 ,, 李玉春, 孔庆鹏,, 张亚平中国科学院昆明动物研究所,遗传资源与进化国家重点实验室,昆明 650223
,, 李玉春, 孔庆鹏,, 张亚平中国科学院昆明动物研究所,遗传资源与进化国家重点实验室,昆明 650223The origin and evolution history of East Asian populations from genetic perspectives
Jiaoyang Tian,, Yuchun Li, Qingpeng Kong,, Yaping ZhangState Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China通讯作者:
编委: 谢小冬
收稿日期:2018-07-16修回日期:2018-09-25网络出版日期:2018-10-20
| 基金资助: |
Received:2018-07-16Revised:2018-09-25Online:2018-10-20
| Fund supported: |
作者简介 About authors
田娇阳,博士,助理研究员,研究方向:东亚人群源流历史E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (462KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
田娇阳, 李玉春, 孔庆鹏, 张亚平. 遗传学视角下东亚人群的起源和演化[J]. 遗传, 2018, 40(10): 814-824 doi:10.16288/j.yczz.18-202
Jiaoyang Tian, Yuchun Li, Qingpeng Kong, Yaping Zhang.
东亚位于欧亚大陆东部,连接东南亚、中亚和北亚地区,是研究解剖学意义上现代人(anatomically modern human, AMH)迁徙扩散和演化的重要地带之一。该地区幅员辽阔、地形地貌复杂多样,有超过16亿的人口数量,大约占全世界人口总数的1/4。同时,环境资源丰富、民族文化众多等背景条件,使得东亚成为研究人类起源演化、民族族源历史和语言农业文化传播等的重要地区之一。
在过去的几十年中,对东亚人群的遗传学研究主要集中在线粒体DNA(mitochondrial DNA, mtDNA)和Y染色体(Y chromosome)方面。在mtDNA研究方面,从最初的限制性片段长度多态(restriction fragment length polymorphism, RFLP)[1],到高变区(hypervariable segments, HVS)和部分编码区测序[2, 3],再到mtDNA全基因组测序[4, 5],mtDNA研究已经成为揭示东亚人群起源演化的主要手段之一。尤其是中国科学院昆明动物研究所张亚平研究员团队基于高分辨率的mtDNA全基因组数据构建的东亚人群系统发育树[4, 5],以及由此发展而出的用于甄别人为重组(artificial recombination)等质量问题的方法[6],在揭示东亚人群的源流历史及民族演化中发挥了重要作用。Y染色体的研究目前则以非重组区(non- recombination regions of Y chromosome, NRY)的单核苷酸多态性(single nucleotide polymorphism, SNP)以及短串联重复序列(short tandem repeat, STR)为主。基于Y染色体遗传多态性等信息,复旦大学金力教授团队从父系遗传角度系统探讨东亚人群的起源、迁徙和遗传结构,对东亚人群是否起源于本土等重大问题提供了新的认识,奠定了东亚人群起源、迁徙历史和遗传结构的框架基础[7,8,9,10]。近年来,随着mtDNA和Y染色体系统发育树的不断完善[11,12,13],东亚人群的遗传历史也得到了更为精细的解析。
随着技术的发展,大量基因组数据得以获得和积累,使得研究人员可以更加详尽地研究东亚人群的遗传结构和进化历史。2003年人类基因组计划(human genome project, HGP)首次完成了人类基因组全图的绘制,随后“国际人类基因组单体型图谱计划(international hapMap project)”和“人类基因组多样性计划(human genome diversity project, HGDP)”对人类全基因组结构进一步深入研究。2008年由中国华大基因、英国Sanger研究所及美国国立人类基因组研究所联合启动的“千人基因组计划(1000 genomes project, 1KG)”,更深层次地绘制出人类基因组多态性分布水平以及稀有突变,是大规模基因组测序时代的一个里程碑。这些公共数据集对全面深入了解东亚人群遗传结构提供了重要的数据支撑和参照[14]。此外,基于东亚人群全基因组SNP[15, 16]、全外显子组数据[17]以及重测序数据[18]等开展的研究,也取得了一些重要的进展。近年来,古DNA研究,包括现代人古DNA以及早期智人古DNA的测序,则为全面揭示东亚人群的遗传结构演变历史提供了新的线索。2017年中国科学院古脊椎动物与古人类研究所的付巧妹团队对北京房山出土的4万年前的田园洞人骨骼化石进行了全基因组测序[19]。这是东亚第一个古人类全基因组分析,也是整个东亚地区最古老的人类基因组,填补了东亚古DNA研究在空间和时间尺度上的空白。
基于不同的分子标记的研究,人们对东亚人群的遗传学研究主要集中在起源、迁徙、民族遗传格局形成等方面。本文将主要从以上这几个方面系统地探讨东亚人群的起源和演化历史。
1 本地起源与非洲起源
关于现代人群的起源,主要有两种对立的假说:“本地起源说(multiregional hypothesis)”与“非洲起源说(recent African origin model)”。这两种假说均认为,早期智人(early homo sapiens或archaic humans)和晚期智人(即现代人,homo sapiens)的共同祖先直立人(homo erectus)起源于非洲,并在200万年前走出非洲扩散到世界其他地区。不同之处在于,前者认为现代人源于本土的早期智人;而后者认为,现居世界各地的现代人均来自20万年前起源于非洲的晚期智人,约在10万年前走出非洲,进而扩散到达欧亚大陆。由于东亚出土了大量形态学上连续的古人类化石,因此考古学认为东亚人群是从本地的早期智人(如许昌人[20]、马鹿洞人[21]等,图1)演化而来,即“本地起源”[22]。近年来,在中国湖南省道县发现的极其古老的现代人化石,由于其年代(8~12万年前)比“非洲起源说”认为的现代人到达东亚的时间(5~6万年前)要早得多[23],因而被看作是支持“本地起源”的最有力的直接证据。然而,由于分子钟估算的走出非洲的时间本身还存在很大争议,例如利用较慢的分子钟推测的欧亚人与非洲人的分歧时间约为10万年左右[24],与该化石样本的时间极为相似。最近在以色列史前洞穴发现的智人化石(17.7~19.4万年),则将人类走出非洲的时间推前了至少约5.5万年[25]。而基于12万年前的尼安德特人(Neanderthal)化石样本mtDNA测序数据所推测的现代人走出非洲的时间更早,约为47万到22万年前[26]。因此,该化石所代表的古老群体也有可能在10万年前或更早的时间与现代欧亚人祖先一起走出非洲到达东亚。另外,如果该古老群体确实从东亚古人类演化而来,那么他们究竟是进一步演化成东亚的现代人群,还是被后来走出非洲的东亚人祖先完全替代,还需要分子遗传学的证据进一步核实[27]。
图1
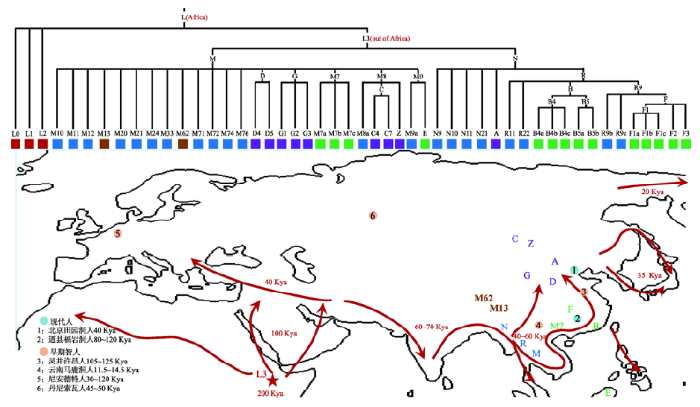 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1走出非洲路线图及东亚人群mtDNA系统发育树
现代人祖先走出非洲的时间和路线分别在地图上用红色数字和线条标出,时间单位千年(Kya, kilo years ago)。地图上标注出了一些东亚人群常见mtDNA单倍型类群,与系统发育树色块相一致。代表性的现代人遗址和早期智人遗址在地图上分别用不同颜色和序号做了标记。
Fig. 1Out-of-Africa migration routes and mtDNA phylogenetic tree of East Asian populations
