 ,1, 袁海明2
,1, 袁海明2Application of chromosomal microarray analysis for a cohort of 2600 Chinese patients with miscarriage
Jiping Peng,1, Haiming Yuan2编委: 卢大儒
收稿日期:2018-06-29修回日期:2018-08-3网络出版日期:2018-09-20
Editorial board:
Received:2018-06-29Revised:2018-08-3Online:2018-09-20
作者简介 About authors
彭继苹,硕士研究生,研究方向:分子遗传学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (343KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
彭继苹, 袁海明. 染色体微阵列分析技术在2600例流产物中的应用[J]. 遗传, 2018, 40(9): 779-788 doi:10.16288/j.yczz.18-120
Jiping Peng, Haiming Yuan.
自然流产是指妊娠不到28周、胎儿体重不足1000 g、胎儿及其附属物脱离母体而妊娠自行终止者。妊娠12周之内终止者称为早期流产,临床上自然流产多表现为胎儿发育的停止。据统计,自然流产的发生率为15%~40%,而其中80%以上为发生在12周之内的早期流产[1]。复发性流产(recurrent spontaneous abortion,RSA)是指与同一性伴侣连续遭受两次或两次以上的自然流产。并且复发性自然流产约占妊娠总数的1%[1]。引起自然流产的因素大致分为胚胎因素、母体因素、环境因素和免疫功能异常,以及其他一些尚不明确的自然流产原因。胚胎因素中染色体异常是最常见的自然流产原因,其中50%~ 60%流产与胚胎的染色体异常相关[2, 3]。染色体异常中有86%为染色体数目异常(包括非整倍体和多倍体),6%为结构异常(包括染色体断裂、重复、缺失和易位等),8%为嵌合体等其他染色体异常[4]。目前发现染色体结构异常高达1万多种,除性染色体异常或部分染色体三体患者能存活下来,其他染色体数目异常患者均以死胎及流产告终,而染色体片段易位、缺失、重复为导致新生儿缺陷的重要因素,其也可导致胎儿的自然流产[5]。为区别基因突变,一般将染色体结构变异称为基因拷贝数变异(copy number variants, CNVs),而产前早期诊断和发现CNVs是产前临床诊断的难点和热点[5]。
早期传统的流产物检测手段包括核型分析与荧光原位杂交(fluorescence in situ hybridization, FISH)。核型分析分辨率低,仅可检测大于5 Mb的遗传物质改变,但无法对许多具有致病性的染色体亚显微结构变异(如染色体微缺失和微重复)进行检测。此外,核型分析需要对绒毛组织和羊水进行培养,耗时长且容易被母体细胞污染;并且一些活性较差的细胞生长将受到抑制,或母体细胞过度生长,这些因素均会导致无法得到精准的核型;另外,临床获得的部分流产物由于细胞活性丧失,会导致细胞培养失败,而无法进行核型分析检测[4, 6]。对流产物进行FISH检测尽管可以省去细胞培养,然而因其现有探针仅能对13、18、21、X、Y染色体数目进行检测, 而不能对全基因组进行检测,并且只能检测已知致病基因或可疑位点,不能区分正常和倒位、平衡易位携带者的胚胎;且一次检测,仅能对少数位点进行分析,缺乏整体性,不适用于全基因组的筛查,因此可能存在假阴性[7,8,9]。近年来,随着第二代测序技术和基因芯片技术的快速发展,它们也已经被应用于流产物病因筛查,并显示出明显检测优势,因此传统的检测方法也渐渐被其所替代。
染色体微阵列分析(chromosomal microarray analysis, CMA)技术可在全基因组范围内同时检测染色体的数目异常、结构异常(包括微缺失、微重复等)、嵌合体和纯合区域(regions of homozygosity, ROHs)等染色体异常类型。CMA技术无需进行细胞培养,具有周期短、高通量、高分辨率、高准确性等优点,通过一次杂交实验就能对整个基因组范围内的染色体非平衡变异进行扫描,将某一DNA序列变异准确定位到染色体上,将染色体病的诊断提高到基因水平上,弥补了核型、FISH检测技术的缺陷[10,11,12]。本研究应用CMA技术的3种不同芯片类型(CytoScan HD、CytoScan 750K和CytoScan Optima)对2600例流产物样本进行了检测,在全基因组水平分析引起流产的染色体异常情况,并评估该技术在临床流产中应用价值。
1 研究对象与方法
1.1 研究对象
2600例流产样本(浸泡于生理盐水中保存运输) 送检至广州金域医学检验中心。所有患者均签署了知情同意书。1.2 CMA分析
使用德国QIAGEN公司生产的组织提取试剂盒提取基因组DNA。应用美国Affymetrix公司生产的CytoScan HD芯片(195万CNV探针+75万SNP探针)、CytoScan 750K (55万CNV探针+20万SNP探针)芯片和专注产前领域的CytoScan Optima芯片对流产物样本进行检测,所得原始数据均应用Affymetrix Chromosome Analysis Suite Sofeware 进行分析。参照国际基因组CNVs多态性数据库Decipher、UCSC Genome Browser、OMIM (Online Mendelian Inheritance in Man)、ISCA (International Standards for Cytogenomic Arrays)、DGV (Database of Genomic Variants)等多个权威数据库以及相关文献来评估CNVs的致病性。1.3 CMA结果评估
检测报告严格按照美国医学遗传学会指南[13],将CNVs分为4个等级:(1)致病性CNVs;(2)可能致病性CNVs;(3)临床意义不明CNVs;(4)良性CNVs。2 结果与分析
2.1 3种芯片比较分析
本研究应用3种芯片对流产物样本进行检测,其中CytoScan HD芯片包含270万多个拷贝数分析标记(195万CNVs探针+75万SNP探针),具有无偏向的全基因组覆盖,跨越整个基因组的卓越性能,覆盖了RefSeq、OMIMTM、ClinGEN和DECIPHER/ DDD遗传基因区域以及Sanger癌基因区域,其可用于检测整个人类基因组中已知和新的染色体畸变。CytoScan 750K芯片包含75万多个拷贝数分析标记(55万CNVs探针+20万SNP探针),这些探针都精选于CytoScan HD芯片的探针,同时针对遗传疾病和肿瘤相关基因增加探针密度,另外也覆盖了RefSeq、OMIMTM、ClinGEN和DECIPHER/DDG2P遗传基因区域以及Sanger癌基因区域,使其更适合临床诊断的细胞遗传学检测。虽然这2种芯片含有的探针数不同,但它们均可在遗传性疾病、癌症、干细胞和神经发育领域中研究各类样品的染色体变异。CytoScan Optima芯片中的探针是从CytoScan HD芯片精选出来的,共包括31万多个拷贝数分析标记,除了包含18 018个CNVs探针和148 450个SNP探针外,还重点加密了既往产前和围产期研究相关的396个基因区域的探针,使其更好的应用于产前和围产期诊断。
