 ,, 雷蕾,哈尔滨医科大学组织学与胚胎学教研室,哈尔滨 150081
,, 雷蕾,哈尔滨医科大学组织学与胚胎学教研室,哈尔滨 150081Histone variant H3.3 and its functions in reprogramming
Xingwei Huang, Xiangrong Cheng, Nan Wang, Yuwei Zhang, Chen Liao, Lianhong Jin,, Lei Lei,Department of histology and embryology, Harbin Medical University, Harbin 150081, China通讯作者:
第一联系人:
编委: 王晓群
收稿日期:2017-09-30修回日期:2017-12-15网络出版日期:2018-03-20
| 基金资助: |
Editorial board:
Received:2017-09-30Revised:2017-12-15Online:2018-03-20
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (398KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄星卫, 程香荣, 王楠, 张雨薇, 廖辰, 金连弘, 雷蕾. 组蛋白H3变体H3.3及其在细胞重编程中的作用. 遗传[J], 2018, 40(3): 186-196 doi:10.16288/j.yczz.17-233
Xingwei Huang, Xiangrong Cheng, Nan Wang, Yuwei Zhang, Chen Liao, Lianhong Jin, Lei Lei.
体细胞核移植(somatic cell nuclear transfer, SCNT)和诱导多潜能干细胞(induced pluripotential stem cells, iPSC)技术都是将已分化的体细胞重编程为未分化的状态,涉及到多潜能基因的重新激活、表观遗传改变及染色质的重塑等过程。以SCNT和iPSC为代表的重编程技术在再生医学领域具有巨大的应用前景,但目前重编程机制尚不甚清楚。SCNT和iPSC均存在效率低下、体细胞表观记忆残留等问 题[1,2],极大地制约了这些技术的应用,因此解析重编程机制进而提高其效率成为当前遗传与发育研究领域的热点之一。
核小体是真核生物染色质的基本组成单位,由146 bp的DNA在组蛋白八聚体上缠绕将近两圈形成。组蛋白八聚体包含两套核心组蛋白H2A、H2B、H3、H4,由两个H2A-H2B二聚体和一个H3-H4四聚体组成。核小体的产生不仅使DNA保持固缩状态,维持基因组的稳定性,而且也保证DNA序列可以正确地进行复制、转录、重组和修复。真核细胞通过组蛋白翻译后修饰(post-translational modifications, PTMs),如甲基化、乙酰化、磷酸化、泛素化等,以及各种组蛋白变体来调节核小体的生物过程。除了H4变体组蛋白只在低等真核生物中被发现外,H2A、H2B、H3在大部分真核生物中都有多种的组蛋白变体,变体组蛋白与常规组蛋白的差异从几个到上百个氨基酸不等。其中H3.3与其常规组蛋白原型仅有几个氨基酸的区别,但H3.3整合进入染色质的方式以及功能却发生了很大的变化。越来越多的研究表明这些组蛋白变体在调节染色质稳态以及相应DNA活性的过程中发挥着重要作用。
目前,在哺乳动物中已经发现了5种H3变体:2种常规组蛋白H3变体H3.1(哺乳动物中特异存在)和H3.2,以及3种替换组蛋白变体H3.3、着丝粒特异的变体CenH3(哺乳动物中是CENP-A)和睾丸特异的组蛋白变体H3T[3,4,5]。除此以外,研究还发现在灵长类动物特异存在的组蛋白变体H3.X和H3.Y、在人类生精小管中表达的类人猿特异的组蛋白变体H3.5,以及在人类中存在的组蛋白H3变体H3.6、H3.7和H3.8,但这些组蛋白变体的具体功能尚不清楚[6,7,8]。其中,H3.3是一种重要的母源因子,能够在受精后替换精子中的鱼精蛋白,从而参与雄原核的重编程过程[9]。最初,组蛋白变体H3.3被认为是转录活性的标志,在表观印记的转变过程中具有着重要作用[10],但也有研究表明,H3.3能整合进入端粒和着丝粒旁的串联重复序列等沉默的染色质[11,12]。本文主要对组蛋白H3变体H3.3相关的特性、定位、功能及其在细胞重编程过程中的作用进行了综述。
1 组蛋白变体H3.3与常规组蛋白H3的区别及特点
编码常规组蛋白H3.1和H3.2的基因不含内含子,且多个基因串联集合成簇存在。其编码的mRNA不包含polyA尾,同时需要结合茎环结合蛋白(stem binding protein)并且在3′端结合U7 snRNA (small nuclear RNA)才能进行翻译[13]。正是这种独特的基因构成及其转录调控方式使得在细胞有丝分裂的间期S期时可以得到大量的常规组蛋白H3,从而保证DNA复制时有足够多的新合成的组蛋白整合进入其中。这是一种依赖DNA复制(DNA synthesis- coupled, DSC)的方式。当紫外线照射或者其他因素导致DNA损伤时,即使细胞不处于S期,常规组蛋白H3.1仍然能以DSC的方式整合进入到染色质 中[14]。而编码非常规组蛋白H3.3的基因却只有一个或几个,且散布在基因组中,转录得到含有polyA尾的mRNA。例如:在小鼠(Mus musculus)、人(Homo sapiens)、果蝇(Drosophila melanogaster)中均有两个编码H3.3的基因,即H3.3A和H3.3B,虽然它们的非编码区不同,但编码相同的H3.3蛋白[15,16,17]。这些基因在整个细胞周期都持续表达,使组蛋白变体H3.3替换常规组蛋白H3,并能以一种不依赖DNA复制(DNA synthesis-independent, DSI)的方式整合进入染色质。所有真核细胞中都存在H3.3或H3.3样的保守蛋白,而在哺乳动物中组蛋白H3.3与H3.2仅有4个氨基酸的差异(第31、87、89和90位氨基酸),H3.3与H3.1也仅有5个氨基酸的差异(第31、87、89、90和96位氨基酸)(
新窗口打开|下载CSV
2 H3.3在特定的基因位置富集及作用
利用染色质免疫共沉淀测序技术(chromatin immunoprecipitation with high-throughput sequencing, ChIP-seq),人们确定了真核生物细胞中详细的H3.3全基因组定位图谱。结果表明对于GC含量高的启动子区域,无论基因是激活还是沉默状态,均有H3.3的富集[18]。而H3.3倾向于整合进入激活的染色质中,主要富集在有活性基因的启动子区域、基因内与基因间的调控区域,说明H3.3与基因活性之间存在一定联系[18,19,20,21]。当基因转录时,RNA聚合酶复合物在基因体上会移除原有的核小体,H3.3能被动地以DSC方式整合进入染色质进行补充[22]。而H3.3也能主动在激活状态基因的调控元件区域持续地以DSI方式替换常规组蛋白H3来维持相关复合体的结合,从而激活转录或保持基因的表观遗传记忆[21,23]。Gurdon等[23]发现在非洲爪蟾(Xenopus laevis)中存在一些H3.3相关的表观遗传标志,其在细胞分裂过程中始终存在,而不是依靠每一个细胞周期中重新激活转录来维持激活状态的记忆。在果蝇中敲除H3.3编码基因(His3.3A和His3.3B)会出现H3的过表达,说明其中可能存在部分补偿机制[24]。但在小鼠胚胎干细胞中,敲除Hira的细胞与野生型细胞相比,H3.3在基因内区域的富集减少对于整个细胞的转录影响不大[18]。这表明H3.3可能在这些细胞中的正常转录中并不是必需的,但考虑到它们是全能或多潜能干细胞,与终末分化的细胞相比,可能不需要一个持续的基因激活的记忆,所以应关注H3.3在已分化的哺乳动物细胞中对于激活状态记忆的作用。H3.3除了整合进入激活的染色质中,同时也富集在另一些非激活状态的基因位置中。小鼠胚胎干细胞中存在一些启动子区域同时富集H3K4me3和H3K27me3两种表观修饰的二价基因,它们大多处于静止状态,只有在细胞分化时才激活表达。在正常情况下H3.3会募集多梳蛋白抑制复合体2(polycomb repressive complex 2, PRC2)到二价基因启动子区域参与建立H3K27me3修饰的过程[25]。当NIH3T3细胞受到某些外界条件(如干扰素)刺激后会激活部分基因的表达,此时H3.3会大量富集到这些基因的编码区的末端。如果细胞不处于有丝分裂时期,即使转录结束后,H3.3一段时间内仍会存在于这些基因的编码区[26]。Ahmad和Henikoff[10]发现H3.3在果蝇Kc细胞中富集到大量rDNA重复序列中,或许是因为这些致密重复的rDNA位点有很高的转录活性又或者是为了维持rDNA异染色质的稳定。而在小鼠胚胎干细胞中,H3.3在rDNA等重复序列中均有富集,且保持较稳定的水平,维持基因的平衡状态[21]。另外,在人、果蝇和小鼠中,还发现H3.3在端粒和着丝粒旁的异染色质区域的聚集,而且对于维持此处重复序列的转录抑制状态是必需的[27,28,29]。因此,可以看出H3.3整合进入端粒和着丝粒旁的异染色质区域与基因组稳定性相关。探究H3.3如何整合进入这些特殊的位点,以及介导H3.3整合进入的组蛋白分子伴侣复合物如何发挥作用,对更好地理解H3.3的富集模式和作用很有意义。
图1
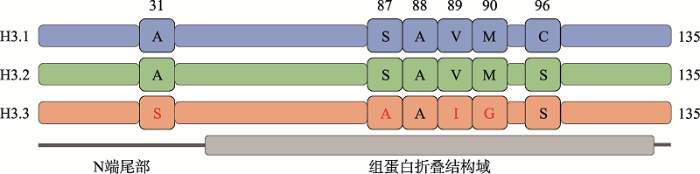 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1组蛋白变体H3.3与常规组蛋白H3(H3.1和H3.2)氨基酸序列的区别
组蛋白变体H3.3与常规组蛋白H3.1和H3.2氨基酸序列仅存在4~5个区别。A:丙氨酸(alanine);S:丝氨酸(serine);V:缬氨酸(valine);M:甲硫氨酸(methionine);I:异亮氨酸(isoleucine);G:甘氨酸(glycine);C:半胱氨酸(cysteine)。
Fig. 1Differences in amino acid sequences between H3 (H3.1 & H3.2) and H3.3
3 H3.3与分子伴侣
核小体的组装需要一系列组蛋白整合进入DNA,而组蛋白变体整合进入的平衡受到组蛋白分子伴侣的影响,这对于细胞命运决定和基因表达程序的稳定很重要。H3.3有很多分子伴侣,一部分特异地与H3.3相互作用,而另一部分则可以与所有的组蛋白H3发生相互作用。在高等动物中,主要由两种特异的组蛋白分子伴侣复合体引导H3.3整合进入染色质:HIRA/UBN1/CABIN1复合体和ATRX/DAXX复合体。3.1 HIRA/UBN1/CABIN1复合体
Almouzni等[30]在分离常规组蛋白H3.1整合进入复合体时发现了染色质合成因子(chromatin assembly factor-1, CAF-1),其包含p150、p60和p50 3个亚基,是在DNA复制和因紫外线照射损伤修复过程中以DSC方式促进核小体合成的分子伴侣原型。在分离H3.3整合进入复合体的研究中,又发现了组蛋白细胞周期调节因子A(histone cell cycle regulator A, HIRA)[31]。该复合体(以下简称HIRA复合体)主要包括HIRA、泛素化核蛋白1(ubinuclein-1, UBN1)、钙依赖磷酸酶结合蛋白1(calcineurin-binding protein 1, CABIN1)(图2)。这是一个进化保守的组装过程,在抗沉默功能蛋白1同源物a (anti-silencing function 1 homolog a, ASF1a)的帮助下,调控H3.3-H4二聚体以DSI方式整合进入或退出染色质[32]。酿酒酵母菌(Saccharomyces cerevisiae)中Hir1p和Hir2p拥有与HIRA同源的结构域,而Hir3和Hpc2分别是CABIN1和UBN1的同源物。其中UBN1能特异性识别H3.3- H4二聚体H3.3第90位上甘氨酸残基,与H3.3高度保守的第87、89和90位的AIG结构域结合。酿酒酵母菌中HIR复合体以DSI方式参与转录调控、延长速度和沉默染色质结构域的建立[33]。在果蝇中,HIRA对于H3.3整合进入解凝集的精子染色质很重要,HIRA突变使卵母细胞中H3.3不能替换精子中鱼精蛋白,精子染色质维持凝集状态,最终导致胚胎致死[34]。然而在出生后的发育阶段,敲除HIRA并不影响果蝇的生长过程,仅雄性果蝇出现不育症状,表明在果蝇中,由HIRA复合体介导的H3.3整合进入核小体并不是生长发育过程必需的,可能还存在其他介导H3.3整合进入核小体或者其他组蛋白变体替代H3.3功能的代偿机制[24]。在非洲爪蟾中,胚胎发育过程中H3.3的整合进入也依赖HIRA复合体,敲减HIRA的蛋白水平会得到与敲减H3.3类似的表型[35]。在哺乳动物中,HIRA复合体介导的H3.3整合进入核小体对于早期胚胎发育和细胞多能性十分重要[36]。在小鼠胚胎干细胞中,HIRA复合体介导H3.3在发育相关基因启动子区域募集PRC2复合体,建立正确的H3K27me3修饰,维持胚胎干细胞中正常的染色质表观修饰标记,这对于分化时的基因调控是必需的[25]。在增殖细胞中,因为HIRA复合体可以控制RNA聚合酶Ⅱ与转录位点和调控元件结合,所以HIRA复合体出现在活跃基因的启动子与基因体等位置,可以调控转录激活基因的功能[37]。最新的研究表明,HIRA复合体介导的H3.3核小体合成依赖复制蛋白A(replication protein A, RPA),RPA是单链DNA结合蛋白,是DNA复制和修复重要的调节因子。如果下调RPA表达水平会影响HIRA复合体的募集和H3.3整合进入调节元件和启动子区域,进而影响基因转录[38,39]。图2
