 ,1,2
,1,2The genetic background and application of Down syndrome mouse models
Xiaowei Meng1, Jie Wang1,2, Qingwen Ma,1,2通讯作者:
第一联系人:
编委: 卢大儒
收稿日期:2017-08-22修回日期:2017-12-5网络出版日期:2018-03-20
| 基金资助: |
Editorial board:
Received:2017-08-22Revised:2017-12-5Online:2018-03-20
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (449KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孟晓伟, 汪洁, 马晴雯. 唐氏综合征小鼠模型的遗传背景和应用. 遗传[J], 2018, 40(3): 207-217 doi:10.16288/j.yczz.17-279
Xiaowei Meng, Jie Wang, Qingwen Ma.
唐氏综合征(Down syndrome, DS)又名21三体综合征或先天愚型,是目前最常见的染色体异常疾病,主要由配子减数分裂时21号染色体不分离造 成。1866年,Langdon Down首次描述了DS的临床表现[1];1959年,Lejeune等[2]首次揭示其发病原因。DS患者表型特征主要包括:外貌异于常人(头颅小而圆、鼻扁平、眼距过宽),智力低下(平均智商20~25之间),无生活自理能力,发育较正常儿童迟缓,男性患者无生育能力,女性患者部分可育。常见的DS伴发疾病主要包括先天性心脏缺陷(congenital heart defects, CHD)[3](如房室间隔缺损(atrentricular septal defects, AVSD)、室间隔缺损(ventricular septal defects, VSD)、房间隔缺损(atrial septal defects, ASD)和法络四联症(tetralogy of Fallot, TOF))、消化器官畸形、急性白血病和早发性阿尔茨海默病等[4]。按核型的不同可以将DS分为3类亚型[5]:典型的21三体(47,+21),该亚型最为常见,约占94%;易位型,主要为罗伯逊易位,约占3%~4%;嵌合型(47,+21/46),约占2%~3%。
目前对DS的研究主要集中在细胞水平和动物模型上,这不仅推进了疾病本身的检测和治疗,同时也为研究其他染色体异常综合征提供了参考。在动物模型选用方面,小鼠(Mus musculus)和人类(Homo sapiens)基因同源性高达95%,是理想的疾病动物模型。理论上,采用合适手段在小鼠体内重复人类21号染色体(human chromosome 21, Hsa21)同源片段,可以在一定程度上模拟DS表型并满足研究需要。自1990年至今,科学家们已陆续制备了多种DS小鼠模型,满足了科学研究和药物测试的需求。本文首先介绍了Hsa21的结构及其小鼠直系同源片段在染色体中的分布,重点介绍了不同时期DS小鼠模型的优势和局限,以期为科研人员在DS研究中对不同小鼠模型的选用提供参考。
1 人类21号染色体及其在小鼠染色体中的同源片段
Hsa21是人类染色体组中最小的常染色体,总长约为46.7 Mb。根据着丝粒位置可将其分为短臂和长臂两部分,分别以21p和21q表示,大小约为10.6 Mb和33.7 Mb(参照公共数据库Ensembl Genome Browser, http://www.ensembl.org/index.html)。21p包含15个蛋白编码基因,其中11个在21q22.3上存在重复拷贝,相对而言21q包含了更多的蛋白编码基因,并且这些基因具有进化保守性。因而DS研究的关键在于分析21q的重复对表型的影响。目前研究表明,21q约含218个蛋白编码基因,其中49个编码角蛋白关联蛋白(keratin associated proteins, KAPs),主要是为毛发纤维提供结构支持并维持其稳定性[6],位于21q22.11和21q22.3区间;164个编码转录因子、智障相关蛋白、阿尔茨海默病相关蛋白、RNA加工蛋白等已知蛋白[7];5个编码结构和功能未知的新蛋白。此外,21q上还包含280个非编码基因,目前对这些基因的研究相对较少。
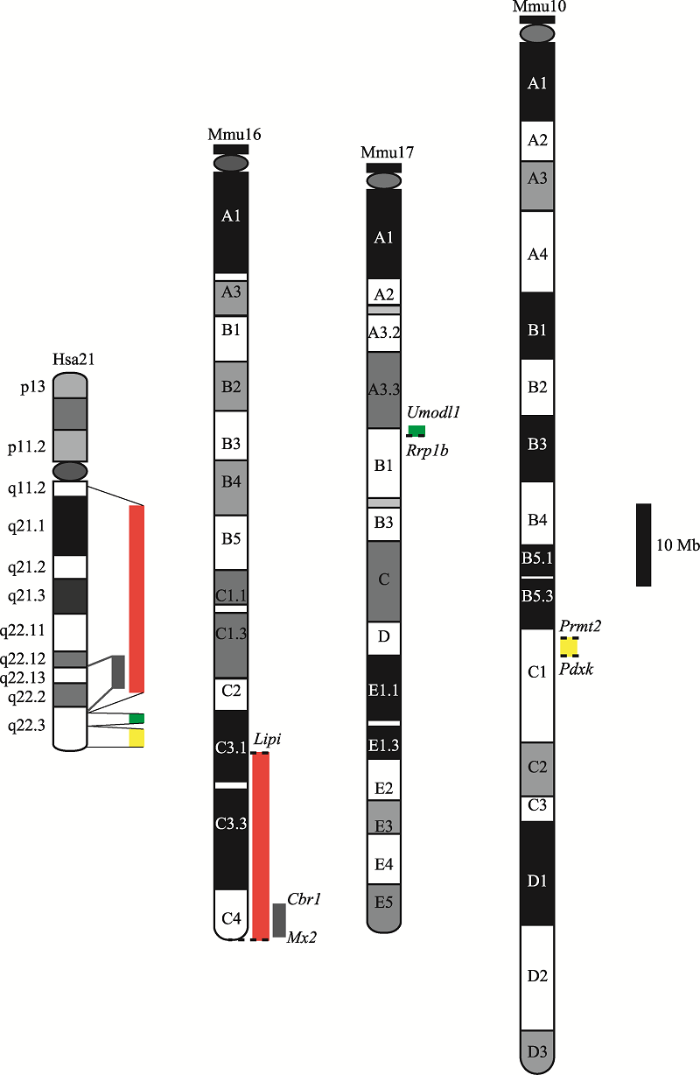
21q在小鼠中的直系同源基因主要分布于16、17、10号染色体上(Mmu16、17、10)。如图1所示,21q在Mmu16上的直系同源基因分布于Lipi基因和端粒之间,区间大小约为27.9 Mb;在Mmu17上的直系同源基因分布于Umodl1和Rrp1b基因之间,区间大小约为1.63 Mb;在Mmu10上的直系同源基因分布于Prmt2和Pdxk基因之间,区间大小约为2.95 Mb[8]。这些直系同源基因包括KAPs的两个基因簇和158个蛋白编码基因:KAPs的两个基因簇分别位于Mmu16和Mmu10上;158个蛋白编码基因中有102个位于Mmu16上,19个位于Mmu17上,37个位于Mmu10上[9]。
21q上的基因和其小鼠直系同源基因的差异在于:(1) 21q上5个新蛋白编码基因和一些非编码基因在小鼠中没有直系同源物[10,11];(2) Mmu16上直系同源基因分布区内的部分基因,如ITGB2L(integrin beta 2-like)蛋白编码基因和3个未知蛋白的编码基因在21q中没有直系同源物。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1人类21号染色体(Hsa21)和小鼠染色体的同源性比较
红色矩形表示Mmu16上的Hsa21直系同源区,位于Lipi基因和端粒之间;绿色矩形表示Mmu17上的Hsa21直系同源区,位于Umodl1和Rrp1b基因之间;黄色矩形表示Mmu10上的Hsa21直系同源区,位于Prmt2和Pdxk基因之间;灰色矩形表示Mmu16上与Hsa21上唐氏综合征关键区(Down syndrome critical region, DSCR)直系同源的区域,位于Cbr1和Mx2基因之间。
Fig.1Human chromosome 21 (Has21) and its orthologs in mouse chromosomes
识别特定基因的三体性对DS表型的贡献是研究人员一直追求的目标,根据小鼠基因与Hsa21的同源性,采用单基因敲除和转基因技术建立重复不同片段的DS小鼠模型,在克服伦理限制的同时也能对特定基因进行针对性研究。但小鼠模型亦有其不足之处:首先,小鼠与人之间存在种间差异,基因组背景并不相同,造成特定基因对表型的影响程度不同;其次,小鼠中一些三体基因的组合会导致死胎情况出现,致使该基因组合的漏缺。因此,在使用DS小鼠模型进行研究前要对不同小鼠模型进行基因型和表型评估。
2 常用DS小鼠模型
自1990年第一只可用的DS小鼠模型——Ts65Dn小鼠建立至今,已发展出数种不同功能特点的DS小鼠模型。每种小鼠模型都具有其独特的遗传学和表型特征(表1),在智障表型模拟、DS相关CHD模拟和认知类药物筛选等方面发挥着各自独特的作用。2.1 Ts65Dn小鼠
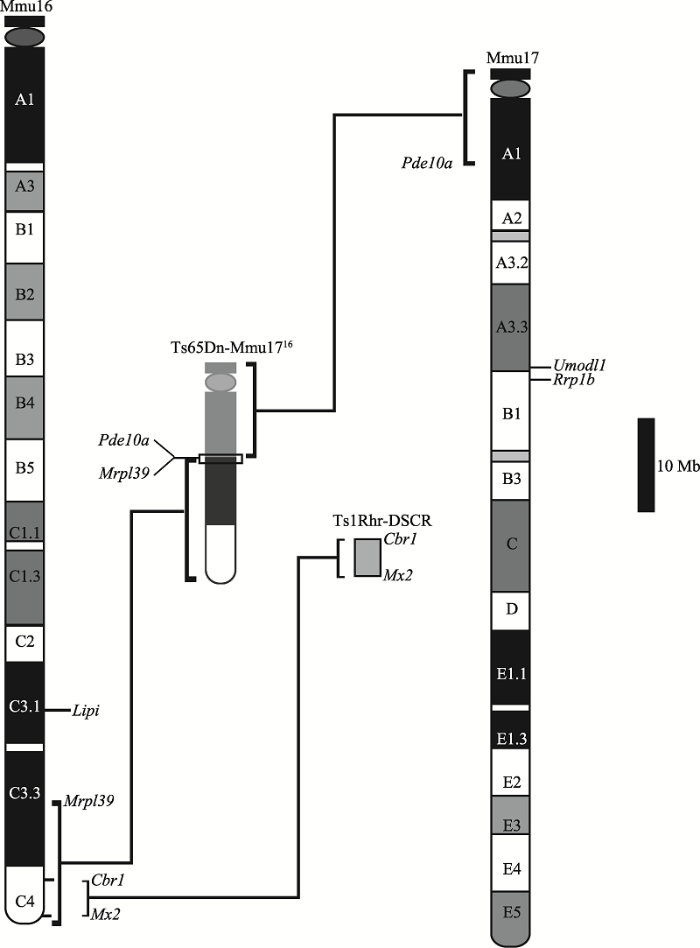