2.2 CMA检出的染色体异常结果
在2600例流产样本中,成功检测2505例,成功率达96.35%。未成功检测的样本主要原因为流产物母体组织污染(每个样本及其母血都先做STR (short tandem repeat)连锁分析)、样本已腐败致无法提取DNA和DNA样本质量太差,导致不能完成CMA检测。经3种芯片检测,2505例流产物样本中有967例检出染色体异常,检出率为38.60%;其中1021例用CytoScan Optima芯片进行检测,有506例检出染色体异常,其检出率为50.00%;1211例用CytoScan 750K芯片进行检测,有388例检出染色体异常,其检出率为32.00%;273例用CytoScan HD芯片进行检测,有73例检出染色体异常,其检出率为26.74% (表1)。根据检出的染色体异常类型,可分为染色体数目异常、染色体结构异常、嵌合体 (图1)和纯合区域4大类。Table 1
表1
表1 不同芯片检出的染色体异常情况
Table 1
| 染色体异常类型 | 具体异常情况 | 3种芯片共 检出例数(%) | CytoScan Optima 芯片检出例数(%) | CytoScan 750K 芯片检出例数(%) | CytoScan HD 芯片检出例数(%) |
|---|---|---|---|---|---|
| 染色体数目异常 | 染色体非整倍体 | ||||
| 染色体三体 | 585 (60.50) | 309 (61.07) | 228 (58.76) | 48 (65.75) | |
| 染色体单体 | 81 (8.38) | 43 (8.50) | 33 (8.51) | 5 (6.85) | |
| 性染色体异常 | 6 (0.62) | 3 (0.59) | 3 (0.77) | 0 (0) | |
| 18四体 | 1 (0.10) | 0 (0) | 1 (0.26) | 0 (0) | |
| 三倍体 | 128 (13.24) | 53 (10.47) | 66 (17.01) | 9 (12.33) | |
| 染色体结构异常 | 染色体部分缺失/重复 | 94 (9.72) | 55 (10.87) | 33 (8.51) | 6 (8.22) |
| 染色体嵌合体 | 嵌合三体 | 48 (4.96) | 30 (5.93) | 17 (4.38) | 1 (1.37) |
| 嵌合单体 | 8 (0.83) | 2 (0.40) | 5 (1.29) | 1 (1.37) | |
| 纯合区域 | 整套染色体单亲二倍体 | 7 (0.72) | 5 (0.99) | 0 (0) | 2 (2.74) |
| 单条染色体单亲二倍体 | 2 (0.21) | 1 (0.20) | 0 (0) | 1 (1.37) | |
| 单条染色体ROHs | 4 (0.41) | 2 (0.40) | 2 (0.52) | 0 (0) | |
| 多条染色体ROHs | 3 (0.31) | 3 (0.59) | 0 (0) | 0 (0) | |
| 总例数 | 967 | 506 | 388 | 73 | |
| 检出率(%) | 38.60 | 50.00 | 32.00 | 26.74 |
新窗口打开|下载CSV
图1
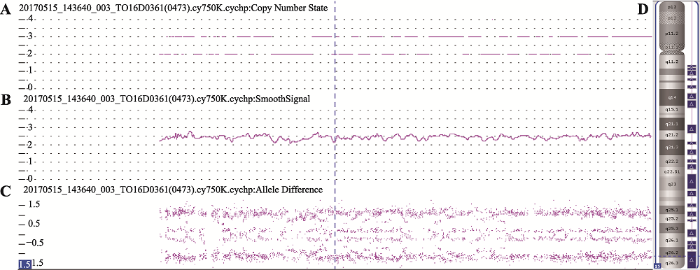 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1流产组织发生嵌合体
A:Copy Number State是CMA检测出的CNV片段的拷贝数,正常常染色体DNA拷贝数是2,当片段拷贝数为1或0时表示发生缺失,当片段拷贝数为3或4时表示发生重复,图中DNA拷贝数是2或3,表示嵌合三体。B:Smooth Signal是拷贝数的真实反映,即患者的15号染色体的DNA拷贝数为2.5。C:Allele Difference是DNA等位基因分布图,正常应该是3条信号带,分别在0、-1、1对应的位置,图中出现了4条信号带,中间2条信号带的位置分别在0~0.5和0~-0.5之间,而发生三体变异类型时中间2条信号带应该在0.5和-0.5处;所以此图表示染色体发生嵌合体时拷贝数和等位基因分布图的具体情况。D:从染色体层面直观反映嵌合重复的现象。
Fig.1Mosaicism of miscarriage specimens
2.3 染色体数目异常
3种CMA芯片在2505例流产物样本中共检出801例染色体数目异常,占所有染色体异常的82.83%;其中CytoScan Optima芯片检出401例,占该芯片检出染色体异常的80.63%;CytoScan 750K芯片检出331例,占该芯片检出染色体异常的85.31%;CytoScan HD芯片检出62例,占该芯片检出染色体异常的84.93%。由此可见,染色体数目异常是导致流产和胚胎停育的主要遗传因素。在801例染色体数目异常中,16三体例数最多,共155例,所占比例高达16.03%;其中CytoScan Optima芯片检出16三体78例;CytoScan 750K芯片检出63例16三体;其中CytoScan HD芯片检出16三体14例。此外,检测过程中也发现了较多的三倍体,共128例,占其中的13.24%;检出81例发生了22三体,所占比例为8.38%;77例为Turner综合征(45,X),所占比例为7.96%;40例发生了多条染色体三体,所占比例为4.14%。在检出的染色体数目异常中,除了1号染色体没有出现三体的外,其余染色体均出现了三体的情况(表2)。Table 2
表2
表2 不同芯片检出的染色体数目异常具体情况
Table 2
| 染色体数目异常 | 染色体 | 3种芯片共 检出例数(%) | CytoScan optima 芯片检出例数(%) | CytoScan 750K 芯片检出例数(%) | CytoScan HD 芯片检出例数(%) |
|---|---|---|---|---|---|
| 单条染色体三体 | Chr.2 | 9 (0.93) | 7 (1.38) | 1 (0.26) | 1 (1.37) |
| Chr.3 | 12 (1.24) | 7 (1.38) | 4 (1.03) | 1 (1.37) | |
| Chr.4 | 9 (0.93) | 6 (1.19) | 2 (0.52) | 1 (1.37) | |
| Chr.5 | 3 (0.31) | 2 (0.40) | 0 (0.00) | 1 (1.37) | |
| Chr.6 | 5 (0.52) | 1 (0.20) | 3 (0.77) | 1 (1.37) | |
| Chr.7 | 13 (1.34) | 5 (0.99) | 7 (1.80) | 1 (1.37) | |
| Chr.8 | 20 (2.07) | 11 (2.17) | 9 (2.32) | 0 (0.00) | |
| Chr.9 | 13 (1.34) | 4 (0.79) | 8 (2.06) | 1 (1.37) | |
