 ,1, 秦俊,1, 汪晖2,3, 陈廖斌1,3
,1, 秦俊,1, 汪晖2,3, 陈廖斌1,3Research progress of epigenetic biomarkers in the early diagnosis and treatment of human diseases
Wei li,1, Jun Qin,1, Hui Wang2,3, Liaobin Chen1,3编委: 朱卫国
收稿日期:2017-10-16修回日期:2018-01-3网络出版日期:2018-01-18
| 基金资助: |
Received:2017-10-16Revised:2018-01-3Online:2018-01-18
| Fund supported: |
作者简介 About authors
作者简介:黎伟,硕士研究生,专业方向:骨关节病E-mail:
通讯作者:秦俊,博士,主治医师,专业方向:骨关节病E-mail:heart.

摘要
关键词:
Abstract
Keywords:
PDF (397KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黎伟, 秦俊, 汪晖, 陈廖斌. 表观遗传生物标志物在人类疾病早期诊治中的研究进展. 遗传[J], 2018, 40(2): 104-115 doi:10.16288/j.yczz.17-220
Wei li, Jun Qin, Hui Wang, Liaobin Chen.
表观遗传是指在不改变DNA序列的前提下通过化学修饰调控基因表达的一种遗传方式,主要包括DNA甲基化、组蛋白修饰以及非编码RNA(non-coding RNA, ncRNA)调控等3种类型。这些调节参与了机体的多项生命活动,任何一种调节异常都可能影响基因表达,进而导致多种疾病。已有的研究证实,表观遗传调节异常与肿瘤[1,2,3,4]、老年性疾病[5,6]和发育源性疾病[7,8,9,10,11]的发生密切相关。同时由于表观遗传修饰改变的稳定性、可逆性和可控性,其作为创新的疾病生物标志物[12,13,14,15],为肿瘤、老年性疾病和发育源性疾病的早期诊断、靶向药物治疗和预防提供了新的机会。
本文对表观遗传学分子调控机制在肿瘤、老年性疾病和发育源性疾病中的研究进展进行综述,以期阐明表观遗传学调控在疾病发生发展中的作用,及疾病相关基因表遗传改变可能作为生物标志物进行疾病诊断,对于寻找疾病早期诊断靶标和改进药物治疗措施提供借鉴和参考。
1 表遗传修饰的主要方式
常见的表遗传修饰方式包括:DNA甲基化、组蛋白修饰及非编码RNA,影响基因的表达和转录,从而调控机体的正常生命活动。1.1 DNA甲基化修饰
DNA甲基化是以S-腺苷甲硫氨酸为甲基供体,由DNA甲基转移酶(DNA methyltransferases, DNMT)催化,将胞嘧啶转变为5-甲基胞嘧啶的反应。其中,CpG岛是甲基化发生的主要区域,这里的CpG岛即指靠近基因启动子区、富含CpG序列并簇集成岛状的区域。催化CpG位点甲基化的DNMT主要有DNMT1、DNMT3A/3B和DNMT2[16,17]。DNMT与一些辅助蛋白共同参与基因表达过程中甲基化模式的维持和调控[18]。过度甲基化可使正常表达的基因沉默[19],甲基化模式通常处于稳定状态且具有可遗传性,同时,甲基化水平可受到饮食及其他环境因素的影响,特定基因的甲基化模式随细胞类型不同而各异。1.2 组蛋白共价修饰
组蛋白是真核生物染色体的基本结构蛋白,是一类小分子碱性蛋白质,有5种类型:H1、H2A、H2B、H3、H4。组蛋白富含带正电荷的碱性氨基酸,能够同DNA中带负电荷的磷酸基团相互作用。组蛋白上的游离氨基端可通过可逆的共价键修饰,如甲基化、乙酰化、磷酸化和泛素化等[20],形成理论上数目繁多的特定的“组蛋白密码(histone code)”来形成“开放”或“关闭”的局部染色质结构,或是决定何种蛋白结合到特定DNA区域,从而调节DNA 功能。组蛋白乙酰化主要受两类作用相反的酶类的调节:组蛋白乙酰基转移酶(histone acetyltransferases, HATs)和组蛋白去乙酰化酶(histone deacetylase, HDACs)。HATs催化组蛋白乙酰化,导致染色质结构松弛,促进基因转录;而HDACs使组蛋白去乙酰化,导致染色质浓缩,抑制基因转录[21]。组蛋白甲基化是由组蛋白甲基转移酶(histone methyltransferases, HMTs)催化完成的,主要发生在H3和H4组蛋白N端精氨酸或赖氨酸或组氨酸残基上。组蛋白甲基化可促进或抑制基因转录,对基因表达调控的作用可以完全相反,某些可以激活基因的转录,而某些则会抑制基因的转录。这与组蛋白甲基化形式和被修饰的氨基酸类型等有关[22]。1.3 非编码RNA
非编码RNA(non-coding RNA, ncRNA)指不编码蛋白质的RNA,包括微小RNA(microRNA, miRNA)和长链非编码RNA(long non-coding RNA, lncRNA)等。miRNA在生物发育过程中发挥着重要作用[23],最早发现于秀丽线虫的生长发育过程中[24]。miRNA通过互补序列与特异性的目标mRNA结合,从而诱导mRNA的分裂、降解或翻译受阻[25]。人类基因组编码超过3400个miRNA[26],部分为组织特异性miRNA,异常表达于多种人类疾病。lncRNA同样作用于多种人类疾病[27,28,29],参与了包括染色体印迹、表观遗传调控和转录过程。转录组学研究发现lncRNA比miRNA更具特异表达谱,在不同的组织、细胞之间以及不同的发育阶段、疾病阶段均具有不同程度的表达[30]。
2 表观遗传与疾病的关系
表观遗传机制在正常发育过程中发挥重要作用,因此当其发生紊乱时,会导致疾病的发生,如肿瘤、老年性疾病和发育源性疾病等。2.1 表观遗传与肿瘤
正常的DNA甲基化对于维持机体正常功能是必须的,如基因组结构的稳定、胚胎的正常发育、细胞分化等;异常的DNA甲基化可能会引发疾病甚至肿瘤的发生。基因启动子区的CpG岛在正常状态下一般是非甲基化的,当其发生甲基化时,常导致基因转录沉寂,使重要基因如抑癌基因、DNA修复基因等丧失功能,从而导致正常细胞的生长分化调控失常以及DNA损伤不能被及时修复。上述异常因素与多种肿瘤形成密切相关,如前列腺癌[1,2]、胃癌[3]、肝脏肿瘤[4]和结肠肿瘤[31,32]。对51例肝细胞癌的研究[33]发现,82%的癌组织出现抑癌基因启动子高甲基化,最常见于APC、SOCS-1、GSTP、E-cadherin和P15基因;同样非小细胞肺癌和结直肠癌的患者中P16/INK4A基因出现高甲基化[34]。多种血液恶性肿瘤P15/INK4A与P16/INK4A基因也呈现高甲基化改变[35]。值得注意的是,研究发现基因的低甲基化也可能与某些癌症的发生有关[36],如人乳腺管癌中PRDM16基因的外显子与CpG岛重叠的区域出现低甲基化改变[37]。总之,基因启动子区的异常甲基化可抑制抑癌基因正常表达或促进原癌基因过度表达,可能导致多种肿瘤的发生。组蛋白修饰可引起核小体结构发生变化,导致染色质重塑,影响各类转录因子与DNA的结合,从而影响基因的转录。组蛋白乙酰化的失衡可引起相应的染色体结构和基因转录水平的改变,并影响细胞周期、分化及凋亡继而导致肿瘤发生。组蛋白乙酰化和甲基化相关酶异常调节可能参与肿瘤的发生[38]。研究表明,HATs可抑制肿瘤,HATs突变易诱导疾病发生,如反应结合蛋白等位基因突变可使HATs失活,导致Rubinstein-Taybi综合征,增加肿瘤发生风险[39]。Okugawa等[40]在结肠癌中发现H3K56和H4K20的乙酰化水平降低,同时组蛋白修饰相关酶失调,如HMTs(EZH2)上调和HATs(P300、CBP和PCAF)改变[41]。肝细胞癌患者癌组织中抑癌基因P16INK4A、RIZ1及RASSF1A的转录沉默与该基因启动子区域组蛋白H3K9及H3K27甲基化水平升高或乙酰化水平降低有关[42]。多种肿瘤发生过程中也存在某些固定组蛋白修饰的改变,如H4K16ac乙酰化水平增加可作为多种肿瘤的表观遗传标志[43]。
miRNA可以调节癌基因或抑癌基因的表达。最早发现miR-15与血液系统肿瘤相关,是导致慢性淋巴细胞白血病发生的重要因素之一[44]。miRNA活性增加或降低可能导致肿瘤形成[45],如肺癌亚型腺癌中miRNA-196-5p和miRNA-21-5p的表达显著增加,胃癌细胞中存在大量miRNA上调或下调[46],DNMT1过表达导致miR-148a基因的超甲基化,导致其失活[47]。Wang等[48]通过细胞证明,miR-26b-5q在肝癌细胞中起负性调节作用,能抑制血管生成及肿瘤细胞转移。此外,TGFβ信号通路在肿瘤发生中发挥关键作用,并且是miR-148家族成员的靶标。通过DNA甲基化灭活miR-148导致TGFβ信号传导增强,导致肿瘤生长和转移[49]。
2.2 表观遗传与老年性疾病
在过去十年中,表观遗传异常与年龄相关疾病发生倍受关注,可能参与了疾病特异性病理过程, 例如阿尔茨海默氏病(Alzheimer°s disease, AD)和亨廷顿病(Huntington disease, HD)。DNA甲基化失调导致各种神经系统相关基因的表达紊乱是神经退行性疾病的发病机制之一。研究表明,AD患者顶叶淀粉样前蛋白基因启动子区的DNA甲基化程度随着年龄的增加而下降,大脑颞中回中甲基化和羟甲基化程度升高[5]。与非AD单卵双胞胎相比,AD患者中皮质神经元细胞核低甲基化[50],同时端粒酶甲基化频率升高[51]和LINE-1基因显著高甲基化[52]。HD是迟发性常染色体显性遗传性中枢神经系统退行性疾病,病变主要累及纹状体和大
脑皮质。研究发现在HD患者大脑皮质,H3K9甲基转移酶ESET上调导致H3K9Me3显著升高[6]。在HD患者和转基因小鼠中,发现腺苷A2a受体表达下调[53],与DNA高甲基化相关。
老年性疾病发生过程中存在组蛋白修饰变化。研究发现,衰老小鼠模型脑组织中H4K20me1、H3K36me3降低和H3K27me3升高[54]。与同卵双胞胎正常成年人相比,AD患者颞叶皮质和海马H3K9me3明显升高,颞叶中组蛋白乙酰化显著降低[55]。在HD小鼠模型中也发现HDAC1表达增加,亨廷顿蛋白直接与组蛋白乙酰化酶家族成员环磷腺苷效应元件结合蛋白结合并使其数量减少,导致组蛋白乙酰化水平降低[56]。组蛋白修饰具有稳定的表达调控,因此对组蛋白修饰酶及下游靶基因的研究,将为某些神经系统功能变异或疾病的早期识别和治疗提供了理论依据。
非编码RNA广泛参与老年性疾病的病理生理过程[57]。如丝氨酸棕榈酰转移酶(serine acyl transferase, SPT)是对神经酰胺合成至关重要的酶,miRNA- 9-29a/b-1、-137和-181c负调节SPT产生,并且其表达水平在AD患者的额叶皮质中降低[58]。ROBERTS等[59]实验发现,AD患者脑组织中lncRNA脑细胞质200(brain cytoplasm 200, BC200)表达显著上调,BC200调节突触后树突结构的变化,导致突触树突损伤退化,进而导致AD形成,且BC200增高程度与病情严重程度成正比。
2.3 表观遗传与发育源性疾病
宫内不良环境特别是暴露在发育可塑时期的不良环境,可以通过表观遗传修饰来编程某些组织特异性基因的表达,导致成年后某些疾病易感性增加,如肥胖、糖尿病、心血管病和神经精神性疾病[60](图1)。图1
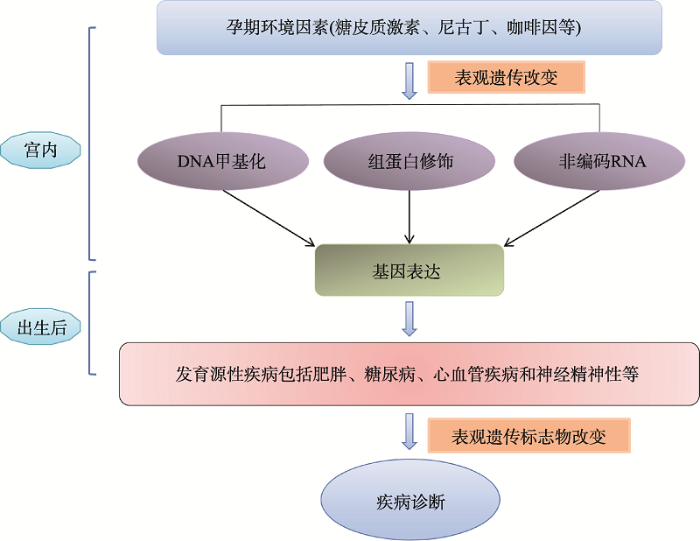 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1宫内发育过程中表观遗传学改变及其相关疾病
Fig. 1Epigenetic regulation and related diseases during intrauterine development
近期研究发现表观遗传调节异常可能在发育源性疾病发生过程起中到关键作用[61, 62]。Xu等[63]动物实验研究发现,宫内营养不良会引起胎鼠神经组织中甲基CpG结合蛋白2 (methylCpG binding protein 2, MeCP2)和溶质载体家族2成员基因的超甲基化,使其表达显著下调,从而影响大脑的学习和记忆功能,导致胎儿出生后易患精神分裂症。母鼠孕期使用丙戊酸钠可以诱导胎鼠脑中Wnt/β-catenin通路的启动子区域去甲基化,导致其目标基因表达增加造成后代自闭症易感[64]。孕期低蛋白饮食会造成胰岛素样生长因子2基因位点超甲基化,使其表达水平下降,同时还伴有肝内DNMT1、DNMT3a及MeCP2表达的增加,从而导致子代的内分泌及代谢功能发育不全[65]。在孕期营养不良羊模型中发现,DNMT活性下降可致胎儿下丘脑皮质激素和糖皮质激素受体基因启动子区域甲基化减少,从而导致长期能量代谢平衡紊乱,进而出现代谢性疾病易感[66]。宫内发育迟缓(IUGR)患儿的HNF4A高甲基化[67],可导致青春期糖尿病的发生[68]。孕期低蛋白饮食可通过诱导胎儿AT1bR基因启动子低甲基化增加肾上腺AT1bR表达,后者介导血管紧张素Ⅱ受体调节血压和血容量,从而导致高血压的发生[69]。宫内雄激素过暴露可能干扰胎儿生殖细胞基因重编程,从而导致子代多囊卵巢综合征易感[70]。因此,胎儿时期DNA甲基化修饰的改变对于成年后疾病的发生提供了重要参考价值。
孕期母体应激、营养供应不足等多种因素导致的胚胎发育不良环境可通过组蛋白修饰调控相关基因的表达,从而提高Ⅱ型糖尿病、肥胖、内分泌相关等代谢疾病的发生率。研究发现组蛋白修饰可调节胰岛β细胞功能,其异常改变可增加Ⅱ型糖尿病的风险[71]。Pdx1是体内β细胞生长关键调节剂,正常条件下,Pdx1基因的近端启动子存在由H3和H4乙酰化标记的未甲基化的染色质以及H3K4me3,然而检测IUGR胎儿胰岛显示Pdx1近端启动子的H3/ H4的乙酰化显著减少,而H3K9me3甲基化显着增加,导致Pdx1基因转录沉默而出现表达下调[72,73]。Pdx1表达降低可以改变β细胞功能,最终导致胰岛素抵抗发生,造成Ⅱ型糖尿病易感[73,74]。Painter等[75]发现,大鼠孕期蛋白限制可致IUGR胎鼠肝胆固醇7α-羟化酶表达降低,伴随着启动子区组蛋白乙酰化修饰下降和H3K9、H3K4甲基化修饰升高,进一步可能导致非酒精性脂肪肝易感[76]。同时,Begum[66]发现孕期营养不良可导致成年后代下丘脑神经元糖皮质激素受体启动子H3K27甲基化降低,以及H3K9乙酰化增加,推测孕妇营养不良可能导致成年后代下丘脑神经元调节能量平衡特定基因的表观遗传修饰变化,从而引起能量代谢性疾病易感。其他研究[77]也报道了母体营养限制致IUGR大鼠肺血管内皮细胞内皮素1基因启动子中组蛋白乙酰化水平升高,此改变可能导致IUGR大鼠对血管压力高度敏感,引起生命晚期缺氧,导致肺动脉高压或肺血管重塑。
以上提示宫内时期胎儿组蛋白修饰改变对成年期相关代谢疾病的发生及进展有显著影响。Chakraborty等[78]研究还发现,miR-143、miR-103、miR-29a、miR-27b等在胎儿生命早期的表达变化与子代成年后T2DM、肥胖症、高脂血症及胰岛素抵抗等的发生密切相关。
3 表观遗传生物标志物在临床疾病早期诊治中的应用
目前,在临床上逐步运用表观遗传分析技术,在患者的血浆或血清标本中检测异常表观遗传修饰,并在疾病早期发现并做出诊断,从而为疾病预防及治疗提供依据。3.1 肿瘤的早期诊治
DNA甲基化改变可作为肿瘤标志物,当癌前组织中异常甲基化程度超过正常组织水平时,通过甲基化特异性PCR方法检测出来。最近Sandoval等[32,79,80]提出表观遗传生物标志物可用于肺癌、结直肠癌和前列腺癌的早期检测。研究认为APC和RASSF1A基因启动子区高甲基化是早期检测乳腺癌的常见表观遗传生物标志物[81];血液的ALX4基因启动子甲基化和血浆中APC、MGMT、RASSF2A和Wif-1基因的异常甲基化可作为早期检测结直肠癌的潜在生物标志物[82,83];早期肺癌患者的血浆、唾液和肿瘤组织中P16/INK42基因启动子甲基化出现异常改变[84]。另外组蛋白乙酰化修饰改变可用于肿瘤治疗后的复发诊断,如低水平的H3K18ac、H3K9Me提示肺癌和肾癌预后较差[85]。表观遗传疗法是基于表观遗传生物标志物治疗癌症的新方法[86]。表观遗传修饰酶抑制剂具有增强宿主抗肿瘤免疫应答和增强免疫治疗剂的功效。目前,几种表观遗传调控药物已得到较好的研究,包括DNA甲基转移酶抑制剂(DNA methyltransferasesinhibitors, DNMTi)和组蛋白去乙酰化酶抑制剂(Histone deacetylase inhibitors, HDACi)等[86]。已发现DNMT抑制剂(阿扎胞苷和地西他滨)可诱导癌细胞上MHC基因[87]和黑素瘤相关抗原1的表达,诱导特异性T细胞杀死癌细胞。对鼠卵巢癌细胞采用去甲基化药物去氧-5-氮胞苷诱导MLH1基因的表达,减少hMLH1基因启动子的甲基化,使肿瘤对顺铂、卡铂等药物敏感[88]。新型HDACi(YCW1)和辐射的组合在体外和原位小鼠模型中下调乳腺癌细胞中的BNIP3基因诱导自噬细胞死亡[89],达到治疗癌症的效果。
3.2 老年性疾病的早期诊治
目前,对于AD和HD,血液中是否存在与疾病相关的表观遗传改变尚不清楚。然而,AD患者血液转录组中的某些变化可反映大脑相关疾病改变。已确定外周单核细胞中染色质状态和miRNA为脑相关病症的潜在诊断标志物[14]。最近研究报道,在AD患者脑和血液中年龄相关的DNA胞嘧啶甲基化下调表达一致,提示血液可能是脑中相关变化的替代物,至少对于一些基因组位点血液中的表观遗传标志物改变可推测大脑中表观遗传学的变化[90]。AD患者血液单核细胞中miRNA研究发现miR-34a和miR-181b上调。尽管仍然需要阐明这些miRNA是否在AD病理学中起重要作用,但有可能作为有价值的预后生物标志物,特别是因为其可以在血液中相对容易检测。在AD发生早期,非症状阶段鉴定miRNA表达改变可能增强AD诊断率。基于表观修饰的可逆性改变,为治疗疾病提供了良好的靶点。旨在确定表观遗传相关药物,以治疗老年性疾病。老年性疾病可能涉及SAM代谢失调,导致全局DNA低甲基化,以及某些关键基因的高甲基化。因此,已开展了旨在增加或减少DNA甲基化的策略研究,可通过SAM作用促进DNA甲基化,叶酸和维生素B12和B6在维持SAM水平发挥重要作用,补充这些维生素以抵抗认知衰退和痴呆发作[91]。另有研究发现多巴胺耗尽导致的疾病与组蛋白H3K4me3的减少有关,而慢性左旋多巴治疗导致组蛋白H4K5、K8、K12和K16的去乙酰化。广泛用作帕金森病的MPTP(1-甲基-4-苯基-1,2,3,6-四氢-四氢吡啶)可诱导H3乙酰化,治疗左旋多巴的减少[92]。事实上,大量的临床前工作认为HDACi同样具有治疗神经系统疾病的潜力[93]。DNMT和HDACi已进行大量动物实验和临床试验,但对于其具体作用机制仍需进一步研究。
3.3 发育源性疾病的早期诊治
基于母体内的胎儿与肿瘤具有相似的生物学特征,Poon等[94, 95]利用表遗传标记物来特异性检测母血浆中的胎儿DNA,标志着胎儿DNA在无创性产前诊断中的进一步应用,有些病症如21三体综合征与DNA甲基化、基因表达之间的相关性越来越明确[96]。最近一项研究表明,脐带血的INS样生长因子-2异常DNA甲基化与超重或肥胖相关[97]。Godfrey等报道,在脐带组织中RXRA核受体的启动子区的单CpG位点的甲基化状态改变与儿童期肥胖密切相关[15]。在脐带组织视黄醇X受体特定的CpG甲基化和新生儿健康内皮型一氧化氮合酶与儿童脂肪量有关。有报道,在临床IUGR胎儿脐血干细胞中,肝细胞核因子4α基因启动子区发生DNA甲基化修饰改变,该基因是导致幼年速发型糖尿病的重要因素[67]。孕早期暴露于不良环境导致PRKCB、PC、NCOR2和Smad3基因DNA甲基化和转录失调,这些Notch信号通路与脂肪合成基因可能成为肝脏相关的慢性病出生后易感的早期诊断标志物[98]。DNA甲基化差异是跨组织高度一致[99,100],如果早期生活环境诱导基因表观遗传调控改变,那么这些改变的表观遗标志物可以在相应的组织检测出。如hsa-miR-99a参与心脏畸形,并且可以在胎儿发育期间显著上调,可作为监测心肌发育和作为胎儿冠心病的非侵袭性生物标志物[101]。近期研究探索表观遗传改变,以及异常表观遗传在子宫内或早期发育过程中的可逆性。Weaver等[102]发现了大鼠的母性grooming行为会导致新生儿行为改变,由于糖皮质激素基因启动子甲基化状态改变,用影响甲基化的药物治疗可以逆转这种表型。哺乳期为母鼠提供足量的叶酸饮食,可逆转孕期低蛋白质饮食所造成的胎肝GR及PPAR基因启动子区DNA甲基化修饰改变[103]。Lillycrop等[104]表明,在母体蛋白质限制大鼠模型,补充甘氨酸或叶酸的限制饮食可防止胎肝GR和PPARα低甲基化。母体暴露于营养素限制的新生大鼠,产后瘦素治疗后逆转启动子甲基化和肝脏增加PPARα和GR表达[105]。已发现组蛋白乙酰化酶HATs和组蛋白去乙酰化酶HDACs在组蛋白乙酰化作用以及染色质结构维持和调控基
因转录中起关键性的作用[106],组蛋白脱乙酰酶抑制已被证明在染色质修饰因子治疗心血管疾病(Cardiac Vascular Disease, CVD)和其他疾病上具有潜在希 望[107]。这些从天然产物中发现的表观遗传抑制剂相关酶可能是发育性疾病治疗中表观遗传调节的潜在靶标[108, 109]。
总之,表观遗传学进展拓宽了通过母体血浆中存在的胎儿DNA诊断疾病的应用领域,随着表观遗传学分析技术的不断发展,此种利用胎儿表观遗传学标记物的产前诊断技术将会得到更多的推广,应用于临床工作中。
4 结语与展望
虽然表观遗传修饰的在临床诊治上的应用目前尚较局限,但是肿瘤基因甲基化治疗的有效性已在许多体外及动物实验中初步得到证实,部分临床试验也取得了较满意的结果。随着人类生活压力和环境污染的不断增加,胎儿面临的宫内不良环境因素也日益增多。这些不良的宫内环境因素通过表观遗传学修饰影响胚胎编程效应,进一步导致其成年后罹患某些慢性疾病的风险大大增加。在疾病发生的早期,通过检测血液或者组织标志基因表观遗传修饰的改变,用于疾病早期诊断。早期确定高危人群能接受的营养或生活方式的干预,提供更有效的预防性治疗策略,这样既能提高生活质量并减少与目前的治疗策略相关的经济负担。有理由相信,随着表观遗传学研究的不断发展,表观遗传生物标志物研究可能为疾病的早期诊断和治疗提供指导,必将成为人类疾病治疗的一条全新的途径。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:28728135 [本文引用: 2]

