 ,西北农林科技大学动物医学院,杨凌 712100
,西北农林科技大学动物医学院,杨凌 712100Expression specificity and compensation effect of Ash2l-1/Ash2l-2 in mouse embryonic stem cells
Jing Xie, Chen Fan, Jinglong Zhang, Shiqiang Zhang,College of Veterinary Medicine, Northwest A&F University, Yangling 712100, China通讯作者:
第一联系人:
编委: 谷峰
收稿日期:2017-08-28修回日期:2017-12-21网络出版日期:2018-03-20
| 基金资助: |
Editorial board:
Received:2017-08-28Revised:2017-12-21Online:2018-03-20
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2036KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
谢晶, 范辰, 张景龙, 张仕强. Ash2l-1/Ash2l-2在小鼠胚胎干细胞中的表达特异性及互补效应. 遗传[J], 2018, 40(3): 237-249 doi:10.16288/j.yczz.17-284
Jing Xie, Chen Fan, Jinglong Zhang, Shiqiang Zhang.
胚胎干细胞(embryonic stem cells, ESCs)是一类具有自我更新能力的细胞,具有无限增殖和多向分化潜能。由于这种特性,ESCs已成为再生医学和细胞治疗的重要种子细胞[1,2]。表观遗传修饰可以在不改变碱基序列的情况下,通过改变染色质的结构调节基因的表达,调控ESCs的自我更新[3]。组蛋白氨基末端残基的翻译后修饰对于调节基因的表达具有重要作用,如甲基化、乙酰化、磷酸化、泛素化及ADP核糖基化等。组蛋白赖氨酸甲基化主要由组蛋白赖氨酸甲基转移酶完成,在组蛋白赖氨酸残基上主要发生单甲基化、双甲基化和三甲基化修饰。通常H3K4、H3K36及H3K79甲基化与基因激活相关,而H3K9、H3K27及H4K20甲基化与基因沉默有关。在小鼠胚胎干细胞(mouse embryonic stem cells, mESCs)中,染色质存在高水平的H3K4三甲基化(H3K4 trimethylation, H3K4me3)和H3K4乙酰化修饰,同时存在低水平的H3K9me3修饰[4,5,6]。
H3K4me3主要分布在常染色质区转录活化基因的启动子区,可与组蛋白乙酰化酶、去乙酰化酶等相互作用改变邻近组蛋白甲基化修饰状态,调控基因转录[4,5,6]。在酵母(Saccharomyces cerevisiae)中,H3K4me3由Set1调节;在哺乳动物中,主要由MLL (mixed lineage leukemia)家族蛋白(包括MLL1、MLL2、MLL3、MLL4、SET1A和SET1Bt)调节[7]。ASH2L(absent, small, homeotic-2-like)是果蝇ASH2 (absent, small, homeotic discs)的同源物[8],其与RBBP5(retinoblastoma binding protein 5)、DPY30 (dumpy 30 protein)及WDR5(WD repeat containing protein 5)共同组成了MLL甲基化转移酶复合体的核心亚单位[9,10,11]。有研究表明,一旦ASH2L缺失,MLL复合体不能进行H3K4me3修饰[12]。Ash2l缺失的小鼠会在妊娠期死亡,揭示Ash2l在小鼠胚胎发展过程中具有重要作用[13]。Wan等[14]发现Ash2l可通过调节H3K4me3修饰影响染色质的结构,调控mESCs自我更新能力。
CRISPR/Cas9(clustered regularly interspaced short palindromic repeats/CRISPR-associated 9)是一种新型的基因组定点编辑技术,常用的Ⅱ型CRISPR系统由负责靶向的导向RNA(guide RNA, gRNA)与负责切割的Cas9核酸酶组成。当基因组上PAM(protospacer adjacent motif)区5′端的前体间隔序列与gRNA中的引导序列互补时,Cas9核酸酶可以对DNA双链进行切割,引发DNA双链断裂,激活细胞内非同源末端连接和同源重组两种DNA修复机制[15]。目前,CRISPR/Cas9基因组编辑技术已被应用于制备多种动物以及细胞的基因敲除[16,17,18]。
本文研究发现,ASH2L在mESCs中存在2个异构体:ASH2L-1和ASH2L-2,且ASH2L-2具有多能干细胞表达特异性。目前,Ash2l-1和Ash2l-2在mESCs中的作用尚不清楚。本文进一步利用CRISPR/Cas9基因组编辑技术建立了Ash2l-1-/-和Ash2l-2-/- mESCs,并分析了分别缺失Ash2l两个变异体对mESCs的影响,为进一步研究Ash2l-1和Ash2l-2对mESCs的作用机制奠定了基础。
1 材料和方法
1.1 mESCs培养
本文使用3株mESCs,分别是TT2(加拿大Matthew Lorincz实验室馈赠,品系为C57BL/6 X CBA)、J1(购自美国模式培养集存库ATCC,品系为129S4/SvJae)及E14TG2a(购自中国科学院干细胞库,品系为129/Ola)。mESCs培养基成分为DMEM (HyClone,美国)、15%胎牛血清(Gibco,美国)、100× GlutaMAX(Gibco,美国)、100×NEAA(Gibco,美国)、100×青霉素/链霉素(Hyclone,美国)、1000 U/mL LIF(Millipore,美国)、1000×β-Mercaptoetanol(Gibco,美国)。在37℃、5% CO2的培养箱中培养。培养皿预先用0.1%明胶于37℃培养箱中包被2 h。每24 h更换新鲜培养基,当细胞密度达80%时,用0.25% (W/V)胰蛋白酶消化传代。1.2 Western blotting检测ASH2L和H3K4me3的表达
Western blotting检测ASH2L表达时,按照以下方法提取蛋白:取5~10×106个细胞,用预冷的PBS漂洗2遍。每106个细胞加入100 μL的RIPA裂解液,冰上放置20 min。于4℃、1 400 g离心15 min;收集上清,用BCA试剂盒(晶彩生物,西安)测定蛋白质浓度。Western blotting检测H3K4me3表达时,按照以下方法提取组蛋白:每106个细胞加入500 μL的0.5% NP-40,冰上放置10 min。于4℃、5000 g离心5 min。加入0.2 mol/L盐酸重悬细胞,4℃过夜。于4℃、20 000 g离心5 min;收集上清,加入1.5 mol/L Tris(pH 8.8)中和。每孔蛋白上样20~35 μg,进行SDS-PAGE凝胶(5%浓缩胶;ASH2L为10%分离胶;H3K4me3为15%分离胶)电泳。电泳条件分别为:80 V,30 min;120 V,1 h 30 min。电泳结束后采用PVDF膜进行转膜 (15 V,45 min)。然后加入一抗于4℃过夜孵育。ASH2L抗体(Cell Signaling,美国)的稀释比例为1∶2000;GAPDH抗体(三箭生物,天津)的稀释比例为1∶5000。H3K4me3抗体(Active Motif,美国)的稀释比例为1∶1000;组蛋白 H3(Sino biological,北京)的稀释比例为1∶1000。一抗孵育后使用TBST漂洗3遍,每次10 min;再加入羊抗兔二抗(三箭生物,天津,稀释比例为1∶5000)和羊抗鼠二抗(三箭生物,天津,稀释比列为1∶5000),室温孵育1 h。TBST漂洗3遍,每次10 min;最后用ECL试剂盒曝光检测。采用Image J软件对3次蛋白结果进行分析,以GAPDH和组蛋白H3为内参,计算目的蛋白的相对表达量。