Ts65Dn小鼠是第一个用于DS研究的小鼠模型,由Davisson等[12]于1990年通过电离辐射DBA/2J雄性小鼠,随后选育包含Mmu16易位产物子代而建立的,是使用最广泛的DS小鼠模型,同时也是目前唯一用于临床认知类药物测试的小鼠模型[21]。在表型模拟方面,Ts65Dn小鼠具有认知障碍、先天性心脏缺陷、颅面部畸形和记忆功能损伤等DS特征表型;在基因型方面,Ts65Dn小鼠携带额外的易位染色体Mmu1716。如图2所示,Mmu1716由Mmu16上Mrpl39基因前端断裂点到端粒约13.4 Mb片段和Mmu17上Pde10a基因后端断裂点到着丝粒约9.4 Mb片段融合而成,其中Mmu16重复区与Hsa21同源,包含130个直系同源基因,而Mmu17重复区则与人类6号染色体(human chromosome 6, Hsa6)同源,包含60个基因,其中35个为蛋白编码基因[22]。Ts65Dn小鼠成为目前应用最广泛的DS小鼠模型,其主要原因在于:(1) Ts65Dn小鼠能较好地模拟人类DS患者出现的认知障碍和记忆功能缺陷等表型;(2) Ts65Dn小鼠具有的颅面部畸形、体重减轻等特点,可方便地与普通小鼠区分。但Ts65Dn小鼠所携带的部分Mmu16和Mmu17重复片段中缺失了一部分Hsa21直系同源基因,同时又重复了一部分Hsa6同源基因,由此带来两个问题:(1)未重复的Hsa21直系同源基因是否对DS表型有潜在的影响;(2)重复的Hsa6同源基因是否对DS表型有干扰。Table 1
表1
表1 常见DS小鼠模型一览表
Table 1
| 编号 | 简称 | 建立时间(年) | 重复片段(Mb) | 主要表型 | 参考文献 |
|---|---|---|---|---|---|
| 1 | Ts65Dn | 1990 | 22.8 | 体重减轻、颅面异常、记忆损伤、认知障碍、先天性心脏缺陷 | [12] |
| 2 | Ts1Cje | 1998 | 10.3 | 颅面异常、认知障碍 | [13] |
| 3 | Ts1Rhr | 2004 | 3.9 | — | [14] |
| 4 | Tc1 | 2005 | 42 | 体重减轻、短期记忆损伤、认知障碍、先天性心脏缺陷 | [15] |
| 5 | Dp(16)1Yu | 2007 | 22.9 | 先天性心脏缺陷、胃肠道异常 | [16] |
| 6 | Dp(16)2Yey | 2011 | 5.4 | 先天性心脏缺陷(发病率较低) | [17] |
| 7 | Dp(16)4Yey | 2014 | 3.7 | 先天性心脏缺陷(发病率较低) | [18] |
| 8 | Dp(10)1Yey | 2010 | 2.3 | — | [19] |
| 9 | Dp(17)1Yey | 2010 | 1.1 | — | [19] |
| 10 | TTS | 2010 | 32.5 | 脑积水、先天性心脏缺陷、肌张力低下 | [19] |
| 11 | Dp1Tyb | 2016 | 23 | 先天性心脏缺陷 | [20] |
| 12 | Dp2Tyb | 2016 | 2.5 | 先天性心脏缺陷(发病率较低) | [20] |
| 13 | Dp3Tyb | 2016 | 4.9 | 先天性心脏缺陷 | [20] |
| 14 | Dp4Tyb | 2016 | 1.5 | — | [20] |
| 15 | Dp5Tyb | 2016 | 1.8 | — | [20] |
| 16 | Dp6Tyb | 2016 | 1.6 | — | [20] |
| 17 | Dp9Tyb | 2016 | 15.6 | — | [20] |
新窗口打开|下载CSV
诸多证据表明,Ts65Dn小鼠中未重复的Hsa21直系同源基因对DS表型具有潜在影响。例如:热休克蛋白HSPA13(heat shock 70-kDa protein member 13)在泛素途径中扮演着重要角色,具有调节神经元细胞pH的作用[23],可能与认知功能相关;核受体相互作用蛋白NRIP1(nuclear receptor interacting protein 1,亦称RIP140, receptor-interacting protein 140)参与糖皮质激素和雌激素等受体转录活性的调控,同时也与肌细胞的新陈代谢和脂肪细胞功能调节相关[24];神经细胞粘附分子NCAM2(neural cell adhesion molecule 2)可以调节神经细胞轴突和树突间的相互作用[25]。Ts65Dn小鼠中过表达的非Hsa21直系同源基因对DS表型的影响也很明显,如:SNX9(sorting nexin 9)参与网格蛋白介导的胞吞作用,过表达会造成海马区神经元突触小泡内吞失调[26];DYNLT1(dynein light chains Tctex-type 1)主要参与信号分子的逆向转导,过表达会改变信号分子的组成并降低信号转导效率[27]。此外,雄性Ts65Dn小鼠存在减数分裂异常和不育问题,其种系的保持依赖正常二倍体雄性和雌性Ts65Dn小鼠的杂交,因此DS母体对胚胎发育具有潜在影响。
2.2 Ts1Rhr小鼠
Ts1Rhr小鼠是Olson等[14]于2004年首次使用Cre重组酶介导不同染色体LoxP位点间重组得到的DS小鼠模型。如图2所示,该小鼠模型重复了Mmu16上Cbr1和Mx2基因间约3.9 Mb片段,即DSCR同源序列区。DSCR位于Hsa21上分子标记D21S55和MX1基因之间约5.4 Mb大小区域[14,28]。DSCR假说认为DS的主要表型是由少数剂量敏感型基因调控的,而这些基因所在区域即DSCR区。为验证DSCR假说,Olson团队建立了Ts1Rhr小鼠,使用相同生长周期的Ts65Dn小鼠作为DS表型对照,整倍体小鼠作为正常对照,与Ts1Rhr小鼠进行体型、下颌骨大小和颅面形态对比,结果发现:(1) Ts1Rhr小鼠出生至成年阶段,平均体型大于整倍体正常对照,Ts65Dn小鼠则小于整倍体对照,该结果与DSCR假说的DSCR区是DS患者矮小身材的关键调控区相反;(2) Ts65Dn小鼠下颌骨小于正常对照组,而Ts1Rhr小鼠下颌骨大于正常对照组,该结果亦与DSCR假说相悖;(3)比较Ts1Rhr小鼠和正常对照组的颅面部形态发现,虽然Ts1Rhr小鼠颅骨大于正常小鼠,但颅面部整体比例正常,这个结果也显示仅重复DSCR同源区无法影响小鼠颅面部形态。与此同时,Ts1Rhr小鼠相较于Ts65Dn小鼠也并没有显示出高比例的CHD[17],进一步说明DSCR理论的局限性。上述基于Ts1Rhr小鼠模型的表型比对,表明不能将DS表型简单归结于特定基因的重复,非连续基因间的相互作用对DS表型同样重要;单独某一基因的重复不足以产生明显剂量效应,与其他重复基因相互作用后才有可能表现出相应表型。
2.3 Tc1小鼠
Tc1小鼠由O’Doherty等[15]于2005年首次建立。该团队利用辐射微细胞介导的染色体转移(irradiation microcell-mediated chromosome transfer)技术将人类细胞系HT1080来源的21号染色体分离并转移到129S2小鼠胚胎干细胞系中,获得多出一条Hsa21的129S2小鼠胚胎干细胞。利用该细胞系制作的DS小鼠模型重复的即是HT1080中21号染色体,命名为Tc1小鼠。Tc1小鼠具有海马区介导的认知障碍[29]、CHD[30]和记忆功能损伤等明显的DS表型,但该小鼠仅表现出短期识别记忆能力的损伤,而长期记忆能力基本无损[31]。由于Tc1小鼠模型是在小鼠的染色体背景中多出一条人类染色体,因此该模型也被用来研究小鼠转录因子和人类启动子的结合以及异种生物染色体相互作用等[32]。尽管Tc1小鼠充分再现了人类DS患者的表型并可用作异种生物基因、染色体和蛋白间相互作用的研究模型,但其亦有不足之处。Tc1小鼠制作过程中人源染色体的转移(本文将转移的Hsa21记为Tc1-Hsa21)需要经过γ射线照射,因而存在随机断裂和重排的可能。经检测,Tc1-Hsa21经过γ射线照射后端粒依旧完整,但着丝粒移至原Hsa21的短臂中部,整条染色体共有41个位点发生断裂和重排,且
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Ts65Dn小鼠易位染色体Mmu1716和Ts1Rhr小鼠重复片段简图
Ts65Dn-Mmu1716为Ts65Dn小鼠中重复片段,包含Mmu16上Mrpl39基因前端断裂点到端粒约13.4 Mb和Mmu17上Pde10a基因后端断裂点到着丝粒约9.4 Mb片段。Ts1Rhr-DSCR为Ts1Rhr小鼠中重复片段,包含Mmu16上Cbr1和Mx2基因间约3.9 Mb片段,与人类染色体中DSCR同源。
