 ,南京大学生命科学学院,医药生物技术国家重点实验室,南京 210023
,南京大学生命科学学院,医药生物技术国家重点实验室,南京 210023Regulation of γ-globin gene expression and its clinical applications
Junyi Ju, Quan Zhao,State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing 210023, China第一联系人:
编委: 徐湘民
收稿日期:2018-04-17修回日期:2018-05-24网络出版日期:2018-06-20
| 基金资助: |
Received:2018-04-17Revised:2018-05-24Online:2018-06-20
| Fund supported: |
作者简介 About authors
鞠君毅,博士,助理研究员,研究方向:基因转录调控E-mail:

赵权,南京大学生命科学学院教授,曾获美国库利(Cooley’s)贫血协会医学研究基金奖并入选教育部“新世纪优秀人才支持计划”1988年毕业于南京大学生物化学系,获学士学位;2002年毕业于澳大利亚墨尔本LaTrobe大学生物化学系,获博士学位2006年受聘于南京大学生命科学学院,主要从事基因转录与表观遗传学调控机理研究多年来在红系细胞基因表达调控机理及其在遗传性血液病治疗基础应用方面开展研究并取得重要进展以人珠蛋白基因簇为研究模型,应用生物化学及表观遗传学等方法,发现并解析多个转录因子和表观修饰分子及其复合体对人珠蛋白基因表达的精细调控机理,提出镰刀型细胞贫血(SCD)和β-地中海贫血治疗新靶标,为临床应用打下良好基础相关研究成果发表在NatureStructural&MolecularBiology、NatureCommunications、Blood、NucleicAcidsResearch、CancerResearch、Haematologica、TheJournalofBiologicalChemistry等重要国际期刊, E-mail:qzhao@nju.edu.cn

摘要
关键词:
Abstract
Keywords:
PDF (610KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
鞠君毅, 赵权. γ-珠蛋白基因表达调控机制与临床应用. 遗传[J], 2018, 40(6): 429-444 doi:10.16288/j.yczz.18-021
Junyi Ju, Quan Zhao.
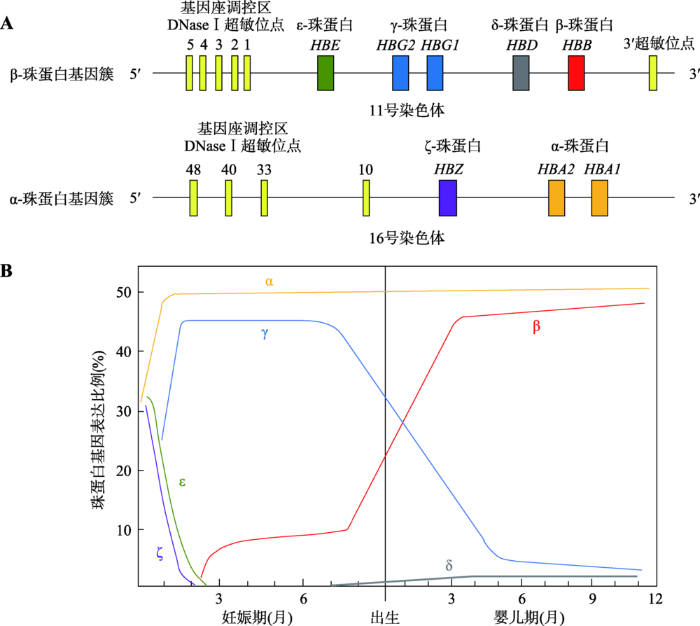
负责人体内氧气运输的血红蛋白是红细胞内最主要的蛋白质,由2个α-珠蛋白亚基(141个氨基酸)和2个β-珠蛋白亚基(146个氨基酸)以及亚基内部结合的血红素分子组成。血红蛋白包含两种珠蛋白亚基,分别由α-珠蛋白基因簇和β-珠蛋白基因簇表达产生。人类α-珠蛋白基因簇位于第16号染色体,包含2个串联的α-珠蛋白基因(HBA2和HBA1)和胚胎期表达的类α-珠蛋白基因(ζ-珠蛋白基因,HBZ);β-珠蛋白基因簇位于第11号染色体,包含β-珠蛋白基因(HBB)以及其他4种类β-珠蛋白基因:胚胎期表达的ε-珠蛋白基因(HBE1),胎儿期表达的γ-蛋白基因(HBG2和HBG1)和成年期低量表达的δ-珠蛋白基因(HBD) (图1A)。这些珠蛋白基因的表达调控精密,表现出高度的发育时序性。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1珠蛋白基因簇及发育过程中珠蛋白基因表达变化
A:人类β-珠蛋白基因簇和α-珠蛋白基因簇。人类β-珠蛋白基因簇位于11号染色体,依次分布基因座调控区、ε-珠蛋白基因、γ-珠蛋白基因、δ-珠蛋白基因和β-珠蛋白基因。α-珠蛋白基因簇位于16号染色体,依次分布基因座调控区、ζ-珠蛋白基因、α-珠蛋白基因。B:人类发育过程中珠蛋白基因表达转换过程。β-珠蛋白基因簇的第一次转换发生在怀孕后6周左右,ε-珠蛋白基因表达降低,γ-珠蛋白基因开始表达;第二次转换发生在出生后,γ-珠蛋白基因表达降低,β-珠蛋白基因开始表达。不同时期血红蛋白组成为胚胎血红蛋白:ζ2ε2, α2ε2, ζ2γ2;胎儿血红蛋白:α2γ2;成人血红蛋白:α2β2, α2δ2。
Fig. 1Human globin gene loci and globin gene expression during development
从受孕到妊娠5周内,胚胎血红蛋白(embryonic hemoglobin,包括ζ2ε2、α2ε2和ζ2γ2)主要在卵黄囊中合成,ε-珠蛋白基因在这段时期表达 [1,2]。随后,β-珠蛋白基因簇上的基因表达会发生两次重要的切换。第一次基因表达切换发生在怀孕后6周左右:ε-珠蛋白基因表达被逐渐关闭,γ-珠蛋白基因表达开启,此时的胎儿血红蛋白(fetal hemoglobin, HbF)由2个α亚基和2个γ亚基(α2γ2)组成,主要在胎儿的肝脏和脾脏中合成[3]。从怀孕后12周到胎儿出生这段时间,HbF是胎儿红细胞中最主要的血红蛋白。第二次基因表达切换发生在出生后:γ-珠蛋白基因表达被逐渐关闭,β-珠蛋白基因开始表达。成人血红蛋白(adult hemoglobin, HbA)由2个α亚基和2个β亚基组成(α2β2),主要在骨髓中合成[4](图1B)。出生6个月后,胎儿血红蛋白HbF会下降到血红蛋白总量的1%以下,成人血红蛋白HbA达到97%左右;另外2%为HbA2,由2个α亚基和2个δ亚基组成(α2δ2)[5]。
β-地中海贫血(β-thalassemia)和镰刀型细胞贫血(sickle-cell disease, SCD)是两种最常见的由于β-珠蛋白基因发生突变所引发的常染色体隐性遗传疾 病[6]。据统计,全世界约有5%~7%的人口受其影 响[6,7,8]。β-地中海贫血病人的β-珠蛋白基因部分表达(β+)或表达完全受限(β0),导致血红蛋白四聚体生成异常,过剩的α-珠蛋白以不稳定的单体存在,容易发生氧化并沉积下来形成包涵体,破坏红细胞引发溶血性贫血。这种疾病主要发生在地中海、中东及东南亚等地区,在我国南方各省(如四川、云南、贵州、福建、广东和广西等)也较为常见[9]。镰刀型细胞贫血是由于病人体内的β-珠蛋白基因发生点突变(GAG→GTG),导致第6位的谷氨酸突变为缬氨酸。由此产生的血红蛋白S(sickle hemoglobin, HbS, α2βS2)溶解度下降,在缺氧时会异常聚集,导致红细胞发生镰变。镰刀型细胞贫血主要发生非洲和美洲的黑人人群中[10]。以上两种贫血病人体内突变的血红蛋白会导致慢性溶血、贫血、血管阻塞以及疼痛等症状,同时还会累及其他组织器官引起并发症,病情严重者需要终身接受输血、药物治疗或进行骨髓移植。目前临床用药主要是通过激活γ-珠蛋白基因表达,以替代病人体内突变的β-珠蛋白与α-珠蛋白形成HbF抑制血红蛋白的异常聚集,降低溶血,提高血红蛋白总量,延长红细胞的寿命,从而减轻病人的临床症状[6,11]。
1948年,Watson[12]发现患有镰刀型细胞贫血病的新生儿直到出生6个月后才会出现贫血症状。这一现象提示:与成人相比,新生儿的红细胞不易发生镰变。1975年,Stamatoyannopoulos等[13]发现同时患有镰刀型细胞贫血和遗传性胎儿血红蛋白持续存在综合征(hereditary persistence of fetal hemoglobin, HPFH)的病人并没有表现出贫血的症状。HPFH是由于β-珠蛋白基因簇上的DNA序列缺失或γ-珠蛋白启动子区域发生点突变导致的成人红细胞中持续存在过量的HbF的遗传性血红蛋白病。HPFH患者血液学检查正常,无临床症状,不需要治疗[14]。这些发现表明提高HbF含量可以治疗镰刀型细胞贫血和地中海贫血。随后,在镰刀型细胞贫血和地中海贫血病人中的研究也证实,提高γ-珠蛋白基因表达具有良好的临床疗效[15]。因此,深入研究发育过程中γ-珠蛋白基因表达切换的分子机制,对寻找能够重新激活成人体内被沉默的γ-珠蛋白基因表达的药物治疗镰刀型细胞贫血和β-地中海贫血具有重要意义。本文主要从分子遗传学和表观遗传学的角度,综述了γ-珠蛋白基因调控的分子机制以及相关转录因子和表观遗传修饰分子,介绍了目前临床上通过激活γ-珠蛋白基因表达治疗镰刀型细胞贫血和β-地中海贫血的药物以及基因治疗的研究进展。
1 γ-珠蛋白基因的表达调控
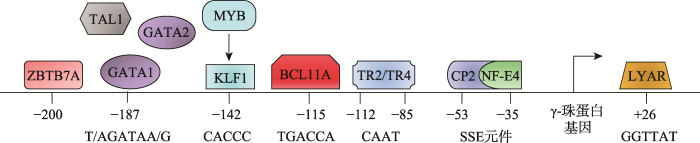
类β-珠蛋白在发育过程中的时序性表达受到非常精细的调控,由基因簇上的顺式作用元件和反式作用因子共同参与完成。目前已经发现了多个参与γ-珠蛋白基因调控的特异性转录因子。这些转录因子可以结合到珠蛋白基因启动子区域的特定序列上,招募相应的具有激活或阻遏作用的辅助因子,并通过染色质重塑过程,实现对γ-珠蛋白基因表达的精细调控(图2)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2γ-珠蛋白基因启动子区域转录因子
γ-珠蛋白基因近端启动子区域的转录因子以及对应DNA结合位点。
Fig. 2Transcription factors binding on the γ-globin gene promoter
1.1 基因座控制区(locus control region, LCR)
位于ε-珠蛋白基因上游6~20 kb区域内的β-珠蛋白基因簇的基因座调控区LCR对于整个基因簇内珠蛋白基因的表达调控非常重要。LCR区内包含5个DNaseⅠ超敏位点(DNase Ⅰ hypersensitive sites, HSs)。其中HS1~HS4内含有多个转录因子的结合位点,可以发挥增强子的作用,而HS5被认为作为隔离子发挥作用[16,17,18]。目前,最被广为接受的能够解释LCR区对类β-珠蛋白基因调控方式的是looping模型:LCR区向基因簇内部弯折,与各个β-类珠蛋白基因启动子区域的顺式作用元件以及转录因子和调节因子协同作用,调控珠蛋白基因的开启和关闭,实现珠蛋白基因的时序性表达[18]。1.2 转录因子
GATA-1是最早被发现的参与调控珠蛋白基因转换过程的转录因子,其DNA结合结构域包含两个高度保守的锌指结构,可以特异性地识别T/A (GATA) A/G序列[19,20]。人类β-珠蛋白基因的启动子区域含有多个这样的位点。GATA-1在早期红系细胞、巨核细胞、嗜酸性粒细胞和嗜碱性粒细胞及肥大细胞中具有特异性表达[21, 22]。GATA-1参与许多红系细胞分化过程中重要基因的调控,包括人类α和β-珠蛋白、亚铁血红素生物合成酶、促红细胞生成素及其受体、以及红系相关转录因子和细胞周期相关基因等[21]。GATA-1基因的缺失会引发红系细胞发育异常,从而导致小鼠在胚胎期10.5~11.5天死亡[23]。GATA-1及其辅助因子FOG-1结合到珠蛋白基因的启动子区域后,将SWI/SNF染色体重塑复合物(switch/sucrose non-fermentable, SWI/SNF,可通过改变核小体结构影响基因转录活性)招募过来,通过染色质重塑使得LCR区与启动子区域结合,激活β-珠蛋白基因的表达[24,25]。GATA-1和辅助因子OCT-1结合到γ-珠蛋白基因启动子区域则会抑制γ-珠蛋白基因的表达[26]。此外,还有研究发现GATA-1可以与FOG-1结合并招募NuRD复合物(nucleoside remodeling and deacetylase, NuRD,可改变核小体结构、催化组蛋白去乙酰化抑制基因表达),参与沉默γ-珠蛋白基因的表达[25, 27, 28]。GATA-2的DNA结合基序与GATA-1一样,但是表达GATA-2与GATA-1的细胞类型不同。GATA-2主要在造血干细胞和祖细胞中表达,调控造血干细胞的增殖和分化[20]。GATA-2缺失的小鼠会因为造血功能衰竭而致死[29]。在造血干细胞中过表达GATA-2会抑制其增殖和分化;而抑制GATA-2的表达,造血细胞的增殖分化功能则会恢复[30]。在K562细胞中过表达GATA-1会抑制GATA-2的表达水平,而过表达GATA-2可以提高γ-珠蛋白基因的表达,表明GATA-1和GATA-2表达水平的比例变化在珠蛋白表达的转换过程中发挥了调控作用[31]。
TAL1是红系细胞发育过程中的重要转录因子。过表达TAL1可以刺激红系祖细胞的分化过程。TAL1可以与GATA-1、LMO1、LDB1和ETO2组成复合物,调节LCR区向珠蛋白基因启动子区域的弯折,从而激活β-珠蛋白基因的表达[32]。此外有研究表明,在K562细胞中抑制TAL1会减少LCR区与γ-珠蛋白基因启动子区域的结合,导致γ-珠蛋白基因的表达下降[33]。
NF-E2是红系细胞特有的转录因子,在红系细胞成熟和珠蛋白调控过程中发挥着重要的作用。NF-E2由一个45 kDa并含有激活结构域的亚基(p45 NF-E2)和一个18 kDa并含有DNA结合结构域的亚基(p18 MAFK蛋白)组成异源二聚体[34],其特异性地结合到β-珠蛋白基因簇LCR区域HS2上的类AP-1位点(TCAT/C),帮助将RNA聚合酶Ⅱ招募到珠蛋白的启动子区域激活珠蛋白基因的表达[35]。
NF-E4作为另一个重要的红系特异性转录因子,可以与CP2形成异源二聚体发育阶段特异蛋白(stage selector protein, SSP)。SSP可以特异性地识别γ-珠蛋白启动子区域的发育阶段特异元件(stage selector element, SSE),并介导LCR区与γ-珠蛋白启动子相互作用,从而激活γ-珠蛋白基因的表达,突变SSE区域则会影响早期γ-珠蛋白的表达[36, 37]。在K562细胞中,过表达NF-E4会激活γ-珠蛋白基因的表达;而在从脐带血中分离得到的红系祖细胞中过表达NF-E4,不仅可以提高γ-珠蛋白基因的表达,同时还会抑制β-珠蛋白基因的表达[38, 39]。在携带人源β-珠蛋白基因簇的YAC转基因小鼠中,过表达NF-E4会导致γ-珠蛋白与β-珠蛋白相对表达比例的升高,从而延迟γ-珠蛋白基因表达向β-珠蛋白基因表达的切换过程。而NF-E4的截短亚型p14 NF-E4,可以通过与全长NF-E4竞争结合CP2,阻止SSP结合到γ-珠蛋白启动子区域的SSE上,从而抑制γ-珠蛋白基因表达[40]。
KLF1是Sp1家族的Krüppel样红系细胞特异性转录因子,它可以与β-珠蛋白启动子区域的CACCC序列结合,并促进LCR区的HS3位点与β-珠蛋白基因启动子区域结合,从而激活β-珠蛋白的表达,该结合位点发生突变会导致β-地中海贫血[41, 42]。对中国南方β-地中海贫血病人的大数据分析表明KLF1基因突变可以减轻病人的症状[43]。KLF1在成人红系细胞中的表达水平明显高于胎儿红系细胞,其与β-珠蛋白基因启动子的结合能力远高于γ-珠蛋白基因启动子[44],在K562细胞中过表达KLF1可以明显上调β-珠蛋白基因的表达。此外,KLF1还可以直接激活BCL11A从而抑制γ-珠蛋白基因的表达[45]。然而,有研究发现在英国HPFH个体中,发生在γ-珠蛋白基因启动子区域-198位点的T→C突变可以被KLF1识别,进而激活γ-珠蛋白基因表达[46]。这些现象表明KLF1在γ-珠蛋白向β-珠蛋白表达的转换过程中发挥着重要作用。对KLF1基因敲除小鼠和细胞学相关研究表明KLF1还参与了红细胞成熟过程中重要基因的调控,包括亚铁血红素合成酶、血型抗原和α-血红蛋白稳定蛋白等[47]。
BCL11A是BCL家族的Krüppel样锌指转录因子,早先研究发现BCL11A主要参与淋巴细胞生成和神经发育过程。通过全基因组关联分析(genome- wide association studies, GWAS)发现,位于2号染色体的BCL11A基因的rs11886868位点单核苷酸多态性与地中海贫血病人体内HbF异常表达高度相关[48]。进一步研究表明BCL11A的主要作用是抑制γ-珠蛋白基因的表达[49]。在红系祖细胞中将BCL11A表达干扰后,γ-珠蛋白基因的表达显著升高。BCL11A敲除后会影响胚胎发育过程中γ-珠蛋白基因的沉默[50]。在镰刀型细胞贫血病模型小鼠的红系细胞中敲除BCL11A可以缓解贫血症状[51]。此前研究表明BCL11A并不直接结合到γ-珠蛋白和β-珠蛋白基因的启动子区域,而是主要结合在β-珠蛋白基因簇上的基因间区和LCR区的HS3位点。它可以与GATA-1、FOG1和SOX6相互作用,并通过Mi-2/NuRD复合物重塑染色质的构象,从而导致γ-珠蛋白基因的沉默[52]。而最近发表在Cell和Nature Genetics上的研究使用最新的用于探究蛋白质结合序列的CUT& RUN技术证实了BCL11A直接结合在γ-珠蛋白基 因启动子区域-115位点的TGACCA序列上发挥功能[53, 54]。
全基因组关联分析还发现位于染色体6q23的HBS1L与MYB基因间区域的SNP同样会直接影响γ-珠蛋白基因的表达水平[55, 56],功能研究表明SNP会影响该区域折叠形成的染色质激活枢纽结构,抑制下游MYB基因的表达,从而影响红系细胞发育过程和HbF的水平。研究表明MYB可以通过以下两种机制参与调控HbF:低表达的MYB可以加速红系分化过程,导致释放出更多的表达HbF的早期红系祖细胞;MYB可以激活KLF1等其他γ-珠蛋白基因抑制子的表达[57, 58]。
ZBTB7A是POK(BTB/POZ和Krüppel)家族的含有C2H2锌指结构域的转录因子[59]。ZBTB7A在多种红系细胞中表达,参与B细胞、T细胞、破骨细胞和红系细胞的发育过程[60]。最新研究表明在红系祖细胞中ZBTB7A可以直接结合到γ-珠蛋白基因上抑制其表达,在CD34+的造血干细胞中干扰ZBTB7A的表达可以显著提高HbF的水平[61]。ZBTB7A的缺失还会延迟造血干细胞的红系分化过程。ZBTB7A基因敲除会导致小鼠胚胎严重贫血致死,在成年小鼠造血干细胞中条件性敲除ZBTB7A基因则会导致EPO-无反应的大细胞性贫血[62]。最新研究表明ZBTB7A结合在γ-珠蛋白基因启动子区域-200处,该区域发生突变会导致γ-珠蛋白基因表达升高[54]。
