Application of medical cases in general genetics teaching in universities
Zhumei He1, Linsai Bie1, Wei Li2第一联系人:
收稿日期:2017-08-7修回日期:2017-08-22网络出版日期:--
| 基金资助: |
Received:2017-08-7Revised:2017-08-22Online:--
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (396KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
贺竹梅, 别林赛, 李蔚. 医学病例在高校普通遗传学教学中的运用. 遗传[J], 2018, 40(1): 75-85 doi:10.16288/j.yczz.17-267
Zhumei He, Linsai Bie, Wei Li.
遗传学一直是生命科学中的核心学科,也是21世纪生命科学领域中最活跃的学科之一,这从到目前为止已有80多位科学家获得与遗传学研究相关的诺贝尔奖可略见一斑。
医学是生物学的应用学科,是通过科学和技术手段处理人体各种疾病或病变的科学,其中遗传学原理和技术在医学中的应用相当广泛。因此,在遗传学教学和教材编写中,各种医学病例的运用和阐述对于较为深奥、抽象和繁琐的遗传学知识的理解起到了重要的作用。然而,在常规遗传学教学中,由于课时限制、书籍排版要求、学生知识背景覆盖度等众多因素,许多医学病例在课堂或教科书中只能作为简单的引入,这在某种程度上制约了学生的学习积极性和学习效率。本文以《现代遗传学教程》第3版[1]为基础,阐述医学病例在高校普通遗传学教学中的作用,特别是将病例渗透到遗传学教学中的重要知识点及各知识点的连接中,提高学生的综合学习能力,让学生更加深刻理解遗传学知识及其应用。
1 医学病例对遗传学发展的影响
18世纪中叶法国数学家和生物学家Maupertius(1698~1759年)研究并发现多指(趾)和白化病的遗传方式,开启了人类医学与遗传学交叉认知的萌芽[2]。当然,遗传学序幕的真正拉开是1866年Mendel (孟德尔)发表豌豆杂交研究结果[3],特别是1900年孟德尔定律的重新发现[4]以及1911年Morgan(摩尔根)对连锁遗传的发现[5]和1917年《基因论》的发表[6]。在这之前,也已有一些人类遗传学的初步研究,如Galton于1875年曾提出用孪生子来分析人类的遗传性状,并主张用统计学方法来研究人类的遗传[7]。20世纪以来,细胞生物学、生物化学、分子生物学等实验技术的不断革新,促进了遗传学的发展,也使得遗传学在医学上得到广泛的应用,从而出现了大量的医学案例。例如,1900年Landsteiner[8]发现ABO血型的遗传后,Bernstein于1924年证明ABO血型受一组复等位基因控制,这是孟德尔定律在医学应用上的第一个例证[9];1902年,Garrod[10]通过对黑尿症(alkaptonuria)的研究解释了黑尿症的遗传方式,并提出了先天缺陷病的概念;1908年,Hardy[11]和Weinberg[12]分别研究了人类群体中的基因频率变化,提出了遗传平衡定律,为群体遗传学奠定了基础;1908年,Nilsson-Ehle[13]提出多基因遗传,为各种人类数量性状遗传的解释作出贡献;1949年Pauling[14]在发现镰状细胞贫血病(sickle-cell disease)患者的血红蛋白与正常血红蛋白的电泳特性不同后,提出了分子病的概念,之后Ingram[15]发现这种差异是由于血红蛋白β链第6位的谷氨酸替换为缬氨酸所致;1952年,Cori和Cori[16]发现糖原贮积病I型(glycogen storage disease type I)是由于葡萄糖-6-磷酸酶(G6Pase)缺陷所致的遗传性代谢病,是首次发现由于酶缺陷所致疾病;1953年,Jervis[17]发现苯丙酮尿症(phenylketonuria)是由于苯丙氨酸羟化酶(PAH)缺欠所致;1959年,Lejeune等[18]发现三体综合征(trisomy syndrome)等异常核型疾病并提出了“染色体病”的概念;同年,Vogel[19]提出药物遗传学概念,并在近年内迅速发展为药物基因组学和个体化医学;1960年Nowell等[20]发现肿瘤Ph染色体(Philadelphia chromosome)并由此开启了肿瘤细胞遗传学研究的新纪元;1978年Kan等[21]对地中海贫血(thalassemia)出生前的诊断创立了首例DNA诊断技术。1990年,人类基因组计划的启动,使得各种疾病相关基因得到发现,基因诊断和基因筛查技术、个体医学等得到发展,医学病例的发展发生了翻天覆地的变化。近年来,随着基因组编辑技术的发展,遗传学教学中的医学病例更是层出不穷[22]。
2 高校普通遗传学教学中的医学病例
2.1 经典孟德尔遗传分析中的医学病例
三位植物学家(Tschermak,De Vries和Correns)对孟德尔遗传规律的重新发现(1900年),McClung等(1902年)对性染色体的发现,Sutton和Boeri(1903年)的遗传染色体学说的提出以及摩尔根(1910年)连锁遗传定律的发现等为遗传学的发展奠定了夯实的基础[1]。遗传学的学习通常是从认识孟德尔遗传规律开始的,孟德尔遗传分析仍然是目前人类遗传病分析的重要方法;反过来,引起人类遗传病的基因突变和表型变化又能很好地帮助理解等位基因、显隐性、基因互作等较为抽象的遗传学概念。2.1.1 常染色体遗传医学病例
常染色体遗传病是指致病基因位于常染色体上,由于等位基因发生突变所引起的疾病,它又分为常染色体显性遗传病[如家族性高胆固醇血症(familial hypercholesterolemia, FH)、亨廷顿舞蹈症(Huntington's disease)、多指/趾症(polydactylism)、多囊肾病(polycystic kidney disease)、家族性腺瘤性息肉(familial ademomatous polyposis, FAP)等]和常染色体隐性遗传病[如白化病(albinsim)、先天性聋哑(congenital deafness)、囊性纤维化(cystic fibrosis)、半乳糖血症(galactosemia)、苯丙酮尿症(phenylketonuria)、黑尿病(alcaptonuria)等]。下面以家族性高胆固醇血症(FH)的遗传特点为例,描述常染色体病的遗传特点及其在教学中对理解遗传学中一些基本概念的扩展。
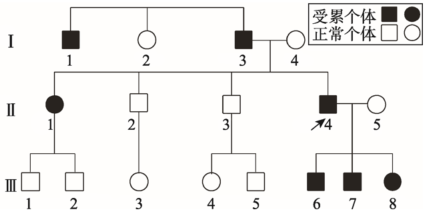
FH是由于患者的低密度脂蛋白受体(LDL-R)基因突变,导致细胞表面LDL-R蛋白功能缺陷,血浆低密度脂蛋白大幅度增高所引起的遗传病[23],表现为早发冠心病,是最早被明确临床表现和基因特征的脂代谢紊乱疾病之一。FH表现为典型的单基因显性遗传(图1):只要带有该致病基因即发病,双亲之一受累的子代平均1/2发病,未受累双亲的后代无人发病[24]。该病虽为典型的孟德尔显性遗传,但具有异质性,由此我们可以用于诠释外显率和表现度等遗传学概念。
在图1的家系中,II-4是一位广告商,表现为典型的FH病征,存在广泛的冠状动脉疾病和严重的高胆固醇血症,青年时开始反复发作胸痛,中年时患心肌梗塞死亡,而他的姐姐(II-1)无典型症状但患有高胆固醇血症,其父亲(I-3)与伯父(I-1)都死于心肌梗塞,这位广告商的孩子跟他们的姑姑一样都只是患有高胆固醇血症[24]。这在遗传学上的概念即为外显率和表现度不同。