Fig. 2The trisomic segments represented in Ts65Dn and Ts1Rhr mouse models
断裂位点处存在随机删除现象,导致Tc1-Hsa21出现很多重复和缺失片段。与人类细胞系HT1080中的21号染色体相比,Tc1-Hsa21中有大约8.7%的片段被删除、10%的片段被重复,导致了至少45个基因的完全或部分删除以及18个基因的重复[33](表2)。此外,Tc1-Hsa21中存在的重排现象会破坏至少11个基因的正常表达。例如,人类淀粉样前体蛋白(amyloid precursor protein, APP)基因末端外显子在Tc1-Hsa21中发生重排,导致完整长度的APP得不到表达而发生自发降解,因而在Tc1小鼠大脑中检测不到完整长度的APP蛋白[34]。断裂重排还会造成融合基因的产生,虽然大部分融合基因并不能正常表达,但仍有小部分可以表达并产生融合蛋白。例如,NDUFV3(NADH:ubiquinone oxidoreductase subunit V3)第一个外显子和PCBP3(poly(RC) binding protein 3)最后9个外显子一起表达可以产生融合蛋白,其转录本已通过RT-PCR确定[33]。
此外,由于Tc1小鼠是在小鼠背景中插入了一条人类染色体,该染色体在小鼠发育过程中会随机丢失,因而Tc1小鼠各组织中存在随机嵌合(Tc1小鼠细胞和丢失了Tc1-Hsa21的细胞随机嵌合)的可能,同时雌性小鼠将Tc1-Hsa21传递给下一代的能力也会随着年龄的增长而下降,老龄化的雌性Tc1小鼠传递Tc1-Hsa21的能力会下降3倍左右[35]。
2.4 Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠
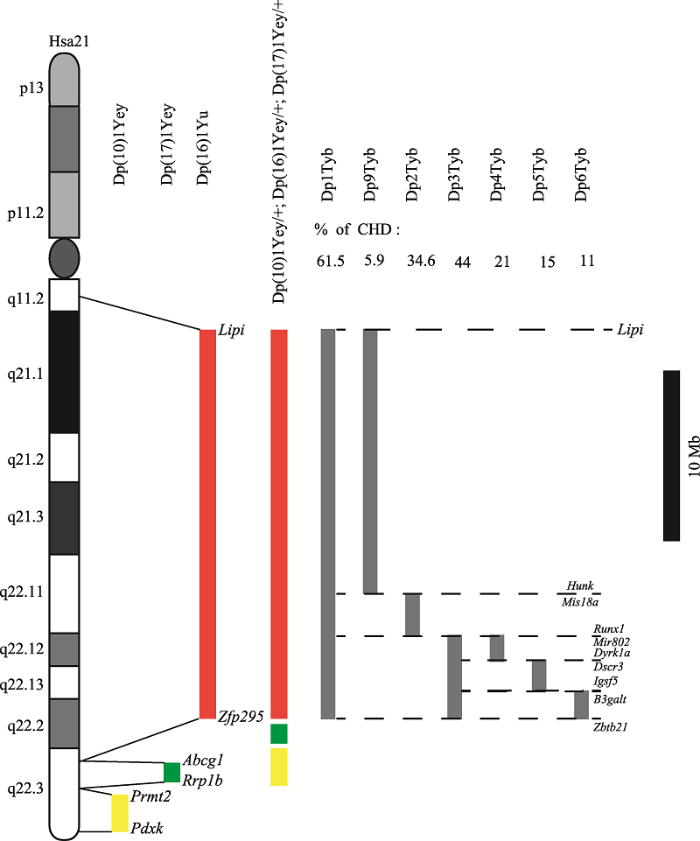
为了更好地应用小鼠模型研究DS的致病机制,Yu等[19]于2010年首次报道利用Cre/LoxP重组酶系统结合杂交手段构建了重复所有Hsa21直系同源基因的小鼠模型,并命名为Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠,其重复的同源片段分别来自于Dp(16)1Yu、Dp(17)1Yey和Dp(10)1Yey3种小鼠(图3),它们分别重复了Mmu16、17、10上对应的Hsa21同源区。Dp(16)1Yu小鼠是由Li等[16]于2007年首次报道构建的DS小鼠模型,该小鼠模型的建立主要利用Cre/LoxP重组酶系统,在小鼠胚胎干细胞系中人工诱导Lipi~Zfp295基因间22.9 Mb的片段(包含113个Hsa21直系同源基因)发生重组,使该片段产生重复。发生重组的胚胎干细胞用于嵌合体小鼠的制备,随后将雄性嵌合体与129SV雌性小鼠杂交,进行选择育种。由于生殖系嵌合发生的概率较低,因而Dp(16)1Yu小鼠的选育过程较为耗时。直到2010年,Yu和Li团队再次报道建立了另外两种小鼠模型Dp(10)1Yey和Dp(17)1Yey[19]。Dp(10)1Yey和Dp(17) 1Yey小鼠模型的建立沿用了Dp(16)1Yu小鼠的构建方法(图3),它们分别重复了Mmu10上Prmt2~Pdxk基因间约2.3 Mb的片段和Mmu17上Abcg1~Rrp1b基因间约1.1 Mb的片段,包含的直系同源基因数分别为41个和19个。在上述3种小鼠模型建立完成后,该团队利用两两杂交方式,首先得到包含两个不同重复片段的突变体小鼠,而后与第3种模型小鼠杂交,利用分子生物学手段鉴别两次杂交的子代小鼠以获得包含全部3个重复片段的突变体,即Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠。
Dp(16)1Yu和Dp(10)1Yey/+; Dp(16)1Yey/+; Dp (17)1Yey/+小鼠的表型特征较为相似,遗传学和生理学检测显示:(1) Dp(16)1Yu小鼠中重复基因的表达水平发生不同程度的升高,其表型特征包括有明显的CHD(如:ASD, VSD, AVSD, TOF)和胃肠道异常;(2) Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠出生后死亡率约为26%,且生长过程中体重明显低于正常对照小鼠;部分具有DS相关的CHD;6.8%的存活小鼠在6~8周龄出现脑积水现象,颅骨扩大变圆,最终死亡。对Dp(10)1Yey/+; Dp(16)1Yey/+; Dp (17)1Yey/+小鼠的认知行为学测试显示,其海马区介导的学习和记忆能力受损并且具有认知能力障碍等特点。不过也有研究指出Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠在自发性活动测试、焦虑性测试和重复动作行为测试中与正常对照组无差异,并不能模拟出DS特征表型[36]。这表明不同物种间基因组背景的差异对基因-表型关联性的影响较大,即使建立了对所有相关直系同源基因重复的小鼠也未必可以对人类疾病表型进行准确模拟。
Table 2
表2
表2 Tc1-Hsa21中重复和删除的基因
Table 2
| 删除/重复基因 | 基因名称 |
|---|---|
| 完全删除基因 | BTG3, C21orf91, NCRNA00157, CHODL, C21orf45, MRAP, URB1, SNORA80, C21orf119, C21orf63, TCP10L, C21orf59, SYNJ1, GCFC1, C21orf49, C21orf62, OLIG2, OLIG1, C21orf54, IFNAR2, IL10RB, IFNAR1, IFNGR2, TMEM50B, DNAJC28, GART, SON, DONSON, CRYZL1, ITSN1, ATP5O, MRPS6, SLC5A3, C21orf82, KCNE2, FAM165B, KCNE1, RCAN1, CLIC6, NCRNA00160, SLC19A1 |
| 部分删除基因 | TMPRSS15, RUNX1, COL18A1, PCBP3 |
| 完全重复基因 | BAGE5, BAGE4, BAGE2, BAGE3, BAGE, RBM11, ABCC13, HSPA13, SAMSN1, NRIP1, USP25, S100B, PRMT2, C21orf131 |
| 部分重复基因 | LIPI, C21orf34, DIP2A, NCAM2 |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠和DS先天性心脏病小鼠重复片段简图
红色、绿色和黄色矩形分别代表Mmu16、17、10中的Hsa21同源区,同时也代表了小鼠模型Dp(16)1Yu、Dp(17)1Yey和Dp(10)1Yey中的重复片段;Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠重复了Hsa21在小鼠染色体中的所有直系同源基因;灰色矩形代表不同DS先天性心脏病小鼠模型中的重复片段范围。
Fig. 3The trisomic segments represented in Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+ and DS mouse models associated with congenital heart defects
2.5 DS相关CHD小鼠模型
