 ,浙江大学生命科学学院植物生物学研究所,杭州 310058
,浙江大学生命科学学院植物生物学研究所,杭州 310058Identifying T-DNA insertion site(s) of transgenic plants by whole-genome resequencing
Jiming Xu, Han Hu, Wenxuan Mao, Chuanzao Mao,Institute of Plant Biology, College of Life Science, Zhejiang University, Hangzhou 310058, China通讯作者:
编委: 吴为人
收稿日期:2018-04-2修回日期:2018-06-11网络出版日期:2018-08-16
| 基金资助: |
Received:2018-04-2Revised:2018-06-11Online:2018-08-16
| Fund supported: |
作者简介 About authors
徐纪明,博士,助理研究员,研究方向:作物磷高效分子机制E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (724KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
徐纪明, 胡晗, 毛文轩, 毛传澡. 利用重测序技术获取转基因植物T-DNA插入位点[J]. 遗传, 2018, 40(8): 676-682 doi:10.16288/j.yczz.18-080
Jiming Xu, Han Hu, Wenxuan Mao, Chuanzao Mao.
T-DNA插入位点是指转基因植物中载体T-DNA序列插入到染色体的位置。在植物功能基因组学研究中,明确T-DNA插入位点的信息对相关研究的顺利开展非常重要。不论是使用T-DNA激活标签群体还是T-DNA插入失活群体筛选材料,获得目标材料后,最关键的一步就是通过得到T-DNA插入位点明确目标基因;而在转基因生产应用中,T-DNA插入位点是每份转基因新材料的标签,转基因材料的筛选鉴定、转基因材料的安全性评估、环境释放申请等都要求提供每份转基因材料的T-DNA插入位点。目前获取T-DNA插入位点的方法很多,根据原理可以分为3类:反向PCR,外源接头介导PCR,半随机引物PCR (如Tail-PCR)。另外,其他方法如质粒拯救法(plasmid rescue)、PCR-walking法等,都已经被广泛用于插入位点的鉴定及侧翼序列的获取。但是,这些方法除了操作步骤复杂、消耗时间长等缺点外,还具有很大的不确定性,特异性比较差,无法获得很高的成功率[1,2,3,4,5]。因此,建立一种简单、快速、成功率高的T-DNA插入位点获取方法非常必要。
近年来,随着测序技术的不断发展,全基因组重测序技术渐趋成熟。全基因组重测序的时间与成本大幅度下降,使得全基因组重测序的应用越来越广泛,除了用于测定不同物种全基因组序列、构建全基因组图谱外,还被用于突变体基因克隆等方面。Nordström等[6]使用全基因组重测序技术进行突变基因的克隆,不但缩短了图位克隆的时间,也使得很多杂交后表型难以分离的突变体基因得到克隆。Hu等[7]利用全基因组重测序对多个相同表型的突变体直接鉴定突变基因,省去杂交创制分离群体的步骤。为了探讨利用基因组重测序技术是否能够进行T-DNA插入位点的鉴定,本文对3份转基因材料基因组DNA打包后,利用全基因组重测序技术,并使用载体T-DNA序列作为模板进行比对分析,成功获得了3份转基因材料的T-DNA插入位点。
1 材料和方法
1.1 转基因材料
3份转基因材料分别为PT8A-1、PT8A-2和PT8A-3,均为本实验室利用农杆菌介导的转基因方法,使用草甘膦作为抗性筛选标记得到的转基因株系,受体亲本为日本晴(Oryza sative L. ssp. Japonica cv. Nipponbare,本实验室保存)。转基因过程参照Chen等[8]方法,转基因载体为35S-1300-G9A-PT8A (图1)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1PT8A转基因载体35S-1300-G9A-PT8A示意图
