 ,, 杨瑞馥, 崔玉军军事医学科学院微生物流行病研究所,病原微生物生物安全国家重点实验室,北京 100071
,, 杨瑞馥, 崔玉军军事医学科学院微生物流行病研究所,病原微生物生物安全国家重点实验室,北京 100071Bacterial genome-wide association study: methodologies and applications
Chao Yang,, Ruifu Yang, Yujun CuiState Key Laboratory of Pathogen and Biosecurity, Institute of Microbiology and Epidemiology, Beijing 100071, China第一联系人:
收稿日期:2017-09-13修回日期:2017-10-30网络出版日期:--
| 基金资助: |
Received:2017-09-13Revised:2017-10-30Online:--
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (368KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨超, 杨瑞馥, 崔玉军. 细菌全基因组关联研究的方法与应用. 遗传[J], 2018, 40(1): 57-65 doi:10.16288/j.yczz.17-303
Chao Yang, Ruifu Yang, Yujun Cui.
全基因组关联研究(genome-wide association study, GWAS)是一种从全基因组水平筛选与某表型(phenotype)显著相关的遗传变异,进而阐明表型遗传机制的方法[1]。相较于传统的分子遗传学方法,GWAS并不对表型产生的遗传机制做任何假设;而是直接从表型出发,设置合理的对照组,通过大样本的数据统计分析找到与表型关联的遗传变异,因此该研究方法可以应用于复杂表型研究。GWAS极大增进了人们对复杂表型的认识,在人类复杂疾病研究中取得了丰硕成果[2,3,4,5]。自2005年第一项有关黄斑病变的人类GWAS[6]发表以来,目前公开发表的人类GWAS工作已达到2982项,累计报道了36948个与疾病/表型相关的单核苷酸多态性位点(single nucleotide polymorphism,SNP)[7],为人类复杂疾病的预防治疗指明了道路。
GWAS同样可用于细菌研究,为宿主适应性、毒力等复杂表型的遗传机制探索提供新思路[8,9]。然而受限于早期相对匮乏的全基因组数据,细菌GWAS(bacterial GWAS, BGWAS)开展相对较晚。随着近年来高通量测序技术的发展,细菌测序成本快速下降,全基因组序列也得以迅速积累,目前NCBI数据库中已有近10万个细菌样本的全基因组序列,为BGWAS工作奠定了基础。自2013年空肠弯曲杆菌(Campylobacter jejuni)宿主适应性的BGWAS文章发表以来[10],目前已有10余项BGWAS工作被发表(表1)。这些研究揭示与细菌宿主适应性、耐药性及毒力等重要表型相关的基因组变异[10,11,12,13,14,15,16,17,18,19,20,21,22,23,24],极大加深了人们对细菌遗传、进化和传播等的认识。
相较于人类GWAS,BGWAS存在天然的“优势”。例如:较小的基因组使得测序成本、计算资源需求和计算时间减少,降低了研究门槛;另外,细菌更容易通过分子生物学实验对所发现的表型相关遗传变异进行验证[8]。同时,BGWAS也面临着特有的挑战,如细菌的分裂繁殖特性所导致的克隆种群结构、物种间重组率差异造成的多样化的连锁不平衡模式、以及基因的高频率获得缺失等[8,15,25]。针对这些问题,已有的BGWAS工作提出了不同的解决方案。本文梳理了当前BGWAS的研究方法,并对取得的成果及存在的问题进行了总结,以期为微生物学领域开展BGWAS研究提供参考。
Table 1
表1
表1 细菌全基因组关联研究(BGWAS)示例
Table 1
| 物种名 | 重组率 | 样本量 | 表型 | 基因型 | 显著相关 | 软件 | 发表时间 | 参考文献 |
|---|---|---|---|---|---|---|---|---|
| 空肠弯曲杆菌 (Campylobacter jejuni) | 高 | 192 | 宿主适应性 | k-mer | 7307 k-mer (7个基因) | - | 2013 | [10] |
| 102 | 生物膜形成 | k-mer | 1657 kmer (46个基因) | - | 2015 | [16] | ||
| 600 | 存活力 | k-mer | 3382 k-mer (20个基因) | - | 2016 | [24] | ||
| 166 | 诊断标记 | 基因 | 25个非核心基因 | - | 2017 | [21] | ||
| 结核杆菌 (Mycobacterium tuberculosis) | 低 | 123 | 耐药性 | SNP | 50 SNPs | phyC | 2013 | [11] |
| 123 | SNP | 133 SNPs | PLINK | 2015 | [15] | |||
| 498 | SNP, indel | 12 SNPs | Bayes Traits | 2016 | [18] | |||
| 金黄色葡萄球菌 (Staphylococcus aureus) | 低 | 75 | 耐药性 | SNP | 1 SNP | ROADTRIPS | 2014 | [12] |
| 90 | 毒力 | SNP, indel | 121 SNPs, indels | PLINK | 2014 | [14] | ||
| 肺炎链球菌 (Streptococcus pneumoniae) | 高 | 3701 | 耐药性 | SNP, indel | 301 SNPs | PLINK | 2014 | [13] |
| 1680 | SNP,基因 | 426 SNPs | PLINK | 2017 | [23] | |||
| 2175 | 体内运输 | k-mer | 2 SNPs, 424 kmer | fast-LMM, SEER | 2017 | [22] | ||
| 猪链球菌 (Streptococcus suis) | 高 | 191 | 宿主适应性 | SNP,基因,k-mer | 0 | PLINK | 2015 | [17] |
| 单增李斯特菌 (Listeria monocytogenes) | 低 | 104 | 毒力 | 基因 | 43个基因 | - | 2016 | [19] |
| 鲍曼不动杆菌 (Acinetobacter baumannii) | 高 | 122 | 耐药性 | k-mer | 469 k-mer | bugwas | 2016 | [20] |
新窗口打开|下载CSV
1 BGWAS的研究方法与工具
BGWAS的研究方法从人类GWAS发展而来,并在研究过程中开发了特有的思路和工具。BGWAS主要可以分成以下4个步骤:表型选取及采样,表型及基因型(genotype)测定、相关性检验及实验验证(图1)。1.1 表型选取及采样
选择合适的表型是BGWAS的第一步。表型通常可分为连续性数据(如细菌细胞的尺寸)和二分类数据(case/control),分别对应不同的相关性检验方法。尽管连续性数据的检验效力更高,但其数据难以获得、统计检验更复杂,因此目前仅有两项样本量较小的BGWAS研究使用了连续性表型数据[12,14]。相对而言,易于通过高通量方法获取大样本量数据的二分类表型是BGWAS研究的首选。选定表型后需要进行样本采集。采样时需注意采样方式和样本量的问题。采样方式可分为连续采样(time-coursed)和横断面采样(cross-sectional)两 种[26]。连续采样的样本,如分离同一病人的不同时期菌株及实验室进化菌株等获取难度较大,难以满足BGWAS的样本量需求。横断面采样通过收集一定时间内大量相关样本,如病人诊断样本或公共卫生监测样本等,能够快速的获得更综合全面的信息,是BGWAS的首选采样方式[26]。此外,采样时可以采用Farhat等[26]提出的成对采样策略,即优先选择遗传距离近、表型不同的菌株对。通过模拟计算发现成对采样不仅能有效降低种群结构带来的假阳性,还能显著提高统计检验效力。实际应用中可先用脉冲场凝胶电泳(pulsed field gel electrophoresis,PFGE)或多位点序列分型(multilocus sequence typing, MLST)等传统的快速分型方法对样本进行初步分型,然后根据分型结果筛选成对菌株进行测序用于BGWAS。