| Chr.10 | 14 (1.45) | 10 (1.98) | 4 (1.03) | 0 (0.00) | |
| Chr.11 | 7 (0.72) | 4 (0.79) | 2 (0.52) | 1 (1.37) | |
| Chr.12 | 7 (0.72) | 2 (0.40) | 5 (1.29) | 0 (0.00) | |
| Chr.13 | 31 (3.21) | 16 (3.16) | 11 (2.84) | 4 (5.48) | |
| Chr.14 | 21 (2.17) | 9 (1.78) | 10 (2.58) | 2 (2.74) | |
| Chr.15 | 46 (4.76) | 23 (4.55) | 18 (4.64) | 5 (6.85) | |
| Chr.16 | 155 (16.03) | 78 (15.24) | 63 (16.24) | 14 (19.18) | |
| Chr.17 | 6 (0.62) | 4 (0.79) | 2 (0.52) | 0 (0.00) | |
| Chr.18 | 26 (2.69) | 12 (2.37) | 11 (2.84) | 3 (4.11) | |
| Chr.19 | 3 (0.31) | 1 (0.20) | 2 (0.52) | 0 (0.00) | |
| Chr.20 | 12 (1.24) | 10 (1.98) | 1 (0.26) | 1 (1.37) | |
| Chr.21 | 52 (5.38) | 29 (5.73) | 19 (4.90) | 4 (1.37) | |
| Chr.22 | 81 (8.38) | 42 (8.30) | 33 (8.51) | 6 (8.22) | |
| 多条染色体三体 | 40 (4.14) | 26 (5.14) | 13 (3.35) | 1 (1.37) | |
| 性染色体异常 | 47, XXX | 1 (0.10) | 1 (0.20) | 0 (0.00) | 0 (0.00) |
| 47, XXY | 3 (0.31) | 2 (0.40) | 1 (0.26) | 0 (0.00) | |
| 47, XYY | 1 (0.10) | 0 (0.00) | 1 (0.26) | 0 (0.00) | |
| 48, XYYY | 1 (0.10) | 0 (0.00) | 1 (0.26) | 0 (0.00) | |
| 染色体单体 | 45, X | 77 (7.96) | 40 (7.91) | 32 (8.25) | 5 (6.85) |
| 21单体 | 4 (0.41) | 3 (0.59) | 1 (0.26) | 0 (0.00) | |
| 18四体 | 1 (0.10) | 0 (0.00) | 1 (0.16) | 0 (0.00) | |
| 三倍体 | 128 (13.24) | 53 (10.47) | 66 (17.01) | 9 (12.33) | |
| 数目异常总例数 | 801 (82.83) | 408 (80.63) | 331 (85.31) | 62 (84.93) | |
| 检出率(%) | 31.98 | 39.96 | 27.33 | 22.71 |
新窗口打开|下载CSV
2.4 染色体结构异常
在967例染色体异常中有94例是染色体结构异常,占其中的9.72% (94/967),其中68例在染色体末端发生缺失和或重复,其长度介于1.0~104 Mb之间,涉及大量功能基因,可导致胚胎严重发育迟缓、多发畸形、先天性心脏病等,且多为致死性。临床资料显示这些样本的孕妇多发生反复流产,提示夫妻双方可能发生相应片段的染色体平衡易位,再发风险高。在26例流产样本中发现了致病性染色体微缺失、微重复,检出率为1.04% (26/2505),其中有4种再发性CNVs,包括22q11.2微缺失、7q11.23微缺失、17q11.23微缺失和22q13.3微缺失,其中22q11.2微缺失可导致Di-George综合征,7q11.23微缺失与Williams-Beuren综合征有关,22q13.3微缺失常出现在神经发育障碍的患者中。
2.5 染色体嵌合体
2505例流产物样本中共检出56例染色体嵌合体,占所有染色体异常的5.79%,其中46例发生了单条染色体嵌合三体,22嵌合三体最多,共10例;8例为嵌合单体,均为X嵌合单体;还有2例同时发生2条染色体嵌合,包括1例携带18和22嵌合三体,另1例携带15和22嵌合三体(表3)。Table 3
表3
表3 不同芯片检出的染色体嵌合体类型
Table 3
| 染色体嵌合体 | 染色体 | 3种芯片共 检出例数 | CytoScan optima 芯片检出例数 | CytoScan 750K 芯片检出例数 | CytoScan HD 芯片检出例数 |
|---|---|---|---|---|---|
| 单条染色体嵌合三体 | Chr.2 | 4 | 1 | 3 | 0 |
| Chr.3 | 1 | 1 | 0 | 0 | |
| Chr.4 | 3 | 3 | 0 | 0 | |
| Chr.5 | 1 | 1 | 0 | 0 | |
| Chr.7 | 3 | 1 | 2 | 0 | |
| Chr.8 | 1 | 0 | 1 | 0 | |
| Chr.9 | 1 | 1 | 0 | 0 | |
| Chr.10 | 1 | 0 | 1 | 0 | |
| Chr.11 | 1 | 1 | 0 | 0 | |
| Chr.13 | 1 | 1 | 0 | 0 | |
| Chr.14 | 1 | 0 | 1 | 0 | |
| Chr.15 | 3 | 2 | 1 | 0 | |
| Chr.16 | 5 | 4 | 0 | 1 | |
| Chr.17 | 1 | 1 | 0 | 0 | |
| Chr.18 | 1 | 0 | 1 | 0 | |
| Chr.19 | 1 | 1 | 0 | 0 | |
| Chr.20 | 2 | 1 | 1 | 0 | |
| Chr.21 | 2 | 0 | 2 | 0 | |
| Chr.22 | 10 | 8 | 2 | 0 | |
| Chr.X | 3 | 3 | 0 | 0 | |
| 多条染色体嵌合三体 | Chr.18 & 22 | 1 | 0 | 1 | 0 |
| Chr.15 & 22 | 1 | 0 | 1 | 0 | |
| 嵌合单体 | (X)×1-2 | 7 | 1 | 5 | 1 |
| (X)×1-2, (Y)×1 | 1 | 1 | 0 | 0 | |
| 嵌合体总例数 | 56 | 32 | 22 | 2 | |
| 检出率(%) | 2.24 | 3.13 | 1.82 | 0.73 |
新窗口打开|下载CSV
2.6 检出的染色体纯合区域