“非洲起源说”的证据则主要来自分子遗传学的研究。1987年,Cann等[1]过对全球范围内147例个体的mtDNA的RFLP进行分析,结果发现非洲以外的个体均属于非洲特有类群(L3)的衍生支系。东亚人群mtDNA及Y染色体的遗传变异研究,也进一步支持了东亚人群起源于非洲的观点。中国科学院昆明动物研究所孔庆鹏等[5, 11]先后系统地分析了数千个东亚个体的mtDNA基因组序列数据,结果揭示东亚所有已鉴定的母系世系均源于非洲(图1),未发现任何源自本地直立人的母系遗传贡献。在Y染色体研究方面,复旦大学金力教授团队也发现东亚现代人中普遍存在的3个变异位点(M89、M130和YAP)均源自非洲古老突变M168[7,8,9],与“非洲起源说”一致。利用全基因组重测序技术,随后对来自东亚的YH (汉族个体)、SJK(韩国人)等个体的研究,也进一步支持东亚人群起源于非洲[14]。
2 源自早期智人的遗传贡献
尽管东亚人群的主体遗传类型来自非洲,越来越多的全基因组证据表明,欧亚大陆上的早期智人可能对走出非洲的现代人有一定程度的遗传贡献[28,29,30]。2010年,Green等[28]对克罗地亚尼安德特人的骨遗骸样本进行了全基因组测序,结果发现,包括东亚人群在内的所有欧亚人群中,约有1%~4%的遗传组分与尼安德特人相似。进一步研究也提示东亚人群中确实存在与尼安德特人相似的遗传组分,并且这种组分在东亚人群(9.6%)中要高于欧洲人群(6.4%)[31]。另外,在亚洲东部的现代人群中还发现了与西伯利亚地区的古人类丹尼索瓦人(Denisova)相似的遗传组分[29, 30]。该遗传组分主要分布在大洋洲(约3%~6%)[30],随后也被证明在东亚人群中有少量分布(0.2%)[32]。对这种遗传共享现象,目前的解释主要有两种模型:一种认为现代人走出非洲后与当地的早期智人发生了遗传交流[29, 30],即“后期基因交流模型(archaic introgression model)”;另一种则认为,现代人与当地早期智人共享的遗传组分可能是他们的共同祖先就已存在遗传分化,由于同源关系导致部分现代人(如欧亚人群)保留了与早期智人相似的基因片段,即“早期非洲子群体模型(ancient population structure model)”[33]。由于共享的古老基因片段发生重组的时间存在差异,因此在后期基因交流模型下,共享的基因片段长度比早期非洲子群体模型下的要长[34]。而根据尼安德特人和现代人共享基因片段的长度,研究者推测二者发生基因交流的时间是4.7~6.5万年前[35],比早期智人与现代人发生分化的时间(27.5~38.3万年前)晚的多[36],从而支持“后期基因交流模型”。同样,丹尼索瓦人与大洋洲及东亚人群的遗传共享,也被证明是后期基因交流的结果[29, 30]。还有研究认为,藏族人群中的高原适应基因EPAS1也可能源于丹尼索瓦人[37],不过也有研究对该观点提出了质疑[38]。
由此可见,尽管东亚人群的mtDNA、Y染色体以及核基因的主体遗传组分来自非洲,但后期也有可能接受了来自早期智人的遗传贡献,因此其进化历史可能更为复杂。而这些可能源自早期智人的遗传组分,对东亚人群的起源扩散历史,更是有着非常重要的提示作用。
3 从非洲到东亚的迁徙
3.1 迁徙路线
越来越多的遗传学研究表明,现代人在更新世晚期走出非洲并且快速扩散到欧亚大陆以及大洋洲等地区[1, 39~41]。进一步的研究表明现代人祖先通过一条“南部”迁徙路线从非洲撒哈拉以南沿海岸线扩散(图1),大约在5~6万年前到达东亚南部[42],并且该扩散是一个相当快速的过程[43, 44]。东亚人群祖先到达东亚后,开始了向东亚内陆的大规模迁徙,进而扩散到整个东亚地区。这些迁徙主要发生在末次盛冰期(last glacial maximum, LGM)[45]之后,可能是由于气候变暖所导致[7, 46]。冰期后的迁徙路线有多条,包括从中国南部向西延展到印度东北部和喜马拉雅以南地区的扩散[47],以及经河流从东南亚进入东亚地区的迁徙[48]。此外,Y染色体的研究还表明,在冰期之后,还有少部分现代东亚人群是来自中亚和西部欧亚的遗传输入(约7%)[49]。由此看来,最初走出非洲的现代人主要是沿海岸线迁徙,而随后(主要是冰期后)主要是沿内陆迁徙路线以及北方路线扩散进入东亚。
3.2 迁徙次数
关于人类走出非洲的次数,目前也存在很大争议,争议的焦点主要是东亚人群是否与澳洲人群来自同一次扩散。早期基于mtDNA的研究认为,澳洲及欧亚人群中的3个超类群(M、N和R)的溯祖时间很相似,由此推测这些地区的人群可能来自走出非洲的同一次迁徙[43, 50]。而全基因组的研究则得出了不同的结论。通过对澳洲土著人的全基因组研究发现,澳洲土著人群和欧亚人群是分别是来自7.5~6.2万年前以及3.8~2.5万年前的不同迁徙事件[51]。这一观点得到了来自丹尼索瓦人基因组研究的支持。由于丹尼索瓦人的遗传组分主要分布于大洋洲等地区,因此Reich等[30]提出,现代人走出非洲可能有两次:第一次走出的人群即大洋洲人群的祖先,可能与丹尼索瓦人发生了基因交流;而第二次走出的人群主要演化成了现代的欧亚人群。值得注意的是,与欧亚人群类似,该澳洲土著人也携带有尼安德特人的遗传组分,这其实是与单扩散模型更加吻合的[31]。因此,在多扩散模型下,对该现象的可能的解释有两种:一种是人类走出非洲确有两次,而两次不同时间的扩散均与尼安德特人发生了基因交流;第二种是现代人与尼安德特人的基因交流可能早在10万年前就已发生,之后这部分现代人又返回非洲[52],继而分别在7万年前以及3万年前发生两次不同的扩散。到目前为止,这两种解释还都存在一定的不确定性,因此东亚人群与澳洲人是否来自不同的迁徙事件,业界仍然没有定论。而该问题的解决,很大程度上依赖于更多全基因组数据的深入研究,以及更精确的时间估算。
4 东亚人群的遗传分布格局与民族形成机制
东亚人群经历了复杂的历史过程和文化演变,形成了现今语言、文化多样的民族群体。那么,在这些多样的民族形成过程中,哪些因素起着重要作用?民族的形成是否仅仅是文化扩散的产物?不同族源历史的民族之间是否存在遗传分化和遗传交流?遗传学的研究为这些问题的解答提供了重要的思路和线索。4.1 早期定居与民族遗传背景
尽管民族的形成是相对近期的事件,但在不同的民族人群中,仍然保留了早期定居东亚的人群遗传印记。例如在藏族人群中,就发现了其特有的古老组分(mtDNA单倍型类群M62b),该组分可追溯至旧石器晚期[53]。这表明,藏族人群除了新石器时期来自中国北方的遗传组分以外[53,54,55],还保留着古老的、代表早期定居青藏高原东亚人祖先的遗传组分[53]。随后的全基因组研究也表明藏族的起源时间十分古老,大约可以追溯至6.2~3.8万年前[18]。此外,在中国南方少数民族人群中,也发现了大量的mtDNA基部类群,如M74、M75、M76、N10和N11等,这些类群从欧亚超类群M和N直接衍生出来,其年代也非常古老(5~6万年前),因此很有可能是早期定居东亚的人群遗传印记[11]。对汉族人群的mtDNA研究也表明,汉族人群的母系遗传结构更多地保留了冰期后的遗传组分[3]。值得注意的是,这些古老的遗传组分在不同的民族人群中也存在特异分布。如M62b局限分布在藏族人群中[53],M74等类群则主要分布在中国西南的少数民族中[11]。这说明,这些史前时期的古老组分对民族遗传背景、乃至民族遗传结构的形成也起到了相当重要的作用。
4.2 文化扩散与人口扩散
同一个民族往往有着相同的文化,而民族的形成究竟是文化同化的产物,还是由人群的迁徙所主导?基于东南亚壮侗语系(Tai-Kadai)人群的mtDNA数据,Kutannan等[56]比较了人口扩散(demic diffusion model)、文化扩散(culture diffusion model)以及混合模型(admixture model)。结果表明,该语系从中国扩散至东南亚地区遵循人口扩散模式。也就是说,来自中国南方的人群迁徙在东南亚壮侗语系民族的形成中起着十分重要的作用[56]。相比之下,汉族的形成则比较复杂。来自mtDNA以及全基因组的证据均表明汉族存在着明显的南北分化[3, 57, 58],而这一分化提示了汉族的形成在很大程度上是对南方土著人群的文化征服和同化[3],即文化扩散模式。而基于Y染色体的研究,文波等[59]进一步发现汉族Y染色体的南北分化并不显著,因此推测汉文化向南扩散其实是伴随着大规模人口迁徙的,只不过这一过程是由男性所主导。因此,汉文化的扩张模式存在明显的性别差异,这也提示了文化扩散和人口扩散的相互作用可能在汉族的形成中发挥了重要作用。