图1
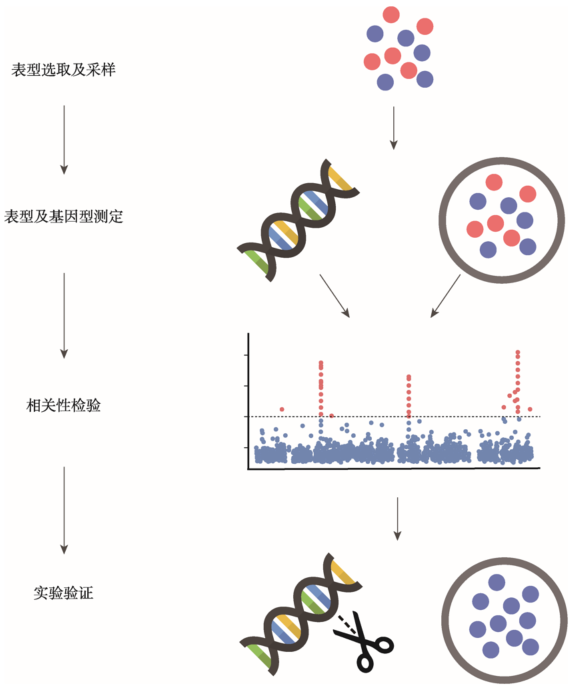 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1细菌全基因组关联研究(BGWAS)分析流程图
Fig. 1Flow of bacterial genome-wide association studies (BGWAS)
样本量大小也是BGWAS研究设计中的重要问题,较大的样本量能够增加统计检验效力,但同时会增加研究成本。影响分析时所需样本量的主要因素是效应量(effect size),即遗传变异能够解释的表型变异量。效应量是表型与基因型相关性的度量单位,其取值范围从0到1,1表示遗传变异能够完全解释表型[27]。对于效应量较大的变异,如抗生素抗性相关变异等,仅需要相对少量样本即可鉴定出相关性;而低效应量的变异则需要较大的样本量来保证检验的统计效力。当前BGWAS主要致力于高效应量变异鉴定,绝大多数样本量相对较小(表1)。理论上低效应量变异在细菌中一定存在并在细菌进化和生存中发挥重要作用;随着样本量的累积增大及BGWAS技术的进一步发展,相信将来会有越来越多的研究关注这类变异。
1.2 表型及基因型测定
实验室检测是获得细菌表型信息的最主要方式。此外,许多数据库整理保存了多种细菌表型信息,如PATRIC(https://www.patricbrc.org/)整合了多种细菌的基因组及对应耐药性等表型信息,NCBI的Pathogen Detection(https://www.ncbi.nlm.nih.gov/pathogens/)整合了病原菌基因组及对应的分离时间、地点、宿主等背景信息等。这些数据库提供的海量信息将极大的促进BGWAS的发展。随着测序技术的发展及细菌变异分析软件的完善,获得细菌基因组变异及相应的基因型信息已经越来越快速且准确[28,29,30]。细菌基因型数据可分为SNP、插入缺失(indel)、非核心基因获得缺失及k-mer等类型(表1)。SNP具有高分辨率、易鉴定等优势,是当前BGWAS最常用的基因型数据。SNP用于分析之前通常要进行质量控制,当前常用的质控标准为:位点测序质量值大于20,支持的reads数大于5条,最小等位基因频率(minor allele frequency, MAF) 大于1%或5%。此外,细菌SNP鉴定中会出现3态甚至4态SNP,但为了便于计算,实际应用中通常只有2态SNP用于关联分析。SNP变异的局限性是仅能反映细菌核心基因组信息,而许多细菌具有开放型泛基因组[31],这些菌株的非核心基因组变异也与细菌表型密切相关。因此,部分BGWAS研究也整合了indel、基因获得缺失信息等非核心基因组变异信息。此外,越来越多的研究使用k-mer来研究基因组变异(表1)。k-mer是指将全基因组序列切分成的长度为几十到上百个碱基的短片段。通过使用基于图论等的算法计算k-mer在不同样本间的存在与否,可以同时综合分析SNP变异及基因获得缺失等信息,从而能够更全面的探索表型的遗传变异机制,这类分析方法正越来越得到科研工作者的青睐。
1.3 相关性检验
获得各样本的表型以及基因型数据后,需要对两类数据之间进行相关性检验,但是在BGWAS中直接对两者做相关性检验容易造成假阳性结果。细菌的种群结构是造成假阳性相关的主要因素[8,15,25]。当研究对象可分成不同种群时,同一种群内部个体之间的遗传距离相对种群之间遗传距离更近,造成等位基因频率的非随机分布;当某一种群仅集中于对照组或者实验组时,BGWAS会鉴定出许多与分群相关而不是与表型相关的变异,进而导致假阳性的产生。降低种群结构所致假阳性的最直接方法是:采样时尽可能选取遗传异质性低的样本作为研究对象(如人类GWAS研究通常选择在同一人种甚至同一民族内进行),或者使用上文提到的Farhat成对采样策略[26]。但是即使选择同一种群的细菌样本,仍可能存在更精细的亚群结构。为了消除种群结构影响,部分研究沿用了人类GWAS工作中建立的软件进行分析(表1),如PLINK[32]、ROADTRIPS[33]、fast-LMM[34]等。此外,针对细菌自身特点的新算法也被不断开发出来,如结合系统发育信息和蒙特卡罗模拟的系统发育校正方法[10,35]以及基于线性混合模型的聚类法等[36,37,38]。这些方法已被整合到BGWAS分析工具中,并得到实际应用(表2)。Table 2
表2
表2 细菌全基因组关联研究(BGWAS)工具
Table 2
| 研究工具 | 发表时间 | 特点 | 种群结构处理 | 适用性 | 应用 | 下载链接 | 参考文献 |
|---|---|---|---|---|---|---|---|
| phyC | 2013 | 通过检测趋同进化鉴定表型相关位点 | 系统发育校正 | 低、中重组率细菌 | 结核杆菌(Mycobacterium tuberculosis) | - | [11] |
| bugwas | 2016 | 基于k-mer,同时检测表型相关位点及家系(lineage) | 线性混合模型校正 | 所有细菌 | 结核杆菌(Mycobacterium tuberculosis),金黄色葡萄球菌(Staphylococcus aureus),大肠杆菌(Escherichia coli),肺炎克雷伯菌(Klebsiella pneumoniae),鲍曼不动杆菌(Acinetobacter baumannii) | https://github.com/jessiewu/bacterialGWAS | [36] |
| SEER | 2016 | 无需参考序列,可变k-mer长度,支持连续表型数据 | 多维尺度变换 | 所有细菌 | 肺炎链球菌(Streptococcus pneumonia),酿脓链球菌(Streptococcus pyogenes) | https://github.com/johnlees/seer | [38] |
| Scoary | 2016 | 针对非核心基因,简单快速 | 两两比对及置换检验 | 所有细菌 | 肺炎链球菌(Streptococcus pneumoniae), 表皮葡萄糖菌(Staphylococcus epidermidis) | https://github.com/AdmiralenOla/Scoary | [37] |
| treeWAS | 2017 | 整合重组及表型聚类信息,支持连续表型 | 系统发育校正 | 低、中重组率细菌 | 脑膜炎双球菌(Neisseria meningitidis) | https://github.com/caitiecollins/treeWAS | [35] |
新窗口打开|下载CSV
由于重组率的物种间差异,不同细菌的连锁不平衡模式多种多样[39],这也为BGWAS带来假阳性问题。对于重组率较低的细菌,变异之间相互连锁,难以区分“搭车”变异与真正与表型相关的变异。目前对该问题的常用解决方法是:综合变异位点特性(如同义/非同义突变)及所在基因的功能注释进行深入推测,缩小靶标范围,并进而通过分子生物学实验以验证靶标变异是否确实与某种表型相关。对于高频重组细菌,变异之间连锁程度低,此类样本更适用于使用传统GWAS方法进行分析。
此外,自然选择可以对细菌种群结构形成非常大的影响,如抗生素选择压力可能在自然界中筛选出特定的病原菌克隆群。能够检测正向选择引起的趋同变异的phyC[11]及整合家系效果(lineage effect)检测的bugwas[36]能够有效解决这类问题。
除了种群结构和重组率的影响,多重检验带来的假阳性也是GWAS不可避免的问题。GWAS通常涉及数以万计的相关性检验,那么按照常用的显著性阈值P<0.05时,理论上会随机产生数百个假阳性结果。如此高的假阳性率显然无法接受,因此需要对多重检验进行校正。目前BGWAS主要沿用了人类GWAS中常用多重检验校正方法,如Bonferoni 校正(显著性阈值=0.05/N,N为变异位点数)及假发现率校正(false discovery rate correction)等[40],能够显著降低假阳性结果的数量。