本研究检出7例样本发生全染色体单亲二倍体(uniparental disomy, UPD),可能是导致其胎儿停止发育而流产的原因,但是目前并没有充分的证据证明整条染色体的单亲二倍体(UPD)会导致胚胎的停育或流产,只能提示胎儿隐性遗传病的发病风险增加。2例出现单条染色体的单亲二倍体——UPD(3)和UPD(4);4例检出单条染色体大片段纯合区域(ROHs):3p13q21.3区域发生56.7 Mb大小的ROHs,7p14.3q11.22区域发生37.2 Mb大小的ROHs,17q12q25.1区域发生37 Mb大小的ROHs,1p36.33p36.21和1q32.1q43区域分别发生了13.0 Mb、37.7 Mb大小的ROHs;3例在染色体多处发生ROHs,其总长度分别为129.8 Mb、132.4 Mb、241.9 Mb,均大于常染色体总长度的4.6%,提示夫妻双方为四级亲缘关系以上,这增加了隐性遗传病的发病风险。3 讨 论
CMA技术是近几年兴起的分子遗传学诊断技术,其最大的特点在于一次检测就能够对整个基因组拷贝数进行高通量、高分辨率、高敏感性扫描和分析,能精确定位变异区段的位置,清楚显示目标片段内的基因含量,并能够对基因型—表型关系进行分析,在基因水平上解释患者的临床表现和评估预后,在染色体非平衡变异检测中具有其他染色体分析技术所无法比拟的优越性[14,15,16,17]。本文用CMA技术的3种类型芯片对2600例自然流产样本进行了检测,成功率高达96.3%,这远高于传统核型分析对流产物检测的成功率[18, 19],从而可对更多不明原因的自然流产和停育做出明确诊断。染色体异常是自然流产的常见原因,占早期流产原因的50%左右[2, 3, 20, 21]。本研究用CMA技术的3种芯片共检出967例染色体异常,总阳性率为38.60%;其中CytoScan Optima芯片的阳性率为50.00%,CytoScan 750K芯片的阳性率为32.00%;CytoScan HD芯片检出73例染色体异常,其阳性率为26.74%,CytoScan 750K和CytoScan HD芯片的阳性率比既往相关研究的检出率偏低(50.1%、55.1%)[6, 22],这可能是由芯片探针设计公司不同、芯片类型不同、芯片探针分布不同、数据分析软件不同等多种技术因素,或者是既往相关研究的样本量(500左右)比本研究的少很多,而造成阳性率的差异。本研究中CytoScan Optima芯片的阳性率与之基本相符(50.1%、55.1%)[6, 22],这可能与CytoScan Optima芯片是专门针对产前和围产期诊断设计的芯片类型有关。本研究中CytoScan 750K和CytoScan HD芯片的阳性率比CytoScan Optima芯片偏低的重要原因可能是前两种芯片并不是用于流产物检测的最佳方法。
967例染色体异常中有801例是染色体数目异常,占其中的82.83%,与既往文献报道(86%)基本一致[4, 23],也证实了染色体数目异常是导致胚胎流产的主要原因[4, 24, 25]。绝大多数染色体非整倍体具有胚胎期致死性,其中16三体所占比例最高(16.3%),其次是三倍体(13.24%),Turner综合征(45,X)和13、15、21、22三体也占较大的比例,而其他染色体三体所占比例较低。绝大多数染色体非整倍体为新生突变,而D组(13、14、15号)染色体和G组(21、22号)染色体易发生罗伯逊易位[26],再发风险高,故建议夫妻双方做核型分析,以排除罗伯逊易位,对再发风险进行评估。大多数人认为孕妇年龄为染色体数目异常的高危因素,而本研究结果显示,各个年龄段的孕妇均有发生染色体数目异常导致流产的情况,801例染色体数目异常的流产物中有92例来自16~25岁的孕妇,281例来自26~30岁的孕妇,175例来自31~35岁的孕妇,179例来自36~40岁的孕妇,74例来自40岁以上的孕妇,由此可见对染色体数目异常的筛查不应只关注高年龄孕妇,各个年龄段的孕妇均需要染色体非整倍体的筛查。2007年美国妇产科医师学会提出,所有年龄段的孕妇均应进行非整倍体筛查[27]。然而,对于流产物染色体异常分析,孕妇年龄和孕周是两个最重要因素,但本研究由于各种原因未能获得全部孕妇年龄和孕周的相关资料,因此不能完成孕妇年龄和孕周的分层分析,从而不能对孕妇年龄和孕周与自然流产的关系进行分析,这是本研究的不足和遗憾之处。
染色体结构变异也是自然流产的重要因素[4, 21],本研究共检出94例染色体结构异常,占9.72%,高于既往相关研究(6%)[4],这可能是由于传统核型分析分辨率低以及FISH只限于靶向检测,而对染色体结构异常的检出率低于CMA。其中有68例在染色体末端发生缺失和/或重复,其长度介于1.0~104 Mb之间,涉及大量功能基因,这些可导致胚胎严重发育迟缓、多发畸形、先天性心脏病等,且多为致死性。临床资料显示,这些样本的孕妇多发生在孕早期,且为反复流产患者,提示夫妻双方可能发生相应片段的染色体平衡易位,再发风险高[4, 28, 29]。而然由于各种原因,未能完成这些夫妻双方是否发生染色体片段的平衡易位的检测。既往文献报道显示,亚显微结构拷贝数变异存在于0.6%和0.78%的自然流产样本中[30, 31],本研究在26例流产样本中发现了致病性染色体微缺失、微重复,检出率为1.04% (26/2505),其中有4种再发性CNVs,包括22q11.2微缺失、7q11.23微缺失、17q11.23微缺失和22q13.3微缺失。22q11.2微缺失可导致Di-George综合征,其主要临床表现包括先天性心脏病,多发畸形,宫内发育迟缓和神经发育障碍等[32]。本研究共有5例流产样本携带22q11.2微缺失,检出率为0.20% (5/2505),这与既往流产物相关研究检出率相符(0.05%~0.8%)[30, 32, 33],但这比正常出生婴儿中22q11.2微缺失发生率(0.013%)高很多[34]。本文的研究结果也进一步证实22q11.2微缺失可能与自然流产有关,22q11.2微缺失导致胎儿心血管系统畸形可能是其致流产的潜在机制。7q11.23微缺失与Williams- Beuren综合征有关,其主要表型为面部畸形、心血管异常、结缔组织异常、高钙血症和一种独特的神经行为表现等[35]。同时,本研究在1例样本中还发现了17q11.23微缺失,检出率为0.04% (1/2505),这与既往研究结果基本相符(0.05%)[30],但比正常出生婴儿中17q11.23微缺失发生率(0.013%)高[36],这表明17q11.23微缺失可能与流产有关。22q13.3微缺失常在神经发育障碍的患者中被检出,本研究中其检出率为0.04% (1/2505),这远远低于既往神经发育障碍相关研究的检出率(1.7%)[37],该变异在既往流产物相关研究中并未被发现过,这提示22q13.3微缺失与流产的相关性有待进一步研究。本研究中发现的其他亚微观的CNVs,之前均没有报道过的,其与流产的相关性现在还不确定,需要进行更大规模的研究来证实这些CNVs是否为流产的原因。染色体微缺失/重复的胚胎致死性没有染色体数目异常强,仅在表型比较严重时才会导致胎儿致死,这是受到遗传外显率和表现度不同的影响。传统核型分析无法检测此类微缺失/重复,而CMA则能够更有效地检出由于染色体结构异常导致的流产,并通过对夫妻双方相应位点的检测,可对再生育进行遗传风险评估。