1.3 gRNA/Cas9打靶载体的构建
在UCSC网站(http://genome.ucsc.edu/)查询Ash2l-1和Ash2l-2的启动子元件,在Optimized CRISPR Design网站(http://crispr.mit.edu)分别设计靶向Ash2l-1-CpG岛和Ash2l-2-RLTR12B的gRNA,序列见表1。hPuro2- hU6-gRNA-CBh-hCas9载体(Matthew Lorincz实验室馈赠)可以同时表达gRNA和Cas9蛋白。用高保真DNA聚合酶复性合成gRNA,PCR扩增条件为:96℃ 3 min,96℃ 30 s,50℃ 30 s,72℃ 3 min,3个循环;72℃ 5 min,10℃保温。用DNA纯化回收试剂盒(TIANGEN,北京)进行胶回收。用SapⅠ(NEB,美国)和XbaⅠ(NEB,美国)限制性内切酶酶切gRNA和骨架载体,经连接、转化感受态细胞、挑取细菌单克隆、摇菌,用质粒提取试剂盒(TIANGEN,北京)抽提质粒。用NdeⅠ(NEB,美国)和SapⅠ限制性内切酶对质粒进行酶切,最后测序鉴定。Table 1
表1
表1 靶向Ash2l-1-CpG岛和Ash2l-2-RLTR12B的gRNAs序列信息
Table 1
| 名称 | 序列(5′→3′) | PAM | Tm(℃) | GC(%) |
|---|---|---|---|---|
| Ash2l-1-CpG岛-5′-gRNA-1 | ACTTTGAGTCCGCCGCTTTA | AGG | 58 | 50 |
| Ash2l-1-CpG岛-5′-gRNA-2 | AAAACGCCTGCTCATAGTCG | TGG | 57 | 50 |
| Ash2l-1-CpG岛-3′-gRNA-1 | TGCACTGGGCTAACGACGGG | AGG | 63 | 65 |
| Ash2l-1-CpG岛-3′-gRNA-2 | GACAAATGTGTTCGCGGCCC | AGG | 61 | 60 |
| Ash2l-2-RLTR12B-5′gRNA-1 | CTAATTACTGCGACAGTTGC | AGG | 53 | 45 |
| Ash2l-2-RLTR12B-5′-gRNA-2 | GAGAACCGCGATAGCAGTCC | AGG | 59 | 60 |
| Ash2l-2-RLTR12B-3′-gRNA-1 | AAATGCGTGTGCGCGCGCTG | GGG | 67 | 65 |
| Ash2l-2-RLTR12B-3′-gRNA-2 | TGCCAGCAAACCCGAGGATC | TGG | 61 | 60 |
新窗口打开|下载CSV
1.4 有效转染组合的筛选
当TT2细胞密度达到50%时,将gRNA/Cas9打靶载体组成4种组合(表2),分别使用Lipofectamine 3000(Invitrogen,美国)转染TT2。24 h后,换成含嘌呤霉素(2 μg/mL)的mESCs培养基进行筛选。用嘌呤霉素处理48 h后,更换为正常的mESCs培养基继续培养。当细胞密度达到80%时,提取细胞基因组DNA,进行PCR鉴定。针对DNA切割靶位点的上下游设计PCR扩增引物(引物序列见表3),根据PCR产物大小确定有效的转染组合。在鉴定Ash2l-2-RLTR12B敲除实验中,PCR引物位置示意图见结果2.2部分C图。第1、2、3对引物组合分别为1F+1R、2F+2R和1F+2R。当无敲除发生时,第1、2、3对引物分别扩增出大小为500 bp、509 bp和2120 bp的条带。当双等位基因敲除发生时,第1、2对引物扩增不出条带,第3对引物扩增出大小为809 bp的条带。当单等位基因敲除发生时,第1、2对引物扩增出大小为500 bp和509 bp的条带,第3对引物扩增出大小分别为2120 bp和809 bp的条带。
Table 2
表2
表2 gRNA/Cas9载体的转染组合
Table 2
| 靶点 | gRNA/Cas9载体的转染组合 |
|---|---|
| Ash2l-1-CpG岛 | 组合1:5-gRNA-1/Cas9与3′-gRNA-1/Cas9 |
| 组合2:5′-gRNA-1/Cas9与3′-gRNA-2/Cas9 | |
| 组合3:5′-gRNA-2/Cas9与3′-gRNA-1/Cas9 | |
| 组合4:5′-gRNA-2/Cas9与3′-gRNA-2/Cas9 | |
| Ash2l-2-RLTR12B | 组合1:5′-gRNA-1/Cas9与3′-gRNA-1/Cas9 |
| 组合2:5′-gRNA-1/Cas9与3′-gRNA-2/Cas9 | |
| 组合3:5′-gRNA-2/Cas9与3′-gRNA-1/Cas9 | |
| 组合4:5′-gRNA-2/Cas9与3′-gRNA-2/Cas9 |
新窗口打开|下载CSV
Table 3
表3
表3 PCR鉴定引物序列信息
Table 3
| 引物名称 | 引物序列(5′→3′) | Tm(℃) | GC(%) |
|---|---|---|---|
| Ash2l-1-CpG岛-1 | F: AGTGTCTCCGGTACCTTCCT | 58 | 55 |
| R: GTGTGAGCCAAGAGCGTTTC | 58 | 55 | |
| Ash2l-1-CpG岛-2 | F: CCTTACCCACTTTCGGCCTC | 59 | 60 |
| R: TGCTCCGCACAGTGTAAAGG | 59 | 55 | |
| Ash2l-1-CpG岛-3 | F: CCCACACTGCAACTCCACTTCTCCCAACTGCCGCCCAT | 75 | 61 |
| R: CCACCCACAACTACCTCGGGCTCTCTGGAATCAGACCT | 72 | 58 | |
| Ash2l-2-RLTR12B-1 | F: GTCCCCCACTGACATGGTTT | 58 | 55 |
| R: TCAAGGGCTGCGAATTGTCT | 58 | 50 | |
| Ash2l-2-RLTR12B-2 | F: CCTTAGACCTTGCTTGGTGACA | 58 | 50 |
| R: AGCAAGAATGGCTCATGGTAGA | 57 | 45 |
新窗口打开|下载CSV
在鉴定Ash2l-1-CpG岛敲除实验中,PCR引物位置示意图见结果2.2部分E图。第1、2对引物组合分别为1F+1R和2F+2R。由于CpG岛中的GC含量高,第3对引物组合1F+2R无法扩增出对应的长片段。针对此问题,在1F和2R的位置附近设计引物3F和3R作为最后选用的第3对引物组合见结果2.2部分E图。当无敲除发生时,第1、2、3对引物分别扩增出大小为500 bp、538 bp和1505 bp的条带。当双等位基因敲除发生时,第1、2对引物扩增不出条带,第3对引物扩增出大小为701 bp的条带。当单等位基因敲除发生时,第1、2对引物分别扩增出大小为500 bp和538 bp的条带,第3对引物扩增出大小分别为1505 bp和701 bp的条带。
1.5 单克隆获取与基因型鉴定
当确定有效的gRNA/Cas9打靶载体组合后,采用上述转染和筛选方法,挑取单克隆至96孔板中。当细胞密度达80%时,取一半细胞接种于24孔板,继续培养,冻存;取一半细胞接种于96孔板,提取基因组DNA。以基因组DNA为模板,采用上述PCR鉴定方法,初步鉴定发生双等位基因敲除的单克隆。然后将PCR产物连接到pGEM-T Easy载体(Promega,美国)进行测序验证。1.6 碱性磷酸酶(alkaline phosphatase, AP)染色
