因治疗等。这些基因组编辑技术研究平台极大推动了精准医学研究领域的发展。本文对基因组编辑技术在精准医学领域的基础研究、转化应用、目前存在的问题以及未来发展的方向进行了讨论。
URL [本文引用: 1]
URLPMID:25894090 [本文引用: 1]
Genome editing tools such as the clustered regularly interspaced short palindromic repeat (CRISPR)-associated system (Cas) have been widely used to modify genes in model systems including animal zygotes and human cells, and hold tremendous promise for both basic research and clinical applications. To date, a serious knowledge gap remains in our understanding of DNA repair mechanisms in human early embryos, and in the efficiency and potential off-target effects of using technologies such as CRISPR/Cas9 in human pre-implantation embryos. In this report, we used tripronuclear (3PN) zygotes to further investigate CRISPR/Cas9-mediated gene editing in human cells. We found that CRISPR/Cas9 could effectively cleave the endogenous 尾-globin gene ( HBB ). However, the efficiency of homologous recombination directed repair (HDR) of HBB was low and the edited embryos were mosaic. Off-target cleavage was also apparent in these 3PN zygotes as revealed by the T7E1 assay and whole-exome sequencing. Furthermore, the endogenous delta-globin gene ( HBD ), which is homologous to HBB , competed with exogenous donor oligos to act as the repair template, leading to untoward mutations. Our data also indicated that repair of the HBB locus in these embryos occurred preferentially through the non-crossover HDR pathway. Taken together, our work highlights the pressing need to further improve the fidelity and specificity of the CRISPR/Cas9 platform, a prerequisite for any clinical applications of CRSIPR/Cas9-mediated editing.
URLPMID:28942539 [本文引用: 1]
β-Thalassemia is a global health issue, caused by mutations in theHBBgene. Among these mutations,HBB6128 (A>G) mutations is one of the three most common mutations in China and Southeast Asia patients with β-thalassemia. Correcting this mutation in human embryos may prevent the disease being passed onto future generations and cure anemia. Here we report the first study using base editor (BE) system to correct disease mutant in human embryos. Firstly, we produced a 293T cell line with an exogenousHBB6128 (A>G) mutant fragment for gRNAs and targeting efficiency evaluation. Then we collected primary skin fibroblast cells from a β-thalassemia patient with HBB 6128 (A>G) homozygous mutation. Data showed that base editor could precisely correctHBB6128 (A>G) mutation in the patient’s primary cells. To model homozygous mutation disease embryos, we constructed nuclear transfer embryos by fusing the lymphocyte or skin fibroblast cells with enucleatedin vitromatured (IVM) oocytes. Notably, the gene correction efficiency was over 23.0% in these embryos by base editor. Although these embryos were still mosaic, the percentage of repaired blastomeres was over 20.0%. In addition, we found that base editor variants, with narrowed deamination window, could promote G-to-A conversion atHBB6128 site precisely in human embryos. Collectively, this study demonstrated the feasibility of curing genetic disease in human somatic cells and embryos by base editor system. The online version of this article (doi:10.1007/s13238-017-0475-6) contains supplementary material, which is available to authorized users.
URLPMID:29326244 [本文引用: 1]
Abstract After almost 30 years of promise tempered by setbacks, gene therapies are rapidly becoming a critical component of the therapeutic armamentarium for a variety of inherited and acquired human diseases. Gene therapies for inherited immune disorders, hemophilia, eye and neurodegenerative disorders, and lymphoid cancers recently progressed to approved drug status in the United States and Europe, or are anticipated to receive approval in the near future. In this Review, we discuss milestones in the development of gene therapies, focusing on direct in vivo administration of viral vectors and adoptive transfer of genetically engineered T cells or hematopoietic stem cells. We also discuss emerging genome editing technologies that should further advance the scope and efficacy of gene therapy approaches.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}