LYAR最早是从小鼠的T细胞白血病细胞系中克隆得到的一个编码含有锌指结构蛋白的基因,LYAR蛋白主要定位在细胞核仁,并且与细胞增殖相关[63]。本课题组首次发现LYAR作为转录因子参与了γ-珠蛋白基因的沉默。在K562细胞和人红系祖细胞中干扰LYAR的表达可以上调γ-珠蛋白基因的表达水平,进一步研究发现LYAR可以特异性地结合到γ-珠蛋白基因+26的GGTTAT位点,并招募蛋白质精氨酸甲基转移酶PRMT5,沉默γ-珠蛋白基因的表达[64]。多个课题组通过对家族谱系及超过1000例β-地中海贫血病人的研究发现,LYAR结合位点GGTTAT的单核苷酸多态性(rs368698783,+25G→A)会导致β-地中海贫血病人HbF水平明显升高[65,66,67]。此外,该位点单核苷酸多态性与γ-珠蛋白基因XmnⅠ单核苷酸多态性(rs7482144,-158C→T)存在连锁不平衡现象,此前报道的由于γ-珠蛋白基因XmnⅠ多态性而出现的γ-珠蛋白基因表达升高很可能是由于rs368698783多态性使得LYAR无法结合而导致的[67]。
Table 1
表1
表1 参与γ-珠蛋白表达调控的主要转录因子
Table 1
| 转录因子 | 结合序列及在γ-珠蛋白基因启动子区定位 | 功能 | 参考文献 |
|---|---|---|---|
| GATA1 | T/AGATAA/G,-187位点 | 抑制γ-珠蛋白基因表达 | [25~28] |
| GATA2 | T/AGATAA/G,-187位点 | 激活γ-珠蛋白基因表达 | [31] |
| TAL1 | 与GATA-1、LMO1、LDB1和ETO1形成复合物结合到γ-珠蛋白基因启动子区域 | 帮助LCR区向珠蛋白启动子弯折,激活γ-珠蛋白基因表达 | [33] |
| NF-E4 | 与CP2形成异源二聚体结合在γ-珠蛋白基因启动子-53到-35位点的SSE元件上 | p22 NF-E4激活γ-珠蛋白基因,抑制γ-珠蛋白向β-珠蛋白表达的转换;p14 NF-E4抑制γ-珠蛋白基因 | [38~40] |
| KLF1 | CACCC,-142位点 | 促进γ-珠蛋白向β-珠蛋白表达的转换;激活BCL11A抑制γ-珠蛋白基因表达 | [44, 45] |
| BCL11A | TGACCA,-115位点 | 抑制γ-珠蛋白基因表达 | [49, 50, 52~54] |
| MYB | 激活KLF1抑制γ-珠蛋白基因表达 | [57, 58] | |
| ZBTB7A | -200位点 | 调节染色质结构,抑制γ-珠蛋白基因表达 | [54, 61] |
| LYAR | GGTTAT,+26位点 | 抑制γ-珠蛋白基因表达 | [65~67] |
| TR2/TR4 | 形成DRED复合物(direct repeat erythroid definitive, DRED,具有抑制基因表达的功能)结合在-112和-85位点的CAAT序列 | 沉默γ-珠蛋白基因表达;过表达后可激活γ-珠蛋白基因表达 | [68~70] |
新窗口打开|下载CSV
TR2/TR4可以特异性地结合到胚胎期的类β-珠蛋白启动子区域,并招募包括DNMT1、NuRD复合物、LSD1/CoREST、HDAC3和TIF1b在内的多种转录抑制子介导胚胎期类β-珠蛋白基因的沉默[68, 69]。然而有趣的是,过表达TR2/TR4却可以诱导γ-珠蛋白基因的表达,研究者推测有可能是由于过量的TR2/TR4稀释了结合到γ-珠蛋白基因启动子上的转录抑制子而导致的[70]。
2 表观遗传与γ-珠蛋白基因表达
表观遗传是指DNA序列不变的情况下,基因表达水平发生的可遗传的变化。表观遗传学研究内容主要包括DNA修饰(如甲基化)、组蛋白修饰、染色体重塑和非编码RNA调控。其中组蛋白修饰包括甲基化修饰、乙酰化修饰、磷酸化修饰和泛素化修饰等。本文简要介绍DNA甲基化和组蛋白修饰在γ-珠蛋白基因表达调控中的作用。细胞内的DNA甲基化由DNA甲基转移酶家族负责催化,该家族包括从头甲基转移酶DNMT3A和DNMT3B以及维持甲基转移酶DNMT1。在珠蛋白基因表达转换过程中,启动子区域的DNA甲基化修饰水平会发生变化,DNA甲基化水平升高会导致珠蛋白基因沉默[71]。此外,识别DNA甲基化修饰的甲基胞嘧啶结合蛋白(methyl cytosine binding domain, MBD)家族也参与了珠蛋白基因的调控。MBD2倾向于结合在高甲基化的CpG岛区域,并招募NuRD复合物,抑制基因表达[72]。在携带人源β-珠蛋白基因簇的YAC转基因小鼠和人红系祖细胞中,干扰MBD2均可以激活γ-珠蛋白基因的表达[73, 74]。
组蛋白乙酰化修饰可以通过电荷的作用使得组蛋白与DNA之间的结合变得松散,使转录因子更容易结合DNA,因此通常与基因的激活相关[75]。蛋白质乙酰化修饰由乙酰基转移酶催化,包括CBP/p300家族、GNAT家族和MYST家族等[76]。与乙酰基转移酶功能相反的是组蛋白去乙酰化酶HDAC家族[77]。当γ-珠蛋白基因表达升高时,基因附近的组蛋白乙酰化修饰水平也会升高,因此抑制组蛋白去乙酰化酶的活性可以激活γ-珠蛋白基因的表达[78, 79]。有趣的是,有报道称同属于组蛋白去乙酰化酶的SIRT家族的SIRT1可以激活γ-珠蛋白基因的表达[80]。此外,组蛋白去乙酰化酶还可通过影响转录因子和其它非组蛋白的乙酰化水平调控珠蛋白基因的表达。GATA-1的乙酰化修饰水平会影响其与DNA结合的能力[81]。本课题组发现乙酰化修饰会影响NF-E4蛋白的稳定性[82]。研究表明,组蛋白去乙酰化酶抑制剂可以通过调控STAT5、cAMP和p38 MAPK信号通路激活γ-珠蛋白基因的表达[83,84,85]。
组蛋白甲基化修饰包括精氨酸甲基化和赖氨酸甲基化,分别由精氨酸甲基转移酶(protein arginine methyltransferases, PRMT)和赖氨酸甲基转移酶家族(分含SET结构域和不含SET结构域两类)催化。本课题组发现精氨酸甲基转移酶PRMT5可以与CK2α、SUV4-20h1和HDAC1等组成抑制复合物,参与γ-珠蛋白基因的沉默[86]。此外,深入研究表明PRMT5催化产生的组蛋白H4第三位精氨酸对称性双甲基化修饰(H4R3me2s)可以招募DNA甲基转移酶DNMT3A,催化γ-珠蛋白基因启动子区域的CpG位点发生甲基化,从而导致γ-珠蛋白基因的沉默[87]。在此基础上,本课题组发现甲基转移酶抑制剂Adox (adenosine-2’, 3’-dialdehyde)可以抑制PRMT5的活性,激活γ-珠蛋白基因表达[88]。精氨酸甲基转移酶家族的另一重要成员PRMT1可以通过其相关蛋白FoP (friend of PRMT1)参与γ-珠蛋白基因沉默,干扰FoP可以提高人红系细胞中γ-珠蛋白基因的表达[89]。此外,PRMT1可以促进组蛋白乙酰化激活β-珠蛋白基因的表达[90]。
在对TR2/TR4的研究过程中,有报道发现赖氨酸去甲基化酶LSD1参与了γ-珠蛋白基因的沉默。LSD1可以与珠蛋白基因调控的重要转录因子BCL11A组成复合物,增强BCL11A对γ-珠蛋白基因的沉默作用[91]。干扰LSD1可以提高携带人源β-珠蛋白基因簇的YAC小鼠和人红系祖细胞中γ-珠蛋白基因的表达[92]。
Table 2
表2
表2 表观遗传修饰在γ-珠蛋白表达调控中的作用
Table 2
| 表观遗传修饰酶 | 表观修饰 | γ-珠蛋白基因表达 | 参考文献 |
|---|---|---|---|
| DNMT1 | DNA甲基化 | 抑制 | [71, 93~95] |
| DNMT3A | DNA甲基化 | 抑制 | [87] |
| PRMT5 | H4R3对称性甲基化 | 抑制 | [86] |
| SUV4-20h1 | H4K20三甲基化 | 抑制 | [86] |
| CK2α | H4S1磷酸化 | 抑制 | [86] |
| HDAC1/2 | H3、H4去乙酰化 | 抑制 | [69, 82, 86] |
| LSD1 | H3K4/9去甲基化 | 抑制 | [69, 92] |
| EHMT1/2 | H3K9二甲基化 | 抑制 | [96] |
| CBP/p300 | H3、H4乙酰化 | 激活 | [97, 98] |
| WDR5 | H3K4三甲基化 | 抑制 | [99] |
| SIRT1 | H4去乙酰化 | 激活 | [80] |
新窗口打开|下载CSV
随着表观遗传学研究的迅速发展,研究人员已经筛选出多个靶向表观遗传修饰酶的小分子候选药物。深入研究珠蛋白基因的表观遗传调控机制为这些药物应用于镰刀型细胞贫血和β-地中海贫血的临床治疗提供了理论基础。
3 γ-珠蛋白基因表达诱导药物
由于提高γ-珠蛋白基因的表达可以减轻镰刀型细胞贫血和β-地中海贫血的症状,相应地激活γ-珠蛋白基因表达的药物不断出现。这些药物可以大致分为以下几类:以羟基脲(Hydroxyurea)为代表的细胞周期特异性药物、以5-氮杂胞苷(5-azacytidine)为代表的DNA甲基化酶抑制剂(DNMT inhibitors)、组蛋白去乙酰化酶抑制剂(HDAC inhibitors)和哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)信号通路抑制剂等。3.1 羟基脲
羟基脲是一种具有细胞毒性和抗代谢作用的抗肿瘤药物,可以抑制合成和修复DNA所需的核糖核苷酸还原酶的活性,从而将细胞阻滞在G1期和S期。羟基脲被发现可以诱导HbF后,迅速被用于治疗镰刀型细胞贫血,并于1996年成为美国食品药品监督管理局(FDA)的批准用药[100]。目前羟基脲诱导HbF的确切机制还不是很清楚,推测可能是由于细胞毒性带来的红细胞生成压力,导致伴随的HbF的产生。β-地中海贫血病人经过羟基脲治疗后,γ-珠蛋白mRNA的水平可以提高2~9倍,产生的γ-珠蛋白可以与多余的α-珠蛋白形成血红蛋白,生成有功能的红细胞[101]。羟基脲治疗可以明显减少病人的输血次数,提高病人的生活质量。镰刀型细胞贫血病治疗统计结果显示,羟基脲除了可以提高HbF的水平,还可以通过改变红细胞的形态和可塑性,降低白细胞和网织细胞的数量,减少溶血,释放NO刺激血管等途径改善病人的临床症状。然而羟基脲激活γ-珠蛋白基因的同时,还会激活β-珠蛋白基因。某些β-地中海贫血病人接受羟基脲治疗后,反而出现α/β-珠蛋白比例的升高,导致治疗效果降低。有报道称大约有30%的镰刀型细胞贫血病人对羟基脲反应较差或无反应[102]。此外,长期接受羟基脲治疗可能会导致病人患癌风险提高[103]。3.2 5-氮杂胞苷
表观遗传学研究表明DNA甲基化修饰会导致基因沉默。5-氮杂胞苷是胞嘧啶类似物,可以掺入DNA,抑制DNA甲基化。研究人员发现成人红系细胞和胎儿红系细胞中γ-珠蛋白基因上DNA甲基化水平存在差异:成人中高水平的DNA甲基化导致了γ-珠蛋白基因的沉默,而胎儿的γ-珠蛋白基因上的DNA甲基化水平极低[104]。5-氮杂胞苷可以特异性地抑制γ-珠蛋白基因启动子区域的甲基化水平开启γ-珠蛋白基因的表达。DeSimone等[105]在灵长类中检测5-氮杂胞苷的作用,发现该化合物能非常有效地提高HbF的水平。随后Ley等[106]将5-氮杂胞苷用于治疗β-地中海贫血患者。治疗7天后病人体内的γ-珠蛋白水平提高了7倍,使得病人体内珠蛋白比例失衡的情况得到改善,血红蛋白水平从80 g/L提高到108 g/L。随后更多类似的使用5-氮杂胞苷治疗β-地中海贫血和镰刀型细胞贫血的案例被报道。然而由于该化合物具有严重的副作用(细胞毒性、诱发突变、免疫抑制导致病毒感染),因此被限制仅用于传统疗法难以实施的重症病人中。地西他滨(decitabine, 5-aza-2’-deoxycytidine)是5-氮杂胞苷的衍生物,同样可以通过去甲基化作用激活γ-珠蛋白基因表达。多个研究表明,地西他滨可以提高大部分镰刀型细胞贫血病人体内HbF和总体血红蛋白的水平[107]。在低剂量时,地西他滨更加安全,不会导致严重的DNA损伤和细胞毒性,副作用仅仅是提高血小板的数量。但由于其去甲基化作用的非特异性,长期服用仍需考虑其潜在的致癌风险。
3.3 组蛋白去乙酰化酶抑制剂
组蛋白可以发生甲基化、乙酰化、磷酸化和泛素化等多种修饰,这些修饰会改变染色质的结构,从而调控基因表达。组蛋白去乙酰化酶可以移除组蛋白赖氨酸上的乙酰基团,导致染色质聚集,抑制基因表达。抑制组蛋白去乙酰化酶的活性会导致组蛋白H3和H4的乙酰化水平升高,使染色质处于开放的状态,便于转录因子的结合开启基因表达[108, 109]。研究表明组蛋白去乙酰化酶抑制剂,如丁酸钠、曲古抑菌素A、apicidin等可以诱导HbF的合成[84, 110]。口服丁酸盐类药物可以有效提高β-地中海贫血和镰刀型细胞贫血病人体内HbF的水平。有研究表明,除了可以通过抑制组蛋白去乙酰化酶的活性,使得转录因子可以结合到γ-珠蛋白基因启动子区域的丁酸盐反应元件(butyrate response elements, BREs)上开启γ-珠蛋白基因表达外,丁酸盐类药物还可以提高γ-珠蛋白基因mRNA的翻译效率[111, 112]。然而该类药物存在使用剂量大、价格昂贵等缺点,临床应用较少。3.4 哺乳动物雷帕霉素靶蛋白信号通路抑制剂
哺乳动物雷帕霉素靶蛋白是一种丝氨酸/苏氨酸蛋白激酶,参与细胞增殖、细胞存活、蛋白合成和基因转录等生物学过程[113]。雷帕霉素可以通过抑制哺乳动物雷帕霉素靶蛋白信号通路发挥免疫抑制、抗真菌和抗肿瘤的作用。雷帕霉素处理红系祖细胞可以提高γ-珠蛋白基因的表达,效果甚至要优于羟基脲。此外,雷帕霉素也可以提高来自于β-地中海贫血病人的红系细胞中HbF的水平[114, 115]。4 基因治疗
传统药物只能减轻镰刀型细胞贫血和β-地中海贫血病人的症状,不能完全治愈,且存在副作用,病人仍需要接受输血治疗。异体造血干细胞移植是目前唯一有效根治镰刀型细胞贫血和β-地中海贫血的方法。在移植过程中,首先使用化疗药物破坏病人自身的造血系统,再使用供体的正常造血干细胞进行替换,无事件生存率高达95%以上[116]。但是由于骨髓配型困难以及患者数量众多等原因,只有一小部分病人能够接受这种治疗。由此衍生的通过基因治疗改造自体同源的造血干细胞可以很好地改善供体不足的情况,同时还可以避免异体移植带来的免疫排斥反应[117]。基因治疗的大致流程:首先从病人体内提取造血干细胞,使用能够表达正常β-珠蛋白基因或γ-珠蛋白基因的病毒载体感染造血干细胞,将正常珠蛋白基因整合到造血干细胞基因组中,最后将扩增的造血干细胞回输到病人体内。其中病毒载体的选择至关重要,要求:(1)能够高效、稳定感染造血干细胞;(2)能够持续在红系细胞中表达正常珠蛋白基因;(3)低细胞毒性。在过去的20年中,研究人员不断改进病毒载体上的插入片段,包括添加LCR区、添加绝缘子、使用锚蛋白的启动子等方式驱动γ-珠蛋白基因表达,或利用HPFH病人体内发现的突变增强子来增强γ-珠蛋白基因表达,使用抗镰β-珠蛋白基因等。同时病毒载体也从最初的逆转录病毒换成了慢病毒载体。2000年,May等[118]使用TNS9载体成功修复了地中海贫血模型小鼠体内的突变基因。2001年,Pawliuk等[119]在两种镰刀型细胞贫血模型小鼠中使用抗镰β-珠蛋白基因进行基因治疗实验,取得了部分疗效。2010年,Cavazzana-Calvo等[120]报道了首个β-地中海贫血临床基因治疗案例,该案例中的病人目前已经不再需要接受输血治疗。2012年,北京生命科学研究所的高绍荣课题组从一位患有β-地中海贫血的男孩体内提取了成纤维细胞,并将其转化成诱导性多能干细胞(induced pluripotent stem cells , iPSCs),随后通过同源重组的方式修复了突变的β-珠蛋白基因。将修复后的iPSCs分化得到的造血祖细胞移植到经过亚致死辐射处理的SCID小鼠中,小鼠体内检测到人类β-珠蛋白基因的表达。该研究为基因改造iPSCs应用于临床治疗β-地中海贫血奠定了基础[121]。2017年,Ribeil等[122]成功使用慢病毒在自体造血干细胞中表达抗镰β-珠蛋白基因用于基因治疗镰刀型细胞贫血。
随着对珠蛋白基因表达调控机制理解的深入,科研人员也开始尝试通过改变珠蛋白相关转录因子的水平,从而间接调节病人珠蛋白基因的表达。BCL11A在珠蛋白表达调控中发挥着非常重要的作用,条件敲除BCL11A的小鼠不会发生HbF向HbA的转换[123],该小鼠与镰刀型细胞贫血模型小鼠杂交的后代的红细胞不会发生镰变,血液各项指标明显提升,表明BCL11A的缺失能够改善镰刀型细胞贫血的症状,可以作为潜在的基因治疗靶标[51]。此外,Bauer等[124]通过基因组学手段在红系细胞中发现了调控BCL11A基因表达的红系特异性增强子,利用基因组编辑技术破坏该增强子可以特异性地抑制红系细胞中BCL11A的表达,提高γ-珠蛋白基因的表达。这种方法由于不靶向淋巴细胞中BCL11A基因,因此不会影响淋巴细胞的功能。但是该方法仍然需要解决临床上实现BCL11A双等位基因敲除的技术难题以及BCL11A基因染色体易位所带来的安全性问题[125]。
5 结语与展望
据2017年Nature的一则新闻报道,珠蛋白基因曾是20世纪研究最热门的基因之一[126]。Pauling等[127]发现镰刀型细胞贫血是由β-珠蛋白基因突变引发;Perutz等[128]使用X射线晶体学解析血红蛋白结构;DNA测序技术解析了珠蛋白基因序列并鉴定出大量珠蛋白基因簇上的突变;第一个红系转录因子GATA-1被鉴定[129];携带有完整人β-珠蛋白基因簇的YAC转基因小鼠的出现[130];基因治疗被开始应用于β-地中海贫血;利用高通量测序技术对大量临床样本进行全基因组关联分析发现了参与调控γ-珠蛋白基因转换过程的重要转录因子BCL11A[48];永生化的人脐带血红系祖细胞HUDEP-2代替了原先广泛使用的K562和MEL细胞成为有力的研究珠蛋白基因表达的工具[131];CRISPR/Cas9等基因组编辑技术被用于解析珠蛋白基因的调控模型[53, 54, 132],并加速了镰刀型细胞贫血和β-地中海贫血基因治疗的进程。实验技术的每一次突破都会为珠蛋白基因研究带来新的发展,BCL11A等珠蛋白基因关键调控因子的鉴定结合基因组编辑技术在临床应用中的发展已经为患者带来了曙光。随着实验技术的不断进步,科学家们终将能够完全揭示人类珠蛋白基因的表达调控过程,并实现相关疾病临床治愈的梦想。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:5948690 [本文引用: 1]

Hemoglobin Gower 1, the structure of which is thought to be $\epsilon _{4}$, is the predominant hemoglobin in early human embryonic life. This finding suggests that the production of ε-chains initially exceeds that of other known (α, β, γ, and δ) chains.
