 ,, 芒来,内蒙古农业大学动物科学学院,内蒙古自治区马属动物遗传育种与繁殖重点实验室,呼和浩特 010018
,, 芒来,内蒙古农业大学动物科学学院,内蒙古自治区马属动物遗传育种与繁殖重点实验室,呼和浩特 010018Overview of the genetic control of horse coat color patterns
Ruoyang Zhao, Yiping Zhao, Bei Li, Gerelchimeg Bou, Xinzhuang Zhang, Togtokh Mongke, Tugeqin Bao, Shurenchimeg Gereliin, Tsimbai Gereltuuin, Chao Li, Dongyi Bai,, Manglai Dugarjaviin,Inner Mongolia Key Laboratory of Equus Genetics, Breeding and Reproduction, Hohhot 010018, China通讯作者:
编委: 吴东东
收稿日期:2017-11-9修回日期:2018-04-25网络出版日期:2018-05-20
| 基金资助: |
Editorial board:
Received:2017-11-9Revised:2018-04-25Online:2018-05-20
| Fund supported: |
作者简介 About authors
赵若阳,在读博士生,专业方向:动物遗传育种与繁殖E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (15822KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
赵若阳, 赵一萍, 李蓓, 格日乐其木格, 张心壮, 陶克涛, 图格琴, 旭仁其木格, 青柏, 李超, 白东义, 芒来. 马毛色遗传机理研究进展. 遗传[J], 2018, 40(5): 357-368 doi:10.16288/j.yczz.17-371
Ruoyang Zhao, Yiping Zhao, Bei Li, Gerelchimeg Bou, Xinzhuang Zhang, Togtokh Mongke, Tugeqin Bao, Shurenchimeg Gereliin, Tsimbai Gereltuuin, Chao Li, Dongyi Bai, Manglai Dugarjaviin.
马(Equus caballus)属于马科,马属,草食性动物。据2016年联合国家畜多样性信息系统(DAD-IS)数据显示,全世界约有800多个马品种。5000~6000年前,马被人类驯服,在古代农业生产、交通运输和军事活动中发挥了关键作用,随着科技的进步,马役用价值被机器取代,现今逐渐用于休闲骑乘、观光旅游等。动物毛色多样性由于人类正向选择而产生[1],是品种登记不可或缺的内容。对史前马化石的相关研究表明,许多影响马毛色的等位基因驯化前就已经在祖先群体中表现出多态性,驯化过程引发了不同等位基因频率的改变,产生了种类繁多的马毛色表型[2],单从表型无法准确判別马毛色,使马品种登记混乱。于是,究竟哪些基因参与决定马的毛色以及表达过程如何被调控引起了科研人员极大的兴趣。此外,马毛色与健康状况相关联,对马品种选育也有着重要意义。某些特殊毛色马存活率不 高[3],如何在选育工作中利用基因筛查淘汰此类个体同样也是关注的焦点。因此,毛色相关候选基因的筛选与确定是马毛色研究在遗传学领域的关键,研究其在生物学通路中的作用与疾病的相关性可为马匹选育工作提供依据。
1 马的毛色分类
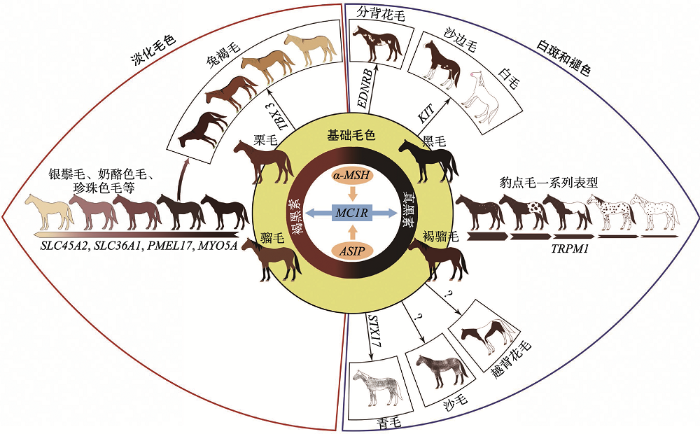
马毛分三类,即被毛、长毛和触毛。被毛是指马体表面的短毛,一年更换两次,春夏短而稀,秋冬长而密;长毛有鬃毛、尾毛等;触毛分布于嘴唇、鼻孔、眼睛周围。马的毛色主要指被毛和长毛的颜色[4]。以往的毛色分类学中,人们通常只关注马被毛多样的颜色表型,并未深究其形成的分子机理。马毛色的命名直接以肉眼观察为主,或简单地以首次发现这类毛色马的人名或主产地来命名,会导致关于马毛色的词汇在不同的语言中无法统一,在马种选育登记时造成混淆。科研人员发现,动物繁复的毛色从基因角度分类有章可循。马的毛色并不符合人们固有的认知:白色作为底色,其他颜色叠加形成;恰好相反,是由若干较深且单一的颜色被影响色素合成的修饰基因和突变位点作用而形成[5]。修饰基因对主基因有不同程度的影响,但是单独存在时不发挥作用[6]。研究表明,哺乳动物中存在着决定毛色呈黑色的主基因,而且有多种修饰基因存在,可抑制色素合成形成灰色、褐色或黄色[7]。这些修饰基因能降低色素表达量,但当主基因决定白化时,任何修饰基因都不起作用。另外,某些修饰基因还可抑制主基因的表达,如鸡(Gallus gallus)中存在能抑制色素合成的显性修饰基因[8],并且对主基因有上位效应。因此,从遗传机理的角度对马的毛色进行分类是相对科学的方法。将马的毛色分为基础毛色、淡化毛色及白斑和褪色(包括白色)3大类型。
基础毛色(base color)在马群中比较普遍,可分为骝毛(bay)、褐骝毛(seal brown)、栗毛(chestnut)和黑毛(black) (图1:A~D)。马群体中常见的颜色为骝色,骝毛马躯干颜色不一,由浅黄色到黑褐色,马鬃、马尾、耳尖和腿的下半部分为黑色。与骝毛马相比,褐骝毛马躯干偏红褐色,另外一些褐色区域分布于肋部和腿的上半部、鼻口部和眼周,其他部位为黑色。栗毛马通体为红褐色。黑毛马全身皆为黑色,胎毛褪去后出现永久黑色,没有白斑。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1马的毛色分类
A:骝毛;B:褐骝毛;C:栗毛;D:黑毛;E:银鬃毛;F:奶酪色毛;G:珍珠色毛;H:香槟色毛;I:银斑毛;J:黄兔褐毛;K:淡粉色毛;L-1~5:兔褐毛及别征;M:分背花毛;N:越背花毛;O:越背花毛;P:沙边毛;Q:白毛;R:青毛;S~W:豹点毛;X:沙毛。以上图片均引自https://en.wikipedia.org/wiki,符合CC BY 2.0、CC BY 2.5和CC BY-SA 3.0开放版权协议或属于公共领域。
Fig. 1Classification of horse coat colors
有一类修饰基因可以不同程度地抑制皮肤或毛囊中真黑素、褐黑素合成。这些基因自由组合,独立或共同发挥作用形成了马的淡化毛色(dilution),包括奶酪色毛(cream)、珍珠色毛(pearl)、香槟色毛(champagne)、银斑毛(silver)和深兔褐毛(dun)等[5]。奶酪色毛马近似白色,通常眼睛为蓝色。珍珠色毛马起源于利比里亚,躯体由浅粉色到杏黄色,伴有蓝色眼睛,大量出现于美国花马(Paints)和夸特马(Quarter)中。香槟色毛马被毛和皮肤变淡呈金色,通常有着琥珀色的眼睛。银斑毛马毛色呈银色或巧克力色,通常四肢与尾毛的颜色比身体浅(图1:E~K)。兔褐毛马是一种野生毛色,其特征是背部有一条引人注目的深色背线(图1:L-1~5)。
另一类修饰基因可在基础毛色及淡化毛色之上进行调控形成更为特殊的毛色表型,如白斑(white spotting)、褪色(depigmentation)等[9,10,11]。分背花毛马的白斑只出现在肋中部、颈部及腹侧,而不越过背中线(图1 M)。越背花毛马则相反,躯干有横贯背部与四肢的白色斑块(图1:N,O),大约占皮肤的10%。沙边毛马四肢有白斑并向上延伸,面部白斑穿过眼睛,且白斑周围有不规则的边界(图1 P)。白毛马被毛通体为白色,皮肤呈粉色,眼睛为黑色或棕色(图1 Q)。豹点毛马属于一类特殊的白斑模型,全身或局部具有大小不一的圆形或椭圆形斑点,斑点含有色素或为白色,分布没有规律(图1:S~W),多个马种皆有此类表型,如阿帕卢萨马(Appaloosa)、诺里克马(Noriker)、美国微型马(American miniature horse)、英国斑点矮马(British spotted pony)和美国矮马(pony of the Americas)[12, 13]。随年龄增加,青毛马白色被毛逐渐增多,最终颜色趋近于白色(图1R)。以往的分类将青毛马(grey)按表型直接归入白斑表型,但因其色素合成是随时间推移而衰减的,准确分类应将其归到褪色表型中。沙毛马(roan)通常表型为白毛与有色毛混杂均匀地分布于全身,头部、身体、尾部和四肢的下半部分没有白毛(图1 X)。少数个体携带可产生腿部白斑表型的基因后与沙边毛马表型极为相似,难以用肉眼区分[4,7]。与青毛马相比,典型的沙毛马白色毛的数量不会随着年龄增长而改变。还有一类特殊白斑表型出现在马的四肢或头部,因白色斑块形状特别,被称作马的别征,在马品种毛色登记时单独作为一项进行登记。白斑出现于头部,称为“额刺毛”或“星”;出现于四肢则按斑块大小和位置命名,如“踏雪”、“管白”等等[4]。
2 马毛色形成的遗传机理
2.1 产生基础毛色表型的遗传机理
基础毛色由两个基因座extension(E)和agouti(A)相互作用,最终影响黑色素细胞的功能形成[14,15]。黑色素小体(包括真黑素小体和棕黑素小体)是色素生成的细胞器,不同的基因座决定着真黑、棕黑素小体是否产生相应的色素,即真黑素(黑色、棕色相关色素)或棕黑素(红色、黄色相关色素)。E基因座编码黑素细胞皮质激素受体1 (melanochortin-1 receptor, MC1R),而A基因座编码MC1R的拮抗剂——刺鼠信号蛋白(agouti-signalling protein, ASIP)。MC1R结合α-促黑色素(α-melanocyte stimulating hormone,α-MSH)诱导剂产生真黑素,通过与拮抗剂ASIP竞争结合产生棕黑素,二者的相互作用决定合成真黑素与棕黑素的含量[16]。最新研究表明,E和A基因座不仅影响马的毛色,同时还影响马的行为[17]。骝毛马由基因座A、E共同决定,显性遗传,位于A基因座的agouti基因限制真黑色素只在马的腿部下半部、鬃、耳尖和尾部合成。若A基因座上的A等位基因突变为At(At/-)则产生了褐骝毛表型[5]。E基因座上野生型等位基因E隐性突变为e (e/e),MC1R基因功能缺失产生栗毛马[18]。Wagner等[19]发现,在黑森林马品种(Black Forest Breed)中还存在另一类隐性等位基因ea (e/ea),此突变导致该基因编码的84位氨基酸由天冬氨酸变为天冬酰胺。E基因座不同类型的基因型(e/e或e/ea)均可产生栗色表型,但可能影响栗色的深浅[20]。栗色表型对于其他基础毛色类型具有上位性,因此非栗色基础毛色马的E基因座上一定存在一个野生型等位基因E (E/-)。马22号染色体agouti基因外显子2上11 bp的缺失(g.2174-2184del)产生移码突变,导致ASIP蛋白变性失去功能,A等位基因隐性突变为a导致产生黑毛马(a/a)[20,21],基因型表型详见表1。
Table 1
表1
表1 马毛色基因型及表型
Table 1
| 毛色类别 | 基因座 | 等位基因 | 等位基因型及表型 |
|---|---|---|---|
| 基础毛色 | E | E | E/-:在皮肤及毛发中有黑色素合成 |
| e | e/e:栗毛(chestnut) | ||
| A | A | A/-:加E/-产生骝毛(bay) | |
| a | At/-:加E/-产生褐骝毛(seal Brown) | ||
| At | a/a:加E/-产生黑毛(black) | ||
| 淡化毛色 | Cr | Cr | C/C:无此类表型 |
| C | C/Cr:银鬃毛(palomino),黄兔褐毛(buckskin) | ||
| Cr/Cr:奶酪色毛(cremello),淡粉色毛(perlino) | |||
| Prl | Prl | Prl/-:无此类表型 | |
| prl | prl/prl:珍珠色毛(pearl) | ||
| Ch | Ch | CH/-:香槟色毛(champagne) | |
| ch | ch/ch:无此类表型 | ||
| Z | Z | Z/-:银斑毛(silver) | |
| z | z/z:无此类表型 | ||
| D | D | D/-:兔褐毛(dun) | |
| d | d/d:无此类表型 | ||
| 白斑和褪色 | O | O | O/o:分背花毛(overo) |
| o | o/o:无此类表型 | ||
| O/O:致死 | |||
| TO | TO | TO/-:越背花毛(tobiano) | |
| to | to/to:无此类表型 | ||
| Sb | Sb | Sb/-:沙边毛(sabino) | |
| sb | sb/sb:无此类表型 | ||
| W | W | W/W:致死 | |
| w | W/w:白毛(white) | ||
| w/w无此类表型 | |||
| LP | Lp | Lp/Lp:少量白斑 | |
| lp | Lp/lp:豹点毛(leopard complex spotting) | ||
| lp/lp:无此类表型 | |||
| G | G | G/-:青毛(grey) | |
| g | g/g:无此类表型 | ||
| Rn | Rn | Rn/Rn:致死 | |
| rn | Rn/rn:沙毛(roan) | ||
| rn/rn:无此类表型 |
新窗口打开|下载CSV
2.2 淡化毛色表型形成的遗传机理
多种毛色淡化相关基因表达不同程度地抑制了马基础毛色的色素合成途径,因而形成奶酪色毛、珍珠色毛、香槟色毛、银斑毛等(图2)。抑制色素合成的途径主要有两种,即直接抑制色素合成通路或间接抑制色素小体的运输,可以分别或同时影响真黑素和棕黑素的合成。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2基础毛色、淡化毛色及白斑和褪色表型形成的遗传机理
Fig. 2The genetic mechanism of horse coat color development
Cream (Cr)基因座决定了多类淡化毛色,半显性遗传。杂合子中棕黑素合成被抑制,毛色由棕红变黄,产生了以下毛色:银鬃毛(palomino),在帕洛米诺马中常见,浅栗色被毛伴有淡黄色或奶油色的长毛;黄兔褐毛(buckskin),被毛呈浅金色。显性纯合子颜色更浅,如奶酪色毛(cremello)和淡粉色毛(perlino),若真黑素合成同时受阻,还会产生烟酪色毛(smoky cream)[4,5]。研究表明,溶质载体45家族A2(solute carrier 45 family A2, SLC45A2)是该表型的候选基因,可编码膜相关转运蛋白(membrane-associated transporter protein, MATP),SLC45A2基因外显子2中第457个碱基G替换为A,导致编码的MATP蛋白功能缺失产生了这类表型[22,23]。但该基因的确切功能目前尚不明确。
珍珠色毛由Pearl(Prl)基因座决定,研究表明,产生此类表型的候选基因也是SLC45A2,具体突变位点目前尚未确定[24]。与奶酪色毛不同,珍珠色毛隐性遗传[5],只有在隐性纯合子中真黑素与褐黑素合成才会受阻,被毛颜色变浅;在杂合子中,被毛颜色不变,但皮肤会出现若干浅色斑点。