同样,这种相互作用在中国西北以及邻近的中亚地区尤为普遍。越来越多的研究表明,中亚人群是欧亚东西部人群在各自扩张过程中发生文化交流和基因融合的结果。例如,在mtDNA研究方面,姚永刚等[60]详细分析了新疆地区5个少数民族(维吾尔族、乌孜别克族、哈萨克族、蒙古族和回族)和中亚人群的mtDNA遗传结构,发现这些群体都可以划分到东亚或者欧洲特有的世系类型,从遗传学上支持欧亚人群之间的遗传融合假说。另外,不同民族群体中欧洲和东亚组分比例各不相同,这些遗传差异可能是不同群体的族源历史、迁徙历史、婚俗习惯等不同的群体历史事件造成的[60]。近期冯启迪等[16]对维吾尔族人群的全基因组SNP数据的研究,则更加细致地重建了维吾尔族人群的混合模型,并且揭示了东西方的遗传交流始自青铜时代,这与小河墓地所发现的木乃伊古DNA研究结果吻合[61]。
4.3 族源历史与遗传分化
在中国少数民族古老的历史记载中,有着氐羌、九黎、三苗、百越等古老的部落。百越部落是古代北方中原诸国对南方楚越地区众多古老部落的泛称,其中“百”是多数的意思。早在2000~3000年前,百越部落广泛分布在中国东南沿海一带,现今主要分布在广西的壮族、贵州的水族以及云南的傣族等都可追溯到古老的百越部落[62]。氐羌部落发源于甘肃、宁夏和青海等地,在4000~5000年前就陆续地向中国西南地区迁徙,并与当地的土著人群发生融合,如今分布在云南地区的彝族、傈僳族、白族、纳西族和拉祜族等民族都是这些古老氐羌部落的后代[62]。族源历史和语言也呈现对应关系,例如百越、九黎、氐羌、百濮等族源的人群,分属于壮侗语系、苗瑶语系、藏缅语族和南亚语系等。那么这些不同族源历史的少数民族在遗传结构上是否也有相应的差异?文化上的差异性和相似性是否留下了遗传上的印记?研究人员对氐羌、百越以及九黎部落等族源的民族开展了mtDNA和Y染色体的遗传结构分析,不同的研究均表明,族源不同的群体间存在遗传差异,族源印记或多或少在群体遗传结构上得到了保留[63, 64]。例如,氐羌部落民族有着更多的源自中国北方的遗传组分,与他们源自中国北方的历史记载吻合[63]。百越民族人群保留了较多的南方土著组分,他们的群体扩张时间也相对的更为古老[63]。苗瑶语系人群的母系遗传组分更可能来自南方;苗族与东亚北方的群体有着更多的遗传交流,这与历史记录相符[64]。同样,中国东北部少数民族(达斡尔族、鄂温克族、朝鲜族、蒙古族和鄂伦春族)的mtDNA主要单倍型类群都是北方群体常见类群D、G、C和Z等,与他们北方起源的历史记载一致[65]。随后基于全基因组SNP分型数据的研究,也表明东亚人群的遗传结构与语言结构,而非地理位置,有很好的对应,表明不同族源的人群在遗传结构上确实存在一定的分化[15]。
另外,不同地区的同一民族群体间也存在一定的遗传差异,可能是这些群体对古老遗传印记不同程度的保留以及后期各自经历了不同的群体历史事件所致。例如,氐羌部落在向南迁徙的过程中,父系方面更多地保留了北方移民的组分,而母系方面则主要源自南方原住居民[66]。一些氐羌民族,例如拉祜族和怒族,在南迁的过程中可能经历了建群者效应[63]。
5 结语与展望
从遗传学角度研究东亚人群的遗传结构,有助于解决东亚人群的起源和迁徙等方面的争议,为东亚人群的迁徙和演化历史提供新的线索。这不仅在人类进化遗传学方面意义显著,也为医学遗传学、生物信息学、法医学等研究提供了大规模遗传信息数据平台,同时也为东亚人群适应性进化机制研究提供了一个极为良好的参照系。采用不同分辨率的mtDNA、Y染色以及全基因组数据,前期的研究对东亚人群的源流历史做了细致的分析,很大程度上解决了考古学、历史学等领域长期以来存在的分歧。然而,由于技术的局限,前期研究或多或少地受到了来自样本量以及数据分辨率的限制。因此,以往研究得出的结论还有待大覆盖度、高分辨率的遗传学数据(尤其是Y染色体及全基因组测序数据)的进一步验证。随着DNA测序技术的发展进步和全基因组测序技术的推广,群体全基因组学的工作将是未来遗传学研究的主流。千人基因组数据的公布以及“中国十万人基因组计划”的启动,为人们从全基因组的角度来探讨东亚人群的起源迁徙历史提供了新的契机。现今测序手段的进步和大数据的出现有望展现出关于人群迁移扩散历史更多的细节,帮助我们更全面地了解现代人的历史。
作为考古学与遗传学学科交叉产生的古DNA研究已成为追溯人类历史的一个重要手段。在现代人群遗传结构的研究基础上,对古人遗骸进行DNA的研究分析,为不同地区、不同时间的古代人群的遗传多样度和演化进程提供了更为直接的证据支持。将考古文化与古DNA测序,与现代人的遗传结构相结合,将会描绘出东亚波澜壮阔的人类演化历史。欧美地区的古DNA遗传学研究日臻成熟,目前已经有众多突破性研究成果[67,68,69,70]。然而,在东亚地区,关于古代DNA的研究仍十分有限[19, 71],我们还有很长一段路要走。随着东亚地区古DNA研究的逐步开展,相信东亚人群的起源演化研究将会取得更多令人瞩目的研究成果。
(责任编委: 谢小冬)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 3]
URL [本文引用: 1]
URL [本文引用: 4]
URL [本文引用: 2]
URL [本文引用: 3]
URLPMID:18714389 [本文引用: 1]

Background Large-scale genome sequencing poses enormous problems to the logistics of laboratory work and data handling. When numerous fragments of different genomes are PCR amplified and sequenced in a laboratory, there is a high immanent risk of sample confusion. For genetic markers, such as mitochondrial DNA (mtDNA), which are free of natural recombination, single instances of sample mix-up involving different branches of the mtDNA phylogeny would give rise to reticulate patterns and should therefore be detectable. Methodology/Principal Findings We have developed a strategy for comparing new complete mtDNA genomes, one by one, to a current skeleton of the worldwide mtDNA phylogeny. The mutations distinguishing the reference sequence from a putative recombinant sequence can then be allocated to two or more different branches of this phylogenetic skeleton. Thus, one would search for two (or three) near-matches in the total mtDNA database that together best explain the variation seen in the recombinants. The evolutionary pathway from the mtDNA tree connecting this pair together with the recombinant then generate a grid-like median network, from which one can read off the exchanged segments. Conclusions We have applied this procedure to a large collection of complete human mtDNA sequences, where several recombinants could be distilled by our method. All these recombinant sequences were subsequently corrected by de novo experiments fully concordant with the predictions from our data-analytical approach.