Fig. 1Map of 35S-1300-G9A-PT8A vectors used for PT8A transgenes
1.2 DNA提取
转基因材料PT8A-1、PT8A-2、PT8A-3和野生型品种日本晴种子萌发露白后,在正常水稻培养液[9]中培养30 d,取叶片用液氮磨碎后,参照陈昆松等[10]方法,提取3份转基因材料基因组DNA(DNA总量至少1 μg)。用含有溴化乙锭的1%琼脂糖凝胶电泳检测DNA的纯度和完整性,并利用Nanodrop测定DNA浓度。根据转基因载体中草甘膦抗性基因G9A序列,使用Oligo7软件设计引物,利用PCR方法确定3份转基因材料为转基因阳性株系。引物由华大基因公司合成,引物信息见表1。Table 1
表1
表1 引物序列信息
Table 1
| 引物名称 | 引物序列(5′→3′) |
|---|---|
| G9A | F: CCCATCTCGCGGAGCACGTT |
| R: CGCCTCCCGATCTCCGTGTCC | |
| Chr.2 | F: ATGAACTGATTGATTCGGTCT |
| R: TTTCATTTGGAGAGAACACGG | |
| Chr.7 | F: TAGAGGATCTACCATGGCCAC |
| R: AGCTGCCTTTTGAATGTCAC | |
| Chr.9 | F: GGCTTCGCTCTTAAACCCCTC |
| R: CAGTACTAAAATCCAGATCCCC | |
| Chr.11 | F: CTGCTCTAGCCAATACGCAAA |
| R: ATGCCCATTTTAACCGGAT |
新窗口打开|下载CSV
1.3 基因组重测序
将3份基因组DNA进行浓度测定后,等量混合。由于重测序需要至少2 μg基因组DNA,因此3份转基因材料的DNA等量混合时要保证DNA总量>2 μg。利用非接触式超声破碎仪把DNA打断为200~ 300 bp片段,按照Nextera DNA文库制备试剂盒(Illumina公司,美国)说明书的方法进行建库,并使用Illumina Hiseq2500进行常规基因组重测序(由杭州谷禾信息技术有限公司完成),每个Reads长度150 bp,得到至少12 Gb数据,并保证数据质量指标Q30≥80% (即:测序错误率大于0.1%的碱基所占的比例低于20%)。1.4 数据分析
根据基因组重测序结果,利用Bowtie2软件以转基因载体T-DNA序列作为模板,与测序得到的全部序列进行序列同源性比较筛选(Bowtie2, http:// bowtie-bio.sourceforge.net/bowtie2/index.shtml,默认设置)。将筛选得到的序列进一步拼装筛选,将序列全部为载体序列的Reads去掉,最终得到一类Reads序列,其特点是一半为基因组序列,另一半为载体序列。根据得到的基因组序列,在水稻基因组网站(http://rice.plantbiology.msu.edu/standalone_blast.shtml)进行Blast序列比对,得到该基因组序列在基因组上的具体位置,即为可能的插入位点。1.5 PCR验证
根据数据分析得到的T-DNA插入位点基因组DNA序列信息,以及测序得到的载体序列信息,使用Oligo7软件设计引物(表1),上游引物设计在载体序列上,下游序列设计在插入位点附近的基因组序列上,PCR扩增产物长度预期在250~500 bp之间,对3份转基因材料基因组DNA进行PCR验证,确定每份转基因材料的插入位点是否正确。引物由华大基因公司合成。1.6 Southern blot验证