香槟色毛由Champagne (CH)基因座决定,显性遗传。纯合或杂合个体中,真黑素和棕黑素合成均可受到抑制,因此单从表型无法判别是纯合子还是杂合子。CH基因座定位于马14号染色体,候选基因溶质载体36A家族成员1 (solute carrier 36 family A1, SLC36A1)外显子2中的一个单核苷酸突变位点,导致编码的溶酶体氨基酸转运蛋白(lysosomal amino acid transporter 1, LYAAT1/PAT1) 第63位氨基酸由苏氨酸替换为精氨酸,从而产生了香槟色毛[25]。有研究者认为SLC36A1基因协助调节胞内pH,并参与黑色素小体的成熟。徐苹等[26]通过对中国家马的研究,发现SLC36A1基因有9种单倍型,但仍未确定突变位点与香槟色毛之间的确切关系。
银斑毛马由Silver (Z)基因座决定,真黑素合成受抑制,褐黑素合成不受影响,马毛色变淡呈现“斑驳纹”,显性遗传,显性纯合子的颜色比杂合子更 浅[27]。马Z基因座定位于6号染色体4.9 Mb区间 内[28],分别编码功能性候选基因前黑素小体蛋白17 (pre-melanosomal protein 17, PMEL17)和SILV (silver homolog)[27,29]。该毛色的形成是由于PMEL17基因外显子11上第1457个碱基由C替换为T,导致编码的第618位氨基酸由精氨酸错义突变为半胱氨 酸[27],影响了前黑素小体的合成[30,31]。此外,马先天性瞳孔异常(multiple congenital ocular anomalies, MCOA)与银斑毛表型相关。普遍认为,MCOA的主要表型为虹膜睫状体囊肿[32,33]。Andersson等[31]认为,马为杂合子时,MCOA表现为虹膜睫状体囊肿;为纯合子时,表现为瞳孔异常[34,35]。
兔褐毛马由Dun (D) 基因座决定,显性遗传。不同于其他毛色淡化相关基因的作用,色素合成受阻后,马背中线凸显出一道黑色条纹和并伴随出现其他原始别征[36]。与CH基因座类似,D基因座可同时抑制真黑素和褐黑素合成,纯合子与杂合子无法从表型上进行区分。Imsland等[37]研究发现,T-box 3(TBX3)基因是此表型的候选基因,兔褐毛马背中线处毛囊中局部高表达TBX3转录因子,导致毛囊黑色素细胞分布不均匀,使色素在毛囊中不对称沉积,毛干中分布了大量黑色素细胞和色素颗粒,背中线颜色深于体侧从而产生兔褐毛表型[37]。
除上述几种较为常见的淡化毛色外,还存在一些特殊的毛色淡化类型。如Coat Color Dilution Lethal (CCDL),又称Lavender Foal syndrome (LFS),是一种隐性遗传病,纯合子个体色素合成减少产生一类较柔和的毛色,如浅灰色、青灰色、浅栗色,有时为淡紫色。此类毛色个体在出生后可能由于神经障碍疾病致死[38]。受此疾病影响的马驹通常存在各式各样的神经性问题,如强直性癫痫、角弓反张、躯干僵硬和眼球震颤等[3],同时还经常伴随着轻度白细胞减少症[3, 39]。这些神经上的损伤致使马驹不能正常站立与饲喂,如果出生后没有立即死亡,也会对其采取安乐死。据报道,LFS最常出现于阿拉伯马的埃及亚群中[38]。Brooks等[40]于2010年完成了对LFS基因的定位与突变检测,结果表明马1号染色体上肌球蛋白VA (myosin VA, MYO5A)基因外显子30缺失(g.138235715del),产生移码突变,导致转录提前终止,致使翻译的蛋白羧基端缺失,球状尾部的促分泌囊泡-特异结合域受损伤,阻碍了转运细胞器(位于囊泡分泌特异性结合位点)结合到正确的受体上[41,42],最终囊泡运输功能异常,树突细胞如黑色素细胞和神经元细胞等无法发挥作用。
2.3 引起白斑及褪色表型的遗传机理
对马匹白斑性状遗传规律的研究开展较早[43]。白斑表型可出现在任何一种基础毛色或淡化毛色马上(图2)。这类表型的成因一般是早期胚胎发育时黑色素细胞分化与增值缺陷,影响了色素合成[44]。而褪色表型大多是由于马匹衰老过程中,色素细胞不断凋亡,色素合成持续减少所致[45]。一些白斑和褪色表型相似,仅以外表判断并不准确,在生产活动中会经常导致马毛色登记出现错误,因此DNA检测可在育种登记时可作为准确鉴定马毛色的有效手 段[46]。马体上白斑类型多种多样,而大多数表型是修饰基因导致的[44]。与其他物种类似,不同的白斑表型伴随着相应的有害多效性效应[46, 47]。分背花毛由Overo(O)基因座决定,显性纯合致死,这种现象称lethal white foal syndrome (LWFS或OLWS)。患该疾病的马驹生下来毛色接近白色并伴有肠神经节细胞缺乏症,肠道功能受阻导致其出生不久即死亡[48,49]。在夸特马(Quarter Horse)群体中具此表型的马可能还伴随着失聪等症状[50]。利用候选基因法证实此现象是由于B型内皮素受体(endothelin receptor type B,EDNRB)外显子1上第353、354个二核苷酸AG发生错义突变,导致编码的第118位氨基酸由异亮氨酸变为赖氨酸,破坏了黑色素细胞和肠神经节细胞的功能导致[51,52,53]。目前准确调控机理尚不明确,但利用DNA方法可以检测出此 突变[54]。
越背花毛存在于多个马品系,由Tobiano (TO)基因座决定,显性遗传。通常纯合子体表的白斑中散布着含色素的杂点,被称作“ink spots”或“paw prints”,但这一表型特征不能直接作为判别纯合或杂合个体的依据[5]。Smith等[55]在矮种马家族首次发现TO基因座与血蛋白基因座(Albumin, ALB)连锁。进一步研究表明,此表型与马3号染色体上保守的单倍型——Albumin(ALB)-B和Vitamin Dbinding factor (GC)-S相关联[56]。Bowling等[56]最早提出一种假设,由于马3号染色体上发生了倒位现象,导致这个区域无法发生重组,以此解释发生于TO、ALB和GC基因座之间的强连锁不平衡现象。然而20世纪末,Raudsepp等[57]的研究并未检测到这样的倒位。随后,Brooks等[10]利用FISH (fluorescence in situ hybridization)技术证实越背花毛马3号染色体上确实存在臂间倒位。倒位发生在原癌基因KIT (与多个物种的白斑表型相关)上,跨度大约47 Mb,但未影响任何编码序列[10]。小鼠中相似的染色体倒位却可以导致某些受体异常表达,最终导致黑色素细胞迁移受阻[58,59]。因此这一倒位现象的研究一直以来颇具争议,后续在美洲和欧洲马种中的研究表明,倒位与越背花毛表型相关[60]。目前产生这一表型的确切分子机理仍无定论。
沙边毛由Sabino(SB)基因座决定,其中Sabino-1 (SB-1)型是目前可阐明机理的一种突变类型,不完全显性遗传。杂合子有着典型的沙边毛表型,纯合子为白色或接近白色,只有背部有少量色素沉积。在美国,很多浅色马种均为SB-1型,尤其是Tennessee Walking马和Missouri Foxtrotter马[5]。KIT基因为该表型的候选基因,对其基因组DNA进行测序揭示了内含子16上第1037个碱基单碱基替换导致其转录时发生外显子跳读,外显子17缺失,合成的蛋白功能受损[9]。外显子跳读并不完全时虽也能合成一些正常的产物,但削弱了3′端竞争性位点的剪接力,仍可以影响蛋白功能。含同类突变的小鼠品系造血功能、配子发生及色素合成均受到影响,而SB-1型马却没有生理缺陷[61]。沙边毛马具有遗传多样性,SB-1型的分子机理不能完全解释所有的沙边毛表型[9]。KIT基因座上的其他突变位点,或另外一些基因座也可导致产生沙边毛马。例如,Clydesdale马和Shire draft马虽从外表鉴定属于沙边毛表型,但对这些群体中抽样检测并没有发现SB-1型突变位点[9]。
白毛由White(W) 基因座决定,显性遗传,具有家系特性,在美国内布拉斯加州的马种中首次发现了显性纯合致死现象[62]。Mau等[63]于2004年确定了瑞士Franches-Montagnes马KIT基因的遗传图距,随后在KIT基因中发现存在许多新的突变位点共同影响白毛表型[11,42]。在不同马品种中,共计有12种W位点突变类型,这些突变可使表型从只有少量白色斑点出现过渡到全身白色,每种突变发生的概率基本一致[44]。因此,DNA检测不适用于白毛表型。迄今为止,没有在任何马个体中发现同时存在两个显性等位基因,这也证实1969年Pulos和Hutt提出的显性纯合致死的假设是成立的[62]。在只含有一个显性等位基因的马个体中并没有发现造血功能异常等多效性效应[13]。
豹点毛是少数古老的毛色表型之一,起源于欧洲西部和东部野生型半家养马种,由Leopard complex spotting (LP)基因座决定,在距今25 000年左右的马DNA样品中能够被检测到[64]。数个修饰基因参与可使被毛从出现少量斑点过渡到全白(Few spots, Snowcap, Leopard complex spotting, Blanket) (图1:S~W)。在这些修饰基因中,与典型豹点毛(图1 U)相关的基因型称为PATN1型,定位于马3号染色体[65]。除了被毛图案,LP基因座也可决定花斑皮肤、条纹马蹄和白色巩膜等其他表型。在阿帕卢萨马群体中,显性纯合LP基因型与先天静止性夜盲症(congenital stationary night blindness, CSNB)相关[66],这类毛色马在弱光条件下表现出视觉障碍。LP基因座最初定位于马1号染色体上,瞬间受体势M亚基1 (transient receptor potential melastatin-1,TRPM1)基因被作为后续研究的功能候选基因[67,68]。利用实时荧光定量PCR分析和精准物理图谱皆证明TRPM1基因是LP和CSNB表型的候选基因[69,70]。目前在已知编码区内未检测到确切突变位点,但此表型能够在TRPM1基因内或相邻的3个SNP位点(g.108281765T C, g.108288853C T和g.108337089T G)进行DNA检测验证[71]。Thomas等[65]研究表明,TRPM1基因是不完全显性遗传,在所研究对象中有97%的豹点毛马是杂合子,且含至少一个显性等位基因。另外,在马3号染色体RFWD3 (RING finger and WD repeat domain 3)基因上的3°-UTR区,一个多肽位点(ECA3:23 658 447T>G)与PATN1型相关联可能产生豹点毛表型[65]。
青毛马由Grey (G)基因座决定,褪色的比例和部位在不同个体中皆不同。基因座定位于马25号染色体内[72,73,74]。遗传图谱最终确定褪色产生的原因是突触融合蛋白17基因(syntaxin 17, STX17)内含子6上有一个4.6 kb的重复[45,75,76]。这一重复作为顺式作用元件调控STX17和核受体NR4A家族成员3基因(nuclear receptor subfamily 4, group A, member3, NR4A3或nur77)表达上调,导致黑色素干细胞的异常增生和持续损耗[5,71]。纯合子中发生的上述序列重复还与褪色速率加快、被毛色素含量减少、皮肤褪色面积增多和黑色素瘤发生率升高密切相关[21,72,77]。
沙毛马虽然表型与沙边毛相近,但遗传机理不同。Roan (RN)基因座定位于马3号染色体上,与KIT基因相邻[48,78]。与W基因座类似,显性纯合致死[7]。精确的突变位点尚未找到,所以目前只能进行纯合性检测[46]。
此外,马头部或腿部的白色别征由多种基因和复杂的遗传过程参与决定,在Franches-Montagnes马种中发现这类表型与KIT基因具相关性[21]。
3 展 望
迄今为止,马遗传学研究中发现的候选基因多与毛色相关,育种过程中获得了大量毛色变异个体,因表型稀有具观赏性,其种质资源得以保留,这为后续的理论研究提供了非常好的样本。但事实表明,这类表型伴随着许多对马匹健康不利的遗传多效性效应,并不符合选育标准。阐明这些表型的遗传机理可为马种选育提供可靠的依据,现今已可以运用已知的毛色遗传机理制定更加合理的育种方案。国外的一些实验室已经开展了相关工作,并用于家畜疾病筛查诊断(http://www.horsetesting.com),可以在配种前对亲本马匹进行毛色相关基因检测,鉴定并推测出不同亲本交配后代的基因型或比例,进而优化配种方案,尽量排除致病基因的纯合,筛选得到更加健康的后代[46]。随着科学技术的飞速发展,不断挖掘马毛色相关新基因及突变位点,通过马基因组测序技术,基因图谱确定复杂性状有望成为现实。当越来越多毛色表型的遗传机理得以阐明,进一步解析色素相关基因之间的联系以及与其他生物学通路的关联,人们对基因功能间相互关系的理解也将更加深入。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:19148282 [本文引用: 1]

Abstract Despite having only begun approximately 10,000 years ago, the process of domestication has resulted in a degree of phenotypic variation within individual species normally associated with much deeper evolutionary time scales. Though many variable traits found in domestic animals are the result of relatively recent human-mediated selection, uncertainty remains as to whether the modern ubiquity of long-standing variable traits such as coat color results from selection or drift, and whether the underlying alleles were present in the wild ancestor or appeared after domestication began. Here, through an investigation of sequence diversity at the porcine melanocortin receptor 1 (MC1R) locus, we provide evidence that wild and domestic pig (Sus scrofa) haplotypes from China and Europe are the result of strikingly different selection pressures, and that coat color variation is the result of intentional selection for alleles that appeared after the advent of domestication. Asian and European wild boar (evolutionarily distinct subspecies) differed only by synonymous substitutions, demonstrating that camouflage coat color is maintained by purifying selection. In domestic pigs, however, each of nine unique mutations altered the amino acid sequence thus generating coat color diversity. Most domestic MC1R alleles differed by more than one mutation from the wild-type, implying a long history of strong positive selection for coat color variants, during which time humans have cherry-picked rare mutations that would be quickly eliminated in wild contexts. This pattern demonstrates that coat color phenotypes result from direct human selection and not via a simple relaxation of natural selective pressures.
