 ,, 张婷婷,, 马磊,石河子大学生命科学学院,石河子 832000
,, 张婷婷,, 马磊,石河子大学生命科学学院,石河子 832000Structural characteristics of natural chimeric genes and their implications for gene design
Yingxia Li,, Tingting Zhang,, Lei Ma,College of Life Science, Shihezi University, Shihezi 832000, China编委: 张勇
收稿日期:2017-08-8修回日期:2017-12-22网络出版日期:2017-12-27
| 基金资助: |
Received:2017-08-8Revised:2017-12-22Online:2017-12-27
| Fund supported: |
作者简介 About authors
作者简介:李迎侠,硕士研究生,专业方向:遗传学E-mail:
张婷婷,硕士,讲师,研究方向:分子遗传学E-mail:
通讯作者:马磊,博士,副教授,研究方向:生物信息学与分子遗传学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (383KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李迎侠, 张婷婷, 马磊. 天然嵌合基因的结构特性及其对基因设计的启示. 遗传[J], 2018, 40(2): 135-144 doi:10.16288/j.yczz.17-268
Yingxia Li, Tingting Zhang, Lei Ma.
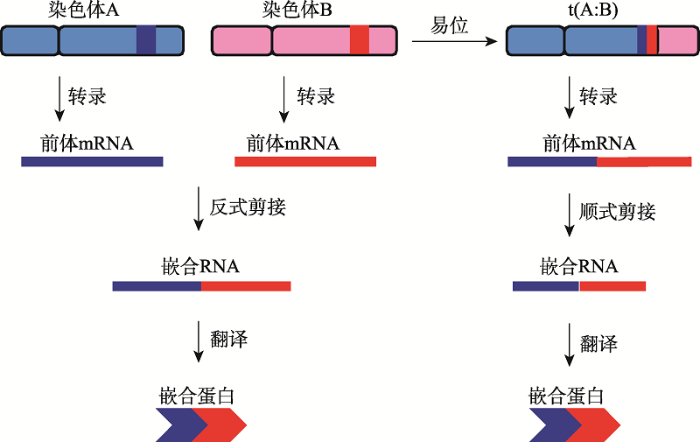
天然嵌合基因(natural chimeric gene)是指两个或两个以上的独立基因(即亲本基因),通过染色体重排、相邻基因转录通读、反式剪接等方式天然融合而成的新基因,又称融合基因。天然嵌合基因的亲本基因可以位于不同染色体,也可位于同一染色体的不同DNA链,亦可在同一条DNA链,但嵌合基因与亲本基因的外显子排列顺序不同[1]。
在人类癌症研究过程中,诸多的嵌合基因参与其中。其致癌方式主要表现为:(1)原癌基因与强启动子基因融合,激活肿瘤转化功能;(2)异源基因的结构域融合,编码致癌功能蛋白;(3)融合导致肿瘤抑制基因失活。
嵌合基因在医学上具有良好的应用前景和重要的研究价值,可作为细胞癌变的分子标记,如在软组织肿瘤、上皮癌、前列腺癌、非白血性白血病、急性白血病、非小细胞肺癌等病变中,发现了许多可作为分子诊断标记的嵌合基因[2,3,4,5]。同时,嵌合基因也可作为治疗癌症的药物靶标,如抗癌药Gleevec能与ATP竞争性结合BCR-ABL1络氨酸激酶催化域上的ATP结合位点,从而抑制BCR-ABL1激酶上的磷酸基团转移,致使络氨酸激酶信号传导通路中断,癌细胞停止分裂增殖死亡[6]。
随着深度测序技术和基因融合比对算法的迅速发展,各种类型的嵌合数据海量般的产生,目前已经在8个物种中发现了约34 900条嵌合转录本[7],已引起越来越多研究者的关注,其结构和功能的特点逐渐显露。本文基于嵌合基因生物信息学方面的相关研究,从天然嵌合基因的形成方式、融合特点、转录、调控,以及融合蛋白的结构域组合形式和功能等方面进行了相关综述。
1 天然嵌合基因的形成方式
天然嵌合基因的形成可以分为两个层次即DNA水平和RNA水平(图1)。嵌合基因DNA水平的形成主要包括染色体内的重组和染色体间的重组,重组可以导致基因的融合。1960年,美国宾夕法尼亚大学Peter Nowell教授和 Fox Chase癌症中心的David Hungerford研究员首次发现染色体变异可导致基因嵌合[8,9]。1973年,美国芝加哥大学Janet Rowley教授发现费城染色体的畸变由染色体易位所致,并在白血病中发现了第一个融合基因[10]。染色体重排事件分为染色体的缺失、重复、插入、翻转、易位和环状染色体等,这些结构性的变异都可形成嵌合基因,并与肿瘤疾病相关[11,12]。由融合基因导致的疾病涉及各种组织,包括乳腺[13,14,15]、肺[16]、前列腺[17,18,19,20]、胃[21]、淋巴[22]和软组织[2]等。另外,融合基因在不同癌症中的患病率也有很大差别,例如,90%的慢性髓细胞性白血病是由BCR-ABL1基因融合产生[23];79%的前列腺癌患者是由TMPRSS2-ETS融合基因导致[24];33%的软组织疾病是由融合基因导致[16]。RNA层次的融合即RNA嵌合,是一种非共线型的RNA编辑。mRNA前体分子(pre-mRNA)的反式剪接是产生嵌合RNA的主要方式,也是真核细胞RNA转录后加工的一种重要机制。1984年,在锥虫(Trypanosoma)中首先发现反式剪接的现象[25]。此后,陆续在其他一些生物,如烟草(tobacco)[26]和衣藻(Chlamydomonas rainhardii)[27]中也证实存在反式剪接。哺乳动物中也发现了反式剪接转录本[28],如在大鼠(Rattus norvegicus)中发现的两个基因,肉毒碱辛基转移酶和中链酞辅基A合成酶,是较早发现的由反式剪接加工合成的嵌合基因[29,30,31]。利用第二代深度测序技术研究发现,在一些原本认为反式剪接发生很稀少的高等真核生物中,也存在大量的嵌合RNA,如人类ENCODE计划指出约有65%的基因可能参与了嵌合RNA的形成。由RNA嵌合产生的嵌合蛋白,同样可以导致肿瘤或非肿瘤的疾病,例如,DNAJB1-PRKACA嵌合蛋白就与纤维板层肝细胞癌的形成有关[32]。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1嵌合基因的形成方式
Fig. 1The formation of chimeric genes
由此可见,嵌合基因的形成不仅丰富了转录组和蛋白质组的多样性和复杂性,而且具有重要的生物学功能。
2 天然嵌合基因的融合位点特征及翻译特点
RNA剪接可产生许多具有功能的mRNA异构体,对生物的发育及进化至关重要。真核细胞pre- mRNA剪接位点处存在一定的序列保守性,其内含子5°端(供体位点)和3°端(受体位点)的碱基常为GU和AG,因此被称为GU/AG规则。嵌合RNA融合位点也遵循GU/AG的规则[33]。在癌症中,产生嵌合基因的染色体断点位置,具有非随机性和复发性的特点,易受细胞核中邻近染色体的空间位置和DNA序列特征的影响,如碱基序列的重复、脆性位点和酶识别位点等[34]。
研究发现98%的染色体断接,会保留基因的阅读框(reading frame),即嵌合基因与原亲本基因的阅读框一致,未产生移码突变[35],这种融合被称为框内融合(in-frame fusion)。然而,对13种由基因融合导致肿瘤的相关研究发现,只有36%的融合转录是框内转录[36]。前列腺癌中融合基因的相关研究,发现可转录的融合基因较少,多为阅读框移位,即嵌合基因与亲本基因阅读框错位[17]。
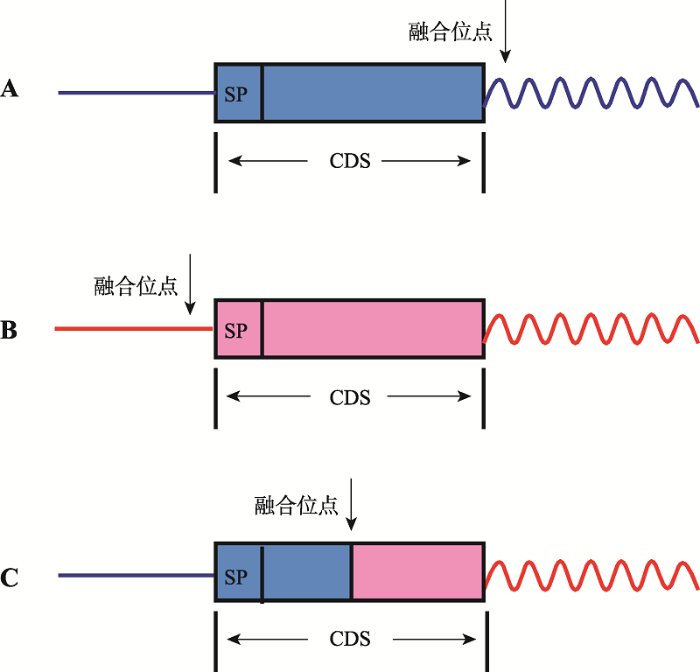
为进一步探究嵌合基因的翻译阅读特性,本课题组曾对猪的嵌合转录本的融合位点进行了探讨(图2),发现嵌合基因的融合位点可在:(1)上游亲本的阅读框终止密码子之后(图2A),此时下游亲本成为融合基因的非翻译区,形成CDS–3°UTR形式;(2)上游亲本起始密码子之前(图2B),此时上游亲本成为融合基因的非翻译区,形成5°UTR–CDS;(3)融合位点在两亲本的编码区之间(图2C),形成CDS–CDS形式。
3 融合蛋白的结构和功能特性
3.1 融合蛋白的结构域组合特性
同一种结构域,可以在不同的融合事件中反复出现[35]。在融合蛋白中反复出现的结构域,包括络氨酸激酶、EWS活性结构域和Runt结构域。首先,Frenkel-Morgenstern等[37]首先,对人类嵌合序列与随机产生的嵌合序列进行结构域的预测,发现人类嵌合基因包含的结构域显著高于随机产生的嵌合序列;其次,又将预测的融合蛋白结构域种类与人类非融合蛋白结构域种类进行分析,研究发现融合蛋白具有69%的蛋白结构域类型,而且一些结构域在嵌合蛋白中出现的频率,显著高于其亲本蛋白,包括AT hooks(涉及转录调控)、MHC(膜蛋白)和受体酪氨酸激酶催化结构域(受体蛋白)等;最后,将融合蛋白的结构域组合模式与非融合蛋白结构域组合模式进行对比,发现了一些正常蛋白中不常见的结构域新组合,如HLH (helix-loop-helix)和GTP_ EFTU (GTP-binding domain)、Hydrolase_3 domain和Polyprenyl_synt、coiled_coil domain和ZnF_C2C2等。
为了深入了解嵌合基因的结构域特征,本课题组曾对1 007条猪的嵌合RNA及其亲本,进行了结构域及其组合模式的分析[37]。通过SMART预测,获得584种嵌合结构域,与ENSEMBL上猪已知亲本的蛋白结构域对比,发现嵌合结构域的来源为:(1)嵌合蛋白结构域可仅来自于上游亲本,下游亲本为其3°UTR,形成CDS–3°UTR模式;(2)嵌合结构示意图呈现了嵌合基因的基因结构,其中A为CDS–3°UTR形式,B为5°UTR–CDS形式,C为CDS–CDS形式。直线:5°UTR,方框:阅读框,波浪线:3°UTR。浅灰色为上游亲本基因片段,深灰色为下游亲本基因片段。SP:可能具有的信号肽结构(signal peptide)。垂直箭头:为上下游亲本基因片段的融合位点(fusion site)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2融合位点的位置
Fig. 2The location of the fusion sites
域也可仅来自于下游亲本,上游亲本为其5°UTR,形成5°UTR–CDS模式;(3)嵌合结构域可来自于两亲本的组合,即形成CDS–CDS模式;(4)在两亲本的基础上,嵌合RNA可形成新的结构域。
嵌合转录的产物可与亲本蛋白竞争底物,形成多聚体或与DNA结合,对抗正常蛋白,从而在癌细胞中出现显著负效应。当融合涉及转录激活因子或抑制因子时,与亲本蛋白的竞争倾向性更强,如在急性髓系白血病中发现的RUNX1-ETO融合蛋白[38~40][。RUNX1是造血干细胞分化的关键调节因子,RUNX1- ETO融合蛋白保留RUNX1转录因子的DNA结合Runt同源结构域,可继承结合RUNX1靶基因的功能;同时,该融合蛋白还含有转录阻遏因子ETO的大部分结构域,进而其还具有转录抑制的功能。当RUNX1-ETO融合蛋白结合到RUNX1靶基因时,会抑制RUNX1靶基因的转录,干扰正常功能,阻断分化,产生显著的负效应,引发白血病[38]。
嵌合基因的结构域并不是两亲本结构域的简单组合,可在两亲本结构域的基础上,形成新的结构域组合,甚至是新的结构域,增加转录组和蛋白质组的多样性。因此,揭示嵌合蛋白结构域的组成模式和特点、嵌合蛋白的功能以及其在细胞分子网络中的作用,对于防治因嵌合基因所致的疾病,具有重要意义。