2016年1月,Lana-Elola等[20]报道了一组用于定位DS相关CHD基因的小鼠模型,该组模型一共7个品系,分别重复了Mmu16上不同的Hsa21直系同源区(图3):Dp1Tyb小鼠的重复片段为Lipi~Zbtb21基因间约23 Mb大小区域,基本包含了Mmu16上所有Hsa21直系同源基因;Dp9Tyb (Lipi到Hunk)、Dp2Tyb (Mis18a到Runx1)和Dp3Tyb (Mir802到Zbtb21)小鼠将Dp1Tyb小鼠的重复片段分为3段,大小分别为15.6 Mb、2.5 Mb和4.9 Mb;Dp4Tyb (Mir802到Dscr3)、Dp5Tyb (Dyrk1a到B3galt5)和Dp6Tyb (Igsf5到Zbtb21) 小鼠又将Dp3Tyb小鼠的重复区划分为3段,大小分别为1.5 Mb、1.8 Mb和1.6 Mb,目的是进一步缩小DS相关CHD效应基因的范围。对E14.5天小鼠的解剖学实验显示:对照组野生型小鼠无明显CHD症状;Dp1Tyb小鼠中62%具有VSD症状,25%具有AVSD症状;Dp9Tyb和Dp2Tyb小鼠相比同窝出生的野生型小鼠,CHD比例无明显升高;Dp3Tyb小鼠中CHD比例明显升高,严重程度近似Dp1Tyb小鼠;Dp4Tyb、Dp5Tyb和Dp6Tyb小鼠亦未出现高比例CHD现象。该实验在建立了一系列小鼠模型后仅对CHD的发生概率进行了检测和统计学分析,并未涉及其他DS表型的观察。
该系列小鼠将CHD关键区限制在Mir802~Zbtb21间4.9 Mb的区间内,并且证明单独的Dp4Tyb、Dp5Tyb和Dp6Tyb小鼠重复区无法造成CHD症状。在此基础上,后续可以利用Dp4Tyb、Dp5Tyb和Dp6Tyb这3种小鼠的两两杂交,进一步缩小DS相关CHD关键区范围。
3 结 语
DS作为最常见的常染色体异常综合征,具有遗传背景复杂、表型多样等特点,利用小鼠模型进行DS机理研究,在避免伦理问题的同时也为深入了解DS发病机制和评估基因-表型关联性提供了可能。自1990年第一只DS小鼠模型——Ts65Dn小鼠建立至今,已经发展出一系列具有不同特点和功能的DS小鼠模型,在疾病模拟、机理研究和药物筛选等方面发挥着重要的作用。目前还没有一种小鼠模型可以做到对人类DS表型和基因型的完全模拟,每种模型都具有各自的优势和不足。以Ts65Dn小鼠和Dp(10)1Yey/+; Dp (16)1Yey/+; Dp(17)1Yey/+小鼠为例:Ts65Dn小鼠充分模拟了人类DS表型,但对Hsa21直系同源基因的重复不完全且携带部分非同源基因;Dp(10)1Yey/+; Dp(16)1Yey/+; Dp(17)1Yey/+小鼠重复了所有Hsa21直系同源基因,但未完全重现人类DS患者中出现的表型。因而在对特定表型和相关基因进行研究前,需要充分了解DS小鼠模型的特点并选择合适小鼠进行研究。值得一提的是,Tc1小鼠是在小鼠染色体组中插入了一条Hsa21,使用时需要考虑小鼠和人类染色体背景间的差异。目前对DS的研究,包括智障机理研究和认知类药物的筛选,主要选择Ts65Dn小鼠,因为Ts65Dn小鼠研究历史最长,智障特点最为突出。在研究CHD、肌张力等DS表型与基因型间关系时,可根据实际情况选用其他小鼠模型。
有****认为,评估人类疾病小鼠模型的首要因素应当是遗传机制的一致性而非片面追求表型相似程度[37, 38],遗传机制的改变不仅可以影响疾病表型的模拟程度,同时也能干扰治疗药物的作用效果。本文就人类-小鼠基因同源性和常用DS小鼠模型的基因型、表型特征两方面进行综述,希望为科研人员在DS研究中选用不同小鼠模型提供参考。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:7707939 [本文引用: 1]

ABSTRACT This reprint of an 1866 essay by the physician who identified Down's syndrome describes the physical and behavioral patterns that characterize individuals with mental retardation who come from different ethnic groups. This evidence of degeneration crossing racial divisions is seen as support for the idea that the human family has a common origin. (JDD)
[本文引用: 1]
URLPMID:2521249 [本文引用: 1]
The Baltimore-Washington Infant Study is a population-based case-control study that seeks to identify risk factors for cardiovascular malformations. Between 1981 and 1986, a total of 2102 infants with cardiovascular malformations were ascertained, among whom 271 (12.9%) also had a chromosome abnormality. Among 2328 random control subjects, only two had a chromosome abnormality. Down syndrome with cardiovascular maiformations had a maternal age-adjusted regional prevalence of 4.33/10,000 for the white population and 3.70/10,000 for the nonwhite population. Endocardial cushion defect, the predominant cardiac abnormality in Down syndrome (60.1%), rarely occurred as an isolated cardiac lesion (2.8%). The absence of transpositions and the rarity of heterotaxias and of right- and left-sided obstructive lesions in trisomies indicate that there may be a genetic influence on specific embryologic mechanisms. Allmentary tract lesions were more common in Down syndrome than among euploid patients with heart disease and more severe than in control subjects. Urinary tract lesions also occurred in excess of the rate in control subjects. The coexistence of these major malformations with heart disease raises the possibility of incomplete expression of the VA(C)TER (vertebral, anal, cardiac, fracheal, esophageal renal) association. The selective association of chromosome abnormalities with certain cardiovascular defects is now beginning to be explained by reported embryologic studies on cellular characteristics. An explanation of the negative association with transposition and obstructive lesions requires further multidisciplinary studies on genetic and epigenetic factors.