URLPMID:19390039 [本文引用: 1]
The study was conducted to determine the dynamics and place of horse domestication. DNA sequence polymorphisms responsible for coat colour variation in fossil horses were analysed. Eight mutations in six genes that were responsible for coat colour variation in 89 ancient samples were successfully typed. Coat colour variation of predomestic horses were analysed using the bones of wild horses fro...
URL [本文引用: 3]
First page of article
[本文引用: 4]
3rd ed.
[本文引用: 8]
URLPMID:3443021 [本文引用: 1]
Background Greying with age in horses is an autosomal dominant trait, associated with loss of hair pigmentation, melanoma and vitiligo-like depigmentation. We recently identified a 4.665kb duplication in STX17 to be associated with the phenotype. The aims of this study were to investigate if the duplication in Grey horses shows copy number variation and to exclude that any other polymorphism is uniquely associated with the Grey mutation. Results We found little evidence for copy number expansion of the duplicated sequence in blood DNA from Grey horses. In contrast, clear evidence for copy number expansions was indicated in five out of eight tested melanoma tissues or melanoma cell lines. A tendency of a higher copy number in aggressive tumours was also found. Massively parallel resequencing of the ~35065kb Grey haplotype did not reveal any additional mutations perfectly associated with the phenotype, confirming the duplication as the true causative mutation. We identified three SNP alleles that were present in a subset of Grey haplotypes within the 35065kb region that shows complete linkage disequilibrium with the causative mutation. Thus, these three nucleotide substitutions must have occurred subsequent to the duplication, consistent with our interpretation that the Grey mutation arose more than 2,00065years before present. Conclusions These results suggest that the mutation acts as a melanoma-driving regulatory element. The elucidation of the mechanistic features of the duplication will be of considerable interest for the characterization of these horse melanomas as well as for the field of human melanoma research.
URLPMID:17247465 [本文引用: 3]
We explore racial-ethnic identity in three ways: (1) by assessing the importance of racial-ethnic identity for self-concept; (2) by comparing the importance of racial-ethnic identity in different settings (at home, in one's neighborhood, in public, at work); and (3) by contrasting the importance of racial-ethnic identity with other identities (gender, age, occupation, marital status, and social class). Data were gathered in a random national telephone survey. Results indicate that for blacks racial-ethnic identity is a more important component of self-concept than it is for multiracials and whites (both of whom say they place little importance on it). Other findings show that the importance of racial-ethnic identity varies across settings (e.g., it is most important for blacks at work and least important at home) and that in general the multiracials are not highly distinguishable from both blacks and whites. Moreover, an interesting and unexpected finding is that for all three groups, the single most important identity is sex/gender.
URL [本文引用: 1]
Birds of the White Leghorn, Barred Plymouth Rock, Rhode Island Red and New Hampshire breeds were reared and kept under comparable conditions, the diet being a standard laying mash supplemented with grain. Blood samples were taken at 3 different times, from different individuals each time. Ascorbic acid was estimated with a Beckman spectrophotometer by the method of Roe and Kuether (Abst. 261, V...
[本文引用: 4]
URLPMID:18253033 [本文引用: 3]
Tobiano is a white spotting pattern in horses caused by a dominant gene, Tobiano(TO). Here, we report TO associated with a large paracentric chromosome inversion on horse chromosome 3. DNA sequences flanking the inversion were identified and a PCR test was developed to detect the inversion. The inversion was only found in horses with the tobiano pattern, including horses with diverse genetic backgrounds, which indicated a common genetic origin thousands of years ago. The inversion does not interrupt any annotated genes, but begins approximately 100 kb downstream of the KIT gene. This inversion may disrupt regulatory sequences for the KIT gene and cause the white spotting pattern. This manuscript is accompanied by supplemental figures S1, S2 and S3, as well as supplemental Tables S1 and S2 (www.karger.com/doi/10.1159/000112065). The DNA sequence generated in this work has been submitted to GenBank under the following accession number: EF442014.
URLPMID:21554354 [本文引用: 2]
White coat colour in horses is inherited as a monogenic autosomal dominant trait showing a variable expression of coat depigmentation. Mutations in the KIT gene have previously been shown to cause white coat colour phenotypes in pigs, mice and humans. We recently also demonstrated that four independent mutations in the equine KIT gene are responsible for the dominant white coat colour phenotype in various horse breeds. We have now analysed additional horse families segregating for white coat colour phenotypes and report seven new KIT mutations in independent Thoroughbred, Icelandic Horse, German Holstein, Quarter Horse and South German Draft Horse families. In four of the seven families, only one single white horse, presumably representing the founder for each of the four respective mutations, was available for genotyping. The newly reported mutations comprise two frameshift mutations (c.1126_1129delGAAC; c.2193delG), two missense mutations (c.856G>A; c.1789G>A) and three splice site mutations (c.338-1G>C; c.2222-1G>A; c.2684+1G>A). White phenotypes in horses show a remarkable allelic heterogeneity. In fact, a higher number of alleles are molecularly characterized at the equine KIT gene than for any other known gene in livestock species.