本研究共检出嵌合体56例,占所有染色体异常的5.79% (56/967),比以往研究结果(8%)偏低[4],其中22嵌合三体和X嵌合体所占比例较多,分别占17.86% (10/56)和14.29% (8/56)。值得注意的是,有2例样本同时携带2条染色体嵌合三体:1例为18和22嵌合三体,另1例为15和22嵌合三体,较为罕见。同时还检出7例全染色体组UPD,这是由孤雄生殖或孤雌生殖导致的。母系UPD将表现为畸胎瘤,而父系UPD则表现为葡萄胎,二者均可导致流产[38, 39]。3例样本出现多条染色体多处大片段ROHs,其总长度均大于常染色体总长度的4.6%,提示夫妻双方为四级亲缘关系或以上,这导致隐性遗传病的发病风险增加[40],然而由ROHs所致流产的病例未被报道过,核型分析和FISH均无法检测该类变异,从而显示出CMA技术独特的优势。
综上所述,CMA是一种全新的现代化分子核型分析技术,其有效克服了传统染色体诊断技术的缺点,能够快速、准确地对流产物全基因组进行分析,从而检测出常规核型分析能发现的非平衡染色体变异和数目异常,以及核型分析技术无法检测到的CNVs (<5 Mb)和ROHs,将染色体病的诊断水平提高到基因层面上。CMA是一种可以应用于临床流产物遗传学诊断的可靠、稳定、高分辨的技术,检测结果能够对再生育风险评估提供指导。
致谢
感谢广州金域医学检验中心有限公司的陈婉华、张玉华、唐荣熹、卜晓玲、杨娟等CMA平台技术人员为本研究提供高质量的检测数据。
(责任编委: 卢大儒)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:12456605 [本文引用: 2]

BACKGROUND: The aim of this study was to investigate the changes in circulating levels and the clinical use of inhibin A, activin A and follistatin as endocrine markers of early pregnancy loss. METHODS: Blood samples were collected from women presenting with a sporadic missed miscarriage (n = 10), and controls having pregnancy termination at 8-12 weeks (n = 15) and from women with a history of unexplained recurrent miscarriages (n = 12) at 6-12 weeks gestation. All samples were assayed for inhibin A, inhibin B, activin A, follistatin, hCG, estradiol and progesterone. RESULTS: Serum inhibin A, hCG, estradiol and progesterone levels were significantly ( approximately 2-3 fold) decreased in sporadic miscarriages compared with controls. In the recurrent miscarriage group, time dependent changes in plasma inhibin A and hCG levels were significantly (P < 0.05) altered in the group that had a subsequent miscarriage compared with those who had a live birth. At 6-7 weeks gestation, plasma inhibin A ( approximately 4 fold, P < 0.01), hCG ( approximately 4 fold, P < 0.01) and estradiol ( approximately 2 fold, P < 0.001) levels were significantly lower in women who went on to have another miscarriage than those with a live birth. Inhibin B levels were near the detection limit of the assay. CONCLUSIONS: Our findings suggest that inhibin A is a specific marker of early pregnancy loss before the onset of the clinical symptoms of recurrent miscarriage. There is a high degree of association between levels of inhibin A and hCG in cases of miscarriage, indicating that these two proteins could be used in combination to predict future pregnancy outcome.
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 8]
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 3]
URL [本文引用: 1]
URLPMID:24646446 [本文引用: 1]
Spontaneous abortion (SA) is the loss of the conceptus before 22 weeks of gestation when fetal weight is less than 500g. The genetic etiology accounts for more than two third of SA, and autosomal aneuploidies alone account for up to 70% fetal loss. The aim of this study was to highlight the most common chromosomal causes of fetal loss. In this study, 220 products of abortion and in utero fetal death were analyzed by using FISH (AneuVysion鈩) on interphase nuclei from chorionic villus and by using MLPA (SALSA P036, P070 and P245 kits) on DNA extracted from fetal tissues. The gestational age ranged from the 7th to the 38th week of gestation. Of a total of 