1.4 实验确认
尽管目前采用多种策略来降低GWAS结果的假阳性,但假阳性问题仍然难以完全避免。为此,人类GWAS通常需要重复研究来确认表型相关变异[41]。得益于细菌易实验操纵的特点,BGWAS中鉴定的靶标变异可通过实验室验证的方法来排除假阳性。Falkow[42]在1988年提出了分子科赫法则,即“基 因失活造成表型消失,重建则表型恢复”。这为实验确认GWAS鉴定的相关变异提供了标准。另外,基因敲除/重组技术的进步及突变体文库的完善极大的方便了BGWAS结果确认,多项BGWAS通过实验对相关位点进行了验证,确认了变异与表型的相关性[10,11,14,18,19,24]。1.5 BGWAS的研究工具
目前已有多种工具被开发出来,用于解决BGWAS分析所面临的问题(表2)。Farhat等[11]开发了通过检测趋同进化来鉴定表型相关变异的软件phyC。该软件能够显著降低假阳性,适用于强选择性状相关变异的检测[15]。Earle等[36]利用线性混合模型整合样本相关性来校正种群结构,开发出了能够同时检测表型相关位点及家系效果的软件bugwas,成功应用于3000多株不同重组率细菌的耐药性研究。Lees[38]利用“Scale-mining”算法,开发出了高计算效率、支持可变长度k-mer的SEER。Brynildsrud等[37]针对细菌泛基因组,开发出了能够快速检测表型相关基因获得/缺失的Scoary。Collins等[35]通过整合重组及表型聚类信息开发出了基于系统发育校正的treeWAS软件。除了专门针对细菌非核心基因组的Scoary软件外,bugwas、SEER等都支持k-mer运算,能够同时捕捉核心及非核心基因组变异信息。由于BGWAS软件开发仍处于萌芽阶段(到目前为止,多数BGWAS软件发表不到一年),这些工具的实际应用价值还有待实践检验。在实际应用中,可以根据基因型数据类型,选择多个软件同时进行数据分析,对运算结果做交叉验证。2 BGWAS研究的应用进展
通过BGWAS研究,多种重要表型与遗传因子之间的相关性被建立起来。目前半数以上BGWAS研究是针对细菌耐药性开展的[11-13,15,18,23]。如Farhat等[11]通过检测趋同进化来筛选结核杆菌(Mycobacterium tuberculosis)的耐药相关变异,除了找到了过去已知的全部耐药位点外,还发现了39个新的耐药相关区域。第一项大样本的BGWAS同样关注于耐药性问题,Chewapreecha等[13]通过分析3701株肺炎链球菌(Streptococcus pneumoniae),找到了与β内酰胺类抗性相关的301个SNP位点。部分BGWAS研究关注宿主适应性、毒力等细菌与宿主相互作用的表型。例如,最早的BGWAS研究通过分析192株来源于不同宿主的空肠弯曲杆菌(Campylobacter jejuni),发现并验证了一组维生素B5合成相关基因与宿主饮食适应相关,既而导致某些基因型的细菌倾向于生活在特定种类的宿主中[10]。Laabei等[14]通过分析90株毒力不同的金黄色葡萄球菌(Staphylococcus aureus),发现了121个毒力相关因子,并通过实验验证了4个毒力因子,进一步增进了人们对细菌毒力的认识。此外,BGWAS还被应用于生物膜形成[16]、存活力[24]、体内运输[22]等多种细菌生理相关的表型研究,为解析这些表型的遗传机制提供了新的数据。通过BGWAS发现的表型相关变异可以很好的促进细菌表型预测研究。Laabei等[14]利用BGWAS在金黄色葡萄球菌(Staphylococcus aureus)中鉴定出的50个毒力相关变异,结合“随机森林”机器学习算法,建立了该病原的毒力预测模型,其预测准确率高达85%以上。Mobegi等[23]利用类似的方法建立了肺炎链球菌(Streptococcus pneumoniae)耐药性预测模型,能够根据基因组序列定量评估分离株耐药性的强弱。表型预测模型的建立进一步拓展了BGWAS的用途,随着算法和模型的完善,将极大加速细菌表型信息的获取,对基因的功能研究以及细菌性病原的监测和控制等领域具有重要意义。
值得关注的是,BGWAS研究还可以应用于开发新的临床诊断标记。Buchanan等[21]通过对166株空肠弯曲杆菌(Campylobacter jejuni)的泛基因组序列BGWAS分析,发现25个非核心基因的获得缺失与弯曲杆菌病发病相关。这些遗传标记可通过PCR等方法实现快速检测,因而可以方便的应用于对细菌病原所致疾病的临床诊断和治疗中。
3 结语与展望
尽管只经历了短短几年发展历史,BGWAS研究已经取得了丰硕成果。通过BGWAS分析,人们揭示了细菌多种重要表型的相关遗传因子,极大加深了人们对细菌遗传机制、适应性进化及传播等领域的认识,并为医学临床诊断、治疗和公共卫生领域的进步提供了新的思路。BGWAS本质上是在不同数据组之间建立关联。数据组的种类、获取难度、累积数量与关联算法决定了BGWAS的应用前景。(1)未来的BGWAS研究将更加全面,不只着眼于耐药性等效应量大的表型,也会增加对效应量小、相关性较弱的表型与变异的关注,从全局角度重新认识功能基因对细菌表型的影响。(2) BGWAS会与宿主基因组数据整合分析,通过细菌基因组、表型与宿主基因组的综合性关联
分析,进一步增进人们对细菌变异是否受宿主影响这一问题的认识。此外,这种细菌—宿主相互作用研究将帮助人们识别细菌的靶标蛋白,进而促进药物及疫苗等的开发[25]。(3) BGWAS可以与宏基因组数据相结合,通过多种细菌之间的基因组关联分析,进一步增进人们对细菌协作、竞争等行为的认识,为宏基因组研究提供新的手段。(4) BGWAS结果易于进行实验室验证的特点,将促进该领域在理论上得到迅速发展,从而可以对人类GWAS研究的基础理论和算法提供支持,为多基因连锁控制某一性状等难题提供新的解决思路。(5) BGWAS在临床细菌检验与疾病诊断中有着广阔的发展前景。临床检验实验如最小抑菌浓度检验等,能为BGWAS提供海量的重要表型信息,而BGWAS能利用这些信息来鉴定相关变异,进而建立和优化细菌表型预测模型,极大改善临床检验与诊断的速度及准确性。相信随着测序成本的进一步降低和研究工具的持续更新,会使BGWAS的深厚发展潜力得以爆发。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:22243964 [本文引用: 1]

The past five years have seen many scientific and biological discoveries made through the experimental design of genome-wide association studies (GWASs). These studies were aimed at detecting variants at genomic loci that are associated with complex traits in the population and, in particular, at detecting associations between common single-nucleotide polymorphisms (SNPs) and common diseases such as heart disease, diabetes, auto-immune diseases, and psychiatric disorders. We start by giving a number of quotes from scientists and journalists about perceived problems with GWASs. We will then briefly give the history of GWASs and focus on the discoveries made through this experimental design, what those discoveries tell us and do not tell us about the genetics and biology of complex traits, and what immediate utility has come out of these studies. Rather than giving an exhaustive review of all reported findings for all diseases and other complex traits, we focus on the results for auto-immune diseases and metabolic diseases. We return to the perceived failure or disappointment about GWASs in the concluding section.