提取转基因材料PT8A-1、PT8A-2、PT8A-3和野生型品种日本晴的基因组DNA[10],每份材料各取20 μg,使用Hind Ⅲ单酶切后,利用G9A引物扩增出的DNA片段作为探针,按照Zhou等[11]的方法进行Southern blot分析。2 结果与分析
2.1 基因组DNA提取与阳性鉴定
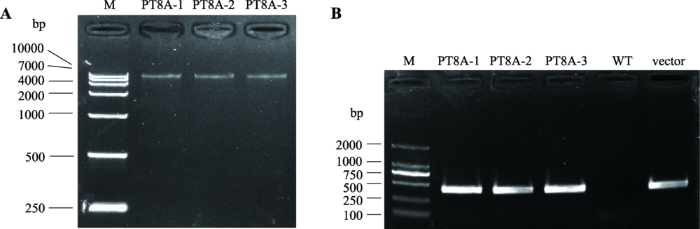
分别提取转基因材料PT8A-1、PT8A-2、PT8A-3的基因组DNA,并进行电泳检测。3份转基因材料提取的DNA结构完整,没有发生降解(图2A)。对3份转基因材料进行草甘膦抗性基因PCR分析,结果显示3份转基因材料均能扩增出条带大小与阳性对照一致的条带,而野生型材料没有任何条带(图2B),说明PT8A-1、PT8A-2和PT8A-3基因组中都含有草甘膦抗性基因,为转基因阳性株系。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图23份转基因材料DNA和抗性基因PCR产物电泳检测结果
A:转基因株系PT8A-1、PT8A-2、PT8A-3基因组DNA电泳图。M:DL10000 marker。B:转基因株系PT8A-1、PT8A-2、PT8A-3 PCR产物电泳图。M:DL2000 marker。WT:野生型阴性对照;vector:35S-1300-G9A-PT8A阳性对照。
Fig. 2Electrophoresis of the DNA extracted from three transgenic plants and PCR analysis of the resistant gene
2.2 T-DNA插入位点分析
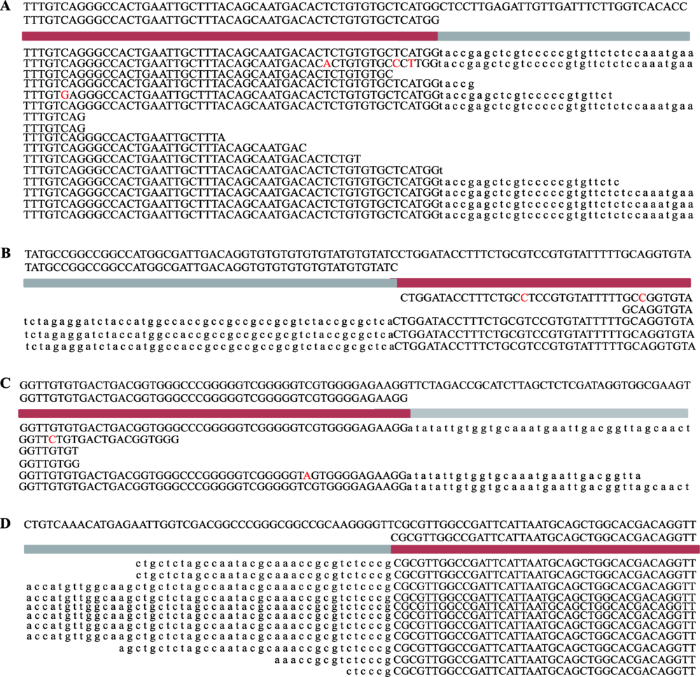
对3份转基因材料基因组DNA等量混合后进行全基因组重测序,共获得总碱基数约为26 589 Mb的数据,84.40%的数据可以比对到水稻基因组上,其中有10倍以上覆盖的区域占水稻基因组的89.24%,1倍以上覆盖的区域占水稻基因组的97.01%,Q30=84%,Mean Quality Score=36.16,满足Q30≥ 80%的高通量测序质量要求[12]。以35S-1300-G9A- PT8A载体T-DNA序列作为参考模板,使用Bowtie2软件(默认设置)对测序数据进行分析,得到4个候选T-DNA插入位点,分别为2号染色体2 078 135 bp处、7号染色体22 297 220 bp处、9号染色体18 991 276 bp处和11号染色体12 123 278 bp处(图3),说明至少有一份转基因材料是双拷贝的。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基因组重测序数据比对分析后得到的插入位点序列信息
A:2号染色体插入位点;B:7号染色体插入位点;C:9号染色体插入位点;D:11号染色体插入位点。大写字母为基因组序列,小写字母为载体序列。
Fig. 3Sequence information of insertion sites obtained from the analysis of genome re-sequencing data
2.3 插入位点PCR验证
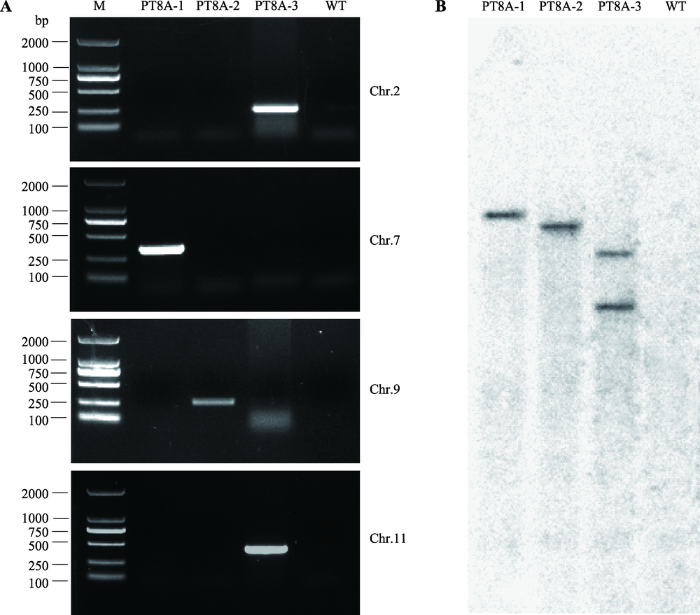
为确定3份转基因材料的T-DNA插入位点,分别用设计的4对引物对3份转基因材料基因组DNA进行PCR扩增。结果显示,PT8A-1材料用Chr.2位点引物可以扩增出PCR产物,PT8A-2材料用Chr.9位点引物可以扩增出PCR产物,而PT8A-3材料用Chr.7位点和 Chr.11位点引物可以扩增出PCR产物,而且所有PCR产物大小都与预测一致(图4A),说明PT8A-1材料在7号染色体有T-DNA插入,PT8A-2材料在9号染色体有T-DNA插入,PT8A-3材料在2号染色体和11号染色体有T-DNA插入,为双拷贝插入。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4转基因株系T-DNA插入位点验证分析