URLPMID:26651341 [本文引用: 1]
Chromosome 21, triplicated in Down Syndrome, contains several genes that are thought to play a critical role in the development of AD neuropathology. The overexpression of the gene for the amyloid precursor protein (APP), on chromosome 21, leads to early onset beta-amyloid (A0205) plaques in DS. In addition to A0205 accumulation, middle-aged people with DS develop neurofibrillary tangles, cerebrovascular pathology, white matter pathology, oxidative damage, neuroinflammation and neuron loss. There is also evidence of potential compensatory responses in DS that benefit the brain and delay the onset of dementia after there is sufficient neuropathology for a diagnosis of AD. This review describes some of the existing literature and also highlights gaps in our knowledge regarding AD neuropathology in DS. It will be critical in the future to develop networked brain banks with standardized collection procedures to fully characterize the regional and temporal pathological events associated with aging in DS. As more information is acquired regarding AD evolution in DS, there will be opportunities to develop interventions that are age-appropriate to delay AD in DS.
URLPMID:26400113 [本文引用: 1]
Abstract Down syndrome (DS) is one of the more commonly occurring genetic disorders, where mental retardation is combined with nutritional diseases. It is caused by having a third copy of chromosome 21, and there exist 3 forms; Simple Trisomy 21, Translocation Trisomy and Mosaic Trisomy. Symptoms include intellectual disability/mental retardation, early onset of Alzheimer's disease and the appearance of various phenotypic features such as narrow slanted eyes, flat nose and short stature. In addition, there are other health problems throughout the body, consisting in part of cardiac defects and thyroid function abnormalities along with nutritional disorders (ie. overweight, obesity, hypercholesterolemia and deficiencies of vitamins and minerals). Those suffering DS have widespread body frame abnormalities and impaired brain development and function; the latter leading to impaired intellectual development. Many studies indicate excessive or deficient nutrient uptakes associated with making inappropriate foodstuff choices, food intolerance, (eg. celiac disease) or malabsorption. DS persons with overweight or obesity are linked with a slow metabolic rate, abnormal blood leptin concentrations and exhibit low levels of physical activity. Vitamin B group deficiencies and abnormal blood homocysteine levels decrease the rate of intellectual development in DS cases. Zinc deficiencies result in short stature, thyroid function disorders and an increased appetite caused by excessive supplementation. Scientific advances in the research and diagnosis of DS, as well as preventing any associated conditions, have significantly increased life expectancies of those with this genetic disorder. Early dietary interventions by parents or guardians of DS children afford an opportunity for decreasing the risk or delaying some of the DS associated conditions from appearing, thus beneficially impacting on their quality of life.
URL [本文引用: 1]
URLPMID:11802376 [本文引用: 1]
Nature is the international weekly journal of science: a magazine style journal that publishes full-length research papers in all disciplines of science, as well as News and Views, reviews, news, features, commentaries, web focuses and more, covering all branches of science and how science impacts upon all aspects of society and life.
URLPMID:21400203 [本文引用: 1]
A comprehensive representation of the gene content of the long arm of human chromosome 21 (Hsa21q) remains of interest for the study of Down syndrome, its associated phenotypic features, and mouse models. Here we compare transcript catalogs for Hsa21q, chimpanzee chromosome 21 (Ptr21q), and orthologous regions of mouse chromosomes 16, 17, and 10 for open reading frame (ORF) characteristics and conservation. The Hsa21q and mouse catalogs contain 552 and 444 gene models, respectively, of which only 162 are highly conserved. Hsa21q transcripts were used to identify orthologous exons in Ptr21q and assemble 533 putative transcripts. Transcript catalogs for all three organisms are searchable for nucleotide and amino acid sequence features of ORF length, repeat content, experimental support, gene structure, and conservation. For human and mouse comparisons, three additional summaries are provided: (1) the chromosomal distribution of novel ORF transcripts versus potential functional RNAs, (2) the distribution of species-specific transcripts within Hsa21q and mouse models of Down syndrome, and (3) the organization of sense–antisense and putative sense–antisense structures defining potential regulatory mechanisms. Catalogs, summaries, and nucleotide and amino acid sequences of all composite transcripts are available and searchable at http://67gfuncpathdb.67ucdenver.67edu/67iddrc/67chr21/67home.67php . These data sets provide comprehensive information useful for evaluation of candidate genes and mouse models of Down syndrome and for identification of potential functional RNA genes and novel regulatory mechanisms involving Hsa21q genes. These catalogs and search tools complement and extend information available from other gene annotation projects.
URLPMID:27538963 [本文引用: 1]
Down syndrome (DS), trisomy of human chromosome 21 (Hsa21), is challenging to model in mice. Not only is it a contiguous gene syndrome spanning 3502Mb of the long arm of Hsa21, but orthologs of Hsa21 g
[本文引用: 1]
URLPMID:8406465 [本文引用: 1]
A partial cDNA (D21S418E) whose nucleotide sequence has no significant homologies with known mammalian DNA sequences was isolated from a human placental library. The cDNA hybridized with a 10-kb transcript present in term placenta. Messages of 10 and 7.5 kb were induced in BeWo and JEG-3 choriocarcinoma cells by treatment with 8-Br-cAMP. The mRNA was not detected in human brain, liver, lung, kidney, pancreas, heart, skeletal muscle, or myometrium. The D21S418E locus was assigned to a 3.5-Mb region of chromosome 21q22.3.
URLPMID:2147289 [本文引用: 1]
Prog Clin Biol Res. 1990;360:263-80. Research Support, U.S. Gov't, P.H.S.
URLPMID:9600952
A mouse model for Down syndrome, Ts1Cje, has been developed. This model has made possible a step in the genetic dissection of the learning, behavioral, and neurological abnormalities associated with segmental trisomy for the region of mouse chromosome 16 homologous with the so-called "Down syndrome region" of human chromosome segment 21q22. Tests of learning in the Morris water maze and assessment of spontaneous locomotor activity reveal distinct learning and behavioral abnormalities, some of which are indicative of hippocampal dysfunction. The triplicated region in Ts1Cje, from Sod1 to Mx1, is smaller than that in Ts65Dn, another segmental trisomy 16 mouse, and the learning deficits in Ts1Cje are less severe than those in Ts65Dn. In addition, degeneration of basal forebrain cholinergic neurons, which was observed in Ts65Dn, was absent in Ts1Cje.