从FH的这种异质性,我们还可以理解许多遗传学知识如遗传背景、复等位基因、多因一效等。我们知道,基因的作用不是孤立的,发生同一位点突变的不同个体,由于其他相关基因的差异,临床表现可能差异较大,这是遗传背景的作用;其次,从分子遗传学水平上理解,突变可以遍布整个LDLR基因,不同位点突变导致不同的等位基因,其表型也可能存在差异,这是所谓的复等位基因;三是相同的临床表现不一定是相同的病因,即多因一效,现已证明,临床诊断的FH患者并非都是由LDLR基因缺陷所致,其它基因突变也可导致严重的FH样表型(FH-like phenotype)。目前发现有约1/3的FH患者并没有LDLR基因的突变,因此遗传方式也就可能多样化[25]。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1一常染色体显性遗传的家族性高胆固醇血症系谱
参考文献[24]修改绘制,箭头示先征者。
Fig. 1Familial hypercholesterolemia: an autosomal dominant trait
又例如,对于雄激素源性秃发(androgenetic alopecia, AGA)的遗传,通常认为是常染色体显性遗传,由一对等位基因控制。按常理,杂合体无论男女都会是受累者,但由于受到雄激素的影响,杂合体男性表现为秃发表型,而女性则为正常表型,表现为从性显性[26]。
许多看起来简单的遗传病,但实际上其遗传机理仍存在争议。关于AGA也有人认为是X连锁遗传病甚至更多的是多基因病,并鉴定出了一些候选基因,如X染色体上的AR/EDA2R、20号染色体上的PAX1/FOXA、7号染色体上的HDAC9,表观遗传也可能参与秃发过程[27]。类似有争议的例子还有很多,如下面要讲到的人类外耳道多毛症的遗传等。
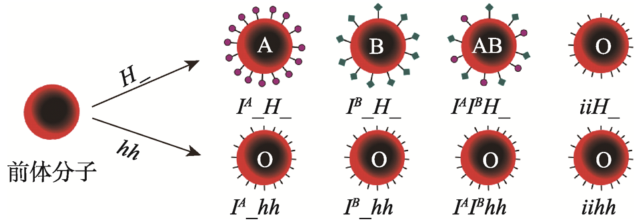
上位效应(epistasis)是孟德尔遗传分析中较难理解的一部分,但如果以常见的人类疾病/性状为例加以说明,就更容易吸引学生的注意。例如,人的血型是临床医学上经常要涉及的内容[28],孟买血型(Bombay phenotype)是由于两对等位基因I和H的作用产生的,并使ABO血型的表型比例发生改变。只要有一个H基因存在,就可以展示ABO血型的各种表型,当基因型为hh时(hh纯合子个体形成所谓“孟买血型”),由于没有H抗原结合到A和/或B抗原上,结果血型测试时,不管什么基因型都显示为O型[29](图2)。这个例子还可以进一步扩展到群体遗传学中的“奠基者效应”,因为人群中h基因纯合的机会很少,但马达加斯加的留尼汪岛(Reunion island)上的居民是个例外,这可能是由于这个岛上的居民由少数基因型为hh或Hh的祖先繁衍而来的[26]。
2.1.2 X连锁遗传病例
X连锁疾病是由于X染色体上的基因发生突变所造成的,X连锁疾病可当作经典家系遗传分析、剂量补偿效应以及群体遗传学教学的例子。由于男女X染色体数目的差异,使得群体中X连锁隐性遗传的男性患者明显多于女性,而在X连锁显性遗传中,女性的发病率明显高于男性。
目前已知有400多种X连锁遗传病,其中典型的X连锁隐性遗传有红绿色盲(red-green color blindness)、甲型血友病(haemophilia A)、Duchenne型肌营养不良症(Duchenne muscular dystrophy)、Wiskott- Aldrich综合征(Wiskott-Aldrich syndrome)、鱼鳞病(ichthyosis)、眼白化病(ocular albinism)和慢性肉芽肿病(chronic granulomatous disease, CGD)等;典型的X连锁显性遗传病有抗维生素D性佝偻病(familial vitamin D resistant rickets)、色素失调症(incontinentia pigmenti, IP)、先天性全身多毛症(congenital generalized hypertrichosis)、鸟氨酸氨甲酰转移酶缺乏症(ornithine transcarbamylase deficiency, OTCD)和口面指综合征I型(oral-facial-digital syndrome type I, OFDS)等。
在教学中,英国维多利亚女王家族的血友病遗传分析是典型案例。甲型血友病是由于体内一种凝血因子即FⅧ因子的缺失,表现为“自发性”关节出血和深部位组织出血,目前发现已有近400种FⅧ基因缺陷导致甲型血友病[30]。其实早在公元前4世纪左右对血友病的遗传规律已有所了解,在犹太法典中规定,婴儿割礼后,由于出血不止而死亡时,其母亲及母亲姐妹生育的所有男孩必须免除割礼,但母亲兄弟生育的男孩可不受法典条文限制[31]。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2ABO血型和孟买血型的遗传
Fig. 2Inheritance of the ABO blood type and Bombay blood type
很有意思的是,我们可以利用甲型血友病这样一种典型的X连锁隐性遗传病,更好的理解遗传学中的剂量补偿效应。由于血友病是X连锁隐性遗传病,按照孟德尔遗传,基因型为杂合子的女性本不会发病,但是,由于人类X染色体存在剂量补偿效应,即女性中的两条X染色体有一条失活,如果在肝脏组织中,刚好携带正常凝血因子编码基因的X染色体失活,这时即使是杂合子基因型的女性也就会患血友病了[30]。
2.1.3 Y连锁遗传病例
人类Y连锁遗传病的致病因子位于Y染色体上,该遗传病的所有受累者仅限于男性,父子相传,它既可以说是伴性遗传,也可以说是限性遗传。到目前为止,Y连锁遗传病/性状仅发现10余种,如性腺发育不全(germinal aplasia)、人类外耳道多毛症(auricular hypertrichosis)等。其中人类外耳道多毛症是这类遗传方式的代表。
外耳道多毛症多发于印第安人中,在高加利索人、澳洲土著人、日本人、尼日利亚人中也有发现。患者初生时外耳道即有绒毛状褐色霭毛,到5岁左右时绒毛颜色开始转变为黑色,在15岁左右患者外耳道部位长出黑色的硬毛,长度2~20 mm不等。此症状全部为对称性分布,多毛常见于耳轮缘、外耳道口处,并且只出现在耳廓之前,其中最长的耳毛可达4.5 cm,老年时的耳毛密而长。虽然耳部未见其他疾患或畸形,但有研究表明,外耳道多毛现象可能与冠心病相关联,可作为冠心病的一个遗传标志[32]。
虽然Y连锁这类遗传病/性状的遗传分析较为简单,但正如上面所分析的,任何性状都可能被用作遗传标记,来发现其他疾病/性状的遗传机理。同时这也提示我们,在学习或做研究的时候,要学会寻找各知识点间的相关性,适当发散思维或许会有新的启发。虽然外耳道多毛症是一种简单的性状,但它同人类秃发遗传一样,其遗传机理也仍存在争议。如Rao[33]提出,毛耳是由2个不同位点的相互作用产生的,1个在Y和X的同源部分,1个在Y染色体的非同源区域;Lee等[34]利用Y染色体DNA的双单体型标记,证明一群印度南部毛耳男性携带的Y染色体来源于一产生了许多独立突变的Y种系单倍群,研究人员进一步对毛耳男性和在地理上匹配的非毛耳男性进行对比发现,Y单倍型频率并没有明显差异,由此他们认为,在印度南部这种毛耳特性并不是Y连锁遗传的,而且在任何人群中这种特征都不太可能是Y连锁的。因此,我们在学习这类貌似简单的经典遗传现象时,可以继续跟踪文献以及运用现代遗传学研究手段进行深入探究。
2.2 染色体畸变中的医学病例