3.2 信号肽对嵌合蛋白细胞定位的影响
在真核细胞中,信号序列通常为15~30个氨基酸长度,可在内质网上引导蛋白质运输。在ChimerDB数据库中的7 224条人类嵌合转录本[41],32%包含信号肽;在Li等[42]的嵌合基因数据集中,34%的嵌合转录本包含信号肽;175条由RNA-seq数据鉴定的嵌合转录本[43],29%包含信号肽。然而,在人类基因组中,只有22%的基因包含信号肽,显示信号肽显著富集于嵌合转录本中[43]。为了探索嵌合基因信号肽的分布情况,本课题组曾利用SignalP 4.1分析了1 007条猪嵌合基因的信号肽,其中13%含有信号肽。同时以猪的整个基因组为背景,分析了25 882条猪的基因,12.5%含有信号肽。在猪的基因组中,未发现嵌合基因信号肽与非嵌合基因的信号肽存在差异。本课题组还进一步分析了猪嵌合基因和它的一对亲本基因的信号肽,发现嵌合基因的信号肽有4种来源(可参见图2):(1)上游亲本基因有信号肽,而下游亲本基因没有,信号肽来自上游亲本。融合到嵌合蛋白中的下游亲本结构域,可能会因上游亲本的信号肽,而改变细胞定位;(2)下游亲本有信号肽,上游亲本没有信号肽,信号肽来自下游亲本,形成5°UTR–CDS形式;(3)一对亲本都有信号肽,根据融合位点与起始密码子和终止密码子之间的关系,嵌合信号肽即可能来自上游亲本也有可能来自下游亲本;(4)一对亲本基因都没有信号肽,嵌合蛋白却含信号肽,其可能来自移码突变。嵌合转录本在翻译的过程中存在移码框突变,例如,PML/RARalpha可以产生移码框转录[44]。
此外,在不含信号肽的嵌合基因中,本课题组还发现嵌合基因可能会因为以下情况失去原亲本基因的信号肽:(1)一些上游亲本含信号肽,但嵌合基因发生了读框移位或上游亲本成为5°UTR,信号肽消失;(2)上游亲本无信号肽,虽然下游亲本含信含肽,但基因融合时丢失;(3)一对亲本基因都有信号肽,但发生读框移位,致使信号肽消失。
3.3 跨膜结构域对嵌合蛋白细胞定位的影响
在ChimerDB数据库中的7 224条人类嵌合转录本[41],51%包含跨膜结构域;在175条由RNA-seq鉴定的嵌合转录本中[43],50%包含跨膜结构域;在Li等[42]的嵌合基因数据集中,55%包含跨膜结构域[43]。在人类基因中,23%包含跨膜结构域,跨膜结构域显著富集于嵌合基因中[43]。为深入探讨嵌合蛋白的跨膜结构域,本课题组利用TMHMM 2.0鉴定了1 007条猪嵌合基因的跨膜结构域,其中19.9%含有跨膜结构域。同时以猪的整个基因组为背景,分析了25 882条猪的基因,25%含有跨膜结构域。在猪的基因组中,跨膜结构域显著富集于非嵌合基因中,与人类上述结果存在差异。本课题组还进一步对比分析了猪嵌合基因的跨膜结构域和它的一对亲本基因的跨膜结构域。在含有跨膜结构域的嵌合蛋白中,跨膜结构域也有4种来源:(1)上游亲本基因有跨膜结构域,下游亲本没有,跨膜结构域来自上游亲本基因。此时,融合到嵌合蛋白中的下游亲本结构域,有可能因上游亲本的跨膜结构域而改变细胞定位;(2)下游亲本有跨膜结构域,上游亲本没有跨膜结构域,跨膜结构域来自下游亲本;(3)一对亲本都有跨膜结构域,嵌合跨膜结构域即可能来自上游亲本,也有可能来自下游亲本;(4)一对亲本都没有跨膜结构域,嵌合蛋白的跨膜结构域来自读框移位。此外,在不含跨膜结构域的嵌合蛋白中,发现虽然上下游亲本均有跨膜结构域,或两亲本其中之一包含跨膜结构域,但嵌合基因发生了读框移位或结构域异常,失去跨膜结构域。
人和猪嵌合蛋白的信号肽和跨膜结构域的分布情况不同,是否不同的物种之间有不同的嵌合规律,或者由于进化的不同,导致物种之间的差异性,都有待进一步的研究。
3.4 上游亲本和下游亲本对嵌合产物的影响
通过对融合蛋白结构域和蛋白相互作用的分析,发现上游亲本和下游亲本存在结构性质上的差异[45]。首先,虽然上游亲本和下游亲本,都存在DNA结合结构域和蛋白相互作用结构域;然而在下游亲本中,鲜见激酶和组蛋白修饰结构域。上游亲本和下游亲本结构域的组合,对嵌合功能存在着至关重要的影响,例如,蛋白质相互作用域与DNA结合域和激酶结构域,不成比例地共存时,会形成信号传导缺陷的组合[46]。其次,二者的结构域保留于嵌合基因的模式也有差异,下游倾向于保留结构域的重要组成部分和相互作用界面,而上游则倾向于保留UTR区域。3.5 长链非编码RNA参与嵌合基因的形成
目前研究已证实长链非编码RNAs(long no-coding RNAs, lncRNAs)参与融合基因的形成。例如,ETV1与前列腺癌前列腺特异性lncRNA之间的融合[47],以及B细胞淋巴瘤患者中BCL6原癌基因与非编码GAS5基因的融合[48]。这些lncRNA可异常调节与其融合的致癌基因,而本身不具有致癌功能[49]。另外,在亚洲人群前列腺癌的研究中,也发现了几种涉及lncRNA的新型融合物[50],其中包括USP9Y蛋白酶和TTTY15 ncRNA之间的基因融合。这种融合丧失了USP9Y的相关功能。USP9Y-TTTY15融合基因,已成为预测前列腺的生物标志物[51]。lncRNA可以通过细胞核的高级结构,调控真核基因表达[52]。由lncRNA参与的基因融合可能具有一定的生物学功能,但目前鉴定非编码基因融合的方法仍然匮乏,可能系统地忽略实质性的有用信息。
总之,融合蛋白在结构上是多样的,且富含某些结构域,如涉及激酶和DNA结合活性的结构域。基因融合倾向于保留框内翻译,上游亲本和下游亲本会为融合蛋白提供不同的结构元件。
4 融合蛋白的表达调控
目前融合蛋白表达和调控的机制仍不清楚,但现有的研究已经开始初步揭示融合蛋白调控的特征。Frenkel-Morgenstern等[43]通过对人类嵌合基因表达量的研究,发现融合蛋白的表达量低于其他蛋白质,而且比其他蛋白质具有更强的组织特异性。前列腺癌中转录通读的研究表明,融合转录本的表达与亲本基因的表达相似[53]。相比之下,前列腺癌中顺式拼接融合,只有一半的融合物相对于亲本基因显著表达[54]。这些结果表明,不同机制所致融合,可能影响融合转录物和蛋白质的表达水平。
4.1 融合亲本对融合蛋白表达调控的影响
对血液癌症中易位基因的计算证实,融合蛋白倾向于低表达和具有组织特异性,但也有报道融合蛋白的亲本表达量水平高于基因组平均值[45]。5°端易位亲本通常有强启动子,3°端亲本有稳定的3°端UTR。融合亲本之一(通常为5°基因)主要促进融合蛋白的过度表达,而不是提供结构特征(如结构域)。因此,5°端亲本促进表达,3°端亲本提供功能性蛋白质片段和保持稳定性,协同促进融合蛋白的表达。鉴于这些趋势,融合转录和蛋白质表达水平理应很高,然而在实践中,融合转录和蛋白质表达水平却很低,其机理尚不清楚[55]。但在融合转录检测中,会有假阳性的现象[56,57],其可通过增加假定的融合转录物的数量,低估融合蛋白表达。
4.2 正常组织中融合蛋白的表达调控
融合蛋白的表达不仅局限于癌组织,也存在于正常细胞[58,59,60,61]。例如,在TCGA数据库364个正常组织样本中,鉴定出192个基因融合体[36]。融合蛋白在健康组织中的功能尚不清楚,目前认为可以增加蛋白质组的多样性和复杂性[1,62]。另外,某些看似正常的融合蛋白在其高表达后也与癌症相关。例如,JAZF1-JJAZ1融合蛋白在正常组织中表达水平很低,但当表达水平升高时就会与子宫内膜间质肉瘤的产生有关联[63]。同样,在前列腺癌和良性前列腺组织,都检测到了SLC45A3- ELK4融合转录,但在癌状态下表达水平高[64]。
融合表达的机制可能更复杂。例如,在42%的结肠直肠癌样品中,发现了复发性VTI1A-TCF7L2融合物,而在正常结肠粘膜样品中也有29%的VTI1A- TCF7L2融合物,来自其他器官的正常组织中也存在25%的VTI1A-TCF7L2融合物[65]。因此,融合蛋白在正常组织与癌变组织中表达水平的区别与联系,需要进一步的探索。
5 天然嵌合基因对基因设计的启示
1993年,华人进化生物学家龙漫远教授用实验方法发现并解析了第一个由两个不同基因片段嵌合的新基因[66]。嵌合基因设计是指按照研究者的意愿,设计和制造出自然界不存在的、具有特定生物学功能的全新蛋白质编码基因。这一技术在新能源、新材料、人工生命、核酸疫苗、生物医药、疾病诊断与治疗中有很大的应用潜能。尽管新基因设计技术具有广阔的应用前景,但是设计成功率不高、蛋白活性较低、合成成本高等问题。生命体系历经几十亿年的自然进化,创造了无数丰富多彩的功能基因[67]。例如,天然嵌合基因,通过重组由来源与功能不同的基因序列,成为了自然界新基因产生的源泉。因而,利用天然嵌合规律,依据已知的蛋白功能机理和结构特性,进行全新的基因设计与合成,可进一步开发新基因设计的潜力,创作出丰富的基因资源。例如,利用天然融合位置规律,当融合位点位于非编码区域时,可以在不改变蛋白功能的情况下增强或抑制蛋白的表达,当基因的融合位点位于编码区域时,可以通过融合不同基因,设计与合成各种功能的新基因;利用融合蛋白对目的基因进行示踪,研究其表达调控特征及生理功能;利用融合基因,增强目的基因的表达;利用融合蛋白改变目的蛋白的免疫原性,产生无毒疫苗;相同或相似功能的基因融合,增强基因的功能。
因此,天然嵌合基因的发现与应用,给基因设计带来了很大的启示作用,突破了新基因自然进化缓慢的局限,加快了新生物功能基因的产生速度,同时按照人类需求快速创建新功能基因,可极大提高自然生物功能改造与创新的速度。
6 结语与展望
经典分子生物学认为一个基因只能表达一种蛋白,实现一种主要功能。嵌合基因的发现打破了这一认知。基因融合可以转录翻译出功能特异的蛋白质,增加转录组和蛋白质组的多样性和复杂性,甚至阻碍正常的信号通路,起始或激活癌细胞生长。嵌合基因在人类医学领域已显现了极其重要的生物学意义,可以作为疾病诊断的生物学标记以及药物作用的靶标。然而,目前针对嵌合基因导致疾病的治疗还处于起步阶段,仍有很多问题需要我们探究,如嵌合基因形成的具体原因,不同的物种以及同一物种的不同组织嵌合形成的机制是否相同,嵌合基因与亲本基因功能与表达量的具体区别和联系等,只有深入了解嵌合基因的各种特性,才有助于我们进一步对其引起的疾病进行防御和治疗。
随着生物信息学研究的不断深入和分子生物学的不断加深,尤其是新算法的开发和实验技术的建立,将有助于进一步揭示生物体内某些特殊生理现象的分子基础,揭开相应基因表达调控的分子机制,更全面地了解嵌合基因的生物学意义。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
URLPMID:5012284 [本文引用: 2]