151 samples analyzed by using FISH, 10 chromosomal abnormalities were observed: four trisomies 21 (one of them was mosaic), a trisomy 18, a trisomy 13, three triploidies and one monosomy X (Turner). From the additional 69 samples analyzed by using MLPA, two anomalies were found: two monosomies X (Turner). FISH and MLPA are simple, rapid and sensitive tools for the detection of chromosomal aneuploidies. Avoiding the cell culture step necessary for karyotyping, they represent very interesting alternative methods to diagnose genomic disorders in products of abortion in which poor sample quality often leads to cell culture failure.
[本文引用: 1]
[本文引用: 1]
URLPMID:23215556 [本文引用: 1]
BACKGROUND Genetic abnormalities have been associated with 6 to 13% of stillbirths, but the true prevalence may be higher. Unlike karyotype analysis, microarray analysis does not require live cells, and it detects small deletions and duplications called copy-number variants. METHODS The Stillbirth Collaborative Research Network conducted a population-based study of stillbirth in five geographic catchment areas. Standardized postmortem examinations and karyotype analyses were performed. A single-nucleotide polymorphism array was used to detect copy-number variants of at least 500 kb in placental or fetal tissue. Variants that were not identified in any of three databases of apparently unaffected persons were then classified into three groups: probably benign, clinical significance unknown, or pathogenic. We compared the results of karyotype and microarray analyses of samples obtained after delivery. RESULTS In our analysis of samples from 532 stillbirths, microarray analysis yielded results more often than did karyotype analysis (87.4% vs. 70.5%, P<0.001) and provided better detection of genetic abnormalities (aneuploidy or pathogenic copy-number variants, 8.3% vs. 5.8%; P=0.007). Microarray analysis also identified more genetic abnormalities among 443 antepartum stillbirths (8.8% vs. 6.5%, P=0.02) and 67 stillbirths with congenital anomalies (29.9% vs. 19.4%, P=0.008). As compared with karyotype analysis, microarray analysis provided a relative increase in the diagnosis of genetic abnormalities of 41.9% in all stillbirths, 34.5% in antepartum stillbirths, and 53.8% in stillbirths with anomalies. CONCLUSIONS Microarray analysis is more likely than karyotype analysis to provide a genetic diagnosis, primarily because of its success with nonviable tissue, and is especially valuable in analyses of stillbirths with congenital anomalies or in cases in which karyotype results cannot be obtained.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:22467164 [本文引用: 1]
Genomic microarrays are now widely used diagnostically for the molecular karyotyping of patients with intellectual disability, congenital anomalies and autistic spectrum disorder and have more recently been applied for the detection of genomic imbalances in prenatal genetic diagnosis. We present an overview of the different arrays, protocols used and discuss methods of genomic array data analysis. 2012 John Wiley & Sons, Ltd.