野生型mESCs、Ash2l-1-/-和Ash2l-2-/- mESCs分别用PBS漂洗2次,加入4%多聚甲醛,室温固定20 min。PBS漂洗3次,加入AP染色液进行染色。AP染色液配制方法:称取5 mg的Fast Red TR(Sigma,美国)和2 mg的α-磷酸萘酚钠盐(Sigma,美国),溶解于5 mL的0.1 mol/L Tris-HCl(pH9.0)中。10~20 min后阳性细胞呈现红色,PBS漂洗以终止反应,在显微镜下观察照相。1.7 免疫荧光染色检测多能性调控转录因子的表达
野生型mESCs、Ash2l-1-/-和Ash2l-2-/- mESCs分别用PBS漂洗2次,加入4%多聚甲醛,室温固定20 min。PBS漂洗3次,加入0.25% Triton-X,室温处理30 min。PBS漂洗3次,加入1% BSA室温封闭1 h;4℃过夜孵育一抗(Oct4,1:200,Santa Cruz,美国;Sox2,1:200,Santa Cruz,美国;Nanog,1:200,Abcam,英国)。PBS漂洗3次,加入二抗(Alexa Fluor 488 donkey anti-mouse IgG(H+L),1:500,Life Technologies,美国;Alexa Fluor 596 donkey anti-rabbit IgG(H+L),1:500,Life Technologies,美国),室温孵育1 h。PBS漂洗2次后,DAPI(1:1000,Roche,瑞士)染色2 min,PBS漂洗2次,在荧光显微镜下观察照相。1.8 拟胚体(embryoid body, EB)的培养和qRT-PCR
当野生型mESCs、Ash2l-1-/-和Ash2l-2-/- mESCs的细胞密度达80%时,用0.25%(W/V)胰蛋白酶消化,吹打为单细胞。140 g离心5 min,去除上清,用撤去LIF的mESCs分化培养基重悬细胞。收集细胞(2×105细胞/mL)到细菌培养皿中培养。悬浮培养8 d后,将EB转移至铺有0.1%明胶的培养皿中进行贴壁培养。贴壁培养8 d后,PBS漂洗1次,加入RZ裂解液,提取总RNA(TIANGEN,北京)。使用反转录试剂盒(TransGen,北京)将RNA反转录为cDNA,进行qRT-PCR,检测三胚层标志基因的表达。实时定量PCR扩增体系:2 μL的cDNA,12.5 μL的TransStart Tip Green qPCR SuperMix(TransGen,北京),各0.3 μmol/L的上下游引物,加水补充到25 μL。以Gapdh为内参,以2-ΔΔCt相对定量进行分析。所有数据均用3次独立实验的平均值±标准偏差表示,并经One-Way ANOVA检验差异是否具有统计学意义。1.9 启动子区转录因子结合位点的预测
利用JASPAR数据库(http://jaspar.genereg.net/)分别预测Ash2l-1-CpG岛和Ash2l-2-RLTR12B启动子区转录因子的结合位点。在DAVID数据库(https:// david.ncifcrf.gov/)中进行KEGG(Kyoto Encyclopedia of Genes and Genomes)分析。2 结果与分析
2.1 mESCs中ASH2L-1和ASH2L-2的表达
通过Western blotting检测3株mESCs (TT2、J1、E14)和小鼠胚胎成纤维细胞(mouse embryonic fibroblast,MEF)中ASH2L的表达。Western blotting和定量分析结果显示,ASH2L在mESCs中有2个异构体:ASH2L-1(80 kDa)和ASH2L-2(65 kDa),且以ASH2L-2的表达为主。而在MEF中,只有ASH2L-1表达(图1:A,B)。这表明ASH2L蛋白在mESCs中存在ASH2L-1和ASH2L-2两个异构体,且ASH2L-2具有干细胞表达特异性。经NCBI网站查询可知,ASH2L-1由623个氨基酸组成,ASH2L-2由534个氨基酸组成。ASH2L-1除了在N端比ASH2L-2多89个氨基酸外,其他序列与ASH2L-2一样,二者都包含PHD、WH和SPRY结构域(图1C)。图1
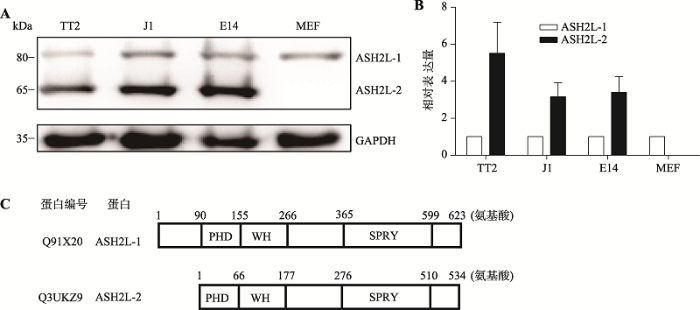 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1ASH2L-1和ASH2L-2的表达
A, B:Western blotting检测ASH2L-1和ASH2L-2的表达及定量分析;C:ASH2L-1和ASH2L-2氨基酸序列结构示意图。
Fig. 1Expression of ASH2L-1 and ASH2L-2
2.2 gRNA/Cas9打靶载体的构建和有效转染组合的筛选
在UCSC网站查询发现,ASH2L-1由CpG岛启动元件驱动表达,而ASH2L-2由长末端重复序列RLTR12B启动元件驱动表达。由此,分别在二者的启动元件两边设计gRNA,其测序结果与设计的序列一致(图2:A,B),表明hPuro2-hU6-gRNA-CBh- hCas9载体构建成功。在鉴定Ash2l-2-RLTR12B敲除实验中,转染组合3可以有效引导gRNA/Cas9打靶载体敲除Ash2l- 2-RLTR12B。PCR引物位置示意图见图2C,凝胶电泳结果见图2D。对照组为野生型mESCs,第1、2、3对引物分别扩增出大小为500 bp、509 bp和2120 bp的条带。实验组分别为发生双等位和单等位基因敲除的单克隆A和B。对于单克隆A,第1、2对引物扩增不出条带,第3对引物扩增出大小为809 bp的条带。对于单克隆B,第1、2对引物分别扩增出大小为500 bp和509 bp的条带,第3对引物扩增出大小分别为2120 bp和809 bp的条带。
在鉴定Ash2l-1-CpG岛敲除实验中,转染组合4可以有效引导gRNA/Cas9打靶载体敲除Ash2l-1- CpG岛。PCR引物位置示意图见图2E,凝胶电泳结果见图2F。对照组为野生型mESCs,第1、2、3对引物分别扩增出大小为500 bp、538 bp和1505 bp的条带。实验组为分别发生双等位和单等位基因敲除的单克隆C和D。对于单克隆C,第1、2对引物扩增不出条带,第3对引物扩增出大小为701 bp的条带。对于单克隆D,第1、2对引物分别扩增出大小为500 bp和538 bp的条带,第3对引物扩增出大小分别为1505 bp和701 bp的条带。
本文统计分析了gRNA/Cas9打靶载体的敲除效率。在挑取的靶向敲除Ash2l-1-CpG岛的48个单克隆中,野生型单克隆有26个;发生单等位基因敲除的单克隆有14个,敲除效率为29.2%;发生双等位基因敲除的单克隆有8个,敲除效率为16.7%。在挑取的靶向敲除Ash2l-2-RLTR12B的32个单克隆中,野生型单克隆有22个;发生单等位基因敲除的单克隆有7个,敲除效率为21.9%;发生双等位基因敲除的单克隆有3个,敲除效率为9.4%。统计结果见表4。
图2
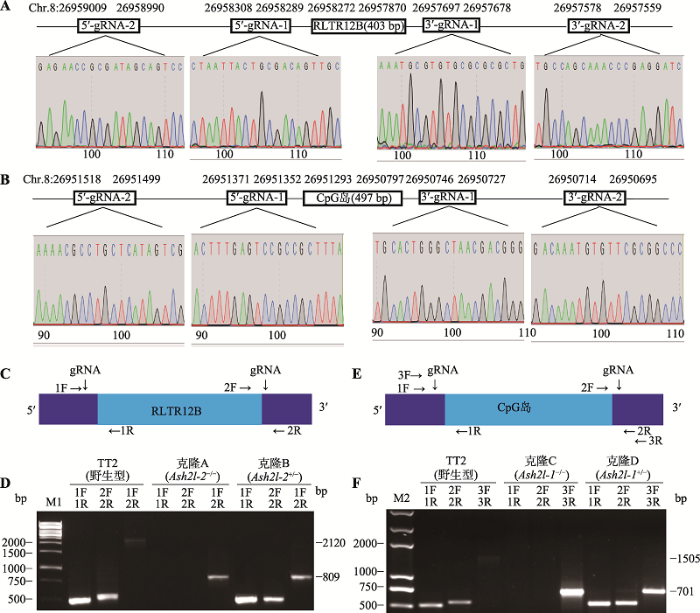 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2gRNA测序和单克隆鉴定