URLPMID:4019810 [本文引用: 2]
Abstract The "Down syndrome critical region" (DSCR) is a chromosome 21 segment purported to contain genes responsible for many features of Down syndrome (DS), including craniofacial dysmorphology. We used chromosome engineering to create mice that were trisomic or monosomic for only the mouse chromosome segment orthologous to the DSCR and assessed dysmorphologies of the craniofacial skeleton that show direct parallels with DS in mice with a larger segmental trisomy. The DSCR genes were not sufficient and were largely not necessary to produce the facial phenotype. These results refute specific predictions of the prevailing hypothesis of gene action in DS.
URLPMID:1378183 [本文引用: 1]
Aneuploidies are common chromosomal defects that result in growth and developmental deficits and high levels of lethality in humans. To gain insight into the biology of aneuploidies, we manipulated mouse embryonic stem cells and generated a trans-species aneuploid mouse line that stably transmits a freely segregating, almost complete human chromosome 21 (Hsa21). This "transchromosomic" mouse line, Tc1, is a model of trisomy 21, which manifests as Down syndrome (DS) in humans, and has phenotypic alterations in behavior, synaptic plasticity, cerebellar neuronal number, heart development, and mandible size that relate to human DS. Transchromosomic mouse lines such as Tc1 may represent useful genetic tools for dissecting other human aneuploidies.
URLPMID:17412756 [本文引用: 1]
Down syndrome is caused by a genomic imbalance of human chromosome 21 which is mainly observed as trisomy 21. The regions on human chromosome 21 are syntenically conserved in three regions on mouse chromosomes 10, 16 and 17. Ts65Dn mice, the most widely used model for Down syndrome, are trisomic for approximately 56.5% of the human chromosome 21 syntenic region on mouse chromosome 16. To generate a more complete trisomic mouse model of Down syndrome, we have established a 22.9 Mb duplication spanning the entire human chromosome 21 syntenic region on mouse chromosome 16 in mice using Cre/loxP-mediated long-range chromosome engineering. The presence of the intact duplication in mice was confirmed by fluorescent in situ hybridization and BAC-based array comparative genomic hybridization. The expression levels of the genes within the duplication interval reflect gene-dosage effects in the mutant mice. The cardiovascular and gastrointestinal phenotypes of the mouse model were similar to those of patients with Down syndrome. This new mouse model represents a powerful tool to further understand the molecular and cellular mechanisms of Down syndrome.
URLPMID:21442329 [本文引用: 1]
Human trisomy 21, the chromosomal basis of Down syndrome (DS), is the most common genetic cause of heart defects. Regions on human chromosome 21 (Hsa21) are syntenically conserved with three regions located on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17. In this study, we have analyzed the impact of duplications of each syntenic region on cardiovascular development in mice and have found that only the duplication on Mmu16, i.e., Dp(16)1Yey, is associated with heart defects. Furthermore, we generated two novel mouse models carrying a 5.43-Mb duplication and a reciprocal deletion between Tiam1 and Kcnj6 using chromosome engineering, Dp(16Tiam1-Kcnj6)Yey/+ and Df(16Tiam1-Kcnj6)Yey/+, respectively, within the 22.9-Mb syntenic region on Mmu16. We found that Dp(16Tiam1-Kcnj6)Yey/+, but not Dp(16)1Yey/Df(16Tiam1-Kcnj6)Yey, resulted in heart defects, indicating that triplication of the Tiam1-Knj6 region is necessary and sufficient to cause DS-associated heart defects. Our transcriptional analysis of Dp(16Tiam1-Kcnj6)Yey/+ embryos confirmed elevated expression levels for the genes located in the Tiam-Kcnj6 region. Therefore, we established the smallest critical genomic region for DS-associated heart defects to lay the foundation for identifying the causative gene(s) for this phenotype.
URLPMID:4024075
Abstract Trisomy 21 (Down syndrome, DS) is the most common human genetic anomaly associated with heart defects. Based on evolutionary conservation, DS-associated heart defects have been modeled in mice. By generating and analyzing mouse mutants carrying different genomic rearrangements in human chromosome 21 (Hsa21) syntenic regions, we found the triplication of the Tiam1-Kcnj6 region on mouse chromosome 16 (Mmu16) resulted in DS-related cardiovascular abnormalities. In this study, we developed two tandem duplications spanning the Tiam1-Kcnj6 genomic region on Mmu16 using recombinase-mediated genome engineering, Dp(16)3Yey and Dp(16)4Yey, spanning the 2.1 Mb Tiam1-Il10rb and 3.7 Mb Ifnar1-Kcnj6 regions, respectively. We found that Dp(16)4Yey/+, but not Dp(16)3Yey/+, led to heart defects, suggesting the triplication of the Ifnar1-Kcnj6 region is sufficient to cause DS-associated heart defects. Our transcriptional analysis of Dp(16)4Yey/+ embryos showed that the Hsa21 gene orthologs located within the duplicated interval were expressed at the elevated levels, reflecting the consequences of the gene dosage alterations. Therefore, we have identified a 3.7 Mb genomic region, the smallest critical genomic region, for DS-associated heart defects, and our results should set the stage for the final step to establish the identities of the causal gene(s), whose elevated expression(s) directly underlie this major DS phenotype.
URL [本文引用: 2]
URLPMID:4764572 [本文引用: 1]
Abstract Down syndrome (DS), caused by trisomy of human chromosome 21 (Hsa21), is the most common cause of congenital heart defects (CHD), yet the genetic and mechanistic causes of these defects remain unknown. To identify dosage-sensitive genes that cause DS phenotypes, including CHD, we used chromosome engineering to generate a mapping panel of 7 mouse strains with partial trisomies of regions of mouse chromosome 16 orthologous to Hsa21. Using high-resolution episcopic microscopy and three-dimensional modeling we show that these strains accurately model DS CHD. Systematic analysis of the 7 strains identified a minimal critical region sufficient to cause CHD when present in 3 copies, and showed that it contained at least two dosage-sensitive loci. Furthermore, two of these new strains model a specific subtype of atrio-ventricular septal defects with exclusive ventricular shunting and demonstrate that, contrary to current hypotheses, these CHD are not due to failure in formation of the dorsal mesenchymal protrusion.
URLPMID:25552901 [本文引用: 1]
Abstract Down syndrome (DS), also known as trisomy 21, is the most common genetic cause of intellectual disability (ID). Although ID can be mild, the average intelligence quotient is in the range of 40-50. All individuals with DS will also develop the neuropathology of Alzheimer's disease (AD) by the age of 30-40 years, and approximately half will display an AD-like dementia by the age of 60 years. DS is caused by an extra copy of the long arm of human chromosome 21 (Hsa21) and the consequent elevated levels of expression, due to dosage, of trisomic genes. Despite a worldwide incidence of one in 700-1,000 live births, there are currently no pharmacological treatments available for ID or AD in DS. However, over the last several years, very promising results have been obtained with a mouse model of DS, the Ts65Dn. A diverse array of drugs has been shown to rescue, or partially rescue, DS-relevant deficits in learning and memory and abnormalities in cellular and electrophysiological features seen in the Ts65Dn. These results suggest that some level of amelioration or prevention of cognitive deficits in people with DS may be possible. Here, we review information from the preclinical evaluations in the Ts65Dn, how drugs were selected, how efficacy was judged, and how outcomes differ, or not, among studies. We also summarize the current state of human clinical trials for ID and AD in DS. Lastly, we describe the genetic limitations of the Ts65Dn as a model of DS, and in the preclinical testing of pharmacotherapeutics, and suggest additional targets to be considered for potential pharmacotherapies.