URL [本文引用: 3]
URLPMID:11253652 [本文引用: 2]
East Asia is one of the few regions in the world where a relatively large number of human fossils have been unearthed -- a discovery that has been taken as evidence for an independent local origin of modern humans outside of Africa. However, genetic studies conducted in the past ten years, especially using Y chromosomes, have provided unequivocal evidence for an African origin of East Asian populations. The genetic signatures present in diverse East Asian populations mark the footsteps of prehistoric migrations that occurred tens of thousands of years ago.
URLPMID:11349147 [本文引用: 2]
To test the hypotheses of modern human origin in East Asia, we sampled 12,127 male individuals from 163 populations and typed for three Y chromosome biallelic markers (YAP, M89, and M130). All the individuals carried a mutation at one of the three sites. These three mutations (YAP+, M89T, and M130T) coalesce to another mutation (M168T), which originated in Africa about 35,000 to 89,000 years ago. Therefore, the data do not support even a minimal in situ hominid contribution in the origin of anatomically modern humans in East Asia.
[本文引用: 1]
[本文引用: 4]
URLPMID:4350590 [本文引用: 1]
Phylogenetically informative Y chromosomal single-nucleotide polymorphisms (Y-SNPs) integrated in DNA chips have not been sufficiently explored in most genome-wide association studies (GWAS). Herein, we introduce a pipeline to retrieve Y-SNP data. We introduce the software YTool (http://mitotool.org/ytool/) to handle conversion, filtering, and annotation of the data. Genome-wide SNP data from populations in Myanmar are used to construct a haplogroup tree for 117 Y chromosomes based on 369 high-confidence Y-SNPs. Parallel genotyping and published resequencing data of Y chromosomes confirm the validity of our pipeline. We apply this strategy to the CEU HapMap data set and construct a haplogroup tree with 107 Y-SNPs from 39 individuals. The retrieved Y-SNPs can discern the parental genetic structure of populations. Given the massive quantity of data from GWAS, this method facilitates future investigations of Y chromosome diversity.
URLPMID:24166809 [本文引用: 1]
Abstract During the last few decades, a wealth of studies dedicated to the human Y chromosome and its DNA variation, in particular Y-chromosome single-nucleotide polymorphisms (Y-SNPs), has led to the construction of a well-established Y-chromosome phylogeny. Since the recent advent of new sequencing technologies, the discovery of additional Y-SNPs is exploding and their continuous incorporation in the phylogenetic tree is leading to an ever higher resolution. However, the large and increasing amount of information included in the "complete" Y-chromosome phylogeny, which now already includes many thousands of identified Y-SNPs, can be overwhelming and complicates its understanding as well as the task of selecting suitable markers for genotyping purposes in evolutionary, demographic, anthropological, genealogical, medical, and forensic studies. As a solution, we introduce a concise reference phylogeny whereby we do not aim to provide an exhaustive tree that includes all known Y-SNPs but, rather, a quite stable reference tree aiming for optimal global discrimination capacity based on a strongly reduced set that includes only the most resolving Y-SNPs. Furthermore, with this reference tree, we wish to propose a common standard for Y-marker as well as Y-haplogroup nomenclature. The current version of our tree is based on a core set of 417 branch-defining Y-SNPs and is available online at http://www.phylotree.org/Y. 2013 WILEY PERIODICALS, INC.