正常情况下染色体有稳定的形态结构和数目,染色体结构或数目的变异往往会给人类带来疾病,例如,唐氏综合征(Down's syndrome)、13和18三体综合征(13 and 18 trisomic syndrome)、Edwards综合征(Edwards syndrome)、猫叫综合征(cri du chat syndrome, CDCS)、克氏综合征(Klinefelter's syndrome, KS)、特纳氏综合征(Turner's syndrome)、慢性骨髓性白血病(Chronic myelogenous leukemia)、费城染色体(philadelphia chromosome)、染色体断裂综合征(chromosomal breakage syndrome)等。染色体畸变一直是医学遗传学和临床检查中的重头戏,在遗传学研究和应用中有重要的作用。在染色体疾病研究中,不得不提及Lejeune所作出的重要贡献,他于1958年和1963年分别发现造成唐氏综合症和猫叫综合症的原因,1966年描述了18q-综合征(Distal 18q-)是因为18号染色体长臂末端的部分丢失,他还发现了Dr表型(ring chromosome 13 syndrome,一个环状染色体替代13号染色体而引起的畸形症状),并在1970年和1971年分别确定了9号染色体和8号染色体的三体性(trisomy 9 or 8)[35]。关于染色体病产生的原因目前还并不十分清楚,研究发现与患者母亲的生育年龄密切相关,如40岁的生育者中出生唐氏综合征患儿的概率高达1/84[36]。染色体疾病临床上多表现为先天性智力低下、发育滞后及多发畸形,目前对于染色体疾病的治疗并没有理想的手段,主要依靠产前染色体检查来进行预防。通过染色体疾病来学习染色体畸变,一方面可增加相关知识的掌握如染色体结构、细胞分裂中的染色体行为等,另一方面必然会增加学生的学习积极性。
2.2.1 染色体结构改变与遗传病
染色体结构变异类型主要分为重复、缺失、倒位、易位4种。
人类猫叫综合征(CDCS)因新生儿患者出生后,哭声似猫叫而得名,但随着患儿年龄的增长,该症状表现会逐渐减弱,2岁后基本消失。CDCS大部分源自新发突变,在外界某种因素或环境的影响下,形成了含有5号染色体短臂部分缺失的配子。其主要核型是5号染色体短臂1区4带(5p14)或是l区5带(5p15)缺失;或是在合子开始卵裂的G1期之前,一条5号染色体在其短臂发生断裂后未能重新连接所致。据不完全统计,CDCS的群体发病率约为1/50,000,女性多于男性(约为4∶3)[37]。
2.2.2 染色体数目改变与遗传病
染色体数目变异主要可分为整倍体变异和非整倍体变异两类。在人类疾病中,主要是非整倍体变异,没有发现整倍体变异的新生儿。
唐氏综合征(DS)又名21-三体综合征,是造成小儿先天智障的最主要原因之一。Down在统计了患者们发病特点的基础上于1866年发表[38],1959年,Lejeune证实此病由小型具有端着丝粒的21号染色体出现三体性所引起[39]。三体的产生主要是由于减数分裂染色体不分离所致。
克氏综合征(KS)最早由Klinefelter等于1942年发现,是一种先天性睾丸发育不全性疾病,在临床上常表现为睾丸小且硬、睾酮生成速度慢、血睾酮的浓度偏低等[40]。KS最常见的核型是47,XXY,占80%;其次为46,XY/47,XXY嵌合型;其余罕见核型还包括48,XXYY、48,XXXY和49,XXXXY等。KS不易被确诊,因为患者与正常人在儿童时期并无明显差别,直到青春期才开始表现出特别的症状[41]。
染色体数目异常往往与癌症的发展有关,事实上,所有的人类癌症都存在异常染色体数目。现已证明,非整倍体或染色体结构的变化可导致癌症的发生[42]。更有甚者,现代研究表明,染色体畸变可引起表观遗传修饰的改变,从而导致基因表达的改变[43]。因此,随着基因组水平遗传学研究的发展,染色体畸变这一经典的遗传学内容与现代遗传学在新的水平上发生了连接。
2.3 基因突变中的医学病例
基因突变是指基因组DNA分子中的核苷酸发生了可遗传的变异。由于基因是生物体一切生命活动的基础,因此可以说基因突变是所有疾病的根源。通过对基因突变所导致的病例分析,可以更好的理解孟德尔遗传规律,如孟德尔的圆形和皱形豌豆种子就是由于淀粉分支酶基因中的一个DNA片段的插入失活造成的[44]。值得强调的是,基因突变不止是由于编码序列中的核苷酸发生变化,在内含子、启动子区、剪接部位等非编码区的核苷酸发生改变,同样也能引起基因突变的表型和疾病,这样我们可以更好地认识到DNA非编码序列的重要性。2.3.1 自发突变与人类疾病
自发突变即在自然条件下发生的DNA复制错误、脱氨基作用、转座因子作用等突变结果。
脆性X染色体综合征(fragile X syndrome, FXS)是自发突变导致发生的人类疾病的典型。在减数分裂中,FMR1基因5°端非翻译区的CGG三核苷酸发生重复数目扩展。正常个体中CGG重复序列拷贝数约为6~50,当重复序列在50~200区间时,临床上通常表现为卵巢早衰、焦虑、抑郁等症状,称为前突变型FXS;当重复序列大于200时,通常会伴随着CpG岛高度的甲基化从而使FMR1表达异常,患者主要表现为孤独症和智力低下,称为全突变型FXS[45]。在男性人群中,全突变类型发病率为1/4000,女性为1/8000。由于FXS可在家族中扩大,因此孕前进行FXS遗传咨询非常必要。
2.3.2 诱发突变与人类疾病
诱发突变即在外界条件如紫外线、X线、热辐射、化学污染、毒素等的作用下,提高了基因突变率所产生的基因突变。实际上,我们每天都暴露在各种诱变剂当中,由诱发突变引起各式各样的癌症正危害着人类的健康。了解诱发突变与人类疾病的关系,就可以更好的保护人类自身的健康。
皮肤癌(skin cancers)是其中典型的病例之一,指在表皮角质形成细胞的恶性增生,主要有基底细胞皮肤癌(basal-cell skin cancer)、鳞状细胞皮肤癌(squamous-cell skin cancer)、皮肤黑素瘤(melanoma)等,90%以上的皮肤癌是由于暴露于太阳紫外线所致[46],患者中男性发病率高于女性。由于皮肤癌的发生涉及到多个基因的变异,是损伤的长期积累结果,所以此病多发于老年人群体。
2.3.3 修复机制问题与人类疾病
着色性干皮病(xeroderma pigmentosum, XP)是人类的一种罕见常染色体隐性遗传病,是由于切除修复酶的缺陷所造成的,其机体内缺乏切除修复机制,患者暴露部分易发生色素沉着,皮肤萎缩,患皮肤癌的概率高,且大部分患者在30岁前死于皮肤癌[47]。
2.3.4 表观遗传变异对人类健康的影响
表观遗传变异指在DNA序列不发生改变的情况下,生物体细胞中的基因表达发生了可遗传的改变,同样也能引起生物体表型的变化和一系列的生理疾病,但幸运的是许多表观遗传变异是动态性的和可逆性的。表观遗传变异主要包括组蛋白修饰、基因组印记、DNA甲基化、非编码RNA作用等[48]。
Prader Willi综合征(Prader-Willi syndrome, PWS)与Angelman综合征(Angelman syndrome, AS)是第一个报导的印记疾病例子,同时也是第一个证明表观遗传在人类疾病中起作用的明确证据来源。在人类中,PWS与AS有不同的临床表现,但却具有相同的病因:15号染色体长臂q11-13上有5~6 Mb的微缺失或典型的印迹基因异常甲基化。不同的是若微缺失来自父亲则表现为PWS,若来自母亲则为AS,这是由于表观遗传造成不同亲本基因差异性表达的结果[49]。对于那些已有一个患儿的父母,其再发风险低于1%[50]。
2.4 多基因遗传中的医学病例
数量性状是相对于质量性状而言的,它受多基因控制,同时其遗传效应受周围环境因素的影响较为明显。在人类中,有很多的疾病或性状属于数量性状,例如肤色、智力、身高、高脂蛋白血症、抑郁症、高血压、糖尿病、唇裂等。抑郁症(major depressive disorder, MDD)是多基因遗传中的典型医学病例之一[51],属于心境障碍的一种,具有情绪障碍、认知损伤、行为改变等症状的精神类疾病,在世界范围内有2亿多人正遭受着抑郁症的困扰和折磨[52]。目前关于抑郁症的发病机制尚不明确,导致难以开展有针对性的治疗。抑郁症通常被认为是由于遗传、环境和心理因素所导致。
通过分子遗传学和全基因组关联研究,已经定位了数十种与抑郁症相关的候选基因,其中5-羟色胺系统基因、多巴胺系统基因、神经营养因子基因等在当前研究最为丰富,并发现多种候选基因不仅直接影响抑郁的发生发展,而且能够调节环境因素与抑郁之间的关联[53]。在遗传学教学中,我们实际上一直强调基因与环境的相互作用,这种作用在对多基因疾病的认知中尤为重要。因此,通过这种病例,我们能更好的理解遗传学的中心命题:基因型+环境=表型[54]。