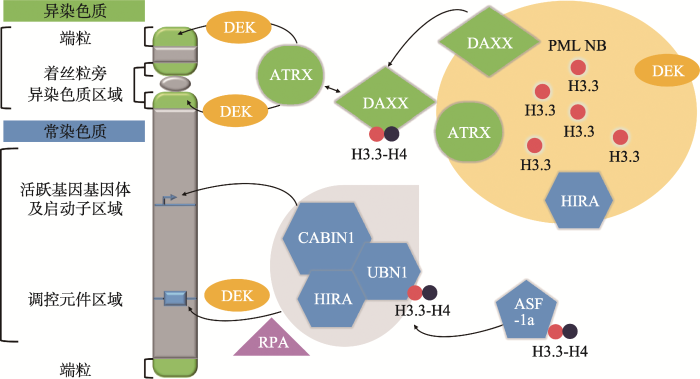 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2H3.3在不同区域的富集及特异的分子伴侣复合体
H3.3由HIRA/UBN1/CABIN1复合体介导整合进入激活基因的基因体和启动子区域以及调控元件区域。而ATRX/DAXX复合体介导H3.3整合进入端粒和着丝粒旁异染色质区域。PML小体形成一个H3.3“仓库”,里面容纳未整合进入染色质的H3.3以及HIRA、ATRX、DAXX和DEK等分子伴侣,在整合进入染色质之前调控H3.3与分子伴侣之间的相互作用。ASF-1a为HIRA/UBN1/CABIN1复合体提供H3.3-H4二聚体,DEK和RPA各自调控H3.3整合进入调控元件区域。PML NB: 早幼粒白血病蛋白小体(promyelocytic leukemia nuclear bodies)。
Fig. 2H3.3 enrichment pattern and specific chaperone complexes
3.2 ATRX/DAXX复合体
除了HIRA复合体外,另一个包含两个蛋白的复合体也参与了H3.3核小体的合成[40],该复合体包含的两个蛋白分别是α地中海贫血/智力低下X连锁综合症蛋白(the alpha-thalassemia/mental retardation X-linked syndrome protein, ATRX)和死亡结构域相关蛋白(the death domain-associated protein, DAXX) (图2)。ATRX属于SNF2相关ATP酶家族,是一个ATP依赖的染色质重构因子。与其他真核生物SWI/ SNF多蛋白复合体的其他螺旋酶亚基一样,ATRX会介导H3.3整合进入特定的靶点位置改变核小体组成。ATRX定位在端粒和着丝粒旁的异染色质区域,表明可能有维持染色质沉默状态的作用[41]。DAXX和ATRX一起介导H3.3整合进入染色质[40],与HIRA复合体类似,DAXX可以识别H3.3中第87~90位氨基酸的AAIG模体,与H3.3-H4二聚体结合[42]。DAXX和ATRX相互作用,引起ATP依赖的染色质重构以及H3.3以DSI方式整合进入特定的基因区域[40,43]。有研究表明,ATRX能通过自身的染色质结合结构域识别H3K9me3和未被修饰的H3K4,与异染色质蛋白1(heterochromatin protein 1, HP1)结合[44]。这会促进macroH2A1退出基因和基因间区域,因为在缺乏ATRX的人源细胞中发现macroH2A1在端粒旁区域聚集[45]。在分裂细胞和分化细胞中,DAXX与ATRX一同控制H3.3整合进入着丝粒旁异染色质、端粒以及转录起始区域[18,40,46~48]。尽管H3.3常与激活基因的启动子和调控区域相关[49,50],但ATRX/DAXX复合体控制H3.3整合进入H3K9me3等甲基化的沉默基因区域,维持表观遗传修饰[48],这也许可以阻止表观遗传记忆的丢失以及异染色质区域的异常基因表达。在小鼠胚胎干细胞中,ATRX/DAXX复合体对于H3.3整合进入内源转座元件(endogenous retroviral elements, ERVs)很重要,例如:H3.3依赖的核小体替换通过募集KRAB相关蛋白1(KRAB-associated protein-1, KAP1)维持H3K9me3标记和ERVs的沉默状态[51,52]。近期有研究发现EB病毒(epstein-barr virus, EBV)可以利用ATRX/DAXX复合体整合进宿主基因组中,并维持潜伏状态[53]。这些结果表明ATRX/DAXX复合体介导的H3.3替换对于维持基因组稳定性和组织中体细胞异质性十分重要。3.3 其他的分子伴侣
其他一些蛋白也被发现与H3.3整合进入特定位置相关,一些能特异地与H3.3相互作用,而另一部分则与所有H3或者所有组蛋白相关。例如:FACT介导组蛋白整合进入与交换,NASP1可以保证新合成的组蛋白不被降解。在果蝇中,染色体结构域螺旋酶DNA结合蛋白1(chromodomain helicase DNA- binding protein 1, CHD1)与HIRA互作,介导H3.3在受精后整合进入精子DNA解凝集的雄性染色质中[54]。早幼粒白血病(promyelocytic leukemia, PML)蛋白形成空腔的PML小体(PML nuclear bodies, PML NB),包含H3.3及HIRA、DAXX、ATRX等分子伴侣,PML小体调控H3.3与分子伴侣之间的相互作用,使之整合进入PML蛋白相关的异染色质区域[55]。在人和果蝇中发现另一个蛋白DEK,是染色质结合原癌基因产物,也被认为是H3.3的一个分子伴侣,介导H3.3整合进入调控元件区域并增强转录[56]。有研究发现DEK聚集在PML小体中,调控H3.3与ATRX/DAXX复合体相互作用,整合进入端粒和异染色质区域,维持端粒和异染色质区域的稳定[57] (图2)。综上所述,这些结果表明H3.3整合进入染色质需要特殊的分子伴侣复合体参与,但可能还存在一些其他的H3.3分子伴侣或者整合进入途径,可以在缺失H3.3特异分子伴侣的时候代偿发挥作用。4 H3.3与细胞重编程
在雄性哺乳动物生殖细胞进入第一次减数分裂前期时,H3.3的整合进入伴随所有染色体中的核小体的替换,发生在减数分裂性染色体失活(meiotic sex chromosome inactivation, MSCI)过程中[58]。这与雄性哺乳动物生殖细胞系中性染色质基因沉默的表观遗传重编程相关。在大部分有性繁殖的动物中,还涉及另外一个主要的重组过程,即在精子发生过程中,组蛋白被鱼精蛋白替换,这对于维持精子细胞基因组凝集状态以及转录抑制状态十分重要[59]。当进入卵母细胞之后,精子的细胞核会经历一系列保守的过程被重编程为雄原核。精子内主要经历鱼精蛋白被组蛋白替换和染色质解凝集。同济大学高绍荣课题组等发现受精后,卵母细胞中的重编程因子更多地进入到雄原核之中[60]。在果蝇和小鼠中,受精后精子中鱼精蛋白被母源H3.3替换,而不是H3,这种状态一直持续到第一次DNA复制之前[9,34]。这说明H3.3整合进入对精子染色质重编程具有重要作用。SCNT是利用去核的卵母细胞将供体细胞核重编程为胚胎样细胞核,从而具有全能性。核移植胚胎激活后4 h内,母源H3.3会逐渐替换供体细胞内原有的H3,类似受精过程中鱼精蛋白被母源H3.3替换的过程,首先是常染色质区域,最后是异染色质区域[61],母源H3.3会重新激活供体细胞核中的多潜能基因Oct4,降低基因组中H3K27me3水平。而敲除母源H3.3后,关键的多潜能基因转录水平下降,体细胞核不能被完全重编程,SCNT胚胎将不能正常发育[62,63]。注入外源H3.3 mRNA而不是H3.2 mRNA可以挽救这种缺陷,说明H3.3在体细胞重编程过程中十分重要。而iPSC是另一种将体细胞重编程为全能性细胞的方式,通过转入外源的转录因子达到体细胞重编程的目的。但H3.3在iPSC重编程过程中的作用机制尚不清楚。小鼠SCNT后代理论上可以无限次通过SCNT得到后代[64],但iPSC后代进行连续iPSC在第六代后会因非同义的单核苷酸突变(single-nucleotide variations, SNVs)累积某些致病基因,造成心肾发育缺陷导致胚胎死亡。且在第一代iPSC小鼠体细胞构建的第二代iPSC细胞系中即可检测到逆转录转座子元件的缺失,如无法检测到长散在核元件(long interspersed nucleotide elements, LINEs)和长末端重复序列(long terminal repeats, LTRs)的存在[65](图3)。已有文献报道,H3.3对于维持H3K9me3标记和逆转录元件的沉默状态十分重要[51,52]。对比这两种重编程过程,在敲除端粒酶Terc后,SCNT胚胎来源的ntESC中端粒比iPSC中端粒更长且更稳定[66],而H3.3在维持端粒稳定性方面也有重要作用[11,46,57]。考虑到iPSC重编程过程中没有卵母细胞参与,缺乏母源H3.3替换,自身H3.3可能不足以维持端粒和着丝粒旁重复序列异染色质的稳定性,这也许是连续iPSC重编程不能无限连续进行的原因之一。过表达外源H3.3或者其特异的分子伴侣是否能提高iPSC的重编程效率,使之更接近正常的ESC还有待进一步研究。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3SCNT和iPSC两种体细胞重编程过程中H3.3的变化情况
SCNT重编程过程中卵母细胞含有大量的母源H3.3,会替换体细胞核中原有的H3.3,而iPSC重编程过程中没有母源H3.3参与。如果进行连续iPSC重编程,从第二代iPSC起可检测到LINEs和LTRs的缺失,到第六代时,四倍体补偿的iPSC胎儿会因非同义的SNVs累积致病基因而死亡[65]。SCNT:体细胞核移植(somatic cell nuclear transfer);iPSC:诱导多潜能干细胞(induced pluripotent stem cell);SNVs:单核苷酸突变(single-nucleotide variations);LINE:长散在核元件(long interspersed nucleotide elements);LTRs:长末端重复序列(long terminal repeats)。
Fig. 3Comparison of H3.3 in somatic cell reprogramming during SCNT and iPSC
rDNA是编码核糖体的基因,在受精过程中最先被激活,以保证受精卵的蛋白合成。其中一部分rDNA具有转录活性,而另一部分则以异染色质形式存在。有文献报道,在SCNT过程中,rDNA并没有被完全激活[67],且核移植效率与rDNA启动子区域甲基化程度相关,而在iPSC建系过程中提高rDNA的表达水平,可以提高iPSC的建系效率[68]。在果蝇Kc细胞中,H3.3富集到大量rDNA重复序列中[10]。在小鼠受精卵中,HIRA介导的H3.3整合进入rDNA区域,保证rDNA的转录[69]。这些结果证明H3.3与rDNA活性相关。目前只在非洲爪蟾中研究表明SCNT重编程过程中HIRA会介导H3.3整合进入rDNA区域,促进rDNA的表达,从而促进供体细胞核重编程[70]。在其他物种中暂无相关报道,但rDNA对于核糖体生物合成至关重要,因此研究H3.3在SCNT和iPSC重编程过程中是否整合进入rDNA区域,对于提高SCNT和iPSC重编程效率有重要意义。
5 结语和展望
H3.3与常规组蛋白H3之间只有几个氨基酸不同,但它们能够驱使H3.3与特异的分子伴侣相互作用。H3.3至少包含两种特异的分子伴侣复合体,HIRA复合体和ATRX/DAXX复合体,与不同的分子伴侣相互作用可以使H3.3富集到基因组中的不同位置。HIRA复合体介导H3.3整合进入转录激活基因的启动子和调控元件区域,促进基因的转录表达。而ATRX/DAXX复合体介导H3.3整合进入端粒或着丝粒旁异染色质区域,维持该区域的抑制性表观遗传标记和基因组稳定性。H3.3是重要的母源因子,在正常受精后精子的重编程以及体细胞核移植后供体细胞核的重编程过程中起重要作用。在正常受精过程中,H3.3能替换精子中的鱼精蛋白,将其重编程为雄原核。而在SCNT过程中,母源H3.3也能替换供体细胞核内原有的H3.3,将其重编程成为具有全能性的胚胎。rDNA是编码核糖体的基因,在受精过程中最先被激活,以保证受精卵的蛋白合成。在SCNT过程中,重编程效率与rDNA启动子区域甲基化程度相关[67],而在iPSC建系过程中提高rDNA的表达水平,可以提高iPSC的建系效率[68]。在果蝇Kc细胞、非洲爪蟾和小鼠受精卵中,H3.3整合进入rDNA区域,保证rDNA的转录[10,69,70]。这些结果证明H3.3与rDNA活性相关。在爪蟾SCNT重编程过程中HIRA介导H3.3进入rDNA区域,促进rDNA的表达,从而促进供体细胞核重编程[70],但在其他物种中尚未见相关报道。那么在其他物种SCNT过程中H3.3是否也会整合进入rDNA区域并参与rDNA重编程过程?如果H3.3确实在SCNT过程中参与rDNA重编程,那么又是由何种特异的分子伴侣介导?是整合进入激活的rDNA区域来维持rDNA的表达或沉默rDNA基因?还是整合进入沉默的rDNA区域,激活rDNA转录或者继续维持rDNA的异染色质结构稳定?调控H3.3的分子伴侣的含量能否促进体细胞重编程效率?关于这些分子机制的研究对于改善SCNT和iPSC的重编程效率低下、消除体细胞表观记忆残留等问题将很有帮助。未来这一领域的研究将会为利用体细胞重编程技术构建正常的全能性干细胞用于细胞替代治疗、组织器官移植提供巨大的应用前景。(责任编委: 王晓群)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLMagsci [本文引用: 1]