A,B:靶向Ash2l-1-CpG岛和Ash2l-2-RLTR12B启动子的gRNA位置示意图及测序结果。C:Ash2l-2敲除鉴定中PCR引物位置示意图。D:Ash2l-2敲除鉴定。M1为AL10000 Plus DNA Marker。第1对引物(1F+1R)(针对野生型等位基因)产物为500 bp,第2对引物(2F+2R)(针对野生型等位基因)产物为509 bp,第3对引物(1F+2R)(针对野生型等位基因)产物为2120 bp,第3对引物(1F+2R)(针对双等位基因敲除)产物为809 bp。E:Ash2l-1敲除鉴定中PCR引物位置示意图。F:Ash2l-1敲除鉴定。M2为Trans2K plus2 DNA marker。第1对引物(1F+1R)(针对野生型等位基因)产物为500 bp,第2对引物(2F+2R)(针对野生型等位基因)产物为538 bp,第3对引物(3F+3R)(针对野生型等位基因)产物为1505 bp,第3对引物(3F+3R)(针对双等位基因敲除)产物为701 bp。
Fig. 2Sanger sequencing of gRNAs and colonies identification
Table 4
表4
表4 CRISPR/Cas9组合3和组合4的敲除效率
Table 4
| 敲除靶点 | 克隆总数 | 野生型(+/+) | 单等位基因敲除(+/-) | 双等位基因敲除(-/-) |
|---|---|---|---|---|
| Ash2l-1-CpG岛 | 48 | 26(54.1%) | 14(29.2%) | 8(16.7%) |
| Ash2l-2-RLTR12B | 32 | 22(68.7%) | 7(21.9%) | 3 (9.4%) |
新窗口打开|下载CSV
2.3 Ash2l-1-/-和Ash2l-2-/- mESCs的鉴定
为了建立Ash2l-1-/-和Ash2l-2-/- mESCs,经PCR鉴定,初步分析单克隆中的基因编辑情况。在鉴定Ash2l-2-RLTR12B敲除实验中,PCR引物位置示意图见图2C,凝胶电泳结果见图3A。对照组为野生型mESCs,第1、2、3对引物分别扩增出大小为500 bp、509 bp和2120 bp的条带。实验组为挑取的单克隆7和19,第1、2对引物扩增不出条带,第3对引物扩增出大小为809 bp的条带,由此初步判断这两株克隆发生了双等位基因敲除。在鉴定Ash2l-1-CpG岛敲除实验中,PCR引物位置示意图见图2E,凝胶电泳结果见图3B。对照组为野生型mESCs,第1、2、3对引物分别扩增出大小为500 bp、538 bp和1505 bp的条带。实验组为挑取的单克隆4和20,第1、2对引物扩增不出条带,第3对引物扩增出大小为701 bp的条带,由此初步判断这两株克隆发生了双等位基因敲除。图3
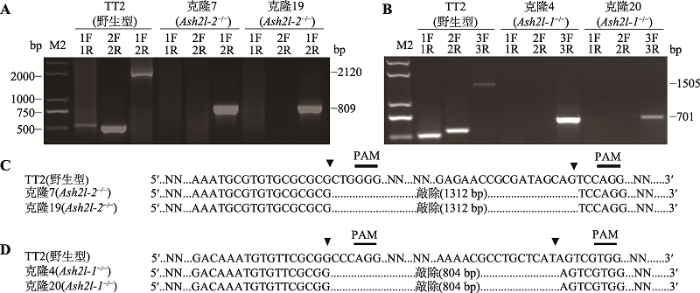 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Ash2l-1-/-和Ash2l-2-/- mESCs的鉴定
A:单克隆7和19中Ash2l-2敲除鉴定。第1对引物(1F+1R)(针对野生型等位基因)产物为500 bp,第2对引物(2F+2R)(针对野生型等位基因)产物为509 bp,第3对引物(1F+2R)(针对野生型等位基因)产物为2120 bp,第3对引物(1F+2R)(针对双等位基因敲除)产物为809 bp。B:单克隆4和20中Ash2l-1敲除鉴定。第1对引物(1F+1R)(针对野生型等位基因)产物为500 bp,第2对引物(2F+2R)(针对野生型等位基因)产物为538 bp,第3对引物(3F+3R)(针对野生型等位基因)产物为1505 bp,第3对引物(3F+3R)(针对双等位基因敲除)产物为701 bp。C, D:单克隆7和19、单克隆4和20的测序结果。
Fig. 3Identification of Ash2l-1-/- and Ash2l-2-/- mESCs
通过以上结果初步判定单克隆7和19、单克隆4和20分别发生了Ash2l-2-RLTR12B和Ash2l-1-CpG岛双等位基因敲除。测序分析表明,切割位点发生在基因组PAM序列上游3~5个碱基处。单克隆7和19敲除了长度为1312 bp的Ash2l-2-RLTR12B(图3C),单克隆4和20敲除了长度为804 bp的Ash2l-1- CpG岛(图3D)。以上结果表明,本文通过CRISPR/ Cas9技术成功获得Ash2l-1-/-和Ash2l-2-/- mESCs。
2.4 Ash2l-1-/-和Ash2l-2-/- mESCs多能性鉴定
野生型mESCs、Ash2l-1-/- mESCs(单克隆4和20)和Ash2l-2-/- mESCs(单克隆7和19)多数呈边缘紧凑的岛状或巢状,在克隆形态和碱性磷酸酶的表达上未有明显差异(图4A)。免疫荧光染色和qRT-PCR结果显示,上述5株细胞中Oct4、Sox2和Nanog的表达没有显著差异(图4:B,C)。EB分化实验表明,相比于野生型EB,Ash2l-1-/- EB中的Snai2(外胚层标志基因)和Gata4(内胚层标志基因)的表达显著降低(P<0.01),Mixl1(中胚层标志基因)的表达没有显著差异。而Ash2l-2-/- EB中,Snai2、Mixl1和Gata4的表达均无显著差异(图4D)。以上结果提示,Ash2l-1-/- mESCs可能在外胚层和内胚层起源的器官形成方面具有发育缺陷。图4
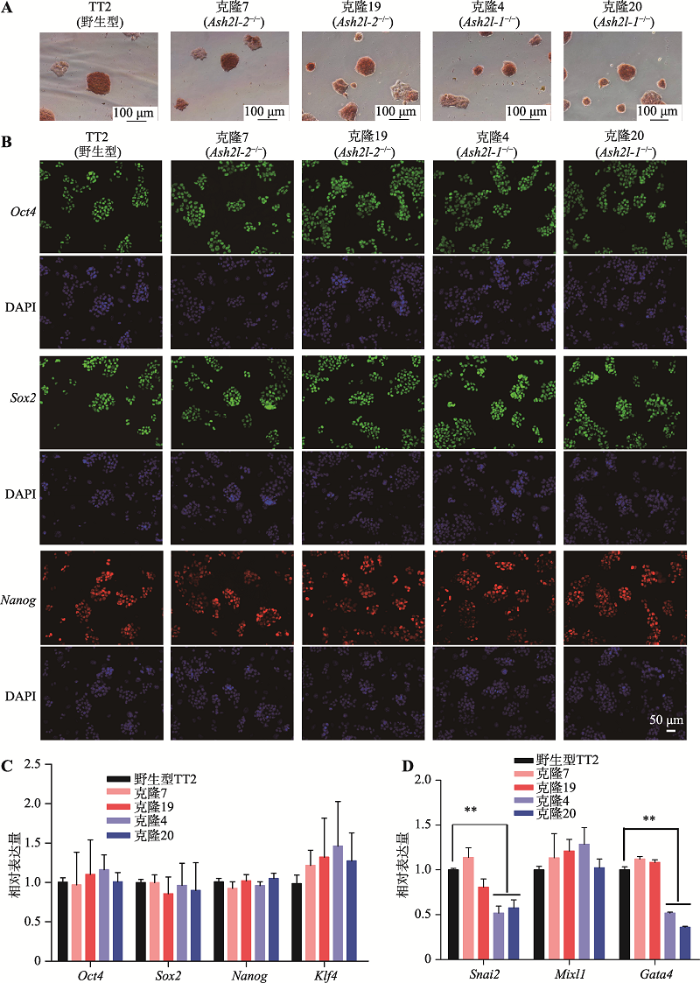 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4Ash2l-1-/-和Ash2l-2-/- mESCs多能性鉴定结果
A:TT2、单克隆7和19、单克隆4和20的AP染色结果(比例尺:100 μm);B:Oct4、Sox2、Nanog的免疫荧光染色(比例尺:50 μm,DAPI为细胞核染色);C:qRT-PCR检测Oct4、Sox2、Nanog和Klf4的表达;D:qRT-PCR检测Snai2、Mixl1和Gata4的表达。**表示P<0.01。
Fig. 4Pluripotency characterization of Ash2l-1-/- and Ash2l-2-/- mESCs
2.5 Ash2l-1和Ash2l-2之间的补偿效应
qRT-PCR结果表明:与野生型mESCs相比,Ash2l-2-/- mESCs中Ash2l-2的mRNA表达量下降,Ash2l-1的表达极显著上调(P<0.01)。Ash2l-1-/- mESCs中,Ash2l-1的mRNA表达量下降,Ash2l-2的表达显著上调(P<0.05)(图5A)。Western blotting结果表明:与野生型mESCs相比,Ash2l-2-/- mESCs中ASH2L-2的表达下降,ASH2L-1的表达极显著上调(P<0.01)。Ash2l-1-/- mESCs中ASH2L-1表达下降,ASH2L-2的表达极显著上调(P<0.01)(图5:B,C)。Ash2l-1-/-和Ash2l-2-/- mESCs中H3K4me3的表达没有显著变化(图5:D,E)。由此推测,Ash2l-1和Ash2l-2之间存在补偿效应。JASPAR数据库预测结果显示,分别有826和1008个转录因子结合在Ash2l-1-CpG岛和Ash2l-2- RLTR12B启动子区。KEGG分析表明,在Ash2l-1- CpG岛启动子区,有3个转录因子(HAND1、SMAD2和SMAD3)参与调节干细胞多能性(图5F)。在Ash2l-2-RLTR12B启动子区,有16个转录因子(HAND1、SMAD2、SMAD3、SMAD4、MEIS1、POU5F1、SOX2、KLF4、HESX1、ESX1、STAT3、ID2、ID4、OTX1、TCF3和NEUROG1)参与调节干细胞多能性(图5G)。以上结果提示,Ash2l-1和Ash2l-2之间的补偿效应可能是通过若干多能性转录因子介导实现的。
图5
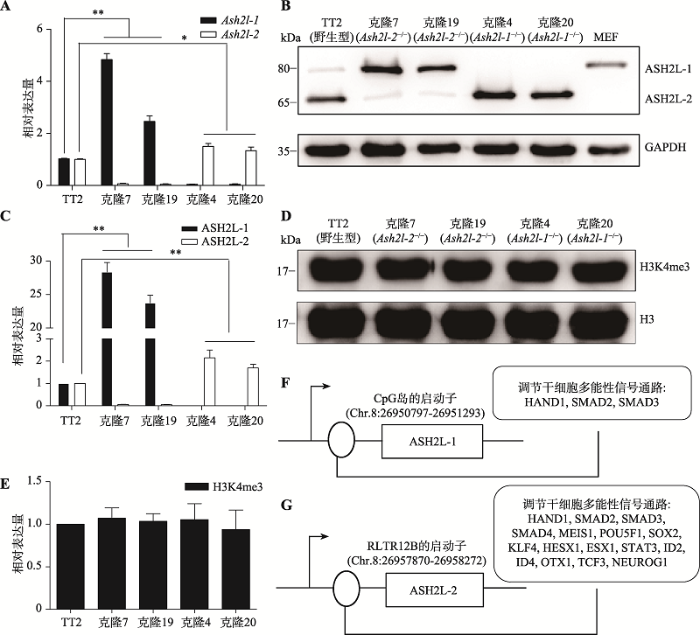 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5Ash2l-1和Ash2l-2之间的补偿效应