2.5 细胞质遗传中的医学病例
细胞质遗传是由细胞质中的细胞器基因控制的。若线粒体DNA(mtDNA)发生点突变或缺失突变都能使线粒体中的代谢酶出现异常,影响线粒体的正常功能,继而引发出一系列严重的人类疾病,如母系遗传性耳聋(matrilineal hereditary deafness)、Leber遗传性视神经病变(Leber’s hereditary optic neuropathy, LHON)、其他神经肌肉疾病等母系遗传病以及如癌症、帕金森病(Parkinson's disease)、糖尿病(diabetes)、高血压(hypertension)等复杂性疾病。1871年,德国科学家Leber报道了Leber遗传性视神经病变(LHON)[55]。LHON的高发年龄段为15~30岁,一开始表现为视力模糊,接下来视力会急剧下降直至演变成无痛性的近乎或完全性失明状态,且经常伴随多种并发症,如痉挛性截瘫、耳聋、痴呆等。至1988年Wallace等揭示线粒体基因组点突变(m.11778G>A)导致LHON为止,已充分证明LHON为一种线粒体疾病[56],这也是发现的第一个人类细胞质遗传疾病。
现已发现18个与LHON相关的mtDNA突变,其中包含3个原发突变(primary mutations)(m.3460 G>A、m.14484 T>C、m.11778 G>A)与许多继发突变位点(OMIM, https://omim.org/entry/535000),这些位点编码的NADH脱氢酶参与线粒体的氧化磷酸化正常功能。通过对LHON的分析,能更充分的理解线粒体遗传的纯质性(homoplasmy)和异质性(heteroplasmy)概念,例如,人的细胞中有100~1000个线粒体,每个线粒体中有1~2个mtDNA分子,在绝大部分LHON家庭中,原发突变是同质的,只有10%~15%是异质的(阀值为60%)[57]。
更有意思的是,LHON虽然表现为母系遗传,但男性是此病的主要受累人群,此外,携带相同mtDNA突变的不同家系和同一家系的不同成员之间的外显率和表现度具有显著性差异。这些都提示,mtDNA原发突变可能与继发突变、单体型、核修饰基因(X染色体或常染色体)以及环境因素等协同作用。如Bu等[58]通过家系分析,提出双基因假说,即LHON发病是由mtDNA突变和X连锁基因相互作用所致,并由于女性X染色体的失活可能造成表现度的差异;又如,根据有些LHON家系表现出不完全外显但家系母系成员中男女发病率无明显差异,而提出常染色体上的核修饰基因对LHON的作用,并且已经鉴定出一些相关基因[59]。
从上面的LHON病例分析,我们可以看到多种遗传现象,如细胞质遗传、核质互作、剂量补偿效应、性染色体遗传、外显率、表现度、环境因子作用等[57]。这样可以加强我们对遗传学知识的灵活运用。
2.6 遗传伦理中的医学病例
随着遗传学的发展,特别是20世纪70年代重组DNA技术的出现,与遗传学相关的生命伦理问题一直引起国际社会的广泛关注,如转基因、基因组研究、基因编辑、基因检测、合成生命、辅助生殖等[60]。近年来比较热门的医学伦理案例有好莱坞电影明星Angelina Jolie由于带有BRAC1变异基因,于是选择切除双侧乳房及卵巢和输卵管。这种由于家庭乳腺癌史而进行乳房切除手术的案例近年在国内有上升的趋势,然而目前在东方女性中,还没有关于BRCA1和BRCA2这两个易感基因突变阳性的大规模人群流行性病学调查确切数据。因此,基于家庭病史或基因检测结果而作出的乳房切除手术决定是否是最为合理的和可接受的,仍存在争议。此外,像人类胚胎基因编辑技术的应用等也是热门的讨论话题,自2012年CRISPR/Cas9发展以来,该技术已被广泛用于从胚胎到成体的遗传病处理甚至是对健康个体的“增强”处理[61,62]。
在遗传学教学中,引入这类遗传伦理学问题,主要是让学生了解最新研究动态和培养学生树立正确的科学伦理观与人生观,顺便还可以介绍生命伦理学的基本原则,使学生在今后的生活中能够更好的思考相关科学问题。
3 医学病例在遗传学教学应用中的优势
遗传学是一门发展迅速的学科,基本概念繁多及要求较强的遗传分析能力[63]。由于在遗传学教学过程中,涉及到大量以动植物和微生物为材料所发现的遗传规律,学生在学习的过程中可能会觉得冗杂,因此,从已有理论出发分析医学病例的特点,找到知识点的相关性及内在联系,并将学科最新研究进展与已有知识有机结合,这样既能使学生更好的掌握遗传学基本理论、新的研究方向和完整的遗传学知识体系,同时可以活跃课堂氛围和加深学生对所学内容的印象。医学病例在遗传学教学中运用表现出的优势在于:(1)激发学习兴趣,拓展学生的思维和综合运用能力。人对自己感兴趣的事情更容易学好,这是自然属性。教师在教学中利用这一属性,运用与生活和个人息息相关的医学病例/性状,可以激发学生的学习兴趣,让学生更轻松地的理解和记忆深奥的遗传学专业知识。由于教材的系统性要求,很难完全揭示实际病例/性状的多样性,为了让学生对某部分知识有一个系统的学习,同时深化教材内容,在课堂上进行恰当的病例引导可以促进学生进行拓展思考,主动提出问题。另一方面,一个看似简单的病例,但实际上可能包含许多知识点,通过病例的教学,可以使学生对遗传学知识有一个全面的掌握和提高综合运用的能力。
(2)有利于理论与实际相结合,促进教学效果。课本中某个具体知识点囊括的往往是一大类案例的基本原理,具有高度的概括性,当学生脱离课本面对具体案例时,也许就会感到棘手,理论与实际发生脱节。如果教师在日常教学中引入病例来阐释理论,一方面可以由一个病例衍生出一系列的学科知识,从简单的理论上升到更为宽广和复杂的知识层面,从而拓展学生的知识面;另一方面,正如前面所讲的,在对一个病例进行分析时,往往要求学生对相应知识有一个综合性的理解,这样在病例的分析过程中,就考察了学生对这部分知识的整体把握度和对理论的灵活运用情况。
(3)有利于学生未来生活和专业的选择,提供人生帮助。遗传学是一个综合性的学科,包含了动物遗传学、植物遗传学、微生物遗传学、细胞遗传学、医学遗传学、肿瘤遗传学等众多子学科,学生们在学习的过程中可能对其中某一个子学科有更大的兴趣。若通过医学病例的教学,起到抛砖引玉的作用,可能为对医学遗传学有兴趣的同学做一个铺垫。此外,在每个人今后的人生生活中,包括健康、婚姻、生育、孩子成长等,都需要用到遗传学知识,虽然具体的概念在若干年后可能会模糊,但在通过病例教学法中所掌握的对病例的遗传分析思维方法,可以轻易地找到答案。
4 结 语
从当代大学生们的学习特点出发,在遗传学教学中运用医学病例,对遗传学基础理论的学习和最前沿科研成果的展示将起到重要的作用。当然对医学病例的选择需要经过仔细斟酌,针对不同的教学目的选择不同的病例。例如,选择典型性病例能充分说明某个规律的真实性,使学生理解某些基本遗传规律;选择具有综合性的病例,能激起同学们对遗传学原理综合运用的兴趣,而不只是局限于对某一遗传现象的认知;第三类病例可选择一些具有复杂机理或机理有争议的病例,这样可使学生意识到遗传分析的复杂性和遗传学研究的深奥性。总之,病例的选择需要很好的与遗传学知识有机结合。由于医学病例中的理论需要运用遗传学的整体知识及包含许多遗传学课本以外的知识,这就要求同学们上课时高度集中听讲,认真思考遗传学知识与病例的相关性。此外,在教学中,还可指导学生利用期刊杂志、网络资源等查阅自己感兴趣的病例,总结归纳,甚至以小组为单位作课堂演讲进行交流讨论,这样对锻炼他们的综合学习能力具有极大的好处。
遗传学教学中病例的运用是理论联系实际,再从实际延伸理论,对理论知识进行更深层次解剖和综合的过程,这对于提高学生们的综合学习能力和探究思维能力是有益的,也将为今后的人生增加更多的财富。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