URLPMID:28686856 [本文引用: 1]
Abstract Application of the experimental design of genome-wide association studies (GWASs) is now 10 years old (young), and here we review the remarkable range of discoveries it has facilitated in population and complex-trait genetics, the biology of diseases, and translation toward new therapeutics. We predict the likely discoveries in the next 10 years, when GWASs will be based on millions of samples with array data imputed to a large fully sequenced reference panel and on hundreds of thousands of samples with whole-genome sequencing data. Copyright 脗漏 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
URLMagsci [本文引用: 1]
<P>2005年, Science杂志首次报道了有关人类年龄相关性黄斑变性的全基因组关联研究, 此后有关肥胖、2型糖尿病、冠心病、阿尔茨海默病等一系列复杂疾病的全基因组关联研究被陆续报道, 这一阶段被称为人类全基因组关联研究的第一次浪潮。文章分别介绍了全基因组关联研究统计分析的方法、软件和应用实例; 比较了关联分析中多重检验的P值调整方法, 包括Bonferroni、递减的Bonferroni校正法、模拟运算法和控制错误发现率的方法; 还讨论了人群混杂对关联分析结果可能产生的影响及原理, 以及全基因组关联研究中控制人群混杂的方法的研究进展和应用实例。在全基因组关联研究的第一次浪潮中, 应用经典的遗传统计方法发现了许多基因-表型之间的关联并且能够对这些关联做出解释, 其中包括许多基因组中的未知基因和染色体区域。然而, 全基因组关联研究的继续发展需要进一步阐述基因组内基因之间相互作用、基因-基因之间的复杂作用网络与环境因素的相互作用在复杂疾病发生中的作用, 现有的统计分析方法肯定不能满足需要, 开发更为高级的统计分析方法势在必行。最后, 文章还给出了全基因组关联研究统计分析软件的相关网站信息。</P>
URLMagsci [本文引用: 1]
<P>2005年, Science杂志首次报道了有关人类年龄相关性黄斑变性的全基因组关联研究, 此后有关肥胖、2型糖尿病、冠心病、阿尔茨海默病等一系列复杂疾病的全基因组关联研究被陆续报道, 这一阶段被称为人类全基因组关联研究的第一次浪潮。文章分别介绍了全基因组关联研究统计分析的方法、软件和应用实例; 比较了关联分析中多重检验的P值调整方法, 包括Bonferroni、递减的Bonferroni校正法、模拟运算法和控制错误发现率的方法; 还讨论了人群混杂对关联分析结果可能产生的影响及原理, 以及全基因组关联研究中控制人群混杂的方法的研究进展和应用实例。在全基因组关联研究的第一次浪潮中, 应用经典的遗传统计方法发现了许多基因-表型之间的关联并且能够对这些关联做出解释, 其中包括许多基因组中的未知基因和染色体区域。然而, 全基因组关联研究的继续发展需要进一步阐述基因组内基因之间相互作用、基因-基因之间的复杂作用网络与环境因素的相互作用在复杂疾病发生中的作用, 现有的统计分析方法肯定不能满足需要, 开发更为高级的统计分析方法势在必行。最后, 文章还给出了全基因组关联研究统计分析软件的相关网站信息。</P>
URLMagsci [本文引用: 1]
在过去的5年中, 全基因组关联研究(Genome-wide association study, GWAS)方法已被证明是研究复杂疾病和性状遗传易感变异的一种有效手段。目前, 各国科学家在多种复杂疾病和性状中开展了大量的GWAS, 对肿瘤、糖尿病、心脏病、神经精神疾病、自身免疫及免疫相关疾病等复杂疾病以及一些常见性状(如身高、体重、血脂、色素等)的遗传易感基因研究取得了重大成果。截止到2010年9月11日, 运用GWAS开展了对近200种复杂疾病/性状的研究, 发现了3 000多个疾病相关的遗传变异。文章就GWAS的发展及其在复杂疾病/性状中的应用做一综述。
URLMagsci [本文引用: 1]
在过去的5年中, 全基因组关联研究(Genome-wide association study, GWAS)方法已被证明是研究复杂疾病和性状遗传易感变异的一种有效手段。目前, 各国科学家在多种复杂疾病和性状中开展了大量的GWAS, 对肿瘤、糖尿病、心脏病、神经精神疾病、自身免疫及免疫相关疾病等复杂疾病以及一些常见性状(如身高、体重、血脂、色素等)的遗传易感基因研究取得了重大成果。截止到2010年9月11日, 运用GWAS开展了对近200种复杂疾病/性状的研究, 发现了3 000多个疾病相关的遗传变异。文章就GWAS的发展及其在复杂疾病/性状中的应用做一综述。
[本文引用: 1]
URLPMID:3965119 [本文引用: 1]
The National Human Genome Research Institute (NHGRI) Catalog of Published Genome-Wide Association Studies (GWAS) Catalog provides a publicly available manually curated collection of published GWAS assaying at least 100,000 single-nucleotide polymorphisms (SNPs) and all SNP-trait associations with P <1 脳 10(-5). The Catalog includes 1751 curated publications of 11 912 SNPs. In addition to the SNP-trait association data, the Catalog also publishes a quarterly diagram of all SNP-trait associations mapped to the SNPs' chromosomal locations. The Catalog can be accessed via a tabular web interface, via a dynamic visualization on the human karyotype, as a downloadable tab-delimited file and as an OWL knowledge base. This article presents a number of recent improvements to the Catalog, including novel ways for users to interact with the Catalog and changes to the curation infrastructure.