诱导性多能干细胞(Induced pluripotent stem cells, iPSCs)是采用特定转录因子,将体细胞重编程为具有多能性的干细胞。iPSCs已成功由多种体细胞诱导出来,不仅具有发育多能性还能避免胚胎干细胞(Embryonic stem cells, ESCs)的伦理道德问题,已成为生命科学领域不可或缺的研究工具,具有广阔的应用前景。但获得高质量、遗传稳定的iPSCs是当前亟须解决的问题。文章对iPSCs重编程机制和遗传稳定性的研究进展进行了综述,以期为提高iPSCs的诱导效率、降低诱导成本、掌握iPSCs质量控制的关键点提供参考,从而推进多能性干细胞临床应用的发展。
URLMagsci [本文引用: 1]
诱导性多能干细胞(Induced pluripotent stem cells, iPSCs)是采用特定转录因子,将体细胞重编程为具有多能性的干细胞。iPSCs已成功由多种体细胞诱导出来,不仅具有发育多能性还能避免胚胎干细胞(Embryonic stem cells, ESCs)的伦理道德问题,已成为生命科学领域不可或缺的研究工具,具有广阔的应用前景。但获得高质量、遗传稳定的iPSCs是当前亟须解决的问题。文章对iPSCs重编程机制和遗传稳定性的研究进展进行了综述,以期为提高iPSCs的诱导效率、降低诱导成本、掌握iPSCs质量控制的关键点提供参考,从而推进多能性干细胞临床应用的发展。
Magsci [本文引用: 1]
体细胞核移植(Somatic cell nuclear transfer, SCNT)是指将高度分化的体细胞移入到去核的卵母细胞中发育并最终产生后代的技术。然而, 体细胞克隆的总体效率仍然处于一个较低的水平, 主要原因之一是由于体细胞供体核不完全的表观遗传重编程, 包括DNA甲基化、组蛋白乙酰化、基因组印记、X染色体失活和端粒长度等修饰出现的异常。使用一些小分子化合物以及Xist基因的敲除或敲低等方法能修复表观遗传修饰错误, 辅助供体核的重编程, 从而提高体细胞克隆效率, 使其更好地应用于基础研究和生产实践。文章对体细胞核移植后胚胎发育过程中出现的异常表观遗传修饰进行了综述, 并着重论述了近年来有关修复表观遗传错误的研究进展。
Magsci [本文引用: 1]
体细胞核移植(Somatic cell nuclear transfer, SCNT)是指将高度分化的体细胞移入到去核的卵母细胞中发育并最终产生后代的技术。然而, 体细胞克隆的总体效率仍然处于一个较低的水平, 主要原因之一是由于体细胞供体核不完全的表观遗传重编程, 包括DNA甲基化、组蛋白乙酰化、基因组印记、X染色体失活和端粒长度等修饰出现的异常。使用一些小分子化合物以及Xist基因的敲除或敲低等方法能修复表观遗传修饰错误, 辅助供体核的重编程, 从而提高体细胞克隆效率, 使其更好地应用于基础研究和生产实践。文章对体细胞核移植后胚胎发育过程中出现的异常表观遗传修饰进行了综述, 并着重论述了近年来有关修复表观遗传错误的研究进展。
URLPMID:846573 [本文引用: 1]
THE genome of eukaryotes is organised in nucleoprotein fibres formed by the interaction of the DNA with small basic proteins, the histones, and by specific interactions between certain histones. To gain more insight into the molecular organisation of chromosomes it is necessary to determine in detail the complexity and variability of the histones. The chromosomes of eukaryotes contain five major types of histones. One of these, the very lysine-rich H1, consists of a small number of poly-peptides which differ slightly in primary structure and which are present in different relative amounts in different tissues. The other four histones, which are involved in histone-histone interactions, were considered homogeneous and invariable until it became possible to resolve H2a, H2b and H3 into variants which exhibit tissue-specific variation by polyacrylamide gel electrophoresis in presence of non-ionic detergents(Fig. 1). We report here on the primary structure of the variants of mammalian histones 2a, 2b and 3.
[本文引用: 1]
URLPMID:8986613 [本文引用: 1]
We have investigated the expression of a recently described, solitary human H3 histone gene. Using RNase protection assays, the corresponding mRNA could only be detected in RNA preparations from human testis, whereas several human cell lines and somatic tissues did not exhibit expression of this gene. In situ hybridization of sections from human testis revealed expression to be confined to primary spermatocytes. In addition to H1t, this novel H3 gene, which is located on chromosome 1, is the second tissue-specific human histone gene that has been found to be expressed solely in the testis.
URLPMID:20819935 [本文引用: 1]
Nucleosomal incorporation of specialized histone variants is an important mechanism to generate different functional chromatin states. Here, we describe the identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. Their messenger RNAs are found in certain human cell lines, in addition to several normal and malignant human tissues. In keeping with their primate specificity, H3.X and H3.Y are detected in different brain regions. Transgenic H3.X and H3.Y proteins are stably incorporated into chromatin in a similar fashion to the known H3 variants. Importantly, we demonstrate biochemically and by mass spectrometry that endogenous H3.Y protein exists in vivo, and that stress stimuli, such as starvation and cellular density, increase the abundance of H3.Y-expressing cells. Global transcriptome analysis revealed that knockdown of H3.Y affects cell growth and leads to changes in the expression of many genes involved in cell cycle control. Thus, H3.Y is a novel histone variant involved in the regulation of cellular responses to outside stimuli.
URLPMID:21274551 [本文引用: 1]
The incorporation of histone variants into chromatin plays an important role for the establishment of particular chromatin states. Six human histone H3 variants are known to date, not counting CenH3 variants: H3.1, H3.2, H3.3 and the testis-specific H3.1t as well as the recently described variants H3.X and H3.Y. We report the discovery of H3.5, a novel non-CenH3 histone H3 variant. H3.5 is encoded on human chromosome 12p11.21 and probably evolved in a common ancestor of all recent great apes (Hominidae) as a consequence of H3F3B gene duplication by retrotransposition. H3.5 mRNA is specifically expressed in seminiferous tubules of human testis. Interestingly, H3.5 has two exact copies of ARKST motifs adjacent to lysine-9 or lysine-27, and lysine-79 is replaced by asparagine. In the Hek293 cell line, ectopically expressed H3.5 is assembled into chromatin and targeted by PTM. H3.5 preferentially colocalizes with euchromatin, and it is associated with actively transcribed genes and can replace an essential function of RNAi-depleted H3.3 in cell growth.