URLPMID:13716547 [本文引用: 1]
IN a previous communication 1 we reported the results of a study of the h03moglobin of 26 human f04tuses and reviewed the relevant literature. In 25 f04tuses from pregnancies of 10 weeks or more we were unable to find a major h04moglobin component which differed from f04tal h03moglobin (Hb–F) or adult h03moglobin (Hb03A). In a 9-week f04tus of 3.5 cm. crown–rump length, however, the largest h03moglobin component was found to differ from Hb–F and Hb–A; its rate of alkali denaturation was intermediate, as also was its electrophoretic mobility in agar at p H 6.2 and it was assumed to be the ‘embryonic’ h03moglobin of Drescher and Künzer 2 . The present communication describes the finding of two new h03moglobin variants in a human f04tus of only 3.4 cm. crown–rump length from a 1005-week pregnancy. For convenience these will be referred to as h03moglobins Gower 1 and Gower 2.
URLPMID:1520882 [本文引用: 1]
The alpha-globin gene cluster contains four functional globin genes, zeta, alpha 2, alpha 1, and theta. The developmental regulation of the embryonic zeta and fetal/adult alpha 2- and alpha 1-globin genes is well characterized at the level of protein synthesis. The developmental pattern of the theta-globin gene is not well characterized due to the inability to detect its encoded protein. Direct analysis of the globin switching at the steady-state messenger RNA (mRNA) level has been hampered by the difficulty in obtaining quantities of embryonic and early fetal mRNA sufficient for analysis. We analyzed the relative levels of the steady-state zeta-, alpha-, and theta-globin mRNAs in yolk sac in 5-, 6-, 7-, and 8-week postconception embryonic liver, and in cord and adult blood reticulocytes. We show that the switch in the alpha-globin gene cluster from the embryonic to fetal/adult pattern of expression begins at 5 to 6 weeks of gestation. Both the theta- and alpha-globin genes show similar patterns of developmental control that are reciprocal to zeta. alpha-globin RNA is barely detectable or undetectable at 5 weeks, and increases in the 6- to 8-week period, while theta-globin mRNA shows a parallel increase at 5 to 8 weeks postconception and is expressed in cord blood and adult reticulocytes. These data show that the theta-globin gene represents a fetal/adult gene, albeit expressed at a low level.
URLPMID:15730849 [本文引用: 1]
Abstract Extensive studies during the last 30 years have led to considerable understanding of cellular and molecular control of hemoglobin switching. Cell biology studies in the 1970s defined the control of globin genes during erythroid differentiation and led to development of therapies for sickle cell disease. Molecular investigations of the last 20 years have delineated the two basic mechanisms that control globin gene activity during development--autonomous silencing and gene competition. Studies of hemoglobin switching have provided major insights on the control of gene loci by remote regulatory elements. Research in this field has an impact on understanding regulatory mechanisms in general and is of particular importance for eventual development of molecular cures for sickle cell disease and beta thalassemia.
URLPMID:8916973 [本文引用: 1]
Hemoglobin A2 (HbA2; alpha 2 delta 2) is a powerful inhibitor of HbS (alpha 2 beta 2(3)) polymerization. However, HbA2 levels are normally low in sickle cell patients. We show that a major reason for low delta-globin gene expression is the defective CACCC box at -90 in the delta-globin promoter. When the CACCC box defect in delta is corrected, expression of an HS2 delta /Luciferase reporter is equivalent to HS2 beta /Luciferase. Erythroid Krupple-like factor (EKLF), which binds to the CACCC box of the beta-globin gene and activates high-level expression, does not bind to the normal delta-globin promoter. Our goal is to design a modified EKLF that binds to the defective delta-globin promoter and enhances delta-globin gene expression. To test the feasibility of this strategy, we inserted the beta-globin CACCC box at -90 of the delta-globin gene promoter to produce an HS2 delta CAC-beta construct and quantitated human delta- and beta-globin mRNA in stably transformed murine erythroleukemia (MEL) cells. delta- Globin mRNA in these cells was 22.0% +/- 9.0% of total human globin mRNA (delta/delta + beta) as compared with 3.0% +/- 1.3% in the HS2 delta-beta control. In a second set of experiments a GAL4 DNA-binding site was inserted at -90 of the delta-globin gene to produce an HS2 delta GAL4-beta construct. This construct and a GAL4(1-147)/EKLF expression vector were stably transfected into MEL cells. delta-Globin mRNA in these cells was 27.8% +/- 7.1% of total human globin mRNA as compared with 9.9% +/- 2.5% in the HS2 delta GAL4-beta plus GAL4(1-147) control. These results show that delta-globin gene expression can be significantly increased by a modified EKLF. Based on these results, we suggest that modified EKLFs, which contain zinc fingers designed to bind specifically to the defective delta-globin CACCC box, may be useful in gene therapy approaches to increase HbA2 levels and inhibit HbS polymerization.
URL [本文引用: 3]
URLPMID:18568278 [本文引用: 1]
To demonstrate a method for using genetic epidemiological data to assess the needs for equitable and cost-effective services for the treatment and prevention of haemoglobin disorders. We obtained data on demographics and prevalence of gene variants responsible for haemoglobin disorders from online databases, reference resources, and published articles. A global epidemiological database for haemoglobin disorders by country was established, including five practical service indicators to express the needs for care (indicator 1) and prevention (indicators 2-5). Haemoglobin disorders present a significant health problem in 71% of 229 countries, and these 71% of countries include 89% of all births worldwide. Over 330,000 affected infants are born annually (83% sickle cell disorders, 17% thalassaemias). Haemoglobin disorders account for about 3.4% of deaths in children less than 5 years of age. Globally, around 7% of pregnant women carry b or a zero thalassaemia, or haemoglobin S, C, D Punjab or E, and over 1% of couples are at risk. Carriers and at-risk couples should be informed of their risk and the options for reducing it. Screening for haemoglobin disorders should form part of basic health services in most countries.
URL [本文引用: 1]
URLPMID:15113860 [本文引用: 1]
Abstract AIM: Thalassaemia is a good candidate disease for control by preventive genetic programmes in developing countries. Accurate population frequency data are needed for planning the control of thalassaemia in the high risk Guangdong Province of southern China. METHODS: In total, 13397 consecutive samples from five geographical areas of Guangdong Province were analysed for both haematological and molecular parameters. RESULTS: There was a high prevalence of carriers of alpha thalassaemia (8.53%), beta thalassaemia (2.54%), and both alpha and beta thalassaemia (0.26%). Overall, 11.07% of the population in this area were heterozygous carriers of alpha and beta thalassaemia. The mutation spectrum of alpha and beta thalassaemia and its constitution were fully described in this area. This study reports the true prevalence of silent alpha thalassaemia in the southern China population for the first time. In addition, two novel mutations that give rise to alpha thalassaemia, one deletion resulting in beta thalassaemia, and a rare deletion (--(THAI) allele) previously unreported in mainland China were detected. The frequency of the most common mutation, the Southeast Asian type of deletion (--(SEA), accounting for 48.54% of all alpha thalassaemias) was similar to the total of two alpha(+) thalassaemia deletions (-alpha(3.7) and -alpha(4.2), accounting for 47.49% of alpha thalassaemia). CONCLUSION: Both alpha and beta thalassaemia are widely distributed in Guangdong Province of China. The knowledge gained in this study will enable the projected number of pregnancies at risk to be estimated and a screening strategy for control of thalassaemia to be designed in this area.
URLPMID:23103089 [本文引用: 1]
Reliable estimates of populations affected by diseases are necessary to guide efficient allocation of public health resources. Sickle haemoglobin (HbS) is the most common and clinically significant haemoglobin structural variant, but no contemporary estimates exist of the global populations affected. Moreover, the precision of available national estimates of heterozygous (AS) and homozygous (SS) neonates is unknown. We aimed to provide evidence-based estimates at various scales, with uncertainty measures. Using a database of sickle haemoglobin surveys, we created a contemporary global map of HbS allele frequency distribution within a Bayesian geostatistical model. The pairing of this map with demographic data enabled calculation of global, regional, and national estimates of the annual number of AS and SS neonates. Subnational estimates were also calculated in data-rich areas. Our map shows subnational spatial heterogeneities and high allele frequencies across most of sub-Saharan Africa, the Middle East, and India, as well as gene flow following migrations to western Europe and the eastern coast of the Americas. Accounting for local heterogeneities and demographic factors, we estimated that the global number of neonates affected by HbS in 2010 included 56447664000 (IQR 56429164000–56467964000) AS neonates and 31264000 (29464000–33064000) SS neonates. These global estimates are higher than previous conservative estimates. Important differences predicted at the national level are discussed. HbS will have an increasing effect on public health systems. Our estimates can help countries and the international community gauge the need for appropriate diagnoses and genetic counselling to reduce the number of neonates affected. Similar mapping and modelling methods could be used for other inherited disorders. The Wellcome Trust.
URLPMID:25742458 [本文引用: 1]
Anemia is a major source of morbidity and mortality worldwide. Here we review recent insights into how red blood cells (RBCs) are produced, the pathogenic mechanisms underlying various forms of anemia, and novel therapies derived from these findings. It is likely that these new insights, mainly arising from basic scientific studies, will contribute immensely to both the understanding of frequently debilitating forms of anemia and the ability to treat affected patients. Major worldwide diseases that are likely to benefit from new advances include the hemoglobinopathies ( -thalassemia and sickle cell disease); rare genetic disorders of RBC production; and anemias associated with chronic kidney disease, inflammation, and cancer. Promising new approaches to treatment include drugs that target recently defined pathways in RBC production, iron metabolism, and fetal globin-family gene expression, as well as gene therapies that use improved viral vectors and newly developed genome editing technologies.
URLPMID:18107723 [本文引用: 1]
The rarity of sickle cell disease in infancy is surprising in view of its frequency in later childhood, which suggests that the potentiality for sickling is incompletely developed at birth. Of 226 negro mothers, 18 (8 per cent.) showed sicklaemia, and in those affected the percentage of red cells sickling was usually 100 and exceeded 84 in every case. Of their 226 newborn infants, 19 (8.4 per c...
URLPMID:1174703 [本文引用: 1]
A new form of hereditary persistence of fetal () producing 3%-8% Hb F in heterozygotes and an elevation of F-cell counts as measured by both the Kleihauer test and an antibody fluorescent procedure was found during the study of a black family. Individuals with this anomaly also had . A sickle cell homozygote who had apparently inherited the determinant had 20.3% Hb F. Both types of gamma-chains were present in equal proportions in the Hb F of these individuals. A population study revealed other AS individuals with increased Hb F synthesis, three of whom were sibs. The presence of this previously unrecognized form of might explain the mild clinical manifestations and the phenotypes of sickle cell homozygotes with unusual elevations of Hb F.
URL [本文引用: 1]
In two black families with the hereditary persistence of fetal hemoglobin (HPFH) gene there are eight A-F heterozygotes and two double heterozygotes for sickle cell trait and HPFH. These patients are clinically asymptomatic and have homogeneous acid elution smears. Measurement of globin chain synthesis in peripheral blood demonstrates balanced production of a alpha and non-alpha (beta plus gamma) chains. In these patients, the balance is achieved by increased gamma globin production and increased activity of the remaining beta globin allele. In two patients, one A-F and the other S-F there is also balanced globin synthesis in the bone marrow. In a double heterozygote for HPFH and beta-thalassemia, anemia (Hb: 11.5 g/100 ml) is associated with a moderate degree of globin chain imbalance. There is a correlation between balanced globin chain synthesis and the absence of anemia in patients with HPFH.
URLPMID:7993409 [本文引用: 1]
Original Article from The New England Journal of Medicine — Mortality In Sickle Cell Disease -- Life Expectancy and Risk Factors for Early Death
URLPMID:12426570 [本文引用: 1]
Nat Genet. 2002 Dec;32(4):623-6. Epub 2002 Nov 11. Research Support, Non-U.S. Gov't
URL [本文引用: 1]
[本文引用: 2]
URLPMID:359949 [本文引用: 1]
GATA-binding proteins constitute a family of transcription factors that recognize a target site conforming to the consensus WGATAR (W = A or T and R = A or G). Here we have used the method of polymerase chain reaction-mediated random site selection to assess in an unbiased manner the DNA-binding specificity of GATA proteins. Contrary to our expectations, we show that GATA proteins bind a variety of motifs that deviate from the previously assigned consensus. Many of the nonconsensus sequences bind protein with high affinity, equivalent to that of conventional GATA motifs. By using the selected sequences as probes in the electrophoretic mobility shift assay, we demonstrate overlapping, but distinct, sequence preferences for GATA family members, specified by their respective DNA-binding domains. Furthermore, we provide additional evidence for interaction of amino and carboxy fingers of GATA-1 in defining its binding site. By performing cotransfection experiments, we also show that transactivation parallels DNA binding. A chimeric protein containing the finger domain of areA and the activation domains of GATA-1 is capable of activating transcription in mammalian cells through GATA motifs. Our findings suggest a mechanism by which GATA proteins might selectively regulate gene expression in cells in which they are coexpressed.
URLPMID:1638017 [本文引用: 2]
Blood. 1992 Aug 1;80(3):575-81. Research Support, Non-U.S. Gov't; Research Support, U.S. Gov't, P.H.S.; Review
URL [本文引用: 2]
URLPMID:18625887 [本文引用: 1]
Abstract The transcription factor GATA1 coordinates timely activation and repression of megakaryocyte gene expression. Loss of GATA1 function results in excessive megakaryocyte proliferation and disordered terminal platelet maturation, leading to thrombocytopenia and leukemia in patients. The mechanisms by which GATA1 does this are unclear. We have used in vivo biotinylated GATA1 to isolate megakaryocyte GATA1-partner proteins. Here, several independent approaches show that GATA1 interacts with several proteins in the megakaryocyte cell line L8057 and in primary megakaryocytes. They include FOG1, the NURD complex, the pentameric complex containing SCL/TAL-1, the zinc-finger regulators GFI1B and ZFP143, and the corepressor ETO2. Knockdown of ETO2 expression promotes megakaryocyte differentiation and enhances expression of select genes expressed in terminal megakaryocyte maturation, eg, platelet factor 4 (Pf4). ETO2-dependent direct repression of the Pf4 proximal promoter is mediated by GATA-binding sites and an E-Box motif. Consistent with this, endogenous ETO2, GATA1, and the SCL pentameric complex all specifically bind the promoter in vivo. Finally, as ETO2 expression is restricted to immature megakaryocytes, these data suggest that ETO2 directly represses inappropriate early expression of a subset of terminally expressed megakaryocyte genes by binding to GATA1 and SCL.
URLPMID:1987478 [本文引用: 1]
Abstract The zinc-finger transcription factor GATA-1 (previously known as GF-1, NF-E1 or Eryf 1 binds to GATA consensus elements in regulatory regions of the alpha- and beta-globin gene clusters and other erythroid cell-specific genes. Analysis of the effects of mutations in GATA-binding sites in cell culture and in binding assays in vitro, as well as transactivation studies with GATA-1 expression vectors in heterologous cells, have provided indirect evidence that this factor is involved in the activation of globin and other genes during erythroid cell maturation. GATA-1 is also expressed in megakaryocytes and mast cells, but not in other blood cell lineages or in non-haemopoietic cells. To investigate the role of this factor in haematopoiesis in vivo, we disrupted the X-linked GATA-1 gene by homologous recombination in a male (XY) murine embryonic stem cell line and tested the GATA-1-deficient cells for their ability to contribute to different tissues in chimaeric mice. The mutant embryonic stem cells contributed to all non-haemopoietic tissues tested and to a white blood cell fraction, but failed to give rise to mature red blood cells. This demonstrates that GATA-1 is required for the normal differentiation of erythroid cells, and that other GATA-binding proteins cannot compensate for its absence.
URL [本文引用: 1]
Cis elements that mediate transcription factor binding are abundant within genomes, but the rules governing occupancy of such motifs in chromatin are not understood. The transcription factor GATA-1 that regulates red blood cell development binds with high affinity to GATA motifs, and initial studies suggest that these motifs are often unavailable for occupancy in chromatin. Whereas GATA-2 regulates the differentiation of all blood cell lineages via GATA motif binding, the specificity of GATA-2 chromatin occupancy has not been studied. We found that conditionally active GATA-1 (ER-GATA-1) and GATA-2 occupy only a small subset of the conserved GATA motifs within the murine -globin locus. Kinetic analyses in GATA-1-null cells indicated that ER-GATA-1 preferentially occupied GATA motifs at the locus control region (LCR), in which chromatin accessibility is largely GATA-1-independent. Subsequently, ER-GATA-1 increased promoter accessibility and occupied the major promoter. ER-GATA-1 increased erythroid Kr ppel-like factor and SWI/SNF chromatin remodeling complex occupancy at restricted LCR sites. These studies revealed three phases of -globin locus activation: GATA-1-independent establishment of specific chromatin structure features, GATA-1-dependent LCR complex assembly, and GATA-1-dependent promoter complex assembly. The differential utilization of dispersed GATA motifs therefore establishes spatial/temporal regulation and underlies the multistep activation mechanism.