Abstract BACKGROUND: Previous studies have uncovered heightened prostatic susceptibility to hormone-induced neoplasia from early-life exposure to low-dose bisphenol A (BPA). However, significant data gaps remain that are essential to address for biological relevance and necessary risk assessment. OBJECTIVES: A complete BPA dose-response analysis of prostate lesions across multiple prostatic lobes was conducted that included internal BPA dosimetry, progression to adenocarcinoma with aging and mechanistic connections to epigenetically reprogramed genes. METHODS: Male neonatal Sprague-Dawley rats were briefly exposed to 0.1 to 5,000 0204g090009BPA/kg090009BW on postnatal days (PND) 1, 3, and 5. Individual prostate lobes plus periurethral prostatic ducts were evaluated at 7 mo or 1 y of age without or with adult testosterone plus estradiol (T+E) to promote carcinogenesis. DNA methylation of five genes was quantified by bisulfite genomic sequencing in d-200 dorsal prostates across BPA doses. Serum free-BPA and BPA-glucuronide were quantitated in sera of individual PND 3 pups collected 1 hr postexposure utilizing ultra-high-pressure tandem mass spectrometry (UHPLC-MS-MS). RESULTS: The lowest BPA dose initiated maximal hormonal carcinogenesis in lateral prostates despite undetectable free BPA 1 hr postexposure. Further, prostatic intraepithelial neoplasia (PIN) progressed to carcinoma in rats given neonatal low-dose BPA with adult T+E but not in rats given adult T+E alone. The dorsal and ventral lobes and periurethral prostatic ducts exhibited a nonmonotonic dose response with peak PIN, proliferation and apoptotic values at 10090009100 0204g/kg BW. This was paralleled by nonmonotonic and dose-specific DNA hypomethylation of genes that confer carcinogenic risk, with greatest hypomethylation at the lowest BPA doses. CONCLUSIONS: Developmental BPA exposures heighten prostate cancer susceptibility in a complex dose- and lobe-specific manner. Importantly, elevated carcinogenic risk is found at doses that yield undetectable serum free BPA. Dose-specific epigenetic modifications of selected genes provide a mechanistic framework that may connect early-life BPA to later-life predisposition to prostate carcinogenesis. https://doi.org/10.1289/EHP1050.
URLPMID:5025578 [本文引用: 2]
Prostate cancer is one of the most common non-cutaneous malignancies among men worldwide. Epigenetic aberrations, including changes in DNA methylation patterns and/or histone modifications, are key drivers of prostate carcinogenesis. These epigenetic defects might be due to deregulated function and/or expression of the epigenetic machinery, affecting the expression of several important genes. Remarkably, epigenetic modifications are reversible and numerous compounds that target the epigenetic enzymes and regulatory proteins were reported to be effective in cancer growth control. In fact, some of these drugs are already being tested in clinical trials. This review discusses the most important epigenetic alterations in prostate cancer, highlighting the role of epigenetic modulating compounds in pre-clinical and clinical trials as potential therapeutic agents for prostate cancer management.
URLPMID:28871451 [本文引用: 2]
Abstract DNA methylation of leukocyte DNA has been proposed to be a biomarker for cancer that can be used to target patients for appropriate clinical implementation. We investigated IGF2 DMR and LINE1 methylation in the leukocyte DNA and their association with clinicopathological features and prognosis of gastric cancer (GC) patients. Methylation status of IGF2 DMR and LINE1 in the leukocyte DNA was quantified using bisulfite pyrosequencing in 207 GC patients. Methylation of both IGF2 DMR and the LINE1 was significantly higher in the undifferentiated histologic type compared to the differentiated histologic type (both P02=020.0002). Hypermethylation of both the IGF2 DMR and the LINE1 was associated with more aggressive features of GC such as advanced stage (IGF2 DMR, P02=020.0002; LINE1, P02<020.0001), lymphatic invasion positive (IGF2 DMR, P02=020.004; LINE1, P02=020.002), venous invasion positive (IGF2 DMR, LINE1, both P02=020.03), lymph node metastasis positive (IGF2 DMR, P02=020.01; LINE1, P02=020.001), peritoneal dissemination positive (IGF2 DMR, P02=020.04; LINE1, P02=020.002), liver metastasis positive (IGF2 DMR, P02=020.008; LINE1, P02=020.001), and other distant metastasis positive (IGF2 DMR, P02=020.04). Our data suggest that high LINE1 and IGF2 DMR methylation status would be a phenomenon that is observed with the progression of GC, supporting their potential utility as a biomarker in GC patients.
URL [本文引用: 2]
Several polymorphic gene variants within one-carbon metabolism, an essential pathway for nucleotide synthesis and methylation reactions, are related to cancer risk. An aberrant DNA methylation is a common feature in cancer but whether the link between one-carbon metabolism variants and cancer occurs through an altered DNA methylation is yet unclear. Aims of the study were to evaluate the frequency of one-carbon metabolism gene variants in hepatocellular-carcinoma, cholangiocarcinoma and colon cancer, and their relationship to cancer risk together with global DNA methylation status. Genotyping for BHMT 716A>G, DHFR 19bp ins/del, MTHFD1 1958G>A, MTHFR 677C>T, MTR 2756A>G, MTRR 66A>G, RFC1 80G>A, SHMT1 1420C>T, TCII 776C>G and TS 2rpt-3rpt was performed in 102 cancer patients and 363 cancer-free subjects. Methylcytosine (mCyt) content was measured by LC/MS/MS in peripheral blood mononuclear cells (PBMCs) DNA. The MTHFD1 1958AA genotype was significantly less frequent among cancer patients as compared to controls (p = 0.007) and related to 63% reduction of overall cancer risk (p = 0.003) and 75% of colon cancer risk (p = 0.006). When considering PBMCs mCyt content, carriers of the MTHFD1 1958GG genotype showed a lower DNA methylation as compared to carriers of the A allele (p = 0.048). No differences were highlighted by evaluating a possible relationship between the other polymorphisms analyzed with cancer risk and DNA methylation. The MTHFD1 1958AA genotype is linked to a significantly reduced cancer risk. The 1958GG genotype is associated to PBMCs DNA hypomethylation as compared to the A allele carriership that may exert a protective effect for cancer risk by preserving from DNA hypomethylation.
URLPMID:24101602 [本文引用: 2]
The hallmark of Alzheimer's disease (AD) pathology is an accumulation of amyloid β (Aβ) and phosphorylated tau, which are encoded by the amyloid precursor protein (APP) and microtubule-associated protein tau (MAPT) genes, respectively. Less than 5% of all AD cases are familial in nature, i.e. caused by mutations in APP, PSEN1 or PSEN2. Almost all mutations found in them are related to an overproduction of Aβ1-42, which is prone to aggregation. While these genes are mutation free, their function, or those of related genes, could be compromised in sporadic AD as well. In this study, pyrosequencing analysis of post-mortem brains revealed aberrant CpG methylation in APP, MAPT and GSK3B genes of the AD brain. These changes were further evaluated by a newly developed in vitro-specific DNA methylation system, which in turn highlighted an enhanced expression of APP and MAPT. Cell nucleus sorting of post-mortem brains revealed that the methylation changes of APP and MAPT occurred in both neuronal and non-neuronal cells, whereas GSK3B was abnormally methylated in non-neuronal cells. Further analysis revealed an association between abnormal APP CpG methylation and apolipoprotein E ε4 allele (APOE ε4)-negative cases. The presence of a small number of highly methylated neurons among normal neurons contribute to the methylation difference in APP and MAPT CpGs, thus abnormally methylated cells could compromise the neural circuit and/or serve as 'seed cells' for abnormal protein propagation. Our results provide a link between familial AD genes and sporadic neuropathology, thus emphasizing an epigenetic pathomechanism for sporadic AD.
URLPMID:18319327 [本文引用: 2]
Abstract Chromatin remodeling is tightly controlled under physiological conditions. Alterations in chromatin structure are involved in the pathogenesis of neuronal systems. We found that the monoallelic deletion of CREB binding protein (CBP) results in the induction of ERG-associated protein with SET domain (ESET) and increases trimethylation of histone H3 (K9) and condensation of pericentromeric heterochromatin structure in neurons. Nested deletion and mutational analysis of the ESET promoter further demonstrated that the Ets-2 transcription factor regulates transcriptional activity of the ESET gene. In CBP+/- mice, Ets-2 occupancy in the ESET promoter DNA was markedly elevated. Our results suggest that CBP is a transcriptional repressor of ESET gene expression by limiting Ets-2 transcriptional activity, while CBP siRNA enhances basal and Ets-2-dependent ESET transcriptional activity. Altered expression of the ESET gene and hypertrimethylation of H3 (K9) correlate with striatal neuron atrophy and dysfunction in CBP+/- mice. These results establish an alternative pathway that loss of CBP leads to the pericentric heterochromatin condensation through ESET expression and trimethylation of H3 (K9).