URLPMID:20264553 [本文引用: 1]

GLASS D.
[本文引用: 1]
[本文引用: 1]
URLPMID:17817675 [本文引用: 1]
Morgan TH.
URL [本文引用: 1]
URLPMID:11700679 [本文引用: 1]
In 1875 Francis Galton was the first to study twins as a test of the relative strenght of heredity and environment. This paper examines Galton's work on twins, using his surviving working papers. It shows that his enquiry was larger and more systematic than previously realized. Galton issued several hundred questionnaires to parents of twins, with the aim of establishing how far the similarities and differences between twins were affected by their life experiences. The paper also discusses Galton's study in relation to his understanding of the physiology of twinning and his theory of heredity. The modern concept of monozygotic twins had not yet been established, and the similarity between Galton's work and modern twin studies should not be overstated. While Galton's work was important as a pioneering study, in some respects his conclusions went beyond his evidence. The paper finally examines whether Galton's twin studies influenced his position on the links between social class, heredity and social mobility, and surveys the evidence for his views on these issues.
URL [本文引用: 1]
react-text: 474 Zusammenfassung Dibenamin hat eine bedeutende Schutzwirkung gegenüber dem durch Histaminvergiftung verursachten Plasmaverlust und Bluteindickung. Das kreisende Plasmavolumen wird durch Dibenamin, aber auch im allgemeinen durch Sympathicusblockade erh02ht. Bei den mit Dibenamin behandelten Tieren wurde au08erdem noch eine Zunahme der Lymphstr02mung, Verz02gerung des Proteinaustausches zwischen Blut... /react-text react-text: 475 /react-text [Show full abstract]
URLPMID:8417988 [本文引用: 1]
Author information: (1)Genetics Department, University of Wisconsin, Madison 53706.
URLPMID:2588790 [本文引用: 1]
PMCID: PMC2588790
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
Not Available
URL [本文引用: 1]
URLPMID:13022673 [本文引用: 1]
Extract: Peripheral uptake of triglyceride from plasma was investigated by intravenous fat tolerance tests and by postheparin lipoprotein lipase measurements in children with different types of glycogen storage disease.The patients with a glucose 6-phosphatase deficiency were characterized by a significantly diminished triglyceride elimination rate (5.79 ± 2.78 % /min) and 5-min postheparin lipoprotein lipase activity (40.2 ± 23 μEq fatty acid (FA)/liter/min). The patients with a deficiency of debranching enzyme showed a significantly diminished triglyceride elimination rate (4.84 ± 1.61 % /min) whereas the 5-min postheparin lipoprotein lipase activities did not significantly differ from the control values (49.6 ± 27.7 μEq FA/liter/min).The patients with a deficiency of the phosphorylase system showed neither a significantly diminished triglyceride elimination rate (7.34 ± 2.65 % /min) nor a diminished 5-min postheparin lipoprotein lipase activity (61.7 ± 30.1 μEq FA/liter/ min).Triglyceride elimination rates were correlated positively with plasma lipoprotein lipase activities (r = 0.55, P < 0.05).Speculation: The dietary treatment of hyperlipidemia in hepatic glycogenosis might be based on the outcome of the intravenous fat tolerance test, a low fat clearance being an indication for a low fat, high carbohydrate diet, a normal fat tolerance for a high fat low carbohydrate diet.