A:qRT-PCR检测Ash2l-1和Ash2l-2的mRNA表达。*表示P<0.05,**表示P<0.01;B, C:Western blotting检测ASH2L-1和ASH2L-2的蛋白表达量和蛋白结果的定量分析;D,E:Western blotting检测H3K4me3的蛋白表达量和蛋白结果的变量分析;F,G:参与调节干细胞多能性的转录因子在Ash2l-1-CpG岛和Ash2l-2-RLTR12B上的结合位点。
Fig. 5The compensation effect between Ash2l-1 and Ash2l-2
3 讨 论
组蛋白甲基化修饰是表观遗传的重要内容,参与染色质形成、基因印记、基因转录调控等多种重要生命活动,对mESCs自我更新具有重要影响。ASH2L是MLL复合体的核心成分,影响H3K4甲基转移酶活性,对mESCs多能性的维持十分重 要[3,12~14]。一旦Ash2l的表达降低,mESCs分化,与mESCs自我更新和多能性相关的转录因子,如Nanog、Oct4、Esrrb及Rex1的表达下降。Ash2l可通过作用于Cdh7、c-Myc和Kdm4c,影响染色质重构、转录调控和H3K9me3去甲基化过程,进而调节染色质开放状态,维持mESCs自我更新能力[14,19~21]。本研究发现,在mESCs中ASH2L存在ASH2L-1和ASH2L-2两个异构体,且以ASH2L-2的表达为主。而在MEF中,只表达ASH2L-1。ASH2L-1由CpG岛驱动表达,而ASH2L-2由长末端重复序列RLTR12B驱动表达。RLTR12B属于内源性逆转录病毒(endogenous retroviruses, ERVs),ERVs可参与ESC多能性的维持[22]。MuERV-L在mESCs中受REX1调控表达,与mESCs多能性有关[23]。而HERV-H和HERV-K的表达受NANOG、OCT4调节,与人类早期胚胎的发育密切联系[24]。ASH2L-2在mESCs中特异表达,是ERVs参与mESCs多能性调控的表现。
在体外分化过程中,Ash2l-1-/- mESCs诱导产生的EB在外胚层标记基因Snai2和内胚层标记基因Gata4的表达上显著低于野生型mESCs产生的EB。由此推断,Ash2l-1-/- mESCs可能会在神经系统、皮肤上皮组织[25,26,27]及内胚层起源的器官(如肝脏、胰腺、肺等)形成方面具有发育缺陷[28,29,30]。Ash2l-1-/-和Ash2l-2-/- mESCs具有多能性,在碱性磷酸酶、多能性调控转录因子(Oct4、Nanog、Sox2和Klf4)的表达上与野生型对照并无显著差异。Ash2l-1和Ash2l-2对基因组H3K4me3没有影响。Wan等[14]用RNAi干扰Ash2l表达,发现mESCs的克隆形态无法维持,多能性丧失,整体H3K4me3水平降低。MLL复合体中其他核心亚单位(RBBP5、DPY30或WDR5)的单独敲除,也会导致整体H3K4me3水平下调[9,10,11]。由此推测,Ash2l-1和Ash2l-2在功能上存在互补效应,这与qRT-PCR和Western blotting检测的结果相吻合(图5:A~C)。有研究表明,Tet家族中的Tetl和Tet2在mESCs中均有表达,具有将DNA甲基化催化为羟甲基化的能力,二者在功能上可以互补。单独敲低Tetl或者Tet2的mESCs仍具有多能性,Nanog和Oct4的表达水平几乎没有变化,但当这两个基因同时被敲低时,Nanog和Oct4表达下降,中胚层分化受阻。这些结果表明补偿效应是细胞内部的一种保守机制[31,32]。
生物信息学预测表明,在Ash2l-1-CpG岛启动子区,有3个转录因子参与调节干细胞多能性(图5F)。其中,HAND1对心血管的正常发育具有关键作用,敲除HAND1可导致小鼠胚胎早期死亡[33,34]。由此推测,Ash2l-2的敲除可能会影响HAND1在Ash2l-1- CpG岛启动子区的结合,影响小鼠胚胎早期的发育。在Ash2l-2-RLTR12B启动子区,有16个转录因子参与调节干细胞多能性(图5G)。其中,MEIS1在胚胎造血系统发育、白血病发生发展及神经系统发育中发挥重要作用[35,36]。经STRING(https://string-db.org/)数据库预测,MEIS1可能与ASH2L相互作用(结果未列出)。由此推测,Ash2l-1敲除可能会影响MEIS1在Ash2l-2-RLTR12B启动子区的结合,影响MEIS1的功能。所以,Ash2l-1和Ash2l-2之间的补偿效应可能是通过这些多能性调控转录因子实现的。这种补偿效应参与mESCs的自我更新能力的维持和基因组H3K4me3的调控,但Ash2l-1-/-和Ash2l-2-/- mESCs的发育缺陷可能在细胞分化过程中才能体现出来。本文为进一步深入研究Ash2l-1和Ash2l-2对mESCs的影响奠定了基础。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:18295582 [本文引用: 1]

Abstract The potential to generate virtually any differentiated cell type from embryonic stem cells (ESCs) offers the possibility to establish new models of mammalian development and to create new sources of cells for regenerative medicine. To realize this potential, it is essential to be able to control ESC differentiation and to direct the development of these cells along specific pathways. Embryology has offered important insights into key pathways regulating ESC differentiation, resulting in advances in modeling gastrulation in culture and in the efficient induction of endoderm, mesoderm, and ectoderm and many of their downstream derivatives. This has led to the identification of new multipotential progenitors for the hematopoietic, neural, and cardiovascular lineages and to the development of protocols for the efficient generation of a broad spectrum of cell types including hematopoietic cells, cardiomyocytes, oligodendrocytes, dopamine neurons, and immature pancreatic beta cells. The next challenge will be to demonstrate the functional utility of these cells, both in vitro and in preclinical models of human disease.