URLPMID:21753753 [本文引用: 2]
The history of human population size is important to understanding human evolution. Heng Li and Richard Durbin use complete genome sequences from Chinese, Korean, European and Yoruban (West African) individuals to estimate population sizes between 10,000 and 1 million years ago. They infer that European and Chinese populations had very similar size histories until about 10,00020,000 years ago. The European, Chinese and African populations all had an elevated effective population between 60,000 and 250,000 years ago. Genomic analysis suggests that the differentiation of genetically modern humans may have started as early as 100,000120,000 years ago.
URL [本文引用: 2]
URLPMID:28595347 [本文引用: 2]
The Uyghur people residing in Xinjiang,a territory located in the far west of China and crossed by the Silk Road,are a key ethnic group for understanding the history of human dispersion in Eurasia.Here we assessed the genetic structure and ancestry of 951 Xinjiang's Uyghurs(XJU) representing 14 geographical subpopulations.We observed a southwest and northeast differentiation within XJU,which was likely shaped jointly by the Tianshan Mountains,which traverses from east to west as a natural barrier,and gene flow from both east and west directions.In XJU we identified 4 major ancestral components that were potentially derived from two earlier admixed groups:one from the West,harboring European(25%-37%) and South Asian ancestries(12%-20%),and the other from the East,with Siberian(15%-17%) and East Asian(29%-47%) ancestries.By using a newly developed method,MultiWaver,the complex admixture history of XJU was modeled as a two-wave admixture.An ancient wave was dated back to ~3,750 years ago(ya),which is much earlier than that estimated by previous studies,but fits within the range of dating of mummies that exhibited European features that were discovered in the Tarim basin,which is situated in southern Xinjiang(4,000-2,000 ya);a more recent wave occurred around 750 ya,which is in agreement with the estimate from a recent study using other methods.We unveiled a more complex scenario of ancestral origins and admixture history in XJU than previously reported,which further suggests Bronze Age massive migrations in Eurasia and EastWest contacts across the Silk Road.
URLPMID:20595611 [本文引用: 1]
Abstract Residents of the Tibetan Plateau show heritable adaptations to extreme altitude. We sequenced 50 exomes of ethnic Tibetans, encompassing coding sequences of 92% of human genes, with an average coverage of 18x per individual. Genes showing population-specific allele frequency changes, which represent strong candidates for altitude adaptation, were identified. The strongest signal of natural selection came from endothelial Per-Arnt-Sim (PAS) domain protein 1 (EPAS1), a transcription factor involved in response to hypoxia. One single-nucleotide polymorphism (SNP) at EPAS1 shows a 78% frequency difference between Tibetan and Han samples, representing the fastest allele frequency change observed at any human gene to date. This SNP's association with erythrocyte abundance supports the role of EPAS1 in adaptation to hypoxia. Thus, a population genomic survey has revealed a functionally important locus in genetic adaptation to high altitude.
URL [本文引用: 2]
URL [本文引用: 2]
URLPMID:28254945 [本文引用: 1]
Abstract Two early Late Pleistocene (~105,000- to 125,000-year-old) crania from Lingjing, Xuchang, China, exhibit a morphological mosaic with differences from and similarities to their western contemporaries. They share pan-Old World trends in encephalization and in supraorbital, neurocranial vault, and nuchal gracilization. They reflect eastern Eurasian ancestry in having low, sagittally flat, and inferiorly broad neurocrania. They share occipital (suprainiac and nuchal torus) and temporal labyrinthine (semicircular canal) morphology with the Neandertals. This morphological combination reflects Pleistocene human evolutionary patterns in general biology, as well as both regional continuity and interregional population dynamics. Copyright 2017, American Association for the Advancement of Science.
URLPMID:4683062 [本文引用: 1]
The number of Late Pleistocene hominin species and the timing of their extinction are issues receiving renewed attention following genomic evidence for interbreeding between the ancestors of some living humans and archaic taxa. Yet, major gaps in the fossil record and uncertainties surrounding the age of key fossils have meant that these questions remain poorly understood. Here we describe and compare a highly unusual femur from Late Pleistocene sediments at Maludong (Yunnan), Southwest China, recovered along with cranial remains that exhibit a mixture of anatomically modern human and archaic traits. Our studies show that the Maludong femur has affinities to archaic hominins, especially Lower Pleistocene femora. However, the scarcity of later Middle and Late Pleistocene archaic remains in East Asia makes an assessment of systematically relevant character states difficult, warranting caution in assigning the specimen to a species at this time. The Maludong fossil probably samples an archaic population that survived until around 14,000 years ago in the biogeographically complex region of Southwest China.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:26466566 [本文引用: 1]
Abstract The hominin record from southern Asia for the early Late Pleistocene epoch is scarce. Well-dated and well-preserved fossils older than 09080445,000 years that can be unequivocally attributed to Homo sapiens are lacking. Here we present evidence from the newly excavated Fuyan Cave in Daoxian (southern China). This site has provided 47 human teeth dated to more than 80,000 years old, and with an inferred maximum age of 120,000 years. The morphological and metric assessment of this sample supports its unequivocal assignment to H. sapiens. The Daoxian sample is more derived than any other anatomically modern humans, resembling middle-to-late Late Pleistocene specimens and even contemporary humans. Our study shows that fully modern morphologies were present in southern China 30,000-70,000 years earlier than in the Levant and Europe. Our data fill a chronological and geographical gap that is relevant for understanding when H. sapiens first appeared in southern Asia. The Daoxian teeth also support the hypothesis that during the same period, southern China was inhabited by more derived populations than central and northern China. This evidence is important for the study of dispersal routes of modern humans. Finally, our results are relevant to exploring the reasons for the relatively late entry of H. sapiens into Europe. Some studies have investigated how the competition with H. sapiens may have caused Neanderthals' extinction (see ref. 8 and references therein). Notably, although fully modern humans were already present in southern China at least as early as 09080480,000 years ago, there is no evidence that they entered Europe before 09080445,000 years ago. This could indicate that H. neanderthalensis was indeed an additional ecological barrier for modern humans, who could only enter Europe when the demise of Neanderthals had already started.
URLPMID:22965354 [本文引用: 1]
It is now possible to make direct measurements of the mutation rate in modern humans using next-generation sequencing. These measurements reveal a value that is approximately half of that previously derived from fossil calibration, and this has implications for our understanding of demographic events in human evolution and other aspects of population genetics. Here, we discuss the implications of a lower-than-expected mutation rate in relation to the timescale of human evolution.
URLPMID:29371468 [本文引用: 1]
Abstract To date, the earliest modern human fossils found outside of Africa are dated to around 90,000 to 120,000 years ago at the Levantine sites of Skhul and Qafzeh. A maxilla and associated dentition recently discovered at Misliya Cave, Israel, was dated to 177,000 to 194,000 years ago, suggesting that members of the Homo sapiens clade left Africa earlier than previously thought. This finding changes our view on modern human dispersal and is consistent with recent genetic studies, which have posited the possibility of an earlier dispersal of Homo sapiens around 220,000 years ago. The Misliya maxilla is associated with full-fledged Levallois technology in the Levant, suggesting that the emergence of this technology is linked to the appearance of Homo sapiens in the region, as has been documented in Africa.