URL [本文引用: 1]
URLPMID:19617844 [本文引用: 1]
About 50% of spontaneous abortions are caused by fetal chromosome abnormalities. Identification of these abnormalities helps to estimate recurrence risks in future pregnancies. However, due to culture failures or maternal contamination often no fetal karyotype can be obtained. Array comparative genomic hybridization can overcome some of these limitations. In this study, we analyzed 103 miscarriages by both T-banding and 1-Mb array comparative genomic hybridization. We found an overall abnormality rate of 35% (34 of 96). In a comparison of 70 samples that were successfully analyzed by both techniques, 54 (77%) had identical karyotypes (42 normal, 12 abnormal) and 16 (23%) cases showed discrepancies. Most of these differences were due to maternal contamination during cell culture, which resulted erroneously in a normal female karyotype. These results demonstrate the improved diagnostic yield of array comparative genomic hybridization as compared with conventional karyotyping. Therefore, we implemented this technique in the diagnostic workup of miscarriages.
URLPMID:20105165 [本文引用: 1]
Please cite this paper as: Choy K, Setlur S, Lee C, Lau T. The impact of human copy number variation on a new era of genetic testing. BJOG 2010;117:391 397. Cytogenetic studies have demonstrated that duplications or deletions of entire chromosomes or microscopically visible aberrations are associated with specific congenital disorders. The subsequent development and application of microarray-based assays have established the importance of copy number variants (CNV) as a substantial source of genetic diversity in the human genome. Pathogenic CNVs are associated not only with birth defects and cancers, but also with neurodevelopmental disorders at birth or neurodegenerative diseases in adulthood. Unfortunately, the limited knowledge of the phenotypic effects of most CNVs has led to the classification of many CNVs as genomic imbalances of unknown clinical significance. This has caused many clinicians to resist the introduction of microarray technologies in detecting CNVs in a genome-wide manner for prenatal applications. This review summarises our current understanding of CNVs, the common detection methods, and the implications for human health and prenatal diagnosis.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 2]
目的:探讨自然流产与染色体异常的关系以及染色体微阵列(chromosomal microarray analysis,CMA)技术在自然流产病因诊断中的应用价值。方法收集自然流产样本440例,对其进行 CMA检测。结果在440例流产样本中,成功检测417例,成功率为94.7%。染色体异常的样本为209例(50.1%),其中染色体数目异常129例(61.7%),结构异常40例(19.1%),嵌合体38例(18.1%),纯合子区域2例(1.0%)。结论相较于染色体核型分析,CMA 可为流产物检测提供更全面的信息,为患者的病因诊断和再生育风险评估提供指导。
URL [本文引用: 2]
目的:探讨自然流产与染色体异常的关系以及染色体微阵列(chromosomal microarray analysis,CMA)技术在自然流产病因诊断中的应用价值。方法收集自然流产样本440例,对其进行 CMA检测。结果在440例流产样本中,成功检测417例,成功率为94.7%。染色体异常的样本为209例(50.1%),其中染色体数目异常129例(61.7%),结构异常40例(19.1%),嵌合体38例(18.1%),纯合子区域2例(1.0%)。结论相较于染色体核型分析,CMA 可为流产物检测提供更全面的信息,为患者的病因诊断和再生育风险评估提供指导。
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:17197615 [本文引用: 1]
In the last decade, numerous markers and strategies for Down syndrome screening have been developed. Algorithms that combine ultrasound and serum markers in the first and second trimesters have been evaluated. Furthermore, the practice of using age cutoffs to determine whether women should be offered screening or invasive diagnostic testing has been challenged. The purpose of this document is to 1) present and evaluate the best available evidence for the use of ultrasonographic and serum markers for selected aneuploidy screening in pregnancy and 2) offer practical recommendations for implementing Down syndrome screening in practice.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 3]
URL [本文引用: 1]
URLPMID:3591861 [本文引用: 2]
Velocardiofacial and DiGeorge syndromes, also known as 22q11.2 deletion syndrome (22q11DS), are congenital-anomaly disorders caused by a de novo hemizygous 22q11.2 deletion mediated by meiotic nonallelic homologous recombination events between low-copy repeats, also known as segmental duplications. Although previous studies exist, each was of small size, and it remains to be determined whether there are parent-of-origin biases for the de novo 22q11.2 deletion. To address this question, we genotyped a total of 389 DNA samples from 22q11DS-affected families. A total of 219 (56%) individuals with 22q11DS had maternal origin and 170 (44%) had paternal origin of the de novo deletion, which represents a statistically significant bias for maternal origin (p = 0.0151). Combined with many smaller, previous studies, 465 (57%) individuals had maternal origin and 345 (43%) had paternal origin, amounting to a ratio of 1.35 or a 35% increase in maternal compared to paternal origin (p = 0.000028). Among 1,892 probands with the de novo 22q11.2 deletion, the average maternal age at time of conception was 29.5, and this is similar to data for the general population in individual countries. Of interest, the female recombination rate in the 22q11.2 region was about 1.6 1.7 times greater than that for males, suggesting that for this region in the genome, enhanced meiotic recombination rates, as well as other as-of-yet undefined 22q11.2-specific features, could be responsible for the observed excess in maternal origin.