URLPMID:3224224 [本文引用: 1]
Down syndrome (DS) is the most frequent genetic disorder leading to intellectual disabilities and is caused by three copies of human chromosome 21. Mouse models are widely used to better understand the physiopathology in DS or to test new therapeutic approaches. The older and the most widely used mouse models are the trisomic Ts65Dn and the Ts1Cje mice. They display deficits similar to those observed in DS people, such as those in behavior and cognition or in neuronal abnormalities. The Ts65Dn model is currently used for further therapeutic assessment of candidate drugs. In both models, the trisomy was induced by reciprocal chromosomal translocations that were not further characterized. Using a comparative genomic approach, we have been able to locate precisely the translocation breakpoint in these two models and we took advantage of this finding to derive a new and more efficient Ts65Dn genotyping strategy. Furthermore, we found that the translocations introduce additional aneuploidy in both models, with a monosomy of seven genes in the most telomeric part of mouse chromosome 12 in the Ts1Cje and a trisomy of 60 centromeric genes on mouse chromosome 17 in the Ts65Dn. Finally, we report here the overexpression of the newly found aneuploid genes in the Ts65Dn heart and we discuss their potential impact on the validity of the DS model.
URLPMID:23303189 [本文引用: 1]
Regulation of intracellular pH is critical for the maintenance of cell homeostasis in response to stress. We used yeast two-hybrid screening to identify novel interacting partners of the pH-regulating transporter NBCe1-B. We identified Hsp70-like stress 70 protein chaperone (STCH) as interacting with NBCe1-B at the N-terminal (amino acids 96-440) region. Co-injection of STCH and NBCe1-B cRNA into Xenopus oocytes significantly increased surface expression of NBCe1-B and enhanced bicarbonate conductance compared with NBCe1-B cRNA alone. STCH siRNA decreased the rate of Na(+)-dependent pHi recovery from NH4(+) pulse-induced acidification in an HSG (human submandibular gland ductal) cell line. We observed that in addition to NBCe1-B, Na(+)/H(+) exchanger (NHE)-dependent pHi recovery was also impaired by STCH siRNA and further confirmed the interaction of STCH with NHE1 but not plasma membrane Ca(2+) ATPase. Both NBCe1-B and NHE1 interactions were dependent on a specific 45-amino acid region of STCH. In conclusion, we identify a novel role of STCH in the regulation of pHi through site-specific interactions with NBCe1-B and NHE1 and subsequent modulation of membrane transporter expression. We propose STCH may play a role in pHi regulation at times of cellular stress by enhancing the recovery from intracellular acidification.
URLPMID:18023280 [本文引用: 1]
The control of physiological processes requires the regulation and coordination of many different signals and is determined in part by the activation and repression of expression of specific target genes. RIP140 is a ligand dependent coregulator of many nuclear receptors that influence such diverse processes as muscle metabolism, adipocyte and hepatocyte function, and reproduction. Recent evidence has shown that the ability of RIP140 to regulate nuclear receptor function is determined by the relative level of RIP140 expression in comparison with other cofactors, by post-translational modifications and by interactions with additional transcription factors. As a result it is becoming apparent that RIP140, via its interplay with other coregulators, plays a fundamental role in determining both the normal and pathogenic physiological state.
URLPMID:22155300 [本文引用: 1]
The protein exists in a transmembrane and a lipid-anchored isoform, and has an ectodomain consisting of five immunoglobulin modules and two fibronectin type 3 homology modules. Structural models of the NCAM2 ectodomain reveal that it facilitates cell adhesion through reciprocal interactions between the membrane-distal immunoglobulin modules. There are no known heterophilic NCAM2 binding partners, and NCAM2 is not glycosylated with polysialic acid, a posttranslational modification known to be a major modulator of NCAM1-mediated processes. This suggests that NCAM2 has a function or mode of action distinctly different from that of NCAM1. NCAM2 is primarily expressed in the brain, where it is believed to stimulate neurite outgrowth and to facilitate dendritic and axonal compartmentalization.
URLPMID:17681954 [本文引用: 1]
Sorting nexin 9 (SNX9) is a member of the sorting nexin family of proteins, each of which contains a characteristic Phox homology domain. SNX9 is widely expressed and plays a role in clathrin-mediated endocytosis, but it is not known if it is present in neuronal cells. We report that SNX9 is expressed in the presynaptic compartment of cultured hippocampal neurons, where it binds to dynamin-1 and N-WASP. Overexpression of full-length SNX9 or a C-terminal truncated version caused severe defects in synaptic vesicle endocytosis during, as well as after, stimulation. Knockdown of SNX9 with short interfering RNA also reduced synaptic vesicle endocytosis, and the W39A mutation of SNX9 abolished the inhibitory effect of SNX9 on endocytosis. Rescue experiments showed that most of the effect of SNX9 on endocytosis results from its interaction with dynamin 1, although its interaction with N-WASP contributes in some degree. We further showed that SNX9 dimerizes through its C-terminal domain, suggesting that it may interact simultaneously with dynamin 1 and N-WASP. We propose that SNX9 interacts with dynamin-1 and N-WASP in presynaptic terminals, where it links actin dynamics and synaptic vesicle endocytosis.
URLPMID:22028875 [本文引用: 1]
Orexins (OX-A, OX-B) are neuropeptides involved in the regulation of the sleep-wake cycle, feeding and reward, via activation of orexin receptors 1 and 2 (OX1R, OX2R). The loss of orexin peptides or functional OX2R has been shown to cause the sleep disorder, narcolepsy. Since the regulation of orexin receptors remains largely undefined, we searched for novel protein partners of the intracellular tail of orexin receptors. Using a yeast two-hybrid screening strategy in combination with co-immunoprecipitation experiments, we found interactions between OX1R and the dynein light chains Tctex-type 1 and 3 (Dynlt1, Dynlt3). These interactions were mapped to the C-terminal region of the dynein light chains and to specific residues within the last 10 amino acids of OX1R. Hence, we hypothesized that dynein light chains could regulate orexin signaling. In HEK293 cells expressing OX1R, stimulation with OX-A produced a less sustained extracellular signal-regulated kinases 1/2 (ERK1/2) activation when Dynlt1 was co-expressed, while it was prolonged under reduced Dynlt1 expression. The amount of OX1R located at the plasma membrane as well as the kinetics and extent of OX-A-induced internalization of OX1R (disappearance from membrane) were not altered by Dynlt1. However, Dynlt1 reduced the localization of OX1R in early endosomes following initial internalization. Taken together, these data suggest that Dynlt1 modulates orexin signaling by regulating OX1R, namely its intracellular localization following ligand-induced internalization.
URLPMID:8055322 [本文引用: 1]
To determine which regions of chromosome 21 are involved in the pathogenesis of specific features of Down syndrome, we analysed, phenotypically and molecularly, 10 patients with partial trisomy 21. Six minimal regions for 24 features were defined by genotype-phenotype correlations. Nineteen of these features could be assigned to just 2 regions: short stature, joint hyperlaxity, hypotonia, major contribution to mental retardation and 9 anomalies of the face, hand and foot to the region D21S55, or Down syndrome chromosome region (DCR), located on q22.2 or very proximal q22.3, and spanning 0.4-3 Mb; 6 facial and dermatoglyphic anomalies to the region D21S55-MX1, including the DCR and spanning a maximum of 6 Mb on q22.2 and part of q22.3. Thus, the complex phenotype that constitutes Down syndrome may in large part simply result from the overdosage of only one or a few genes within the DCR and/or region D21S55-MX1.
URLPMID:4552261 [本文引用: 1]
Hippocampal pathology is likely to contribute to cognitive disability in Down syndrome (DS), yet the neural network basis of this pathology and its contributions to different facets of cognitive impairment remain unclear. Here, we report dysfunctional connectivity between dentate gyrus (DG) and CA3 networks in the transchromosomic Tc1 mouse model of DS, demonstrating that ultrastructural abnormalities and impaired short-term plasticity at DG-CA3 excitatory synapses culminate in impaired coding of novel spatial information in CA3 and CA1 and disrupted behaviourin vivo. These results highlight the vulnerability of DG-CA3 networks to aberrant human chromosome 21 gene expression, and delineate hippocampal circuit abnormalities likely to contribute to distinct cognitive phenotypes in DS.