A:PCR验证分析结果;B:Southern blot 验证分析结果。M:DL2000 marker。
Fig. 4Verification of T-DNA insertion sites for transgenic plants
2.4 Southern blot验证分析
为了确定重测序分析结果是否得到了3份转基因株系全部插入位点,本文对3份转基因材料进行Southern blot验证分析。结果表明,野生型(WT)没有检测到任何杂交条带,PT8A-1、PT8A-2材料基因组DNA各有一条杂交条带,而PT8A-3材料基因组DNA有2条杂交条带(图4B),说明PT8A-1、PT8A-2材料为单拷贝插入,PT8A-3材料为双拷贝插入。Southern blot分析结果与测序分析结果一致,说明3份转基因材料的基因组DNA打包后进行重测序分析,能够得到全部的T-DNA插入位点信息。3 讨 论
T-DNA插入位点的获得在基因克隆、转基因安全等方面有着非常重要的应用。目前,获得插入位点比较常用的方法是Tail-PCR方法,由于Tail-PCR方法使用的是简并引物,在实际操作过程中效率并不高。本课题组在使用Tail-PCR方法获得插入位点时,大多数转基因材料都不能顺利得到插入位点,而且如果是多拷贝转基因株系,成功率更低。而其他方法,如反向PCR、接头-PCR、SON-PCR等[2,13,14],都需要复杂的操作,成功率也很低。本文通过提取3份转基因材料基因组DNA,等量混合后,进行全基因组重测序,使用载体序列作为参考模板进行分析筛选,顺利得到了3份转基因材料的T-DNA插入位点,而且其中有一份是双拷贝插入。本文利用重测序技术建立了一种简单、可靠、高效的获得转基因植物T-DNA插入位点的方法,本方法有以下优点:(1) 操作方法简单。本方法中使用的DNA提取、PCR分析等都是实验室常用的分子生物学技术,而且随着基因组重测序技术的发展,基因组重测序的成本下降,时间加快,数据质量进一步提高,使得基因组重测序已经成为了大多数实验室常规操作;(2) 分析工作量小。目前在拟南芥(Arabidopsis thaliana)、大豆(Glycine max)等转基因植株中有利用基因组重测序获得T-DNA插入位点的研究[12, 15],但是使用的是全基因组和载体序列同时进行比对,分析工作量大、消耗时间长。本方法使用载体序列与重测序得到的序列直接进行分析比对,不需要使用全基因组序列信息,减少了基因组组装及分析比对的工作量,减少了数据分析的时间,而且在一些没有得到全基因组序列的物种上,使用本方法也可以快速地获得插入位点信息。因此,本方法相比于已有的T-DNA插入位点获得方法,效率更高,几乎所有分子生物学实验室都可以使用,并能获得很好的结果。
本方法在重测序时,要求把DNA打断为长度200~300 bp片段,并且每个Reads测序长度为150 bp,保证2个Reads拼接后能得到单个片段全部序列信息。在得到基因组重测序数据后,使用载体序列进行比对分析时,会得到2种比对类型:第一种,两端Reads均为载体序列,此类序列在获得插入位点时不能提供有用信息;第二种,单条Read本身包含了T-DNA和基因组序列,此类序列就是有用的序列类型,此时根据序列信息就明确得到T-DNA的插入位置,后续经过PCR验证,就可以得到相应株系的插入位点。
基因组建库和测序深度是决定本方法能否成功的关键因素,均匀建库是基础,而测序深度决定着能否得到所有材料的插入位点,混合的材料越多,对测序深度要求越高。Polko等[15]将4份拟南芥转基因材料混合进行全基因组重测序,由于测序深度不够,导致有1份材料未得到插入位点信息。本文将3份转基因材料的基因组DNA混合后进行重测序,共得到大约25 Gb的数据,经分析,3份转基因材料都得到了准确的插入位点。因此,多份转基因材料等比例混合后测序,在建库均匀和保证测序深度的情况下都可以得到插入位点。Polko等[15]认为多至20份材料混合后也可以成功得到插入位点信息。但是,如果混合的材料过多,会不会由于目前方法的局限性,导致建库不均匀,进而导致得不到插入位点或者多拷贝株系的插入位点信息不完整,还需要进一步的研究去验证。