Gene fusions have been described in approximately one-third of soft tissue tumors (STT); of the 142 different fusions that have been reported, more than half are recurrent in the same histologic subtype. These gene fusions constitute pivotal driver mutations, and detailed studies of their cellular effects have provided important knowledge about pathogenetic mechanisms in STT. Furthermore, most fusions are strongly associated with a particular histotype, serving as ideal molecular diagnostic markers. In recent years, it has also become apparent that some chimeric proteins, directly or indirectly, constitute excellent treatment targets, making the detection of gene fusions in STT ever more important. Indeed, pharmacological treatment of STT displaying fusions that activate protein kinases, such as ALK and ROS1, or growth factors, such as PDGFB, is already in clinical use. However, the vast majority (52/78) of recurrent gene fusions create structurally altered and/or deregulated transcription factors, and a small but growing subset develops through rearranged chromatin regulators. The present review provides an overview of the spectrum of currently recognized gene fusions in STT, and, on the basis of the protein class involved, the mechanisms by which they exert their oncogenic effect are discussed.
URLPMID:23201468 [本文引用: 1]
TMPRSS2-ERG had independent additional predictive value to PCA3 and the ERSPC risk calculator parameters for predicting PCa. TMPRSS2-ERG had prognostic value, whereas PCA3 did not. Implementing the novel urinary biomarker panel PCA3 and TMPRSS2-ERG into clinical practice would lead to a considerable reduction of the number of prostate biopsies.
URLPMID:23499337 [本文引用: 1]
ALK gene rearrangements in advanced non-small cell lung carcinomas (NSCLC) are an indication for targeted therapy with crizotinib. Fluorescence in situ hybridization (FISH) using a recently approved companion in vitro diagnostic class FISH system commonly assesses ALK status. More accessible IHC is challenged by low expression of ALK-fusion transcripts in NSCLC. We compared ultrasensitive automated IHC with FISH for detecting ALK status on 318 FFPE and 40 matched ThinPrep specimens from 296 patients with advanced NSCLC. IHC was concordant with FFPE-FISH on 229 of 231 dual-informative samples (31 positive and 198 negative) and with ThinPrep-FISH on 34 of 34 samples (5 positive and 29 negative). Two cases with negative IHC and borderline-positive FFPE-FISH (15% and 18%, respectively) were reclassified as concordant based on negative matched ThinPrep-FISH and clinical data consistent with ALK-negative status. Overall, after including ThinPrep-FISH and amending the falsepositive FFPE-FISH results, IHC demonstrated 100% sensitivity and specificity (95% CI, 0.86 to 1.00 and 0.97 to 1.00, respectively) for ALK detection on 249 dual-informative NSCLC samples. IHC was informative on significantly more samples than FFPE-FISH, revealing additional ALK-positive cases. The high concordance with FISH warrants IHC's routine use as the initial component of an algorithmic approach to clinical ALK testing in NSCLC, followed by reflex FISH confirmation of IHC-positive cases.
URLPMID:26684754 [本文引用: 1]
Enabled by high-throughput sequencing approaches, epithelial cancers across a range of tissue types are seen to harbor gene fusions as integral to their landscape of somatic aberrations. Although many gene fusions are found at high frequency in several rare solid cancers, apart from fusions involving the ETS family of transcription factors which have been seen in approximately 50 % of prostate cancers, several other common solid cancers have been shown to harbor recurrent gene fusions at low frequencies. On the other hand, many gene fusions involving oncogenes, such as those encoding ALK, RAF or FGFR kinase families, have been detected across multiple different epithelial carcinomas. Tumor-specific gene fusions can serve as diagnostic biomarkers or help define molecular subtypes of tumors; for example, gene fusions involving oncogenes such as ERG, ETV1, TFE3, NUT, POU5F1, NFIB, PLAG1, and PAX8 are diagnostically useful. Tumors with fusions involving therapeutically targetable genes such as ALK, RET, BRAF, RAF1, FGFR1???4, and NOTCH1???3 have immediate implications for precision medicine across tissue types. Thus, ongoing cancer genomic and transcriptomic analyses for clinical sequencing need to delineate the landscape of gene fusions. Prioritization of potential oncogenic ???drivers??? from ???passenger??? fusions, and functional characterization of potentially actionable gene fusions across diverse tissue types, will help translate these findings into clinical applications. Here, we review recent advances in gene fusion discovery and the prospects for medicine.