URLPMID:16782339 [本文引用: 4]
Abstract Bacteria display many interesting phenotypes such as virulence, tissue specificity and host range, for which it would be useful to know the genetic basis. Association mapping involves identifying causal variants by showing that particular genotypes are statistically associated with a phenotypic trait in a sample of strains taken from a natural population. With the advent of high-throughput genotyping, association mapping is becoming an increasingly powerful approach. However, until recently, association studies had not been used in bacteria because of their strong population structure, which can produce false positives and/or loss of statistical power unless elucidated and taken into account in analyses. Here, we describe how association mapping could be successfully applied to bacteria and outline the necessary sampling and genotyping strategies.
URLPMID:27572652 [本文引用: 1]
Abstract A linear-mixed modelling genome-wide association approach for detecting genes and genetic variants underlying antibiotic resistance in bacterial pathogens heralds a new era for microbial genome-wide association studies.
URLPMID:23818615 [本文引用: 5]
Genome-wide association studies have the potential to identify causal genetic factors underlying important phenotypes but have rarely been performed in bacteria. We present an association mapping method that takes into account the clonal population structure of bacteria and is applicable to both core and accessory genome variation. Campylobacter is a common cause of human gastroenteritis as a consequence of its proliferation in multiple farm animal species and its transmission via contaminated meat and poultry. We applied our association mapping method to identify the factors responsible for adaptation to cattle and chickens among 192 Campylobacter isolates from these and other host sources. Phylogenetic analysis implied frequent host switching but also showed that some lineages were strongly associated with particular hosts. A seven-gene region with a host association signal was found. Genes in this region were almost universally present in cattle but were frequently absent in isolates from chickens and wild birds. Three of the seven genes encoded vitamin B-5 biosynthesis. We found that isolates from cattle were better able to grow in vitamin B-5-depleted media and propose that this difference may be an adaptation to host diet.
[本文引用: 6]
[本文引用: 2]
URL [本文引用: 3]
URLPMID:24717264 [本文引用: 5]
Microbial virulence is a complex and often multifactorial phenotype, intricately linked to a pathogen's evolutionary trajectory. Toxicity, the ability to destroy host cell membranes, and adhesion, the ability to adhere to human tissues, are the major virulence factors of many bacterial pathogens, including Staphylococcus aureus. Here, we assayed the toxicity and adhesiveness of 90 MRSA (methicillin resistant S. aureus) isolates and found that while there was remarkably little variation in adhesion, toxicity varied by over an order of magnitude between isolates, suggesting different evolutionary selection pressures acting on these two traits. We performed a genome-wide association study (GWAS) and identified a large number of loci, as well as a putative network of epistatically interacting loci, that significantly associated with toxicity. Despite this apparent complexity in toxicity regulation, a predictive model based on a set of significant single nucleotide polymorphisms (SNPs) and insertion and deletions events (indels) showed a high degree of accuracy in predicting an isolate's toxicity solely from the genetic signature at these sites. Our results thus highlight the potential of using sequence data to determine clinically relevant parameters and have further implications for understanding the microbial virulence of this opportunistic pathogen.
URL [本文引用: 5]
Significant advances in sequencing technologies and genome-wide association studies (GWAS) have revealed substantial insight into the genetic architecture of human phenotypes. In recent years, the application of this approach in bacteria has begun to reveal the genetic basis of bacterial host preference, antibiotic resistance, and virulence. Here, we consider relevant differences between bacterial and human genome dynamics, apply GWAS to a global sample of Mycobacterium tuberculosis genomes to highlight the impacts of linkage disequilibrium, population stratification, and natural selection, and finally compare the traditional GWAS against phyC, a contrasting method of mapping genotype to phenotype based upon evolutionary convergence. We discuss strengths and weaknesses of both methods, and make suggestions for factors to be considered in future bacterial GWAS.
URL [本文引用: 2]
Multicellular biofilms are an ancient bacterial adaptation that offers a protective environment for survival in hostile habitats. In microaerophilic organisms such asCampylobacter, biofilms play a key role in transmission to humans as the bacteria are exposed to atmospheric oxygen concentrations when leaving the reservoir host gut. Genetic determinants of biofilm formation differ between species, but little is known about how strains of the same species achieve the biofilm phenotype with different genetic backgrounds. Our approach combines genome‐wide association studies with traditional microbiology techniques to investigate the genetic basis of biofilm formation in 102Campylobacter jejuniisolates. We quantified biofilm formation among the isolates and identified hotspots of genetic variation in homologous sequences that correspond to variation in biofilm phenotypes. Thirteen genes demonstrated a statistically robust association including those involved in adhesion, motility, glycosylation, capsule production and oxidative stress. The genes associated with biofilm formation were different in the host generalistST‐21 andST‐45 clonal complexes, which are frequently isolated from multiple host species and clinical samples. This suggests the evolution of enhanced biofilm from different genetic backgrounds and a possible role in colonization of multiple hosts and transmission to humans.