URLPMID:18347053 [本文引用: 2]
Abstract Autonomous silencing of gamma-globin transcription is an important developmental regulatory mechanism controlling globin gene switching. An adult stage-specific silencer of the (A)gamma-globin gene was identified between -730 and -378 relative to the mRNA start site. A marked copy of the (A)gamma-globin gene inserted between locus control region 5' DNase I-hypersensitive site 1 and the epsilon-globin gene was transcriptionally silenced in adult beta-globin locus yeast artificial chromosome (beta-YAC) transgenic mice, but deletion of the 352-bp region restored expression. This fragment reduced reporter gene expression in K562 cells, and GATA-1 was shown to bind within this sequence at the -566 GATA site. Further, the Mi2 protein, a component of the NuRD complex, was observed in erythroid cells with low gamma-globin levels, whereas only a weak signal was detected when gamma-globin was expressed. Chromatin immunoprecipitation of fetal liver tissue from beta-YAC transgenic mice demonstrated that GATA-1, FOG-1, and Mi2 were recruited to the (A)gamma-globin -566 or (G)gamma-globin -567 GATA site when gamma-globin expression was low (day 18) but not when gamma-globin was expressed (day 12). These data suggest that during definitive erythropoiesis, gamma-globin gene expression is silenced, in part, by binding a protein complex containing GATA-1, FOG-1, and Mi2 at the -566/-567 GATA sites of the proximal gamma-globin promoters.
URL [本文引用: 1]
URLPMID:23284307 [本文引用: 1]
Abstract Activation of 0206-globin gene expression in adults is known to be therapeutic for sickle cell disease. Thus, it follows that the converse, alleviation of repression, would be equally effective, since the net result would be the same: an increase in fetal hemoglobin. A GATA-1-FOG-1-Mi2 repressor complex was recently demonstrated to be recruited to the -566 GATA motif of the (A)0206-globin gene. We show that Mi20205 is essential for 0206-globin gene silencing using Mi20205 conditional knockout 0205-YAC transgenic mice. In addition, increased expression of (A)0206-globin was detected in adult blood from 0205-YAC transgenic mice containing a T>G HPFH point mutation at the -566 GATA silencer site. ChIP experiments demonstrated that GATA-1 is recruited to this silencer at day E16, followed by recruitment of FOG-1 and Mi2 at day E17 in wild-type 0205-YAC transgenic mice. Recruitment of the GATA-1-mediated repressor complex was disrupted by the -566 HPFH mutation at developmental stages when it normally binds. Our data suggest that a temporal repression mechanism is operative in the silencing of 0206-globin gene expression and that either a trans-acting Mi20205 knockout deletion mutation or the cis-acting -566 (A)0206-globin HPFH point mutation disrupts establishment of repression, resulting in continued 0206-globin gene transcription during adult definitive erythropoiesis.
URLPMID:20439494 [本文引用: 1]
Abstract The human beta-globin genes are expressed in a developmentally controlled fashion. Studies on the molecular mechanisms underlying the stage-specific regulation of globin genes have been fueled by the clinical benefit of elevated fetal gamma-globin expression in patients with sickle cell anemia and thalassemia. Recent reports suggested a role of the hematopoietic transcription factor GATA-1, its cofactor FOG-1, and the associated chromatin remodeling complex NuRD in the developmental silencing of HBG1 and HBG2 gene expression. To examine whether FOG-1 via NuRD controls HBG1 and HBG2 silencing in vivo, we created mice in which the FOG-1/NuRD complex is disrupted (A. Miccio et al., EMBO J. 29:442-456, 2010) and crossed these with animals carrying the entire human beta-globin gene locus as a transgene. We found that the FOG-1/NuRD interaction is dispensable for the silencing of human HBG1 and HBG2 expression. In addition, mutant animals displayed normal silencing of the endogenous embryonic globin genes. In contrast, a significant reduction of adult-type human and murine globin gene expression was found in adult bone marrows of mutant animals. These results suggest that, unexpectedly, NuRD is required for FOG-1-dependent activation of adult-type globin gene expression but is dispensable for human gamma-globin silencing in vivo.
URL [本文引用: 1]
URLPMID:9885210 [本文引用: 1]
The zinc finger transcription factor GATA-2 is highly expressed in immature hematopoietic cells and declines with blood cell maturation. To investigate its role in normal adult hematopoiesis, a bicistronic retroviral vector encoding GATA-2 and the green fluorescent protein (GFP) was used to maintain the high levels of GATA-2 that are normally present in primitive hematopoietic cells. Coexpression of the GFP marker facilitated identification and quantitation of vector-expressing cells. Bone marrow cells transduced with the GATA-2 vector expressed GFP as judged by flow cytometry and GATA-2 as assessed by immunoblot analysis. A 50% to 80% reduction in hematopoietic progenitor-derived colony formation was observed with GATA-2/GFP-transduced marrow, compared with marrow transduced with a GFP-containing vector lacking the GATA-2 cDNA. Culture of purified populations of GATA-2/GFP-expressing and nonexpressing cells confirmed a specific ablation of the colony-forming ability of GATA-2/GFP-expressing progenitor cells. Similarly, loss of spleen colony-forming ability was observed for GATA-2/GFP-expressing bone marrow cells. Despite enforced GATA-2 expression, marrow cells remained viable and were negative in assays to evaluate apoptosis. Although efficient transduction of primitive Sca-1(+) Lin- cells was observed with the GATA-2/GFP vector, GATA-2/GFP-expressing stem cells failed to substantially contribute to the multilineage hematopoietic reconstitution of transplanted mice. Additionally, mice transplanted with purified, GATA-2/GFP-expressing cells showed post-transplant cytopenias and decreased numbers of total and gene-modified bone marrow Sca-1(+) Lin- cells. Although Sca-1(+) Lin- bone marrow cells expressing the GATA-2/GFP vector were detected after transplantation, no appreciable expansion in their numbers occurred. In contrast, control GFP-expressing Sca-1(+) Lin- cells expanded at least 40-fold after transplantation. Thus, enforced expression of GATA-2 in pluripotent hematopoietic cells blocked both their amplification and differentiation. There appears to be a critical dose-dependent effect of GATA-2 on blood cell differentiation in that downregulation of GATA-2 expression is necessary for stem cells to contribute to hematopoiesis in vivo.
URL [本文引用: 1]
URLPMID:18082606 [本文引用: 1]
Abstract Long-range interactions between distant regulatory elements, such as enhancers, and their target genes underlie the specificity of gene expression in many developmentally regulated gene families. NLI/Ldb1, a widely expressed nuclear factor, is a potential mediator of long-range interactions. Here, we show that NLI/Ldb1 and erythroid-binding partners GATA-1/SCL/LMO2 bind in vivo to the beta-globin locus control region (LCR). The C-terminal LIM interaction domain of NLI is required for formation of the complex on chromatin. Loss of the LIM domain converts NLI into a dominant-negative inhibitor of globin gene expression, and knockdown of NLI by using shRNA results in failure to activate beta-globin expression. Kinetic studies reveal that the NLI/GATA-1/SCL/LMO2 complex is detected at the beta-globin promoter coincident with RNA Pol II recruitment, beta-globin transcription, and chromatin loop formation during erythroid differentiation, providing evidence that NLI facilitates long-range gene activation.
URLPMID:3985645 [本文引用: 1]
Abstract TAL1 is a key hematopoietic transcription factor that binds to regulatory regions of a large cohort of erythroid genes as part of a complex with GATA-1, LMO2 and Ldb1. The complex mediates long-range interaction between the 0205-globin locus control region (LCR) and active globin genes, and although TAL1 is one of the two DNA-binding complex members, its role is unclear. To explore the role of TAL1 in transcription activation of the human 0206-globin genes, we reduced the expression of TAL1 in erythroid K562 cells using lentiviral short hairpin RNA, compromising its association in the 0205-globin locus. In the TAL1 knockdown cells, the 0206-globin transcription was reduced to 35% and chromatin looping of the (G)0206-globin gene with the LCR was disrupted with decreased occupancy of the complex member Ldb1 and LMO2 in the locus. However, GATA-1 binding, DNase I hypersensitive site formation and several histone modifications were largely maintained across the 0205-globin locus. In addition, overexpression of TAL1 increased the 0206-globin transcription and increased interaction frequency between the (G)0206-globin gene and LCR. These results indicate that TAL1 plays a critical role in chromatin loop formation between the 0206-globin genes and LCR, which is a critical step for the transcription of the 0206-globin genes.
URL [本文引用: 1]
Abstract Top of page Abstract Introduction Results Discussion Materials and methods Acknowledgements References Members of the small Maf family of transcription factors play important roles in hematopoiesis. Using transgenic assays, we discovered a tissue-specific enhancer 3 to the mafK gene. This enhancer directs mafK transcription in hematopoietic as well as in developing cardiac muscle cells, and was thus designated the h ematopoietic and c ardiac e nhancer of maf K (HCEK). Only two of four GATA consensus motifs identified within HCEK contributed to enhancer activity, and both of these sites were required for both cardiac and hematopoietic transcriptional activation. The expression profile of MafK significantly overlapped that of GATA-1 in hematopoietic cells and of GATA-4/-6 in cardiac tissues. Each of these GATA factors bound with high specificity to both of the critical GATA sites in HCEK. Hence, the mafK gene is regulated by different GATA proteins in the hematopoietic and cardiac compartments through the same two GATA-binding sites in HCEK. These data provide the first in vivo demonstration that distinct members of a related transcription factor family activate the tissue-specific expression of a single target gene using the same cis -regulatory element.
URLPMID:11517325 [本文引用: 1]
Abstract The mouse beta-globin gene locus control region (LCR), located upstream of the beta-globin gene cluster, is essential for the activated transcription of genes in the cluster. The LCR contains multiple binding sites for transactivators, including Maf-recognition elements (MAREs). However, little is known about the specific proteins that bind to these sites or the time at which they bind during erythroid differentiation. We have performed chromatin immunoprecipitation experiments to determine the recruitment of the erythroid-specific transactivator p45 NF-E2/MafK (p18 NF-E2) heterodimer and small Maf proteins to various regions in the globin gene locus before and after the induction of murine erythroleukemia (MEL) cell differentiation. We report that, before induction, the LCR is occupied by small Maf proteins, and, on erythroid maturation, the NF-E2 complex is recruited to the LCR and the active globin promoters, even though the promoters do not contain MAREs. This differentiation-coupled recruitment of NF-E2 complex correlates with a greater than 100-fold increase in beta-major globin transcription, but is not associated with a significant change in locus-wide histone H3 acetylation. These findings suggest that the beta-globin gene locus exists in a constitutively open chromatin conformation before terminal differentiation, and we speculate that recruitment of NF-E2 complex to the LCR and active promoters may be a rate-limiting step in the activation of beta-globin gene expression.
URLPMID:359778000180160 [本文引用: 1]
The human gamma-globin gene promoter contains a stage selector element (SSE) responsible for preferential interaction of the promoter with a powerful erythroid-specific enhancer in the fetal developmental stage (S.M. Jane, P.A. Ney, E.F. Vanin, D.L. Gumucio, and A.W. Nienhuis. EMBO J. 11:2691-2699, 1992). The element binds two proteins, the ubiquitous activator Sp1 and a protein previously known as -50 gamma and now named the stage selector protein (SSP). Binding of the second protein correlates with SSE activity in transient-transfection assays. We now report that a de novo binding site for the SSP is created by the -202(C-->G) mutation that causes hereditary persistence of fetal hemoglobin (HPFH). This site functions in an analogous manner to the SSE in hybrid beta-promoter/reporter gene constructs transfected into K562 cells. In contrast, the wild-type -202 sequence, which fails to bind the SSP, is incapable of activating the beta-gene promoter. Both the -50 and -202 HPFH sites for SSP binding overlap a consensus sequence for the transcriptional regulator Sp1. In addition, both sites contain CpG dinucleotides that are contact bases for SSP. Since the gamma promoter is known to be hypomethylated in fetal cells but fully methylated at CpG residues in adult erythroid cells, we examined the effects of this DNA modification on protein binding to the two regions. Gel mobility shift assays with nuclear extract from K562 cells (which contain both Sp1 and SSP) demonstrate preferential binding of SSP to the SSE and HPFH sites under conditions in which probe was limiting. Methylation of the CpG residues reverses this preference only in the SSE site, with a marked increase in the binding of Sp1 at the expense of the SSP. Purified Sp1 binds with 10-fold higher affinity to the methylated than to the nonmethylated -50 probe but with the same affinity to the -202 HPFH probe. The methylation-induced preferential binding of Sp1 to the SSE at the expense of SSP may be part of the mechanism by which the gamma genes are repressed in normal adult erythroid cells. In cells containing the -202 HPFH mutation, the inability of Sp1 to displace SSP in the methylated state may explain the persistence of gamma-promoter activity and gamma-gene expression observed in adults with this mutation.
URLPMID:7828600 [本文引用: 1]
Abstract The human stage selector protein (SSP) has been implicated in the developmental regulation of the globin genes. Binding of SSP to the stage selector element (SSE) in the proximal gamma-globin promoter is integral to the competitive silencing of a linked beta-promoter in embryonic/fetal stage erythroleukemia (K562) cells. We now report the biochemical purification of SSP from K562 cell nuclear extract and demonstrate that the ubiquitously expressed transcription factor CP2 is pivotal to, but not sufficient for, SSP binding activity. Although addition of anti-CP2 antiserum disrupts the formation of the SSP-SSE complex in the electrophoretic mobility shift assay (EMSA), recombinant CP2 fails to bind to the SSE. Binding of CP2 to the SSE requires a heterodimeric partner present in K562 cells. We have defined the molecular weight of the partner protein as 40-45 kDa in UV and protein cross-linking experiments. An element analogous to the human SSE has previously been demonstrated in the chicken beta A-gene-promoter. The effects of this element are dependent on the binding of the chicken stage selector protein, NF-E4. Comparative studies between human CP2 and chicken NF-E4 demonstrate homology between the protein complexes. SSP binds to the chicken SSE and formation of this complex is ablated by the addition of anti-CP2 antiserum or a monoclonal antibody to NF-E4. Western analysis of partially purified NF-E4 using anti-CP2 antiserum or the NF-E4 monoclonal antibody both demonstrate a dominant band at 66 kDa. Similarly, the NF-E4 antibody recognizes the 66 kDa human CP2 protein in Western analysis of the SSP-SSE complex.(ABSTRACT TRUNCATED AT 250 WORDS)
URLPMID:86334 [本文引用: 1]
The stage selector protein (SSP) is a heteromeric complex involved in preferential expression of the human gamma-globin genes in fetal-erythroid cells. We have previously identified the ubiquitous transcription factor CP2 as a component of this complex. Using the protein dimerization domain of CP2 in a yeast two-hybrid screen, we have cloned a novel gene, NF-E4, encoding the tissue-restricted component of the SSP. NF-E4 and CP2 coimmunoprecipitate from extract derived from a fetal-erythroid cell line, and antiserum to NF-E4 ablates binding of the SSP to the gamma promoter. NF-E4 is expressed in fetal liver, cord blood, and bone marrow and in the K562 and HEL cell lines, which constitutively express the fetal globin genes. Enforced expression of NF-E4 in K562 cells and primary erythroid progenitors induces endogenous fetal globin gene expression, suggesting a possible strategy for therapeutic intervention in the hemoglobinopathies.
URLPMID:15084587 [本文引用: 1]
The human stage selector protein, a complex containing the ubiquitous transcription factor CP2 and the erythroid-specific factor p22 NF-E4, facilitates the interaction of the γ-globin genes with the locus control region in fetal erythroid cells. Enforced expression of p22 NF-E4 in K562 cells and human cord blood progenitors increases fetal globin gene expression, and in progenitors, reduces β-globin expression. To examine the role of NF-E4 in an model of hemoglobin switching, we enforced the expression of p22 NF-E4 in transgenic mice carrying the human β-globin locus yeast artificial chromosome. Although murine erythropoiesis and globin gene expression is unaffected in these mice, the expression profile of the human globin genes is altered. All three transgenic lines displayed an increased γ:β-globin ratio in E12.5–14.5 fetal liver, resulting in a delay in the fetal/adult switch. At E12.5, this is primarily due to a reduction of β-gene expression, whereas at E14.5, the increased γ:β ratio is due to enhanced γ-gene expression. Despite this, the switch in globin subtype is fully completed in the adult bone marrow. These findings indicate that p22 NF-E4 is capable of influencing human globin gene expression but is incapable of overriding the intrinsic mechanisms governing γ-gene silencing in this context.
URLPMID:16263792 [本文引用: 1]
Abstract Binding of the stage selector protein (SSP) to the stage selector element (SSE) in the human gamma-globin promoter contributes to the preferential expression of the gamma-gene in fetal erythroid cells. The SSP contains the transcription factor CP2 and an erythroid-specific partner, NF-E4. The NF-E4 gene encodes a 22-kDa polypeptide employing a non-AUG initiation codon. Antisera specific to NF-E4 detects this species and an additional 14 kDa protein, which initiates from an internal methionine. Enforced expression of p14 NF-E4 in the K562 fetal/erythroid cell line, and in primary erythroid cord blood progenitors, results in repression of gamma-gene expression. Biochemical studies reveal that p14 NF-E4 interacts with CP2, resulting in diminished association of CP2 with the SSE in chromatin immunoprecipitation assays. p45 NF-E2 recruitment to the gamma-promoter is also lost, resulting in a reduction in RNA polymerase II and TBP binding and a fall in promoter transcriptional activity. This effect is specific, as enforced expression of a mutant form of p14 NF-E4, which fails to interact with CP2, also fails to repress gamma-gene expression in K562 cells. These findings provide one potential mechanism that could contribute to the autonomous silencing of the human gamma-genes in adult erythroid cells.
URL [本文引用: 1]
URLPMID:12582237 [本文引用: 1]
The overall structure of the DNase I hypersensitive sites (HSs) that comprise the beta-globin locus control region (LCR) is highly conserved among mammals, implying that the HSs have conserved functions. However, it is not well understood how the LCR HSs, either individually or collectively, activate transcription. We analyzed the interactions of HS2, HS3 and HS4 with the human epsilon- and beta-globin genes in chromatinized episomes in fetal/embryonic K562 cells. Only HS2 activates transcription of the epsilon-globin gene, while all three HSs activate the beta-globin gene. HS3 stimulates the beta-globin gene constitutively, but HS2 and HS4 transactivation requires expression of the transcription factor EKLF, which is not present in K562 cells but is required for beta-globin expression in vivo. To begin addressing how the individual HSs may interact with one another in a complex, we linked the beta-globin gene to both the HS2 and HS3. HS2 and HS3 together resulted in synergistic stimulation of beta-globin transcription. Unexpectedly, mutated, inactive forms of HS2 impeded the activation of the beta-globin gene by HS3. Thus, there appear to be distinct interactions among the HSs and between the HSs and the globin genes. These preferential, non-exclusive interactions may underlie an important structural and functional cooperativity among the regulatory sequences of the beta-globin locus in vivo.