[本文引用: 1]
URLPMID:19362501 [本文引用: 2]
The KIT receptor protein–tyrosine kinase plays an important role during embryonic development. Activation of KIT is crucial for the development of various cell lineages such as melanoblasts, stem cells of the haematopoietic system, spermatogonia and intestinal cells of Cajal. In mice, many mutations in the Kit gene cause pigmentation disorders accompanied by pleiotropic effects on blood cells and male fertility. Previous work has demonstrated that dominant white Franches–Montagnes horses carry one copy of the KIT gene with the p.Y717X mutation. The targeted breeding of white horses would be ethically questionable if white horses were known to suffer from anaemia or leukopenia. The present study demonstrates that no statistically significant differences in peripheral blood parameters are detectable between dominant white and solid-coloured Franches–Montagnes horses. The data indicate that KIT mutations may have different effects in mice, pigs, and horses. The KIT p.Y717X mutation does not have a major negative effect on the haematopoietic system of dominant white horses.
URLPMID:8948501 [本文引用: 1]
Abstract Top of page Abstract REFERENCES Important regulatory controls of melanogenesis that operate at the subcellular level to modulate the structural and/or the functional nature of the melanins and melanin granules produced in melanocytes are reviewed. Melanocyte stimulating hormone and agouti signal protein have antagonistic roles and possibly opposing mechanisms of action in the melanocyte. In the mouse, melanocyte stimulating hormone promotes melanogenic enzyme function and elicits increases in the amount of eumelanins produced, while agouti signal protein reduces total melanin production and elicits the synthesis of pheomelanin rather than eumelanin. We are now beginning to understand the complex controls involved in regulating this switch at the molecular and biochemical levels. The quality and quantity of melanins produced by melanocytes have important physiological consequences for melanocyte function and undoubtedly play important roles in the various functions of the melanins per se, including hair and skin coloration and photoprotection.
URLPMID:14616056 [本文引用: 1]
Differences in skin and hair color are principally genetically determined and are due to variation in the amount, type, and packaging of melanin polymers produced by melanocytes secreted into keratinocytes. Pigmentary phenotype is genetically complex and at a physiological level complicated. Genes determining a number of rare Mendelian disorders of pigmentation such as albinism have been identified, but only one gene, the melanocortin 1 receptor (MCR1), has so far been identified to explain variation in the normal population such as that leading to red hair, freckling, and sun-sensitivity. Genotype-phenotype relations of the MC1R are reviewed, as well as methods to improve the phenotypic assessment of human pigmentary status. It is argued that given advances in model systems, increases in technical facility, and the lower cost of genotype assessment, the lack of standardized phenotype assessment is now a major limit on advance.
URLPMID:5506451 [本文引用: 1]
Horses have substantial variation in coat color, and the genetic loci responsible for the coat color variations have been well investigated. It has been believed that some color variations should follow a single-locus Mendelian law. Examples include the Gray locus that causes the gray phenotype and the Extension locus that specifies the chestnut phenotype. We reevaluated the roles of the Gray and Extension loci by using a large number of mating records of Thoroughbred racing horses. We showed that the data indeed fits the Mendelian law extremely well for the two loci. Furthermore, we demonstrated that the Extension and Agouti loci might have an additional role in determining the degree of melanin that should distinguish bay, dark bay, and brown.
URLPMID:26884605 [本文引用: 1]
Abstract Shared signaling pathways utilized by melanocytes and neurons result in pleiotropic traits of coat color and behavior in many mammalian species. For example, in humans polymorphisms at MC1R cause red hair, increased heat sensitivity, and lower pain tolerance. In deer mice, rats, and foxes, ASIP polymorphisms causing black coat color lead to more docile demeanors and reduced activity. Horse (Equus caballus) base coat color is primarily determined by polymorphisms at the Melanocortin-1 Receptor (MC1R) and Agouti Signaling Protein (ASIP) loci, creating a black, bay, or chestnut coat. Our goal was to investigate correlations between genetic loci for coat color and temperament traits in the horse. We genotyped a total of 215 North American Tennessee Walking Horses for the 2 most common alleles at the MC1R (E/e) and ASIP (A/a) loci using previously published PCR and RFLP methods. The horses had a mean age of 10.5 years and comprised 83 geldings, 25 stallions, and 107 mares. To assess behavior, we adapted a previously published survey for handlers to score horses from 1 to 9 on 20 questions related to specific aspects of temperament. We utilized principle component analysis to combine the individual survey scores into 4 factors of variation in temperament phenotype. A factor component detailing self-reliance correlated with genotypes at the ASIP locus; black mares (aa) were more independent than bay mares (A_) (P = 0.0063). These findings illuminate a promising and novel animal model for future study of neuroendocrine mechanisms in complex behavioral phenotypes.
URLPMID:8995760 [本文引用: 1]
The melanocyte-stimulating hormone receptor gene (MCIR) is the major candidate gene for the chestnut coat color in horses since it is assumed to be controlled by an allele at the extension locus. MCIR sequences were PCR amplified from chestnut (e/e) and non-chestnut (EI-) horses. A single-strand conformation polymorphism was found that showed a complete association to the chestnut coat color among 144 horses representing 12 breeds. Sequence analysis revealed a single missense mutation (83Ser 鈫 Phe) in the MCIR allele associated with the chestnut color. The substitution occurs in the second transmembrane region, which apparently plays a key role in the molecule since substitutions associated with coat color variants in mice and cattle as well as red hair and fair skin in humans are found in this part of the molecule. We propose that the now reported mutation is likely to be the causative mutation for the chestnut coat color. The polymorphism can be detected with a simple PCR-RFLP test, since the mutation creates a TaqI restriction site in the chestnut allele.
URL [本文引用: 1]
No abstract is available for this article.
URLMagsci [本文引用: 2]
毛色不仅是马品种和个体识别的重要依据, 而且还可以作为某些疾病筛查的有力工具和手段。马的毛色主要由黑色素细胞产生的真黑素和褐黑素两种黑色素的分布及比例所决定, 许多基因对黑色素的产生和分布的调控起着重要的作用, 各基因相互间共同作用最终形成各种单毛色和复毛色, 这些基因主要包括<em>MC1R、ASIP、KIT、TYRP</em>和<em>EDNRB</em>。另外<em>STX17、MATP</em>和<em>PMEL17</em>也在马毛色形成过程具有重要的作用, 同时还发现个别毛色基因与黑色素瘤疾病有关。文章对近年来马主要毛色候选基因的作用机理、DNA序列多态性与毛色性状及黑色素瘤疾病的关系等研究进行了详细的阐述, 为今后马匹育种工作和疾病防治提供重要理论依据。
URLMagsci [本文引用: 2]
毛色不仅是马品种和个体识别的重要依据, 而且还可以作为某些疾病筛查的有力工具和手段。马的毛色主要由黑色素细胞产生的真黑素和褐黑素两种黑色素的分布及比例所决定, 许多基因对黑色素的产生和分布的调控起着重要的作用, 各基因相互间共同作用最终形成各种单毛色和复毛色, 这些基因主要包括<em>MC1R、ASIP、KIT、TYRP</em>和<em>EDNRB</em>。另外<em>STX17、MATP</em>和<em>PMEL17</em>也在马毛色形成过程具有重要的作用, 同时还发现个别毛色基因与黑色素瘤疾病有关。文章对近年来马主要毛色候选基因的作用机理、DNA序列多态性与毛色性状及黑色素瘤疾病的关系等研究进行了详细的阐述, 为今后马匹育种工作和疾病防治提供重要理论依据。
URLPMID:18296388 [本文引用: 3]
Abstract White markings and spotting patterns in animal species are thought to be a result of the domestication process. They often serve for the identification of individuals but sometimes are accompanied by complex pathological syndromes. In the Swiss Franches-Montagnes horse population, white markings increased vastly in size and occurrence during the past 30 years, although the breeding goal demands a horse with as little depigmented areas as possible. In order to improve selection and avoid more excessive depigmentation on the population level, we estimated population parameters and breeding values for white head and anterior and posterior leg markings. Heritabilities and genetic correlations for the traits were high (h(2) > 0.5). A strong positive correlation was found between the chestnut allele at the melanocortin-1-receptor gene locus and the extent of white markings. Segregation analysis revealed that our data fit best to a model including a polygenic effect and a biallelic locus with a dominant-recessive mode of inheritance. The recessive allele was found to be the white trait-increasing allele. Multilocus linkage disequilibrium analysis allowed the mapping of the putative major locus to a chromosomal region on ECA3q harboring the KIT gene.
URLPMID:11736803 [本文引用: 1]
The colour locus historically referred to as C in the horse is linked to microsatellites markers on horse chromosome 21. Preliminary results demonstrated linkage of C cr, thought to be the cream dilution variant of the C locus, to HTG10 . An analysis of horse chromosome 21 using additional families confirmed and established a group of markers linked to C cr. This work also improved the resolution of previously reported linkage maps for this chromosome. Linkage analysis unambiguously produced the map order: SGCV16 -(19.102cM)- HTG10- (3.802cM)- LEX60/COR73- (1.302cM)- COR68- (4.502cM)- C cr-(11.902cM)- LEX31 . Comparative and synteny data suggested that the horse C locus is not tyrosinase ( TYR ).
URL [本文引用: 1]
URLPMID:27194311 [本文引用: 1]
Abstract Considering the hidden mode of inheritance of some coat-color-associated alleles, we investigated the presence/absence of coat-color-associated alleles in 1093 domestic horses of 55 breeds and 20 specimens of Przewalski's horse. For coat-color genotyping, allele specific PCR, pyrosequencing and Li-Cor analyses were conducted on 12 coat-color-associated alleles of five genes. Our data provide deep insight into the distribution of coat-color-associated alleles within breeds. We found that the alleles for the basic colorations (bay, black, and chestnut) are widely distributed and occur in nearly all breeds. Alleles leading to dilutions or patterns are rare in domestic breeds and were not found in Przewalski's horse. Higher frequencies of these alleles are only found in breeds that are selected for their expressed phenotypes (e.g., Kinsky horse, Lewitzer, Tinker). Nevertheless, our study produced strong evidence that molecular testing of the coat color is necessary for well-defined phenotyping to avoid unexpected colorations of offspring that can result in legal action.
URLPMID:2535566 [本文引用: 1]
Abstract Champagne coat color in horses is controlled by a single, autosomal-dominant gene (CH). The phenotype produced by this gene is valued by many horse breeders, but can be difficult to distinguish from the effect produced by the Cream coat color dilution gene (CR). Three sires and their families segregating for CH were tested by genome scanning with microsatellite markers. The CH gene was mapped within a 6 cM region on horse chromosome 14 (LOD = 11.74 for theta = 0.00). Four candidate genes were identified within the region, namely SPARC [Secreted protein, acidic, cysteine-rich (osteonectin)], SLC36A1 (Solute Carrier 36 family A1), SLC36A2 (Solute Carrier 36 family A2), and SLC36A3 (Solute Carrier 36 family A3). SLC36A3 was not expressed in skin tissue and therefore not considered further. The other three genes were sequenced in homozygotes for CH and homozygotes for the absence of the dilution allele (ch). SLC36A1 had a nucleotide substitution in exon 2 for horses with the champagne phenotype, which resulted in a transition from a threonine amino acid to an arginine amino acid (T63R). The association of the single nucleotide polymorphism (SNP) with the champagne dilution phenotype was complete, as determined by the presence of the nucleotide variant among all 85 horses with the champagne dilution phenotype and its absence among all 97 horses without the champagne phenotype. This is the first description of a phenotype associated with the SLC36A1 gene.
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
URLPMID:2653074 [本文引用: 1]
No abstract available.