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
URLPMID:5124939 [本文引用: 1]
Abstract Carbapenems are a class of last-resort antibiotics; thus, the increase in bacterial carbapenem-resistance is a serious public health threat. Acinetobacter baumannii is one of the microorganisms that can acquire carbapenem-resistance; it causes severe nosocomial infection, and is notoriously difficult to control in hospitals. Recently, a machine-learning approach was first used to analyze the genome sequences of hundreds of susceptible and resistant A. baumannii strains, including those carrying commonly acquired resistant mechanisms, to build a classifier that can predict strain resistance. A complementary approach is to explore novel genetic elements that could be associated with the antimicrobial resistance of strains, independent of known mechanisms. Therefore, we carefully selected A. baumannii strains, spanning various genotypes, from public genome databases, and conducted the first genome-wide association study (GWAS) of carbapenem resistance. We employed a recently developed method, capable of identifying any kind of genetic variation and accounting for bacterial population structure, and evaluated its effectiveness. Our study identified a surface adhesin gene that had been horizontally transferred to an ancestral branch of A. baumannii, as well as a specific region of that gene that appeared to accumulate multiple individual variations across the different branches of carbapenem-resistant A. baumannii strains.
URLPMID:5492696 [本文引用: 2]
Campylobacter jejuni is a leading human enteric pathogen worldwide and despite an improved understanding of its biology, ecology, and epidemiology, limited tools exist for identifying strains that are likely to cause disease. In the current study, we used subtyping data in a database representing over 24,000 isolates collected through various surveillance projects in Canada to identify 166 representative genomes from prevalent C. jejuni subtypes for whole genome sequencing. The sequence data was used in a genome-wide association study (GWAS) aimed at identifying accessory gene markers associated with clinically-related C. jejuni subtypes. Prospective markers (n=28) were then validated against a large number (n=3,902) of clinically-associated and non-clinically-associated genomes from a variety of sources. A total of 25 genes, including six sets of genetically linked genes, were identified as robust putative diagnostic markers for clinically-related C. jejuni subtypes. Although some of the genes identified in this study have been previously shown to play a role in important processes such as iron acquisition and vitamin B5 biosynthesis, others have unknown function or are unique to the current study and warrant further investigation. As few as four of these markers could be used in combination to detect up to 90% of clinically-associated isolates in the validation dataset, and such markers could form the basis for a screening assay to rapidly identify strains that pose an increased risk to public health. The results of the current study are consistent with the notion that specific groups of C. jejuni strains of interest are defined by the presence of specific accessory genes.
URLPMID:5576492 [本文引用: 2]
Streptococcus pneumoniaeis a leading cause of invasive disease in infants, especially in low-income settings. Asymptomatic carriage in the nasopharynx is a prerequisite for disease, but variability in its duration is currently only understood at the serotype level. Here we developed a model to calculate the duration of carriage episodes from longitudinal swab data, and combined these results with whole genome sequence data. We estimated that pneumococcal genomic variation accounted for 63% of the phenotype variation, whereas the host traits considered here (age and previous carriage) accounted for less than 5%. We further partitioned this heritability into both lineage and locus effects, and quantified the amount attributable to the largest sources of variation in carriage duration: serotype (17%), drug-resistance (9%) and other significant locus effects (7%). A pan-genome-wide association study identified prophage sequences as being associated with decreased carriage duration independent of serotype, potentially by disruption of the competence mechanism. These findings support theoretical models of pneumococcal competition and antibiotic resistance. Microorganisms live in most parts of our body, including the inside of our nose. Most of the microbes are harmless and can even be beneficial to our health. However, some microbes can cause diseases 鈥 although they often go unnoticed, as our immune system can remove them before we show any symptoms. For example, the bacteriumStreptococcus pneumoniaecan cause diseases such as pneumonia and meningitis, but generally, it lives harmlessly in the nose, and is particularly common in children and the elderly. The longer the bacteria live in the nose before being killed by the immune system, the more likely they are to be transmitted to another person. The amount of time it takes for the immune system to clear the bacteria depends on various factors, such as the age of the person or the bacterium鈥檚 defense mechanism and its genetic material. A particularly important aspect is to what subtype, also known as serotype, a bacterium belongs to, which is characterized by differences in the structure of the sugar coating that surrounds the microbe. However, until now, it was not known how much each of these factors contributes. Now, Lees et al. have developed a mathematical model to calculate how long the bacteria are carried in the nose before they are cleared away, and compared it with the genomic data of the bacteria. For this, over 14,000 nose swabs from almost 600 children were collected over a two-year period. In their model, Lees et al. calculated that the bacteria鈥檚 genetics explained over 60% of the variability in survival time. They also found that the serotype was the most important individual factor that influenced how long a bacterium could survive. The age of the child was less important and only accounted for 5%. In addition, Lees et al. also found that when viruses infected someS. pneumoniae, the bacteria died sooner. A next step will be to confirm the effect of a viral infection on the bacteria鈥檚 survival time in a controlled model system, and also replicate the findings in separate population study.Understanding how long people can carry bacteria and transmit them to others may help to develop new vaccination or treatment strategies to control infections. Moreover, the discovery that viruses can negatively affect how long a bacterium lives, could motivate studies to investigate these findings further.
URLPMID:5311915 [本文引用: 3]
Advances in genome sequencing technologies and genome-wide association studies (GWAS) have provided unprecedented insights into the molecular basis of microbial phenotypes and enabled the identification of the underlying genetic variants in real populations. However, utilization of genome sequencing in clinical phenotyping of bacteria is challenging due to the lack of reliable and accurate approaches. Here, we report a method for predicting microbial resistance patterns using genome sequencing data. We analyzed whole genome sequences of 1,680Streptococcus pneumoniaeisolates from four independent populations using GWAS and identified probable hotspots of genetic variation which correlate with phenotypes of resistance to essential classes of antibiotics. With the premise that accumulation of putative resistance-conferring SNPs, potentially in combination with specific resistance genes, precedes full resistance, we retrogressively surveyed the hotspot loci and quantified the number of SNPs and/or genes, which if accumulated would confer full resistance to an otherwise susceptible strain. We name this approach the 鈥榙istance to resistance鈥. It can be used to identify the creep towards complete antibiotics resistance in bacteria using genome sequencing. This approach serves as a basis for the development of future sequencing-based methods for predicting resistance profiles of bacterial strains in hospital microbiology and public health settings.
URLPMID:27883255 [本文引用: 3]
Campylobacter jejuni is a major cause of bacterial gastroenteritis worldwide, primarily associated with the consumption of contaminated poultry. C. jejuni lineages vary in host range and prevalence in human infection, suggesting differences in survival throughout the poultry processing chain. From 7,343 MLST-characterised isolates, we sequenced 600 C. jejuni and C. coli isolates from various stages of poultry processing and clinical cases. A genome-wide association study (GWAS) in C. jejuni ST-21 and ST-45 complexes identified genetic elements over-represented in clinical isolates that increased in frequency throughout the poultry processing chain. Disease-associated SNPs were distinct in these complexes, sometimes organised in haplotype blocks. The function of genes containing associated elements was investigated, demonstrating roles for cj1377c in formate metabolism, nuoK in aerobic survival and oxidative respiration, and cj1368-70 in nucleotide salvage. This work demonstrates the utility of GWAS for investigating transmission in natural zoonotic pathogen populations and provides evidence that major C. jejuni lineages have distinct genotypes associated with survival, within the host specific niche, from farm to fork. This article is protected by copyright. All rights reserved.