URLPMID:13047448 [本文引用: 1]
Proc Soc Exp Biol Med. 1953 Mar;82(3):514-5.
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
react-text: 476 Ama04: Kemik ili06indeki hematopoietik hücrelerin anormal birikimiyle karakterize olan kronik myeloid l02semi (KML) i04in t(9;22) kromozomal translokasyonu tan03sal bir belirte04tir. Philadelphia kromozomu olu06umuyla sonu04lanan translokasyon, KML olgular03nda geli06mekte ve hastalar03n03n %95’inde g02rülmektedir ve akut lenfoblastik l02semi (ALL) ve akut myeloid l02semi (AML)’de daha k02tü bir prognozun... /react-text react-text: 477 /react-text [Show full abstract]
URLPMID:74407 [本文引用: 1]
Hemoglobin H disease was diagnosed prior to the twenty-second week of gestation in a pregnancy at risk for homozygous alpha-thalassemia using the technique of DNA-DNA hybridization. Fetal DNA was obtained from amniotic fluid fibroblasts obtained during the thirteenth week of gestation and grown in culture. The fetal fibroblast DNA was hybridized to radioactive alpha-globin cDNA. The number of alpha-globin genes present in the fetus was determined by comparing results of hybridization studies on the fetal DNA to similar studies on subjects with well-defined alpha-thalassemia syndromes and with normal subjects. The diagnosis of hemoglobin H disease was confirmed at birth by studies of the cord blood. This study confirms the ability of DNA-DNA hybridization techniques to distinguish the three-gene defect of hemoglobin H disease from the lethal four-gene defect of homozygous alpha-thalassemia.
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 3]
[本文引用: 3]
URLMagsci [本文引用: 1]
家族性高胆固醇血症(FH)是由于低密度脂蛋白受体(LDL-R)基因突变,致使细胞表面LDL-R蛋白功能缺陷,导致血浆低密度脂蛋白(LDL)大幅度增高,并可导致早发冠心病。“FH”已经成为携带LDL-R基因突变患者的同意词,但日益增多的研究证实,其他6种基因突变也可通过不同机制导致FH样表型。这些致病基因的发现,促进胆固醇代谢的研究进入新领域,有助于深入探讨胆固醇代谢的调节机制,并将为FH样表型的诊断和治疗提供新的理论依据。本文就有关FH样表型遗传异质性的分子基础研究的近况作一简要综述, 以引起人们的关注。
URLMagsci [本文引用: 1]
家族性高胆固醇血症(FH)是由于低密度脂蛋白受体(LDL-R)基因突变,致使细胞表面LDL-R蛋白功能缺陷,导致血浆低密度脂蛋白(LDL)大幅度增高,并可导致早发冠心病。“FH”已经成为携带LDL-R基因突变患者的同意词,但日益增多的研究证实,其他6种基因突变也可通过不同机制导致FH样表型。这些致病基因的发现,促进胆固醇代谢的研究进入新领域,有助于深入探讨胆固醇代谢的调节机制,并将为FH样表型的诊断和治疗提供新的理论依据。本文就有关FH样表型遗传异质性的分子基础研究的近况作一简要综述, 以引起人们的关注。
[本文引用: 2]
[本文引用: 1]
URLMagsci [本文引用: 1]
<p>血型是人类日常生活中非常常见的一种遗传表型, 拥有丰富的遗传学内涵。随着科技的发展, 其内涵不断得到新的揭示, 新的研究结果不断补充, 持续吸引着人们对血型遗传机制的探索。血型遗传案例除了与孟德尔遗传和连锁遗传、基因突变和染色体畸变四大内容关联外, 还涉及到其他多方面的遗传学知识点。在教学中, 依据遗传学的知识脉络, 贯穿以ABO血型作为经典案例, 结合拓展的白细胞血型, 孟买、Rh、MN等血型的遗传规律及其应用, 并且开展相关的实验教学, 理论联系实际, 增强了学生的兴趣, 提高了教学效果。在遗传学实验教学中, 有80%的学生选择ABO血型鉴定这个自选实验, 并表示出对这个实验的浓厚兴趣。在讲授相关知识点时, 用恰当的血型案例为引导, 设计相关的讨论主题, 开展PPT展示性讨论和辩论式讨论, 所有的学生都积极主动参与进来, 与现实生活相结合, 引导学生思考问题, 使学生的思维在辨析中得到操练, 提高分析问题和解决问题的能力, 深刻理解遗传学基本理论知识。</p>
URLMagsci [本文引用: 1]
<p>血型是人类日常生活中非常常见的一种遗传表型, 拥有丰富的遗传学内涵。随着科技的发展, 其内涵不断得到新的揭示, 新的研究结果不断补充, 持续吸引着人们对血型遗传机制的探索。血型遗传案例除了与孟德尔遗传和连锁遗传、基因突变和染色体畸变四大内容关联外, 还涉及到其他多方面的遗传学知识点。在教学中, 依据遗传学的知识脉络, 贯穿以ABO血型作为经典案例, 结合拓展的白细胞血型, 孟买、Rh、MN等血型的遗传规律及其应用, 并且开展相关的实验教学, 理论联系实际, 增强了学生的兴趣, 提高了教学效果。在遗传学实验教学中, 有80%的学生选择ABO血型鉴定这个自选实验, 并表示出对这个实验的浓厚兴趣。在讲授相关知识点时, 用恰当的血型案例为引导, 设计相关的讨论主题, 开展PPT展示性讨论和辩论式讨论, 所有的学生都积极主动参与进来, 与现实生活相结合, 引导学生思考问题, 使学生的思维在辨析中得到操练, 提高分析问题和解决问题的能力, 深刻理解遗传学基本理论知识。</p>
URLPMID:27834485 [本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:4669458 [本文引用: 1]
****
URLPMID:15367914 [本文引用: 1]
The human Hairy Ears phenotype has traditionally been regarded as the only Y-linked heritable trait. Here, we use Y-chromosomal DNA binary-marker haplotyping to show that a cohort of southern Indian Hairy-Eared males carries Y chromosomes from many haplogroups of the Y-phylogeny, which, under a hypothesis of Y linkage, would require multiple independent mutations within a single population. We further show that there is no significant difference between the Y-haplogroup spectrum in Hairy-Eared males and that in a geographically matched control sample of unaffected males. The trait cannot, therefore, be Y-linked in southern Indians, and by extension, is unlikely to be so in any population.
URL [本文引用: 1]
URLPMID:11943789 [本文引用: 1]
Objectives: To revise the estimates of maternal age specific live birth prevalence of Down's syndrome in the absence of antenatal screening and selective termination using newly available data. Setting and Design: Data were used from the National Down Syndrome Cytogenetic Register (NDSCR), which contains information on nearly all antenatally or postnatally diagnosed cases of Down's syndrome in which a karyotype was confirmed between 1989 and 1998 in England and Wales. It is the largest single series of data on the prevalence of Down's syndrome. Results and conclusion: The prevalence does not continue increasing at an increasing rate with age above age 45 as has been previously assumed. Above this age the rate of increase declines with increasing age. The overall age pattern is sigmoidal. A new logit logistic model is proposed which fits the data well. The risk of a Down's syndrome live birth is given by: risk=1/(1+exp(7.330-4.211/(1+exp(-0.282x(age-37.23))))).