URLPMID:19217274 [本文引用: 1]
As an inhibitor of the tyrosine kinase activity of the BCR-Abl oncoprotein, imatinib sets a new paradigm for the treatment of cancer with molecularly targeted therapies. Subsequent structural studies have provided in depth knowledge of how this antileukaemia drug interacts with the catalytic site of the enzyme and allowed the rationalisation of mechanisms of drug-resistance which can lead to patient relapse. This understanding has facilitated the design of new inhibitors of BCR-Abl, as well as the discovery of inhibitors of many other kinases. As structural information accumulates for more of the 518 kinases encoded within the human genome, the design of many more highly selective, well-tolerated kinase inhibitors should be possible.
URLPMID:52105852 [本文引用: 1]
Abstract Discovery of chimeric RNAs, which are produced by chromosomal translocations as well as the joining of exons from different genes by trans-splicing, has added a new level of complexity to our study and understanding of the transcriptome. The enhanced ChiTaRS-3.1 database (http://chitars.md.biu.ac.il) is designed to make widely accessible a wealth of mined data on chimeric RNAs, with easy-to-use analytical tools built-in. The database comprises 34 922: chimeric transcripts along with 11 714: cancer breakpoints. In this latest version, we have included multiple cross-references to GeneCards, iHop, PubMed, NCBI, Ensembl, OMIM, RefSeq and the Mitelman collection for every entry in the 'Full Collection'. In addition, for every chimera, we have added a predicted chimeric PROTEIN-PROTEIN INTERACTION CHIPPI NETWORK: , which allows for easy visualization of protein partners of both parental and fusion proteins for all human chimeras. The database contains a comprehensive annotation for 34 922: chimeric transcripts from eight organisms, and includes the manual annotation of 200: sense-antiSense (SaS) chimeras. The current improvements in the content and functionality to the ChiTaRS database make it a central resource for the study of chimeric transcripts and fusion proteins. 漏 The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
URL [本文引用: 1]
react-text: 476 Ama04: Kemik ili06indeki hematopoietik hücrelerin anormal birikimiyle karakterize olan kronik myeloid l02semi (KML) i04in t(9;22) kromozomal translokasyonu tan03sal bir belirte04tir. Philadelphia kromozomu olu06umuyla sonu04lanan translokasyon, KML olgular03nda geli06mekte ve hastalar03n03n %95’inde g02rülmektedir ve akut lenfoblastik l02semi (ALL) ve akut myeloid l02semi (AML)’de daha k02tü bir prognozun... /react-text react-text: 477 /react-text [Show full abstract]
[本文引用: 1]
URLPMID:4126434 [本文引用: 1]
CELLS from nine consecutive patients with chronic myelogenous leukaemia (CML) have been analysed with quinacrine fluorescence and various Giemsa staining techniques. The Philadelphia (Ph) chromosome in all nine patients represents a deletion of the long arm of chromosome 22 (22q-). An unsuspected abnormality in all cells from the nine patients has been detected with these new staining techniques. It consists of the addition of dully fluorescing material to the end of the long arm of one chromosome 9 (9q+). In Giemsa-stained preparations, this material appears as an additional faint terminal band in one chromosome 9. The amount of additional material is approximately equal to the amount missing from the Ph(22q-) chromosome, suggesting that there may be a hitherto undetected translocation between the long arm of 22 and the long arm of 9, producing the 9q+ chromosome.
URLPMID:20033038 [本文引用: 1]
Multiple somatic rearrangements are often found in cancer genomes; however, the underlying processes of rearrangement and their contribution to cancer development are poorly characterized. Here we use a paired-end sequencing strategy to identify somatic rearrangements in breast cancer genomes. There are more rearrangements in some breast cancers than previously appreciated. Rearrangements are more frequent over gene footprints and most are intrachromosomal. Multiple rearrangement architectures are present, but tandem duplications are particularly common in some cancers, perhaps reflecting a specific defect in DNA maintenance. Short overlapping sequences at most rearrangement junctions indicate that these have been mediated by non-homologous end-joining DNA repair, although varying sequence patterns indicate that multiple processes of this type are operative. Several expressed in-frame fusion genes were identified but none was recurrent. The study provides a new perspective on cancer genomes, highlighting the diversity of somatic rearrangements and their potential contribution to cancer development.
[本文引用: 1]
URLPMID:26227178 [本文引用: 1]
Relatively few recurrent gene fusion events have been associated with breast cancer to date. In an effort to uncover novel fusion transcripts, we performed whole-transcriptome sequencing of 120 fresh-frozen primary breast cancer samples and five adjacent normal breast tissues using the Illumina HiSeq2000 platform. Three different fusion-detecting tools (deFuse, Chimerascan, and TopHatFusion) were used, and the results were compared. These tools detected 3,831, 6,630 and 516 fusion transcripts (FTs) overall. We primarily focused on the results obtained using the deFuse software. More FTs were identified from HER2 subtype breast cancer samples than from the luminal or triple-negative subtypes ( P
URLPMID:22101766 [本文引用: 1]
Breast cancer is a heterogeneous disease that has a wide range of molecular aberrations and clinical outcomes. Here we used paired-end transcriptome sequencing to explore the landscape of gene fusions in a panel of breast cancer cell lines and tissues. We observed that individual breast cancers have a variety of expressed gene fusions. We identified two classes of recurrent gene rearrangements involving genes encoding microtubule-associated serine-threonine kinase (MAST) and members of the Notch family. Both MAST and Notch-family gene fusions have substantial phenotypic effects in breast epithelial cells. Breast cancer cell lines harboring Notch gene rearrangements are uniquely sensitive to inhibition of Notch signaling, and overexpression of MAST1 or MAST2 gene fusions has a proliferative effect both in vitro and in vivo. These findings show that recurrent gene rearrangements have key roles in subsets of carcinomas and suggest that transcriptome sequencing could identify individuals with rare, targetable gene fusions.
URLPMID:21247443 [本文引用: 1]
Abstract BACKGROUND: Until recently, chromosomal translocations and fusion genes have been an underappreciated class of mutations in solid tumors. Next-generation sequencing technologies provide an opportunity for systematic characterization of cancer cell transcriptomes, including the discovery of expressed fusion genes resulting from underlying genomic rearrangements. RESULTS: We applied paired-end RNA-seq to identify 24 novel and 3 previously known fusion genes in breast cancer cells. Supported by an improved bioinformatic approach, we had a 95% success rate of validating gene fusions initially detected by RNA-seq. Fusion partner genes were found to contribute promoters (5' UTR), coding sequences and 3' UTRs. Most fusion genes were associated with copy number transitions and were particularly common in high-level DNA amplifications. This suggests that fusion events may contribute to the selective advantage provided by DNA amplifications and deletions. Some of the fusion partner genes, such as GSDMB in the TATDN1-GSDMB fusion and IKZF3 in the VAPB-IKZF3 fusion, were only detected as a fusion transcript, indicating activation of a dormant gene by the fusion event. A number of fusion gene partners have either been previously observed in oncogenic gene fusions, mostly in leukemias, or otherwise reported to be oncogenic. RNA interference-mediated knock-down of the VAPB-IKZF3 fusion gene indicated that it may be necessary for cancer cell growth and survival. CONCLUSIONS: In summary, using RNA-sequencing and improved bioinformatic stratification, we have discovered a number of novel fusion genes in breast cancer, and identified VAPB-IKZF3 as a potential fusion gene with importance for the growth and survival of breast cancer cells.
URLPMID:22975805 [本文引用: 2]
All cancers harbor molecular alterations in their genomes. The transcriptional consequences of these somatic mutations have not yet been comprehensively explored in lung cancer. Here we present the first large scale RNA sequencing study of lung adenocarcinoma, demonstrating its power to identify somatic point mutations as well as transcriptional variants such as gene fusions, alternative splicing events, and expression outliers. Our results reveal the genetic basis of 200 lung adenocarcinomas in Koreans including deep characterization of 87 surgical specimens by transcriptome sequencing. We identified driver somatic mutations in cancer genes including EGFR, KRAS, NRAS, BRAF, PIK3CA, MET, and CTNNB1. Candidates for novel driver mutations were also identified in genes newly implicated in lung adenocarcinoma such as LMTK2, ARID1A, NOTCH2, and SMARCA4. We found 45 fusion genes, eight of which were chimeric tyrosine kinases involving ALK, RET, ROS1, FGFR2, AXL, and PDGFRA. Among 17 recurrent alternative splicing events, we identified exon 14 skipping in the proto-oncogene MET as highly likely to be a cancer driver. The number of somatic mutations and expression outliers varied markedly between individual cancers and was strongly correlated with smoking history of patients. We identified genomic blocks within which gene expression levels were consistently increased or decreased that could be explained by copy number alterations in samples. We also found an association between lymph node metastasis and somatic mutations in TP53. These findings broaden our understanding of lung adenocarcinoma and may also lead to new diagnostic and therapeutic approaches.
URLPMID:24488012 [本文引用: 2]
Abstract Gene fusions, mainly between TMPRSS2 and ERG, are frequent early genomic rearrangements in prostate cancer (PCa). In order to discover novel genomic fusion events, we applied whole-genome paired-end sequencing to identify structural alterations present in a primary PCa patient (G089) and in a PCa cell line (PC346C). Overall, we identified over 3800 genomic rearrangements in each of the two samples as compared with the reference genome. Correcting these structural variations for polymorphisms using whole-genome sequences of 46 normal samples, the numbers of cancer-related rearrangements were 674 and 387 for G089 and PC346C, respectively. From these, 192 in G089 and 106 in PC346C affected gene structures. Exclusion of small intronic deletions left 33 intergenic breaks in G089 and 14 in PC346C. Out of these, 12 and 9 reassembled genes with the same orientation, capable of generating a feasible fusion transcript. Using PCR we validated all the reliable predicted gene fusions. Two gene fusions were in-frame: MPP5-FAM71D in PC346C and ARHGEF3-C8ORF38 in G089. Downregulation of FAM71D and MPP5-FAM71D transcripts in PC346C cells decreased proliferation; however, no effect was observed in the RWPE-1-immortalized normal prostate epithelial cells. Together, our data showed that gene rearrangements frequently occur in PCa genomes but result in a limited number of fusion transcripts. Most of these fusion transcripts do not encode in-frame fusion proteins. The unique in-frame MPP5-FAM71D fusion product is important for proliferation of PC346C cells.