URLPMID:17257181 [本文引用: 1]
In horses, a pigment dilution acting only on black eumelanin is the so-called silver coat colour, which is characterized by a chocolate-to-reddish body with a white mane and tail. Using information from other species, we focused our study on SILV as a possible candidate gene for the equine silver phenotype. A 1559-bp genomic fragment was sequenced in 24 horses, and five SNPs were detected. Two of the five SNPs (DQ665301:g.697A>T and DQ665301:g.1457C>T) were genotyped in 112 horses representing eight colour phenotypes. Both mutations were completely associated with the silver phenotype: all eumelanin-producing horses (blacks and bays) with atypical white mane and tail were carriers of the [g.697T; g.1457T] haplotype. We identified this haplotype as well as the silver phenotype only in Shetland ponies and Icelandic horses. Horses without eumelanin (chestnuts) were carriers of the [g.697T; g.1457T] haplotype, but they showed no phenotypic effect. The white or flaxen mane often detected in chestnuts is presumably based on another SILV mutation or on polymorphisms in other genes.
URLPMID:15695812 [本文引用: 1]
Abstract More than 125 genes that regulate pigmentation have been identified to date. Of those, MART-1 has been widely studied as a melanoma-specific antigen and as a melanosome-specific marker. Whereas the functions of other melanosomal proteins, such as tyrosinase, tyrosinase-related protein-1, dopachrome tautomerase, and Pmel17, are known, the function of MART-1 in melanogenesis, is unclear. A role for MART-1 in pigmentation is expected because its expression pattern and subcellular distribution is quite similar to the other melanosomal proteins and usually correlates with melanin content. We investigated the function of MART-1 using a multidisciplinary approach, including the use of siRNA to inhibit MART-1 function and the use of transfection to re-express MART-1 in MART-1-negative cells. We show that MART-1 forms a complex with Pmel17 and affects its expression, stability, trafficking, and the processing which is required for melanosome structure and maturation. We conclude that MART-1 is indispensable for Pmel17 function and thus plays an important role in regulating mammalian pigmentation.
URL [本文引用: 2]
URLPMID:10768120 [本文引用: 1]
Anterior segment dysgenesis syndrome occurs frequently in Rocky Mountain horses and has two distinct ocular phenotypes: (1) large cysts originating from the temporal ciliary body or peripheral retina and (2) multiple anterior segment anomalies including ciliary cysts, iris hypoplasia, iridocorneal adhesions and opacification, nuclear cataract, and megalocornea. To determine if anterior segment dysgenesis syndrome is heritable in horses we performed ophthalmic examinations and collected pedigree information on horses (n = 516) in an extended Rocky Mountain horse pedigree. Logistic regressive segregation analysis of a subset of animals (n = 337) in which the ocular phenotypes of progeny and both parents were known indicated that the codominant inheritance model best fit the data. This model predicted cyst phenotype expression in heterozygous animals and multiple anterior segment anomalies in homozygous animals. Several cases of nonpenetrance of the cyst phenotype were detected in one lineage. The close resemblance between the inheritance and lesions observed in Small eye mice and rats, humans with congenital aniridia or anterior segment malformation, and horses with anterior segment dysgenesis syndrome supported the conclusion that anterior segment dysgenesis syndrome in the horse may be homologous to similar ophthalmic anomalies in other species.
URLPMID:18827844 [本文引用: 1]
ABSTRACT Multiple congenital ocular anomalies in purebred and crossbred Rocky and Kentucky Mountain horses in Canada are frequently diagnosed with biomicroscopic and indirect ophthalmoscopic examination. In order of frequency detected, these include temporal ciliary epithelial cysts; iridal hypoplasia; prominent corneas; focal temporal retinal degeneration related to ciliary cysts; and, rarely, retinal detachment. A pedigree analysis confirms a dominant mode of inheritance with incomplete penetrance and with a linkage to coat color.
URLPMID:3781063 [本文引用: 1]
Equine Multiple Congenital Ocular Anomalies (MCOA) syndrome is a heritable eye disorder mainly affecting silver colored horses. Clinically, the disease manifests in two distinct classes depending on the horse genotype. Horses homozygous for the mutant allele present with a wide range of ocular defects, such as iris stromal hypoplasia, abnormal pectinate ligaments, megaloglobus, iridociliary cysts and cataracts. The phenotype of heterozygous horses is less severe and predominantly includes iridociliary cysts, which occasionally extend into the temporal retina. In order to determine the genetic cause of MCOA syndrome we sequenced the entire previously characterized 208 kilobase region on chromosome 6 in ten individuals; five MCOA affected horses from three different breeds, one horse with the intermediate Cyst phenotype and four unaffected controls from two different breeds. This was performed using Illumina TruSeq technology with paired-end reads. Through the systematic exclusion of all polymorphisms barring two SNPs in PMEL, a missense mutation previously reported to be associated with the silver coat colour and a non-conserved intronic SNP, we establish that this gene is responsible for MCOA syndrome. Our finding, together with recent advances that show aberrant protein function due to the coding mutation, suggests that the missense mutation is causative and has pleiotrophic effect, causing both the horse silver coat color and MCOA syndrome.
URL [本文引用: 1]
SummaryMultiple congenital ocular anomalies (MCOA) and their relation to coat colour genotype have not yet been described in Comtois horses, unlike in Rocky Mountain Horses. The objectives of the study were to describe prevalence, nature and severity of congenital ocular anomalies relating to the PMEL17 (Silver) mutation in Comtois horses. Seventy-four purebred Comtois and one half-cross Comtois horses, aged 10 days to 18 years, were examined by transillumination, direct ophthalmoscopy and ultrasonography. Hair samples were collected from 34 horses for coat colour genotyping. Sixty-six horses (88%) revealed cysts (65 horses) or abnormal thickness (one horse) of the ciliary bodies, most of them only diagnosed by ultrasonography. Cysts were localised in the nasal part of the eye in 8 horses. All these horses presented the silver phenotype with mane and tail being white or flaxen, or were chestnut with genetic testing confirming PMEL17 mutation. Of these, 39 (58%) showed MCOA-syndrome with iridal hypoplasia (100%), cataract (85%), cornea globosa (56%) and lens luxation (8%). Only 8 bay mature horses (11%) were classified as being disease-free. Genetic testing confirmed that cyst-phenotype horses were heterozygous carriers for the Silver mutation, MCOA-phenotype horses were homozygous carriers, and bay horses were noncarriers. Bay homozygous carriers had significantly lighter coat colour than heterozygous carriers. One foal with heterozygous mutation had normal eyes. Thus, MCOA-syndrome related to PMEL17 mutation is overrepresented in Comtois horses, and should be taken into consideration for breeding purposes. Ultrasonography permitted detection of cysts in all Silver carriers apart from one, some of them being localised in the nasal part of the eye.
URLPMID:731005 [本文引用: 1]
Abstract The coat colors of 161 progeny from matings between 10 yellow dun and 6 blue dun stallions and mares of 8 different colors are described. The results confirm the previous hypothesis that a dominant dilution gene, D, converts bay to yellow dun with dark mane and tail, chestnut to yellow dun and dun mane and tail, and black to blue dun (mouse, grullo). The palomino gene, c cr, on the other hand, is hypostatic to black and blue dun. In heterozygous form, c cr converts bay to buckskin, and chestnut and sorrel to palomino, and results in blue-eyed white when homozygous. No particular effect of D is known in the homozygous state. Altogether 12 progeny were obtained from matings where both parents carried D; all progeny carried D, and no abnormal colors occurred.
URLPMID:4731265 [本文引用: 2]
Abstract Dun is a wild-type coat color in horses characterized by pigment dilution with a striking pattern of dark areas termed primitive markings. Here we show that pigment dilution in Dun horses is due to radially asymmetric deposition of pigment in the growing hair caused by localized expression of the T-box 3 (TBX3) transcription factor in hair follicles, which in turn determines the distribution of hair follicle melanocytes. Most domestic horses are non-dun, a more intensely pigmented phenotype caused by regulatory mutations impairing TBX3 expression in the hair follicle, resulting in a more circumferential distribution of melanocytes and pigment granules in individual hairs. We identified two different alleles (non-dun1 and non-dun2) causing non-dun color. non-dun2 is a recently derived allele, whereas the Dun and non-dun1 alleles are found in ancient horse DNA, demonstrating that this polymorphism predates horse domestication. These findings uncover a new developmental role for T-box genes and new aspects of hair follicle biology and pigmentation.
.
[本文引用: 2]
URLPMID:17186871 [本文引用: 1]
Author information: (1)Department of Companion Animal Clinical Studies Faculty of Veterinary Science, University of Pretoria, Onderstepoort, South Africa.
URLPMID:2855325 [本文引用: 1]
Lavender Foal Syndrome (LFS) is a lethal inherited disease of horses with a suspected autosomal recessive mode of inheritance. LFS has been primarily diagnosed in a subgroup of the Arabian breed, the Egyptian Arabian horse. The condition is characterized by multiple neurological abnormalities and a dilute coat color. Candidate genes based on comparative phenotypes in mice and humans include the ras-associated protein RAB27a (RAB27A) and myosin Va (MYO5A). Here we report mapping of the locus responsible for LFS using a small set of 36 horses segregating for LFS. These horses were genotyped using a newly available single nucleotide polymorphism (SNP) chip containing 56,402 discriminatory elements. The whole genome scan identified an associated region containing these two functional candidate genes. Exon sequencing of theMYO5Agene from an affected foal revealed a single base deletion in exon 30 that changes the reading frame and introduces a premature stop codon. A PCR鈥揵ased Restriction Fragment Length Polymorphism (PCR FLP) assay was designed and used to investigate the frequency of the mutant gene. All affected horses tested were homozygous for this mutation. Heterozygous carriers were detected in high frequency in families segregating for this trait, and the frequency of carriers in unrelated Egyptian Arabians was 10.3%. The mapping and discovery of the LFS mutation represents the first successful use of whole-genome SNP scanning in the horse for any trait. The RFLP assay can be used to assist breeders in avoiding carrier-to-carrier matings and thus in preventing the birth of affected foals. Genetic disorders affect many domesticated species, including the horse. In this study we have focused on Lavender Foal Syndrome, a seizure disorder that leads to suffering and death in foals soon after birth. A recessively inherited disorder, its occurrence is often unpredictable and difficult for horse breeders to avoid without a diagnostic test for carrier status. The recent completion of the horse genome sequence has provided new tools for mapping traits with unprecedented resolution and power. We have applied one such tool, the Equine SNP50 genotyping chip, to a small sample set from horses affected with Lavender Foal Syndrome. A single genetic location associated with the disorder was rapidly identified using this approach. Subsequent sequencing of functional candidate genes in this location revealed a single base deletion that likely causes Lavender Foal Syndrome. From a practical standpoint, this discovery and the development of a diagnostic test for the LFS allele provides a valuable new tool for breeders seeking to avoid the disease in their foal crop. However, this work also illustrates the utility of whole-genome association studies in the horse.
URLPMID:15684027 [本文引用: 1]
The myosin V carboxyl -terminal globular tail domain is essential for the attachment of myosin V to all known cargoes. Previously, the globular tail was viewed as a single, functional entity. Here, we show that the globular tail of the yeast myosin Va homologue, Myo2p, contains two structural subdomains that have distinct functions, namely, vacuole-specific and secretory vesicle-specific movement. Biochemical and genetic analyses demonstrate that subdomain I tightly associates with subdomain II, and that the interaction does not require additional proteins. Importantly, although neither subdomain alone is functional, simultaneous expression of the separate subdomains produces a functional complex in vivo. Our results suggest a model whereby intramolecular interactions between the globular tail subdomains help to coordinate the transport of multiple distinct cargoes by myosin V.