URLPMID:24829204 [本文引用: 1]
Abstract Mutations in human Krüppel-like factor 1 (KLF1) have recently been reported to be responsible for increased fetal hemoglobin (HbF) and hemoglobin A2 (HbA2). Because increased HbF and HbA2 levels are important features of β-thalassemia, we examined whether there is any relationship between KLF1 mutation and β-thalassemia in China. To do this, we first studied the incidence of KLF1 mutations in 2 Chinese populations: 3839 individuals from a thalassemia endemic region in south China and 1190 individuals from a non-thalassemia endemic region in north China. Interestingly, we found that the prevalence of KLF1 mutations is significantly higher in the thalassemia endemic region than that in non-thalassemia endemic region (1.25% vs 0.08%). Furthermore, we identified 7 functional variants including 4 previously reported (p.Gly176AlafsX179, p.Ala298Pro, p.Thr334Arg, and c.913+1G>A) and 3 novel variants (p.His299Asp, p.Cys341Tyr, and p.Glu5Lys) in southern China. The 2 most common mutations, p.Gly176AlafsX179 and p.His299Asp, accounted for 90.6% of the total. We found that zinc-finger mutations in KLF1 were selectively represented in 12 β-thalassemia intermedia patients and resulted in significantly different transfusion-free survival curves. Our findings suggest that KLF1 mutations occur selectively in the presence of β-thalassemia to increase the production of HbF, which in turn ameliorates the clinical severity of β-thalassemia. 08 2014 by The American Society of Hematology.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:28659276 [本文引用: 1]
β-Hemoglobinopathies are among the most common single-locus inherited diseases. In this condition, high fetal hemoglobin (HbF) levels have been found to be beneficial, and boosting HbF expression is seen as an attractive therapy. Naturally occurring mutations in the fetal globin promoter can result
URLPMID:239526 [本文引用: 1]
Abstract Development of red blood cells requires the correct regulation of cellular processes including changes in cell morphology, globin expression and heme synthesis. Transcription factors such as erythroid Kruppel-like factor EKLF (Klf1) play a critical role in erythropoiesis. Mice lacking EKLF die around embryonic day 14 because of defective definitive erythropoiesis, partly caused by a deficit in beta-globin expression. To identify additional target genes, we analyzed the phenotype and gene expression profiles of wild-type and EKLF null primary erythroid progenitors that were differentiated synchronously in vitro. We show that EKLF is dispensable for expansion of erythroid progenitors, but required for the last steps of erythroid differentiation. We identify EKLF-dependent genes involved in hemoglobin metabolism and membrane stability. Strikingly, expression of these genes is also EKLF-dependent in primitive, yolk sac-derived, blood cells. Consistent with lack of upregulation of these genes we find previously undetected morphological abnormalities in EKLF-null primitive cells. Our data provide an explanation for the hitherto unexplained severity of the EKLF null phenotype in erythropoiesis.
URLPMID:18245381 [本文引用: 2]
Abstract beta-Thalassemia and sickle cell disease both display a great deal of phenotypic heterogeneity, despite being generally thought of as simple Mendelian diseases. The reasons for this are not well understood, although the level of fetal hemoglobin (HbF) is one well characterized ameliorating factor in both of these conditions. To better understand the genetic basis of this heterogeneity, we carried out genome-wide scans with 362,129 common SNPs on 4,305 Sardinians to look for genetic linkage and association with HbF levels, as well as other red blood cell-related traits. Among major variants affecting HbF levels, SNP rs11886868 in the BCL11A gene was strongly associated with this trait (P < 10(-35)). The C allele frequency was significantly higher in Sardinian individuals with elevated HbF levels, detected by screening for beta-thalassemia, and patients with attenuated forms of beta-thalassemia vs. those with thalassemia major. We also show that the same BCL11A variant is strongly associated with HbF levels in a large cohort of sickle cell patients. These results indicate that BCL11A variants, by modulating HbF levels, act as an important ameliorating factor of the beta-thalassemia phenotype, and it is likely they could help ameliorate other hemoglobin disorders. We expect our findings will help to characterize the molecular mechanisms of fetal globin regulation and could eventually contribute to the development of new therapeutic approaches for beta-thalassemia and sickle cell anemia.
URL [本文引用: 1]
URLPMID:20712774 [本文引用: 1]
The -thalassemia syndromes are a major global health problem. Increased levels of fetal hemoglobin (HbF) ameliorate the clinical symptoms seen in this disease. By taking advantage of the natural variation in the level of HbF in various populations, we and others identified several common genetic variants in three major loci that regulate HbF levels. One of these variants resides in the gene BCL11A. We have studied the role of this gene product and established that BCL11A maintains silencing of -globin expression in adult erythroid cells and functions as a direct transcriptional regulator of the fetal to adult hemoglobin switch in humans. Moreover, we found that BCL11A plays a central role in the evolutionarily divergent globin gene switches of mammals. As a factor critical for -globin gene silencing, BCL11A should be considered as a therapeutic target to increase HbF in a directed manner in -thalassemia patients.
URLPMID:21998251 [本文引用: 2]
Persistence of human fetal hemoglobin (HbF, α(2)γ(2)) in adults lessens the severity of sickle cell disease (SCD) and the β-thalassemias. Here, we show that the repressor BCL11A is required in vivo for silencing of γ-globin expression in adult animals, yet dispensable for red cell production. BCL11A serves as a barrier to HbF reactivation by known HbF inducing agents. In a proof-of-principle test of BCL11A as a potential therapeutic target, we demonstrate that inactivation of BCL11A in SCD transgenic mice corrects the hematologic and pathologic defects associated with SCD through high-level pancellular HbF induction. Thus, interference with HbF silencing by manipulation of a single target protein is sufficient to reverse SCD.
URL [本文引用: 1]
442.
URLPMID:29606353 [本文引用: 2]
Abstract Fetal hemoglobin (HbF, α 2 γ 2 ) level is genetically controlled and modifies severity of adult hemoglobin (HbA, α 2 β 2 ) disorders, sickle cell disease, and β-thalassemia. Common genetic variation affects expression of BCL11A, a regulator of HbF silencing. To uncover how BCL11A supports the developmental switch from γ- to β- globin, we use a functional assay and protein binding microarray to establish a requirement for a zinc-finger cluster in BCL11A in repression and identify a preferred DNA recognition sequence. This motif appears in embryonic and fetal-expressed globin promoters and is duplicated in γ-globin promoters. The more distal of the duplicated motifs is mutated in individuals with hereditary persistence of HbF. Using the CUT&RUN approach to map protein binding sites in erythroid cells, we demonstrate BCL11A occupancy preferentially at the distal motif, which can be disrupted by editing the promoter. Our02findings reveal that direct γ-globin gene promoter repression by BCL11A underlies hemoglobin switching.
URLPMID:29610478 [本文引用: 3]
Abstract 0205-hemoglobinopathies such as sickle cell disease (SCD) and 0205-thalassemia result from mutations in the adult HBB (0205-globin) gene. Reactivating the developmentally silenced fetal HBG1 and HBG2 (0206-globin) genes is a therapeutic goal for treating SCD and 0205-thalassemia 1 . Some forms of hereditary persistence of fetal hemoglobin (HPFH), a rare benign condition in which individuals express the 0206-globin gene throughout adulthood, are caused by point mutations in the 0206-globin gene promoter at regions residing ~115 and 200090009bp upstream of the transcription start site. We found that the major fetal globin0002gene repressors BCL11A and ZBTB7A (also known as LRF) directly bound to the sites at -115 and -200090009bp, respectively. Furthermore, introduction of naturally occurring HPFH-associated mutations into erythroid cells by CRISPR-Cas9 disrupted repressor binding and raised 0206-globin gene expression. These findings clarify how these HPFH-associated mutations operate and demonstrate that BCL11A and ZBTB7A are major direct repressors of the fetal globin gene.
URLPMID:17592125 [本文引用: 1]
Individual variation in fetal hemoglobin (HbF, alpha(2)gamma(2)) response underlies the remarkable diversity in phenotypic severity of sickle cell disease and beta thalassemia. HbF levels and HbF-associated quantitative traits (e.g., F cell levels) are highly heritable. We have previously mapped a major quantitative trait locus (QTL) controlling F cell levels in an extended Asian-Indian kindred with beta thalassemia to a 1.5-Mb interval on chromosome 6q23, but the causative gene(s) are not known. The QTL encompasses several genes including HBS1L, a member of the GTP-binding protein family that is expressed in erythroid progenitor cells. In this high-resolution association study, we have identified multiple genetic variants within and 5' to HBS1L at 6q23 that are strongly associated with F cell levels in families of Northern European ancestry (P = 10(-75)). The region accounts for 17.6% of the F cell variance in northern Europeans. Although mRNA levels of HBS1L and MYB in erythroid precursors grown in vitro are positively correlated, only HBS1L expression correlates with high F cell alleles. The results support a key role for the HBS1L-related genetic variants in HbF control and illustrate the biological complexity of the mechanism of 6q QTL as a modifier of fetal hemoglobin levels in the beta hemoglobinopathies.
URL [本文引用: 1]
URLPMID:19528534 [本文引用: 1]
HBS1L-MYB intergenic polymorphism (HMIP) on chromosome 6q23 is associated with elevated fetal hemoglobin levels and has pleiotropic effects on several hematologic parameters. To investigate potential regulatory activity in the region, we have measured sensitivity of the sequences to DNase I cleavage that identified 3 tissue-specific DNase I hypersensitive sites in the core intergenic interval. Chromatin immunoprecipitation with microarray (ChIP-chip) analysis showed strong histone acetylation in a defined interval of 65 kb corresponding to the core HBS1L-MYB intergenic region in primary human erythroid cells but not in non YB-expressing HeLa cells. ChIP-chip analysis also identified several potential cis-regulatory elements as strong GATA-1 signals that coincided with the DNase I hypersensitive sites present in MYB-expressing erythroid cells. We suggest that HMIP contains regulatory sequences that could be important in hematopoiesis by controlling MYB expression. This study provides the functional link between genetic association of HMIP with control of fetal hemoglobin and other hematologic parameters. We also present a large-scale analysis of histone acetylation as well as RNA polymerase II and GATA-1 interactions on chromosome 6q, and and globin gene loci. The data suggest that GATA-1 regulates numerous genes of various functions on chromosome 6q.
URLPMID:3973089 [本文引用: 1]
Genetic studies have identified common variants within the intergenic region (HBS1L-MYB) between GTP-binding elongation factor HBS1L and myeloblastosis oncogene MYB on chromosome 6q that are associated with elevated fetal hemoglobin (HbF) levels and alterations of other clinically important human erythroid traits. It is unclear how these noncoding sequence variants affect multiple erythrocyte characteristics. Here, we determined that several HBS1L-MYB intergenic variants affect regulatory elements that are occupied by key erythroid transcription factors within this region. These elements interact with MYB, a critical regulator of erythroid development and HbF levels. We found that several HBS1L-MYB intergenic variants reduce transcription factor binding, affecting long-range interactions with MYB and MYB expression levels. These data provide a functional explanation for the genetic association of HBS1L-MYB intergenic polymorphisms with human erythroid traits and HbF levels. Our results further designate MYB as a target for therapeutic induction of HbF to ameliorate sickle cell and 尾-thalassemia disease severity.
URLPMID:9927193 [本文引用: 1]
Oncogene. 1999 Jan 14;18(2):365-75. Research Support, Non-U.S. Gov't; Research Support, U.S. Gov't, P.H.S.
URLPMID:5001899 [本文引用: 1]
Hematopoietic development is governed by the coordinated expression of lineage- and differentiation stage-specific genes. Transcription factors play major roles in this process and their perturbation...
URLPMID:4778394 [本文引用: 1]
Abstract Genes encoding human 0205-type globin undergo a developmental switch from embryonic to fetal to adult-type expression. Mutations in the adult form cause inherited hemoglobinopathies or globin disorders, including sickle cell disease and thalassemia. Some experimental results have suggested that these diseases could be treated by induction of fetal-type hemoglobin (HbF). However, the mechanisms that repress HbF in adults remain unclear. We found that the LRF/ZBTB7A transcription factor occupies fetal 0206-globin genes and maintains the nucleosome density necessary for 0206-globin gene silencing in adults, and that LRF confers its repressive activity through a NuRD repressor complex independent of the fetal globin repressor BCL11A. Our study may provide additional opportunities for therapeutic targeting in the treatment of hemoglobinopathies. Copyright 0008 2016, American Association for the Advancement of Science.
URLPMID:19853566 [本文引用: 1]
Abstract GATA-1-dependent transcription is essential for erythroid differentiation and maturation. Suppression of programmed cell death is also thought to be critical for this process; however, the link between these two features of erythropoiesis has remained elusive. Here, we show that the POZ-Kr ppel family transcription factor, LRF (also known as Zbtb7a/Pokemon), is a direct target of GATA1 and plays an essential antiapoptotic role during terminal erythroid differentiation. We find that loss of Lrf leads to lethal anemia in embryos, due to increased apoptosis of late-stage erythroblasts. This programmed cell death is Arf and p53 independent and is instead mediated by upregulation of the proapoptotic factor Bim. We identify Lrf as a direct repressor of Bim transcription. In strong support of this mechanism, genetic Bim loss delays the lethality of Lrf-deficient embryos and rescues their anemia phenotype. Thus, our data define a key transcriptional cascade for effective erythropoiesis, whereby GATA-1 suppresses BIM-mediated apoptosis via LRF.
URLPMID:8491376 [本文引用: 1]
A cDNA encoding a novel zinc finger protein has been isolated from a mouse T-cell leukemia line on the basis of its expression of a Ly-1 epitope in a lambda gt11 library. The putative gene was mapped on mouse chromosome 1, closely linked to Idh-1, but not linked to the Ly-1 (CD5) gene. The cDNA is therefore named Ly-1 antibody reactive clone (LYAR). The putative polypeptide encoded by the cDNA consists of 388 amino acids with a zinc finger motif and three copies of nuclear localization signals. Antibodies raised against a LYAR fusion protein reacted with a protein of 45 kD on Western blots and by immunoprecipitation. Immunolocalization indicated that LYAR was present predominantly in the nucleoli. The LYAR mRNA was not detected in brain, thymus, bone marrow, liver, heart, and muscle. Low levels of LYAR mRNA were detected in kidney and spleen. However, the LYAR gene was expressed at very high levels in immature spermatocytes in testis. The LYAR mRNA is present at high levels in early embryos and preferentially in fetal liver and fetal thymus. A number of B- and T-cell leukemic lines expressed LYAR at high levels, although it was not detectable in bone marrow and thymus. During radiation-induced T-cell leukemogenesis, high levels of LYAR were expressed in preleukemic thymocytes and in acute T leukemia cells. Fibroblast cells overexpressing the LYAR cDNA from a retrovirus vector, though not phenotypically transformed in vitro, had increased ability to form tumors in nu/nu mice. Therefore, LYAR may function as a novel nucleolar oncoprotein to regulate cell growth.
URLPMID:4150809 [本文引用: 1]
Human globin gene expression during development is modulated by transcription factors in a stage-dependent manner. However, the mechanisms controlling the process are still largely unknown. In this study, we found that a nuclear protein, LYAR (human homologue of mouse Ly-1 antibody reactive clone) directly interacted with the methyltransferase PRMT5 which triggers the histone H4 Arg3 symmetric dimethylation (H4R3me2s) mark. We found that PRMT5 binding on the proximal γ-promoter was LYAR-dependent. The LYAR DNA-binding motif (GGTTAT) was identified by performing CASTing (cyclic amplification and selection of targets) experiments. Results of EMSA and ChIP assays confirmed that LYAR bound to a DNA region corresponding to the 5'-untranslated region of the γ-globin gene. We also found that LYAR repressed human fetal globin gene expression in both K562 cells and primary human adult erythroid progenitor cells. Thus, these data indicate that LYAR acts as a novel transcription factor that binds the γ-globin gene, and is essential for silencing the γ-globin gene.
[本文引用: 1]
URLPMID:26897028 [本文引用: 1]
Introduction Several DNA polymorphisms have been associated with high production of fetal hemoglobin (HbF), although the molecular basis is not completely understood. In order to identify and...
URLPMID:28669403 [本文引用: 2]
Abstract A delayed fetal-to-adult hemoglobin (Hb) switch ameliorates the severity of 0205-thalassemia and sickle cell disease. The molecular mechanism underlying the epigenetic dysregulation of the switch is unclear. To explore the potential cis-variants responsible for the Hb switching, we systematically analyzed an 80-kb region spanning the 0205-globin cluster using capture-based next-generation sequencing of 1142 Chinese 0205-thalassemia persons and identified 31 fetal hemoglobin (HbF)-associated haplotypes of the selected 28 tag regulatory single-nucleotide polymorphisms (rSNPs) in seven linkage disequilibrium (LD) blocks. A Ly1 antibody reactive (LYAR)-binding motif disruptive rSNP rs368698783 (G/A) from LD block 5 in the proximal promoter of hemoglobin subunit gamma 1 (HBG1) was found to be a significant predictor for 0205-thalassemia clinical severity by epigenetic-mediated variant-dependent HbF elevation. We found this rSNP accounted for 41.6% of 0205-hemoglobinopathy individuals as an ameliorating factor in a total of 2,738 individuals from southern China and Thailand. We uncovered that the minor allele of the rSNP triggers the attenuation of LYAR and two repressive epigenetic regulators DNA methyltransferase 3 alpha (DNMT3A) and protein arginine methyltransferase 5 (PRMT5) from the HBG promoters, mediating allele-biased 0206-globin elevation by facilitating demethylation of HBG core promoter CpG sites in erythroid progenitor cells from 0205-thalassemia persons. The present study demonstrates that this common rSNP in the proximal A 0206-promoter is a major genetic modifier capable of ameliorating the severity of thalassemia major through the epigenetic-mediated regulation of the delayed fetal-to-adult Hb switch and provides potential targets for the treatment of 0205-hemoglobinopathy. Copyright 0008 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
URL [本文引用: 1]
Abstract The TR2 and TR4 orphan nuclear receptors comprise the DNA-binding core of direct repeat erythroid definitive, a protein complex that binds to direct rep
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:17456718 [本文引用: 1]
The mechanisms underlying the human fetal-to-adult β-globin gene switch remain to be determined. While there is substantial experimental evidence to suggest that promoter DNA methylation is involved in this process, most data come from studies in nonhuman systems. We have evaluated human γ- and β-globin promoter methylation in primary human fetal liver (FL) and adult bone marrow (ABM) erythroid cells. Our results show that, in general, promoter methylation and gene expression are inversely related. However, CpGs at 61162 of the γ promoter and 61126 of the β promoter are hypomethylated in ABM and FL, respectively. We also studied γ-globin promoter methylation during in vitro differentiation of erythroid cells. The γ promoters are initially hypermethylated in CD34+ cells. The upstream γ promoter CpGs become hypomethylated during the preerythroid phase of differentiation and are then remethylated later, during erythropoiesis. The period of promoter hypomethylation correlates with transient γ-globin gene expression and may explain the previously observed fetal hemoglobin production that occurs during early adult erythropoiesis. These results provide the first comprehensive survey of developmental changes in human γ- and β-globin promoter methylation and support the hypothesis that promoter methylation plays a role in human β-globin locus gene switching.