URLPMID:27840430 [本文引用: 3]
Abstract The reduced costs of sequencing have led to whole-genome sequences for a large number of microorganisms, enabling the application of microbial genome-wide association studies (GWAS). Given the successes of human GWAS in understanding disease aetiology and identifying potential drug targets, microbial GWAS are likely to further advance our understanding of infectious diseases. These advances include insights into pressing global health problems, such as antibiotic resistance and disease transmission. In this Review, we outline the methodologies of GWAS, the current state of the field of microbial GWAS, and how lessons from human GWAS can direct the future of the field.
URLPMID:25484920 [本文引用: 4]
Whole genome sequencing is increasingly used to study phenotypic variation among infectious pathogens and to evaluate their relative transmissibility, virulence, and immunogenicity. To date, relatively little has been published on how and how many pathogen strains should be selected for studies associating phenotype and genotype. There are specific challenges when identifying genetic associations in bacteria which often comprise highly structured populations. Here we consider general methodological questions related to sampling and analysis focusing on clonal to moderately recombining pathogens. We propose that a matched sampling scheme constitutes an efficient study design, and provide a power calculator based on phylogenetic convergence. We demonstrate this approach by applying it to genomic datasets for two microbial pathogens: Mycobacterium tuberculosis and Campylobacter species.
URLPMID:25593593 [本文引用: 1]
Abstract Genome-wide association studies (GWASs) have become an increasingly important approach for eukaryotic geneticists, facilitating the identification of hundreds of genetic polymorphisms that are responsible for inherited diseases. Despite the relative simplicity of bacterial genomes, the application of GWASs to identify polymorphisms responsible for important bacterial phenotypes has only recently been made possible through advances in genome sequencing technologies. Bacterial GWASs are now about to come of age thanks to the availability of massive datasets, and because of the potential to bridge genomics and traditional genetic approaches that is provided by improving validation strategies. A small number of pioneering GWASs in bacteria have been published in the past 2脗聽years, examining from 75 to more than 3,000 strains. The experimental designs have been diverse, taking advantage of different processes in bacteria for generating variation. Analysis of data from bacterial GWASs can, to some extent, be performed using software developed for eukaryotic systems, but there are important differences in genome evolution that must be considered. The greatest experimental advantage of bacterial GWASs is the potential to perform downstream validation of causality and dissection of mechanism. We review the recent advances and remaining challenges in this field and propose strategies to improve the validation of bacterial GWASs.
URLPMID:4361730 [本文引用: 1]
Since the first two complete bacterial genome sequences were published in 1995, the science of bacteria has dramatically changed. Using third-generation DNA sequencing, it is possible to completely sequence a bacterial genome in a few hours and identify some types of methylation sites along the genome as well. Sequencing of bacterial genome sequences is now a standard procedure, and the information from tens of thousands of bacterial genomes has had a major impact on our views of the bacterial world. In this review, we explore a series of questions to highlight some insights that comparative genomics has produced. To date, there are genome sequences available from 50 different bacterial phyla and 11 different archaeal phyla. However, the distribution is quite skewed towards a few phyla that contain model organisms. But the breadth is continuing to improve, with projects dedicated to filling in less characterized taxonomic groups. The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system provides bacteria with immunity against viruses, which outnumber bacteria by tenfold. How fast can we go? Second-generation sequencing has produced a large number of draft genomes (close to 90聽% of bacterial genomes in GenBank are currently not complete); third-generation sequencing can potentially produce a finished genome in a few hours, and at the same time provide methlylation sites along the entire chromosome. The diversity of bacterial communities is extensive as is evident from the genome sequences available from 50 different bacterial phyla and 11 different archaeal phyla. Genome sequencing can help in classifying an organism, and in the case where multiple genomes of the same species are available, it is possible to calculate the pan- and core genomes; comparison of more than 2000 Escherichia coli genomes finds an E. coli core genome of about 3100 gene families and a total of about 89,000 different gene families. Why do we care about bacterial genome sequencing? There are many practical applications, such as genome-scale metabolic modeling, biosurveillance, bioforensics, and infectious disease epidemiology. In the near future, high-throughput sequencing of patient metagenomic samples could revolutionize medicine in terms of speed and accuracy of finding pathogens and knowing how to treat them.
URLPMID:4493402 [本文引用: 1]
Innovations in sequencing technologies have allowed biologists to make incredible advances in understanding biological systems. As experience grows, researchers increasingly recognize that analyzing the wealth of data provided by these new sequencing platforms requires careful attention to detail for robust results. Thus far, much of the scientific Communit's focus for use in bacterial genomics has been on evaluating genome assembly algorithms and rigorously validating assembly program performance. Missing, however, is a focus on critical evaluation of variant callers for these genomes. Variant calling is essential for comparative genomics as it yields insights into nucleotide-level organismal differences. Variant calling is a multistep process with a host of potential error sources that may lead to incorrect variant calls. Identifying and resolving these incorrect calls is critical for bacterial genomics to advance. The goal of this review is to provide guidance on validating algorithms and pipelines used in variant calling for bacterial genomics. First, we will provide an overview of the variant calling procedures and the potential sources of error associated with the methods. We will then identify appropriate datasets for use in evaluating algorithms and describe statistical methods for evaluating algorithm performance. As variant calling moves from basic research to the applied setting, standardized methods for performance evaluation and reporting are required; it is our hope that this review provides the groundwork for the development of these standards.
URLPMID:26548914 [本文引用: 1]
Twenty years ago, the publication of the first bacterial genome sequence, from Haemophilus influenzae, shook the world of bacteriology. In this Timeline, we review the first two decades of bacterial genome sequencing, which have been marked by three revolutions: whole-genome shotgun sequencing, high-throughput sequencing and single-molecule long-read sequencing. We summarize the social history of sequencing and its impact on our understanding of the biology, diversity and evolution of bacteria, while also highlighting spin-offs and translational impact in the clinic. We look forward to a 'sequencing singularity', where sequencing becomes the method of choice for as-yet unthinkable applications in bacteriology and beyond.
URLPMID:25483351 [本文引用: 1]
Next generation sequencing technologies have engendered a genome sequence data deluge in public databases. Genome analyses have transitioned from single or few genomes to hundreds to thousands of genomes. Pan-genome analyses provide a framework for estimating the genomic diversity of the dataset at hand and predicting the number of additional whole genomes sequences that would be necessary to fully characterize that diversity. We review recent implementations of the pan-genome approach, its impact and limits, and we propose possible extensions, including analyses at the whole genome multiple sequence alignment level.