URLPMID:2175197 [本文引用: 2]
The Saccharomyces cerevisiae myosin-V, Myo2p, is essential for polarized growth, most likely through transport of secretory vesicles to the developing bud. Myo2p is also required for vacuole movement, a process not essential for growth. The globular region of the myosin-V COOH-terminal tail domain is proposed to bind cargo. Through random mutagenesis of this globular tail, we isolated six new single point mutants defective in vacuole inheritance, but not polarized growth. These point mutations cluster to four amino acids in an 11-amino acid span, suggesting that this region is important for vacuole movement. In addition, through characterization of myo2-DeltaAflII, a deletion of amino acids 1,459-1,491, we identified a second region of the globular tail specifically required for polarized growth. Whereas this mutant does not support growth, it complements the vacuole inheritance defect in myo2-2 (G1248D) cells. Moreover, overexpression of the myo2-DeltaAflII globular tail interferes with vacuole movement, but not polarized growth. These data indicate that this second region is dispensable for vacuole movement. The identification of these distinct subdomains in the cargo-binding domain suggests how myosin-Vs can move multiple cargoes. Moreover, these studies suggest that the vacuole receptor for Myo2p differs from the receptor for the essential cargo.
URLPMID:2065884 [本文引用: 1]
White coat color has been a highly valued trait in horses for at least 2,000 years. Dominant white (W) is one of several known depigmentation phenotypes in horses. It shows considerable phenotypic variation, ranging from 50% depigmented areas up to a completely white coat. In the horse, the four depigmentation phenotypes roan, sabino, tobiano, and dominant white were independently mapped to a chromosomal region on ECA 3 harboring theKITgene. KIT plays an important role in melanoblast survival during embryonic development. We determined the sequence and genomic organization of the 82 kb equineKITgene. A mutation analysis of all 21KITexons in white Franches-Montagnes Horses revealed a nonsense mutation in exon 15 (c.2151C>G, p.Y717X). We analyzed theKITexons in horses characterized as dominant white from other populations and found three additional candidate causative mutations. Three almost completely white Arabians carried a different nonsense mutation in exon 4 (c.706A>T, p.K236X). Six Camarillo White Horses had a missense mutation in exon 12 (c.1805C>T, p.A602V), and five white Thoroughbreds had yet another missense mutation in exon 13 (c.1960G>A, p.G654R). Our results indicate that the dominant white color in Franches-Montagnes Horses is caused by a nonsense mutation in theKITgene and that multiple independent mutations within this gene appear to be responsible for dominant white in several other modern horse populations. White horses have always been highly valued by their human owners. Their important role in history is reflected by their widespread use as heraldic animals (e.g., on the flags of the German states of Lower Saxony and North Rhine-Westphalia). In the Swiss Franches-Montagnes Horse population, a completely white mare named Cigale was born out of solid brown parents in 1957. The white phenotype is inherited as an autosomal dominant trait and all living white Franches-Montagnes Horses are descendants of Cigale. We sequenced theKITgene in white and solid-colored Franches-Montagnes Horses and found a mutation that inactivates the gene product and thus leads to a lack of pigment-forming cells in the skin of white horses. We then analyzed white horses from other populations and found three additional independent candidate causative mutations in white Thoroughbreds, Arabians, and Camarillo White Horses. The research thus revealed independent mutation events leading to white coat color in different horse populations. Our findings will allow genetic testing and a more precise classification of horses with white coat color.
URLMagsci [本文引用: 3]
马毛色是品种鉴定和个体识别的重要依据, 也是制定育种方案时必须考虑的重要性状之一。因此, 研究马被毛褪色已成为当今国际马毛色研究领域的重要内容, 试图弄清导致马被毛褪色的真正机理。目前已经发现, 许多马种被毛褪色表型个体中3号染色体上的<em>kit</em>基因存在不同的显著突变。研究结果表明马<em>kit</em>基因的正常表达与否与表皮中黑色素细胞及黑色素的形成密切相关, 从而控制是否出现褪色表型。然而, 研究证明在不同马种间褪色表型个体在该位点上出现的突变存在着较大的种间差异。具有被毛完全褪色表型的马群非常少见, 只是偶尔见于有些马种, 但在内蒙古锡林郭勒盟西乌珠穆沁旗生存着较大数量的被毛褪色表型个体, 被称为蒙古白马。然而, 造成其被毛褪色的机理还没有得到证实, 有趣的是至今为止在蒙古白马<em>kit</em>基因的21个外显子中还没有发现任何典型突变。因此, 文章对近些年国际上对马被毛褪色的分子研究进展做一比较系统的综合叙述, 为蒙古白马毛色形成的机理研究奠定基础, 为今后的马匹毛色研究及其育种工作提供有价值的参考依据。
URLMagsci [本文引用: 3]
马毛色是品种鉴定和个体识别的重要依据, 也是制定育种方案时必须考虑的重要性状之一。因此, 研究马被毛褪色已成为当今国际马毛色研究领域的重要内容, 试图弄清导致马被毛褪色的真正机理。目前已经发现, 许多马种被毛褪色表型个体中3号染色体上的<em>kit</em>基因存在不同的显著突变。研究结果表明马<em>kit</em>基因的正常表达与否与表皮中黑色素细胞及黑色素的形成密切相关, 从而控制是否出现褪色表型。然而, 研究证明在不同马种间褪色表型个体在该位点上出现的突变存在着较大的种间差异。具有被毛完全褪色表型的马群非常少见, 只是偶尔见于有些马种, 但在内蒙古锡林郭勒盟西乌珠穆沁旗生存着较大数量的被毛褪色表型个体, 被称为蒙古白马。然而, 造成其被毛褪色的机理还没有得到证实, 有趣的是至今为止在蒙古白马<em>kit</em>基因的21个外显子中还没有发现任何典型突变。因此, 文章对近些年国际上对马被毛褪色的分子研究进展做一比较系统的综合叙述, 为蒙古白马毛色形成的机理研究奠定基础, 为今后的马匹毛色研究及其育种工作提供有价值的参考依据。
URLPMID:16167981 [本文引用: 2]
Summary Grey horses are born coloured, turn progressively grey and often develop melanomas late in life. Grey shows an autosomal dominant inheritance and the locus has previously been mapped to horse chromosome 25 (ECA25), around the TXN gene. We have now developed eight new single nucleotide polymorphisms (SNPs) associated with genes on ECA25 using information on the linear order of genes on human chromosome 9q, as well as the human and mouse coding sequences. These SNPs were mapped in relation to the Grey locus using more than 300 progeny from matings between two Swedish Warmblood grey stallions and non-grey mares. Grey was firmly assigned to an interval with flanking markers NANS and ABCA1. This corresponds to a region of approximately 6.9Mb on human chromosome 9q. Furthermore, no recombination was observed between Grey , TGFBR1 and TMEFF1 , the last two being 1.4Mb apart in human. There are no obvious candidate genes in this region and none of the genes has been associated with pigmentation disorders or melanoma development, suggesting that the grey phenotype is caused by a mutation in a novel gene.
URLPMID:19912415 [本文引用: 4]
Abstract Colour phenotypes may have played a major role during early domestication events and initial selection among domestic animal species. As coat colours mostly follow a relatively simple mode of Mendelian inheritance, they have been among the first traits to be systematically analysed at the molecular level. As a result of the number of genetic tools developed during the past decade, horse coat colour tests have been designed and are now commercially available for some of the basic phenotypes. These tests enable breeders to verify segregation within particular pedigrees, to select specific colour phenotypes according to market demand or studbook policies and to avoid complex inherited diseases associated with some of the colour patterns. This paper reviews the relevance of the topic, describes all currently available tests for coat colours in horses and addresses also ongoing research in this field.
URL [本文引用: 1]
URLPMID:7096983 [本文引用: 2]
Abstract The equine coat color genes chestnut (e) and roan (Rn) have been tested for linkage to 15 protein and blood group loci. Data showing close or fairly close linkage to the serum albumin locus (Al) and loose linkage to the serum esterase locus (Es) for both e and Rn are presented. This means that three coat color genes (To, e and Rn) and three serum protein loci (Al, Gc, and Es) are linked in the same linkage group. The gene order can tentatively be written Al, Gc, Rn, To-e-Es. The implications of the results for studies on coat color inheritance in horses are discussed. The possibility of using electrophoretic markers when testing hypotheses of allelism between coat color genes is suggested. The linkage of e and Es in the horse is proposed to be homologous to the loose linkage of the extension locus (e) and a cluster of esterase loci on chromosome 8 in the mouse, and on linkage group IV in the rabbit. Designations for the known autosomal linkage groups in the horse are suggested.
URLPMID:2363434 [本文引用: 1]
The lethal white foal syndrome (LWFS) is a congenital abnormality of overo spotted horses which is a model for human aganglionic megacolon or Hirschsprung disease. Foals with LWFS have an all white, or nearly all white, coat. They also present clinically with an intestinal obstruction that proves fatal within the first few days of life. The LWFS involves both melanocytes and intestinal ganglion cells, and appears to result from a genetic defect involving neural crest cells. This report describes pathologic studies of two recent cases of LWFS. Two different hypothetical models of inheritance of LWFS are presented and discussed.
URLPMID:19912043 [本文引用: 1]
To evaluate deafness in American Paint Horses by phenotype, clinical findings, brainstem auditory-evoked responses (BAERs), and endothelin B receptor (EDNBR) genotype.Case series and case-control studies.14 deaf American Paint Horses, 20 suspected-deaf American Paint Horses, and 13 nondeaf American Paint Horses and Pintos.Horses were categorized on the basis of coat color pattern and eye color. Testing for the EDNBR gene mutation (associated with overo lethal white foal syndrome) and BAERs was performed. Additional clinical findings were obtained from medical records.All 14 deaf horses had loss of all BAER waveforms consistent with complete deafness. Most horses had the splashed white or splashed white-frame blend coat pattern. Other patterns included frame overo and tovero. All of the deaf horses had extensive head and limb white markings, although the amount of white on the neck and trunk varied widely. All horses had at least 1 partially heterochromic iris, and most had 2 blue eyes. Ninety-one percent (31/34) of deaf and suspected-deaf horses had the EDNBR gene mutation. Deaf and suspected-deaf horses were used successfully for various performance events. All nondeaf horses had unremarkable BAER results.Veterinarians should be aware of deafness among American Paint Horses, particularly those with a splashed white or frame overo coat color pattern, blend of these patterns, or tovero pattern. Horses with extensive head and limb markings and those with blue eyes appeared to be at particular risk.
URLPMID:9585428 [本文引用: 1]
Lethal White Foal Syndrome is a disease associated with horse breeds that register white coat spotting patterns. Breedings between particular spotted horses, generally described as frame overo, produce some foals that, in contrast to their parents, are all white or nearly all white and die shortly after birth of severe intestinal blockage. These foals have aganglionosis characterized by a lack of submucosal and myenteric ganglia from the distal small intestine to the large intestine, similar to human Hirschsprung Disease. Some sporadic and familial cases of Hirschsprung Disease are due to mutations in the endothelin B receptor gene (EDNRB). In this study, we investigate the role of EDNRB in Lethal White Foal Syndrome. A cDNA for the wild-type horse endothelin-B receptor gene was cloned and sequenced. In three unrelated lethal white foals, the EDNRB gene contained a 2-bp nucleotide change leading to a missense mutation (I118K) in the first transmembrane domain of the receptor, a highly conserved region of this protein among different species. Seven additional unrelated lethal white foal samples were found to be homozygous for this mutation. No other homozygotes were identified in 138 samples analyzed, suggesting that homozygosity was restricted to lethal white foals. All (40/40) horses with the frame overo pattern (a distinct coat color pattern that is a subset of overo horses) that were tested were heterozygous for this allele, defining a heterozygous coat color phenotype for this mutation. Horses with tobiano markings included some carriers, indicating that tobiano is epistatic to frame overo. In addition, horses were identified that were carriers but had no recognized overo coat pattern phenotype, demonstrating the variable penetrance of the mutation. The test for this mutant allele can be utilized in all breeds where heterozygous animals may be unknowingly bred to each other including the Paint Horse, Pinto horse, Quarter Horse, Miniature Horse, and Thoroughbred.