URLPMID:11297506 [本文引用: 1]
Histone deacetylation plays an important role in methylated DNA silencing. Recent studies indicated that the methyl-CpG-binding protein, MBD2, is a component of the MeCP1 histone deacetylase complex. Interestingly, MBD2 is able to recruit the nucleosome remodeling and histone deacetylase, NuRD, to methylated DNA in vitro. To understand the relationship between the MeCP1 complex and the NuRD complex, we purified the MeCP1 complex to homogeneity and found that it contains 10 major polypeptides including MBD2 and all of the known NuRD components. Functional analysis of the purified MeCP1 complex revealed that it preferentially binds, remodels, and deacetylates methylated nucleosomes. Thus, our study defines the MeCP1 complex, and provides biochemical evidence linking nucleosome remodeling and histone deacetylation to methylated gene silencing.
URLPMID:16608912 [本文引用: 1]
The genes of the vertebrate beta-globin locus undergo a switch in expression during erythroid development whereby embryonic/fetal genes of the cluster are sequentially silenced and adult genes are activated. We describe here a role for DNA methylation and MBD2 in the silencing of the human fetal gamma-globin gene. The gamma-globin gene is reactivated upon treatment with the DNA methyltransferase inhibitor 5-azacytidine in the context of a mouse containing the entire human beta-globin locus as a yeast artificial chromosome (betaYAC) transgene. To elucidate the mechanism through which DNA methylation represses the gamma-globin gene in adult erythroid cells, betaYAC/MBD2-/- mice were generated by breeding betaYAC mice with MBD2-/- mice. Adult betaYAC/MBD2-/- mice continue to express the gamma-globin gene at a level commensurate with 5-azacytidine treatment, 10- to 20-fold over that observed with 1-acetyl-2-phenylhydrazine treatment alone. In addition, the level of gamma-globin expression is consistently higher in MBD2-/- mice in 14.5- and 16.5-days postcoitus fetal liver erythroblasts suggesting a role for MBD2 in embryonic/fetal erythroid development. DNA methylation levels are modestly decreased in MBD2-/- mice. MBD2 does not bind to the gamma-globin promoter region to maintain gamma-globin silencing. Finally, treatment of MBD2-null mice with 5-azacytidine induces only a small, nonadditive induction of gamma-globin mRNA, signifying that DNA methylation acts primarily through MBD2 to maintain gamma-globin suppression in adult erythroid cells.
URL [本文引用: 1]
Nucleosome remodeling complexes comprise several large families of chromatin modifiers that integrate multiple epigenetic control signals to play key roles in cell type-specific transcription regulation. We previously isolated a methyl-binding domain protein 2 (MBD2)-containing nucleosome remodeling and deacetylation (NuRD) complex from primary erythroid cells and showed that MBD2 contributes to DNA methylation-dependent embryonic and fetal β-type globin gene silencing during development in vivo. Here we present structural and biophysical details of the coiled-coil interaction between MBD2 and p66α, a critical component of the MBD2–NuRD complex. We show that enforced expression of the isolated p66α coiled-coil domain relieves MBD2-mediated globin gene silencing and that the expressed peptide interacts only with a subset of components of the MBD2–NuRD complex that does not include native p66α or Mi-2. These results demonstrate the central importance of the coiled-coil interaction and suggest that MBD2-dependent DNA methylation-driven gene silencing can be disrupted by selectively targeting this coiled-coil complex.
URLPMID:23463310 [本文引用: 1]
Abstract Chromatin is a dynamic structure that must respond to myriad stimuli to regulate access to DNA, and chemical modification of histones is a major means by which the cell modulates nucleosome mobility and turnover. Histone modifications are linked to essentially every cellular process requiring DNA access, including transcription, replication and repair. Here we consider properties of the major types of histone modification in the context of their associated biological processes to view them in light of the cellular mechanisms that regulate nucleosome dynamics.
URLPMID:11437231 [本文引用: 1]
Abstract Histone acetyltranferase (HAT) enzymes are the catalytic subunit of large multisubunit HAT complexes that acetylate the epsilon-amino group of specific lysine residues on histone tails to promote transcriptional activation. Recent structural and functional studies on the divergent HAT enzymes Gcn5/PCAF, Esa1 and Hat1 have provided new insights into the underlying mechanism of histone binding and acetylation by HAT proteins. The three HAT enzymes contain a structurally conserved core domain that plays a functionally conserved role in binding the coenzyme A cofactor and in harboring the putative general base for catalysis. Structurally variable N- and C-terminal domains appear to contain a related scaffold that mediates histone substrate binding. These data provide a framework for understanding the structure and function of other more divergent HAT proteins such as TAF(II)250 and CBP/p300, and provides a starting point for understanding how HAT proteins may cooperate with other factors within in vivo HAT complexes to promote transcriptional activation.
URLPMID:27599530 [本文引用: 1]
Over the last several decades, it has become clear that epigenetic abnormalities may be one of the hallmarks of cancer. Posttranslational modifications of histones, for example, may play a crucial role in cancer development and progression by modulating gene transcription, chromatin remodeling, and nuclear architecture. Histone acetylation, a well-studied posttranslational histone modification, is controlled by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). By removing acetyl groups, HDACs reverse chromatin acetylation and alter transcription of oncogenes and tumor suppressor genes. In addition, HDACs deacetylate numerous nonhistone cellular substrates that govern a wide array of biological processes including cancer initiation and progression. This review will discuss the role of HDACs in cancer and the therapeutic potential of HDAC inhibitors (HDACi) as emerging drugs in cancer treatment.
URLPMID:6204332 [本文引用: 1]
Adult White Leghorn chickens were rendered anemic by injection with 1-acetyl-2-phenylhydrazine and then treated with parenteral 5-azacytidine, and levels of embryonic globin RNA in circulating reticulocytes were measured. A very small but detectable amount of correctly initiated embryonic ρ -type globin RNA was detected in reticulocytes from birds treated with 5-azacytidine, while none was detected in reticulocytes from those receiving only phenylhydrazine or phenylhydrazine plus 1-β -D-arabinofuranosylcytosine (cytosine arabinonucleoside). An attempt to increase embryonic globin RNA induction by treatment with parenteral sodium butyrate after 7 days of 5-azacytidine administration resulted in a 5- to 10-fold increase in the level of embryonic globin RNA. However, sodium butyrate did not induce embryonic gene expression when given alone or after treatment with cytosine arabinonucleoside. Sodium butyrate treatment also caused a DNase I-hypersensitive site to be exposed at the 5′end of the ρ -globin gene only after 5-azacytidine induced demethylation of several CpG sites in and around the gene. The implications of this model of gene activation in vivo are discussed in the context of multistep gene regulation.
URLPMID:2460870 [本文引用: 1]
The switch from fetal to adult hemoglobin expression is regulated in many mammalian species by a developmental clock-like mechanism and determined by the gestational age of the fetus. Prolonging fetal globin gene expression is of considerable interest for therapeutic potential in diseases caused by abnormal 尾 -globin genes. Butyric acid, which is found in increased plasma concentrations in infants of diabetic mothers who have delayed globin gene switching, was infused into catheterized fetal lambs in utero during the time of the normal globin gene switch period. The globin gene switch was significantly delayed in three of four butyrate-treated fetuses compared with controls and was entirely prevented in one fetus in whom the infusion was begun before the globin switch was under way. These data provide a model for investigating and arresting the biologic clock of hemoglobin switching.
URLPMID:28776729 [本文引用: 1]
High fetal hemoglobin (HbF, α2 γ2 ) levels ameliorate the clinical manifestations of sickle cell disease and β thalassemia. The mechanisms that repress HbF expression and silence γ-globin genes in adults are incompletely characterized and only a single HbF inducer, hydroxyurea, is approved for treatment, and only in patients with sickle cell disease. We identified SIRT1, a protein deacetylase, as a new inducer of γ-globin. SIRT1 knockdown decreased, while SIRT1 ectopic expression upregulated γ-globin gene (HBG) expression in primary human erythroid cells and in K562 cells. The small molecule SIRT1 activators SRT2104 and SRT1720 enhanced HBG expression in cord blood human erythroblasts and reactivated silenced HBG in adult human erythroblasts. Furthermore, SIRT1 binds in the β-globin gene cluster locus control region (LCR) and HBG promoters, promotes the looping of the LCR to HBG promoter, and increases the binding of RNA polymerase II and H4K16Ac in the HBG promoter. SIRT1 suppressed the expression of the HBG suppressors BCL11A, KLF1, HDAC1 and HDAC2. Lastly, SIRT1 did not change the proliferation of human erythroid progenitor cells or the expression of differentiation marker CD235a. These data suggest that SIRT1 activates HBG expression through facilitating LCR looping to the HBG promoter, inhibiting the expression of transcriptional suppressors of HBG, and indirectly increasing histone acetylation in the HBG promoter. SIRT1 is a potential therapeutic target for γ-globin gene induction, and small molecule SIRT1 activators might serve as a lead compound for the development of new HbF inducers.
URLPMID:9859997 [本文引用: 1]
Modification of histones, DNA-binding proteins found in chromatin, by addition of acetyl groups occurs to a greater degree when the histones are associated with transcriptionally active DNA. A breakthrough in understanding how this acetylation is mediated was the discovery that various transcriptional co-activator proteins have intrinsic histone acetyltransferase activity (for example, Gcn5p, PCAF, TAF(II)250 and p300/CBP. These acetyltransferases also modify certain transcription factors (TFIIEbeta, TFIIF, EKLF and p53). GATA-1 is an important transcription factor in the haematopoietic lineage and is essential for terminal differentiation of erythrocytes and megakaryocytes. It is associated in vivo with the acetyltransferase p300/CBP. Here we report that GATA-1 is acetylated in vitro by p300. This significantly increases the amount of GATA-1 bound to DNA and alters the mobility of GATA-1-DNA complexes, suggestive of a conformational change in GATA-1. GATA-1 is also acetylated in vivo and acetylation directly stimulates GATA-1-dependent transcription. Mutagenesis of important acetylated residues shows that there is a relationship between the acetylation and in vivo function of GATA-1. We propose that acetylation of transcription factors can alter interactions between these factors and DNA and among different transcription factors, and is an integral part of transcription and differentiation processes.
URLPMID:15273251 [本文引用: 1]
Acetylation provides one mechanism by which the functional diversity of individual transcription factors can be expanded. This is valuable in the setting of complex multigene loci that are regulated by a limited number of proteins, such as the human -globin locus. We have studied the role of acetylation in the regulation of the transcription factor NF-E4, a component of a protein complex that facilitates the preferential expression of the human -globin genes in fetal erythroid cells. We have shown that NF-E4 interacts directly with, and serves as a substrate for, the acetyltransferase co-activator PCAF. Acetylation of NF-E4 is restricted to a single residue (Lys) in the amino-terminal domain of the protein and results in two important functional consequences. Acetylation of NF-E4 prolongs the protein half-life by preventing ubiquitin-mediated degradation. This stabilization is PCAF-dependent, since enforced expression in fetal/erythroid cells of a mutant form of PCAF lacking the histone acetyltransferase domain (PCAF HAT) decreases NF-E4 stability. Acetylation of Lysalso reduces the interaction between NF-E4 and HDAC1, potentially maximizing the activating ability of the factor at the -promoter. These results provide further demonstration that co-activators, such as PCAF, can influence individual transcription factor properties at multiple levels to alter their effects on gene expression.
URLPMID:4263369 [本文引用: 1]
Current chemotherapeutic and butyrate therapeutics that induce fetal hemoglobin expression generally also suppress erythropoiesis, limiting the production of cells containing fetal hemoglobin (F cells). Recently, selected short-chain fatty acid derivatives (SCFADs) were identified that induce endogenous gamma -globin expression in K562 cells and human burst-forming units-erythroid and that increase proliferation of human erythroid progenitors and a multilineage interleukin-3-dependent hematopoietic cell line. In this report, gamma - globin inducibility by these SCFADs was further demonstrated in mice transgenic for the locus control region and the entire beta -globin gene locus in a yeast artificial chromosome and in 2 globin promoter-reporter assays. Conditioned media experiments strongly suggest that their proliferative activity is a direct effect of the test compounds. Investigation of potential mechanisms of action of these SCFADs demonstrates that these compounds induce prolonged expression of the growth-promoting genes c-myb and c-myc. Both butyrate and specific growth-stimulatory SCFADs induced prolonged signal transducer and activator of transcription (STAT)-5 phosphorylation and activation, and c-cis expression, persisting for more than 120 minutes, whereas with IL-3 alone phosphorylation disappeared within minutes. In contrast to butyrate treatment, the growth-stimulating SCFADs did not result in bulk histone H4 hyperacetylation or induction of p21(Waf/Cip), which mediates the suppression of cellular growth by butyrate. These findings suggest that the absence of bulk histone hyperacetylation and p21 induction, but prolonged induction of cis, myb, myc, and STAT-5 activation, contribute to the cellular proliferation induced by selected SCFADs. (Blood. 2001;97:3259-3267) (C) 2001 by The American Society of Hematology.
URLPMID:12393499 [本文引用: 2]
Pharmacologic stimulation of fetal hemoglobin (HbF) expression may be a promising approach for the treatment of beta-thalassemia. In this study, we have investigated the HbF-inducing activity and molecular mechanisms of specific histone deacetylase (HDAC) inhibitors in human K562 erythroleukemia cells. Apicidin was the most potent agent compared with other HDAC inhibitors (trichostatin A, MS-275, HC-toxin, suberoylanilide hydroxamic acid [SAHA]) and previously tested compounds (butyrate, phenylbutyrate, isobutyramide, hydroxyurea, 5-aza-cytidine), leading to a 10-fold stimulation of HbF expression at nanomolar to micromolar concentrations. Hyperacetylation of histones correlated with the ability of HDAC inhibitors to stimulate HbF synthesis. Furthermore, analysis of different mitogen-activated protein (MAP) kinase signaling pathways revealed that p38 signaling was activated following apicidin treatment of cells and that inhibition of this pathway abolished the HbF-inducing effect of apicidin. Additionally, activation of the Agamma-globin promoter by apicidin could be inhibited by p38 inhibitor SB203580. In summary, the novel HDAC inhibitor apicidin was found to be a potent inducer of HbF synthesis in K562 cells. The present data outline the role of histone hyperacetylation and p38 MAP kinase signaling as molecular targets for pharmacologic stimulation of HbF production in erythroid cells.
URL [本文引用: 1]
URLPMID:20495075 [本文引用: 1]
Abstract Defining the molecular mechanisms underpinning fetal (gamma) globin gene silencing may provide strategies for reactivation of gamma-gene expression, a major therapeutic objective in patients with beta-thalassemia and sickle cell disease (SCD). We have previously demonstrated that symmetric methylation of histone H4 Arginine 3 (H4R3me2s) by the protein arginine methyltransferase PRMT5 is required for recruitment of the DNA methyltransferase DNMT3A to the gamma-promoter, and subsequent DNA methylation and gene silencing. Here we show in an erythroid cell line, and in primary adult erythroid progenitors that PRMT5 induces additional repressive epigenetic marks at the gamma-promoter through the assembly of a multiprotein repressor complex containing the histone modifying enzymes SUV4-20h1, casein kinase 2alpha (CK2alpha), and components of the nucleosome remodeling and histone deacetylation complex. Expression of a mutant form of PRMT5 lacking methyltransferase activity or shRNA-mediated knockdown of SUV4-20h1 resulted in loss of complex binding to the gamma-promoter, reversal of both histone and DNA repressive epigenetic marks, and increased gamma-gene expression. The repressive H4K20me3 mark induced by SUV4-20h1 is enriched on the gamma-promoter in erythroid progenitors from adult bone marrow compared with cord blood, suggesting developmental specificity. These studies define coordinated epigenetic events linked to fetal globin gene silencing, and provide potential therapeutic targets for the treatment of beta-thalassemia and SCD.
URLPMID:19234465 [本文引用: 1]
Mammalian gene silencing is established through methylation of histones and DNA, although the order in which these modifications occur remains contentious. Using the human beta-globin locus as a model, we demonstrate that symmetric methylation of histone H4 arginine 3 (H4R3me2s) by the protein arginine methyltransferase PRMT5 is required for subsequent DNA methylation. H4R3me2s serves as a direct binding target for the DNA methyltransferase DNMT3A, which interacts through the ADD domain containing the PHD motif. Loss of the H4R3me2s mark through short hairpin RNA-mediated knockdown of PRMT5 leads to reduced DNMT3A binding, loss of DNA methylation and gene activation. In primary erythroid progenitors from adult bone marrow, H4R3me2s marks the inactive methylated globin genes coincident with localization of PRMT5. Our findings define DNMT3A as both a reader and a writer of repressive epigenetic marks, thereby directly linking histone and DNA methylation in gene silencing. (c) 2009 Nature America, Inc. All rights reserved.
URLPMID:3599103 [本文引用: 1]
Pharmacologic reactivation of fetal hemoglobin expression is a promising strategy for treatment of sickle cell disease and β-thalassemia. The objective of this study was to investigate the effect of the methyl transferase inhibitor adenosine-2’,3’-dialdehyde (Adox) on induction of human fetal hemoglobin (HbF) in K562 cells and human hematopoietic progenitor cells. Expression levels of human fetal hemoglobin were assessed by northern blot analysis and Real-time PCR. HbF and adult hemoglobin (HbA) content were analyzed using high-performance liquid chromatography (HPLC). DNA methylation levels on human gamma-globin gene promoters were determined using Bisulfite sequence analysis. Enrichment of histone marks on genes was assessed by chromosome immunoprecipitation (ChIP). Adox induced γ-globin gene expression in both K562 cells and in human bone marrow erythroid progenitor cells through a mechanism potentially involving inhibition of protein arginine methyltransferase 5 (PRMT5). The ability of methyl transferase inhibitors such as Adox to efficiently reactivate fetal hemoglobin expression suggests that these agents may provide a means of reactivating fetal globin expression as a therapeutic option for treating sickle cell disease and β-thalassemia.
URLPMID:20688955 [本文引用: 1]
Abstract An estimated 6% to 7% of the earth's population carries a mutation affecting red blood cell function. The 0205-thalassemias and sickle cell disease are the most common monogenic disorders caused by these mutations. Increased levels of 0206-globin ameliorate the severity of these diseases because fetal hemoglobin (HbF; 02±202062) can effectively replace adult hemoglobin (HbA; 02±202052) and counteract polymerization of sickle hemoglobin (HbS; 02±20205(S)2). Therefore, understanding the molecular mechanism of globin switching is of biologic and clinical importance. Here, we show that the recently identified chromatin factor Friend of Prmt1 (FOP) is a critical modulator of 0206-globin gene expression. Knockdown of FOP in adult erythroid progenitors strongly induces HbF. Importantly, 0206-globin expression can be elevated in cells from 0205-thalassemic patients by reducing FOP levels. These observations identify FOP as a novel therapeutic target in 0205-hemoglobinopathies.
URL [本文引用: 1]
Histone modifications play an important role in the process of transcription. However, in contrast to lysine methylation, the role of arginine methylation in chromatin structure and transcription has been underexplored. The globin genes are regulated by a highly organized chromatin structure that juxtaposes the locus control region (LCR) with downstream globin genes. We report here that the targeted recruitment of asymmetric dimethyl H4R3 catalyzed by PRMT1 (protein arginine methyltransferase 1) facilitates histone H3 acetylation on Lys9/Lys14. Dimethyl H4R3 provides a binding surface for P300/CBP-associated factor (PCAF) and directly enhances histone H3 acetylation in vitro. We show that these active modifications are essential for efficient interactions between the LCR and the maj-promoter as well as transcription of the -globin gene. Furthermore, knockdown (KD) of PRMT1 by RNA interference in erythroid progenitor cells prevents histone acetylation, enhancer and promoter interaction, and recruitment of transcription complexes to the active -globin promoter. Reintroducing rat PRMT1 into the PRMT1 KD MEL cells rescues PRMT1 binding, -globin transcription, and erythroid differentiation. Taken together, our data suggest that PRMT1-mediated dimethyl H4R3 facilitates histone acetylation and enhancer/promoter communications, which lead to the efficient recruitment of transcription preinitiation complexes to active promoters.