URLPMID:24064364 [本文引用: 1]
Epidemiological data indicate that an adverse maternal environment during pregnancy predisposes offspring to metabolic syndrome with increased obesity, and type 2 diabetes. The mechanisms are still unclear although epigenetic modifications are implicated and the hypothalamus is a likely target. We hypothesized that maternal undernutrition (UN) around conception in sheep would lead to epigenetic changes in hypothalamic neurons regulating energy balance in the offspring, up to 5 years after the maternal insult. We found striking evidence of decreased glucocorticoid receptor (GR) promoter methylation, decreased histone lysine 27 trimethylation, and increased histone H3 lysine 9 acetylation in hypothalami from male and female adult offspring of UN mothers. These findings are entirely compatible with the increased GR mRNA and protein observed in the hypothalami. The increased GR predicted the decreased hypothalamic proopiomelanocortin expression and increased obesity that we observed in the 5-year-old adult males. The epigenetic and expression changes in GR were specific to the hypothalamus. Hippocampal GR mRNA and protein were decreased in UN offspring, whereas pituitary GR was altered in a sex-specific manner. In peripheral polymorphonuclear leukocytes there were no changes in GR methylation or protein, indicating that this epigenetic analysis did not predict changes in the brain. Overall, these results suggest that moderate changes in maternal nutrition, around the time of conception, signal life-long and tissue-specific epigenetic alterations in a key gene regulating energy balance in the hypothalamus.
URL [本文引用: 1]
Intrauterine growth restriction (IUGR) increases the risk of serious adult morbidities such as hypertension. In an IUGR rat model of hypertension, we reported a persistent decrease in kidney 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2) mRNA and protein levels from birth through postnatal (P) day 21. This enzyme deficiency can lead to hypertension by limiting renal glucocorticoid deactivation. In the present study, we hypothesized that IUGR affects renal 11beta-HSD2 epigenetic determinants of chromatin structure and alters key transcription factor binding to the 11beta-HSD2 promoter in association with persistent downregulation of its mRNA expression. To test this hypothesis, we performed bilateral uterine artery ligation on embryonic day 19.5 pregnant rats and harvested kidneys at day 0 (P0) and P21. Key transcription factors that can affect 11beta-HSD2 expression include transcriptional enhancers specificity protein 1 (SP1) and NF-kappaB p65 and transcriptional repressors early growth response factor (Egr-1) and NF-kappaB p50. Our most important findings were as follows: 1) IUGR significantly decreased SP1 and NF-kappaB (p65) binding to the 11beta-HSD2 promoter in males, while it increased Egr-1 binding in females and NF-kappaB (p50) binding in males; 2) IUGR increased CpG methylation status, as well as modified the pattern of methylation in several CpG sites of 11beta-HSD2 promoter at P0 also in a sex-specific manner; and 3) IUGR decreased trimethylation of H3K36 in exon 5 of 11beta-HSD2 at P0 and P21 in both genders. We conclude that IUGR is associated with altered transcriptional repressor/activator binding in connection with increased methylation in the 11beta-HSD2 promoter region in a sex-specific manner, possibly leading to decreased transcriptional activity. Furthermore, IUGR decreased trimethylation of H3K36 of the 11beta-HSD2 gene in both genders, which is associated with decreased transcriptional elongation. We speculate that alterations in transcription factor binding and chromatin structure play a role in in utero reprogramming.
URLPMID:17272666 [本文引用: 1]
Abstract Clinical and animal studies indicate that intrauterine growth restriction (IUGR) following uteroplacental insufficiency (UPI) reduces nephron number and predisposes toward renal insufficiency early in life and increased risk of adult-onset hypertension. In this study, we hypothesized that the inducible enzyme cyclooxygenase-2 (COX-2), a pivotal protein in nephrogenesis, constitutes a mechanism through which UPI and subsequent glucocorticoid overexposure can decrease nephron number. We further hypothesized that UPI downregulates the key enzyme 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2), which converts corticosterone to inert 11-dehydrocorticosterone, thereby protecting both the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) from the actions of corticosterone. Following bilateral uterine ligation on the pregnant rat, UPI significantly decreased renal COX-2, 11beta-HSD2, and GR mRNA and protein levels, but upregulated expression of MR at birth. At day 21 of life, 11beta-HSD2, GR, and also MR mRNA and protein levels were downregulated. UPI did not affect blood pressures (BP) at day 21 of life but significantly increased systolic BP in both genders at day 140. We conclude that in our animal model, UPI decreases fetal COX-2 expression during a period of active nephrogenesis in the IUGR rat, which is also characterized by decreased nephron number and adult-onset hypertension.
URLPMID:22434078 [本文引用: 1]
Abstract The links between obesity in parents and their offspring and the role of genes and a shared environment are not completely understood. Adipocytokines such as leptin and adiponectin play important roles in glucose and lipid metabolism. Therefore, we examined whether the offspring from dams exposed to a high-fat diet during pregnancy (OH mice) exhibited hypertension, insulin resistance, and hyperlipidemia along with epigenetic changes in the expression of adipocytokine genes. OH mice were significantly heavier than the offspring of dams exposed to a control diet during pregnancy (OC mice) from 14 wk of age after an increased caloric intake from 8 wk. OH mice exhibited higher blood pressure and worse glucose tolerance than the OC mice at 24 wk. Total triglyceride and leptin levels were significantly higher and the adiponectin level was significantly lower in OH compared with OC mice at 12 wk of age. This was associated with changes in leptin and adiponectin expression in white adipose tissue. There were lower acetylation and higher methylation levels of histone H3 at lysine 9 of the promoter of adiponectin in adipose tissues of OH mice at 2 wk of age as well as at 12 and 24 wk of age compared with OC mice. In contrast, methylation of histone 4 at lysine 20 in the leptin promoter was significantly higher in OH compared with OC mice. Thus, exposure to a high-fat diet in utero might cause a metabolic syndrome-like phenomenon through epigenetic modifications of adipocytokine, adiponectin, and leptin gene expression.
URLPMID:23768418 [本文引用: 1]
Ecological evidence suggests that niacin (nicotinamide and nicotinic acid) fortification may be involved in the increased prevalence of obesity and type 2 diabetes, both of which are associated with insulin resistance and epigenetic changes. The purpose of the present study was to investigate nicotinamide-induced metabolic changes and their relationship with possible epigenetic changes. Male rats (5 weeks old) were fed with a basal diet (control group) or diets supplemented with 1 or 4g/kg of nicotinamide for 8 weeks. Low-dose nicotinamide exposure increased weight gain, but high-dose one did not. The nicotinamide-treated rats had higher hepatic and renal levels of 8-hydroxy-2'-deoxyguanosine, a marker of DNA damage, and impaired glucose tolerance and insulin sensitivity when compared with the control rats. Nicotinamide supplementation increased the plasma levels of nicotinamide, N 1-methylnicotinamide and choline and decreased the levels of betaine, which is associated with a decrease in global hepatic DNA methylation and uracil content in DNA. Nicotinamide had gene-specific effects on the methylation of CpG sites within the promoters and the expression of hepatic genes tested that are responsible for methyl transfer reactions (nicotinamide N-methyltransferase and DNA methyltransferase 1), for homocysteine metabolism (betaine-homocysteine S-methyltransferase, methionine synthase and cystathionine $\beta$-synthase) and for oxidative defence (catalase and tumour protein p53). It is concluded that nicotinamide-induced oxidative tissue injury, insulin resistance and disturbed methyl metabolism can lead to epigenetic changes. The present study suggests that long-term high nicotinamide intake (e.g. induced by niacin fortification) may be a risk factor for methylation- and insulin resistance-related metabolic abnormalities.
URLPMID:23628807 [本文引用: 1]
Abstract Urinary bladder cancer is the fifth most common cancer in the Western world. Increasing evidence has shown that DNA methylation in bladder cancer is expansive and is implicated in pathogenesis. Furthermore, distinct methylation patterns have been identified between non-muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC), as well as between FGFR3-mutant and wild-type tumours. Given these distinctions in expression, methylated genes have been proposed as diagnostic and prognostic biomarkers for patients with bladder cancer. Indeed, several studies have revealed that methylated genes--including CDH1, FHIT, LAMC2, RASSF1A, TIMP3, SFRP1, SOX9, PMF1 and RUNX3--are associated with poor survival in patients with MIBC. Further validation of these markers for prognostication as well as surveillance (of patients with NMIBC) is required. Validated markers for progression, diagnosis, survival and BCG response will contribute to clinical decision-making and individualized treatment.
URLPMID:3891681 [本文引用: 1]
Bladder cancer is the fourth most common cancer in men in the United States, and its recurrence rate is highest among all malignancies. The unmet need for improved strategies for early detection, treatment, and monitoring of the progression of this disease continues to translate into high mortality and morbidity. The quest for advanced diagnostic, therapeutic, and prognostic approaches for bladder cancer is a high priority, which can be achieved by understanding the molecular mechanisms of the initiation and progression of this malignancy. Aberrant DNA methylation in single or multiple cancer-related genes/loci has been found in human bladder tumors and cancer cell lines, and urine sediments, and correlated with many clinicopathological features of this disease, including tumor relapse, muscle-invasiveness, and survival. The present review summarizes the published research on aberrant DNA methylation in connection with human bladder cancer. Representative studies are highlighted to set forth the current state of knowledge, gaps in the knowledgebase, and future directions in this prime epigenetic field of research. Identifying the potentially reversible and drugable' aberrant DNA methylation events that initiate and promote bladder cancer development can highlight biological markers for early diagnosis, effective therapy and accurate prognosis of this malignancy.
URLPMID:3560446 [本文引用: 2]
Abstract This study was designed to identify clinically accessible molecular biomarkers of mild traumatic brain injury (mTBI) that could be used to help identify returning Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) Veterans who are suffering from the effects of mTBI. While analyzing the expression profile of small non-coding RNAs in peripheral blood mononuclear cells (PBMCs) from an OEF/OIF veteran study cohort using a high throughput array chip platform, we identified 18 candidate small non-coding RNA biomarkers that are differentially regulated in PBMCs of mTBI compared to non-TBI control cases. Independent quantitative real-time polymerase chain reaction assays confirmed that 13 of these candidate small RNA biomarker species are, indeed, significantly down-regulated in PBMCs of mTBI compared to non-TBI control veteran cases. Based on unsupervised clustering analysis, we identified a 3-biomarker panel which was most able to distinguish mTBI from non-TBI control veteran cases with high accuracy, selectivity and specificity. The majority of mTBI cases in our biomarker study were co-morbid with Post-Traumatic Stress Disorder (PTSD), and thus our non-TBI control cases were selected to match PTSD diagnoses. Therefore, our identified panel of 3 small RNA biomarkers likely represents a biological index selective for mTBI. Outcomes from our studies suggest that additional applications of the clinically accessible small non-coding RNA biomarkers to current diagnostic criteria may lead to improved mTBI detection and more sensitive outcome measures for clinical trials. Future studies exploring the physiological relevance of mTBI biomarkers will also provide a better understanding of the biological mechanisms underlying mTBI and insights into novel therapeutic targets for mTBI.
URLPMID:21471513 [本文引用: 2]
Fixed genomic variation explains only a small proportion of the risk of adiposity. In animal models, maternal diet alters offspring body composition, accompanied by epigenetic changes in metabolic control genes. Little is known about whether such processes operate in humans. Using Sequenom MassARRAY we measured the methylation status of 68 CpGs 5' from five candidate genes in umbilical cord tissue DNA from healthy neonates. Methylation varied greatly at particular CpGs: for 31 CpGs with median methylation ≥5% and a 5-95% range ≥10%, we related methylation status to maternal pregnancy diet and to child's adiposity at age 9 years. Replication was sought in a second independent cohort. In cohort 1, retinoid X receptor-α (RXRA) chr9:136355885+ and endothelial nitric oxide synthase (eNOS) chr7:150315553+ methylation had independent associations with sex-adjusted childhood fat mass (exponentiated regression coefficient [β] 17% per SD change in methylation [95% CI 4-31], P = 0.009, n = 64, and β = 20% [9-32], P < 0.001, n = 66, respectively) and %fat mass (β = 10% [1-19], P = 0.023, n = 64 and β =12% [4-20], P = 0.002, n = 66, respectively). Regression analyses including sex and neonatal epigenetic marks explained >25% of the variance in childhood adiposity. Higher methylation of RXRA chr9:136355885+, but not of eNOS chr7:150315553+, was associated with lower maternal carbohydrate intake in early pregnancy, previously linked with higher neonatal adiposity in this population. In cohort 2, cord eNOS chr7:150315553+ methylation showed no association with adiposity, but RXRA chr9:136355885+ methylation showed similar associations with fat mass and %fat mass (β = 6% [2-10] and β = 4% [1-7], respectively, both P = 0.002, n = 239). Our findings suggest a substantial component of metabolic disease risk has a prenatal developmental basis. Perinatal epigenetic analysis may have utility in identifying individual vulnerability to later obesity and metabolic disease.
In: Karpf A.
URL [本文引用: 1]
[本文引用: 1]
URLPMID:5062992 [本文引用: 1]
In mammals, DNA methylation is introduced by the DNMT1, DNMT3A and DNMT3B methyltransferases, which are all large multi-domain proteins containing a catalytic C-terminal domain and an N-terminal part with regulatory functions. Recently, two novel regulatory principles of DNMTs were uncovered. It was shown that their catalytic activity is under allosteric control of N-terminal domains with autoinhibitory function, the RFT and CXXC domains in DNMT1 and the ADD domain in DNMT3. Moreover, targeting and activity of DNMTs were found to be regulated in a concerted manner by interactors and posttranslational modifications (PTMs). In this review, we describe the structures and domain composition of the DNMT1 and DNMT3 enzymes, their DNA binding, catalytic mechanism, multimerization and the processes controlling their stability in cells with a focus on their regulation and chromatin targeting by PTMs, interactors and chromatin modifications. We propose that the allosteric regulation of DNMTs by autoinhibitory domains acts as a general switch for the modulation of the function of DNMTs, providing numerous possibilities for interacting proteins, nucleic acids or PTMs to regulate DNMT activity and targeting. The combined regulation of DNMT targeting and catalytic activity contributes to the precise spatiotemporal control of DNMT function and genome methylation in cells.
URLPMID:17708532 [本文引用: 1]
DNA methylation is an epigenetic modification which plays an important role in chromatin organization and gene expression. DNA methylation can silence genes and repetitive elements through a process which leads to the alteration of chromatin structure. The mechanisms which target DNA methylation to specific sites in the genome are not fully understood. In this review, we will discuss the mechanisms which lead to the long-term silencing of genes and will survey the progression that has been made in determining the targeted mechanisms for de novo DNA methylation.
[本文引用: 1]
URL [本文引用: 1]
Among the epigenetic changes, histone acetylation has been recognized as a fundamental process that strongly affects gene expression regulation. Disrupt of this phenomenon has been linked to carcinogenesis. In this review, we analysed studies reporting the process of histone modification, the enzymes associated and affected genes concerning human malignancies and histone enzyme inhibitor drugs used in cancer treatment. Variable degrees of expression of HDACs (histone deacetylases) and HATs (histone acetyltransferases) are found in many human malignant tissues and the histones acetylation seems to influence different processes including the progression of cell cycle, the dynamics of chromosomes, DNA recombination, DNA repair and apoptosis. Thus, the control of aberrant activity and/or expression of these proteins have been favorable in treatment of diseases as cancer. HDACi have shown efficacy in clinical trials in solid and hematological malignancies. Therefore, the development and use of HDACs inhibitors are increasing, leading to continue studying these enzyme expressions and behavior, aiming to determine tumors that will respond better to this type of treatment.
URLPMID:22473383 [本文引用: 1]
Organisms require an appropriate balance of stability and reversibility in gene expression programmes to maintain cell identity or to enable responses to stimuli; epigenetic regulation is integral to this dynamic control. Post-translational modification of histones by methylation is an important and widespread type of chromatin modification that is known to influence biological processes in the context of development and cellular responses. To evaluate how histone methylation contributes to stable or reversible control, we provide a broad overview of how histone methylation is regulated and leads to biological outcomes. The importance of appropriately maintaining or reprogramming histone methylation is illustrated by its links to disease and ageing and possibly to transmission of traits across generations.