URLPMID:28374988 [本文引用: 1]
Abstract Non-allelic histone variants are considered as epigenetic factors that regulate genomic DNA functions in eukaryotic chromosomes. In the present study, we identified three new human histone H3 variants (named H3.6, H3.7, and H3.8), which were previously annotated as pseudo-genes. H3.6 and H3.8 conserve the H3.3-specific amino acid residues, but H3.7 shares the specific amino acid residues with H3.1. We successfully reconstituted the nucleosome containing H3.6 in vitro, and determined its crystal structure. In the H3.6 nucleosome, the H3.6-specific Val62 residue hydrophobically contacts the cognate H4 molecule, but its contact area is smaller than that of the corresponding H3.3 Ile62 residue. The thermal stability assay revealed that the H3.6 nucleosome is substantially unstable, as compared to the H3.3 nucleosome. Interestingly, the mutational analysis demonstrated that the H3.6 Val62 residue is fully responsible for the H3.6 nucleosome instability, probably by the reduced hydrophobic interaction with H4. We also reconstituted the nucleosome containing H3.8, but its thermal stability was quite low. In contrast, purified H3.7 failed to form nucleosomes in vitro. The identification and characterization of these novel human histone H3 variants provide important new insights toward understanding the epigenetic regulation of the human genome.
[本文引用: 2]
URLPMID:12086617 [本文引用: 5]
Two very similar H3 histones—differing at only four amino acid positions—are produced in Drosophila cells. Here we describe a mechanism of chromatin regulation whereby the variant H3.3 is deposited at particular loci, including active rDNA arrays. While the major H3 is incorporated strictly during DNA replication, amino acid changes toward H3.3 allow replication-independent (RI) deposition. In contrast to replication-coupled (RC) deposition, RI deposition does not require the N-terminal tail. H3.3 is the exclusive substrate for RI deposition, and its counterpart is the only substrate retained in yeast. RI substitution of H3.3 provides a mechanism for the immediate activation of genes that are silenced by histone modification. Inheritance of newly deposited nucleosomes may then mark sites as active loci.
[本文引用: 2]
URLPMID:20676102 [本文引用: 1]
In mammals, oocyte fertilization by sperm initiates development. This is followed by epigenetic reprogramming of both parental genomes, which involves the de novo establishment of chromatin domains. In the mouse embryo, methylation of histone H3 establishes an epigenetic asymmetry and is predominant in the maternal pronucleus. However, the roles of differential incorporation of histone H3 variants in the parental chromatin, and of modified residues within specific histone variants, have not been addressed. Here we show that the histone variant H3.3, and in particular lysine 27, is required for the establishment of heterochromatin in the mouse embryo. H3.3 localizes to paternal pericentromeric chromatin during S phase at the time of transcription of pericentromeric repeats. Mutation of H3.3 K27, but not of H3.1 K27, results in aberrant accumulation of pericentromeric transcripts, HP1 mislocalization, dysfunctional chromosome segregation and developmental arrest. This phenotype is rescued by injection of double-stranded RNA (dsRNA) derived from pericentromeric transcripts, indicating a functional link between H3.3K27 and the silencing of such regions by means of an RNA-interference (RNAi) pathway. Our work demonstrates a role for a modifiable residue within a histone-variant-specific context during reprogramming and identifies a novel function for mammalian H3.3 in the initial formation of dsRNA-dependent heterochromatin.
URLPMID:12408966 [本文引用: 1]
The multigene family encoding the five classes of replication-dependent histones has been identified from the human and mouse genome sequence. The large cluster of histone genes, HIST1, on human chromosome 6 (6p21 22) contains 55 histone genes, and Hist1 on mouse chromosome 13 contains 51 histone genes. There are two smaller clusters on human chromosome 1: HIST2 (at 1q21), which contains six genes, and HIST3 (at 1q42), which contains three histone genes. Orthologous Hist2 and Hist3 clusters are present on mouse chromosomes 3 and 11, respectively. The organization of the human and mouse histone genes in the HIST1 cluster is essentially identical. All of the histone H1 genes are in HIST1, which is spread over about 2 Mb. There are two large gaps (>250 kb each) within this cluster where there are no histone genes, but many other genes. Each of the histone genes encodes an mRNA that ends in a stemloop followed by a purine-rich region that is complementary to the 5 end of U7 snRNA. In addition to the histone genes on these clusters, only two other genes containing the stem-loop sequence were identified, a histone H4 gene on human chromosome 12 (mouse chromosome 6) and the previously described H2a.X gene located on human chromosome 11. Each of the 14 histone H4 genes encodes the same protein, and there are only three histone H3 proteins encoded by the 12 histone H3 genes in each species. In contrast, both the mouse and human H2a and H2b proteins consist of at least 10 non-allelic variants, making the complexity of the histone protein complement significantly greater than previously thought.
URLPMID:17081972 [本文引用: 1]
Chromatin organization is compromised during the repair of DNA damage. It remains unknown how and to what extent epigenetic information is preserved in vivo. A central question is whether chromatin reorganization involves recycling of parental histones or new histone incorporation. Here, we devise an approach to follow new histone deposition upon UV irradiation in human cells. We show that new H3.1 histones get incorporated in vivo at repair sites. Remarkably we find that H3.1, which is deposited during S phase, is also incorporated outside of S phase. Histone deposition is dependent on nucleotide excision repair (NER), indicating that it occurs at a postrepair stage. The histone chaperone chromatin assembly factor 1 (CAF-1) is directly involved in the histone deposition process in vivo. We conclude that chromatin restoration after damage cannot rely simply on histone recycling. New histone incorporation at repair sites both challenges epigenetic stability and possibly contributes to damage memory.
URLPMID:309673 [本文引用: 1]
Differential hybridization to a cDNA library made from the mRNA of differentiating mouse erythroleukemia (MEL) cells has been used to identify sequences that are induced during the early stages of MEL cell differentiation. One of the differentially expressed genes identified encodes the H3.3 histone subtype. We show here that the three polyadenylated mRNAs produced from the H3.3B gene, as well as the single mRNA produced from the related H3.3A gene, are coordinately induced during the first few hours of MEL cell differentiation and subsequently down regulated as cells undergo terminal differentiation. Nuclear run-on transcription experiments indicate that the accumulation and decay of these mRNAs are controlled at the post-transcriptional level. Unlike the polyadenylated mRNAs of two H1 histone genes that exhibit similar kinetics of induction and decay controlled by c-myc, induction of the H3.3 mRNAs is unaffected by deregulated expression of c-myc.
URLPMID:12909349 [本文引用: 1]
Histones are the major protein component of chromatin. Except H4, all histone classes consist of several subtypes. The H3 family includes two replacement histone genes, H3.3A and H3.3B, which both encode the same protein and are expressed independently from the cell cycle. Since the two genes encode an identical protein, we analyzed whether they are differentially expressed. Therefore we cloned, sequenced and characterized the regulatory structures of the H3.3A gene and compared these with the corresponding regions in the H3.3B gene. In contrast to the H3.3B promoter, the promoter region of the H3.3A gene revealed neither a TATA nor any CCAAT boxes but an initiator element and several SP1 binding sequence motifs within an overall GC-rich sequence. Northern blot analysis of RNA from six human cell lines revealed that every cell line expressed each of the H3 isoform genes H3.1, H3.3A and H3.3B. In contrast, analysis of total RNA from human tissues showed a differential expression of the H3 isoform genes. The H3.3 genes are essentially only expressed in adult tissue, whereas the H3.1 gene is transcribed just in fetal tissue. The functional relevance of the elements identified by sequence analysis was established using a reporter gene assay with deletion constructs of the H3.3A promoter. In this assay a 256 bp fragment was sufficient for the full promoter activity and three promoter segments, each containing SP1 binding motifs, contribute to the H3.3A gene expression. The possible functional relevance of the differences between the two H3.3 genes in structure and expression is discussed.
[本文引用: 1]
URLPMID:20211137 [本文引用: 5]
Abstract The incorporation of histone H3 variants has been implicated in the epigenetic memory of cellular state. Using genome editing with zinc-finger nucleases to tag endogenous H3.3, we report genome-wide profiles of H3 variants in mammalian embryonic stem cells and neuronal precursor cells. Genome-wide patterns of H3.3 are dependent on amino acid sequence and change with cellular differentiation at developmentally regulated loci. The H3.3 chaperone Hira is required for H3.3 enrichment at active and repressed genes. Strikingly, Hira is not essential for localization of H3.3 at telomeres and many transcription factor binding sites. Immunoaffinity purification and mass spectrometry reveal that the proteins Atrx and Daxx associate with H3.3 in a Hira-independent manner. Atrx is required for Hira-independent localization of H3.3 at telomeres and for the repression of telomeric RNA. Our data demonstrate that multiple and distinct factors are responsible for H3.3 localization at specific genomic locations in mammalian cells. (c) 2010 Elsevier Inc. All rights reserved.
[本文引用: 1]
URLPMID:3983652 [本文引用: 1]
Background Nucleosomes are present throughout the genome and must be dynamically regulated to accommodate binding of transcription factors and RNA polymerase machineries by various mechanisms. Despite the development of protocols and techniques that have enabled us to map nucleosome occupancy genome-wide, the dynamic properties of nucleosomes remain poorly understood, particularly in mammalian cells. The histone variant H3.3 is incorporated into chromatin independently of DNA replication and requires displacement of existing nucleosomes for its deposition. Here, we measure H3.3 turnover at high resolution in the mammalian genome in order to present a genome-wide characterization of replication-independent H3.3-nucleosome dynamics. Results We developed a system to study the DNA replication-independent turnover of nucleosomes containing the histone variant H3.3 in mammalian cells. By measuring the genome-wide incorporation of H3.3 at different time points following epitope-tagged H3.3 expression, we find three categories of H3.3-nucleosome turnover in vivo: rapid turnover, intermediate turnover and, specifically at telomeres, slow turnover. Our data indicate that H3.3-containing nucleosomes at enhancers and promoters undergo rapid turnover that is associated with active histone modification marks including H3K4me1, H3K4me3, H3K9ac, H3K27ac and the histone variant H2A.Z. The rate of turnover is negatively correlated with H3K27me3 at regulatory regions and with H3K36me3 at gene bodies. Conclusions We have established a reliable approach to measure turnover rates of H3.3-containing nucleosomes on a genome-wide level in mammalian cells. Our results suggest that distinct mechanisms control the dynamics of H3.3 incorporation at functionally different genomic regions.