URLPMID:365706 [本文引用: 1]
Abstract Data for 331 cri du chat cases, including 34 Danish probands, are reviewed. The incidence nad the prevalence among the mentally retarded population amounted to 1/45,000 and 1.5/1000, respectively. No striking association with prenatal events, parental ages, or birth order could be demonstrated. There was a significant excess of females. Parental translocations were present in slightly more than 10% of the families, while more rare cytogenetic aberrations (mosaicism, rings, and de novo translocations) accounted for less than 10% of all cases. The phenotypically relevant segment has been narrowed down to the midportion of the 5p15 band. Clinical, radiologic, and dermatoglyphic features are summarized and discussed, with special attention to the abnormal cry, which persists in many older probands, and to developmental abnormalities. No obvious correlation could be detected between clinical features and the localization of the deletion. No marker locus has yet been assigned to the short arm of chromosome 5. Treatment and prevention are briefly discussed.
URLPMID:7707939 [本文引用: 1]
ABSTRACT This reprint of an 1866 essay by the physician who identified Down's syndrome describes the physical and behavioral patterns that characterize individuals with mental retardation who come from different ethnic groups. This evidence of degeneration crossing racial divisions is seen as support for the idea that the human family has a common origin. (JDD)
URLPMID:22789577 [本文引用: 1]
Adv Pediatr. 2012;59(1):137-57. doi: 10.1016/j.yapd.2012.04.006. Review
URL [本文引用: 1]
URLPMID:3529433 [本文引用: 1]
Abstract When described more than 40 years ago, Klinefelter's syndrome (small testes, sterility, increased excretion of follicle-stimulating hormone, and usually gynecomastia) was thought to be an endocrine disorder. A second testicular hormone was postulated but has never been isolated. During the ensuing years, the syndrome has been found to be a chromosomal disorder, in which there is an extra X chromosome in 80% of the patients. The disorder occurs once in 500 to 1,000 male births and is best diagnosed by a buccal smear. When there is androgen deficiency, it is treated with testosterone. Gynecomastia is treated surgically because of the potential danger of malignancy or for cosmetic reasons.
URLPMID:28192779 [本文引用: 1]
Abstract Genomic instability is a hallmark of cancer, and it is well-known that in several cancers the karyotype is unstable and rapidly evolving. Molecular cytogenetics has contributed to the description and interpretation of cancer karyotypes, in particular through multicolor FISH approaches which can define even complex chromosome rearrangements. The introduction of genome-wide methods has made available a powerful set of tools with higher resolution than cytogenetics, thus appropriate to comprehend the huge variability of cancer cells. This review focuses on novel findings deriving from the combination of cytogenetic and genomic approaches in cancer research. 脗漏 2017 S. Karger AG, Basel.
URLPMID:27546843 [本文引用: 1]
Abstract Cancer is fundamentally a genetic disease caused by mutational or epigenetic alterations in DNA. There has been a remarkable expansion of the molecular understanding of colonic carcinogenesis in the last 300002years and that understanding is changing many aspects of colorectal cancer care. It is becoming increasingly clear that there are genetic subsets of colorectal cancer that have different risk factors, prognosis, and response to treatment. This article provides a general update on colorectal cancer and highlights the ways that genetics is changing clinical care. Copyright 0008 2016 Elsevier Inc. All rights reserved.
URLPMID:2153053 [本文引用: 1]
We describe the cloning of the r ( rugosus ) locus of pea (Pisum sativum L.), which determines whether the seed is round or wrinkled. Wrinkled ( rr ) seeds lack one isoform of starch-branching enzyme (SBEI), present in round ( RR or Rr ) seeds. A major polymorphism in the SBEI gene between near-isogenic RR and rr lines shows 100% cosegregation with the r locus, establishing that the SBEI gene is at the r locus. An aberrant transcript for SBEI is produced in rr embryos. In rr lines the SBEI gene is interrupted by a 0.8 kb insertion that is very similar to the Ac/Ds family of transposable elements from maize. Failure to produce SBEI has complex metabolic consequences on starch, lipid, and protein biosynthesis in the seed.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
胎盘介于胎儿与母体之间,是维持胎儿宫内生长发育的重要器官。在胎盘的正常发育过程中,子宫正常蜕膜化、滋养层细胞粘附与侵袭、胎盘血管生成与形成、胎盘印记基因表达都受到表观遗传修饰(如DNA甲基化、组蛋白修饰、非编码RNA等)的调控。研究已经证实环境因素如重金属、化合物、现代辅助生殖技术、营养物质均可导致胎盘上多种基因的表观遗传修饰异常。此外,胎盘基因表达存在性别差异也可能与表观遗传修饰有关。目前,在临床上可运用产前DNA甲基化水平分析技术检测异常的表观遗传修饰,并在疾病早期发现并做出诊断,从而为疾病预防及治疗提供依据。本文对胎盘正常发育过程中表观遗传修饰的调控及环境因素所致的胎盘基因表观遗传改变进行了综述,以期对胎盘相关疾病的诊断与治疗提供借鉴和参考。
URL [本文引用: 1]
胎盘介于胎儿与母体之间,是维持胎儿宫内生长发育的重要器官。在胎盘的正常发育过程中,子宫正常蜕膜化、滋养层细胞粘附与侵袭、胎盘血管生成与形成、胎盘印记基因表达都受到表观遗传修饰(如DNA甲基化、组蛋白修饰、非编码RNA等)的调控。研究已经证实环境因素如重金属、化合物、现代辅助生殖技术、营养物质均可导致胎盘上多种基因的表观遗传修饰异常。此外,胎盘基因表达存在性别差异也可能与表观遗传修饰有关。目前,在临床上可运用产前DNA甲基化水平分析技术检测异常的表观遗传修饰,并在疾病早期发现并做出诊断,从而为疾病预防及治疗提供依据。本文对胎盘正常发育过程中表观遗传修饰的调控及环境因素所致的胎盘基因表观遗传改变进行了综述,以期对胎盘相关疾病的诊断与治疗提供借鉴和参考。
URLPMID:7795645 [本文引用: 1]
Abstract A subset of patients with Angelman and Prader-Willi syndrome have apparently normal chromosomes of biparental origin, but abnormal DNA methylation at several loci within chromosome 15q11-13, and probably have a defect in imprinting. Using probes from a newly established 160-kb contig including D15S63 (PW71) and SNRPN, we have identified inherited microdeletions in two AS families and three PWS families. The deletions probably affect a single genetic element that we term the 15q11-13 imprinting centre (IC). In our model, the IC regulates the chromatin structure, DNA methylation and gene expression in cis throughout 15q11-13. Mutations of the imprinting centre can be transmitted silently through the germline of one sex, but appear to block the resetting of the imprint in the germline of the opposite sex.
URL [本文引用: 1]
URLPMID:5051620 [本文引用: 1]
Baker M, Dorzab J, Winokur G, Cadoret R.