URLPMID:22923342 [本文引用: 1]
Atrial tissue fibrosis is often an important component of the atrial fibrillation (AF) substrate. Small noncoding microRNAs are important mediators in many cardiac remodeling paradigms. MicroRNA-21 (miR-21) has been suggested to be important in ventricular fibrotic remodeling by downregulating Sprouty-1, a protein that suppresses fibroblast proliferation. The present study examined the potential role of miR-21 in the atrial AF substrate resulting from experimental heart failure after myocardial infarction (MI).Large MIs (based on echocardiographic left ventricular wall motion score index) were created by left anterior descending coronary artery ligation in rats. Changes induced by MI versus sham controls were first characterized with echocardiography, histology, biochemistry, and in vivo electrophysiology. Additional MI rats were then randomized to receive anti-miR-21 (KD21) or scrambled control sequence (Scr21) injections into the left atrial myocardium. Progressive left ventricular enlargement, hypocontractility, left atrial dilation, fibrosis, refractoriness prolongation, and AF promotion occurred in MI rats versus sham controls. Atrial tissues of MI rats showed upregulation of miR-21, along with dysregulation of the target genes Sprouty-1, collagen-1, and collagen-3. KD21 treatment reduced atrial miR-21 expression levels in MI rats to values in sham rats, decreased AF duration from 417 (69-1595; median [Q1-Q3]) seconds to 3 (2-16) seconds (8 weeks after MI; P<0.05), and reduced atrial fibrous tissue content from 14.4 ± 1.8% (mean ± SEM) to 4.9 ± 1.2% (8 weeks after MI; P<0.05) versus Scr21 controls.MI-induced heart failure leads to AF-promoting atrial remodeling in rats. Atrial miR-21 knockdown suppresses atrial fibrosis and AF promotion, implicating miR-21 as an important signaling molecule for the AF substrate and pointing to miR-21 as a potential target for molecular interventions designed to prevent AF.
URLPMID:14744438 [本文引用: 1]
MicroRNAs (miRNAs) are endogenous 6522 nt RNAs that can play important regulatory roles in animals and plants by targeting mRNAs for cleavage or translational repression. Although they escaped notice until relatively recently, miRNAs comprise one of the more abundant classes of gene regulatory molecules in multicellular organisms and likely influence the output of many protein-coding genes.
URLPMID:22836355 [本文引用: 1]
While microRNAs have been shown to copurify with polysomes, their relative fraction in the translation pool (polysome occupancy) has not yet been measured. Here, we introduce a high-throughput method for quantifying polysome occupancies of hundreds of microRNAs and use it to investigate factors affecting these occupancies. Analysis in human embryonic stem cells (hESCs) and foreskin fibroblasts (hFFs) revealed microRNA-specific preferences for low, medium, or high polysome occupancy. Bioinformatics and functional analysis based on overexpression of endogenous and chimeric microRNAs showed that the polysome occupancy of microRNAs is specified by its mature sequence and depends on the choice of seed. Nuclease treatment further suggested that the differential occupancy of the microRNAs reflects interactions with their mRNA targets. Indeed, analysis of microNRA RNA duplexes showed that pairs involving high occupancy microRNAs exhibit significantly higher binding energy compared to pairs with low occupancy microRNAs. Since mRNAs reside primarily in polysomes, strong interactions lead to high association of microRNAs with polysomes and vice versa for weak interactions. Comparison between hESCs and hFFs data revealed that hESCs tend to express lower occupancy microRNAs, suggesting that cell type-dependent translational features may be affected by expression of a particular set of microRNAs.
URLPMID:15965474 [本文引用: 1]
MicroRNAs are noncoding RNAs of approximately 22 nucleotides that suppress translation of target genes by binding to their mRNA and thus have a central role in gene regulation in health and disease. To date, 222 human microRNAs have been identified, 86 by random cloning and sequencing, 43 by computational approaches and the rest as putative microRNAs homologous to microRNAs in other species. To prove our hypothesis that the total number of microRNAs may be much larger and that several have emerged only in primates, we developed an integrative approach combining bioinformatic predictions with microarray analysis and sequence-directed cloning. Here we report the use of this approach to clone and sequence 89 new human microRNAs (nearly doubling the current number of sequenced human microRNAs), 53 of which are not conserved beyond primates. These findings suggest that the total number of human microRNAs is at least 800.
URLPMID:24296535 [本文引用: 1]
Genomes of multicellular organisms are characterized by the pervasive expression of different types of non-coding RNAs (ncRNAs). Long ncRNAs (lncRNAs) belong to a novel heterogeneous class of ncRNAs that includes thousands of different species. lncRNAs have crucial roles in gene expression control during both developmental and differentiation processes, and the number of lncRNA species increases in genomes of developmentally complex organisms, which highlights the importance of RNA-based levels of control in the evolution of multicellular organisms. In this Review, we describe the function of lncRNAs in developmental processes, such as in dosage compensation, genomic imprinting, cell differentiation and organogenesis, with a particular emphasis on mammalian development.
URLPMID:3651923 [本文引用: 1]
In biology as in real estate, location is a cardinal organizational principle that dictates the accessibility and flow of informational traffic. An essential question in nuclear organization is the nature of the address code--how objects are placed and later searched for and retrieved. Long noncoding RNAs (lncRNAs) have emerged as key components of the address code, allowing protein complexes, genes, and chromosomes to be trafficked to appropriate locations and subject to proper activation and deactivation. lncRNA-based mechanisms control cell fates during development, and their dysregulation underlies some human disorders caused by chromosomal deletions and translocations.
URLPMID:29016735 [本文引用: 1]
Emerging evidence indicates that long non-coding RNAs (lncRNAs) play a vital role in cardiovascular physiology and pathology. Although the lncRNA TUG1 is implicated in atherosclerosis, its function in calcific aortic valve disease (CAVD) remains unknown. In this study, we found that TUG1 was highly expressed in human aortic valves and primary valve interstitial cells (VICs). Moreover, TUG1 knockdown induced inhibition of osteoblast differentiation in CAVD both in vitro and in vivo . Mechanistically, silencing of TUG1 increased the expression of miR-204-5p and subsequently inhibited Runx2 expression at the posttranscriptional level. Importantly, TUG1 directly interacted with miR-204-5p and downregulation of miR-204-5p efficiently reversed the suppression of Runx2 induced by TUG1 short hairpin RNA (shRNA). Thus, TUG1 positively regulated the expression of Runx2, through sponging miR-204-5p, and promoted osteogenic differentiation in CAVD. All together, the evidence generated by our study elucidates the role of lncRNA TUG1 as a miRNA sponge in CAVD, and sheds new light on lncRNA-directed diagnostics and therapeutics in CAVD.
URLPMID:24905165 [本文引用: 1]
In recent years, long noncoding RNAs (lncRNAs) have emerged as an important class of regulators of gene expression. lncRNAs exhibit several distinctive features that confer unique regulatory functions, including exquisite cell- and tissue-specific expression and the capacity to transduce higher-order spatial information. Here we review evidence showing that lncRNAs exert critical functions in adult tissue stem cells, including skin, brain, and muscle, as well as in developmental patterning and pluripotency. We highlight new approaches for ascribing lncRNA functions and discuss mammalian dosage compensation as a classic example of an lncRNA network coupled to stem cell differentiation.
URLPMID:28622390 [本文引用: 1]
Innovative therapies for solid tumors are urgently needed. Recently, therapies that harness the host immune system to fight cancer cells have successfully treated a subset of patients with solid tumors. These responses have been strong and durable but observed in subsets of patients. Work from our group and others has shown that epigenetic therapy, specifically inhibiting the silencing DNA methylation mark, activates immune signaling in tumor cells and can sensitize to immune therapy in murine models. Here we show that colon and ovarian cancer cell lines exhibit lower expression of transcripts involved in antigen processing and presentation to immune cells compared to normal tissues. In addition, treatment with clinically relevant low doses of DNMT inhibitors (that remove DNA methylation) increases expression of both antigen processing and presentation and Cancer Testis Antigens in these cell lines. We confirm that treatment with DNMT inhibitors upregulates expression of the antigen processing and presentation molecules B2M, CALR, CD58, PSMB8, PSMB9 at the RNA and protein level in a wider range of colon and ovarian cancer cell lines and treatment time points than had been described previously. In addition, we show that DNMTi treatment upregulates many Cancer Testis Antigens common to both colon and ovarian cancer. This increase of both antigens and antigen presentation by epigenetic therapy may be one mechanism to sensitize patients to immune therapies.
URLPMID:27385266 [本文引用: 2]
Abstract Since genetic and epigenetic alterations influence the development of colorectal cancer (CRC), huge potential lies in the use of DNA methylation as biomarkers to improve the current diagnosis, screening, prognosis and treatment prediction. Here we performed a systematic review on DNA methylation-based biomarkers published in CRC, and discussed the current state of findings and future challenges. Based on the findings, we then provide a perspective on future studies. Genome-wide studies on DNA methylation revealed novel biomarkers as well as distinct subgroups that exist in CRC. For diagnostic purposes, the most independently validated genes to study further are VIM, SEPT9, ITGA4, OSM4, GATA4 and NDRG4. These hypermethylated biomarkers can even be combined with LINE1 hypomethylation and the performance of markers should be examined in comparison to FIT further to find sensitive combinations. In terms of prognostic markers, myopodin, KISS1, TMEFF2, HLTF, hMLH1, APAF1, BCL2 and p53 are independently validated. Most prognostic markers published lack both a multivariate analysis in comparison to clinical risk factors and the appropriate patient group who will benefit by adjuvant chemotherapy. Methylation of IGFBP3, mir148a and PTEN are found to be predictive markers for 5-FU and EGFR therapy respectively. For therapy prediction, more studies should focus on finding markers for chemotherapeutic drugs as majority of the patients would benefit. Translation of these biomarkers into clinical utility would require large-scale prospective cohorts and randomized clinical trials in future. Based on these findings and consideration we propose an avenue to introduce methylation markers into clinical practice in near future. For future studies, multi-omics profiling on matched tissue and non-invasive cohorts along with matched cohorts of adenoma to carcinoma is indispensable to concurrently stratify CRC and find novel, robust biomarkers. Moreover, future studies should examine the timing and heterogeneity of methylation as well as the difference in methylation levels between epithelial and stromal tissues.
URL [本文引用: 1]
Hepatocellular carcinoma (HCC) is one of the most fatal human malignancies, but the molecular mechanisms of hepatocarcinogenesis remain unclear. Although p53 mutations are frequently observed in Asian HCC, it is not a common event in Western HCC. Recent studies suggest that tumor suppressor genes (TSGs) can also be silenced through epigenetic disruption, such as promoter CpG island methylation, during carcinogenesis. To further understand the molecular mechanism of hepatocarcinogenesis, we have investigated the promoter methylation status of nine TSGs (SOCS-1, GSTP, APC, E-cadherin, RAR-beta, p14, p15, p16, and p73) in 51 cases of HCC using methylation-specific polymerase chain reaction. We found that 82% of HCCs had methylation of at least one TSG promoter. The most frequently methylated TSGs in HCC were: SOCS-1 (65%), GSTP (54%), APC (53%), E-cadherin (49%), and p15 (49%). Methylation of SOCS-1, GSTP, APC, E-cadherin, and p15 was more frequent in HCC than in nontumor liver (P < 0.05). Methylation of SOCS-1, GSTP, and p15 was also significantly more frequent in HCC than cirrhotic liver (P < 0.05). Although methylation of one or two genes could be seen in both nontumor and cirrhotic livers, 53% of the HCC cases had three or more TSG promoters methylated, in comparison to 0% in nontumor liver and 13% in cirrhosis (P = 0.001). Methylation of SOCS-1, APC, and p15 was more frequently seen in hepatitis C virus-positive HCC than hepatitis C virus/hepatitis B virus-negative HCC. Our data suggest that promoter hypermethylation of TSGs is a common event in HCC and may play an important role in hepatocarcinogenesis.
[本文引用: 1]
URLPMID:22475363 [本文引用: 1]
Approximately half of the patients with myelofibrosis (MF) carry mutant JAK2(V617F) proteins. JAK2(V617F) has been recently shown to translocate to the nucleus and modify specific histones, thus regulating transcription. We report on a phase II study testing the activity and tolerability of the histone deacetylase inhibitor pracinostat given at 60 mg every other day for three weeks per month in 22 patients with intermediate or high risk MF. Eight (36%) patients experienced clinical benefit, with 6 (27%) experiencing reductions in splenomegaly (median 3 cm, range 1-4 cm). According to International Working Group criteria, 2 (9%) patients had clinical improvement (anemia response in both cases). The most frequent side effect associated to pracinostat therapy was fatigue, which occurred in 20 (91%) patients (grade 2 in 3 patients). Grade 3-4 neutropenia, anemia, and thrombocytopenia occurred in 13%, 0%, and 21%, respectively. Twenty-one patients permanently discontinued pracinostat, mainly due to lack of efficacy. In conclusion, pracinostat at the dose tested is reasonably tolerated and has modest activity in patients with MF.
URLPMID:22245671 [本文引用: 1]
Hydroquinone (HQ), one of the most important metabolites derived from benzene, is known to be associated with acute myelogenous leukemia (AML) risk, however, its carcinogenic mechanism remains unclear. In this study, the epigenetic mechanism of HQ exposure was investigated. We characterized the epigenomic response of TK6 cells to HQ exposure, and examined the mRNA expression of DNA methyltransferases (DNMTs) including DNMT1, DNMT3a and DNMT3b, methyl-CpG-binding domain protein 2 (MBD2) and six proto-oncogenes (MPL, RAF1, MYB, MYC, ERBB2 and BRAF). Compared to the control cells, HQ exposure (2.5, 5.0, 10.0 and 20.002μM for 4802h) resulted in the decrease of DNMTs and MBD2 expression, the global hypomethylation and increase of MPL at mRNA level. Meanwhile, most of these changes were in dose-dependent manner. Moreover, inhibition of DNMTs induced by 5-aza-2′-deoxycytidine (5-AZA), an identified DNMT inhibitor, caused more induction of MPL expression at mRNA level compared to the HQ (10.002μM) pre-treated group. Furthermore, treatment of HQ potentially led to MPL itself hypomethylation (10.0 and 20.002μM reduced by 47% and 44%, respectively), further revealing that the activation of proto-oncogene MPL was related to hypomethylation in its DNA sequences. In conclusion, hypomethylation, including global and specific hypomethylation, might be involved in the activation of MPL, and the hypomethylation could be induced by decreased DNMTs in TK6 cells exposed to HQ.
In: Karpf A, ed.
URL [本文引用: 1]
URLPMID:26289459 [本文引用: 1]
Abstract The epigenetic landscape is deregulated in cancer due to aberrant activation or inactivation of enzymes maintaining and modifying the epigenome. Histone modifications and global aberrations at the histone level may result in distorted patterns of gene expression, and malfunction of proteins that regulate chromatin modification and remodeling. Recent whole genome studies demonstrated that histones and chaperone proteins harbor mutations that may result in gross alterations of the epigenome leading to genome instability. Glioma development is a multistep process, involving genetic and epigenetic alterations. This review summarizes newly identified mechanisms affecting expression/functions of histone-modifying enzymes and chromatin modifiers in gliomas. We discuss recent approaches to overcome epigenetic alterations with histone-modifying enzyme inhibitors and their prospects for glioma therapy.
URLPMID:24381114 [本文引用: 1]
Rubinstein-Taybi syndrome (RTS) is an incurable genetic disorder with combination of mental retardation and physical features including broad thumbs and toes, craniofacial abnormalities, and growth deficiency. While the autosomal dominant mode of transmission is limitedly known, the majority of cases are attributable to de novo mutations in RTS. The first identified gene associated with RTS is CREB-binding protein (CREBBP/CBP). Alterations of the epigenetic 'histone code' due to dysfunction of the CBP histone acetyltransferase activity deregulate gene transcriptions that are prominently linked to RTS pathogenesis. In this review, we discuss how CBP mutation contributes to modifications of histone and how histone deacetylase inhibitors are therapeutically applicable to epigenetic conditioning in RTS. Since most genetic mutations are irreversible and therapeutic approaches are limited, therapeutic targeting of reversible epigenetic components altered in RTS may be an ideal strategy. Expeditious further study on the role of the epigenetic mechanisms in RTS is encouraged to identify novel epigenetic markers and therapeutic targets to treat RTS.
URLPMID:26216839 [本文引用: 1]
Colorectal cancer (CRC) is a leading cause of cancer deaths worldwide. One of the fundamental processes driving the initiation and progression of CRC is the accumulation of a variety of genetic and epigenetic changes in colonic epithelial cells. Over the past decade, major advances have been made in our understanding of cancer epigenetics, particularly regarding aberrant DNA methylation, microRNA (miRNA) and noncoding RNA deregulation, and alterations in histone modification states. Assessment of the colon cancer "epigenome" has revealed that virtually all CRCs have aberrantly methylated genes and altered miRNA expression. The average CRC methylome has hundreds to thousands of abnormally methylated genes and dozens of altered miRNAs. As with gene mutations in the cancer genome, a subset of these epigenetic alterations, called driver events, are presumed to have a functional role in CRC. In addition, the advances in our understanding of epigenetic alterations in CRC have led to these alterations being developed as clinical biomarkers for diagnostic, prognostic, and therapeutic applications. Progress in this field suggests that these epigenetic alterations will be commonly used in the near future to direct the prevention and treatment of CRC.