URLPMID:25598842 [本文引用: 3]
Abstract BACKGROUND: The histone variant H3.3 plays a critical role in maintaining the pluripotency of embryonic stem cells (ESCs) by regulating gene expression programs important for lineage specification. H3.3 is deposited by various chaperones at regulatory sites, gene bodies, and certain heterochromatic sites such as telomeres and centromeres. Using Tet-inhibited expression of epitope-tagged H3.3 combined with ChIP-Seq we undertook genome-wide measurements of H3.3 dissociation rates across the ESC genome and examined the relationship between H3.3-nucleosome turnover and ESC-specific transcription factors, chromatin modifiers, and epigenetic marks. RESULTS: Our comprehensive analysis of H3.3 dissociation rates revealed distinct H3.3 dissociation dynamics at various functional chromatin domains. At transcription start sites, H3.3 dissociates rapidly with the highest rate at nucleosome-depleted regions (NDRs) just upstream of Pol II binding, followed by low H3.3 dissociation rates across gene bodies. H3.3 turnover at transcription start sites, gene bodies, and transcription end sites was positively correlated with transcriptional activity. H3.3 is found decorated with various histone modifications that regulate transcription and maintain chromatin integrity. We find greatly varying H3.3 dissociation rates across various histone modification domains: high dissociation rates at active histone marks and low dissociation rates at heterochromatic marks. Well- defined zones of high H3.3-nucleosome turnover were detected at binding sites of ESC-specific pluripotency factors and chromatin remodelers, suggesting an important role for H3.3 in facilitating protein binding. Among transcription factor binding sites we detected higher H3.3 turnover at distal cis-acting sites compared to proximal genic transcription factor binding sites. Our results imply that fast H3.3 dissociation is a hallmark of interactions between DNA and transcriptional regulators. CONCLUSION: Our study demonstrates that H3.3 turnover and nucleosome stability vary greatly across the chromatin landscape of embryonic stem cells. The presence of high H3.3 turnover at RNA Pol II binding sites at extragenic regions as well as at transcription start and end sites of genes, suggests a specific role for H3.3 in transcriptional initiation and termination. On the other hand, the presence of well-defined zones of high H3.3 dissociation at transcription factor and chromatin remodeler binding sites point to a broader role in facilitating accessibility.
URL [本文引用: 1]
URLPMID:18066050 [本文引用: 2]
Abstract The remarkable stability of gene expression in somatic cells is exemplified by the way memory of an active gene state is retained when an endoderm cell nucleus is transplanted to an enucleated egg. Here we analyse the mechanism of a similar example of epigenetic memory. We find that memory can persist through 24 cell divisions in the absence of transcription and applies to the expression of the myogenic gene MyoD in non-muscle cell lineages of nuclear transplant embryos. We show that memory is not explained by the methylation of promoter DNA. However, we demonstrate that epigenetic memory correlates with the association of histone H3.3 with the MyoD promoter in embryos that display memory but not in those where memory has been lost. The association of a mutated histone H3.3 (H3.3 E4, which lacks the methylatable H3.3 lysine 4) with promoter DNA eliminates memory, indicating a requirement of H3.3 K4 for memory. We also show that overexpression of H3.3 can enhance memory in transplanted nuclei. We therefore conclude that the association of histone H3.3 with the MyoD promoter makes a necessary contribution to this example of memory. Hence, we suggest that epigenetic memory helps to stabilize gene expression in normal development; it might also help to account for the inefficient reprogramming in some transplanted nuclei.
URLPMID:2783816 [本文引用: 2]
Changes in chromatin composition accompany cellular differentiation in eukaryotes. Although bulk chromatin is duplicated during DNA replication, replication-independent (RI) nucleosome replacement occurs in transcriptionally active chromatin and during specific developmental transitions where the genome is repackaged [1, 2]. In most animals, replacement uses the conserved H3.3 histone variant [3], but02the functions of this variant have not been defined. Using02mutations for the two H3.3 genes in Drosophila, we identify widespread transcriptional defects in H3.3-deficient animals. We show that mutant animals compensate for the lack of H3.302in two ways: they upregulate the expression of the major histone H3 genes, and they maintain chromatin structure by using H3 protein for RI nucleosome replacement at active genes. Rescue experiments show that increased expression of H3 is sufficient to relieve transcriptional defects. In contrast, H3.3 is essential for male fertility, and germline cells specifically require the histone variant. Defects without H3.3 first occur around meiosis, resulting in a failure to condense, segregate, and reorganize chromatin. Rescue experiments with mutated transgenes demonstrate that H3.3-specific residues involved in RI nucleosome assembly—but not major histone modification sites—are required for male fertility. Our results imply that the H3.3 variant plays an essential role in chromatin transitions in the male germline.
URLPMID:3838450 [本文引用: 2]
Polycomb repressive complex 2 (PRC2) regulates gene expression during lineage specification through trimethylation of lysine 27 on histone H3 (H3K27me3). In Drosophila, polycomb binding sites are dynamic chromatin regions enriched with the histone variant H3.3. Here, we show that, in mouse embryonic stem cells (ESCs), H3.3 is required for proper establishment of H3K27me3 at the promoters of developmentally regulated genes. Upon H3.3 depletion, these promoters show reduced nucleosome turnover measured by deposition of de novo synthesized histones and reduced PRC2 occupancy. Further, we show H3.3-dependent interaction of PRC2 with the histone chaperone, Hira, and that Hira localization to chromatin requires H3.3. Our data demonstrate the importance of H3.3 in maintaining a chromatin landscape in ESCs that is important for proper gene regulation during differentiation. Moreover, our findings support the emerging notion that H3.3 has multiple functions in distinct genomic locationsthat are not always correlated with an "active" chromatin state.
URLPMID:19244243 [本文引用: 1]
The H3.3 histone variant is synthesized throughout cell cycle and deposited onto chromatin in a replication-independent manner. It is enriched in transcriptionally active regions of chromatin and is implicated in epigenetic memory. The dynamics of H3.3 deposition during transcriptional activation, however, have not been fully studied so far. Here we examined H3.3 incorporation into interferon (IFN)-stimulated genes in confluent mouse NIH3T3 cells expressing H3.3 fused to the yellow fluorescent protein (YFP). Following IFN stimulation, H3.3-YFP was rapidly incorporated into all four IFN-activated genes tested, with the highest enrichment seen in the distal end of the coding region. Surprisingly, H3.3 enrichment in the coding region continued for an extended period of time, long after transcription ceased. The promoter region, although constitutively enriched with H3.3-YFP, did not show an increase in its deposition in response to IFN stimulation. Further, although H3.3-YFP deposition stably remained in non-dividing cells for days after IFN stimulation, it was rapidly diminished in dividing cells. Lastly, we examined the role of H3.3 in IFN-stimulated transcription by a short hairpin RNA approach and found that IFN-stimulated transcription was significantly impaired in H3.3 knockdown cells. Results indicate that H3.3 plays a role in IFN-mediated transcription, and its deposition leaves a prolonged post-transcriptional mark in these genes.
URLPMID:15851689 [本文引用: 1]
Histones are the fundamental components of the nucleosome. Physiologically relevant variation is introduced into this structure through chromatin remodeling, addition of covalent modifications, or replacement with specialized histone variants. The histone H3 family contains an evolutionary conserved variant, H3.3, which differs in sequence in only five amino acids from the canonical H3, H3.1, and was shown to play a role in the transcriptional activation of genes. Histone H3.3 contains a serine (S) to alanine (A) replacement at amino acid position 31 (S31). Here, we demonstrate by both MS and biochemical methods that this serine is phosphorylated (S31P) during mitosis in mammalian cells. In contrast to H3 S10 and H3 S28, which first become phosphorylated in prophase, H3.3 S31 phosphorylation is observed only in late prometaphase and metaphase and is absent in anaphase. Additionally, H3.3 S31P forms a speckled staining pattern on the metaphase plate, whereas H3 S10 and H3 S28 phosphorylation localizes to the outer regions of condensed DNA. Furthermore, in contrast to phosphorylated general H3, H3.3 S31P is localized in distinct chromosomal regions immediately adjacent to centromeres. These findings argue for a unique function for the phosphorylated isoform of H3.3 that is distinct from its suspected role in gene activation.
[本文引用: 1]
URLPMID:4617904 [本文引用: 1]
Abstract Background The death domain-associated protein (DAXX) collaborates with accessory proteins to deposit the histone variant H3.3 into mouse telomeric and pericentromeric repeat DNA. Pericentromeric repeats are the main genetic contributor to spatially discrete, compact, constitutive heterochromatic structures called chromocentres. Chromocentres are enriched in the H3K9me3 histone modification and serve as integral, functionally important components of nuclear organization. To date, the role of DAXX as an H3.3-specific histone chaperone has been investigated primarily using biochemical approaches which provide genome-wide views on cell populations and information on changes in local chromatin structures. However, the global chromatin and subnuclear reorganization events that coincide with these changes remain to be investigated. Results Using electron spectroscopic imagine (ESI), a specialized form of energy-filtered transmission electron microscopy that allows us to visualize chromatin domains in situ with high contrast and spatial resolution, we show that in the absence of DAXX, H3K9me3-enriched domains are structurally altered and become uncoupled from major satellite DNA. In addition, the structural integrity of nucleoli and the organization of ribosomal DNA (rDNA) are disrupted. Moreover, the absence of DAXX leads to chromatin that is more sensitive, on a global level, to micrococcal nuclease digestion. Conclusions We identify a novel role of DAXX as a major regulator of subnuclear organization through the maintenance of the global heterochromatin structural landscape. As well, we show, for the first time, that the loss of a histone chaperone can have severe consequences for global nuclear organization.
URLPMID:8808624 [本文引用: 1]
DNA repair in the eukaryotic cell disrupts local chromatin organization. To investigate whether the resetting of nucleosomal arrays can be linked to the repair process, we developed model systems, with both Xenopus egg extract and human cell extracts, to follow repair and chromatin assembly in parallel on circular DNA templates. Both systems were able to carry out nucleotide excision repair of DNA lesions. We observed that UV-dependent DNA synthesis occurs simultaneously with chromatin assembly, strongly indicating a mechanistic coupling between the two processes. A complementation assay established that chromatin assembly factor I (CAF1) is necessary for this repair associated chromatin formation.
URLPMID:16117659 [本文引用: 1]
Deposition of the major histone H3 (H3.1) is coupled to DNA synthesis during DNA replication and possibly DNA repair, whereas histone variant H3.3 serves as the replacement variant for the DNA-synthesis-independent deposition pathway. To address how histones H3.1 and H3.3 are deposited into chromatin through distinct pathways, we have purified deposition machineries for these histones. The H3.1 and H3.3 complexes contain distinct histone chaperones, CAF-1 and HIRA, that we show are necessary to mediate DNA-synthesis-dependent and -independent nucleosome assembly, respectively. Notably, these complexes possess one molecule each of H3.1/H3.3 and H4, suggesting that histones H3 and H4 exist as dimeric units that are important intermediates in nucleosome formation. This finding provides new insights into possible mechanisms for maintenance of epigenetic information after chromatin duplication.
URLPMID:27871933 [本文引用: 1]
Incorporation of variant histone sequences, in addition to post-translational modification of histones, serves to modulate the chromatin environment. Different histone chaperone proteins mediate the storage and chromatin deposition of variant histones. Although the two non-centromeric histone H3 variants, H3.1 and H3.3, differ by only five amino acids, replacement of histone H3.1 with H3.3 can modulate transcription for highly expressed and developmentally required genes, lead to the formation of repressive heterochromatin, or aid in DNA and chromatin repair. The human HIRA complex composed of HIRA, UBN1, CABIN1 and transiently ASF1a, forms one of two complexes that binds and deposit H3.3/H4 into chromatin. A number of recent biochemical and structural studies have revealed important details underlying how these proteins assemble and function together as a multi-protein H3.3-specific histone chaperone complex. Here we present a review of existing data and present a new model for assembly of the HIRA complex and for HIRA-mediated incorporation of H3.3/H4 into chromatin.