URLPMID:9530628 [本文引用: 1]
Abstract Overo lethal white syndrome (OLWS) is an inherited syndrome of foals born to American Paint Horse parents of the overo coat-pattern lineage. Affected foals are totally or almost totally white and die within days from complications due to intestinal aganglionosis. Related conditions occur in humans and rodents in which mutations in the endothelin receptor B (EDNRB) gene are responsible. EDNRB is known to be involved in the developmental regulation of neural crest cells that become enteric ganglia and melanocytes. In this report we identify a polymorphism in the equine EDNRB gene closely associated with OLWS. This Ile to Lys substitution at codon 118 is located within the first transmembrane domain of this seven-transmembrane domain G-protein-coupled receptor protein. All 22 OLWS-affected foals examined were homozygous for the Lys118 EDNRB allele, while all available parents of affected foals were heterozygous. All but one of the parents also had an overo white body-spot phenotype. Solid-colored control horses of other breeds were homozygous for the Ile118 EDNRB allele. Molecular definition of the basis for OLWS in Paint Horses provides a genetic test for the presence of the Lys118 EDNRB allele and adds to our understanding of the basis for coat color patterns in the horse.
URLPMID:9580670 [本文引用: 1]
Lethal white foal syndrome (LWFS) is a congenital anomaly of horses characterized by a white coat colour and aganglionosis of the bowel, which is similar to Hirschsprung disease (HSCR). We decided to investigate possible mutations of the endothelin-B receptor gene ( EDNRB ) in LWFS as recent studies in mutant rodents and some patients have demonstrated EDNRB defects. First, we identified a full-length cDNA for horse EDNRB . This cDNA fragment contained a 1329 bp open reading frame which encoded 443 amino acid residues. The predicted amino acid sequence was 89, 91 and 85% identical to human, bovine and mouse as well as rat EDNRB respectively, but only 55% identical to the human, bovine and rat endothelin A receptor (EDNRA). Secondly, sequence analysis, together with allele-specific PCR and the amplification-created restriction site (ACRS) technique, revealed a dinucleotide TC-->AG mutation, which changed isoleucine to lysine in the predicted first transmembrane domain of the EDNRB protein. This was associated with LWFS when homozygous and with the overo phenotype when heterozygous.
URLPMID:27039359 [本文引用: 1]
61Different methods have been described to detect theEDNRBIle118Lys mutation, including AS-PCR and PIRA-PCR.61These tests, which have been carried out by polyacrylamide gel electrophoresis (PAGE), are time-consuming techniques.61In our study, we developed a new method based on mutagenically separated PCR (MS-PCR).61Our method provides a fast and simple analysis of theEDNRBhorse gene.
URLPMID:569673 [本文引用: 1]
Genetic segregation patterns among blood type markers and various phenotypically observed traits were studied in a small herd of ponies. The herd consisted of 10 mares without white spotting and a single stallion with the dominant pattern of tobiano spotting. Comparison of segregation patterns at loci for which the stallion was heterozygous showed tight linkage for the -B and tobiano markers. In 17 cases in which the contribution of the sire could be determined, all 10 foals that inherited from him were tobiano spotted, and all 7 non-spotted foals inherited his . The use of the symbol To is proposed for dominantly inherited tobiano spotting linked to the albumin.
URLPMID:3624845 [本文引用: 2]
Abstract Blood type analysis of 29 foals in a paternal half-sib family verified linkage of five LGII loci (Es, E, To, Gc, Al). Population and parentage data from other tobiano-spotted horses suggested conservation of a tightly linked (To:GcS:AlB) marker complex.
URLPMID:10051324 [本文引用: 1]
The melanocortin 1 receptor ( MC1R ), mast/stem cell growth factor receptor ( KIT ), and platelet-derived growth factor receptor 伪 ( PDGFRA ) are loci that all belong to equine linkage group 2 (LG2). Of these, KIT was fluorescent in situ hybridization (FISH) mapped to ECA3q21 with equine cDNA and heterologous porcine BAC probes, while MC1R was localized to ECA3p12 and PDGFRA to ECA3q21 with heterologous porcine BAC probes. A three-step comparison between ECA3 and donkey chromosomes was carried out. First, microdissected ECA3 painting probe was used on donkey chromosomes, which showed disruption of the equine synteny. Next, human (HSA) Chromosomes (Chrs) 16q and 4 specific paints, known to be homologous to ECA3p and 3q, respectively, were applied to detect homologous chromosomal segment(s) in donkey. Finally, four genes ( MC1R, ALB, PDGFRA, KIT ) and two equine microsatellite markers ( SGCV18 and SGCV33 ) located on ECA3 were FISH mapped to donkey chromosomes. The findings refined the cross species painting homology results and added six new markers to the nascent donkey gene map. The hypothesis that Tobiano coat color in horses may be associated with a chromosomal inversion involving genes within LG2 was tested by G-banding-based cytogenetic analysis and ordering of four loci鈥 KIT, PDGFRA, albumin ( ALB ), and MC1R 鈥攊n Tobiano and non-tobiano (homozygous as well as heterozygous) horses. However, no difference either in banding patterns or location/relative order of the genes was observed in the three classes. The study highlights successful FISH mapping of BAC probes across evolutionarily diverged species, viz., pig and horse/donkey, and represents the first use of large-sized individual clones across distantly related farm animals.
URLPMID:8589683 [本文引用: 1]
Abstract In the mouse, mutations in the c-Kit proto-oncogene, a member of the receptor tyrosine kinase (RTK) gene family, have pleiotropic effects on hematopoiesis, pigmentation and fertility (dominant spotting, W). However, in the Wsh allele the defect is confined to abnormal pigmentation caused by the disruption of 5' regulatory sequences of Kit leaving an intact structural gene. In this report, the previously published physical map around the Pdgfra-Kit-Flk1 RTK loci is extended by mapping the loci encoding the GABAA (gamma-aminobutyric acid) receptor subunit beta 1, Gabrb1 and a cytoplasmic kinase (Tec) 3 Mb proximal to Kit. PFGE analysis of the wild-type (C57BL/6J) chromosome demonstrates the following gene order: cen-Gabrb1-Tec-Pdgfra-Kit, whereas the analysis of Wsh/Wsh DNA is consistent with the order: cen-Gabrb1-Pdgfra-Tec-Kit. This altered physical map can be explained by an inversion on the Wsh chromosome located proximally to the Kit locus and spanning the 2.8 Mb Pdgfra-Tec chromosomal segment. This high resolution physical mapping study identifies large DNA fragments that span the two inversion breakpoints and potentially carry Kit upstream regulatory elements involved in the control of Kit expression during embryonic development.
URL [本文引用: 1]
URLPMID:18410476 [本文引用: 1]
The tobiano white-spotting pattern is one of several known depigmentation phenotypes in horses and is desired by many horse breeders and owners. The tobiano spotting phenotype is inherited as an autosomal dominant trait. Horses that are heterozygous or homozygous for the tobiano allele ( To ) are phenotypically indistinguishable. A SNP associated with To had previously been identified in intron 13 of the equine KIT gene and was used for an indirect gene test. The test was useful in several horse breeds. However, genotyping this sequence variant in the Lewitzer horse breed revealed that 14% of horses with the tobiano pattern did not show the polymorphism in intron 13 and consequently the test was not useful to identify putative homozygotes for To within this breed. Speculations were raised that an independent mutation might cause the tobiano spotting pattern in this breed. Recently, the putative causative mutation for To was described as a large chromosomal inversion on equine chromosome 3. One of the inversion breakpoints is approximately 70 kb downstream of the KIT gene and probably disrupts a regulatory element of the KIT gene. We obtained genotypes for the intron 13 SNP and the chromosomal inversion for 204 tobiano spotted horses and 24 control animals of several breeds. The genotyping data confirmed that the chromosomal inversion was perfectly associated with the To allele in all investigated horses. Therefore, the new test is suitable to discriminate heterozygous To/+ and homozygous To/To horses in the investigated breeds.
URLPMID:7274658 [本文引用: 1]
Characterization of the pleiotropic effects of ten new putative W locus mutations, nine co-isogenic and one highly congenic with the C57BL/6J strain, reveals a wide variety of influences upon pigmentation, blood formation and gametogenesis. None of the putative alleles, each of which is closely linked to Ph, a gene 0.1 cM from W, gave evidence of complementation with W39, a new allele previously shown to be allelic to Wv. All W/W39 genotypes resulted in black-eyed-white anemics with reduced gametogenic activity. Homozygotes for seven of these mutations are lethal during perinatal life; anemic embryos have been identified in litters produced by intercross matings involving each of these alleles.--Phenotypes of mice of several mutant genotypes provide exceptions to the frequent observation that a double dose of dominant W alleles (e.g., W/Wv or W/W) results in defects of corresponding severity in each of the three affected tissues. One viable homozygote has little or no defect in blood formation, and another appears to have normal fertility. The phenotypes of these homozygotes support the conclusion that the three tissue defects are not dependent on each other for their appearance and probably do not result from a single physiological disturbance during the development of the embryo.--Although homozygosity for members of this series results in a wide range of phenotypes, the absence of complementation of any allele with W39, the close proximity of each mutant to Ph, and the fact that all alleles produce detectable (though sometimes marginal) defects in the same tissues affected by W and Wv, support the hypothesis that each new mutant gene is a W allele.
URLPMID:5816567 [本文引用: 2]
J Hered. 1969 Mar-Apr;60(2):59-63.
URL [本文引用: 1]
Summary Dominant white coat colour ( W ) is a depigmentation syndrome, known in miscellaneous species. When homozygous in the horse (similar in mice), the mutation responsible for the white phenotype is lethal in a very early stage of gestation. It seems, that the action of the dominant white allele is not always fully penetrant, resulting occasionally in spotted look alike offspring. These horses resemble a coat colour pattern known as sabino spotting. So far, it is not known whether dominant white ( W ) and sabino spotting ( S ) share a common genetic background. In this study, a pedigree consisting of 87 horses segregating for dominant white ( W ) was used to genetically localize the horse ( W )-locus. Microsatellite ASB23 was found linked to ( W ), which allowed us to map dominant white to a region on horse chromosome 3q22. Tyrosine kinase receptor ( KIT ) was previously mapped to this same chromosome region (3q21鈥22). KIT and its ligand ( KITLG ) are responsible for the normal function of melanogenesis, haematopoiesis and gametogenesis. So far, sequence analysis of different KIT gene fragments did not lead to new polymorphisms, except for a SNP detected in KIT intron 3 ( KITSNPIn3 ). Additional microsatellites from ECA3q ( TKY353 and LEX7 ), together with KITSNPIn3 allowed us to state more precisely the ( W )-mutation. The positional results and comparative functional data strongly suggest that KIT encodes for the horse ( W )-locus.
URLPMID:22065780 [本文引用: 1]
Archaeologists often argue whether Paleolithic works of art, cave paintings in particular, constitute reflections of the natural environment of humans at the time. They also debate the extent to which these paintings actually contain creative artistic expression, reflect the phenotypic variation of the surrounding environment, or focus on rare phenotypes. The famous paintings "The Dappled Horses of Pech-Merle," depicting spotted horses on the walls of a cave in Pech-Merle, France, date back ~25,000 y, but the coat pattern portrayed in these paintings is remarkably similar to a pattern known as "leopard" in modern horses. We have genotyped nine coat-color loci in 31 predomestic horses from Siberia, Eastern and Western Europe, and the Iberian Peninsula. Eighteen horses had bay coat color, seven were black, and six shared an allele associated with the leopard complex spotting (LP), representing the only spotted phenotype that has been discovered in wild, predomestic horses thus far. LP was detected in four Pleistocene and two Copper Age samples from Western and Eastern Europe, respectively. In contrast, this phenotype was absent from predomestic Siberian horses. Thus, all horse color phenotypes that seem to be distinguishable in cave paintings have now been found to exist in prehistoric horse populations, suggesting that cave paintings of this species represent remarkably realistic depictions of the animals shown. This finding lends support to hypotheses arguing that cave paintings might have contained less of a symbolic or transcendental connotation than often assumed.