URL [本文引用: 1]
URLPMID:26868479 [本文引用: 1]
Abstract The present study examined memory function in Tc1 mice, a transchromosomic model of Down syndrome (DS). Tc1 mice demonstrated an unusual delay-dependent deficit in recognition memory. More specifically, Tc1 mice showed intact immediate (30sec), impaired short-term (10-min) and intact long-term (24-h) memory for objects. A similar pattern was observed for olfactory stimuli, confirming the generality of the pattern across sensory modalities. The specificity of the behavioural deficits in Tc1 mice was confirmed using APP overexpressing mice that showed the opposite pattern of object memory deficits. In contrast to object memory, Tc1 mice showed no deficit in either immediate or long-term memory for object-in-place information. Similarly, Tc1 mice showed no deficit in short-term memory for object-location information. The latter result indicates that Tc1 mice were able to detect and react to spatial novelty at the same delay interval that was sensitive to an object novelty recognition impairment. These results demonstrate (1) that novelty detection per se and (2) the encoding of visuo-spatial information was not disrupted in adult Tc1 mice. The authors conclude that the task specific nature of the short-term recognition memory deficit suggests that the trisomy of genes on human chromosome 21 in Tc1 mice impacts on (perirhinal) cortical systems supporting short-term object and olfactory recognition memory. Copyright 2016 The Authors. Published by Elsevier Inc. All rights reserved.
URLPMID:3717767 [本文引用: 1]
Homologous sets of transcription factors direct conserved tissue-specific gene expression, yet transcription factor-binding events diverge rapidly between closely related species. We used hepatocytes from an aneuploid mouse strain carrying human chromosome 21 to determine, on a chromosomal scale, whether interspecies differences in transcriptional regulation are primarily directed by human genetic sequence or mouse nuclear environment. Virtually all transcription factor-binding locations, landmarks of transcription initiation, and the resulting gene expression observed in human hepatocytes were recapitulated across the entire human chromosome 21 in the mouse hepatocyte nucleus. Thus, in homologous tissues, genetic sequence is largely responsible for directing transcriptional programs; interspecies differences in epigenetic machinery, cellular environment, and transcription factors themselves play secondary roles.
URLPMID:3626651 [本文引用: 2]
Down syndrome (DS) is caused by trisomy of chromosome 21 (Hsa21) and presents a complex phenotype that arises from abnormal dosage of genes on this chromosome. However, the individual dosage-sensitive genes underlying each phenotype remain largely unknown. To help dissect genotype phenotype correlations in this complex syndrome, the first fully transchromosomic mouse model, the Tc1 mouse, which carries a copy of human chromosome 21 was produced in 2005. The Tc1 strain is trisomic for the majority of genes that cause phenotypes associated with DS, and this freely available mouse strain has become used widely to study DS, the effects of gene dosage abnormalities, and the effect on the basic biology of cells when a mouse carries a freely segregating human chromosome. Tc1 mice were created by a process that included irradiation microcell-mediated chromosome transfer of Hsa21 into recipient mouse embryonic stem cells. Here, the combination of next generation sequencing, array-CGH and fluorescence in situ hybridization technologies has enabled us to identify unsuspected rearrangements of Hsa21 in this mouse model; revealing one deletion, six duplications and more than 25 de novo structural rearrangements. Our study is not only essential for informing functional studies of the Tc1 mouse but also (1) presents for the first time a detailed sequence analysis of the effects of gamma radiation on an entire human chromosome, which gives some mechanistic insight into the effects of radiation damage on DNA, and (2) overcomes specific technical difficulties of assaying a human chromosome on a mouse background where highly conserved sequences may confound the analysis. Sequence data generated in this study is deposited in the ENA database, Study Accession number: ERP000439.
[本文引用: 1]
URLPMID:4939633 [本文引用: 1]
Down syndrome incidence in humans increases dramatically with maternal age. This is mainly the result of increased meiotic errors, but factors such as differences in abortion rate may play a role as well. Since the meiotic error rate increases almost exponentially after a certain age, its contribution to the overall incidence aneuploidy may mask the contribution of other processes. To focus on such selection mechanisms we investigated transmission in trisomic females, using data from mouse models and from Down syndrome humans. In trisomic females the a-priori probability for trisomy is independent of meiotic errors and thus approximately constant in the early embryo. Despite this, the rate of transmission of the extra chromosome decreases with age in females of the Ts65Dn and, as we show, for the Tc1 mouse models for Down syndrome. Evaluating progeny of 73 Tc1 births and 112 Ts65Dn births from females aged 130/days to 250/days old showed that both models exhibit a 3-fold reduction of the probability to transmit the trisomy with increased maternal ageing. This is concurrent with a 2-fold reduction of litter size with maternal ageing. Furthermore, analysis of previously reported 30 births in Down syndrome women shows a similar tendency with an almost three fold reduction in the probability to have a Down syndrome child between a 20 and 30/years old Down syndrome woman. In the two types of mice models for Down syndrome that were used for this study, and in human Down syndrome, older females have significantly lower probability to transmit the trisomy to the offspring. Our findings, taken together with previous reports of decreased supportive environment of the older uterus, add support to the notion that an older uterus negatively selects the less fit trisomic embryos. The online version of this article (doi:10.1186/s12863-016-0412-3) contains supplementary material, which is available to authorized users.
URL [本文引用: 1]
URLPMID:23954708 [本文引用: 1]
Animal models have historically played a critical role in the exploration and characterization of disease pathophysiology, target identification, and in the in vivo evaluation of novel therapeutic agents and treatments. In the wake of numerous clinical trial failures of new chemical entities (NCEs) with promising preclinical profiles, animal models in all therapeutic areas have been increasingly criticized for their limited ability to predict NCE efficacy, safety and toxicity in humans. The present review discusses some of the challenges associated with the evaluation and predictive validation of animal models, as well as methodological flaws in both preclinical and clinical study designs that may contribute to the current translational failure rate. The testing of disease hypotheses and NCEs in multiple disease models necessitates evaluation of pharmacokinetic/pharmacodynamic (PK/PD) relationships and the earlier development of validated disease-associated biomarkers to assess target engagement and NCE efficacy. Additionally, the transparent integration of efficacy and safety data derived from animal models into the hierarchical data sets generated preclinically is essential in order to derive a level of predictive utility consistent with the degree of validation and inherent limitations of current animal models. The predictive value of an animal model is thus only as useful as the context in which it is interpreted. Finally, rather than dismissing animal models as not very useful in the drug discovery process, additional resources, like those successfully used in the preclinical PK assessment used for the selection of lead NCEs, must be focused on improving existing and developing new animal models.
URLPMID:5451508 [本文引用: 1]
Animal models of concussion, traumatic brain injury (TBI), and chronic traumatic encephalopathy (CTE) are widely available and routinely deployed in laboratories around the world. Effective animal modeling requires careful consideration of four basic principles. First, animal model use must be guided by clarity of definitions regarding the human disease or condition being modeled. Concussion, TBI, and CTE represent distinct clinical entities that require clear differentiation: concussion is aneurological syndrome, TBI is aneurological event, and CTE is aneurological disease. While these conditions are all associated with head injury, the pathophysiology, clinical course, and medical management of each are distinct. Investigators who use animal models of these conditions must take into account these clinical distinctions to avoid misinterpretation of results andcategory mistakes. Second, model selection must be grounded by clarity of purpose with respect to experimental questions and frame of reference of the investigation. Distinguishinginjury context(“inputs”) frominjury consequences(“outputs”) may be helpful during animal model selection, experimental design and execution, and interpretation of results. Vigilance is required to rout out, or rigorously control for, model artifacts with potential to interfere with primary endpoints. The widespread use of anesthetics in many animal models illustrates the many ways that model artifacts can confound preclinical results. Third, concordance between key features of the animal model and the human disease or condition being modeled is required to confirm model biofidelity. Fourth, experimental results observed in animals must be confirmed in human subjects for model validation. Adherence to these principles serves as a bulwark against flawed interpretation of results, study replication failure, and confusion in the field. Implementing these principles will advance basic science discovery and accelerate clinical translation to benefit people affected by concussion, TBI, and CTE.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}