URLPMID:21941277 [本文引用: 1]
Abstract Embryonic stem (ES) cells are pluripotent cells isolated from an early embryo and grown as a cell line in tissue culture. Their discovery came from the conjunction of studies in human pathology, mouse genetics, early mouse embryo development, cell surface immunology and tissue culture. ES cells provided a crucial tool for manipulating mouse embryos to study mouse genetics, development and physiology. They have not only revolutionized experimental mammalian genetics but, with the advent of equivalent human ES cells, have now opened new vistas for regenerative medicine.
URLPMID:16549544 [本文引用: 2]
Stem cells undergo extensive self-renewal and have the capacity to differentiate along multiple cell lineages. Progression from stem cells into differentiated progeny requires long-lasting changes in gene expression. Epigenetic mechanisms, including DNA methylation, histone modifications, and non-coding RNA-mediated regulatory events, are essential to controlling the heritable cellular memory of gene expression during development. Recent studies on cell fate specification of embryonic and adult stem cells/progenitors have highlighted a general and critical role for dynamic epigenetic regulation in stem cell self-renewal and differentiation.
URLPMID:4641500 [本文引用: 2]
Histones are subject to a vast array of posttranslational modifications including acetylation, methylation, phosphorylation, and ubiquitylation. The writers of these modifications play important roles in normal development and their mutation or misregulation is linked with both genetic disorders and various cancers. Readers of these marks contain protein domains that allow their recruitment to chromatin. Interestingly, writers often contain domains which can read chromatin marks, allowing the reinforcement of modifications through a positive feedback loop or inhibition of their activity by other modifications. We discuss how such positive reinforcement can result in chromatin states that are robust and can be epigenetically maintained through cell division. We describe the implications of these regulatory systems in relation to modifications including H3K4me3, H3K79me3, and H3K36me3 that are associated with active genes and H3K27me3 and H3K9me3 that have been linked to transcriptional repression. We also review the crosstalk between active and repressive modifications, illustrated by the interplay between the Polycomb and Trithorax histone-modifying proteins, and discuss how this may be important in defining gene expression states during development.
URLPMID:17320507 [本文引用: 2]
The surface of nucleosomes is studded with a multiplicity of modifications. At least eight different classes have been characterized to date and many different sites have been identified for each class. Operationally, modifications function either by disrupting chromatin contacts or by affecting the recruitment of nonhistone proteins to chromatin. Their presence on histones can dictate the higher-order chromatin structure in which DNA is packaged and can orchestrate the ordered recruitment of enzyme complexes to manipulate DNA. In this way, histone modifications have the potential to influence many fundamental biological processes, some of which may be epigenetically inherited.
URLPMID:20226651 [本文引用: 2]
In search of the mechanisms that govern pluripotency and embryonic stem cell (ESC) self-renewal, a growing list of evidence highlights chromatin as a leading factor, controlling ESC maintenance and differentiation. In-depth investigation of chromatin in ESCs revealed distinct features, including DNA methylation, histone modifications, chromatin protein composition and nuclear architecture. Here we review recent literature describing different aspects of chromatin and genome organization in ESCs. The emerging theme seems to support a mechanism maintaining chromatin plasticity in ESCs but without any dramatic changes in the organization and nuclear positioning of chromosomes and gene loci themselves. Plasticity thus seems to be supported more by different mechanisms maintaining an open chromatin state and less by regulating the location of genomic regions.
URLPMID:2873088 [本文引用: 1]
The fourth lysine of histone H3 is post-translationally modified by a methyl group via the action of histone methyltransferase, and such a covalent modification is associated with transcriptionally active and/or repressed chromatin states. Thus, histone H3 lysine 4 methylation has a crucial role in maintaining normal cellular functions. In fact, misregulation of this covalent modification has been implicated in various types of cancer and other diseases. Therefore, a large number of studies over recent years have been directed towards histone H3 lysine 4 methylation and the enzymes involved in this covalent modification in eukaryotes ranging from yeast to human. These studies revealed a set of histone H3 lysine 4 methyltransferases with important cellular functions in different eukaryotes, as discussed here.
[本文引用: 1]
URLPMID:16878130 [本文引用: 2]
Abstract Histone H3 Lys4 (H3K4) methylation is a prevalent mark associated with transcription activation. A common feature of several H3K4 methyltransferase complexes is the presence of three structural components (RbBP5, Ash2L and WDR5) and a catalytic subunit containing a SET domain. Here we report the first biochemical reconstitution of a functional four-component mixed-lineage leukemia protein-1 (MLL1) core complex. This reconstitution, combined with in vivo assays, allows direct analysis of the contribution of each component to MLL1 enzymatic activity and their roles in transcriptional regulation. Moreover, taking clues from a crystal structure analysis, we demonstrate that WDR5 mediates interactions of the MLL1 catalytic unit both with the common structural platform and with the histone substrate. Mechanistic insights gained from this study can be generalized to the whole family of SET1-like histone methyltransferases in mammals.
URLPMID:3572774 [本文引用: 2]
Abstract Histone H3K4 methylation is associated with active genes and, along with H3K27 methylation, is part of a bivalent chromatin mark that typifies poised developmental genes in embryonic stem cells (ESCs). However, its functional roles in ESC maintenance and differentiation are not established. Here we show that mammalian Dpy-30, a core subunit of the SET1/MLL histone methyltransferase complexes, modulates H3K4 methylation in vitro, and directly regulates chromosomal H3K4 trimethylation (H3K4me3) throughout the mammalian genome. Depletion of Dpy-30 does not affect ESC self-renewal, but significantly alters the differentiation potential of ESCs, particularly along the neural lineage. The differentiation defect is accompanied by defects in gene induction and in H3K4 methylation at key developmental loci. Our results strongly indicate an essential functional role for Dpy-30 and SET1/MLL complex-mediated H3K4 methylation, as a component of the bivalent mark, at developmental genes during the ESC fate transitions. Copyright 2011 Elsevier Inc. All rights reserved.
URLPMID:2147785120 [本文引用: 2]
78 Wdr5 is enriched in ES/iPS cells and is required for ES cell self-renewal 78 Wdr5-Oct4 interaction links core transcriptional network with epigenetic machinery 78 Trithorax and core transcriptional network cooperate in transcriptional activation 78 Wdr5 is required during the initial phases of somatic cell reprogramming
URLPMID:16892064 [本文引用: 2]
MLL complexes are homologs of yeast COMPASS capable of methylating histone H3 Lys4 (H3K4). ASH2L, RbBP5 and WDR5 are conserved subunits of MLL complexes with homology to the Cps40/Cps60, Cps50 and Cps30 subunits of COMPASS, respectively. We report that ASH2L differentially regulates MLL's catalysis of H3K4 trimethylation similarly to Cps40 and Cps60. Furthermore, WDR5 is required to maintain MLL complex integrity, including the stability of ASH2L within the complex. These findings offer insight into the molecular role of ASH2L, and by extension that of WDR5, in proper H3K4 trimethylation.