URLPMID:26159857 [本文引用: 1]
Abstract Histone chaperones bind specific histones to mediate their storage, eviction or deposition from/or into chromatin. The HIRA histone chaperone complex, composed of HIRA, ubinuclein-1 (UBN1) and CABIN1, cooperates with the histone chaperone ASF1a to mediate H3.3-specific binding and chromatin deposition. Here we demonstrate that the conserved UBN1 Hpc2-related domain (HRD) is a novel H3.3-specific-binding domain. Biochemical and biophysical studies show the UBN1-HRD preferentially binds H3.3/H4 over H3.1/H4. X-ray crystallographic and mutational studies reveal that conserved residues within the UBN1-HRD and H3.3 G90 as key determinants of UBN1-H3.3-binding specificity. Comparison of the structure with the unrelated H3.3-specific chaperone DAXX reveals nearly identical points of contact between the chaperone and histone in the proximity of H3.3 G90, although the mechanism for H3.3 G90 recognition appears to be distinct. This study points to UBN1 as the determinant of H3.3-specific binding and deposition by the HIRA complex.
URL [本文引用: 2]
URLPMID:22813747 [本文引用: 1]
Discovering how histone variants affect development is a major challenge in the epigenetics field. Szenker, Lacoste, and Almouzni show a unique requirement of the H3.3 histone variant at late gastrulation during Xenopus development. Depletion of either H3.3 or its chaperone HIRA resulted in similar phenotypes, with comparable losses of H3.3 incorporation into chromatin. These data support the view that a DNA synthesis-independent H3.3 deposition requiring HIRA is critical during early development.
URLPMID:3974909 [本文引用: 1]
The HIRA chaperone complex, comprised of HIRA, UBN1, and CABIN1, collaborates with histone-binding protein ASF1a to incorporate histone variant H3.3 into chromatin in a DNA replication-independent manner. To better understand HIRA's function and mechanism, we integrated HIRA, UBN1, ASF1a, and histone H3.3 chromatin immunoprecipitation sequencing and gene expression analyses. Most HIRA-binding sites colocalize with UBN1, ASF1a, and H3.3 at active promoters and active and weak/poised enhancers. At promoters, binding of HIRA/UBN1/ASF1a correlates with the level of gene expression. HIRA is required for deposition of histone H3.3 at its binding sites. There are marked differences in nucleosome and coregulator composition at different classes of HIRA-bound regulatory sites. Underscoring this, we report physical interactions between the HIRA complex and transcription factors, a chromatin insulator and an ATP-dependent chromatin-remodeling complex. Our results map the distribution of the HIRA chaperone across the chromatin landscape and point to different interacting partners at functionally distinct regulatory sites.
URLPMID:22195966 [本文引用: 1]
Establishment of a proper chromatin landscape is central to genome function. Here, we explain H3 variant distribution by specific targeting and dynamics of deposition involving the CAF-1 and HIRA histone chaperones. Impairing replicative H3.1 incorporation via CAF-1 enables an alternative H3.3 deposition at replication sites via HIRA. Conversely, the H3.3 incorporation throughout the cell cycle via HIRA cannot be replaced by H3.1. ChIP-seq analyses reveal correlation between HIRA-dependent H3.3 accumulation and RNA pol II at transcription sites and specific regulatory elements, further supported by their biochemical association. The HIRA complex shows unique DNA binding properties, and depletion of HIRA increases DNA sensitivity to nucleases. We propose that protective nucleosome gap filling of naked DNA by HIRA leads to a broad distribution of H3.3, and HIRA association with Pol II ensures local H3.3 enrichment at specific sites. We discuss the importance of this H3.3 deposition as a salvage pathway to maintain chromatin integrity.
URLPMID:28107649 [本文引用: 1]
The histone chaperone HIRA is involved in depositing histone variant H3.3 into distinct genic regions, including promoters, enhancers, and gene bodies. However, how HIRA deposits H3.3 to these regions remains elusive. Through a short hairpin RNA (shRNA) screening, we identified single-stranded DNA binding protein replication protein A (RPA) as a regulator of the deposition of newly synthesized H3.3 into chromatin. We show that RPA physically interacts with HIRA to form RPA-HIRA-H3.3 complexes, and it co-localizes with HIRA and H3.3 at gene promoters and enhancers. Depletion of RPA1, the largest subunit of the RPA complex, dramatically reduces both HIRA association with chromatin and the deposition of newly synthesized H3.3 at promoters and enhancers and leads to altered transcription at gene promoters. These results support a model whereby RPA, best known for its role in DNA replication and repair, recruits HIRA to promoters and enhancers and regulates deposition of newly synthesized H3.3 to these regulatory elements for gene regulation.
URLPMID:28126821 [本文引用: 1]
Abstract DNA replication-coupled nucleosome assembly is essential to maintain genome integrity and retain epigenetic information. Multiple involved histone chaperones have been identified, but how nucleosome assembly is coupled to DNA replication remains elusive. Here we show that replication protein A (RPA), an essential replisome component that binds single-stranded DNA, has a role in replication-coupled nucleosome assembly. RPA directly binds free H3-H4. Assays using a synthetic sequence that mimics freshly unwound single-stranded DNA at replication fork showed that RPA promotes DNA-(H3-H4) complex formation immediately adjacent to double-stranded DNA. Further, an RPA mutant defective in H3-H4 binding exhibited attenuated nucleosome assembly on nascent chromatin. Thus, we propose that RPA functions as a platform for targeting histone deposition to replication fork, through which RPA couples nucleosome assembly with ongoing DNA replication. Copyright 2017, American Association for the Advancement of Science.
URL [本文引用: 4]
URL [本文引用: 1]
URLPMID:23075851 [本文引用: 1]
Abstract Histone chaperones represent a structurally and functionally diverse family of histone-binding proteins that prevent promiscuous interactions of histones before their assembly into chromatin. DAXX is a metazoan histone chaperone specific to the evolutionarily conserved histone variant H3.3. Here we report the crystal structures of the DAXX histone-binding domain with a histone H3.3-H4 dimer, including mutants within DAXX and H3.3, together with in vitro and in vivo functional studies that elucidate the principles underlying H3.3 recognition specificity. Occupying 40% of the histone surface-accessible area, DAXX wraps around the H3.3-H4 dimer, with complex formation accompanied by structural transitions in the H3.3-H4 histone fold. DAXX uses an extended 02±-helical conformation to compete with major inter-histone, DNA and ASF1 interaction sites. Our structural studies identify recognition elements that read out H3.3-specific residues, and functional studies address the contributions of Gly09000990 in H3.3 and Glu090009225 in DAXX to chaperone-mediated H3.3 variant recognition specificity.
URLPMID:12953102 [本文引用: 1]
Abstract ATRX syndrome is characterized by X-linked mental retardation associated with alpha-thalassemia. The gene mutated in this disease, ATRX, encodes a plant homeodomain-like finger and a SWI2/SNF2-like ATPase motif, both of which are often found in chromatin-remodeling enzymes, but ATRX has not been characterized biochemically. By immunoprecipitation from HeLa extract, we found that ATRX is in a complex with transcription cofactor Daxx. The following evidence supports that ATRX and Daxx are components of an ATP-dependent chromatin-remodeling complex: (i) Daxx and ATRX can be coimmunoisolated by antibodies specific for each protein; (ii) a proportion of Daxx cofractionates with ATRX as a complex of 1 MDa by gel-filtration analysis; (iii) in extract from cells of a patient with ATRX syndrome, the level of the Daxx-ATRX complex is correspondingly reduced; (iv) a proportion of ATRX and Daxx colocalize in promyelocytic leukemia nuclear bodies, with which Daxx had previously been located; and (v) the ATRX complex displays ATP-dependent activities that resemble those of other chromatin-remodeling complexes, including triple-helix DNA displacement and alteration of mononucleosome disruption patterns. But unlike the previously described SWI/SNF or NURD complexes, the ATRX complex does not randomize DNA phasing of the mononucleosomes, suggesting that it may remodel chromatin differently. Taken together, the results suggest that ATRX functions in conjunction with Daxx in a novel chromatin-remodeling complex. The defects in ATRX syndrome may result from inappropriate expression of genes controlled by this complex.
URLPMID:11882902 [本文引用: 1]
Specific modifications to histones are essential epigenetic markers-heritable changes in gene expression that do not affect the DNA sequence. Methylation of lysine 9 in histone H3 is recognized by heterochromatin protein 1 (HP1), which directs the binding of other proteins to control chromatin structure and gene expression. Here we show that HP1 uses an induced-fit mechanism for recognition of this modification, as revealed by the structure of its chromodomain bound to a histone H3 peptide dimethylated at N味 of lysine 9. The binding pocket for the N-methyl groups is provided by three aromatic side chains, Tyr21, Trp42 and Phe45, which reside in two regions that become ordered on binding of the peptide. The side chain of Lys9 is almost fully extended and surrounded by residues that are conserved in many other chromodomains. The QTAR peptide sequence preceding Lys9 makes most of the additional interactions with the chromodomain, with HP1 residues Val23, Leu40, Trp42, Leu58 and Cys60 appearing to be a major determinant of specificity by binding the key buried Ala7. These findings predict which other chromodomains will bind methylated proteins and suggest a motif that they recognize.
URLPMID:22391447 [本文引用: 1]
Abstract The histone variant macroH2A generally associates with transcriptionally inert chromatin; however, the factors that regulate its chromatin incorporation remain elusive. Here, we identify the SWI/SNF helicase ATRX (02±-thalassemia/MR, X-linked) as a novel macroH2A-interacting protein. Unlike its role in assisting H3.3 chromatin deposition, ATRX acts as a negative regulator of macroH2A's chromatin association. In human erythroleukemic cells deficient for ATRX, macroH2A accumulates at the HBA gene cluster on the subtelomere of chromosome 16, coinciding with the loss of 02±-globin expression. Collectively, our results implicate deregulation of macroH2A's distribution as a contributing factor to the 02±-thalassemia phenotype of ATRX syndrome.
URL [本文引用: 2]
The histone variant H3.3 is implicated in the formation and maintenance of specialized chromatin structure in metazoan cells. H3.3-containing nucleosomes are assembled in a replication-independent manner by means of dedicated chaperone proteins. We previously identified the death domain associated protein (Daxx) and the alpha-thalassemia X-linked mental retardation protein (ATRX) as H3.3-associated proteins. Here, we report that the highly conserved N terminus of Daxx interacts directly with variant-specific residues in the H3.3 core. Recombinant Daxx assembles H3.3/H4 tetramers on DNA templates, and the ATRX-Daxx complex catalyzes the deposition and remodeling of H3.3-containing nucleosomes. We find that the ATRX-Daxx complex is bound to telomeric chromatin, and that both components of this complex are required for H3.3 deposition at telomeres in murine embryonic stem cells (ESCs). These data demonstrate that Daxx functions as an H3.3-specific chaperone and facilitates the deposition of H3.3 at heterochromatin loci in the context of the ATRX-Daxx complex.
URLPMID:22500635
Activity-dependent modifications of chromatin are believed to contribute to dramatic changes in neuronal circuitry. The mechanisms underlying these modifications are not fully understood. The histone variant H3.3 is incorporated in a replication-independent manner into different regions of the genome, including gene regulatory elements. It is presently unknown whether H3.3 deposition is involved in neuronal activity-dependent events. Here, we analyze the role of the histone chaperone DAXX in the regulation of H3.3 incorporation at activity-dependent gene loci. DAXX is found to be associated with regulatory regions of selected activity-regulated genes, where it promotes H3.3 loading upon membrane depolarization. DAXX loss not only affects H3.3 deposition but also impairs transcriptional induction of these genes. Calcineurin-mediated dephosphorylation of DAXX is a key molecular switch controlling its function upon neuronal activation. Overall, these findings implicate the H3.3 chaperone DAXX in the regulation of activity-dependent events, thus revealing a new mechanism underlying epigenetic modifications in neurons.