URL [本文引用: 1]
URLPMID:4771949 [本文引用: 1]
Although it is widely accepted that modem humans (Homo sapiens sapiens) can trace their African origins to 150-200 kilo years ago (kya) (recent African origin model;Henn et al, 2012;Ingman et al, 2000;Poznik et al, 2013;Weaver, 2012), an alternative model suggests that the diverse populations of our species evolved separately on different continents from archaic human forms (multiregional origin model;Wolpoff et al, 2000;Wu, 2006).The recent discovery of 47 teeth from a Fuyan cave in southem China (Liu et al, 2015) indicated the presence of H.s.sapiens in eastern Eurasia during the early Late Pleistocene.Since the age of the Fuyan teeth (80-120 kya) predates the previously assumed out-of-Africa exodus (60 kya) by at least 20 kya, this inconsistency provides some support for the multiregional origin model, and thus may challenge the recent African origin hypothesis.
URLPMID:5100745 [本文引用: 2]
particularly toward the 5-ends of DNA reads, where at the first position ~40% of cytosine residues can appear as thymine residues. The frequency of C to T misincorporations progressively diminishes further into the molecules. At the 3-ends, comple- mentary G to A transitions are seen as a result of the enzymatic fill-in procedure in which blunt DNA ends are created before adaptor ligation (23). We implemented an alignment approach that takes these nucleotide misincorporation patterns into account (SOM Text 3) and aligned the Neandertal sequences to either the reference human genome (UCSC hg18), the reference chimpanzee genome ( panTro2), or the inferred human-chimpanzee common ancestral sequence (SOM Text 3).To estimate the error rate in the Neandertal DNA sequences determined, we compared reads that map to the mitochondrial genomes, which we assembled to 35-, 29- and 72-fold coverage for each of the bones, respectively (15, 45) (SOM Text 4). Although C to T and G to A substitutions, which are caused by deaminated cytosine residues, occur at a rate of 4.5 to 5.9%, other error rates are at most 0.3% (fig. S4). Because we sequence each DNA fragment from both sides, and most frag- ments more than once (49), the latter error rate is substantially lower than the error rate of the Illumina platform itself (48, 50).Number of Neandertal individuals. To assess whether the three bones come from different individuals, we first used their mtDNAs. We have previously determined the complete mtDNA sequences from the bones Vi33.16 and Vi33.25 (15, 45), and these differ at 10 positions. There- fore, Vi33.16 and Vi33.25 come from different Neandertal individuals. For the bone Vi33.26, we assembled the mtDNA sequence (SOM Text 4) and found it to be indistinguishable from Vi33.16, suggesting that it could come from the same in- dividual. We analyzed autosomal DNA sequences from the three bones (SOM Text 4) by asking whether the frequency of nucleotide differences between pairs of bones was significantly higher than the frequency of differences within the bones. We find that the within-bone differences are significantly fewer than the between-bone differ- ences for all three comparisons (P 0.001 in all cases). Thus, all three bones derive from different individuals, although Vi33.16 and Vi33.26 may stem from maternally related individuals.
URLPMID:4306417 [本文引用: 4]
中国科学院机构知识库(CAS IR GRID)以发展机构知识能力和知识管理能力为目标,快速实现对本机构知识资产的收集、长期保存、合理传播利用,积极建设对知识内容进行捕获、转化、传播、利用和审计的能力,逐步建设包括知识内容分析、关系分析和能力审计在内的知识服务能力,开展综合知识管理。
URLPMID:21944045 [本文引用: 6]
It has recently been shown that ancestors of New Guineans and Bougainville Islanders have inherited a proportion of their ancestry from Denisovans, an archaic hominin group from Siberia. However, only a sparse sampling of populations from Southeast Asia and Oceania were analyzed. Here, we quantify Denisova admixture in 33 additional populations from Asia and Oceania. Aboriginal Australians, Near Oceanians, Polynesians, Fijians, east Indonesians, and Mamanwa (a 鈥淣egrito group from the Philippines) have all inherited genetic material from Denisovans, but mainland East Asians, western Indonesians, Jehai (a Negrito group from Malaysia), and Onge (a Negrito group from the Andaman Islands) have not. These results indicate that Denisova gene flow occurred into the common ancestors of New Guineans, Australians, and Mamanwa but not into the ancestors of the Jehai and Onge and suggest that relatives of present-day East Asians were not in Southeast Asia when the Denisova gene flow occurred. Our finding that descendants of the earliest inhabitants of Southeast Asia do not all harbor Denisova admixture is inconsistent with a history in which the Denisova interbreeding occurred in mainland Asia and then spread over Southeast Asia, leading to all its earliest modern human inhabitants. Instead, the data can be most parsimoniously explained if the Denisova gene flow occurred in Southeast Asia itself. Thus, archaic Denisovans must have lived over an extraordinarily broad geographic and ecological range, from Siberia to tropical Asia.
URL [本文引用: 2]
Neanderthals were a group of archaic hominins that occupied most of Europe and parts of Western Asia from similar to 30,000 to 300,000 years ago (KYA). They coexisted with modern humans during part of this time. Previous genetic analyses that compared a draft sequence of the Neanderthal genome with genomes of several modern humans concluded that Neanderthals made a small (1-4%) contribution to the gene pools of all non-African populations. This observation was consistent with a single episode of admixture from Neanderthals into the ancestors of all non-Africans when the two groups coexisted in the Middle East 50-80 KYA. We examined the relationship between Neanderthals and modern humans in greater detail by applying two complementary methods to the published draft Neanderthal genome and an expanded set of high-coverage modern human genome sequences. We find that, consistent with the recent finding of Meyer et al. (2012), Neanderthals contributed more DNA to modern East Asians than to modern Europeans. Furthermore we find that the Maasai of East Africa have a small but significant fraction of Neanderthal DNA. Because our analysis is of several genomic samples from each modern human population considered, we are able to document the extent of variation in Neanderthal ancestry within and among populations. Our results combined with those previously published show that a more complex model of admixture between Neanderthals and modern humans is necessary to account for the different levels of Neanderthal ancestry among human populations. In particular, at least some Neanderthal-modern human admixture must postdate the separation of the ancestors of modern European and modern East Asian populations.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:25963373 [本文引用: 1]
As modern and ancient DNA sequence data from diverse human populations accumulate, evidence is increasing in support of the existence of beneficial variants acquired from archaic humans that may have accelerated adaptation and improved survival in new environments - a process known as adaptive introgression. Within the past few years, a series of studies have identified genomic regions that show strong evidence for archaic adaptive introgression. Here, we provide an overview of the statistical methods developed to identify archaic introgressed fragments in the genome sequences of modern humans and to determine whether positive selection has acted on these fragments. We review recently reported examples of adaptive introgression, grouped by selection pressure, and consider the level of supporting evidence for each. Finally, we discuss challenges and recommendations for inferring selection on introgressed regions.