URLPMID:3017365 [本文引用: 1]
TBX1 encodes a DNA binding transcription factor that is commonly deleted in human DiGeorge syndrome and plays an important role in heart development. Mechanisms of Tbx1 function, such as Tbx1 interacting regulatory proteins and transcriptional target specificity, are largely unknown. Ash2l is the mammalian homolog of Drosophila Ash2 (absent small homeotic 2) and is a core component of a multimeric histone methyltransferase complex that epigenetically regulates transcription via methylation of histone lysine residues. We undertook an unbiased yeast two-hybrid screen to look for functionally relevant Tbx1-interacting proteins and report a physical and functional interaction between Tbx1 and Ash2l. Tbx1 interacts with Ash2l in both yeast and mammalian cells and Ash2l acts as a transcriptional co-activator in luciferase reporter assays. Expression analysis shows that Tbx1 and Ash2l have overlapping mRNA and protein expression patterns during development. By generating an Ash2l knockout mouse utilizing gene-trap technology, we show that although Ash2l heterozygous mice are normal, Ash2l-null embryos die early during gestation. Thus, Ash2l is required for the earliest stages of embryogenesis. Furthermore, our finding of a physical interaction between Tbx1 and Ash2l suggest that at least some functions of Tbx1 may be mediated by direct interactions with a histone methyltransferase complex.
URLPMID:3576105 [本文引用: 4]
Embryonic stem (ES) cells exhibit general characteristics of open chromatin, a state that may be necessary for ES cells to efficiently self-renew while remaining poised for differentiation. Histone H3K4 and H3K9 trimethylation associate as a general rule, with open and silenced chromatin, respectively, for ES cell pluripotency maintenance. However, how histone modifications are regulated to maintain open chromatin in ES cells remains largely unknown. Here, we demonstrate that trithorax protein Ash2l, homologue of the Drosophila Ash2 (absent, small, homeotic-2) protein, is a key regulator of open chromatin in ES cells. Consistent with Ash2l being a core subunit of mixed lineage leukemia methyltransferase complex, RNAiknockdownofAsh2lwassufficienttoreduceH3K4 methylation levels and drive ES cells to a silenced chromatin state with high H3K9 trimethylation. Genome-wide ChIP-seq analysis indicated that Ash2l is recruited to target loci through two distinct modes and enriched at a family of genes implicated in open chromatin regulation, including chromatin remodeler Cdh7, transcription factor c-Myc, and H3K9 demethylase Kdm4c. Our results underscore the importance of Ash2l in open chromatin regulation and provide insight into how the open chromatin landscape is maintained in ES cells.
URL [本文引用: 1]
Functional elucidation of causal genetic variants and elements requires precise genome editing technologies. The type II prokaryotic CRISPR (clustered regularly interspaced short palindromic repeats)/Cas adaptive immune system has been shown to facilitate RNA-guided site-specific DNA cleavage. We engineered two different type II CRISPR/Cas systems and demonstrate that Cas9 nucleases can be directed by short RNAs to induce precise cleavage at endogenous genomic loci in human and mouse cells. Cas9 can also be converted into a nicking enzyme to facilitate homology-directed repair with minimal mutagenic activity. Lastly, multiple guide sequences can be encoded into a single CRISPR array to enable simultaneous editing of several sites within the mammalian genome, demonstrating easy programmability and wide applicability of the RNA-guided nuclease technology.
URLPMID:24315440 [本文引用: 1]
The CRISPR-Cas9 system has been employed to generate mutant alleles in a range of different organisms. However, so far there have not been reports of use of this system for efficient correction of a genetic disease. Here we show that mice with a dominant mutation in Crygc gene that causes cataracts could be rescued by coinjection into zygotes of Cas9 mRNA and a single-guide RNA (sgRNA) targeting the mutant allele. Correction occurred via homology-directed repair (HDR) based on an exogenously supplied oligonucleotide or the endogenous WT allele, with only rare evidence of off-target modifications. The resulting mice were fertile and able to transmit the corrected allele to their progeny. Thus, our study provides proof of principle for use of the CRISPR-Cas9 system to correct genetic disease.
URL [本文引用: 1]
URLPMID:23287722 [本文引用: 1]
Bacteria and archaea have evolved adaptive immune defenses termed clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems that use short RNA to direct degradation of foreign nucleic acids. Here, we engineer the type II bacterial CRISPR system to function with custom guide RNA (gRNA) in human cells. For the endogenous AAVS1 locus, we obtained targeting rates of 10 to 25% in 293T cells, 13 to 8% in K562 cells, and 2 to 4% in induced pluripotent stem cells. We show that this process relies on CRISPR components, is sequence-specific, and upon simultaneous introduction of multiple gRNAs, can effect multiplex editing of target loci. We also compute a genome-wide resource of ~190k unique gRNAs targeting ~40.5% of human exons. Our results establish an RNA-guided editing tool for facile, robust, and multiplexable human genome engineering.
URL [本文引用: 1]
URLPMID:20946988
Abstract c-Myc (Myc) is an important transcriptional regulator in embryonic stem (ES) cells, somatic cell reprogramming, and cancer. Here, we identify a Myc-centered regulatory network in ES cells by combining protein-protein and protein-DNA interaction studies and show that Myc interacts with the NuA4 complex, a regulator of ES cell identity. In combination with regulatory network information, we define three ES cell modules (Core, Polycomb, and Myc) and show that the modules are functionally separable, illustrating that the overall ES cell transcription program is composed of distinct units. With these modules as an analytical tool, we have reassessed the hypothesis linking an ES cell signature with cancer or cancer stem cells. We find that the Myc module, independent of the Core module, is active in various cancers and predicts cancer outcome. The apparent similarity of cancer and ES cell signatures reflects, in large part, the pervasive nature of Myc regulatory networks. Copyright 2010 Elsevier Inc. All rights reserved.
URLPMID:17938240 [本文引用: 1]
Embryonic stem (ES) cells are pluripotent cells with the ability to self-renew indefinitely. These unique properties are controlled by genetic factors and chromatin structure. The exit from the self-renewing state is accompanied by changes in epigenetic chromatin modifications such as an induction in the silencing-associated histone H3 Lys 9 dimethylation and trimethylation (H3K9Me2/Me3) marks. Here, we show that the H3K9Me2 and H3K9Me3 demethylase genes, Jmjd1a and Jmjd2c, are positively regulated by the ES cell transcription factor Oct4. Interestingly, Jmjd1a or Jmjd2c depletion leads to ES cell differentiation, which is accompanied by a reduction in the expression of ES cell-specific genes and an induction of lineage marker genes. Jmjd1a demethylates H3K9Me2 at the promoter regions of Tcl1, Tcfcp211, and Zfp57 and positively regulates the expression of these pluripotency-associated genes. Jmjd2c acts as a positive regulator for Nanog, which encodes for a key transcription factor for self-renewal in ES cells. We further demonstrate that Jmjd2c is required to reverse the H3K9Me3 marks at the Nanog promoter region and consequently prevents transcriptional repressors HP1 and KAP1 from binding. Our results connect the ES cell transcription circuitry to chromatin modulation through H3K9 demethylation in pluripotent cells.
[本文引用: 1]
URLPMID:24567914 [本文引用: 1]
Abstract About half of the mammalian genome is occupied by DNA sequences that originate from transposable elements. Retrotransposons can modulate gene expression in different ways and, particularly retrotransposon-derived long terminal repeats, profoundly shape expression of both surrounding and distant genomic loci. This is especially important in pre-implantation development, during which extensive reprograming of the genome takes place and cells pass through totipotent and pluripotent states. At this stage, the main mechanism responsible for retrotransposon silencing, i.e., DNA methylation, is inoperative. A particular retrotransposon called muERV-L/MERVL is expressed during pre-implantation stages and contributes to the plasticity of mouse embryonic stem cells. This review will focus on the role of MERVL-derived sequences as controlling elements of gene expression specific for pre-implantation development, two-cell stage-specific gene expression, and stem cell pluripotency, the epigenetic mechanisms that control their expression, and the contributions of the pluripotency marker REX1 and the related Yin Yang 1 family of transcription factors to this regulation process.