[本文引用: 1]
URLPMID:21571633 [本文引用: 1]
Transcription-induced chimeric RNAs, possessing sequences from different genes, are expected to increase the proteomic diversity through chimeric proteins or altered regulation. Despite their importance, few studies have focused on chimeric RNAs especially regarding their presence/roles in human cancers. By deep sequencing the transcriptome of 20 human prostate cancer and 10 matched benign prostate tissues, we obtained 1.3 billion sequence reads, which led to the identification of 2,369 chimeric RNA candidates. Chimeric RNAs occurred in significantly higher frequency in cancer than in matched benign samples. Experimental investigation of a selected 46 set led to the confirmation of 32 chimeric RNAs, of which 27 were highly recurrent and previously undescribed in prostate cancer. Importantly, a subset of these chimeras was present in prostate cancer cell lines, but not detectable in primary human prostate epithelium cells, implying their associations with cancer. These chimeras contain discernable 5' and 3' splice sites at the RNA junction, indicating that their formation is mediated by splicing. Their presence is also largely independent of the expression of parental genes, suggesting that other factors are involved in their production and regulation. One chimera, TMEM79-SMG5, is highly differentially expressed in human cancer samples and therefore a potential biomarker. The prevalence of chimeric RNAs may allow the limited number of human genes to encode a substantially larger number of RNAs and proteins, forming an additional layer of cellular complexity. Together, our results suggest that chimeric RNAs are widespread, and increased chimeric RNA events could represent a unique class of molecular alteration in cancer.
URLPMID:25963990 [本文引用: 1]
Fusion transcript formation is one of the fundamental mechanisms that drives the development of prostate cancer. Because of the advance of high-throughput parallel sequencing, many fusion transcripts have been discovered. However, the discovery rate of fusion transcripts specific for prostate cancer is lagging behind the discoveries made on chromosome abnormalities of prostate cancer. Recent analyses suggest that many fusion transcripts are present in both benign and cancerous tissues. Some of these fusion transcripts likely represent important components of normal gene expression in cells. It is necessary to identify the criteria and features of fusion transcripts that are specific for cancer. In this review, we discuss optimization of RNA sequencing depth for fusion transcript discovery and the characteristics of fusion transcripts in normal prostate tissues and prostate cancer. We also propose a new classification of cancer-specific fusion transcripts on the basis of their tail gene fusion protein product and the roles that these fusions may play in cancer development.
URLPMID:24240688 [本文引用: 1]
Gene fusion is involved in the development of various types of malignancies. Recent advances in sequencing technology have facilitated identification of gene fusions and have stimulated the research of this field in cancer. In the present study, we performed next-generation transcriptome sequencing in order to discover novel gene fusions in gastric cancer. A total of 282 fusion transcript candidates were detected from 12 gastric cancer cell lines by bioinformatic filtering. Among the candidates, we have validated 19 fusion transcripts, which are 7 inter-chromosomal and 12 intra-chromosomal fusions. A novel DUS4L-BCAP29 fusion transcript was found in 2 out of 12 cell lines and 10 out of 13 gastric cancer tissues. Knockdown of DUS4L-BCAP29 transcript using siRNA inhibited cell proliferation. Soft agar assay further confirmed that this novel fusion transcript has tumorigenic potential. We also identified that microRNA-coding gene PVT1, which is amplified in double minute chromosomes in SNU-16 cells, is recurrently involved in gene fusion. PVT1 produced six different fusion transcripts involving four different genes as fusion partners. Our findings provide better insight into transcriptional and genetic alterations of gastric cancer: namely, the tumorigenic effects of transcriptional read-through and a candidate region for genetic instability.
URLPMID:3902849 [本文引用: 1]
Abstract Chromosomal translocations are critically involved in the molecular pathogenesis of B-cell lymphomas, and highly recurrent and specific rearrangements have defined distinct molecular subtypes linked to unique clinicopathological features. In contrast, several well-characterized lymphoma entities still lack disease-defining translocation events. To identify novel fusion transcripts resulting from translocations, we investigated two Hodgkin lymphoma cell lines by whole-transcriptome paired-end sequencing (RNA-seq). Here we show a highly expressed gene fusion involving the major histocompatibility complex (MHC) class II transactivator CIITA (MHC2TA) in KM-H2 cells. In a subsequent evaluation of 263 B-cell lymphomas, we also demonstrate that genomic CIITA breaks are highly recurrent in primary mediastinal B-cell lymphoma (38%) and classical Hodgkin lymphoma (cHL) (15%). Furthermore, we find that CIITA is a promiscuous partner of various in-frame gene fusions, and we report that CIITA gene alterations impact survival in primary mediastinal B-cell lymphoma (PMBCL). As functional consequences of CIITA gene fusions, we identify downregulation of surface HLA class II expression and overexpression of ligands of the receptor molecule programmed cell death 1 (CD274/PDL1 and CD273/PDL2). These receptor-ligand interactions have been shown to impact anti-tumour immune responses in several cancers, whereas decreased MHC class II expression has been linked to reduced tumour cell immunogenicity. Thus, our findings suggest that recurrent rearrangements of CIITA may represent a novel genetic mechanism underlying tumour-microenvironment interactions across a spectrum of lymphoid cancers.
URLPMID:12476311 [本文引用: 1]
The Philadelphia chromosome (Ph), a minute chromosome that derives from the balanced translocation between chromosomes 9 and 22, was first described in 1960 and was for a long time the only genetic lesion consistently associated with human cancer. This chromosomal translocation results in the fusion between the 5' part of BCR gene, normally located on chromosome 22, and the 3' part of the ABL gene on chromosome 9 giving origin to a BCR/ABL fusion gene which is transcribed and then translated into a hybrid protein. Three main variants of the BCR/ABL gene have been described, that, depending on the length of the sequence of the BCR gene included, encode for the p190(BCR/ABL), P210(BCR/ABL), and P230(BCR/ABL) proteins. These three main variants are associated with distinct clinical types of human leukemias. Herein we review the data on the correlations between the type of BCR/ABL gene and the corresponding leukemic clinical features. Lastly, drawing on experimental data, we provide insight into the different transforming power of the three hybrid BCR/ABL proteins.
[本文引用: 1]
URLPMID:6091897 [本文引用: 1]
The 35 nucleotide spliced leader (SL) sequence is found on the 5′ end of numerous trypanosome mRNAs, yet the tandemly organized reiteration units encoding this leader are not detectably linked to any of these structural genes. Here we report the presence of a class of discrete small SL RNA molecules that are derived from the genomic SL reiteration units of Trypanosoma brucei, Trypanosoma cruzi, and Leptomonas collosoma. These small SL RNAs are 135, 105, and 95 nucleotides, respectively, and contain a 5′-terminal SL or SL-like sequence. S1 nuclease analyses demonstrate that these small SL RNAs are transcribed from continuous sequence within the respective SL reiteration units. With the exception of the SL sequence and a concensus donor splice site immediately following it, these small RNAs are not well conserved. We suggest that the small SL RNAs may function as a donor of the SL sequence in an intermolecular process that places the SL at the 5′ terminus of many trypanosomatid mRNAs.
[本文引用: 1]
[本文引用: 1]
URLPMID:2395638 [本文引用: 1]
Abstract Several known trans-splicing RNA structures were used to define a canonical trans-splicing structure which was then used to perform a computer search of the EMBL nucleotide database. In addition to most known trans-splicing structures, many putative new trans-splicing sites were detected. These were found in a broad range of organisms including the vertebrates. Control experiments indicate that the search predicts known false positives at a rate of only 20%. Trans-splicing may therefore be a very wide-spread phenomenon.
[本文引用: 1]
[本文引用: 1]
URLPMID:373324 [本文引用: 1]
Exon repetition describes the presence of tandemly repeated exons in mRNA in the absence of duplications in the genome. Its existence challenges our understanding of gene expression, because the linear organization of sequences in apparently normal genes must be subverted during RNA synthesis or processing. It is restricted to a small number of genes in some of which over half of the mRNA contains specific patterns of repetition. Although it is sometimes assumed to arise by trans-splicing, there is no evidence of this and the efficiency is very much higher than for examples of bona fide trans-splicing in mammals. Furthermore, a potentially ubiquitous reaction such as trans-splicing is not consistent with a phenomenon that involves such a high proportion of the products of so few genes. Instead, it seems more probable that exon repetition is caused by a specific trans-acting factor. We have tested this and demonstrate for the two best characterized examples that the property is restricted to specific alleles of the affected genes and is determined in cis. It is not determined by exonic splicing signals, as had been suggested previously. In heterozygotes, RNA transcribed from the two alleles of an affected gene can have fundamentally different fates.
URLPMID:4286414 [本文引用: 1]
Abstract Fibrolamellar hepatocellular carcinoma (FL-HCC) is a rare liver tumor affecting adolescents and young adults with no history of primary liver disease or cirrhosis. We identified a chimeric transcript that is expressed in FL-HCC but not in adjacent normal liver and that arises as the result of a ~400-kilobase deletion on chromosome 19. The chimeric RNA is predicted to code for a protein containing the amino-terminal domain of DNAJB1, a homolog of the molecular chaperone DNAJ, fused in frame with PRKACA, the catalytic domain of protein kinase A. Immunoprecipitation and Western blot analyses confirmed that the chimeric protein is expressed in tumor tissue, and a cell culture assay indicated that it retains kinase activity. Evidence supporting the presence of the DNAJB1-PRKACA chimeric transcript in 100% of the FL-HCCs examined (15/15) suggests that this genetic alteration contributes to tumor pathogenesis.