URLPMID:25865896 [本文引用: 2]
Histone H3.3 is a replication-independent histone variant, which replaces histones that are turned over throughout the entire cell cycle. H3.3 deposition at euchromatin is dependent on HIRA, whereas ATRX/Daxx deposits H3.3 at pericentric heterochromatin and telomeres. The role of H3.3 at heterochromatic regions is unknown, but mutations in the ATRX/Daxx/H3.3 pathway are linked to aberrant telomere lengthening in certain cancers. In this study, we show that ATRX-dependent deposition of H3.3 is not limited to pericentric heterochromatin and telomeres but also occurs at heterochromatic sites throughout the genome. Notably, ATRX/H3.3 specifically localizes to silenced imprinted alleles in mouse ESCs. ATRX KO cells failed to deposit H3.3 at these sites, leading to loss of the H3K9me3 heterochromatin modification, loss of repression, and aberrant allelic expression. We propose a model whereby ATRX-dependent deposition of H3.3 into heterochromatin is normally required to maintain the memory of silencing at imprinted loci.
URLPMID:3125718 [本文引用: 1]
To understand how chromatin structure is organized by different histone variants, we have measured the genome-wide distribution of NCPs (nucleosome core particles) containing the histone variants H3.3 and H2A.Z. We find a special class of NCPs containing both variants, enriched at ‘nucleosome-free regions’ of active promoters, enhancers and insulator regions. We show that previous preparative methods resulted in loss of these unstable double variant NCPs. This instability should facilitate the accessibility of transcription factors to promoters and other regulatory sites in vivo. Other combinations of variants have different distributions, consistent with distinct roles for the histone variants in modulation of gene expression.
URL [本文引用: 1]
Abstract Top of page Abstract Introduction Results And Discussion Methods Acknowledgements References Supporting Information Variant histone H3.3 is incorporated into nucleosomes by a mechanism that does not require DNA replication and has also been implicated as a potential mediator of epigenetic memory of active transcriptional states. In this study, we have used chromatin immunoprecipitation analysis to show that H3.3 is found mainly at the promoters of transcriptionally active genes. We also show that H3.3 combines with H3 acetylation and K4 methylation to form a stable mark that persists during mitosis. Our results suggest that H3.3 is deposited principally through the action of chromatin-remodelling complexes associated with transcriptional initiation, with deposition mediated by RNA polymerase II elongation having only a minor role.
URLPMID:25938714 [本文引用: 2]
Transposable elements (TEs) comprise roughly forty per cent of mammalian genomes1. TEs have played an active role in genetic variation, adaptation, and evolution through the duplication or deletion of genes or their regulatory elements2-4and TEs themselves can act as alternative promoters for nearby genes resulting in non-canonical regulation of transcription5,6. However, TE activity can lead to detrimental genome instability7, and hosts have evolved mechanisms to appropriately silence TE mobility8,9. Recent studies have demonstrated that a subset of TEs, endogenous retroviral elements (ERVs) containing long terminal repeats (LTRs), are silenced through trimethylation of histone H3 on lysine 9 (H3K9me3) by ESET (also known as SETDB1, SET domain bifurcated 1, or KMT1E)10and a co-repressor complex containing KAP1 (KRAB-associated protein 1, also known as tripartite motif-containing protein 28, TRIM28)11in mouse embryonic stem cells (ESCs). Here we show that the replacement histone variant H3.3 is enriched at class I and class II ERVs, notably early transposon (ETn)/MusD and intracisternal A-type particles (IAPs). Deposition at a subset of these elements is dependent upon the H3.3 chaperone complex containing ATRX (alpha thalesemia/mental retardation syndrome X)12and DAXX (Death-associated protein 6)12-14. We demonstrate that recruitment of DAXX, H3.3, and KAP1 to ERVs are co-dependent and upstream of ESET, linking H3.3 to ERV-associated H3K9me3. Importantly, H3K9me3 is reduced at ERVs upon H3.3 deletion, resulting in derepression and dysregulation of adjacent, endogenous genes, along with increased retrotransposition of IAPs. Our study identifies a unique heterochromatin state marked by the presence of both H3.3 and H3K9me3 and establishes an important role for H3.3 in control of ERV retrotransposition in ESCs.
URLPMID:28770848 [本文引用: 2]
On the role of H3.3 in retroviral silencing
URLPMID:27581705 [本文引用: 1]
The histone H3.3 chaperone DAXX is implicated in formation of heterochromatin and transcription silencing, especially for newly infecting DNA virus genomes entering the nucleus. Epstein-Barr virus (EBV) can efficiently establish stable latent infection as a chromatinized episome in the nucleus of infected cells. The EBV tegument BNRF1 is a DAXX-interacting protein required for the establishment of selective viral gene expression during latency. Here we report the structure of BNRF1 DAXX-interaction domain (DID) in complex with DAXX histone-binding domain (HBD) and histones H3.3-H4. BNRF1 DID contacts DAXX HBD and histones through non-conserved loops. The BNRF1-DAXX interface is responsible for BNRF1 localization to PML-nuclear bodies typically associated with host-antiviral resistance and transcriptional repression. Paradoxically, the interface is also required for selective transcription activation of viral latent cycle genes required for driving B-cell proliferation. These findings reveal molecular details of virus reprogramming of an antiviral histone chaperone to promote viral latency and cellular immortalization. The Epstein-Barr virus tegument protein BNRF1 is required for the establishment of selective viral gene expression during latency and interacts with the histone chaperone DAXX. Here the authors provide structural insight into how BNRF1 hijacks the DAXX-histone H3.3-H4 complex.
URLPMID:17717186 [本文引用: 1]
The organization of chromatin affects all aspects of nuclear DNA metabolism in eukaryotes. H3.3 is an evolutionarily conserved histone variant and a key substrate for replication-independent chromatin assembly. Elimination of chromatin remodeling factor CHD1 in Drosophila embryos abolishes incorporation of H3.3 into the male pronucleus, renders the paternal genome unable to participate in zygotic mitoses, and leads to the development of haploid embryos. Furthermore, CHD1, but not ISWI, interacts with HIRA in cytoplasmic extracts. Our findings establish CHD1 as a major factor in replacement histone metabolism in the nucleus and reveal a critical role for CHD1 in the earliest developmental instances of genome-scale, replication-independent nucleosome assembly. Furthermore, our results point to the general requirement of adenosine triphosphate (ATP)-utilizing motor proteins for histone deposition in vivo.
URLPMID:28341773 [本文引用: 1]
Maintenance of chromatin homeostasis involves proper delivery of histone variants to the genome. The interplay between different chaperones regulating the supply of histone variants to distinct chromatin domains is largely undeciphered. We report here a role of promyelocytic leukemia (PML) protein in routing histone variant H3.3 to chromatin and in the organization of megabase-size heterochromatic PML-associated domains which we call PADs. Loss of PML alters the heterochromatic state of PADs by shifting the histone H3 methylation balance from K9me3 to K27me3. Loss of PML impairs deposition of H3.3 by ATRX and DAXX in PADs but preserves the H3.3 loading function of HIRA in these regions. Our results unveil an unappreciated role of PML in the large-scale organization of chromatin and demonstrate a PML-dependent role of ATRX/DAXX in the deposition of H3.3 in PADs. Our data suggest that H3.3 loading by HIRA and ATRX-dependent H3K27 trimethylation constitute mechanisms ensuring maintenance of heterochromatin when integrity of these domains is compromised.
URL [本文引用: 1]
Chromatin reorganization is essential for transcriptional control by sequence-specific transcription factors. However, the molecular link between transcriptional control and chromatin reconfiguration remains unclear. By colocalization of the nuclear ecdysone receptor (EcR) on the ecdysone-induced puff in the salivary gland, Drosophila DEK (dDEK) was genetically identified as a coactivator of EcR in both insect cells and intact flies. Biochemical purification and characterization of the complexes containing fly and human DEKs revealed that DEKs serve as histone chaperones via phosphorylation by forming complexes with casein kinase 2. Consistent with the preferential association of the DEK complex with histones enriched in active epigenetic marks, dDEK facilitated H3.3 assembly during puff formation. In some human myeloid leukemia patients, DEK was fused to CAN by chromosomal translocation. This mutation significantly reduced formation of the DEK complex, which is required for histone chaperone activity. Thus, the present study suggests that at least one histone chaperone can be categorized as a type of transcriptional coactivator for nuclear receptors.
URLPMID:25049225 [本文引用: 2]
Histone variant H3.3 is deposited in chromatin at active sites, telomeres, and pericentric heterochromatin by distinct chaperones, but the mechanisms of regulation and coordination of chaperone-mediated H3.3 loading remain largely unknown. We show here that the chromatin-associated oncoprotein DEK regulates differential HIRA- and DAAX/ATRX-dependent distribution of H3.3 on chromosomes in somatic cells and embryonic stem cells. Live cell imaging studies show that nonnucleosomal H3.3 normally destined to PML nuclear bodies is re-routed to chromatin after depletion of DEK. This results in HIRA-dependent widespread chromatin deposition of H3.3 and H3.3 incorporation in the foci of heterochromatin in a process requiring the DAXX/ATRX complex. In embryonic stem cells, loss of DEK leads to displacement of PML bodies and ATRX from telomeres, redistribution of H3.3 from telomeres to chromosome arms and pericentric heterochromatin, induction of a fragile telomere phenotype, and telomere dysfunction. Our results indicate that DEK is required for proper loading of ATRX and H3.3 on telomeres and for telomeric chromatin architecture. We propose that DEK acts as a "gatekeeper" of chromatin, controlling chromatin integrity by restricting broad access to H3.3 by dedicated chaperones. Our results also suggest that telomere stability relies on mechanisms ensuring proper histone supply and routing.
URL [本文引用: 1]
URLPMID:25142466 [本文引用: 1]
Abstract The histone variant H3.3 is involved in diverse biological processes, including development, transcriptional memory and transcriptional reprogramming, as well as diseases, including most notably malignant brain tumors. Recently, we developed a knockout mouse model for the H3f3b gene, one of two genes encoding H3.3. Here, we show that targeted disruption of H3f3b results in a number of phenotypic abnormalities, including a reduction in H3.3 histone levels, leading to male infertility, as well as abnormal sperm and testes morphology. Additionally, null germ cell populations at specific stages in spermatogenesis, in particular spermatocytes and spermatogonia, exhibited increased rates of apoptosis. Disruption of H3f3b also altered histone post-translational modifications and gene expression in the testes, with the most prominent changes occurring at genes involved in spermatogenesis. Finally, H3f3b null testes also exhibited abnormal germ cell chromatin reorganization and reduced protamine incorporation. Taken together, our studies indicate a major role for H3.3 in spermatogenesis through regulation of chromatin dynamics. 2014. Published by The Company of Biologists Ltd.