URLPMID:4862087 [本文引用: 1]
Transposable elements (TEs) are notable drivers of genetic innovation. Over evolutionary time, TE insertions can supply new promoter, enhancer, and insulator elements to protein-coding genes and establish novel, species-specific gene regulatory networks. Conversely, ongoing TE-driven insertional mutagenesis, nonhomologous recombination, and other potentially deleterious processes can cause sporadic disease by disrupting genome integrity or inducing abrupt gene expression changes. Here, we discuss recent evidence suggesting that TEs may contribute regulatory innovation to mammalian embryonic and pluripotent states as a means to ward off complete repression by their host genome.
URLPMID:7720581 [本文引用: 1]
The earliest sign of the prospective neural crest of Xenopus is the expression of the ectodermal component of Xsna (the Xenopus homologue of snail) in a low arc on the dorsal aspect of stage 11 embryos, which subsequently assumes the horseshoe shape characteristic of the neural folds as the convergence-extension movements shape the neural plate. A related zinc-finger gene called Slug (Xslu) is expressed specifically in this tissue (i.e. the prospective crest) when the convergence extension movements are completed. Subsequently, Xslu is found in pre- and post-migratory cranial and trunk neural crest and also in lateral plate mesoderm after stage 17. Both Xslu and Xsna are induced by mesoderm from the dorsal or lateral marginal zone but not from the ventral marginal zone. From stage 10.5, explants of the prospective neural crest, which is underlain with tissue, are able to express Xslu. However expression of Xsna is not apparently specified until stage 12 and further contact with the inducer is required to raise the level of expression to that seen later in development. Xslu is specified at a later time. Embryos injected with noggin mRNA at the 1-cell stage or with plasmids driving noggin expression after the start of zygotic transcription express Xslu in a ring surrounding the embryo on the ventroposterior side. We suggest this indicates (a) that noggin interacts with another signal that is present throughout the ventral side of the embryo and (b) that Xslu is unable to express in the neural plate either because of the absence of a co-inducer or by a positive prohibition of expression. The ventral co-inducer, in the presence of overexpressed noggin, seems to generate an anterior/posterior pattern in the ventral part of the embryo comparable to that seen in neural crest of normal embryos. We suggest that the prospective neural crest is induced in normal embryos in the ectoderm that overlies the junction of the domains that express noggin and Xwnt-8. In support of this, we show animal cap explants from blastulae and gastrulae, treated with bFGF and noggin express Xslu but not NCAM although the mesoderm marker Xbra is also expressed. Explants treated with noggin alone express NCAM only. An indication that induction of the neural plate border is regulated independently of the neural plate is obtained from experiments using ultraviolet irradiation in the precleavage period. At certain doses, the cranial crest domains are not separated into lateral masses and there is a reduction in the size of the neural plate.
URLPMID:21715424 [本文引用: 1]
Abstract The neural crest is an induced tissue that is unique to vertebrates. In the clawed frog Xenopus laevis, neural crest induction depends on signals secreted from the prospective dorsolateral mesodermal zone during gastrulation. The transcription factors Snail2 (Slug), Snail1 and Twist1 are expressed in this region. It is known that Snail2 and Twist1 are required for both mesoderm formation and neural crest induction. Using targeted blastomere injection, morpholino-based loss of function and explant studies, we show that: (1) Snail1 is also required for mesoderm and neural crest formation; (2) loss of snail1, snail2 or twist1 function in the C2/C3 lineage of 32-cell embryos blocks mesoderm formation, but neural crest is lost only in the case of snail2 loss of function; (3) snail2 mutant loss of neural crest involves mesoderm-derived secreted factors and can be rescued synergistically by bmp4 and wnt8 RNAs; and (4) loss of snail2 activity leads to changes in the RNA levels of a number of BMP and Wnt agonists and antagonists. Taken together, these results identify Snail2 as a key regulator of the signals involved in mesodermal induction of neural crest.
URLPMID:10419699 [本文引用: 1]
An epithelial-mesenchymal cell transformation occurs during the development of the endocardial cushions in the atrioventricular (AV) canal of the heart. We hypothesized that the transcription factor Slug is required for this epithelial-mesenchymal cell transformation since Slug is required for similar transformations during gastrulation and neural crest differentiation in chicken embryos. We found by RT-PCR and immunostaining that the temporal and spatial localization of Slug in the embryonic chicken heart is consistent with a role for Slug in endocardial cushion formation. Moreover, we found that Slug expression by AV canal endothelial cells is induced by a signal provided by AV canal myocardium. Slug appears to be required for epithelial-mesenchymal cell transformation in the chicken heart since treatment of AV canal explants with antisense Slug oligodeoxynucleotides inhibited mesenchymal cell formation in vitro. Antisense Slug oligodeoxynucleotides prevented endothelial cell-cell separation, suggesting that Slug acts early in the transformation pathway.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:3154739 [本文引用: 1]
78 Tet161/61 ESCs show a partial reduction in 5hmC and subtle changes in gene expression 78 Loss of Tet1 is compatible with maintaining pluripotency 78 Tet161/61 ESCs can drive normal embryo development in 4n complementation assays 78 Tet1 mutant mice are viable and fertile with slight variation in body size
URLPMID:27184841 [本文引用: 1]
-Tet enzymes maintains telomere length-Tet enzymes maintain chromosomal stability of ES cells-Dnmt3b and 5hmC are involved in Tet signaling-mediated telomere maintenance-Excessive Zscan4 and Dnmt3b lead to heterogeneous telomere elongation and loss
[本文引用: 1]
URL [本文引用: 1]
URLPMID:25911551 [本文引用: 1]
Hox homeobox transcription factors drive leukemogenesis efficiently only in the presence of Meis or Pbx proteins. Here we show that Pbx3 and Meis1 need to dimerize to support Hox-induced leukemia and we analyze the molecular details of this cooperation. In the absence of Pbx3, Meis1 was highly unstable. As shown by a deletion analysis Meis1 degradation was contingent on a motif coinciding with the Pbx-binding domain. Either deletion of this sequence or binding to Pbx3 prolonged the half-life of Meis1 by preventing its ubiquitination. Meis1 break-down could also be blocked by inhibition of the ubiquitin proteasome system, indicating tight post-transcriptional control. In addition, Meis1 and Pbx3 cooperated genetically as overexpression of Pbx3 induced endogenous Meis1 transcription. These functional interactions translated into in vivo activity. Blocking Meis1/Pbx3 dimerization abrogated the ability to enhance proliferation and colony-forming cell numbers in primary cells transformed by Hoxa9. Furthermore, expression of Meis1 target genes Flt3 and Trib2 was dependent on Pbx3/Meis1 dimerization. This correlated with the requirement of Meis1 to bind Pbx3 in order to form high affinity DNA/Hoxa9/Meis1/Pbx3 complexes in vitro. Finally, kinetics and severity of disease in transplantation assays indicated that Pbx3/Meis1 dimers are rate-limiting factors for Hoxa9-induced leukemia.
URLPMID:24498346 [本文引用: 1]
Abstract Hematopoietic stem cells in the bone marrow have the capacity to both self-renew and to generate all cells of the hematopoietic system. The balance of these two activities is controlled by hematopoietic stem cell-intrinsic regulatory mechanisms as well as extrinsic signals from the microenvironment. Here we demonstrate that Meis1, a TALE family homeodomain transcription factor involved in numerous embryonic developmental processes, is selectively expressed in hematopoietic stem/progenitor cells. Conditional Meis1 knockout in adult hematopoietic cells resulted in a significant reduction in the hematopoietic stem/progenitor cells. Suppression of hematopoiesis by Meis1 deletion appears to be caused by impaired self-renewal activity and reduced cellular quiescence of hematopoietic stem/progenitor cells in a cell autonomous manner, resulting in stem cell exhaustion and defective long-term hematopoiesis. Meis1 deficiency down-regulated a subset of Pbx1-dependent hematopoietic stem cell signature genes, suggesting a functional link between them in the maintenance of hematopoietic stem/progenitor cells. These results show the importance of Meis1 in adult hematopoiesis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}