URLPMID:17339880 [本文引用: 1]
An altered pattern of epigenetic modifications is central to many common human diseases, including cancer. Many studies have explored the mosaic patterns of DNA methylation and histone modification in cancer cells on a gene-by-gene basis; among their results has been the seminal finding of transcriptional silencing of tumour-suppressor genes by CpG-island-promoter hypermethylation. However, recent technological advances are now allowing cancer epigenetics to be studied genome-wide-an approach that has already begun to provide both biological insight and new avenues for translational research. It is time to 'upgrade' cancer epigenetics research and put together an ambitious plan to tackle the many unanswered questions in this field using epigenomics approaches.
URLPMID:2881805 [本文引用: 1]
Abstract Stable epigenetic silencing of p16(INK4a) is a common event in hepatocellular carcinoma (HCC) cells, which is associated with abnormal cell proliferation and liberation from cell cycle arrest. Understanding the early epigenetic events in silencing p16(INK4a) expression may illuminate a prognostic strategy to block HCC development. Toward this end, we created a reprogram cell model by the fusion mouse HCC cells with mouse embryonic stem cells, in which the ES-Hepa hybrids forfeited HCC cell characteristics along with reactivation of the silenced p16(INK4a). HCC characteristics, in terms of gene expression pattern and tumorigenic potential, was restored upon induced differentiation of these reprogrammed ES-Hepa hybrids. The histone methylation pattern relative to p16(INK4a) silencing during differentiation of the ES-Hepa hybrids was analyzed. H3K27 trimethylation at the p16(INK4a) promoter region, occurring in the early onset of p16(INK4a) silencing, was followed by H3K9 dimethylation at later stages. During the induced differentiation of the ES-Hepa hybrids, H3K4 di- and trimethylations were maintained at high levels during the silencing of p16(INK4a), strongly suggesting that H3K4 methylation events did not cause the silencing of p16(INK4a). Our results suggested that the enrichment of H3K27 trimethylation, independent of H3K9 dimethylation, trimethylation, and DNA methylation, was an early event in the silencing of p16(INK4a) during the tumor development. This unique chromatin pattern may be a heritable marker of epigenetic regulation for p16(INK4a) silencing during the developmental process of hepatocellular carcinogenesis.
URL [本文引用: 1]
CpG island hypermethylation and global genomic hypomethylation are common epigenetic features of cancer cells. Less attention has been focused on histone modifications in cancer cells. We characterized post-translational modifications to histone H4 in a comprehensive panel of normal tissues, cancer cell lines and primary tumors. Using immunodetection, high-performance capillary electrophoresis and mass spectrometry, we found that cancer cells had a loss of monoacetylated and trimethylated forms of histone H4. These changes appeared early and accumulated during the tumorigenic process, as we showed in a mouse model of multistage skin carcinogenesis. The losses occurred predominantly at the acetylated Lys16 and trimethylated Lys20 residues of histone H4 and were associated with the hypomethylation of DNA repetitive sequences, a well-known characteristic of cancer cells. Our data suggest that the global loss of monoacetylation and trimethylation of histone H4 is a common hallmark of human tumor cells.
URL [本文引用: 1]
随着分子生物学技术的飞速发展,人们对于长链非编码RNA(Long non-coding RNA,lncRNA)的研究越来越深入。lncRNA 不仅在生物体正常生物活动中不可或缺,还在许多疾病尤其是肿瘤中扮演重要角色。已有的研究表明多种lncRNA与血液系统肿瘤密切相关,具有影响抑癌基因 p15表达、p53蛋白功能,以及与miRNA相互作用参与疾病等功能。本文综述了血液系统肿瘤相关的 lncRNA并着重介绍与 p15、p53、miRNA有关的lncRNA以及它们的相互作用在疾病中发挥的功能,以期能够全面了解血液系统肿瘤相关lncRNA的作用特点,为血液 系统肿瘤的研究、诊断以及治疗提供新的思路。
URL [本文引用: 1]
随着分子生物学技术的飞速发展,人们对于长链非编码RNA(Long non-coding RNA,lncRNA)的研究越来越深入。lncRNA 不仅在生物体正常生物活动中不可或缺,还在许多疾病尤其是肿瘤中扮演重要角色。已有的研究表明多种lncRNA与血液系统肿瘤密切相关,具有影响抑癌基因 p15表达、p53蛋白功能,以及与miRNA相互作用参与疾病等功能。本文综述了血液系统肿瘤相关的 lncRNA并着重介绍与 p15、p53、miRNA有关的lncRNA以及它们的相互作用在疾病中发挥的功能,以期能够全面了解血液系统肿瘤相关lncRNA的作用特点,为血液 系统肿瘤的研究、诊断以及治疗提供新的思路。
URLPMID:28992617 [本文引用: 1]
Iodine may trigger tumorigenesis and development of thyroid carcinoma, but the mechanisms involved remained elusive. MicroRNA (MiRNAs) are known to be involved in each stage of cancer development; however, the role of miRNAs in iodine-induced tumorigenesis of thyroid carcinoma remained unknown. In this study, we aimed at investigating miRNA related signaling pathway in thyroid cancer cells. Levels of miRNAs and mRNAs were determined using RT-qPCR and proteins were quantified by western blotting. Cell migration and proliferation were checked using Transwell assay and CCK8 assay respectively. Tumor xenografts in nude mice were established by subcutaneous injection of cancer cells. Mitogen activated protein kinase 1 (MAPK1) was significantly up-regulated, while miR-422a was down-regulated in thyroid cancer cells cultured with high iodine; miR-422a directly bound to the 3'UTR of MAPK1 mRNA. Moreover, miR-422a negatively regulated MAPK1 expression, and down-regulated miR-422a promoted proliferation and migration of TPC-1 cells. In vivo studies also confirmed that iodine promoted tumor growth by suppressing miR-422a and up-regulating MAPK1. Our study illustrates a new pathway comprising iodine, miRNA and MAPK1, and defines a novel mechanism in thyroid cancer.
URLPMID:5028809 [本文引用: 1]
Alterations in epigenetic control of gene expression play an important role in many diseases, including gastric cancer. Many studies have identified a large number of upregulated oncogenic mi RNAs and downregulated tumour-suppressor mi RNAs in this type of cancer. In this review, we provide an overview of the role of mi RNAs, pointing to their potential to be useful as diagnostic and/or prognostic biomarkers in gastric cancer. Moreover, we discuss the influence of polymorphisms and epigenetic modifications on mi RNA activity.
URL [本文引用: 1]
Gastric cancer is one of the most common malignant diseases worldwide, although much progress has been achieved in recent years, the early diagnosis and treatment for gastric cancer are not yet satisfactory and, thus the prognosis is still poor. MicroRNAs (miRNAs) can regulate a variety of physiological and developmental processes, it has been revealed that many miRNAs contribute the initiation and progression of various cancers. MiR-148a is one of the most important miRNAs in gastric cancer, and the aim of this paper is to provide an overview of various roles of miR-148a in gastric cancer.We searched studies in electronic databases. MiR-148a was down-regulated in gastric cancer tissues and cell lines, which was resulted from the hypermethylation in its promoter region. Furthermore, miR-148a could regulate several different target genes and pathways involving tumor proliferation, invasion and metastasis.MiR-148a may serve as a novel biomarker for the diagnosis and as a new therapeutic target in gastric cancer.
URLPMID:26891666 [本文引用: 1]
MicroRNAs (miRNAs) play vital roles in cell proliferation, differentiation and apoptosis in hepatocellular carcinoma (HCC). miR-26b has been confirmed as an important regulator in carcinogenesis and other pathological processes. miR-26b-5p is one member of the mature miR-26 family, and its functional role in proliferation, angiogenesis and apoptosis in HCC remains unknown. Here, we demonstrate that miR-26b-5p expression was significantly decreased in HCC tissues and HCC cell lines compared with normal liver tissues and liver cells by quantitative real-time polymerase chain reaction (qRT-PCR). The relationships between miR-26b-5p and the clinical characteristics of HCC patients were further analysed, and miR-26b-5p was positively correlated with the differentiation of HCC cells. Computational searches were further used to identify the downstream targets and signalling pathways of miR-26b-5p in HCC cells. Cell viability, proliferation and tube formation abilities were assessed by scrape, 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and three-dimensional culture assays to confirm that miR-26b-5p inhibited HCC cell growth and impaired the tube formation ability of the HCC cells. Both in vitro and in vivo studies showed that miR-26b-5p could suppress vascular mimicry (VM) and angiogenesis by down-regulating the expression of VE-cadherin, Snail and MMP2 and could inhibit the apoptosis of HCC cells. Using mouse models, we revealed that tumours derived from miR-26b-5p-expressing HCC cells displayed a significant decrease in microvessel density compared with those derived from control cells. Therefore, our data provide further insight into the role of miR-26b-5p as a negative regulator of proliferation, angiogenesis, and apoptosis in HCC.
URL [本文引用: 1]
TGF-β is a ubiquitous cytokine that plays an active role in many cellular processes. Nearly every cell type has the ability to secrete TGF-β, as well as the ability to respond to TGF-β via the presence of TGF-β receptors on the cell surface. Consequently, gain or loss of function of the TGF-β pathway and its components are known to lead to a variety of diseases, including cancer. In epithelial cells, TGF-β functions as a tumor suppressor, where it inhibits proliferation, induces apoptosis, and mediates differentiation. Conversely, in other contexts, TGF-β promotes tumor progression through increasing tumor cell invasion and metastasis. Thus, TGF-β can have opposing roles, likely dependent, in part, on whether the cancer is early or late stage. The effects of TGF-β on tumor suppression and promotion are not limited to the tumor cell itself; rather, these effects can also be mediated through the stroma and the immune system. The dichotomous role of TGF-β in cancer highlights our need to understand the contextual effects of this cytokine to better guide patient selection for the use of anti-TGF-β therapies currently in clinical trials.
URLPMID:19672297 [本文引用: 1]
DNA methylation is capable of modulating coordinate expression of large numbers of genes across many different pathways, and may therefore warrant investigation for their potential role between genes and disease phenotype. In a rare set of monozygotic twins discordant for Alzheimer's disease (AD), significantly reduced levels of DNA methylation were observed in temporal neocortex neuronal nuclei of the AD twin. These findings are consistent with the hypothesis that epigenetic mechanisms may mediate at the molecular level the effects of life events on AD risk, and provide, for the first time, a potential explanation for AD discordance despite genetic similarities.
URLPMID:2692619 [本文引用: 1]
An age-related decline in chondrocyte production of osteogenic protein-1 (OP-1) (Bone Morphogenetic Protein-7) may contribute to cartilage loss in osteoarthritis. This study was designed to determine if increased methylation of the OP-1 promoter might serve as a mechanism for the age-related decline in OP-1 expression. Human articular chondrocytes were isolated from cartilage obtained after death from tissue donors (ages 19 86 years) without a known history of arthritis. DNA was obtained from isolated chondrocytes in primary culture and analyzed for OP-1 promoter methylation by polymerase chain reaction (PCR) after bisulfite treatment. Cultured cells were treated with the DNA methyltransferase inhibitor 5-azacytidine and OP-1 production was measured in the media by enzyme-linked immunosorbent assay (ELISA). RNA was isolated to measure expression of insulin-like growth factor-1 (IGF-1), the IGF-1 receptor (IGF-1R), aggrecan, and OP-1 by real-time PCR. Methylation of the OP-1 promoter was detected in chondrocytes isolated from tissue obtained from older adults and there was a positive correlation between age and OP-1 methylation status ( n = 22, R 2 = 0.277, P = 0.014). Inhibition of methylation in cultured cells with 5-azacytidine increased chondrocyte production of OP-1 protein and increased the expression of the IGF-1, the IGF-1R, aggrecan, and OP-1 genes but not GAPDH. Age-related methylation of the OP-1 promoter may contribute to a decrease in OP-1 production in cartilage and a decrease in expression of OP-1 responsive genes such as IGF-1, the IGF-1R, and aggrecan.
URLPMID:18376059 [本文引用: 1]
Longevity related genes were investigated concerning promoter methylation. SIRT3, SMARCA5, HTERT and CDH1 promoters were analyzed in peripheral blood in relation to gender, age and Alzheimer's disease (AD). Methylation Specific PCR assay (MSP) was used. There were no significant differences in methylation frequencies of SIRT3, SMARCA5 and CDH1 among young, elderly and AD groups (p> 0.05), showing no association with aging or AD. On the other hand, HTERT methylation frequency was associated with the aging process, in that AD patients differed from elderly controls (p=0.0086), probably due to telomere and immune dysfunctions involved in AD pathogenesis.
URLPMID:23399643 [本文引用: 1]
Comment on Proc Natl Acad Sci U S A. 2013 Feb 5;110(6):2354-9. Proc Natl Acad Sci U S A. 2013 Feb 5;110(6):2366-70.
URLPMID:19434510 [本文引用: 1]
Histone post-translational modifications (PTMs) are involved in diverse biological processes and methylation was regarded as a long-term epigenetic mark. Though aging represented one of the major risk factors for neurodegenerative diseases, no systematic investigations had correlated the patterns of histone PTMs in the brain with aging and the roles of such concerted histone PTMs in brain aging are still unknown. In this study, enzyme digestion, nano-LC, MALDI-TOF/TOF MS analysis and Western blotting were combined to investigate the defined methylation of core histones (H2A, H2B, H3 and H4) in the brain of 12-month-old senescence accelerated mouse prone 8 (SAMP8). The expression of several modified histones in the brain of 3-, and 12-month-old SAMP8 mice as well as that of the age-matched control senescence accelerated-resistant mouse (SAMR1) was compared. In the brain of 12-month-old SAMP8 mice, seven methylation sites (H3K24, H3K27, H3K36, H3K79, H3R128, H4K20 and H2A R89) were detected and most PTMs sites were located on histone H3. Mono-methylated H4K20 decreased significantly in the brain of 12-month-old SAMP8 mice. Methylated H3K27 and H3K36 coexisted in the aged brain with different methylation multiplicities. Di-methylated H3K79 expressed in the neurons of cerebral cortex and hippocampus. This study showed histone methylation patterns in the aged SAMP8 mice brain and provided the experimental evidences for further research on histone PTMs in the aged brain. We hope these results could initiate a platform for the exchange of comprehensive information concerning aging or neurodegenerative disease and help us interpret the change of gene expression and DNA repair ability at epigenetic level.
URLPMID:22577027 [本文引用: 1]
Abstract The epigenetic remodeling of chromatin histone proteins by acetylation has been the subject of recent investigations searching for biomarkers indicative of late onset cognitive loss. Histone acetylations affect the regulation of gene transcription, and the loss of learning induced deacetylation at specific histone sites may represent biomarkers for memory loss and Alzheimer's disease (AD). Selected-reaction-monitoring (SRM) has recently been advanced to quantitate peptides and proteins in complex biological systems. In this paper, we provide evidence that SRM-based targeted proteomics can reliably quantify specific histone acetylations in both AD and control brain by identifying the patterns of H3 K18/K23 acetylations Results of targeted proteomics assays have been validated by Western blot (WB) analysis. As compared with LC-MS/MS-TMT (tandem-mass-tagging) and WB methods, the targeted proteomics method has shown higher throughput, and therefore promised to be more suitable for clinical applications. With this methodology, we find that histone acetylation is significantly lower in AD temporal lobe than found in aged controls. Targeted proteomics warrants increased application for studying epigenetics of neurodegenerative diseases.
URLPMID:20877454 [本文引用: 1]
Abstract The family of histone deacetylases (HDACs) has recently emerged as important drug targets for treatment of slow progressive neurodegenerative disorders, including Huntington's disease (HD). Broad pharmaceutical inhibition of HDACs has shown neuroprotective effects in various HD models. Here we examined the susceptibility of HDAC targets for drug treatment in affected brain areas during HD progression. We observed increased HDAC1 and decreased HDAC4, 5 and 6 levels, correlating with disease progression, in cortices and striata of HD R6/2 mice. However, there were no significant changes in HDAC protein levels, assessed in an age-dependent manner, in HD knock-in CAG140 mice and we did not observe significant changes in HDAC1 levels in human HD brains. We further assessed acetylation levels of $\beta$-tubulin, as a biomarker of HDAC6 activity, and found it unchanged in cortices from R6/2, knock-in, and human subjects at all disease stages. Inhibition of deacetylase activities was identical in cortical extracts from R6/2 and wild-type mice treated with a class II-selective HDAC inhibitor. Lastly, treatment with class I- and II-selective HDAC inhibitors showed similar responses in HD and wild-type rat striatal cells. In conclusion, our results show that class I and class II HDAC targets are present and accessible for chronic drug treatment during HD progression and provide impetus for therapeutic development of brain-permeable class- or isoform-selective inhibitors.