URLPMID:25846569 [本文引用: 1]
ABSTRACT Objectives To determine the frequency of clinically significant chromosomal abnormalities identified by chromosomal microarray in pregnancy losses at any gestational age and to compare microarray performance with that of traditional cytogenetic analysis when testing pregnancy losses. Methods Among 535 fetal demise specimens of any gestational age, clinical microarray-based comparative genomic hybridization (aCGH) was performed successfully on 515, and a subset of 107 specimens underwent additional single nucleotide polymorphism (SNP) analysis. Results Overall, clinically significant abnormalities were identified in 12.8% (64/499) of specimens referred with normal or unknown karyotypes. Detection rates were significantly higher with earlier gestational age. In the subset with normal karyotype, clinically significant abnormalities were identified in 6.9% (20/288). This detection rate did not vary significantly with gestational age, suggesting that, unlike aneuploidy, the contribution of submicroscopic chromosomal abnormalities to fetal demise does not vary with gestational age. In the 107 specimens that underwent aCGH and SNP analysis, seven cases (6.5%) had abnormalities of potential clinical significance detected by the SNP component, including female triploidy. aCGH failed to yield fetal results in 8.3%, which is an improvement over traditional cytogenetic analysis of fetal demise specimens. Conclusions Both the provision of results in cases in which karyotype fails and the detection of abnormalities in the presence of a normal karyotype demonstrate the increased diagnostic utility of microarray in pregnancy loss. Thus, chromosomal microarray testing is a preferable, robust method of analyzing cases of pregnancy loss to better delineate possible genetic etiologies, regardless of gestational age. Copyright 2015 ISUOG. Published by John Wiley & Sons Ltd.
URLPMID:1717723 [本文引用: 1]
AIMSTo determine the prevalence of submicroscopic deletions within chromosome band 22q11 in infants with significant heart disease and compare this with the prevalence of other chromosomal abnormalities causing significant heart disease. To determine a minimum prevalence of deletions within chromosome band 22q11 in infants in the general population.METHODSChromosome analysis was performed on samples from infants born in the former UK Northern Health Region in 1994 and 1995 who either had significant heart disease or who were suspected to have a chromosome band 22q11 deletion following referral to the Northern Genetics Service. Significant heart disease was defined as major structural malformation or cases where invasive investigation or intervention was required in infancy.RESULTSChromosome band 22q11 deletions were identified in nine infants in a population of 69 129 livebirths, giving a minimum prevalence of 13 per 100 000 (95% confidence interval 4.5 to 21.5). Six cases had significant heart disease, one of whom died before diagnosis. In the same population there were 53 cases of trisomy 21, 15 of whom had significant heart disease.CONCLUSIONThe most common chromosomal cause of significant congenital heart disease remains trisomy 21, while the second most common chromosomal cause is deletion in chromosome band 22q11.
URLPMID:20437059 [本文引用: 1]
Copy number variants (CNVs) of the Williams euren syndrome (WBS) 7q11.23 region are responsible for neurodevelopmental disorders with multi-system involvement and variable expressivity. Typical features of WBS microdeletion comprise a recognizable pattern of facial dysmorphisms, supravalvular aortic stenosis, connective tissue abnormalities, hypercalcemia, and a distinctive neurobehavioral phenotype. Conversely, the phenotype of patients carrying the 7q11.23 reciprocal duplications includes less distinctive facial dysmorphisms and prominent speech delay. The common deletion/duplication ranges in size from 1.5 to 1.8 Mb and encompasses approximately 28 genes. This region is flanked by low copy repeats (LCRs) with greater than ~97% identity, which can mediate non-allelic homologous recombination resulting from misalignment of LCRs during meiosis. A clear genotype henotype correlation has been established in WBS only for the elastin gene, which is responsible for the vascular and connective tissue abnormalities. The molecular substrates underlying the other clinical features of 7q11.23 CNVs, including the neurocognitive phenotypes, are still debated. Recent studies suggest that besides the role of the genes in the deleted/duplicated interval, multiple factors such as regulatory sequences, epigenetic mechanisms, parental origin of the CNV, and nucleotide variations in the non-deleted/duplicated allele may be important in determining the variable expressivity of 7q11.23 CNV phenotypes. Here, we review the clinical and molecular findings and the recent insights on genomic disorders associated with CNVs involving the 7q11.23 region.
URLPMID:12088082 [本文引用: 1]
There are limited population-based data on the occurrence of Williams syndrome. We estimated its prevalence combining data from two investigations. One was an epidemiologic study originally designed to assess the prevalence and etiology of mental retardation among 30,037 Norwegian children born between 1980 and 1985 and living in Akershus County on January 1, 1993. The other investigation was a national survey of Williams syndrome. In the first study, 213 children were referred for evaluation, whereas the second study comprised 57 cases with Williams syndrome born between 1970 and 1992, who were referred for evaluation from all Norwegian counties. The epidemiologic study revealed three children with Williams syndrome, whereas one additional case complying with our demographic criteria was identified in the national survey, thus giving a prevalence of 1 in 7500. In all cases, a typical chromosome 7q11.23 deletion was detected. We also conclude that Williams syndrome is not an uncommon cause of mental retardation, with a prevalence of approximately 6% of patients with genetic etiology. (J Child Neurol 2002;17:269-271).
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]

{kind=link}
{kind=link}