URLPMID:22925561 [本文引用: 1]
Background Gene fusion is ubiquitous over the course of evolution. It is expected to increase the diversity and complexity of transcriptomes and proteomes through chimeric sequence segments or altered regulation. However, chimeric mRNAs in pigs remain unclear. Here we identified some chimeric mRNAs in pigs and analyzed the expression of them across individuals and breeds using RNA-sequencing data. Results The present study identified 669 putative chimeric mRNAs in pigs, of which 251 chimeric candidates were detected in a set of RNA-sequencing data. The 618 candidates had clear trans-splicing sites, 537 of which obeyed the canonical GU-AG splice rule. Only two putative pig chimera variants whose fusion junction was overlapped with that of a known human chimeric mRNA were found. A set of unique chimeric events were considered middle variances in the expression across individuals and breeds, and revealed non-significant variance between sexes. Furthermore, the genomic region of the 5 鈥 partner gene shares a similar DNA sequence with that of the 3 鈥 partner gene for 458 putative chimeric mRNAs. The 81 of those shared DNA sequences significantly matched the known DNA-binding motifs in the JASPAR CORE database. Four DNA motifs shared in parental genomic regions had significant similarity with known human CTCF binding sites. Conclusions The present study provided detailed information on some pig chimeric mRNAs. We proposed a model that trans-acting factors, such as CTCF , induced the spatial organisation of parental genes to the same transcriptional factory so that parental genes were coordinatively transcribed to give birth to chimeric mRNAs.
URLPMID:24691255 [本文引用: 1]
Chromosome translocations are catastrophic genomic events and often play key roles in tumorigenesis. Yet the biogenesis of chromosome translocations is remarkably poorly understood. Recent work has delineated several distinct mechanistic steps in the formation of translocations, and it has become apparent that non-random spatial genome organization, DNA repair pathways and chromatin features, including histone marks and the dynamic motion of broken chromatin, are critical for determining translocation frequency and partner selection.
[本文引用: 2]
URLPMID:4468049 [本文引用: 2]
Transcript fusions as a result of chromosomal rearrangements have been a focus of attention in cancer as they provide attractive therapeutic targets. To identify novel fusion transcripts with the potential to be exploited therapeutically, we analyzed RNA sequencing, DNA copy number and gene mutation data from 4366 primary tumor samples. To avoid false positives, we implemented stringent quality criteria that included filtering of fusions detected in RNAseq data from 364 normal tissue samples. Our analysis identified 7887 high confidence fusion transcripts across 13 tumor types. Our fusion prediction was validated by evidence of a genomic rearrangement for 78 of 79 fusions in 48 glioma samples where whole-genome sequencing data were available. Cancers with higher levels of genomic instability showed a corresponding increase in fusion transcript frequency, whereas tumor samples harboring fusions contained statistically significantly fewer driver gene mutations, suggesting an important role for tumorigenesis. We identified at least one in-frame protein kinase fusion in 324 of 4366 samples (7.4%). Potentially druggable kinase fusions involving ALK, ROS, RET, NTRK and FGFR gene families were detected in bladder carcinoma (3.3%), glioblastoma (4.4%), head and neck cancer (1.0%), low-grade glioma (1.5%), lung adenocarcinoma (1.6%), lung squamous cell carcinoma (2.3%) and thyroid carcinoma (8.7%), suggesting a potential for application of kinase inhibitors across tumor types. In-frame fusion transcripts involving histone methyltransferase or histone demethylase genes were detected in 111 samples (2.5%) and may additionally be considered as therapeutic targets. In summary, we described the landscape of transcript fusions detected across a large number of tumor samples and revealed fusion events with clinical relevance that have not been previously recognized. Our results support the concept of basket clinical trials where patients are matched with experimental therapies based on their genomic profile rather than the tissue where the tumor originated.
URLPMID:3371848 [本文引用: 2]
Motivation:Chimeric RNA transcripts are generated by different mechanisms including pre-mRNA trans-splicing, chromosomal translocations and/or gene fusions. It was shown recently that at least some of chimeric transcripts can be translated into functional chimeric proteins. Results:To gain a better understanding of the design principles underlying chimeric proteins, we have analyzed 7,424 chimeric RNAs from humans. We focused on the specific domains present in these proteins, comparing their permutations with those of known human proteins. Our method uses genomic alignments of the chimeras, identification of the gene鈥揼ene junction sites and prediction of the protein domains. We found that chimeras contain complete protein domains significantly more often than in random data sets. Specifically, we show that eight different types of domains are over-represented among all chimeras as well as in those chimeras confirmed by RNA-seq experiments. Moreover, we discovered that some chimeras potentially encode proteins with novel and unique domain combinations. Given the observed prevalence of entire protein domains in chimeras, we predict that certain putative chimeras that lack activation domains may actively compete with their parental proteins, thereby exerting dominant negative effects. More generally, the production of chimeric transcripts enables a combinatorial increase in the number of protein products available, which may disturb the function of parental genes and influence their protein鈥損rotein interaction network. Availability:our scripts are available upon request. Contact:avalencia@cnio.es Supplementary information:Supplementary dataare available atBioinformaticsonline.
URLPMID:22201794 [本文引用: 1]
RUNX1 is a transcription factor that regulates critical processes in many aspects of hematopoiesis. RUNX1 is also integral in defining the definitive hematopoietic stem cell. In addition, many hematological diseases like myelodysplastic syndrome and myeloproliferative neoplasms have been associated with mutations in RUNX1. Located on chromosomal 21, the RUNX1 gene is involved in many forms of chromosomal translocations in leukemia. t(8;21) is one of the most common chromosomal translocations found in acute myeloid leukemia (AML), where it results in a fusion protein between RUNX1 and ETO. The RUNX1-ETO fusion protein is found in approximately 12% of all AML patients. In this review, we detail the structural features, functions, and models used to study both RUNX1 and RUNX1-ETO in hematopoiesis over the past two decades.
PMID:15156181
A common chromosomal translocation in acute myeloid leukemia (AML) involves the AML1 (acute myeloid leukemia 1, also called RUNX1, core binding factor protein (CBF alpha), and PEBP2 alpha B) gene on chromosome 21 and the ETO (eight-twenty one, also called MTG8) gene on chromosome 8. This translocation generates an AML1-ETO fusion protein. t(8;21) is associated with 12% of de novo AML cases and up to 40% in the AML subtype M2 of the French-American-British classification. Furthermore, it is also reported in a small portion of M0, M1, and M4 AML samples. Despite numerous studies on the function of AML1-ETO, the precise mechanism by which the fusion protein is involved in leukemia development is still not fully understood. In this review, we will discuss structural aspects of the fusion protein and the accumulated knowledge from in vitro analyses on AML1-ETO functions, and outline putative mechanisms of its leukemogenic potential.
URLPMID:3419980
The t(8;21) translocation fuses the DNA-binding domain of the hematopoietic master regulator RUNX1 to the ETO protein. The resultant RUNX1/ETO fusion protein is a leukemia-initiating transcription factor that interferes with RUNX1 function. The result of this interference is a block in differentiation and, finally, the development of acute myeloid leukemia (AML). To obtain insights into RUNX1/ETO-dependant alterations of the epigenetic landscape, we measured genome-wide RUNX1- and RUNX1/ETO-bound regions in t(8;21) cells and assessed to what extent the effects of RUNX1/ETO on the epigenome depend on its continued expression in established leukemic cells. To this end, we determined dynamic alterations of histone acetylation, RNA Polymerase II binding and RUNX1 occupancy in the presence or absence of RUNX1/ETO using a knockdown approach. Combined global assessments of chromatin accessibility and kinetic gene expression data show that RUNX1/ETO controls the expression of important regulators of hematopoietic differentiation and self-renewal. We show that selective removal of RUNX1/ETO leads to a widespread reversal of epigenetic reprogramming and a genome-wide redistribution of RUNX1 binding, resulting in the inhibition of leukemic proliferation and self-renewal, and the induction of differentiation. This demonstrates that RUNX1/ETO represents a pivotal therapeutic target in AML.
URLPMID:19906715 [本文引用: 2]
Chromosome translocations and gene fusions are frequent events in the human genome and have been found to cause diverse types of tumor. ChimerDB is a knowledgebase of fusion genes identified from bioinformatics analysis of transcript sequences in the GenBank and various other public resources such as the Sanger cancer genome project (CGP), OMIM, PubMed and the Mitelman's database. In this updated version, we significantly modified the algorithm of identifying fusion transcripts. Specifically, the new algorithm is more sensitive and has detected 2699 fusion transcripts with high confidence. Furthermore, it can identify interchromosomal translocations as well as the intrachromosomal deletions or inversions of large DNA segments. Importantly, results from the analysis of next-generation sequencing data in the short read archives are incorporated as well. We updated and integrated all contents (GenBank, Sanger CGP, OMIM, PubMed publications and the Mitelman's database), and the user-interface has been improved to support diverse types of searches and to enhance the user convenience especially in browsing PubMed articles. We also developed a new alignment viewer that should facilitate examining reliability of fusion transcripts and inferring functional significance. We expect ChimerDB 2.0, available at http://ercsb.ewha.ac.kr/fusiongene, to be a valuable tool in identifying biomarkers and drug targets.