URLPMID:23576758 [本文引用: 1]
Abstract Reactivation of fetal hemoglobin (HbF) in adults ameliorates the severity of the common 0205-globin disorders. The transcription factor BCL11A is a critical modulator of hemoglobin switching and HbF silencing, yet the molecular mechanism through which BCL11A coordinates the developmental switch is incompletely understood. Particularly, the identities of BCL11A cooperating protein complexes and their roles in HbF expression and erythroid development remain largely unknown. Here we determine the interacting partner proteins of BCL11A in erythroid cells by a proteomic screen. BCL11A is found within multiprotein complexes consisting of erythroid transcription factors, transcriptional corepressors, and chromatin-modifying enzymes. We show that the lysine-specific demethylase 1 and repressor element-1 silencing transcription factor corepressor 1 (LSD1/CoREST) histone demethylase complex interacts with BCL11A and is required for full developmental silencing of mouse embryonic 0205-like globin genes and human 0206-globin genes in adult erythroid cells in vivo. In addition, LSD1 is essential for normal erythroid development. Furthermore, the DNA methyltransferase 1 (DNMT1) is identified as a BCL11A-associated protein in the proteomic screen. DNMT1 is required to maintain HbF silencing in primary human adult erythroid cells. DNMT1 haploinsufficiency combined with BCL11A deficiency further enhances 0206-globin expression in adult animals. Our findings provide important insights into the mechanistic roles of BCL11A in HbF silencing and clues for therapeutic targeting of BCL11A in 0205-hemoglobinopathies.
URLPMID:5512162 [本文引用: 1]
Abstract Enhanced fetal γ-globin synthesis alleviates symptoms of β-globinopathies such as sickle cell disease and β-thalassemia, but current γ-globin-inducing drugs offer limited beneficial effects. We show here that lysine-specific demethylase 1 (LSD1) inhibition by RNAi in human erythroid cells or by the monoamine oxidase inhibitor tranylcypromine in human erythroid cells or β-type globin-transgenic mice enhances γ-globin expression. LSD1 is thus a promising therapeutic target for γ-globin induction, and tranylcypromine may serve as a lead compound for the development of a new γ-globin inducer.
URLPMID:18263785
Comment on Blood. 2008 Jan 1;111(1):411-20.
URLPMID:24371119
Abstract The clinical symptoms of hemoglobin disorders such as 0205-thalassemia and sickle cell anemia are significantly ameliorated by the persistent expression of 0206-globin after birth. This knowledge has driven the discovery of important regulators that silence 0206-globin postnatally. Improved understanding of the 0206- to 0205-globin switching mechanism holds the key to devising targeted therapies for 0205-hemoglobinopathies. To further investigate this mechanism, we used the murine erythroleukemic (MEL) cell line containing an intact 183-kb human 0205-globin locus, in which the (G)0206- and 0205-globin genes are replaced by DsRed and eGFP fluorescent reporters, respectively. Following RNA interference (RNAi)-mediated knockdown of two key transcriptional regulators, Myb and BCL11A, we observed a derepression of 0206-globin, measured by DsRed fluorescence and qRT-PCR (P<0.001). Interestingly, double knockdown of Myb and DNA methyltransferase 1 (DNMT1) resulted in a robust induction of 0208-globin, (up to 20% of total 0205-like globin species) compared to single knockdowns (P<0.001). Conversely, double knockdowns of BCL11A and DNMT1 enhanced 0206-globin expression (up to 90% of total 0205-like globin species) compared to single knockdowns (P<0.001). Moreover, following RNAi treatment, expression of human 0205-like globin genes mirrored the expression levels of their endogenous murine counterparts. These results demonstrate that Myb and BCL11A cooperate with DNMT1 to achieve developmental repression of embryonic and fetal 0205-like globin genes in the adult erythroid environment.
URLPMID:26320100
Abstract Fetal hemoglobin (HbF, 02±202062) induction is a well-validated strategy for sickle cell disease (SCD) treatment. Using a small-molecule screen, we found that UNC0638, a selective inhibitor of EHMT1 and EHMT2 histone methyltransferases, induces 0206-globin expression. EHMT1/2 catalyze mono- and dimethylation of lysine 9 on histone 3 (H3K9), raising the possibility that H3K9Me2, a repressive chromatin mark, plays a role in silencing 0206-globin expression. In primary human adult erythroid cells, UNC0638 and EHMT1 or EHMT2 short hairpin RNA-mediated knockdown significantly increased 0206-globin expression, HbF synthesis, and the percentage of cells expressing HbF. At effective concentrations, UNC0638 did not alter cell morphology, proliferation, or erythroid differentiation of primary human CD34(+) hematopoietic stem and progenitor cells in culture ex vivo. In murine erythroleukemia cells, UNC0638 and Ehmt2 CRISPR/Cas9-mediated knockout both led to a marked increase in expression of embryonic 0205-globin genes Hbb-0208y and Hbb-0205h1. In primary human adult erythroblasts, chromatin immunoprecipitation followed by sequencing analysis revealed that UNC0638 treatment leads to genome-wide depletion in H3K9Me2 and a concomitant increase in the activating mark H3K9Ac, which was especially pronounced at the 0206-globin gene region. In RNA-sequencing analysis of erythroblasts, 0206-globin genes were among the most significantly upregulated genes by UNC0638. Further increase in 0206-globin expression in primary human adult erythroid cells was achieved by combining EHMT1/2 inhibition with the histone deacetylase inhibitor entinostat or hypomethylating agent decitabine. Our data provide genetic and pharmacologic evidence that EHMT1 and EHMT2 are epigenetic regulators involved in 0206-globin repression and represent a novel therapeutic target for SCD. 0008 2015 by The American Society of Hematology.
URL
URLPMID:11121052
We have defined the histone acetylation pattern of the endogenous murine beta-globin domain, which contains the erythroidspecific beta-globin genes. The beta-globin locus control region (LCR) and transcriptionally active promoters were enriched in acetylated histones in fetal liver relative to fetal brain, whereas the inactive promoters were hypoacetylated. In contrast, the LCR and both active and inactive promoters were hyperacetylated in yolk sac. Hypersensitive site two of the LCR was also hyperacetylated in murine embryonic stem cells, whereas beta-globin promoters were hypoacetylated. Thus, the acetylation pattern varied at different developmental stages. Histone deacetylase inhibition selectively increased acetylation at a hypoacetylated promoter in fetal liver, suggesting that active deacetylation contributes to silencing of promoters. We propose that dynamic histone acetylation and deacetylation play an important role in the developmental control of beta-globin gene expression.
URL
BACKGROUND: Histone H3 lysine 4 (K4) methylation has been linked with transcriptional activity in mammalian cells. The WD40-repeat protein, WDR5, is an essential component of the MLL complex that induces histone H3 K4 methylation, but the role of WDR5 in human globin gene regulation has not yet been established. DESIGN AND METHODS: To study the role of WDR5 in human globin gene regulation, we performed knockdown experiments in both K562 cells and primary human bone marrow erythroid progenitor cells (BMC). The effects of WDR5 knockdown on gamma-globin gene expression were determined. Biochemical approaches were also employed to investigate WDR5 interaction molecules. Chromosomal marks in the globin locus were analyzed by ChIP. RESULTS: We found that WDR5 interacted with protein arginine methyltransferase 5 (PRMT5), a known repressor of gamma-globin gene expression, and was essential for generating tri-methylated H3K4 (H3K4me3) at the gamma-globin promoter in K562 cells. Enforced expression of WDR5 in K562 cells reduced gamma-globin gene expression, whereas knockdown of WDR5 increased gamma-globin gene expression in both K562 cells and primary human bone marrow erythroid progenitor cells. Consistent with this, both histone H3 and H4 acetylation at the gamma-globin promoter were increased, while histone H4R3 and H3K9 methylation were decreased, in WDR5 knockdown cells compared to controls. We found that WDR5 interacted with HDAC1 and a PHD domaincontaining protein, ING2 (inhibitor of growth), an H3K4me3 mark reader, to enhance gamma-globin gene transcriptional repression. In human BMC, levels of WDR5 were highly enriched on the gamma-promoter relative to levels on other globin promoters and compared to the gamma-promoter in cord blood erythroid progenitors, suggesting that WDR5 is important in the developmental globin gene expression program. CONCLUSIONS: Our data are consistent with a model in which WDR5 binds the gamma-globin promoter in a PRMT5-dependent manner; H3K4me3 induced at the gamma-globin promoter by WDR5 may result in the recruitment of the ING2-associated HDAC1 component and consequent silencing of gamma-globin gene expression.
URL [本文引用: 1]
URLPMID:15838670 [本文引用: 1]
Pharmacological agents such as hydroxyurea (HU) have been known to cause induction of fetal hemoglobin and possibly may alleviate the symptoms in thalassemia intermedia patients. Thirty-seven patients with β-thalassemia intermedia were enrolled to assess response to HU therapy. Major response was defined as transfusion independence or hemoglobin rise of more than 2002g/l and minor response as rise in hemoglobin of 10–2002g/l or reduction in transfusion frequency by 50%. The median age was 10 years (range: 4–50 years) and median follow-up was 12 months (range: 4–36 months). Twenty-six patients (70.2%) showed response to HU therapy. Seventeen patients (45.9%) were major responders, and nine patients (24.3%) showed minor response. There was no correlation of response with β-thalassemia mutation or Xmn I polymorphism; however, the presence of α 3.7 deletion was associated with major response in three patients. Mean fetal hemoglobin (HbF) levels rose on HU therapy. Older age, low baseline F cell percent, and low baseline HbF levels (below 10%) were predictors of poor response. Response was evident within 1 month of starting HU therapy in the majority of responders. Thus, a short trial of HU therapy can predict durable response.
URL [本文引用: 1]
Hydroxyurea (HU), the first of two drugs approved by the US Food and Drug Administration for treating patients with sickle cell disease (SCD), produces anti-sickling effect by re-activating fetal -globin gene to enhance production of fetal hemoglobin. However, approximately 30% of the patients do not respond to HU therapy. The molecular basis of non-responsiveness to HU is not clearly understood. To address this question, we examined HU-induced changes in the RNA and protein levels of transcription factors NF-Y, GATA-1, -2, BCL11A, TR4, MYB and NF-E4 that assemble the -globin promoter complex and regulate transcription of -globin gene. In erythroblasts cultured from peripheral blood CD34+cells of patients with SCD, we found that HU-induced changes in the protein but not the RNA levels of activator GATA-2 and repressors GATA-1, BCL11A and TR4 correlated with HU-induced changes in fetal hemoglobin (HbF) levels in the peripheral blood of HU high and low responders. However, HU did not significantly induce changes in the protein or RNA levels of activators NF-Y and NF-E4. Based on HU-induced changes in the protein levels of GATA-2, -1 and BCL11A, we calculated an Index of Hydroxyurea Responsiveness (IndexHU-3). Compared to the HU-induced fold changes in the individual transcription factor protein levels, the numerical values of IndexHU-3 statistically correlated best with the HU-induced peripheral blood HbF levels of the patients. Thus, IndexHU-3 can serve as an appropriate indicator for inherent HU responsiveness of patients with SCD.
URLPMID:10828048 [本文引用: 1]
Abstract Hydroxyurea (HU) is an effective therapeutic agent for patients with myeloproliferative disorders (MPDs) or sickle cell disease (SCD). Short-term HU toxicities primarily include transient myelosuppression, but long-term HU risks have not been defined. The mutagenic and carcinogenic potential of HU is not established, although HU has been associated with an increased risk of leukemia in some patients with MPD. In this study, 2 assays were used to quantitate acquired somatic DNA mutations in peripheral blood mononuclear cells (PBMCs) after in vivo HU exposure. The HPRT assay measures hypoxanthine phosphoribosyl transferase (hprt) mutations, while the VDJ assay identifies "illegitimate" T-cell receptor Vgamma-Jbeta interlocus recombination events. PBMCs were analyzed from patients with MPD, adults and children with SCD, and normal controls. MPD patients with prolonged HU exposure had numbers of DNA mutations equivalent to patients with low HU exposure or controls. Similarly, adults with SCD had equivalent numbers of DNA mutations regardless of HU exposure. Children with SCD and 30-month HU exposure had equivalent hprt(-) mutations but significantly more VDJ mutations (1.82 +/- 1.20 events per microg DNA) than children with 7-month HU exposure (1.58 +/- 0.87 events) or no HU exposure (1.06 +/- 0.45 events), P =.04 by analysis of variance. Taken together, these data suggest that the mutagenic and carcinogenic potential of in vivo HU therapy is low. Although increased numbers of illegitimate VDJ recombination events do not directly portend leukemia, young patients with SCD and HU exposure should be monitored serially for increases in DNA mutations.
URLPMID:6883509 [本文引用: 1]
We have studied the effect of DNA methylation on human gamma-globin gene expression, using a novel in vitro DNA methylation technique. With this method we have methylated specific segments of the gamma-globin gene and its 5'-flanking region, which were cloned for this purpose in the bacteriophage vector M13mp8. The vitro methylated DNA was introduced into mouse L-cells by cotransfer using the herpes simplex virus thymidine kinase gene as a selective marker. Transformed cell lines were isolated to examine the inheritance of the segmental methylation pattern and its effect on the expression of the gamma-globin gene. Cells transformed with DNA methylated throughout the M13 vector and gamma-globin DNA sequences do not express the gamma-globin gene. Methyl-C residues in either the M13 DNA sequences or the gamma-globin structural gene have, however, no inhibitory effect on globin transcription. In contrast, methylation in the 5' region of the gamma-globin gene (from nucleotides -760 to +100) prevents transcription. These data indicate that DNA methylation in the 5' region of a gene might play a direct role in the regulation of gene expression.
URLPMID:6181507 [本文引用: 1]
Abstract In an attempt to stimulate Hb F synthesis in baboons by means other than erythropoietic stress, we considered the possibility that an agent that inhibits methylation of CpG sequences in DNA may be effective. 5-Azacytidine, a cytosine analogue that cannot be methylated, is such an agent. Animals whose packed red cell volume was maintained at approximately 20% by bleeding were given 10 daily intravenous injections of the drug (6 mg/kg) in 12 days. Hb F levels in these animals started to increase on day 5 of this regimen and peak levels, which were 6-30 times higher than those produced by bleeding alone, occurred 5-7 days after the last dose of the drug. In animals previously identified as genetically "high" or "low" Hb F responders, the maximal Hb F levels were 70-85% and 35-40% respectively. In dose-response studies 5-azacytidine given daily at 3-4 mg/kg produced maximal Hb F increases. The drug did not correlate the percentage (number) of Hb F-containing cells (F cells) beyond the maximal number achieved by bleeding alone and thus its main effect was to increase Hb F per F cell. The finding that Hb F synthesis can be modulated to such a high degree by a drug may have therapeutic implications--e.g., in sickle cell anemia, in which stimulation of Hb F synthesis may prevent sickling.
URL [本文引用: 1]
URLPMID:11001887 [本文引用: 1]
Abstract Augmentation of the fetal hemoglobin (HbF) levels is of therapeutic benefit in patients with sickle cell anemia. Hydroxyurea (HU), by increasing HbF, lowers rates of pain crisis, episodes of acute chest syndrome, and requirements for blood transfusions. For patients with no HbF elevation after HU treatment, augmentation of HbF levels by 5-aza-2'-deoxycytidine (5-aza-CdR, decitabine) could serve as an alternate mode of treatment. Eight adult patients participated in a dose-escalating phase I/II study with 5-aza-CdR at doses ranging from 0.15 to 0.30 mg/kg given 5 days a week for 2 weeks. HbF, F cell, F/F cell, gamma-globin synthesis ratio, complete blood count, and chemistry were measured. The average gamma-globin synthesis relative to non-alpha-globin synthesis prior to therapy was 3.19% +/- 1.43% and increased to 13.66% +/- 4.35% after treatment. HbF increased from 3.55% +/- 2.47% to 13.45% +/- 3.69%. F cells increased from 21% +/- 14.8% to 55% +/- 13.5% and HbF/F cell increased from 17% to 24%. In the HU nonresponders HbF levels increased from 2.28% +/- 1.61% to 2.6% +/- 2.15% on HU, whereas on 5-aza-CdR HbF increased to 12.70% +/- 1.81%. Total hemoglobin increased by 1 g/dL in 6 of 8 patients with only minor reversible toxicities, and all patients tolerated the drug. Maximum HbF was attained within 4 weeks of treatment and persisted for 2 weeks before falling below 90% of the maximum. Therefore 5-aza-CdR could be effective in increasing HbF in patients with sickle cell anemia who failed to increase HbF with HU. Demonstration of sustained F levels with additional treatment cycles without toxicity is currently being performed.
URLPMID:15204104 [本文引用: 1]
Abstract A number of pharmacological agents are currently available for the induction of the fetal hemoglobin (Hb F) to treat the patients with sickle cell disease and beta-thalassemia. In the present review, we summarized the investigation and development of these Hb F-inducing agents and introduced histone deacetylase inhibitors as the new strategy to induce Hb F to treat the hemoglobin disorders Copyright 2004 Taylor and Francis Ltd
URLPMID:17362198 [本文引用: 1]
Histone acetylation regulates many cellular processes, and specific acetylation marks, either singly or in combination, produce distinct outcomes. For example, the acetylation pattern on newly synthesized histones is important for their assembly into nucleosomes by histone chaperones. Additionally, the degree of chromatin compaction and folding may be regulated by acetylation of histone H4 at lysine 16. Histone acetylation also regulates the formation of heterochromatin; deacetylation of H4 lysine 16 is important for spreading of heterochromatin components, whereas acetylation of this site serves as a barrier to this spreading. Finally, histone acetylation is critical for gene transcription, but recent results suggest that deacetylation of certain sites also plays an important role. There are many histone acetyltransferases (HATs) and deacetylases, with differing preferences for the various histone proteins and for specific sites on individual histones. Determining how these enzymes create distinct acetylation patterns and regulate the functional outcome is an important challenge.
URLPMID:12920038 [本文引用: 1]
Abstract We investigated the induction of human gamma globin gene activity by 3 classes of histone deacetylase inhibitors: amide analogues of trichostatin A, hydroxamic acid analogues of trapoxin, and scriptaid and its analogues. The screening consisted of measuring the effects of these compounds on gamma and beta human gene promoter activity by using cultures of GM979 cells stably transfected with a construct containing a gamma promoter linked to firefly luciferase and a beta promoter linked to renilla luciferase. Compounds belonging to all 3 classes induced gamma gene promoter activity in the screening assay in low micromolar concentrations. Histone deacetylase (HDAC) inhibitors increased acetylation of histone H4 and induced the expression of endogenous murine embryonic genes. They also increased the levels of gamma mRNA and the frequency of fetal hemoglobin-containing erythroblasts in erythroid burst-forming unit (BFUe) cultures from healthy adult individuals. Compounds that displayed very similar degrees of inhibition of the HDAC activity in an HDAC enzymatic assay differed strikingly on their effects on gamma gene promoter activity, raising the possibility of selectivity of HDACs that interact with the gamma globin gene chromatin.