URLPMID:20380815 [本文引用: 1]
Neurodegeneration is characterized by the progressive loss of neuronal cell types in the nervous system. Although the main cause of cell dysfunction and death in many neurodegenerative diseases is not known, there is increasing evidence that their demise is a result of a combination of genetic and environmental factors which affect key signaling pathways in cell function. This view is supported by recent observations that disease-compromised cells in late-stage neurodegeneration exhibit profound dysregulation of gene expression. MicroRNAs (miRNAs) introduce a novel concept of regulatory control over gene expression and there is increasing evidence that they play a profound role in neuronal cell identity as well as multiple aspects of disease pathogenesis. Here, we review the molecular properties of brain cells derived from patients with neurodegenerative diseases, and discuss how deregulated miRNA/mRNA expression networks could be a mechanism in neurodegeneration. In addition, we emphasize that the dysfunction of these regulatory networks might overlap between different cell systems and suggest that miRNA functions might be common between neurodegeneration and other disease entities.
URLPMID:3200297 [本文引用: 1]
Abstract The contribution of mutations in amyloid precursor protein (APP) and presenilin (PSEN) to familial Alzheimer's disease (AD) is well established. However, little is known about the molecular mechanisms leading to amyloid 0205 (A0205) generation in sporadic AD. Increased brain ceramide levels have been associated with sporadic AD, and are a suggested risk factor. Serine palmitoyltransferase (SPT) is the first rate-limiting enzyme in the de novo ceramide synthesis. However, the regulation of SPT is not yet understood. Evidence suggests that it may be posttranscriptionally regulated. Therefore, we investigated the role of miRNAs in the regulation of SPT and amyloid 0205 (A0205) generation. We show that SPT is upregulated in a subgroup of sporadic AD patient brains. This is further confirmed in mouse model studies of risk factors associated with AD. We identified that the loss of miR-137, -181c, -9, and 29a/b-1 increases SPT and in turn A0205 levels, and provides a mechanism for the elevated risk of AD associated with age, high-saturated-fat diet, and gender. Finally, these results suggest SPT and the respective miRNAs may be potential therapeutic targets for sporadic AD.
URLPMID:25135968 [本文引用: 1]
Long non-coding RNAs (lncRNAs) are transcripts with low protein-coding potential that represent a large proportion of the transcriptional output of the cell. Many lncRNAs exhibit features indicative of functionality including tissue-restricted expression, localization to distinct subcellular structures, regulated expression and evolutionary conservation. Some lncRNAs have been shown to associate with chromatin-modifying activities and transcription factors, suggesting that a common mode of action may be to guide protein complexes to target genomic loci. However, the functions (if any) of the vast majority of lncRNA transcripts are currently unknown, and the subject of investigation. Here, we consider the putative role(s) of lncRNAs in neurodevelopment and brain function with an emphasis on the epigenetic regulation of gene expression. Associations of lncRNAs with neurodevelopmental/neuropsychiatric disorders, neurodegeneration and brain cancers are also discussed.
URLPMID:2055548 [本文引用: 1]
Epigenetic changes are heritable modifications that do not involve alterations in the primary DNA sequence. They regulate crucial cellular functions such as genome stability, X-chromosome inactivation, and gene imprinting. Epidemiological and experimental observations now suggest that such changes may also explain the fetal basis of adult diseases such as cancer, obesity, diabetes, cardiovascular disorders, neurological diseases, and behavioral modifications. The main molecular events known to initiate and sustain epigenetic modifications are histone modification and DNA methylation. This review specifically focuses on existing and emerging technologies used in studying DNA methylation, which occurs primarily at CpG dinucleotides in the genome. These include standard exploratory tools used for global profiling of DNA methylation and targeted gene investigation: methylation sensitive restriction fingerprinting (MSRF), restriction landmark genomic scanning (RLGS), methylation CpG island amplification-representational difference analysis (MCA-RDA), differential methylation hybridization (DMH), and cDNA microarrays combined with treatment with demethylating agents and inhibitors of histone deacetylase. The basic operating principals, resource requirements, applications, and benefits and limitations of each methodology are discussed. Validation methodologies and functional assays needed to establish the role of a CpG-rich sequence in regulating the expression of a target or candidate gene are outlined. These include in silico database searches, methylation status studies (bisulfite genomic sequencing, COBRA, MS-PCR, MS-SSCP), gene expression studies, and promoter activity analyses. Our intention is to give readers a starting point for choosing methodologies and to suggest a workflow to follow during their investigations. We believe studies of epigenetic changes such as DNA methylation hold great promise in understanding the early origins of adult diseases and in advancing their diagnosis, prevention, and treatment.
URLPMID:3747760 [本文引用: 1]
Abstract According to the developmental origins of health and disease hypothesis, in utero experiences reprogram an individual for immediate adaptation to gestational perturbations, with the sequelae of later-in-life risk of metabolic disease. An altered gestational milieu with resultant adult metabolic disease has been observed in instances of both in utero constraint (e.g., from famine or uteroplacental insufficiency) and overt caloric abundance (e.g., from a maternal high-fat, caloric-dense diet). The commonality of the adult metabolic phenotype begs the question of how diverse in utero experiences (i.e., reprogramming events) converge on common metabolic pathways and how the memory of these events is maintained across the lifespan. We and others have investigated the molecular mechanisms underlying fetal programming and observed that epigenetic modifications to the fetal and placental epigenome accompany these reprogramming events. Based on several lines of emerging data in human and nonhuman primates, it is now felt that modified epigenetic signature--and the histone code in particular--underlies alterations in postnatal gene expression and metabolic pathways central to accurate functioning and maintenance of health. Because of the tissue lineage specificity of many of these modifications, nonhuman primates serve as an apt model system for the capacity to recapitulate human gene expression and regulation during development. This review summarizes recent epigenetic advances using rodent and primate (both human and nonhuman) models during in utero development and contributing to adult diseases later in life.
URLPMID:16310649 [本文引用: 1]
ABSTRACT The combined epidemiologic, clinical, and animal studies clearly demonstrate that the intrauterine environment influences growth and development of the fetus and the subsequent development of adult diseases. There are critical specific windows during development, often coincident with periods of rapid cell division, during which a stimulus or insult may have long-lasting consequences on tissue or organ function after birth. Birth weight is only one marker of an adverse fetal environment, and confining studies to this population only may lead to erroneous conclusions regarding etiology. Studies using animal models of uteroplacental insufficiency suggest that mitochondrial dysfunction and oxidative stress play an important role in the pathogenesis of the fetal origins of adult disease.
URLPMID:25522397 [本文引用: 1]
Epidemiological studies have identified prenatal exposure to famine as a risk factor for schizophrenia, and animal models of prenatal malnutrition display structural and functional brain abnormalities implicated in schizophrenia.The offspring of the RLP50 rat, a recently developed animal model of prenatal famine malnutrition exposure, was used to investigate the changes of gene expression and epigenetic modifications in the brain regions. Microarray gene expression analysis was carried out in the prefrontal cortex and the hippocampus from 8 RLP50 offspring rats and 8 controls. MBD-seq was used to test the changes in DNA methylation in hippocampus depending on prenatal malnutrition exposure.In the prefrontal cortex, offspring of RLP50 exhibit differences in neurotransmitters and olfactory-associated gene expression. In the hippocampus, the differentially-expressed genes are related to synaptic function and transcription regulation. DNA methylome profiling of the hippocampus also shows widespread but systematic epigenetic changes; in most cases (87%) this involves hypermethylation. Remarkably, genes encoded for the plasma membrane are significantly enriched for changes in both gene expression and DNA methylome profiling screens (p = 2.37$\times$10(-9) and 5.36$\times$10(-9), respectively). Interestingly, Mecp2 and Slc2a1, two genes associated with cognitive impairment, show significant down-regulation, and Slc2a1 is hypermethylated in the hippocampus of the RLP50 offspring.Collectively, our results indicate that prenatal exposure to malnutrition leads to the reprogramming of postnatal brain gene expression and that the epigenetic modifications contribute to the reprogramming. The process may impair learning and memory ability and result in higher susceptibility to schizophrenia.
URLPMID:20734317 [本文引用: 1]
Abstract Top of page Abstract INTRODUCTION MATERIALS AND METHODS RESULTS DISCUSSION LITERATURE CITED Valproate (VPA) has been used for decades in the treatment of epilepsy and migraine. However, maternal administration of VPA during pregnancy increases susceptibility to autism spectrum disorders (ASDs) in the offspring. The aim of this study was to investigate the methylation modification and its effects on the activity of Wnt/$\beta$-catenin pathway in the rat brain prenatally exposed to VPA. We exposed the rats in early pregnancy to VPA and found that the prenatal VPA exposure, in comparison with the prenatal vehicle exposure, induced demethylation in the promoter regions of wnt1 and wnt2 , but not in those of Wnt inhibitory factor-1 and Dickkopf 1 , in the prefrontal cortexes and hippocampi of the offspring. Consequently, both mRNA and protein expression of wnt1 and wnt2 were increased. Furthermore, the activity of Wnt/$\beta$-catenin pathway was upregulated, as indicated by the increased levels of $\beta$-catenin, hence the growing expression of its target genes. This work suggested an epigenetic action via which VPA, when administered in early pregnancy, induced dysregulation of signaling pathway, further facilitating susceptibility to ASDs.
URLPMID:4663595 [本文引用: 1]
Exposure to environmental factors in early life can influence developmental processes and long-term health in humans. Early life nutrition and maternal diet are well-known examples of conditions shown to influence the risk of developing metabolic diseases, including type 2 diabetes mellitus and cardiovascular diseases, in adulthood. It is increasingly accepted that environmental compounds, including nutrients, can produce changes in the genome activity that, in spite of not altering the DNA sequence, can produce important, stable and, in some instances, transgenerational alterations in the phenotype. Epigenetics refers to changes in gene function that cannot be explained by changes in the DNA sequence, with DNA methylation patterns/histone modifications that can make important contributions to epigenetic memory. The epigenome can be considered as an interface between the genome and the environment that is central to the generation of phenotypes and their stability throughout the life course. To better understand the role of maternal health and nutrition in the initiation and progression of diseases in childhood and adulthood, it is necessary to identify the physiological and/or pathological roles of specific nutrients on the epigenome and how dietary interventions in utero and early life could modulate disease risk through epigenomic alteration.
URLPMID:22223754 [本文引用: 2]
Abstract Undernutrition during pregnancy is implicated in the programming of offspring for the development of obesity and diabetes. We hypothesized that maternal programming causes epigenetic changes in fetal hypothalamic pathways regulating metabolism. This study used sheep to examine the effect of moderate maternal undernutrition (60 d before to 30 d after mating) and twinning to investigate changes in the key metabolic regulators proopiomelanocortin (POMC) and the glucocorticoid receptor (GR) in fetal hypothalami. Methylation of the fetal hypothalamic POMC promoter was reduced in underfed singleton, fed twin, and underfed twin groups (60, 73, and 63% decrease, respectively). This was associated with reduced DNA methyltransferase activity and altered histone methylation and acetylation. Methylation of the hypothalamic GR promoter was decreased in both twin groups and in maternally underfed singleton fetuses (52, 65, and 55% decrease, respectively). This correlated with changes in histone methylation and acetylation and increased GR mRNA expression in the maternally underfed singleton group. Alterations in GR were hypothalamic specific, with no changes in hippocampi. Unaltered levels of OCT4 promoter methylation indicated gene-specific effects. In conclusion, twinning and periconceptional undernutrition are associated with epigenetic changes in fetal hypothalamic POMC and GR genes, potentially resulting in altered energy balance regulation in the offspring.
URL [本文引用: 2]
URLPMID:23382021 [本文引用: 1]
Obesity is a leading risk factor for impaired glucose tolerance and type 2 diabetes (T2D). Although the cause of the obesity epidemic is multi-factorial and not entirely clear, the recent acceleration in incidence is too rapid to be accounted for only by genetics, the wide availability of calorie-rich foods, and increasingly sedentary lifestyles. Accumulating data suggest that the important causes of the obesity epidemic may be related to developmental and early life environmental conditions. The concept of the developmental origins of health and disease (DOHaD) suggests that adverse influences early in development, particularly during intrauterine life, may result in permanent changes in the physiology and metabolism of the infant, which in turn result in an increased risk of non-communicable diseases in adulthood. For example, undernutrition during pregnancy and rapid postnatal weight gain are associated with obesity and T2D in the adult offspring. Moreover, increasing evidence suggests that early-life exposure to a wide range of chemicals has a significant impact on the causes of metabolic disorders. Although the underlying molecular mechanisms remain to be determined, these factors can affect epigenetic processes, such as DNA methylation, allowing the developmental environment to modulate gene transcription. The objective of this review article was to summarize recent progress in the biomedical implications of the DOHaD concept, focusing on the pathogenesis of obesity and T2D, and to discuss a future direction for preventive strategies from a public health perspective.
URLPMID:22951100 [本文引用: 1]
An adverse relationship between suboptimal fetal environments and the development of adult diseases, such as hypertension, type II diabetes, and cardiovascular disease, has been reported in numerous studies. The purpose of this study was to investigate the strain difference of offspring response to maternal malnutrition during pregnancy and the involvement of the renin-angiotensin system (RAS) in the development of adult hypertension using C57BL/6J (C57) mice and angiotensin II(Ang II) type 1 receptor-associated protein-transgenic (ATRAP-Tg) mice. Pregnant dams were fed an isocaloric diet containing either 20% (normal protein; NP) or 8% (low protein; LP) protein. Birth weight was significantly reduced in C57-LP offspring, but not in ATRAP-Tg-LP offspring. Arterial blood pressure was higher in C57-LP offspring than in the other groups. In contrast, ATRAP-Tg-LP offspring did not show an increase in blood pressure compared with NP offspring. Renal angiotensin II type 1 (AT 1 ) receptor expression was not altered by maternal malnutrition, whereas angiotensin II type 2receptor expression was significantly decreased in C57-LP offspring. In conclusion, these findings suggest that a suboptimal intrauterine environment induces adult hypertension because of an alteration of expression of RAS components, which waspartly suppressed by sustained ATRAP overexpression via attenuation of the AT 1 receptor-mediated pathological response.
URLPMID:17764855 [本文引用: 1]
Polycystic ovary syndrome (PCOS) is one of the most common, yet heterogeneous and complex, endocrine disorders in women of reproductive age. Although the aetiology of PCOS remains uncertain, emerging evidence has indicated that exposure of the female fetus to the hyperandrogenism milieu in utero may result in PCOS phenotype after birth. Such a phenomenon has been formulated as the fetal origin of PCOS, which intends to give a possible explanation for PCOS aetiology. Given that the epigenetic modifications are usually involved in the development and inheritance of many adult diseases with fetal origin, we propose a hypothesis here referred to as "epigenetic abnormality underlying the fetal origin of PCOS". It states that in utero hyperandrogenism exposure may disturb the epigenetic reprogramming in fetal reproductive tissue, thereby resulting in postnatal POCS phenotype in women of reproductive age. Meanwhile, the incomplete erasure of such epigenetic abnormality in germ cells after fertilization may promote the transgenerational inherence of POCS. Thus, this epigenetic abnormality hypothesis has established a novel mechanism for PCOS development and inheritance. If verified, our hypothesis would open new avenues for the possible intervention at the critical period of prenatal life to prevent PCOS development and inheritance in adult women. Moreover, analysis of the epigenetic phenotypes and identification of specific epigenetic changes may help develop new tools for monitoring fetal development under an in utero hyperandrogenism environment.
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
Magsci [本文引用: 1]
表观遗传学是研究没有DNA序列变化的、可遗传的基因表达改变。表观遗传修饰可以参与多种生命过程, 其在糖脂代谢中也发挥了重要的作用。生物体内的糖脂代谢关系密切, 糖脂代谢紊乱会导致多种代谢性疾病的发生。文章主要从DNA甲基化、组蛋白修饰、非编码RNA调控等方面综述了表观遗传修饰在糖脂代谢中的研究进展。
Magsci [本文引用: 1]
表观遗传学是研究没有DNA序列变化的、可遗传的基因表达改变。表观遗传修饰可以参与多种生命过程, 其在糖脂代谢中也发挥了重要的作用。生物体内的糖脂代谢关系密切, 糖脂代谢紊乱会导致多种代谢性疾病的发生。文章主要从DNA甲基化、组蛋白修饰、非编码RNA调控等方面综述了表观遗传修饰在糖脂代谢中的研究进展。
URLPMID:16914156 [本文引用: 1]
Exposure to famine in utero may reduce IMT. However, it does not reduce the risk of coronary heart disease among famine exposed people.
URLPMID:21097519 [本文引用: 1]
Abstract The effect of in utero exposure to a maternal high-fat diet on the peripheral circadian system of the fetus is unknown. Using mRNA copy number analysis, we report that the components of the peripheral circadian machinery are transcribed in the nonhuman primate fetal liver in an intact phase-antiphase fashion and that Npas2, a paralog of the Clock transcription factor, serves as the rate-limiting transcript by virtue of its relative low abundance (10- to 1000-fold lower). We show that exposure to a maternal high-fat diet in utero significantly alters the expression of fetal hepatic Npas2 (up to 7.1-fold, P0.05). Although the Npas2 promoter remains largely unmethylated, differential Npas2 promoter occupancy of acetylation of fetal histone H3 at lysine 14 (H3K14ac) occurs in response to maternal high-fat diet exposure compared with control diet-exposed animals. Furthermore, we find that disruption of Npas2 is consistent with high-fat diet exposure in juvenile animals, regardless of in utero diet exposure. In summary, the data suggest that peripheral Npas2 expression is uniquely vulnerable to diet exposure.