URLPMID:19089307 [本文引用: 2]
Chimeric RNAs have been reported in varieties of organisms and are conventionally thought to be produced by trans-splicing of two or more distinct transcripts. Here, we conducted a large-scale search for chimeric RNAs in the budding yeast, fruit fly, mouse, and human. Thousands of chimeric transcripts were identified in these organisms except in yeast, in which five chimeric RNAs were observed. RT-PCR experiments for a sample of yeast and fly chimeric transcripts using specific primers show that about one-third of these chimeric RNAs can be reproduced. The results suggest that at least a considerable amount of chimeric RNAs is unlikely from aberrant transcription or splicing, and thus formation of chimeric RNAs is probably a widespread process and can greatly contribute to the complexity of the transcriptome and proteome of organisms. However, only a small fraction (<20%) of these chimeric RNAs has GU-AG at the junction sequences which fits the classical trans-splicing model. In contrast, we observed that about half of the chimeric RNAs have short homologous sequences (SHSs) at the junction sites of the source sequences. Our sequence mutation experiments in yeast showed that disruption of SHSs resulted in the disappearance of the corresponding chimeric RNAs, suggesting that SHSs are essential for generating this kind of chimeric RNA. In addition to the classical trans-splicing model, we propose a new model, the transcriptional slippage model, to explain the generation of those chimeric RNAs synthesized from templates with SHSs.
URLPMID:22588898 [本文引用: 6]
Chimeric RNAs comprise exons from two or more different genes and have the potential to encode novel proteins that alter cellular phenotypes. To date, numerous putative chimeric transcripts have been identified among the ESTs isolated from several organisms and using high throughput RNA sequencing. The few corresponding protein products that have been characterized mostly result from chromosomal translocations and are associated with cancer. Here, we systematically establish that some of the putative chimeric transcripts are genuinely expressed in human cells. Using high throughput RNA sequencing, mass spectrometry experimental data, and functional annotation, we studied 7424 putative human chimeric RNAs. We confirmed the expression of 175 chimeric RNAs in 16 human tissues, with an abundance varying from 0.06 to 17 RPKM (Reads Per Kilobase per Million mapped reads). We show that these chimeric RNAs are significantly more tissue-specific than non-chimeric transcripts. Moreover, we present evidence that chimeras tend to incorporate highly expressed genes. Despite the low expression level of most chimeric RNAs, we show that 12 novel chimeras are translated into proteins detectable in multiple shotgun mass spectrometry experiments. Furthermore, we confirm the expression of three novel chimeric proteins using targeted mass spectrometry. Finally, based on our functional annotation of exon organization and preserved domains, we discuss the potential features of chimeric proteins with illustrative examples and suggest that chimeras significantly exploit signal peptides and transmembrane domains, which can alter the cellular localization of cognate proteins. Taken together, these findings establish that some chimeric RNAs are translated into potentially functional proteins in humans.
URLPMID:17454588 [本文引用: 1]
Acute promyelocytic leukemia (APL) is characterized by increased promyelocytes in the marrow that harbor a t(15;17) and promyelocyte leukemia (PML)/RARalpha fusion gene. The oncogenic gene product is believed to act through disruption of the transcription-modulating function of RARalpha. Differentiation of promyelocytes and remission is achieved with all transretinoic acid (ATRA) therapy usually in combination with chemotherapy. This report describes a patient with the t(15;17) who did not respond typically to ATRA and IDAC induction chemotherapy, although achieved and remains in complete remission five years following induction and one consolidation with high dose cytarabine (HIDAC). RT-PCR and sequencing revealed a novel fusion of RARalpha exon 3 to PML exon 5 that creates a frameshift and premature stop codon in the RARalpha portion of the transcript. Since none of the RARalpha functional domains are maintained, this case highlights the possibility that PML/RARalpha may directly affect promyelocyte differentiation through disruption of PML function.
URLPMID:3516532 [本文引用: 2]
Reciprocal chromosomal translocations (RCTs) leading to the formation of fusion genes are important drivers of hematological cancers. Although the general requirements for breakage and fusion are fairly well understood, quantitative support for a general mechanism of RCT formation is still lacking. The aim of this paper is to analyze available high-throughput datasets with computational and robust statistical methods, in order to identify genomic hallmarks of translocation partner genes (TPGs). Our results show that fusion genes are generally overexpressed due to increased promoter activity of 5′ TPGs and to more stable 3′-UTR regions of 3′ TPGs. Furthermore, expression profiling of 5′ TPGs and of interaction partners of 3′ TPGs indicates that these features can help to explain tissue specificity of hematological translocations. Analysis of protein domains retained in fusion proteins shows that the co-occurrence of specific domain combinations is non-random and that distinct functional classes of fusion proteins tend to be associated with different components of the gene fusion network. This indicates that the configuration of fusion proteins plays an important role in determining which 5′ and 3′ TPGs will combine in specific fusion genes. It is generally accepted that chromosomal proximity in the nucleus can explain the specific pairing of 5′ and 3′ TPGS and the recurrence of hematological translocations. Using recently available data for chromosomal contact probabilities (Hi-C) we show that TPGs are preferentially located in early replicated regions and occupy distinct clusters in the nucleus. However, our data suggest that, in general, nuclear position of TPGs in hematological cancers explains neither TPG pairing nor clinical frequency. Taken together, our results support a model in which genomic features related to regulation of expression and replication timing determine the set of candidate genes more likely to be translocated in hematological tissues, with functional constraints being responsible for specific gene combinations.
URLPMID:17322911 [本文引用: 1]
Signaling pathways in mammalian cells are assembled and regulated by a finely controlled network of protein-protein and protein-interactions, mediated by dedicated signaling domains and their cognate motifs. The domain-based modular architecture of signaling proteins may have facilitated the evolution of complex biological systems, and can be exploited experimentally to generate synthetic signaling pathways and artificial mechanisms of autoregulation. Pathogenic proteins, such as those encoded by and , frequently form ectopic signaling complexes to respecify cellular . In a similar fashion, proteins expressed as a consequence of oncogenic fusions, mutations or amplifications can elicit ectopic protein-protein interactions that re-wire signaling pathways, in a fashion that promotes . Compounds that directly or indirectly reverse these aberrant interactions offer new possibilities for therapy in .
URLPMID:3215093 [本文引用: 1]
The discovery of numerous noncoding (ncRNA) transcripts in species from to has dramatically altered our understanding of cell biology, especially the biology of diseases such as . In , the identification of abundant long ncRNA (lncRNA) >200 bp has catalyzed their characterization as critical components of biology. Recently, roles for lncRNAs as drivers of suppressive and oncogenic functions have appeared in prevalent types, such as breast and . In this review, we highlight the emerging impact of ncRNAs in research, with a particular focus on the mechanisms and functions of lncRNAs.
URLPMID:18406879 [本文引用: 1]
The BCL6 gene is frequently disrupted at its 5′ noncoding region by 3q27 chromosomal translocations in B-cell lymphoma. As a result of translocation, BCL6 is juxtaposed to reciprocal partners, such as the immunoglobulin (Ig) gene family. Besides the Ig loci, multiple non-Ig partners of the BCL6 translocation have been reported. Here we describe the identification of the GAS5 (growth arrest-specific transcript 5) gene as a novel partner of the BCL6 in a patient with diffuse large B-cell lymphoma, harboring the t(1;3)(q25;q27). In this case, the chromosome 1 breakpoint was located within the intronic small nucleolar RNA (snoRNA) sequence of GAS5 and the chromosome 3 breakpoint at 4 kb upstream of BCL6 exon 1a. As the result of chromosomal translocation, the GAS5–BCL6 chimeric transcripts were expressed, in which the 5′-terminal oligopyrimidine (5′TOP) sequence of GAS5 was fused to the whole coding sequence of BCL6. The GAS5 gene on chromosome 1q25 is the second BCL6 partner, to the SNHG5 on 6q15, which is classified as a non-protein-coding multiple snoRNA host and 5′-TOP class gene.
URLPMID:3439828 [本文引用: 1]
The evidence that links classical protein-coding proto-oncogenes and tumor suppressors, such asMYC,RAS,P53, andRB, to carcinogenesis is indisputable. Multiple lines of proof show how random somatic genomic alteration of such genes (e.g., mutation, deletion, or amplification), followed by selection and clonal expansion, forms the main molecular basis of tumor development. Many important cancer genes were discovered using low-throughput approaches in the pre-genomic era, and this knowledge is today solidified and expanded upon by modern genome-scale methodologies. In several recent studies, non-coding RNAs (ncRNAs), such as microRNAs and long ncRNAs (lncRNAs), have been shown to contribute to tumor development. However, in comparison with coding cancer genes, the genomic (DNA-level) evidence is sparse for ncRNAs. The coding proto-oncogenes and tumor suppressors that we know of today are major molecular hubs in both normal and malignant cells. The search for ncRNAs with tumor driver or suppressor roles therefore holds the additional promise of pinpointing important, biologically active, ncRNAs in a vast and largely uncharacterized non-coding transcriptome. Here, we assess the available DNA-level data that links non-coding genes to tumor development. We further consider historical, methodological, and biological aspects, and discuss future prospects of ncRNAs in cancer.
[本文引用: 1]
URLPMID:26008593 [本文引用: 1]
In conclusion, our study explored the potential utility of measuring the TTTY15-USP9Y score in post–digital rectal examination urine samples to predict biopsy outcome and provided the basis for the utility of this novel gene fusion in multicenter and large cohort studies.