URLPMID:15479724 [本文引用: 1]
Fetal hemoglobin (Hb F) levels increase in most patients with sickle cell disease following intermittent butyrate therapy. Although the full effects of butyrate on Hb F levels usually require multiple treatment cycles, in some patients a peak level is achieved after a few days of butyrate therapy. Our investigation of the mechanism(s) responsible for this rapid induction of Hb F by butyrate showed that reticulocyte gamma-globin chain synthesis markedly increased within 24 hours of butyrate exposure, without concomitant changes in reticulocyte gamma-globin mRNA levels. This suggests that butyrate might induce Hb F by increasing the efficiency of translation of gamma-globin mRNA. This hypothesis was confirmed by ribosome loading studies that demonstrated enrichment of the polysomal fraction of reticulocytes with gamma-globin mRNA following butyrate exposure. Thus, the induction of Hb F by butyrate may be mediated by translational effects in addition to its well-known effects on transcription of the gamma-globin genes.
URLPMID:10720693 [本文引用: 1]
Abstract OBJECTIVE: Several agents including hydroxyurea, erythropoietin and butyric acid have been shown to reactivate gamma gene expression during adult stage development by unknown molecular mechanisms. In addition to inhibiting the enzyme histone deacetylase, butyrate may modulate transcription factor binding to specific DNA sequences defined as butyrate response elements (BREs). The purpose of this study was to identify promoter sequences involved in gamma gene activation by butyrate using truncation mutants in stable cell lines. MATERIALS AND METHODS: A detailed analysis of Agamma gene activation in the presence of alpha-aminobutyric acid and sodium butyrate was completed in stable mouse erythroleukemia (MEL) cell pools established with seven Agamma promoter truncation mutants. Functional studies were performed in a transient assay system followed by gel mobility shift assays to define protein binding patterns and to demonstrate transcription factor interactions in the gamma promoter BRE. RESULTS: Agamma promoter analysis in stable MEL cell pools revealed BREs between nucleotide-141 and -201, and nucleotide-822 and -893 (gammaBRE). The gammaBRE required the minimal Agamma promoter (-201 to +36) to stimulate gene expression. We observed a 6.1-fold (p < 0.05) increase in CAT activity for the minimal Agamma promoter alone compared with an 11.5-fold (p < 0.05) increase when the gamma promoter was combined with the -822 to -893 fragment. Protein binding studies demonstrated altered protein-DNA interactions in the gammaBRE after butyrate induction. The pattern for binding observed suggest both negative- and positive-acting transcription factors may interact in this region. CONCLUSION: The data supports the -822 to -893 region as a DNA regulatory element that contributes to Agamma gene inducibility by butyrate.
URLPMID:16550606 [本文引用: 1]
Abstract Top of page Abstract Material and methods Results Discussion Acknowledgements References Curcumin (diferuloylmethane), a polyphenol natural product of the plant Curcuma longa , is undergoing early clinical trials as a novel anticancer agent. However, the anticancer mechanism of curcumin remains to be elucidated. Here we show that curcumin inhibited growth of rhabdomyosarcoma cells (Rh1 and Rh30) (IC 50 = 2–5 μM) and arrested cells in G 1 phase of the cell cycle. Curcumin also induced apoptosis and inhibited the basal or type I insulin-like growth factor-induced motility of the cells. At physiological concentrations (002.5 μM), curcumin rapidly inhibited phosphorylation of the mammalian target of rapamycin (mTOR) and its downstream effector molecules, p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1), in a panel of cell lines (Rh1, Rh30, DU145, MCF-7 and Hela). Curcumin also inhibited phosphorylation of Akt in the cells, but only at high concentrations (>40 μM). The data suggest that curcumin may execute its anticancer activity primarily by blocking mTOR-mediated signaling pathways in the tumor cells. 08 2006 Wiley-Liss, Inc.
URLPMID:15314020 [本文引用: 1]
The evolutionarily conserved checkpoint kinase, TOR (), has emerged as a major effector of and proliferation via the . Work in the last decade clearly demonstrates that TOR controls through a stunning number of downstream targets. Some of the targets are phosphorylated directly by TOR, but many are phosphorylated indirectly. In this review, we summarize some recent developments in this fast-evolving field. We describe both the upstream components of the pathway(s) that activates mammalian TOR () and the downstream targets that affect . We also summarize the roles of in the control of and proliferation, as well as its relevance to and synaptic plasticity.
URLPMID:16939628 [本文引用: 1]
Abstract We studied the effects of rapamycin on cultures of erythroid progenitors derived from the peripheral blood of 10 beta-thalassaemia patients differing widely with respect to their potential to produce foetal haemoglobin (HbF). For this, we employed the two-phase liquid culture procedure for growing erythroid progenitors, high performance liquid chromatography for analysis of HbF production and reverse transcription polymerase chain reaction for quantification of the accumulation of globin mRNAs. The results demonstrated that rapamycin induced an increase of HbF in cultures from all the beta-thalassaemia patients studied and an increase of their overall Hb content/cell. The inducing effect of rapamycin was restricted to gamma-globin mRNA accumulation, being only minor for beta-globin and none for alpha-globin mRNAs. The ability of rapamycin to preferentially increase gamma-globin mRNA content and production of HbF in erythroid precursor cells from beta-thalassaemia patients is of great importance as this agent (also known as sirolimus or rapamune) is already in clinical use as an anti-rejection agent following kidney transplantation. These data suggest that rapamycin warrants further evaluation as a potential therapeutic drug in beta-thalassaemia and sickle cell anaemia.
URLPMID:17606762 [本文引用: 1]
Abstract Allogeneic hematopoietic stem-cell transplantation (HSCT) is the only curative treatment for sickle cell disease (SCD); nevertheless, its use has been limited by the risk of transplantation-related mortality (TRM). Between November 1988 and December 2004, 87 consecutive patients with severe SCD ranging from 2 to 22 years of age received transplants in France. Cerebral vasculopathy was the principal indication for transplantation (55 patients). All the patients received grafts from a sibling donor after a myeloablative conditioning regimen (CR). The only change in the CR during the study period was the introduction of antithymocyte globulin (ATG) in March 1992. The rejection rate was 22.6% before the use of ATG but 3% thereafter. With a median follow-up of 6 years (range, 2.0 to 17.9 years), the overall and event-free survival (EFS) rates were 93.1% and 86.1%, respectively. Graft versus host disease (GVHD) was the main cause of TRM. Importantly, cord blood transplant recipients did not develop GVHD. No new ischemic lesions were detected after engraftment, and cerebral velocities were significantly reduced. The outcome improved significantly with time: the EFS rate among the 44 patients receiving transplants after January 2000 was 95.3%. These results indicate that HLA-identical sibling HSCT after myeloablative conditioning with ATG should be considered as a standard of care for SCD children who are at high risk for stroke.
URLPMID:28377044 [本文引用: 1]
Abstract β-Thalassemia and sickle cell disease (SCD) are the world's two most widely disseminated hereditary hemoglobinopathies. β-Thalassemia originated in the Mediterranean, Middle Eastern, and Asian regions, and SCD originated in central Africa. However, subsequent population migration means that these two diseases are now global and thus constitute a growing health problem in many countries. Despite remarkable improvements in medical care for patients with β-hemoglobinopathies, there is still only one definitive treatment option: allogeneic hematopoietic stem cell (HSC) transplantation. The development of gene therapy for β-hemoglobinopathies has been justified by (1) the limited availability of human leukocyte antigen (HLA)-identical donors, (2) the narrow window of application of HSC transplantation to the youngest patients, and (3) recent advances in HSC-based gene therapy. The huge ongoing efforts in translational medicine and the high number of related publications show that gene therapy has the potential to become the treatment of choice for patients who lack either an HLA genoidentical sibling or an alternative, medically acceptable donor. In this dynamic scientific context, we first summarize the main steps toward clinical translation of02this therapeutic approach and then discuss novel lentiviral- and genome editing-based treatment strategies for β-hemoglobinopathies. Copyright 08 2017 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
URL [本文引用: 1]
Shows that the use of recombinant lentiviruses enables efficient transfer and faithful integration of the human beta-globin gene together with large segments of its locus control region. Speculation that the stable introduction of a functional beta-globin gene in hemotopoietic stem cells could be a powerful approach to treat beta-thalassemia and sickle-cell disease; Findings of experiments with beta-thalassemic mice; Conclusion that such therapy should be of benefit to patients with severe defects in hemoglobin production.
URLPMID:11743206 [本文引用: 1]
Abstract Sickle cell disease (SCD) is caused by a single point mutation in the human betaA globin gene that results in the formation of an abnormal hemoglobin [HbS (alpha2betaS2)]. We designed a betaA globin gene variant that prevents HbS polymerization and introduced it into a lentiviral vector we optimized for transfer to hematopoietic stem cells and gene expression in the adult red blood cell lineage. Long-term expression (up to 10 months) was achieved, without preselection, in all transplanted mice with erythroid-specific accumulation of the antisickling protein in up to 52% of total hemoglobin and 99% of circulating red blood cells. In two mouse SCD models, Berkeley and SAD, inhibition of red blood cell dehydration and sickling was achieved with correction of hematological parameters, splenomegaly, and prevention of the characteristic urine concentration defect.
URLPMID:20844535 [本文引用: 1]
Abstract The 0205-haemoglobinopathies are the most prevalent inherited disorders worldwide. Gene therapy of 0205-thalassaemia is particularly challenging given the requirement for massive haemoglobin production in a lineage-specific manner and the lack of selective advantage for corrected haematopoietic stem cells. Compound 0205(E)/0205(0)-thalassaemia is the most common form of severe thalassaemia in southeast Asian countries and their diasporas. The 0205(E)-globin allele bears a point mutation that causes alternative splicing. The abnormally spliced form is non-coding, whereas the correctly spliced messenger RNA expresses a mutated 0205(E)-globin with partial instability. When this is compounded with a non-functional 0205(0) allele, a profound decrease in 0205-globin synthesis results, and approximately half of 0205(E)/0205(0)-thalassaemia patients are transfusion-dependent. The only available curative therapy is allogeneic haematopoietic stem cell transplantation, although most patients do not have a human-leukocyte-antigen-matched, geno-identical donor, and those who do still risk rejection or graft-versus-host disease. Here we show that, 33 months after lentiviral 0205-globin gene transfer, an adult patient with severe 0205(E)/0205(0)-thalassaemia dependent on monthly transfusions since early childhood has become transfusion independent for the past 21090009months. Blood haemoglobin is maintained between 9 and 10090009g090009dl(-1), of which one-third contains vector-encoded 0205-globin. Most of the therapeutic benefit results from a dominant, myeloid-biased cell clone, in which the integrated vector causes transcriptional activation of HMGA2 in erythroid cells with further increased expression of a truncated HMGA2 mRNA insensitive to degradation by let-7 microRNAs. The clonal dominance that accompanies therapeutic efficacy may be coincidental and stochastic or result from a hitherto benign cell expansion caused by dysregulation of the HMGA2 gene in stem/progenitor cells.
URLPMID:3317563 [本文引用: 1]
The generation of induced pluripotent stem cells (iPSCs) from differentiated somatic cells by over-expression of several transcription factors has the potential to cure many genetic and degenerative diseases currently recalcitrant to traditional clinical approaches. One such genetic disease is β-thalassemia major (Cooley's anemia). This disease is caused by either a point mutation or the deletion of several nucleotides in the β-globin gene, and it threatens the lives of millions of people in China. In the present study, we successfully generated iPSCs from fibroblasts collected from a 2-year-old patient who was diagnosed with a homozygous 41/42 deletion in his β-globin gene. More importantly, we successfully corrected this genetic mutation in the β-thalassemia iPSCs by homologous recombination. Furthermore, transplantation of the genetically corrected iPSCs-derived hematopoietic progenitors into sub-lethally irradiated immune deficient SCID mice showed improved hemoglobin production compared with the uncorrected iPSCs. Moreover, the generation of human β-globin could be detected in the mice transplanted with corrected iPSCs-derived hematopietic progenitors. Our study provides strong evidence that iPSCs generated from a patient with a genetic disease can be corrected by homologous recombination and that the corrected iPSCs have potential clinical uses.
URLPMID:28249145 [本文引用: 1]
Sickle cell disease results from a homozygous missense mutation in the β-globin gene that causes polymerization of hemoglobin S. Gene therapy for patients with this disorder is complicated by the complex cellular abnormalities and challenges in achieving effective, persistent inhibition of polymerization of hemoglobin S. We describe our first patient treated with lentiviral vector-mediated addition of an antisickling β-globin gene into autologous hematopoietic stem cells. Adverse events were consistent with busulfan conditioning. Fifteen months after treatment, the level of therapeutic antisickling β-globin remained high (approximately 50% of β-like-globin chains) without recurrence of sickle crises and with correction of the biologic hallmarks of the disease. (Funded by Bluebird Bio and others; HGB-205 ClinicalTrials.gov number, NCT02151526 .).
URLPMID:19657335 [本文引用: 1]
Abstract The contribution of changes in cis-regulatory elements or trans-acting factors to interspecies differences in gene expression is not well understood. The mammalian beta-globin loci have served as a model for gene regulation during development. Transgenic mice containing the human beta-globin locus, consisting of the linked embryonic (epsilon), fetal (gamma) and adult (beta) genes, have been used as a system to investigate the temporal switch from fetal to adult haemoglobin, as occurs in humans. Here we show that the human gamma-globin (HBG) genes in these mice behave as murine embryonic globin genes, revealing a limitation of the model and demonstrating that critical differences in the trans-acting milieu have arisen during mammalian evolution. We show that the expression of BCL11A, a repressor of human gamma-globin expression identified by genome-wide association studies, differs between mouse and human. Developmental silencing of the mouse embryonic globin and human gamma-globin genes fails to occur in mice in the absence of BCL11A. Thus, BCL11A is a critical mediator of species-divergent globin switching. By comparing the ontogeny of beta-globin gene regulation in mice and humans, we have shown that alterations in the expression of a trans-acting factor constitute a critical driver of gene expression changes during evolution.
URLPMID:24115442 [本文引用: 1]
Abstract Genome-wide association studies (GWASs) have ascertained numerous trait-associated common genetic variants, frequently localized to regulatory DNA. We found that common genetic variation at BCL11A associated with fetal hemoglobin (HbF) level lies in noncoding sequences decorated by an erythroid enhancer chromatin signature. Fine-mapping uncovers a motif-disrupting common variant associated with reduced transcription factor (TF) binding, modestly diminished BCL11A expression, and elevated HbF. The surrounding sequences function in vivo as a developmental stage-specific, lineage-restricted enhancer. Genome engineering reveals the enhancer is required in erythroid but not B-lymphoid cells for BCL11A expression. These findings illustrate how GWASs may expose functional variants of modest impact within causal elements essential for appropriate gene expression. We propose the GWAS-marked BCL11A enhancer represents an attractive target for therapeutic genome engineering for the -hemoglobinopathies.
URL [本文引用: 1]
URLPMID:29168817 [本文引用: 1]
A tour through the most studied genes in biology reveals some surprises.
URL [本文引用: 1]
The erythrocytes of certain individuals possess the capacity to undergo reversible changes in shape in response to changes in the partial pressure of oxygen. When the oxygen pressure is lowered, these cells change their forms from the normal biconcave disk to crescent, holly wreath, and other forms. This process is known as sickling. About 8 percent of American Negroes possess this characteristic; usually they exhibit no pathological consequences ascribable to it. These people are said to have sicklemia, or sickle cell trait. However, about 1 in 40 (4) of these individuals whose cells are capable of sickling suffer from a severe chronic anemia resulting from excessive destruction of their erythrocytes; the term sickle cell anemia is applied to their condition.
URL [本文引用: 1]
URLPMID:3413070 [本文引用: 1]
We have identified a protein present only in erythroid cells that binds to two adjacent sites within an enhancer region of the chicken 尾 -globin locus. Mutation of the sites, so that binding by the factor can no longer be detected in vitro, leads to a loss of enhancing ability, assayed by transient expression in primary erythrocytes. Binding sites for the erythroid-specific factor (Eryf1) are found within regulatory regions for all chicken globin genes. A strong Eryf1 binding site is also present within the enhancer of at least one human globin gene, and proteins from human erythroid cells (but not HeLa cells) bind to both the chicken and the human sites.
URLPMID:8248258 [本文引用: 1]
Abstract Sequential expression of the genes of the human beta-globin locus requires the formation of an erythroid-specific chromatin domain spanning > 200 kb. Regulation of this gene family involves both local interactions with proximal cis-acting sequences and long-range interactions with control elements upstream of the locus. To make it possible to analyze the interactions of cis-acting sequences of the human beta-globin locus in their normal spatial and sequence context, we characterized two yeast artificial chromosomes (YACs) 150 and 230 kb in size, containing the entire beta-globin locus. We have now successfully integrated the 150-kb YAC into the germ line of transgenic mice as a single unrearranged fragment that includes the locus control region, structural genes, and 30 kb of 3' flanking sequences present in the native locus. Expression of the transgenic human beta-globin locus is tissue- and developmental stage-specific and closely follows the pattern of expression of the endogenous mouse beta-globin locus. By using homology-directed recombination in yeast and methods for the purification and transfer of YACs into transgenic mice, it will now be feasible to study the physiological role of cis-acting sequences in specifying an erythroid-specific chromatin domain and directing expression of beta-globin genes during ontogeny.
URLPMID:23533656 [本文引用: 1]
Abstract Transfusion of red blood cells (RBCs) is a standard and indispensable therapy in current clinical practice. In vitro production of RBCs offers a potential means to overcome a shortage of transfusable RBCs in some clinical situations and also to provide a source of cells free from possible infection or contamination by microorganisms. Thus, in vitro production of RBCs may become a standard procedure in the future. We previously reported the successful establishment of immortalized mouse erythroid progenitor cell lines that were able to produce mature RBCs very efficiently. Here, we have developed a reliable protocol for establishing immortalized human erythroid progenitor cell lines that are able to produce enucleated RBCs. These immortalized cell lines produce functional hemoglobin and express erythroid-specific markers, and these markers are upregulated following induction of differentiation in vitro. Most importantly, these immortalized cell lines all produce enucleated RBCs after induction of differentiation in vitro, although the efficiency of producing enucleated RBCs remains to be improved further. To the best of our knowledge, this is the first demonstration of the feasibility of using immortalized human erythroid progenitor cell lines as an ex vivo source for production of enucleated RBCs.
URLPMID:28841410 [本文引用: 1]
Cis-regulatory elements (CREs) are commonly recognized by correlative chromatin features, yet the molecular composition of the vast majority of CREs in chromatin remains unknown. Here, we describe a CRISPR affinity purification in situ of regulatory elements (CAPTURE) approach to unbiasedly identify locus-specific chromatin-regulating protein complexes and long-range DNA interactions. Using an in vivo biotinylated nuclease-deficient Cas9 protein and sequence-specific guide RNAs, we show high-resolution and selective isolation of chromatin interactions at a single-copy genomic locus. Purification of human telomeres using CAPTURE identifies known and new telomeric factors. In situ capture of individual constituents of the enhancer cluster controlling human -globin genes establishes evidence for composition-based hierarchical organization. Furthermore, unbiased analysis of chromatin interactions at disease-associated cis-elements and developmentally regulated super-enhancers reveals spatial features that causally control gene transcription. Thus, comprehensive and unbiased analysis of locus-specific regulatory composition provides mechanistic insight into genome structure and function in development and disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}