URLPMID:26568529 [本文引用: 3]
Summary Leopard complex spotting (LP), the result of an incompletely dominant mutation in TRPM1 , produces a collection of unique depigmentation patterns in the horse. Although the LP mutation allows for expression of the various patterns, other loci are responsible for modification of the extent of white. Pedigree analysis of families segregating for high levels of patterning indicated a single dominant gene, named Pattern-1 ( PATN1 ), as a major modifier of LP . Linkage analysis in two half-sibling families segregating for PATN1 identified a 15-Mb region on ECA3p that warranted further investigation. Whole transcriptome sequencing of skin samples from horses with and without the PATN1 allele was performed to identify genic SNPs for fine mapping. Two Sequenom assays were utilized to genotype 192 individuals from five LP -carrying breeds. The initial panel highlighted a 1.6-Mb region without a clear candidate gene. In the second round of fine mapping, SNP ECA3:230265802447T>G in the 3′-UTR of RING finger and WD repeat domain 3 ( RFWD3 ) reached a significance level of P = 1.06302×02106139. Sequencing of RFWD3 did not identify any coding polymorphisms specific to PATN1 horses. Genotyping of the RFWD3 3′-UTR SNP in 54 additional LP animals and 327 horses from nine breeds not segregating for LP further supported the association ( P = 4.1702×021061115). This variant is a strong candidate for PATN1 and may be particularly useful for LP breeders to select for high levels of white patterning.
URLPMID:17970998 [本文引用: 1]
Abstract OBJECTIVE: To determine the prevalence of congenital stationary night blindness (CSNB) in Appaloosa horses in western Canada, investigate the association with the leopard complex of white spotting patterns, and further characterize the clinical and electroretinographic aspects of CSNB in the Appaloosa. ANIMALS STUDIED: Three groups of 10 Appaloosas were studied based on coat patterns suggestive of LpLp, Lplp, and lplp genotype. PROCEDURES: Neurophthalmic examination, slit-lamp biomicroscopy, indirect ophthalmoscopy, measurement of corneal diameter, streak retinoscopy, scotopic and photopic full-field and flicker ERGs and oscillatory potentials (OPs) were completed bilaterally. RESULTS: All horses in the LpLp group were affected by CSNB, while none in the Lplp or lplp groups was affected. The LpLp and Lplp groups had significantly smaller vertical and horizontal corneal diameters than the lplp group had. Median refractive error was zero for all groups. Scotopic ERGs in the LpLp (CSNB-affected) group were consistent with previous descriptions. The CSNB-affected horses had significantly longer photopic a-wave implicit times, greater a-wave amplitudes, and lower b-wave amplitudes than the Lplp and lplp (normal) groups did. No differences were present in photopic flicker amplitude or implicit times. Scotopic flickers in the CSNB-affected horses were markedly reduced in amplitude and abnormal in appearance. No differences were noted in OP implicit times; however, amplitudes of some OPs were reduced in CSNB-affected horses. There were no differences in scotopic and photopic or flicker ERGs or OPs between the normal groups. CONCLUSIONS: CSNB was present in one-third of horses studied and there was a significant association between CSNB and the inheritance of two Lp alleles. ERG abnormalities support the hypothesis that CSNB is caused by a defect in neural transmission through the rod pathway involving the inner nuclear layer.
[本文引用: 1]
URLPMID:15025575 [本文引用: 1]
Summary A single autosomal dominant locus, leopard complex ( LP ) controls the presence of appaloosa pigmentation patterns in the horse. The causative gene for LP is unknown. This study was undertaken to map LP in the horse. Two paternal half sib families segregating for the LP locus and including a total of 47 offspring were used to perform a genome scan which localized LP to horse chromosome 1 (ECA1). LP was linked to ASB 08 (LOD02=029.99 at Θ 02=020.02) and AHT 21 (LOD02=025.03 at Θ 02=020.14). To refine the map position of LP , eight microsatellite markers on ECA1 ( UM 041, LEX 77, 1CA 41, TKY 374, COR 046, 1CA 32, 1CA 43, and TKY 002) were analysed in the two half sib families. Results from this linkage analysis showed LP was located in the interval between ASB 08 and 1 CA 43. Tight junction protein ( TJP 1), which lies within the LP interval on ECA1, was used to determine the homologous chromosomes in humans (HSA15) and mice (mouse chromosome 7). We propose that the pink eyed dilution ( p ) gene and transient receptor potential cation channel subfamily M, member 1 ( TRPM 1) are positional candidate genes for LP .
[本文引用: 1]
URLPMID:20353955 [本文引用: 1]
Leopard Complex spotting occurs in several breeds of horses and is caused by an incompletely dominant allele (LP). Homozygosity for LP is also associated with congenital stationary night blindness (CSNB) in Appaloosa horses. Previously, LP was mapped to a 6 cm region on ECA1 containing the candidate gene TRPM1 (Transient Receptor Potential Cation Channel, Subfamily M, Member 1) and decreased ex...
URL [本文引用: 2]
No abstract is available for this article.
URLPMID:12370784 [本文引用: 2]
No abstract prepared.
URLPMID:12354140 [本文引用: 1]
Abstract The progressive loss of colour in the hair of grey horses is controlled by a dominantly inherited allele at the Grey locus (GG). In this study, two paternal Quarter Horse (QH) families segregating for the GG allele were genotyped with a set of 101 microsatellite markers spanning the 31 autosomes and the X chromosome. This genome scan demonstrated linkage of Grey to COR018 (RF=0.02, LOD=12.04) on horse chromosome 25 (ECA25). Further chromosome-specific analysis of seven total QH families confirmed the linkage of Grey to a group of ECA25 markers and the map order of NVHEQ43-(0.24)-UCDEQ405-(0.09)-COR080-(0.05)-GREY-(0.14)-UCDEQ464 was produced. Although G was found to be linked to TXN and COR018 in the chromosome-specific analysis, the data were not sufficiently informative to place either marker on our ECA25 map with significant LODs. Our results excluded the equine tyrosinase related protein 1 (TYRP1) and melanocyte protein 17 (Pmel17) genes as possible candidates for the grey phenotype in horses.
URLPMID:12354141 [本文引用: 1]
The dominant grey coat colour gene of horses has been mapped using a whole genome scanning approach. Samples from a large half-sibling pedigree of Thoroughbred horses were utilized in order to map the grey coat colour locus, G . Multiplex groups of microsatellite markers were developed and used to efficiently screen the horse genome at a resolution of approximately 22 cM, based on an estimated map length for the horse genome of 2720 cM. The grey gene was assigned to chromosome 25 (ECA25), one of the smaller acrocentric horse chromosomes. Based on the current state of knowledge of conserved synteny and coat colour genetics in other mammalian species, there are no obvious candidate genes for the grey gene in the region.
URLPMID:18641652 [本文引用: 1]
Gray horses are born colored but gradually lose hair pigmentation and become white, a trait that is transmitted in an autosomal dominant manner. Leif Andersson and colleagues report that the the mutation causing the Gray phenotype is a 4.6-kb duplication in intron 6 of STX17, which promotes overexpression of both STX17 and the neighboring gene NR4A3 in melanomas from Gray horses.
URLPMID:3443021 [本文引用: 1]
Background Greying with age in horses is an autosomal dominant trait, associated with loss of hair pigmentation, melanoma and vitiligo-like depigmentation. We recently identified a 4.665kb duplication in STX17 to be associated with the phenotype. The aims of this study were to investigate if the duplication in Grey horses shows copy number variation and to exclude that any other polymorphism is uniquely associated with the Grey mutation. Results We found little evidence for copy number expansion of the duplicated sequence in blood DNA from Grey horses. In contrast, clear evidence for copy number expansions was indicated in five out of eight tested melanoma tissues or melanoma cell lines. A tendency of a higher copy number in aggressive tumours was also found. Massively parallel resequencing of the ~35065kb Grey haplotype did not reveal any additional mutations perfectly associated with the phenotype, confirming the duplication as the true causative mutation. We identified three SNP alleles that were present in a subset of Grey haplotypes within the 35065kb region that shows complete linkage disequilibrium with the causative mutation. Thus, these three nucleotide substitutions must have occurred subsequent to the duplication, consistent with our interpretation that the Grey mutation arose more than 2,00065years before present. Conclusions These results suggest that the mutation acts as a melanoma-driving regulatory element. The elucidation of the mechanistic features of the duplication will be of considerable interest for the characterization of these horse melanomas as well as for the field of human melanoma research.
URLPMID:8531173 [本文引用: 1]
A study of 57 cutaneous melanocytic tumors from 53 horses revealed 4 distinct clinical syndromes: melanocytic nevus, dermal melanoma, dermal melanomatosis, and anaplastic malignant melanoma. Melanocytic nevus and anaplastic melanoma each had histopathologic features that distinguished them from dermal melanoma and dermal melanomatosis. Dermal melanoma and dermal melanomatosis were histologically similar but could be differentiated by their clinical features. Melanocytic nevi were diagnosed in 29 horses with an average age of 5 years; they were solitary, superficial masses that occurred in both grey and nongrey horses, and in which surgical excision was generally curative. Dermal melanomas were diagnosed in 20 horses with an average age of 13 years; all horses of known coat color were grey. Eight horses with an average age of 7 years had 1 or 2 discrete dermal melanomas. Follow-up information was available for 6 horses; metastases occurred in 2 horses, and surgical excision was apparently curative in 4 horses. Dermal melanomatosis was diagnosed in 12 grey horses with an average age of 17 years; all 6 of these horses evaluated had internal metastases. In 2 aged nongrey horses with anaplastic malignant melanoma, the tumors metastasized within 1 year of diagnosis. Two tumors with features of both melanocytic nevus and dermal melanoma remained unclassified.
URLPMID:10051325 [本文引用: 1]
Abstract The roan coat color in horses is controlled by a dominant allele that is lethal in the homozygous condition. Phenotypic similarities to some pigmentation disorders in human and mouse, combined with comparative mapping data, identified KIT, encoding the mast cell growth factor receptor, as a major candidate gene for the roan locus (Rn). Rn has previously been mapped to equine linkage group (LG) II. In this study, LGII was expanded with KIT and PDGFRA (platelet-derived growth factor receptor alpha) by use of RFLP and linkage analysis. Moreover, highly significant linkage disequilibrium between Rn and a KIT TaqI RFLP, representing a synonymous substitution in exon 19, was revealed. There was a strong KIT-Rn association in most breeds. Almost the complete KIT-encoding sequence was determined by sequence analysis of RT-PCR products. Comparison of horse KIT cDNA sequences, representing three different alleles (two different rn and one Rn), revealed five sequence polymorphisms and several mRNA splice variants, but none of these proved to be specifically associated with Rn. An insertion of a partial (79 bp) LINE1-element between exons 1 and 2, leading to a frameshift, represented about 30% of KIT transcripts in the Belgian roan horse used for the sequence analysis. However, an association between this L1 splice insertion and the roan phenotype was not verified when testing additional unrelated roan and non-roan horses from different breeds. The study strengthens the hypothesis that the roan coat color is controlled by KIT, but further analyses are needed to reveal the causative mutation(s).

{kind=link}
{kind=link}
{kind=link}
{kind=link}