[本文引用: 1]
URLPMID:23886513 [本文引用: 1]
Increasing evidence supports the involvement of both heritable and environmental risk factors in major depression (MD) and suicidal behavior (SB). Studies investigating gene–environment interaction (G02×02E) may be useful for elucidating the role of biological mechanisms in the risk for mental disorders. In the present paper, we review the literature regarding the interaction between genes modulating brain functions and stressful life events in the etiology of MD and SB and discuss their potential added benefit compared to genetic studies only. Within the context of G02×02E investigation, thus far, only a few reliable results have been obtained, although some genes have consistently shown interactive effects with environmental risk in MD and, to a lesser extent, in SB. Further investigation is required to disentangle the direct and mediated effects that are common or specific to MD and SB. Since traditional G02×02E studies overall suffer from important methodological limitations, further effort is required to develop novel methodological strategies with an interdisciplinary approach.
URL [本文引用: 1]
遗传学作为高等院校生命科学及 农林医等相关专业教学中的一门重要基础课程,其知识点和涉及的内容众多,知识体系的构成丰富多样。本文尝试从遗传学发展的阶段性、遗传规律认知的动态性和 遗传研究策略与手段的丰富性三个方面对遗传学知识体系的构建进行探讨。这对于遗传学教学的改善及促进学科的健康有序发展具有一定的参考作用。
URL [本文引用: 1]
遗传学作为高等院校生命科学及 农林医等相关专业教学中的一门重要基础课程,其知识点和涉及的内容众多,知识体系的构成丰富多样。本文尝试从遗传学发展的阶段性、遗传规律认知的动态性和 遗传研究策略与手段的丰富性三个方面对遗传学知识体系的构建进行探讨。这对于遗传学教学的改善及促进学科的健康有序发展具有一定的参考作用。
URL [本文引用: 1]
No Abstract available for this article.
URLPMID:3201231 [本文引用: 1]
Leber's hereditary optic neuropathy is a maternally inherited disease resulting in optic nerve degeneration and cardiac dysrhythmia. A mitochondrial DNA replacement mutation was identified that correlated with this disease in multiple families. This mutation converted a highly conserved arginine to a histidine at codon 340 in the NADH dehydrogenase subunit 4 gene and eliminated an Sfa NI site, thus providing a simple diagnostic test. This finding demonstrated that a nucleotide change in a mitochondrial DNA energy production gene can result in a neurological disease.
[本文引用: 2]
URL [本文引用: 1]
URLPMID:15924081 [本文引用: 1]
Introduction. In 1961, Garcin et al. described a family with several members affected with optic atrophy associated with cataract, and neurological symptoms. The authors believed this condition to be distinct from other diseases known at that time, e.g. the Behr syndrome, Marinesco-Sj 顣廹ren syndrome and Friedreich's ataxia. Method. This family was followed over a period of 40 years and genes known to be responsible for optic atrophy were sequenced. Results. The G277A mutation of OPA3 gene was responsible for this familial disease. Discussion. A new clinical entity is identified: autosomal dominant optic atrophy and cataract, due to a heterozygous mutation of the OPA3 gene, a nuclear gene encoding a mitochondrial protein.
URL [本文引用: 1]
在科学技术迅猛发展的时代,加强对青年学生生命价值、人生意义、人我关系、人与自然关系,以及生与死等问题的教育至关重要.生命伦理学课程将生命科学知识教育与人文精神培养有机结合,对全面提高学生的科学素养和真善美的精神理念是一种积极探索,对培养大众的伦理思考和抉择能力将起到无以替代的作用.本文从历史责任感培养、科学伦理观培养、情感和道德教育等方面阐述了大学生命伦理学课程开设的重要意义和作用,并在综合我国高校生命伦理学课程开设现状的基础上,对该课程建设与发展进行了思考,期待引起我国高校对生命伦理学课程建设的重视.
URL [本文引用: 1]
在科学技术迅猛发展的时代,加强对青年学生生命价值、人生意义、人我关系、人与自然关系,以及生与死等问题的教育至关重要.生命伦理学课程将生命科学知识教育与人文精神培养有机结合,对全面提高学生的科学素养和真善美的精神理念是一种积极探索,对培养大众的伦理思考和抉择能力将起到无以替代的作用.本文从历史责任感培养、科学伦理观培养、情感和道德教育等方面阐述了大学生命伦理学课程开设的重要意义和作用,并在综合我国高校生命伦理学课程开设现状的基础上,对该课程建设与发展进行了思考,期待引起我国高校对生命伦理学课程建设的重视.
URLPMID:28783728 [本文引用: 1]
Nature advance online publication 02 August 2017. doi:10.1038/nature23305Authors: Hong Ma, Nuria Marti-Gutierrez, Sang-Wook Park, Jun Wu, Yeonmi Lee, Keiichiro Suzuki, Amy Koski, Dongmei Ji, T ...
URLPMID:28473627 [本文引用: 1]
The ease and applicability of CRISPR/Cas9--a new and precise gene editing and reproductive technology--have garnered hype and heightened concern about its potential 'unprecedented and horrific consequences' and have led many scientific leaders to call for a moratorium on its research and use. CRISPR appears distinctly more controversial than previous technological innovations (genetic or otherwise), with a greater reach and speed of human treatment and enhancement; however, we have seen similarly inflated hopes and fears in response to other medical innovations for well over a century. One intervention that has both historically and recently incited alarm--vaccines--serves as a pertinent example of what could go wrong if a technology's reach is shortened due to inflated fears. By comparing the vaccine controversy and the CRISPR debate, we can help separate the hype from the realistic potential of these technologies. How our society grapples with such innovations will determine the extent to which their impact on our individual and collective health will be beneficial. We must recognise the need for a tempered approach to CRISPR conversation leading to regulation and ethical application. Although CRISPR's reach will continue expanding with ongoing research, thus requiring continuous evaluation, the lessons we have learned from the vaccine controversy demonstrate that our approach must not be to shut down regulation and application now, but to thoughtfully conjoin productive debate and action so that therapeutic gene editing can alleviate suffering as soon as possible without precipitating social outcomes we would belatedly deplore.
URL [本文引用: 1]
课堂是本科教学的主要场地,充分利用课堂教学培养本科生的科研素质,是一个值得思考的教改问题。本文从系统架构课程内容体系、教学内容反映科研进展、知识点的讲解反映其科研活动、用PPT动画讲解科学原理和实验过程、用双语教学提高专业英语阅读能力、考试考察学生的科研分析能力6个方面介绍了作者多年来在遗传学课堂教学中培养学生科研素质的教改措施与实践经验。这些改革措施有利于激发本科生的学习积极性,培养本科生发现问题、分析问题和解决问题的科学思维能力,提高本科生的科技英语阅读能力和文献查阅能力,为本科生进入科研领域奠定了良好的基础。
URL [本文引用: 1]
课堂是本科教学的主要场地,充分利用课堂教学培养本科生的科研素质,是一个值得思考的教改问题。本文从系统架构课程内容体系、教学内容反映科研进展、知识点的讲解反映其科研活动、用PPT动画讲解科学原理和实验过程、用双语教学提高专业英语阅读能力、考试考察学生的科研分析能力6个方面介绍了作者多年来在遗传学课堂教学中培养学生科研素质的教改措施与实践经验。这些改革措施有利于激发本科生的学习积极性,培养本科生发现问题、分析问题和解决问题的科学思维能力,提高本科生的科技英语阅读能力和文献查阅能力,为本科生进入科研领域奠定了良好的基础。

{kind=link}
{kind=link}
{kind=link}
{kind=link}