综上所述,本文提供了一种简单、可靠、高效的获得转基因插入位点的方法,利用该方法可以快速得到多份转基因株系的插入位点,而且不受插入拷贝数的影响。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLMagsci [本文引用: 1]

扩增基因的侧翼序列在分子生物学研究中具有重要作用。到目前为止,克隆基因侧翼序列的方法主要可以分为3类:反向PCR、外源接头介导PCR、半随机引物PCR。对这些技术的原理以及近期的应用情况进行了较为系统的综述,旨在为研究者选择更可靠、更合理的方法提供依据。
URLMagsci [本文引用: 1]
扩增基因的侧翼序列在分子生物学研究中具有重要作用。到目前为止,克隆基因侧翼序列的方法主要可以分为3类:反向PCR、外源接头介导PCR、半随机引物PCR。对这些技术的原理以及近期的应用情况进行了较为系统的综述,旨在为研究者选择更可靠、更合理的方法提供依据。
[本文引用: 2]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:26842621 [本文引用: 1]
Abstract The Gene Identification via Phenotype Sequencing (GIPS) software considers a range of experimental and analysis choices in sequencing-based forward genetics studies within an integrated probabilistic framework, which enables direct gene cloning from the sequencing of several unrelated mutants of the same phenotype without the need to create segregation populations. GIPS estimates four measurements to help optimize an analysis procedure as follows: 1) the chance of reporting the true phenotype-associated gene, 2) the expected number of random genes that may be reported, 3) the significance of each candidate gene's association with the phenotype, and 4) the significance of violating the Mendelian assumption if no gene is reported or if all candidate genes have failed validation. The usage of GIPS is illustrated with the identification of a rice gene that epistatically suppresses the phenotype of PHO2 mutant from sequencing three unrelated ethyl methanesulfonate mutants. GIPS is available at https://github.com/synergy-zju/gips/wiki with the user manual and an analysis example. 2016 American Society of Plant Biologists. All rights reserved.