URLPMID:24630997 [本文引用: 1]
Abstract It has been demonstrated that reprogramming factors are sequestered in the pronuclei of zygotes after fertilization, because zygotes enucleated at the M phase instead of interphase of the first mitosis can support the development of cloned embryos. However, the contribution of the parental pronucleus derived from either the sperm or the oocyte in reprogramming remains elusive. Here, we demonstrate that the parental pronuclei have asymmetric reprogramming capacities and that the reprogramming factors reside predominantly in the male pronucleus. As a result, only female pronucleus-depleted (FPD) mouse zygotes can reprogram somatic cells to a pluripotent state and support the full-term development of cloned embryos; male pronucleus-depleted (MPD) zygotes fail to support somatic cell reprogramming. We further demonstrate that fusion of an additional male pronucleus into a zygote greatly enhances reprogramming efficiency. Our data provide a clue to further identify critical reprogramming factors in the male pronucleus. Copyright 2014 The Authors. Published by Elsevier Inc. All rights reserved.
URLPMID:22139579 [本文引用: 1]
The genome of differentiated somatic nuclei is remodeled to a totipotent state when they are transplanted into enucleated oocytes. To clarify the mechanism of this genome remodeling, we analyzed changes in the composition of core histone variants in nuclear-transferred embryos, since recent evidence has revealed that chromatin structure can be remodeled as a result of variant histone replacement. We found that the donor cell-derived histone H3 variants H3.1, H3.2, and H3.3, as well as H2A and H2A.Z, were rapidly eliminated from the chromatin of nuclei transplanted into enucleated oocytes. Accompanying this removal, oocyte-stored histone H3 variants and H2A.X were incorporated into the transplanted nuclei, while the incorporation of H2A and H2A.Z was minimal or not detected. The incorporation of these variant histones was DNA replication-independent. These results suggest that most core histone H2A and H3 components are dynamically exchanged between donor nuclei and recipient cytoplasm, which further suggests that replacement of donor cell histones with oocyte-stored histones may play a key role in genome remodeling in nuclear-transferred embryos. In addition, the incorporation patterns of all of the histone variants in the nuclear-transferred embryos were virtually the same as in the fertilized embryos. Only the incorporation pattern of H3.1 differed; it was incorporated into the transplanted donor nuclei, but not in the pronuclei of fertilized embryos. This result suggests that the incorporation of H3.1 has a detrimental effect on the process of genome remodeling and contributes to the low success rate of somatic nuclear cloning.
URLPMID:25482190 [本文引用: 1]
Transfer of a somatic nucleus into an enucleated oocyte is the most efficient approach for somatic cell reprogramming. While this process is known to involve extensive chromatin remodeling of the donor nucleus, the maternal factors responsible and the underlying chromatin-based mechanisms remain largely unknown. Here we discuss our recent findings demonstrating that the histone variant H3.3 plays an essential role in reprogramming and is required for reactivation of key pluripotency genes in somatic cell nuclear transfer (SCNT) embryos. Maternal-derived H3.3 replaces H3 in the donor nucleus shortly after oocyte activation, with the amount of replacement directly related to the differentiation status of the donor nucleus in SCNT embryos. We provide additional evidence to suggest that de novo synthesized H3.3 replaces histone H3 carrying repressive modifications in the donor nuclei of SCNT embryos, and hypothesize that replacement may occur at specific loci that must be reprogrammed for gene reactivation.
URLPMID:24799717 [本文引用: 1]
Transfer of a somatic nucleus into an enucleated oocyte is the most efficient approach for somatic cell reprogramming. While this process is known to involve extensive chromatin remodeling of the donor nucleus, the maternal factors responsible and the underlying chromatin-based mechanisms remain largely unknown. Here we discuss our recent findings demonstrating that the histone variant H3.3 plays an essential role in reprogramming and is required for reactivation of key pluripotency genes in somatic cell nuclear transfer (SCNT) embryos. Maternal-derived H3.3 replaces H3 in the donor nucleus shortly after oocyte activation, with the amount of replacement directly related to the differentiation status of the donor nucleus in SCNT embryos. We provide additional evidence to suggest that de novo synthesized H3.3 replaces histone H3 carrying repressive modifications in the donor nuclei of SCNT embryos, and hypothesize that replacement may occur at specific loci that must be reprogrammed for gene reactivation.
URLPMID:23472871 [本文引用: 1]
Previous studies of serial cloning in animals showed a decrease in efficiency over repeated iterations and a failure in all species after a few generations. This limitation led to the suggestion that repeated recloning might be inherently impossible because of the accumulation of lethal genetic or epigenetic abnormalities. However, we have now succeeded in carrying out repeated recloning in the mouse through a somatic cell nuclear transfer method that includes a histone deacetylase inhibitor. The cloning efficiency did not decrease over 25 generations, and, to date, we have obtained more than 500 viable offspring from a single original donor mouse. The reprogramming efficiency also did not increase over repeated rounds of nuclear transfer, and we did not see the accumulation of reprogramming errors or clone-specific abnormalities. Therefore, our results show that repeated iterative recloning is possible and suggest that, with adequately efficient techniques, it may be possible to reclone animals indefinitely.
URLPMID:25692725 [本文引用: 2]
Although viable mice can be generated from induced pluripotent stem cells (iPSCs), the impact of accumulated mutations on the developmental potential of the resulting iPSCs remains to be determined. Here, we demonstrate that all-iPSC mice generated through tetraploid blastocysts complementation can tolerate the accumulation of somatic mutations for up to six generations using a Tet-on inducible reprogramming system. But, the viability of the all-iPS mice decreased with increasing generations. A whole-genome sequencing survey revealed that thousands of single-nucleotide variations (SNVs), including 44 non-synonymous ones, accumulated throughout the sequential reprogramming process. Subsequent analysis provides evidence that these accumulated SNVs account for the gradual reduction in viability of the resultant all-iPSC mice. Unexpectedly, our present reprogramming system revealed that pluripotent stem cells are heterogeneous in terms of possessing a set of copy-number alterations (CNAs). These CNAs are unique for pluripotent cells and subsequently disappear in the differentiating progenies.
URLPMID:24268696 [本文引用: 1]
正Telomere shortening and mitochondrial dysfunction are closely linked with the degenerative pathogenesis in humans.Reversion of telomere dysfunction and mitochondrial defects of patients has been considered crucial for the acquisition of authentic pluripotency by somatic cell reprogramming.Somatic cell nuclear transfer(SCNT)and induced pluripotent stem cells(iPSCs)
URLPMID:22467869 [本文引用: 2]
The well known and most important function of nucleoli is ribosome biogenesis. However, the nucleolus showed delayed development and malfunction in somatic cell nuclear transfer (NT) embryos. Previous studies indicated that nearly half rRNA genes (rDNA) in somatic cells were inactive and not transcribed. We compared the rDNA methylation level, active nucleolar organizer region (NORs) numbers, nucleolar proteins (upstream binding factor (UBF), nucleophosmin (B23)) distribution, and nucleolar-related gene expression in three different donor cells and NT embryos. The results showed embryonic stem cells (ESCs) had the most active NORs and lowest rDNA methylation level (7.66 and 6.76%), whereas mouse embryonic fibroblasts (MEFs) were the opposite (4.70 and 22.57%). After the donor cells were injected into enucleated MII oocytes, cumulus cells and MEFs nuclei lost B23 and UBF signals in 20 min, whereas in ESC-NT embryos, B23 and UBF signals could still be detected at 60 min post-NT. The embryos derived from ESCs, cumulus cells, and MEFs showed the same trend in active NORs numbers (7.19 versus 6.68 versus 5.77, p < 0.05) and rDNA methylation levels (6.36 versus 9.67% versus 15.52%) at the 4-cell stage as that in donor cells. However, the MEF-NT embryos displayed low rRNA synthesis/processing potential at morula stage and had an obvious decrease in blastocyst developmental rate. The results presented clear evidences that the rDNA reprogramming efficiency in NT embryos was determined by the rDNA activity in donor cells from which they derived.
URLPMID:4981958 [本文引用: 2]
rDNA, the genes encoding ribosomal RNA (rRNA), is highly demanded for ribosome production and protein synthesis in growing cells such as pluripotent stem cells. rDNA transcription activity varies between cell types, metabolism conditions, and specific environmental challenges. Embryonic stem cells (ESCs), partially reprogrammed cells, and somatic cells reveal different epigenetic signatures, including rDNA epigenetic marks. rDNA epigenetic characteristic resetting is not quite clear during induced pluripotent stem cell (iPSC) generation. Little is known that whether the different rDNA epigenetic status in donor cells will result in different rDNA transcription activities, and furthermore affect reprogramming efficiency. We utilized serum starvation-synchronized mouse embryonic fibroblasts (MEFs) to generate S-iPSCs. Both MEFs and serum-refeeding MEFs (S-MEFs) were reprogrammed to a pluripotent state. rDNA-related genes, UBF proteins, and rDNA methylation levels were detected during the MEF and S-MEF cell reprogramming process. We demonstrated that, after transient inhibition, retroviral induced rRNA transcriptional activity was reprogrammed towards a pluripotent state. Serum starvation would stimulate rDNA transcription reactivation during somatic cell reprogramming. Serum starvation improved the methylation status of donor cells at rRNA gene promoter regions. Our results provide insight into regulation of rDNA transcriptional activity during somatic cell reprogramming and allow for comparison of rDNA regulation patterns between iPSCs and S-iPSCs. Eventually, regulation of rDNA transcriptional activity will benefit partially reprogrammed cells to overcome the epigenetic barrier to pluripotency. The online version of this article (doi:10.1186/s13287-016-0369-1) contains supplementary material, which is available to authorized users.
URLPMID:25087892 [本文引用: 2]
Abstract Extensive chromatin reprogramming occurs at fertilization and is thought to be under the control of maternal factors, but the underlying mechanisms remain poorly understood. We report that maternal Hira, a chaperone for the histone variant H3.3, is required for mouse development past the zygote stage. Male pronucleus formation is inhibited upon deletion of Hira due to a lack of nucleosome assembly in the sperm genome. Hira mutant oocytes are incapable of developing parthenogenetically, indicative of a role for Hira in the female genome. Both parental genomes show highly reduced levels of DNA replication and transcription in the mutants. It has long been thought that transcription is not required for zygote development. Surprisingly, we found that Hira/H3.3-dependent transcription of ribosomal RNA is required for first cleavage. Our results demonstrate that Hira-mediated H3.3 incorporation is essential for parental genome reprogramming and reveal an unexpected role for rRNA transcription in the mouse zygote. Copyright 2014 Elsevier Inc. All rights reserved.
URLPMID:23102146 [本文引用: 3]
Background Nuclear reprogramming is potentially important as a route to cell replacement and drug discovery, but little is known about its mechanism. Nuclear transfer to eggs and oocytes attempts to identify the mechanism of this direct route towards reprogramming by natural components. Here we analyze how the reprogramming of nuclei transplanted to Xenopus oocytes exploits the incorporation of the histone variant H3.3. Results After nuclear transplantation, oocyte-derived H3.3 but not H3.2, is deposited on several regions of the genome including rDNA, major satellite repeats, and the regulatory regions of Oct4. This major H3.3 deposition occurs in absence of DNA replication, and is HIRA-and transcription-dependent. It is necessary for the shift from a somatic- to an oocyte-type of transcription after nuclear transfer. Conclusions This study demonstrates that the incorporation of histone H3.3 is an early and necessary step in the direct reprogramming of somatic cell nuclei by oocyte. It suggests that the incorporation of histone H3.3 is necessary during global changes in transcription that accompany changes in cell fate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}