URLPMID:3464203 [本文引用: 1]
Comparisons of DNA sequences between Neandertals and present-day humans have shown that Neandertals share more genetic variants with non-Africans than with Africans. This could be due to interbreeding between Neandertals and modern humans when the two groups met subsequent to the emergence of modern humans outside Africa. However, it could also be due to population structure that antedates the origin of Neandertal ancestors in Africa. We measure the extent of linkage disequilibrium (LD) in the genomes of present-day Europeans and find that the last gene flow from Neandertals (or their relatives) into Europeans likely occurred 37,000–86,000 years before the present (BP), and most likely 47,000–65,000 years ago. This supports the recent interbreeding hypothesis and suggests that interbreeding may have occurred when modern humans carrying Upper Paleolithic technologies encountered Neandertals as they expanded out of Africa. One of the key discoveries from the analysis of the Neandertal genome is that Neandertals share more genetic variants with non-Africans than with Africans. This observation is consistent with two hypotheses: interbreeding between Neandertals and modern humans after modern humans emerged out of Africa or population structure in the ancestors of Neandertals and modern humans. These hypotheses make different predictions about the date of last gene exchange between the ancestors of Neandertals and modern non-Africans. We estimate this date by measuring the extent of linkage disequilibrium (LD) in the genomes of present-day Europeans and find that the last gene flow from Neandertals into Europeans likely occurred 37,000–86,000 years before the present (BP), and most likely 47,000–65,000 years ago. This supports the recent interbreeding hypothesis and suggests that interbreeding occurred when modern humans carrying Upper Paleolithic technologies encountered Neandertals as they expanded out of Africa.
URLPMID:24352235 [本文引用: 1]
Abstract We present a high-quality genome sequence of a Neanderthal woman from Siberia. We show that her parents were related at the level of half-siblings and that mating among close relatives was common among her recent ancestors. We also sequenced the genome of a Neanderthal from the Caucasus to low coverage. An analysis of the relationships and population history of available archaic genomes and 25 present-day human genomes shows that several gene flow events occurred among Neanderthals, Denisovans and early modern humans, possibly including gene flow into Denisovans from an unknown archaic group. Thus, interbreeding, albeit of low magnitude, occurred among many hominin groups in the Late Pleistocene. In addition, the high-quality Neanderthal genome allows us to establish a definitive list of substitutions that became fixed in modern humans after their separation from the ancestors of Neanderthals and Denisovans.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:9326335 [本文引用: 1]
Abstract mtDNA studies support an African origin for modern Eurasians, but expansion events within Africa have not previously been investigated. We have therefore analyzed 407 mtDNA control-region sequences from 13 African ethnic groups. A number of sequences (13%) were highly divergent and coalesced on the "mitochondrial Eve" in Africans. The remaining sequences also ultimately coalesced on this sequence but fell into four major clusters whose starlike phylogenies testify to demographic expansions. The oldest of these African expansions dates to approximately 60,000-80,000 years ago. Eurasian sequences are derived from essentially one sequence within this ancient cluster, even though a diverse mitochondrial pool was present in Africa at the time.
URLPMID:11130070
Abstract The analysis of mitochondrial DNA (mtDNA) has been a potent tool in our understanding of human evolution, owing to characteristics such as high copy number, apparent lack of recombination, high substitution rate and maternal mode of inheritance. However, almost all studies of human evolution based on mtDNA sequencing have been confined to the control region, which constitutes less than 7% of the mitochondrial genome. These studies are complicated by the extreme variation in substitution rate between sites, and the consequence of parallel mutations causing difficulties in the estimation of genetic distance and making phylogenetic inferences questionable. Most comprehensive studies of the human mitochondrial molecule have been carried out through restriction-fragment length polymorphism analysis, providing data that are ill suited to estimations of mutation rate and therefore the timing of evolutionary events. Here, to improve the information obtained from the mitochondrial molecule for studies of human evolution, we describe the global mtDNA diversity in humans based on analyses of the complete mtDNA sequence of 53 humans of diverse origins. Our mtDNA data, in comparison with those of a parallel study of the Xq13.3 region in the same individuals, provide a concurrent view on human evolution with respect to the age of modern humans.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:15890885 [本文引用: 2]
A recent dispersal of modern humans out of Africa is now widely accepted, but the routes taken across Eurasia are still disputed. We show that mitochondrial DNA variation in isolated "relict" populations in southeast Asia supports the view that there was only a single dispersal from Africa, most likely via a southern coastal route, through India and onward into southeast Asia and Australasia. There was an early offshoot, leading ultimately to the settlement of the Near East and Europe, but the main dispersal from India to Australia ~65,000 years ago was rapid, most likely taking only a few thousand years.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:3027199 [本文引用: 1]
Background Archaeological studies have revealed a series of cultural changes around the Last Glacial Maximum in East Asia; whether these changes left any signatures in the gene pool of East Asians remains poorly indicated. To achieve deeper insights into the demographic history of modern humans in East Asia around the Last Glacial Maximum, we extensively analyzed mitochondrial DNA haplogroup M9a'b, a specific haplogroup that was suggested to have some potential for tracing the migration around the Last Glacial Maximum in East Eurasia. Results A total of 837 M9a'b mitochondrial DNAs (583 from the literature, while the remaining 254 were newly collected in this study) pinpointed from over 28,000 subjects residing across East Eurasia were studied here. Fifty-nine representative samples were further selected for total mitochondrial DNA sequencing so we could better understand the phylogeny within M9a'b. Based on the updated phylogeny, an extensive phylogeographic analysis was carried out to reveal the differentiation of haplogroup M9a'b and to reconstruct the dispersal histories. Conclusions Our results indicated that southern China and/or Southeast Asia likely served as the source of some post-Last Glacial Maximum dispersal(s). The detailed dissection of haplogroup M9a'b revealed the existence of an inland dispersal in mainland East Asia during the post-glacial period. It was this dispersal that expanded not only to western China but also to northeast India and the south Himalaya region. A similar phylogeographic distribution pattern was also observed for haplogroup F1c, thus substantiating our proposition. This inland post-glacial dispersal was in agreement with the spread of the Mesolithic culture originating in South China and northern Vietnam.
URLPMID:25826227 [本文引用: 1]
Abstract Given the existence of plenty of river valleys connecting Southeast and East Asia, it is possible that some inland route(s) might have been adopted by the initial settlers to migrate into the interior of East Asia. Here we analyzed mitochondrial DNA (mtDNA) HVS variants of 845 newly collected individuals from 14 Myanmar populations and 5,907 published individuals from 115 populations from Myanmar and its surroundings. Enrichment of basal lineages with the highest genetic diversity in Myanmar suggests that Myanmar was likely one of the differentiation centers of the early modern humans. Intriguingly, some haplogroups were shared merely between Myanmar and southwestern China, hinting certain genetic connection between both regions. Further analyses revealed that such connection was in fact attributed to both recent gene flow and certain ancient dispersals from Myanmar to southwestern China during 25-10鈥卥ya, suggesting that, besides the coastal route, the early modern humans also adopted an inland dispersal route to populate the interior of East Asia.