URLPMID:23406533 [本文引用: 1]
Background Accumulating evidence reveals that intrauterine growth retardation (IUGR) can cause varying degrees of pulmonary arterial hypertension (PAH) later in life. Moreover, epigenetics plays an important role in the fetal origin of adult disease. The goal of this study was to investigate the role of epigenetics in the development of PAH following IUGR. Methods The IUGR rats were established by maternal undernutrition during pregnancy. Pulmonary vascular endothelial cells (PVEC) were isolated from the rat lungs by magnetic-activated cell sorting (MACS). We investigated epigenetic regulation of the endothelin-1 (ET-1) gene in PVEC of 1-day and 6-week IUGR rats, and response of IUGR rats to hypoxia. Results The maternal nutrient restriction increased the histone acetylation and hypoxia inducible factor-1?? (HIF-1??) binding levels in the ET-1 gene promoter of PVEC in IUGR newborn rats, and continued up to 6 weeks after birth. These epigenetic changes could result in an IUGR rat being highly sensitive to hypoxia later in life, causing more significant PAH or pulmonary vascular remodeling. Conclusions These findings suggest that epigenetics is closely associated with the development of hypoxic PAH following IUGR, further providing a new insight for improved prevention and treatment of IUGR-related PAH.
URLPMID:24944010 [本文引用: 1]
The prevalence of type-2 diabetes (T2D) is increasing significantly throughout the globe since the last decade. This heterogeneous and multifactorial disease, also known as insulin resistance, is caused by the disruption of the insulin signaling pathway. In this review, we discuss the existence of various miRNAs involved in regulating the main protein cascades in the insulin signaling pathway that affect insulin resistance. The influence of miRNAs (miR-7, miR-124a, miR-9, miR-96, miR-15a/b, miR-34a, miR-195, miR-376, miR-103, miR-107, and miR-146) in insulin secretion and beta ($\beta$) cell development has been well discussed. Here, we highlight the role of miRNAs in different significant protein cascades within the insulin signaling pathway such as miR-320, miR-383, miR-181b with IGF-1, and its receptor (IGF1R); miR-128a, miR-96, miR-126 with insulin receptor substrate (IRS) proteins; miR-29, miR-384-5p, miR-1 with phosphatidylinositol 3-kinase (PI3K); miR-143, miR-145, miR-29, miR-383, miR-33a/b miR-21 with AKT/protein kinase B (PKB) and miR-133a/b, miR-223, miR-143 with glucose transporter 4 (GLUT4). Insulin resistance, obesity, and hyperlipidemia (high lipid levels in the blood) have a strong connection with T2D and several miRNAs influence these clinical outcomes such as miR-143, miR-103, and miR-107, miR-29a, and miR-27b. We also corroborate from previous evidence how these interactions are related to insulin resistance and T2D. The insights highlighted in this review will provide a better understanding on the impact of miRNA in the insulin signaling pathway and insulin resistance-associated diagnostics and therapeutics for T2D.
URLPMID:23782253 [本文引用: 1]
Abstract Epigenetics has emerged as a new and promising field in recent years. Lifestyle, stress, drugs, physiopathological situations and pharmacological interventions have a great impact on the epigenetic code of the cells by altering the methylome, miRNA expression and the covalent histone modifications. Since there exists a need to find new biomarkers and improve diagnosis for several diseases, the research on epigenetic biomarkers for molecular diagnostics encourages the translation of this field from the bench to clinical practice. In this context, deciphering intricate epigenetic modifications involved in several molecular processes is a challenge that will be solved in the near future. In this review, the authors present an overview of the high-throughput technologies and laboratory techniques available for epigenetic studies, and also discuss which of them are more reliable to be used in a clinical diagnostic laboratory. In addition, the authors describe the most promising epigenetic biomarkers in lung, colorectal and prostate cancer, in which most advances have been achieved. Finally, the authors describe epigenetic biomarkers in some rare diseases; these rare syndromes are paradigms for a specific impaired molecular pathway, thus providing valuable information on the discovery of new epigenetic biomarkers.
URLPMID:25939322 [本文引用: 1]
Abstract Lung cancer is the leading cause of cancer-related deaths worldwide. The initiation and progression of lung cancer is the result of the interaction between permanent genetic and dynamic epigenetic alterations. DNA methylation is the best studied epigenetic mark in human cancers. Altered DNA methylation in cancer was identified in 1983. Within 30 years of this discovery, DNA methylation inhibitors are used clinically to treat a variety of cancers, highlighting the importance of the epigenetic basis of cancer. In addition, histone modifications, nucleosome remodeling, and micro RNA (miRNA)-mediated gene regulation are also fundamental to tumor genesis. Distinct chromatin alterations occur in all stages of lung cancer, including initiation, growth, and metastasis. Therefore, stage-specific epigenetic changes can be used as powerful and reliable tools for early diagnosis of lung cancer and to monitor patient prognosis. Moreover, since epigenetic changes are dynamic and reversible, chromatin modifiers are promising targets for the development of more effective therapeutic strategies against cancer. This review summarizes the chromatin alterations in lung cancer, focusing on the diagnostic and therapeutic approaches targeting epigenetic modifications that could help to reduce the high case-fatality rate of this dreadful disease.
URLPMID:17301249 [本文引用: 1]
Abstract Physical activity reduces breast cancer risk. Promoter hypermethylation of the tumor suppressor genes APC and RASSF1A, which is potentially reversible, is associated with breast cancer risk. We conducted a cross-sectional study in 45 women without breast cancer to determine the association of physical activity with promoter hypermethylation of APC and RASSF1A in breast tissue. We used quantitative methylation-specific PCR to test the methylation status of APC and RASSF1A, and questionnaires to assess study covariates and physical activity (measured in metabolic equivalent hours per week). In univariate analyses, the study covariate, benign breast biopsy number, was positively associated with promoter hypermethylation of APC (P = 0.01) but not RASSF1A. Mulitvariate logistic regression indicated that, although not significant, physical activities for a lifetime [odds ratio (OR), 0.57; 95% confidence interval (95% CI), 0.22-1.45; P = 0.24], previous 5 years (OR, 0.62; 95% CI, 0.34-1.12; P = 0.11), and previous year (OR, 0.72; 95% CI, 0.43-1.22; P = 0.22) were inversely related to promoter hypermethylation of APC but not RASSF1A for all physical activity measures. Univariate logistic regression indicated that physical activities for a lifetime, previous 5 years, and previous year were inversely associated with benign breast biopsy number, and these results were approaching significance for lifetime physical activity (OR, 0.41; 95% CI, 0.16-1.01; P = 0.05) and significant for physical activity in the previous 5 years (OR, 0.57; 95% CI, 0.34-0.94; P = 0.03). The study provides indirect evidence supporting the hypothesis that physical activity is inversely associated with promoter hypermethylation of tumor suppressor genes, such as APC, in nonmalignant breast tissue.
URLPMID:4746937 [本文引用: 1]
Background: To develop a non-invasive screening method for colorectal cancer, we evaluated the methylation of ALX4 gene promoter in serum samples from patients with colorectal cancer (CRC) and equal number of healthy individuals. Materials and Methods: In serum samples from 25 patients with colorectal cancer and 25 healthy control subjects, isolated serum free-floating DNA was treated with sodium bisulfite and analyzed by methylation-specific polymerase chain reaction (MSP) with primers specific for methylated or unmethylated promoter CpG island sequences of the ALX4 gene. Results: Methylation of the ALX4 gene promoter was present in the serum DNA of patients with adenoma and colorectal cancer. A sensitivity of 68% and specificity of 88% were achieved in the detection of promoter methylation in colorectal neoplasia samples. The difference in methylation status of the ALX4 promoter between the patients with colorectal neoplasia and the control group was statistically highly significant (P < 0.001). Conclusions: The results indicate that this serum free DNA test of methylation of the ALX4 gene promoter is a sensitive and specific method. Therefore in combination with other useful markers it seems ALX4 has the potential of a clinically useful test for the early detection of colorectal cancer. Keywords: Colorectal cancer, DNA methylation, Free-floating DNA, non-invasive colorectal cancer diagnosis
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
Cancer cells exhibit alterations in histone modification patterns at individual genes and globally at the level of single nuclei in individual cells. We demonstrated previously that lower global/cellular levels of histone H3 lysine 4 dimethylation (H3K4me2) and H3K18 acetylation (ac) predict a higher risk of prostate cancer recurrence. Here we show that the cellular levels of both H3K4me2 and H3K18ac also predict clinical outcome in both lung and kidney cancer patients, with lower levels predicting significantly poorer survival probabilities in both cancer groups. We also show that lower cellular levels of H3K9me2, a modification associated with both gene activity and repression, is also prognostic of poorer outcome for individuals with either prostate or kidney cancers. The predictive power of these histone modifications was independent of tissue-specific clinicopathological variables, the proliferation marker Ki-67, or a p53 tumor suppressor mutation. Chromatin immunoprecipitation experiments indicated that the lower cellular levels of histone modifications in more aggressive cancer cell lines correlated with lower levels of modifications at DNA repetitive elements but not with gene promoters across the genome. Our results suggest that lower global levels of histone modifications are predictive of a more aggressive cancer phenotype, revealing a surprising commonality in prognostic epigenetic patterns of adenocarcinomas of different tissue origins.
URLPMID:21386836 [本文引用: 2]
Epigenetics is one of the most promising and expanding fields in the current biomedical research landscape. Since the inception of epigenetics in the 1940s, the discoveries regarding its implications in normal and disease biology have not stopped, compiling a vast amount of knowledge in the past decade. The field has moved from just one recognized marker, DNA methylation, to a variety of others, including a wide spectrum of histone modifications. From the methodological standpoint, the successful initial single gene candidate approaches have been complemented by the current comprehensive epigenomic approaches that allow the interrogation of genomes to search for translational applications in an unbiased manner. Most important, the discovery of mutations in the epigenetic machinery and the approval of the first epigenetic drugs for the treatment of subtypes of leukemias and lymphomas has been an eye-opener for many biomedical scientists and clinicians. Herein, we will summarize the progress in the field of cancer epigenetics research that has reached mainstream oncology in the development of new biomarkers of the disease and new pharmacological strategies.
URLPMID:27671170 [本文引用: 1]
Decitabine has been found to have anti-metabolic and anti-tumor activities in various tumor cells.
URLPMID:11085525 [本文引用: 1]
Loss of DNA mismatch repair because of hypermethylation of the hMLH1 gene promoter occurs at a high frequency in a number of human tumors. A role for loss of mismatch repair (MMR) in resistance to a number of clinically important anticancer drugs has been shown. We have investigated whether the demethylating agent 2'-deoxy-5-azacytidine (DAC) can be used in vivo to sensitize MMR-deficient, drug-resistant ovarian (A2780/cp70) and colon (SW48) tumor xenografts that are MLH1 negative because of gene promoter hypermethylation. Treatment of tumor-bearing mice with the demethylating agent DAC at a nontoxic dose induces MLH1 expression. Re-expression of MLH1 is associated with a decrease in hMLH1 gene promoter methylation. DAC treatment alone has no effect on the growth rate of the tumors. However, DAC treatment sensitizes the xenografts to cisplatin, carboplatin, temozolomide, and epirubicin. Sensitization is comparable with that obtained by reintroduction of the hMLH1 gene by chromosome 3 transfer. Consistent with loss of MMR having no effect on sensitivity in vitro to Taxol, DAC treatment has no effect on the Taxol sensitivity of the xenografts. DAC treatment does not sensitize xenografts of HCT116, which lacks MMR because of hMLH1 mutation. Because there is emerging data on the role of loss of MMR in clinical drug resistance, DAC could have a role in increasing the efficacy of chemotherapy for patients whose tumors lack MLH1 expression because of hMLH1 promoter methylation.
[本文引用: 1]
URLPMID:40537331 [本文引用: 1]
Background Several recent studies reported aging effects on DNA methylation levels of individual CpG dinucleotides. But it is not yet known whether aging-related consensus modules, in the form of clusters of correlated CpG markers, can be found that are present in multiple human tissues. Such a module could facilitate the understanding of aging effects on multiple tissues. Results We therefore employed weighted correlation network analysis of 2,442 Illumina DNA methylation arrays from brain and blood tissues, which enabled the identification of an age-related co-methylation module. Module preservation analysis confirmed that this module can also be found in diverse independent data sets. Biological evaluation showed that module membership is associated with Polycomb group target occupancy counts, CpG island status and autosomal chromosome location. Functional enrichment analysis revealed that the aging-related consensus module comprises genes that are involved in nervous system development, neuron differentiation and neurogenesis, and that it contains promoter CpGs of genes known to be down-regulated in early Alzheimer's disease. A comparison with a standard, non-module based meta-analysis revealed that selecting CpGs based on module membership leads to significantly increased gene ontology enrichment, thus demonstrating that studying aging effects via consensus network analysis enhances the biological insights gained. Conclusions Overall, our analysis revealed a robustly defined age-related co-methylation module that is present in multiple human tissues, including blood and brain. We conclude that blood is a promising surrogate for brain tissue when studying the effects of age on DNA methylation profiles.
URLPMID:23224755 [本文引用: 1]
Inheired or acquired hyperhomocysteinemia (HHcy) is associated with several impairments, as certain tumors, deep venous thrombosis, tube neural defects, osteoporosis, early atherosclerosis and vascular acute events (IMA, stroke, PVD), mild cognitive impairments till Alzheimer's disease (AD). But, vascular and neuronal derangements are the most frequent HHcy-manifestations. As far as early atherosclerosis, some clinical trials demonstrated that folates and B6-12 vitamins supplementation is unable to reduce atherosclerotic lesions and cardiovascular events, even if it lowers HHcy levels. Thus, for atherosclerosis and its acute events (IMA, stroke, PVD) HHcy acts as a powerful biomarker rather than a risk factor. For that, the supplementation with folates and B vitamins to lower atherosclerotic lesions-events in hyperhomocysteinemic patients is not recommended. On the contrary, several clinical investigations demonstrated that folates and vitamins administration is able to reduce Hcy serum levels and antagonize some mechanisms favouring neurodegenerative impairments, as mild cognitive impairment, AD and dementia. Thus, contrarily to the atherosclerotic manifestations in hyperhomocysteinemic patients, preventive treatment with folates and B6-12 vitamins reduces Hcy concentration and could prevent or delay cognitive decline and AD.
URLPMID:18410512 [本文引用: 1]
Abstract Top of page Abstract Materials and methods Results Discussion Acknowledgement References Despite recent advances in the treatment of Parkinson disease (PD), levodopa remains the most effective and widely used therapy. A major limitation to the use of levodopa is the development of abnormal involuntary movements, termed levodopa-induced dyskinesia (LDID), following chronic levodopa treatment. Since recent studies have suggested that modifications of chromatin structure may be responsible for many long-lasting changes in brain function, we have examined post-translational modifications of striatal histones in two models of LDID: an acute murine model and a chronic macaque monkey model, both exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). In the primate model, which closely resembles human LDID, we observed that chronic levodopa and the appearance of LDID was associated with marked deacetylation of histone H4, hyperacetylation and dephosphorylation of histone H3, and enhancement of the phosphorylation of extracellular signal-regulated kinase (ERK). In the murine model of acutely rather than chronically induced LDID, dopamine depletion and levodopa treatment also induced deacetylation of histone H4 and phosphorylation of ERK, but histone H3 exhibited decreased trimethylation and reduced rather than enhanced acetylation. These data demonstrate striking changes in striatal histones associated with the induction of LDID in both animal models. The pattern of changes observed, as well as the behavioral features, differed in the two models. However, both models exhibit marked deacetylation of histone H4, suggesting that inhibitors of H4 deacetylation may be useful in preventing or reversing LDID.
URLPMID:23688931 [本文引用: 1]
The vast majority of Alzheimer's disease (AD) are late-onset forms (LOAD) likely due to the interplay of environmental influences and individual genetic susceptibility. Epigenetic mechanisms, including DNA methylation, histone modifications and non-coding RNAs, constitute dynamic intracellular processes for translating environmental stimuli into modifications in gene expression. Over the past decade it has become increasingly clear that epigenetic mechanisms play a pivotal role in aging the pathogenesis of AD. Here, we provide a review of the major mechanisms for epigenetic modification and how they are reportedly altered in aging and AD. Moreover, we also consider how aberrant epigenetic modifications may lead to AD pathogenesis, and we review the therapeutic potential of epigenetic treatments for AD.