URL [本文引用: 1]
长链非编码RNA(long non-coding RNA, lncRNA)是一类转录本长度超过200nt、不编码蛋白质的RNA。近年来,随着染色质构象捕获及转录组测序等技术的发展,lncRNA与染色质构象间的关系越来越受到重视。多项研究表明,lncRNA在基因调控网络中具有重要的作用,可通过影响细胞核高级结构的动态变化来调控真核基因的表达。因其广泛的基因调控功能及在肿瘤发生过程中的重要作用,lncRNA被认为是未来肿瘤临床诊断和预后判定的新型标志物之一。本文旨在介绍lncRNA改变细胞核高级结构从而调控关键基因表达的分子机制,并详细介绍lncRNA在肿瘤治疗中的临床意义。
URL [本文引用: 1]
长链非编码RNA(long non-coding RNA, lncRNA)是一类转录本长度超过200nt、不编码蛋白质的RNA。近年来,随着染色质构象捕获及转录组测序等技术的发展,lncRNA与染色质构象间的关系越来越受到重视。多项研究表明,lncRNA在基因调控网络中具有重要的作用,可通过影响细胞核高级结构的动态变化来调控真核基因的表达。因其广泛的基因调控功能及在肿瘤发生过程中的重要作用,lncRNA被认为是未来肿瘤临床诊断和预后判定的新型标志物之一。本文旨在介绍lncRNA改变细胞核高级结构从而调控关键基因表达的分子机制,并详细介绍lncRNA在肿瘤治疗中的临床意义。
[本文引用: 1]
[本文引用: 1]
URLPMID:19741701 [本文引用: 1]
Deep sequencing of 'transcriptomes'--the collection of all RNA transcripts produced at a given time--from worms to humans reveals that some transcripts are composed of sequence segments that are not co-linear, with pieces of sequence coming from distant regions of DNA, even different chromosomes. Some of these 'chimaeric' transcripts are formed by genetic rearrangements, but others arise during post-transcriptional events. The 'trans-splicing' process in lower eukaryotes is well understood, but events in higher eukaryotes are not. The existence of such chimaeric RNAs has far-reaching implications for the potential information content of genomes and the way it is arranged.
[本文引用: 1]
URLPMID:25053845 [本文引用: 1]
Abstract Global transcriptome investigations often result in the detection of an enormous number of transcripts composed of non-co-linear sequence fragments. Such 'aberrant' transcript products may arise from post-transcriptional events or genetic rearrangements, or may otherwise be false positives (sequencing/alignment errors or in vitro artifacts). Moreover, post-transcriptionally non-co-linear ('PtNcl') transcripts can arise from trans-splicing or back-splicing in cis (to generate so-called 'circular RNA'). Here, we collected previously-predicted human non-co-linear RNA candidates, and designed a validation procedure integrating in silico filters with multiple experimental validation steps to examine their authenticity. We showed that >50% of the tested candidates were in vitro artifacts, even though some had been previously validated by RT-PCR. After excluding the possibility of genetic rearrangements, we distinguished between trans-spliced and circular RNAs, and confirmed that these two splicing forms can share the same non-co-linear junction. Importantly, the experimentally-confirmed PtNcl RNA events and their corresponding PtNcl splicing types (i.e. trans-splicing, circular RNA, or both sharing the same junction) were all expressed in rhesus macaque, and some were even expressed in mouse. Our study thus describes an essential procedure for confirming PtNcl transcripts, and provides further insight into the evolutionary role of PtNcl RNA events, opening up this important, but understudied, class of post-transcriptional events for comprehensive characterization. 脗漏 The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
URLPMID:16344562 [本文引用: 1]
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
URLPMID:16344564 [本文引用: 1]
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
[本文引用: 1]
URLPMID:3251577 [本文引用: 1]
The classic organization of a gene structure has followed the Jacob and Monod bacterial gene model proposed more than 50 years ago. Since then, empirical determinations of the complexity of the transcriptomes found in yeast to human has blurred the definition and physical boundaries of genes. Using multiple analysis approaches we have characterized individual gene boundaries mapping on human chromosomes 21 and 22. Analyses of the locations of the 5鈥 and 3鈥 transcriptional termini of 492 protein coding genes revealed that for 85% of these genes the boundaries extend beyond the current annotated termini, most often connecting with exons of transcripts from other well annotated genes. The biological and evolutionary importance of these chimeric transcripts is underscored by (1) the non-random interconnections of genes involved, (2) the greater phylogenetic depth of the genes involved in many chimeric interactions, (3) the coordination of the expression of connected genes and (4) the close in vivo and three dimensional proximity of the genomic regions being transcribed and contributing to parts of the chimeric RNAs. The non-random nature of the connection of the genes involved suggest that chimeric transcripts should not be studied in isolation, but together, as an RNA network.
URLPMID:23161619 [本文引用: 1]
Three main molecular mechanisms are considered to contribute expanding the repertoire and diversity of proteins present in living organisms: first, at DNA level (gene polymorphisms and single nucleotide polymorphisms); second, at messenger RNA (pre-mRNA and mRNA) level including alternative splicing (also termed differential splicing or cis-splicing); finally, at the protein level mainly driven through PTM and specific proteolytic cleavages. Chimeric mRNAs constitute an alternative source of protein diversity, which can be generated either by chromosomal translocations or by trans-splicing events. The occurrence of chimeric mRNAs and proteins is a frequent event in cells from the immune system and cancer cells, mainly as a consequence of gene rearrangements. Recent reports support that chimeric proteins may also be expressed at low levels under normal physiological circumstances, thus, representing a novel source of protein diversity. Notably, recent publications demonstrate that chimeric protein products can be successfully identified through bottom-up proteomic analyses. Several questions remain unsolved, such as the physiological role and impact of such chimeric proteins or the potential occurrence of chimeric proteins in higher eukaryotic organisms different from humans. The occurrence of chimeric proteins certainly seems to be another unforeseen source of complexity for the proteome. It may be a process to take in mind not only when performing bottom-up proteomic analyses in cancer studies but also in general bottom-up proteomics experiments.
URLPMID:19158498 [本文引用: 1]
Abstract Chimeric gene products, most often resulting from chromosome translocations, have been considered unique features of cancer, or at least of cells at high risk for becoming cancerous. Chimeric JAZF1-JJAZ1 mRNA transcribed from DNA spanning the site of recombination in the (7;17)(p15;q21) chromosomal translocation found in half of endometrial stromal sarcomas and most cases of benign stromal nodules is one such example. The recent finding that chimeric JAZF1-JJAZ1 mRNA can also be detected in normal endometrial stromal cells suggests that chimeric gene products are not limited to cancer or pre-cancerous cells. The JAZF1-JJAZ1 mRNA and the protein encoded by it appear to be identical to that synthesized from the gene fusion in neoplastic cells. In cultured cells, the chimeric protein has anti-apoptotic properties and is pro-proliferative when unrearranged JJAZ1 alleles are silenced, as they are in endometrial stromal sarcomas but not in the stromal nodules. These observations are consistent with the conclusion that chromosomal rearrangements and gene fusions in neoplastic cells may represent mechanisms for the deregulated expression of chimeric gene products that are generated at specific stages in cell development and have physiologic functions in normal cells. Furthermore, it may be possible that other means for abnormal production of chimeric gene products, such as hyperactive transsplicing of RNA, may be another mechanism underlying the neoplastic properties of tumor cells.
[本文引用: 1]
URLPMID:24608966 [本文引用: 1]
Abstract VTI1A-TCF7L2 was reported as a recurrent fusion gene in colorectal cancer (CRC), found to be expressed in three out of 97 primary cancers, and one cell line, NCI-H508, where a genomic deletion joins the two genes [1]. To investigate this fusion further, we analyzed high-throughput DNA and RNA sequencing data from seven CRC cell lines, and identified the gene RP11-57H14.3 (ENSG00000225292) as a novel fusion partner for TCF7L2. The fusion was discovered from both genome and transcriptome data in the HCT116 cell line. By triplicate nested RT-PCR, we tested both the novel fusion transcript and VTI1A-TCF7L2 for expression in a series of 106 CRC tissues, 21 CRC cell lines, 14 normal colonic mucosa, and 20 normal tissues from miscellaneous anatomical sites. Altogether, 42% and 45% of the CRC samples expressed VTI1A-TCF7L2 and TCF7L2-RP11-57H14.3 fusion transcripts, respectively. The fusion transcripts were both seen in 29% of the normal colonic mucosa samples, and in 25% and 75% of the tested normal tissues from other organs, revealing that the TCF7L2 fusion transcripts are neither specific to cancer nor to the colon and rectum. Seven different splice variants were detected for the VTI1A-TCF7L2 fusion, of which three are novel. Four different splice variants were detected for the TCF7L2-RP11-57H14.3 fusion. In conclusion, we have identified novel variants of VTI1A-TCF7L2 fusion transcripts, including a novel fusion partner gene, RP11-57H14.3, and demonstrated detectable levels in a large fraction of CRC samples, as well as in normal colonic mucosa and other tissue types. We suggest that the fusion transcripts observed in a high frequency of samples are transcription induced chimeras that are expressed at low levels in most samples. The similar fusion transcripts induced by genomic rearrangements observed in individual cancer cell lines may yet have oncogenic potential as suggested in the original study by Bass et al.
URLPMID:7682012 [本文引用: 1]
The origin of new genes includes both the initial molecular events and subsequent population dynamics. A processed Drosophila alcohol dehydrogenase (Adh) gene, previously thought to be a pseudogene, provided an opportunity to examine the two phases of the origin of a new gene. The sequence of the processed Adh messenger RNA became part of a new functional gene by capturing several upstream exons and introns of an unrelated gene. This novel chimeric gene, jingwei, differs from its parent Adh gene in both its pattern of expression and rate of molecular evolution. Natural selection participated in the origin and subsequent evolution of this gene.
URLPMID:11073452 [本文引用: 1]
Abstract Gene duplication has generally been viewed as a necessary source of material for the origin of evolutionary novelties, but it is unclear how often gene duplicates arise and how frequently they evolve new functions. Observations from the genomic databases for several eukaryotic species suggest that duplicate genes arise at a very high rate, on average 0.01 per gene per million years. Most duplicated genes experience a brief period of relaxed selection early in their history, with a moderate fraction of them evolving in an effectively neutral manner during this period. However, the vast majority of gene duplicates are silenced within a few million years, with the few survivors subsequently experiencing strong purifying selection. Although duplicate genes may only rarely evolve new functions, the stochastic silencing of such genes may play a significant role in the passive origin of new species.

{kind=link}
{kind=link}
{kind=link}
{kind=link}