URL [本文引用: 1]
[本文引用: 1]
URLMagsci [本文引用: 2]
<br><br>根据多年生植物组织富含多酚、多糖的具体特性,对现有的DNA提取方法进行了改进。通过增加提取缓冲液中b-巯基乙醇用量,简化氯仿/异戊醇抽提液步骤,改用经-20℃预冷异丙醇沉淀DNA等,对CTAB法加以改进。改进后方法具有以下优点:(1)获得的DNA质量良好,提取过程无明显的DNA降解,基本上排除了多酚物质的干扰;(2)用获得的DNA进行Southern杂交,可得到理想的杂交信号,可满足相关的分子研究要求;(3)操作简便。Abstract It is a difficult problem to isolate high quality DNA from plants containing a high contents of polyphenolics and polysaccharose, such as Actinidia plant. The protocol described in this paper is a modified CTAB (hexadecyltrimethylammonium bromide) method. High quality genomic DNA can be isolated from Actinidia plant using the improved method. The DNA is good enough for Southern blot and other uses in DNA research. The protocol is also efficient for quick and macro-DNA extraction. <br>
URLMagsci [本文引用: 2]
<br><br>根据多年生植物组织富含多酚、多糖的具体特性,对现有的DNA提取方法进行了改进。通过增加提取缓冲液中b-巯基乙醇用量,简化氯仿/异戊醇抽提液步骤,改用经-20℃预冷异丙醇沉淀DNA等,对CTAB法加以改进。改进后方法具有以下优点:(1)获得的DNA质量良好,提取过程无明显的DNA降解,基本上排除了多酚物质的干扰;(2)用获得的DNA进行Southern杂交,可得到理想的杂交信号,可满足相关的分子研究要求;(3)操作简便。Abstract It is a difficult problem to isolate high quality DNA from plants containing a high contents of polyphenolics and polysaccharose, such as Actinidia plant. The protocol described in this paper is a modified CTAB (hexadecyltrimethylammonium bromide) method. High quality genomic DNA can be isolated from Actinidia plant using the improved method. The DNA is good enough for Southern blot and other uses in DNA research. The protocol is also efficient for quick and macro-DNA extraction. <br>
URL [本文引用: 1]
[本文引用: 2]
URL [本文引用: 1]
URLPMID:16914272 [本文引用: 1]
We describe here a nested PCR-based strategy for genome walking to extend a known sequence region to its uncharacterized flanking regions. This technique involves the use of a partially degenerate primer as a walker primer and a set of nested specific primers to perform two to three successive rounds of nested PCR. To increase the success rate of genome walking, four different walker primers were designed to allow the setup of parallel reactions. This technique was applied to amplify flanking sequences of known genomic loci of two highly divergent photosynthetic organisms, Rhodobacter capsulatus and Heliophilum fasciatum. Specific products were preferentially amplified using this strategy, which were verified using DNA sequencing. The extremely high success rate of extension of genomic regions in these two organisms suggests that this technique can be applied to a wide range of genomes.
[本文引用: 3]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}