URLPMID:11751536 [本文引用: 1]
Background: Fetal DNA has been detected in maternal plasma by the use of genetic differences between mother and fetus. We explore the possibility of using epigenetic markers for the specific detection of fetal DNA in maternal plasma. Methods: A differentially methylated region in the human IGF2-H19 locus and a single-nucleotide polymorphism in this region were chosen for the study. The methylation status in this region is maintained in such a way that the paternal allele is methylated and the maternal allele is unmethylated. The single-nucleotide polymorphism was typed by direct sequencing of PCR products. The methylation status of this region was ascertained by bisulfite conversion and methylation-specific PCR. Differentially methylated fetal alleles were detected in maternal plasma by direct sequencing and a primer-extension assay. Results: Women in the second (n = 21; 17-21 weeks) and third (n = 18; 37-42 weeks) trimesters of pregnancy were recruited. Among these 39 volunteers, the 16 who were heterozygous for the single-nucleotide polymorphism were chosen for further analysis. In 11 of these 16 cases, paternally inherited methylated fetal alleles were different from the methylated alleles of the respective mothers. Using direct sequencing, we detected paternally inherited methylated fetal DNA in 6 of 11 (55%) cases. In 8 of the 16 heterozygous cases, the fetuses possessed an unmethylated maternally inherited allele that was different from the unmethylated allele of the mother. Using a primer-extension assay, we detected fetal-derived maternally inherited alleles in maternal plasma of four of eight (50%) cases. Conclusions: These results represent the first use of fetal epigenetic markers in noninvasive prenatal analysis. These data may also have implications for the investigation of other types of chimerism.
URLPMID:3035115 [本文引用: 1]
Abstract The presence of fetal DNA in the plasma of pregnant women has opened up new possibilities for noninvasive prenatal diagnosis. Over the past decades, different types of fetal markers have been developed, initially based on discriminative genetic markers such as male-specific signals or paternally-inherited polymorphisms, and gradually evolved to the detection of fetal-specific transcripts or epigenetic signatures. This development has extended the coverage of the application of cell-free fetal DNA to essentially all pregnancies, regardless of the gender of the fetus or its polymorphic status. In this review, we present an overview of the development of noninvasive prenatal diagnosis through epigenetics. We introduce the basis of how fetal DNA could be detected from a large background of maternal DNA in maternal plasma based on fetal-specific DNA methylation patterns. We evaluate the methodologies involved and discuss the factors that affect the robustness of the detection. We review the progress in adopting fetal epigenetic markers for noninvasive prenatal assessment of fetal chromosomal aneuploidies and pregnancy-associated disorders. We conclude with comments on the future directions regarding the search for new fetal epigenetic markers and the clinical implementation of epigenetic approaches for noninvasive prenatal diagnosis.
URL [本文引用: 1]
胎盘介于胎儿与母体之间,是维持胎儿宫内生长发育的重要器官.在胎盘的正常发育过程中,子宫正常蜕膜化、滋养层细胞粘附与侵袭、胎盘血管生成与形成、胎盘印记基因表达都受到表观遗传修饰(如DNA甲基化、组蛋白修饰、非编码RNA等)的调控.研究已经证实环境因素如重金属、化合物、现代辅助生殖技术、营养物质均可导致胎盘上多种基因的表观遗传修饰异常.此外,胎盘基因表达存在性别差异也可能与表观遗传修饰有关.目前,在临床上可运用产前DNA甲基化水平分析技术检测异常的表观遗传修饰,并在疾病早期发现并做出诊断,从而为疾病预防及治疗提供依据.本文对胎盘正常发育过程中表观遗传修饰的调控及环境因素所致的胎盘基因表观遗传改变进行了综述,以期对胎盘相关疾病的诊断与治疗提供借鉴和参考.
URL [本文引用: 1]
胎盘介于胎儿与母体之间,是维持胎儿宫内生长发育的重要器官.在胎盘的正常发育过程中,子宫正常蜕膜化、滋养层细胞粘附与侵袭、胎盘血管生成与形成、胎盘印记基因表达都受到表观遗传修饰(如DNA甲基化、组蛋白修饰、非编码RNA等)的调控.研究已经证实环境因素如重金属、化合物、现代辅助生殖技术、营养物质均可导致胎盘上多种基因的表观遗传修饰异常.此外,胎盘基因表达存在性别差异也可能与表观遗传修饰有关.目前,在临床上可运用产前DNA甲基化水平分析技术检测异常的表观遗传修饰,并在疾病早期发现并做出诊断,从而为疾病预防及治疗提供依据.本文对胎盘正常发育过程中表观遗传修饰的调控及环境因素所致的胎盘基因表观遗传改变进行了综述,以期对胎盘相关疾病的诊断与治疗提供借鉴和参考.
URLPMID:22341586 [本文引用: 1]
Our findings suggest that IGF2 plasticity may be mechanistically important in early childhood overweight or obese status. If confirmed in larger studies, these findings suggest aberrant DNA methylation at sequences regulating imprinted genes may be useful identifiers of children at risk for the development of early obesity.
URLPMID:27470058 [本文引用: 1]
Summary Adverse environmental exposures of mothers during fetal period predispose offspring to a range of age-related diseases earlier in life. Here, we set to determine whether a deregulated epigenetic pattern is similar in young animals whose mothers nutrition was modulated during fetal growth to that acquired during normal aging in animals. Using a rodent model of maternal undernutrition (UN) or overnutrition (ON), we examined cytosine methylation profiles of liver from young female offspring and compared them to age-matched young controls and aged (20-month-old) animals. HELP-tagging, a genomewide restriction enzyme and sequencing assay demonstrates that fetal exposure to two different maternal diets is associated with nonrandom dysregulation of methylation levels with profiles similar to those seen in normal aging animals and occur in regions mapped to genes relevant to metabolic diseases and aging. Functional consequences were assessed by gene expression at 9 weeks old with more significant changes at 6 months of age. Early developmental exposures to unfavorable maternal diets result in altered methylation profiles and transcriptional dysregulation in Prkcb, Pc, Ncor2, and Smad3 that is also seen with normal aging. These Notch pathway and lipogenesis genes may be useful for prediction of later susceptibility to chronic disease.
URLPMID:24445802 [本文引用: 1]
Abstract Differences in methylation across tissues are critical to cell differentiation and are key to understanding the role of epigenetics in complex diseases. In this investigation, we found that locus-specific methylation differences between tissues are highly consistent across individuals. We developed a novel statistical model to predict locus-specific methylation in target tissue based on methylation in surrogate tissue. The method was evaluated in publicly available data and in two studies using the latest IlluminaBeadChips: a childhood asthma study with methylation measured in both peripheral blood leukocytes (PBL) and lymphoblastoid cell lines; and a study of postoperative atrial fibrillation with methylation in PBL, atrium and artery. We found that our method can greatly improve accuracy of cross-tissue prediction at CpG sites that are variable in the target tissue [R(2) increases from 0.38 (original R(2) between tissues) to 0.89 for PBL-to-artery prediction; from 0.39 to 0.95 for PBL-to-atrium; and from 0.81 to 0.98 for lymphoblastoid cell line-to-PBL based on cross-validation, and confirmed using cross-study prediction]. An extended model with multiple CpGs further improved performance. Our results suggest that large-scale epidemiology studies using easy-to-access surrogate tissues (e.g. blood) could be recalibrated to improve understanding of epigenetics in hard-to-access tissues (e.g. atrium) and might enable non-invasive disease screening using epigenetic profiles.
URLPMID:3446315 [本文引用: 1]
Background Dynamic changes to the epigenome play a critical role in establishing and maintaining cellular phenotype during differentiation, but little is known about the normal methylomic differences that occur between functionally distinct areas of the brain. We characterized intra- and inter-individual methylomic variation across whole blood and multiple regions of the brain from multiple donors. Results Distinct tissue-specific patterns of DNA methylation were identified, with a highly significant over-representation of tissue-specific differentially methylated regions (TS-DMRs) observed at intragenic CpG islands and low CG density promoters. A large proportion of TS-DMRs were located near genes that are differentially expressed across brain regions. TS-DMRs were significantly enriched near genes involved in functional pathways related to neurodevelopment and neuronal differentiation, including BDNF, BMP4, CACNA1A, CACA1AF, EOMES, NGFR, NUMBL, PCDH9, SLIT1, SLITRK1 and SHANK3. Although between-tissue variation in DNA methylation was found to greatly exceed between-individual differences within any one tissue, we found that some inter-individual variation was reflected across brain and blood, indicating that peripheral tissues may have some utility in epidemiological studies of complex neurobiological phenotypes. Conclusions This study reinforces the importance of DNA methylation in regulating cellular phenotype across tissues, and highlights genomic patterns of epigenetic variation across functionally distinct regions of the brain, providing a resource for the epigenetics and neuroscience research communities.
URLPMID:26623032 [本文引用: 1]
The current standard for prenatal screening is mostly based on biochemical marker tests and the use of ultrasonography. There is no secure stand-alone screening marker for congenital heart (CHDs). MicroRNAs (miRNAs) that are associated with cardiogenesis enter the maternal peripheral bloodstream during pregnancy and allow non-invasive prenatal testing (NIPT). The present study investigated the plasma expression profile of fetal hsa-miR-99a in maternal blood. Peripheral blood samples were collected from 39 pregnant patients, comprising 22 with CHD-positive fetuses and 17 with CHD-free controls. miRNAs were isolated from the maternal serum and -quantitative polymerase chain reaction was carried out to determine the expression of hsa-miR-99a. While the miRNA concentrations were almost identical among the affected and control groups (5.54 vs. 6.40 ng/08l), significantly upregulated hsa-miR-99a levels were identified in the affected group (1.78×10(-2)±3.53×10(-2) vs. 1.09×10(-3)±3.55×10(-3) ng/08l, P=0.038). In conclusion, according to the present study, hsa-miR-99a is involved in cardiac malformation and may serve as a biomarker during fetal development, and therefore presents as a candidate for monitoring cardiomyogenesis and potential use as a NIPT-biomarker for fetal CHD.
[本文引用: 1]
URLPMID:20388836 [本文引用: 1]
Studies in and suggest that intrauterine retardation (IUGR) permanently resets the hypothalamic-pituitary-adrenal (HPA) axis. HPA axis reprogramming may involve persistently altered expression of the hippocampal (hpGR), an important regulator of HPA axis reactivity. Persistent alteration of , long after the inciting event, is thought to be mediated by epigenetic mechanisms that affect mRNA and mRNA variant expression. GR mRNA variants in both and include eleven 5'-end variants and GRalpha, the predominant 3'-end variant. The 3'-end variants associated with in (GRbeta, GRgamma, GRA, and ) have not been reported in . We hypothesized that in the hippocampus IUGR would decrease total GR mRNA, increase GRbeta, GRgamma, GRA, and , and affect epigenetics of the GR gene at birth (D0) and at 21 days of life (D21). IUGR increased hpGR and exon 1.7 hpGR mRNA in males at D0 and D21, associated with increased trimethyl H3/K4 at exon 1.7 at both time points. IUGR also increased hpGRgamma in males at D0 and D21, associated with increased H3/K9 at exon both time points. hpGRA increased in female IUGR at D0 and D21. In addition, our data support the existence of hpGRbeta and hpGRP in the . IUGR has sex-specific, persistent effects on GR expression and its code. We speculate that postnatal changes in hippocampal GR variant and total mRNA expression may underlie IUGR-associated HPA axis reprogramming.
URLPMID:15930441 [本文引用: 1]
Environmental constraints during early life result in phenotypic changes that can be associated with increased disease risk in later life. This suggests persistent alteration of gene transcription. DNA methylation, which is largely established in utero, provides a causal mechanism by which unbalanced prenatal nutrition results in such altered gene expression. We investigated the effect of unbalanced maternal nutrition on the methylation status and expression of the glucocorticoid receptor (GR) and peroxisomal proliferator-activated receptor (PPAR) genes in rat offspring after weaning. Dams were fed a control protein (C; 180 g/kg protein plus 1 mg/kg folic acid), restricted protein (R; 90 g/kg casein plus 1 mg/kg folic acid), or restricted protein plus 5 mg/kg folic acid (RF) diet throughout pregnancy. Pups were killed 6 d after weaning (= 10 per group). Gene methylation was determined by methylation-sensitive PCR and mRNA expression by semiquantitative RT-PCR. PPAR gene methylation was 20.6% lower (< 0.001) and expression 10.5-fold higher in R compared with C pups. GR gene methylation was 22.8% lower (< 0.05) and expression 200% higher (< 0.01) in R pups than in C pups. The RF diet prevented these changes. PPAR methylation status and expression did not differ among the groups. Acyl-CoA oxidase expression followed that of PPAR. These results show that unbalanced prenatal nutrition induces persistent, gene-specific epigenetic changes that alter mRNA expression. Epigenetic regulation of gene transcription provides a strong candidate mechanism for fetal programming.
URLPMID:17646663 [本文引用: 1]
Developmental plasticity in response to environmental cues can take the form of polyphenism, as for the discrete morphs of some insects, or of an apparently continuous spectrum of phenotype, as for most mammalian traits. The metabolic phenotype of adult rats, including the propensity to obesity, hyperinsulinemia, and hyperphagia, shows plasticity in response to prenatal nutrition and to neonatal administration of the adipokine leptin. Here, we report that the effects of neonatal leptin on hepatic gene expression and epigenetic status in adulthood are directionally dependent on the animal's nutritional status in utero. These results demonstrate that, during mammalian development, the direction of the response to one cue can be determined by previous exposure to another, suggesting the potential for a discontinuous distribution of environmentally induced phenotypes, analogous to the phenomenon of polyphenism.
URLPMID:11250138 [本文引用: 1]
Histone acetyltransferases (HATs) directly link chromatin modification to gene activation. Recent structure/function studies provide insights into HAT catalysis and histone binding, and genetic studies suggest cross-talk between acetylation and other histone modifications. Developmental aberrations in mice and certain human cancers are associated with HAT mutations, further highlighting the importance of these enzymes to normal cell growth and differentiation.
URLPMID:22711276 [本文引用: 1]
BACKGROUND: Epigenetic programming, dynamically regulated by histone acetylation, is a key mechanism regulating cell proliferation and survival. Little is known about the contribution of histone deacetylase (HDAC) activity to the development of pulmonary arterial hypertension, a condition characterized by profound structural remodeling of pulmonary arteries and arterioles. METHODS AND RESULTS: HDAC1 and HDAC5 protein levels were elevated in lungs from human idiopathic pulmonary arterial hypertension and in lungs and right ventricles from rats exposed to hypoxia. Immunohistochemistry localized increased expression to remodeled vessels in the lung. Both valproic acid, a class I HDAC inhibitor, and suberoylanilide hydroxamic acid (vorinostat), an inhibitor of class I, II, and IV HDACs, mitigated the development of and reduced established hypoxia-induced pulmonary hypertension in the rat. Both valproic acid and suberoylanilide hydroxamic acid inhibited the imprinted highly proliferative phenotype of fibroblasts and R-cells from pulmonary hypertensive bovine vessels and platelet-derived growth factor-stimulated growth of human vascular smooth muscle cells in culture. Exposure to valproic acid and suberoylanilide hydroxamic acid was associated with increased levels of p21 and FOXO3 and reduced expression of survivin. The significantly higher levels of expression of cKIT, monocyte chemoattractant protein-1, interleukin-6, stromal-derived factor-1, platelet-derived growth factor-b, and S100A4 in R-cells were downregulated by valproic acid and suberoylanilide hydroxamic acid treatment. CONCLUSIONS: Increased HDAC activity contributes to the vascular pathology of pulmonary hypertension. The effectiveness of HDAC inhibitors, valproic acid, and suberoylanilide hydroxamic acid, in models of pulmonary arterial hypertension supports a therapeutic strategy based on HDAC inhibition in pulmonary arterial hypertension.
URLPMID:25487010 [本文引用: 1]
Histone deacetylases (HDACs) have recently emerged as key elements in epigenetic control of gene expression. Due to the implication of HDACs in a variety of diseases ranging from cancer to neurodegenerative disorder, HDAC inhibitors have received increased attention in recent years. Over the last few decades, a myriad of HDAC inhibitors containing a wide variety of structural features have been identified from natural sources. Here, we review the discovery, synthesis, biological properties, and modes of action of these naturally occurring HDAC inhibitors and consider their implications for future research.
URLPMID:25581683 [本文引用: 1]
Abstract Zinc-dependent histone deacetylases (HDACs), a family of hydrolases that remove acetyl groups from lysine residues, play an important role in the regulation of multiple processes, from gene expression to protein activity. The dysregulation of HDACs is associated with many diseases including cancer, neurological disorders, cellular metabolism disorders, and inflammation. Molecules that act as HDAC inhibitors (HDACi) exhibit a variety of related bioactivities. In particular, HDACi have been applied clinically for the treatment of cancers. Inhibition through competitive binding of the catalytic domain of these enzymes has been achieved by a diverse array of small-molecule chemotypes, including a number of natural products. This review provides a systematic introduction of natural HDACi, with an emphasis on their enzyme inhibitory potency, selectivity, and biological activities, highlighting their various binding modes with HDACs.

{kind=link}
{kind=link}