URL [本文引用: 1]
URLPMID:15101581 [本文引用: 1]
Modern DNA, in particular maternally inherited mitochondrial DNA (mtDNA), is now routinely used to trace ancient migration routes and to obtain absolute dates for genetic prehistory. The errors on absolute genetic dates are often large (50% or more) and depend partly on the inherent evolutionary signal in the DNA data, and partly on our imperfect knowledge of the DNA mutation rate. Despite their imprecision, the genetic dates do provide an independent, consistent and global chronology linking living with their ancestors. Combining this chronology with archaeological and climatological data, most of our own mtDNA studies during the past decade strongly imply a major role for palaeoclimate in determining conditions for prehistoric migrations and demographic expansions. This paper summarizes our interpretation of the genetic findings, covering the initial and modest spread of within Africa more than 100 ka, the striking re-expansion within Africa 60-80 ka, leading ultimately to the out-of-Africa migration of a single, small group which settled in Australia, Eurasia and America during windows of opportunity at least partly dictated by fluctuations in sea-levels and climatic conditions.
URLPMID:21940856 [本文引用: 1]
We present an Aboriginal Australian genomic sequence obtained from a 100-year-old lock of hair donated by an Aboriginal man from southern Western Australia in the early 20th century. We detect no evidence of European admixture and estimate contamination levels to be below 0.5%. We show that Aboriginal Australians are descendants of an early human dispersal into eastern Asia, possibly 62,000 to 75,000 years ago. This dispersal is separate from the one that gave rise to modern Asians 25,000 to 38,000 years ago. We also find evidence of gene flow between populations of the two dispersal waves prior to the divergence of Native Americans from modern Asian ancestors. Our findings support the hypothesis that present-day Aboriginal Australians descend from the earliest humans to occupy Australia, likely representing one of the oldest continuous populations outside Africa.
URLPMID:16920617 [本文引用: 1]
The recent publication of three old Neandertal mitochondrial sequences shows that the genetic diversity of the Neandertals has been largely underestimated. It suggests that the Neandertal population was extensively subdivided geographically, and that its genetic diversity changed markedly over time.
URL [本文引用: 4]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 2]
URLPMID:19944401 [本文引用: 1]
Population stratification is a potential problem for genome-wide association studies (GWAS), confounding results and causing spurious associations. Hence, understanding how allele frequencies vary across geographic regions or among subpopulations is an important prelude to analyzing GWAS data. Using over 350,000 genome-wide autosomal SNPs in over 6000 Han Chinese samples from ten provinces of China, our study revealed a one-dimensional “north-south” population structure and a close correlation between geography and the genetic structure of the Han Chinese. The north-south population structure is consistent with the historical migration pattern of the Han Chinese population. Metropolitan cities in China were, however, more diffused “outliers,” probably because of the impact of modern migration of peoples. At a very local scale within the Guangdong province, we observed evidence of population structure among dialect groups, probably on account of endogamy within these dialects. Via simulation, we show that empirical levels of population structure observed across modern China can cause spurious associations in GWAS if not properly handled. In the Han Chinese, geographic matching is a good proxy for genetic matching, particularly in validation and candidate-gene studies in which population stratification cannot be directly accessed and accounted for because of the lack of genome-wide data, with the exception of the metropolitan cities, where geographical location is no longer a good indicator of ancestral origin. Our findings are important for designing GWAS in the Chinese population, an activity that is expected to intensify greatly in the near future.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
[本文引用: 2]
URLPMID:11953946 [本文引用: 4]
The origin and demographic history of the ethnic populations of China have not been clearly resolved. In this study, we examined the hypervariable segment I sequences (HVSI) of the mitochondrial DNA control region in 372 individuals from nine Chinese populations and one northern Thai population. A relatively high percentage of individuals was found to share sequences with those from other populations of the same ethnogenesis. In general, the populations of southern or Pai-Yuei tribal origin showed high haplotype diversity and nucleotide diversity compared with the populations of northern or Di-Qiang tribal origin. Mismatch distributions from these populations showed concordant features. All except the northern groups Nu, Lisu, Tibetan, and Mongolian showed typical signatures of ancient population expansions in the mismatch distributions and neutrality tests. Episodes of extreme size reduction in the past are one of the likely explanations for the absence of evidence of expansion in northern populations. Small sample sizes as well as samples from isolated subpopulations contributed to the bumpy mismatch distributions observed. Phylogenetic analysis and haplotype sharing among populations suggest that current mtDNA variation in these ethnic populations could reveal their ethnohistory to some extent, but in general, linguistic and geographic classifications of the populations did not agree well with classification by mtDNA variation. Am J Phys Anthropol 118:63-76, 2002. ? 2002 Wiley-Liss, Inc.
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:25455029 [本文引用: 1]
Abstract Understanding the peopling of the Americas remains an important and challenging question. Here, we present (14)C dates, and morphological, isotopic and genomic sequence data from two human skulls from the state of Minas Gerais, Brazil, part of one of the indigenous groups known as 'Botocudos'. We find that their genomic ancestry is Polynesian, with no detectable Native American component. Radiocarbon analysis of the skulls shows that the individuals had died prior to the beginning of the 19th century. Our findings could either represent genomic evidence of Polynesians reaching South America during their Pacific expansion, or European-mediated transport. Copyright 2014 Elsevier Ltd. All rights reserved.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:4943878 [本文引用: 1]
Modern humans arrived in Europe ~45,000 years ago, but little is known about their genetic composition before the start of farming ~8,500 years ago. We analyze genome-wide data from 51 Eurasians from ~45,000-7,000 years ago. Over this time, the proportion of Neanderthal DNA decreased from 3鈥6% to around 2%, consistent with natural selection against Neanderthal variants in modern humans. Whereas the earliest modern humans in Europe did not contribute substantially to present-day Europeans, all individuals between ~37,000 and ~14,000 years ago descended from a single founder population which forms part of the ancestry of present-day Europeans. A ~35,000 year old individual from northwest Europe represents an early branch of this founder population which was then displaced across a broad region, before reappearing in southwest Europe during the Ice Age ~19,000 years ago. During the major warming period after ~14,000 years ago, a new genetic component related to present-day Near Easterners appears in Europe. These results document how population turnover and migration have been recurring themes of European pre-history.
URLPMID:4753769 [本文引用: 1]
Abstract We present the high-quality genome sequence of a 09080445,000-year-old modern human male from Siberia. This individual derives from a population that lived before-or simultaneously with-the separation of the populations in western and eastern Eurasia and carries a similar amount of Neanderthal ancestry as present-day Eurasians. However, the genomic segments of Neanderthal ancestry are substantially longer than those observed in present-day individuals, indicating that Neanderthal gene flow into the ancestors of this individual occurred 7,000-13,000 years before he lived. We estimate an autosomal mutation rate of 0.400020103000210(-9) to 0.600020103000210(-9) per site per year, a Y chromosomal mutation rate of 0.700020103000210(-9) to 0.900020103000210(-9) per site per year based on the additional substitutions that have occurred in present-day non-Africans compared to this genome, and a mitochondrial mutation rate of 1.800020103000210(-8) to 3.200020103000210(-8) per site per year based on the age of the bone.

{kind=link}
{kind=link}