URL [本文引用: 1]
URLPMID:20137780 [本文引用: 1]
Genome-wide association studies are routinely conducted to identify genetic variants that influence complex disorders. It is well known that failure to properly account for population or pedigree structure can lead to spurious association as well as reduced power. We propose a method, ROADTRIPS, for case-control association testing in samples with partially or completely unknown population and pedigree structure. ROADTRIPS uses a covariance matrix estimated from genome-screen data to correct for unknown population and pedigree structure while maintaining high power by taking advantage of known pedigree information when it is available. ROADTRIPS can incorporate data on arbitrary combinations of related and unrelated individuals and is computationally feasible for the analysis of聽genetic studies with millions of markers. In simulations with related individuals and population structure, including admixture, we demonstrate that ROADTRIPS provides a substantial improvement over existing methods in terms of power and type 1 error. The ROADTRIPS method can be used across a variety of study designs, ranging from studies that have a combination of unrelated individuals and small pedigrees to studies of isolated founder populations with partially known or completely unknown pedigrees. We apply the method to analyze two data sets: a study of rheumatoid arthritis in small UK pedigrees, from Genetic Analysis Workshop 15, and data from the Collaborative Study of the Genetics of Alcoholism on alcohol dependence in a sample of moderate-size pedigrees of European descent, from Genetic Analysis Workshop 14. We detect genome-wide significant association, after Bonferroni correction, in both studies.
URLPMID:21892150 [本文引用: 1]
We describe factored spectrally transformed linear mixed models (FaST-LMM), an algorithm for genome-wide association studies (GWAS) that scales linearly with cohort size in both run time and memory use. On Wellcome Trust data for 15,000 individuals, FaST-LMM ran an order of magnitude faster than current efficient algorithms. Our algorithm can analyze data for 120,000 individuals in just a few hours, whereas current algorithms fail on data for even 20,000 individuals (http://mscompbio.codeplex.com/).
URL [本文引用: 2]
Genome-Wide Association Studies (GWAS) in microbial organisms have the potential to vastly improve the way we understand, manage, and treat infectious diseases. Yet, GWAS methods established thus far remain insufficiently able to capitalise on the growing wealth of bacterial and viral genetic sequence data. Facing clonal population structure and homologous recombination, existing GWAS methods struggle to achieve both the precision necessary to reject spurious findings and the power required to detect associations in microbes. In this paper, we introduce a novel phylogenetic approach that has been tailor-made for microbial GWAS, which is applicable to organisms ranging from purely clonal to frequently recombining, and to both binary and continuous phenotypes. Our approach is robust to the confounding effects of both population structure and recombination, while maintaining high statistical power to detect associations. Thorough testing via application to simulated data provides strong support for the power and specificity of our approach and demonstrates the advantages offered over alternative cluster-based and dimension-reduction methods. Two applications to Neisseria meningitidis illustrate the versatility and potential of our method, confirming previously-identified penicillin resistance loci and resulting in the identification of both well-characterised and novel drivers of invasive disease. Our method is implemented as an open-source R package called treeWAS which is freely available at https://github.com/caitiecollins/treeWAS.
URLPMID:5049680 [本文引用: 3]
Abstract Bacteria pose unique challenges for genome-wide association studies because of strong structuring into distinct strains and substantial linkage disequilibrium across the genome(1,2). Although methods developed for human studies can correct for strain structure(3,4), this risks considerable loss-of-power because genetic differences between strains often contribute substantial phenotypic variability(5). Here, we propose a new method that captures lineage-level associations even when locus-specific associations cannot be fine-mapped. We demonstrate its ability to detect genes and genetic variants underlying resistance to 17 antimicrobials in 3,144 isolates from four taxonomically diverse clonal and recombining bacteria: Mycobacterium tuberculosis, Staphylococcus aureus, Escherichia coli and Klebsiella pneumoniae. Strong selection, recombination and penetrance confer high power to recover known antimicrobial resistance mechanisms and reveal a candidate association between the outer membrane porin nmpC and cefazolin resistance in E. coli. Hence, our method pinpoints locus-specific effects where possible and boosts power by detecting lineage-level differences when fine-mapping is intractable.
URLPMID:5124306 [本文引用: 2]
Genome-wide association studies (GWAS) have become indispensable in human medicine and genomics, but very few have been carried out on bacteria. Here we introduce Scoary, an ultra-fast, easy-to-use, and widely applicable software tool that scores the components of the pan-genome for associations to observed phenotypic traits while accounting for population stratification, with minimal assumptions about evolutionary processes. We call our approach pan-GWAS to distinguish it from traditional, single nucleotide polymorphism (SNP)-based GWAS. Scoary is implemented in Python and is available under an open source GPLv3 license athttps://github.com/AdmiralenOla/Scoary. The online version of this article (doi:10.1186/s13059-016-1108-8) contains supplementary material, which is available to authorized users.
URL [本文引用: 2]
Bacterial genomes vary extensively in terms of both gene content and gene sequence. This plasticity hampers the use of traditional SNP-based methods for identifying all genetic associations with phenotypic variation. Here we introduce a computationally scalable and widely applicable statistical method (SEER) for the identification of sequence elements that are significantly enriched in a phenotype of interest. SEER is applicable to tens of thousands of genomes by counting variable-length k-mers using a distributed string-mining algorithm. Robust options are provided for association analysis that also correct for the clonal population structure of bacteria. Using large collections of genomes of the major human pathogensStreptococcus pneumoniaeandStreptococcus pyogenes, SEER identifies relevant previously characterized resistance determinants for several antibiotics and discovers potential novel factors related to the invasiveness ofS. pyogenes. We thus demonstrate that our method can answer important biologically and medically relevant questions. Plasticity and clonal population structure in bacterial genomes can hinder traditional SNP-based genetic association studies. Here, Corander and colleagues present a method to identify variable-length sequence elements enriched in a phenotype of interest, and demonstrate its use in human pathogens.
URLPMID:20452218 [本文引用: 1]
Genetic exchange plays a defining role in the evolution of many bacteria. The recent accumulation of nucleotide sequence data from multiple members of diverse bacterial genera has facilitated comparative studies that have revealed many features of this process. Here we focus on genetic exchange that has involved homologous recombination and illustrate how nucleotide sequence data have furthered our understanding of: (i) the frequency of recombination; (ii) the impact of recombination in different parts of the genome; and (iii) patterns of gene flow within bacterial populations. Summarizing the results obtained for a range of bacteria, we survey evidence indicating that the extent and nature of recombination vary widely among microbiological species and often among lineages assigned to the same microbiological species. These results have important implications in studies ranging from epidemiological investigations to examination of the bacterial species problem.
URL [本文引用: 1]
[本文引用: 1]
URLPMID:3055197 [本文引用: 1]
Microbial genetics and molecular cloning now permit us to routinely isolate specific genes from a variety of microbial pathogens. Obviously not all genes from pathogenic microorganisms play a role in pathogenicity or virulence. Just as Koch's postulates were formulated to identify the causal relationship between an organism and a specific disease, the notion is presented here that a form of molecular Koch's postulates is needed when examining the potential role of genes and their products in the pathogenesis of infection and disease.

{kind=link}
{kind=link}