 ,1,2
,1,2miR-362 regulates the proliferation and apoptosis of porcine immature Sertoli cells through targeting the ZNF644 gene
Maoliang Ran1,2, Lianhua Dong1,2, Bo Weng1,2, Rong Cao1,2, Fuzhi Peng1,2, Hu Gao1,2, Hui Luo1,2, Bin Chen,1,2第一联系人:
编委: 李明洲
收稿日期:2018-01-29修回日期:2018-03-21网络出版日期:2018-07-20
| 基金资助: |
Received:2018-01-29Revised:2018-03-21Online:2018-07-20
| Fund supported: |
作者简介 About authors
冉茂良,博士,讲师,研究方向:猪的分子遗传学E-mail:
董莲花,硕士研究生,专业方向:动物遗传育种E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1191KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
冉茂良, 董莲花, 翁波, 曹蓉, 彭馥芝, 高虎, 罗荟, 陈斌. miR-362靶向ZNF644基因调控猪未成熟支持细胞的增殖和凋亡. 遗传[J], 2018, 40(7): 572-584 doi:10.16288/j.yczz.18-028
Maoliang Ran, Lianhua Dong, Bo Weng, Rong Cao, Fuzhi Peng, Hu Gao, Hui Luo, Bin Chen.
睾丸中的支持细胞是位于曲细精管管壁上的一种体细胞,通过细胞间紧密连接形成血睾屏障为精子生长发育提供有利环境,分泌雄激素结合蛋白,同时也为精子生长发育提供多种营养因子,从而在精子生长发育过程中扮演着重要的角色[1,2,3]。研究表明,成熟支持细胞不具有增殖能力[4],因此未成熟支持细胞的增殖能力决定着成熟支持细胞的数量,进而制约着成年雄性动物的精子生成能力。研究显示,未成熟支持细胞的增殖受到多种因素的调控,例如蛋白编码基因[5]、非编码基因[6]和信号通路[5]等。
MicroRNA (miRNA)是一类长约22 nt的非编码小RNA,通过与靶基因3′非翻译区(un-translated region, UTR)的一个或多个位点不紧密的配对结合形成双链RNA,从而诱导靶mRNA降解或抑制其翻译过程。miRNA广泛参与调控细胞的多种生物学过程,例如增殖、凋亡、分化、自噬、迁移等[7,8,9,10]。本课题组前期采用RNA-seq技术在不同发育阶段的猪睾丸组织中鉴定出374个miRNA[11],随后其他课题组相继证实多个miRNA参与调控支持细胞的增殖和凋亡。如miR-133b[12]、miR-762[13]、miR-1285[14]、miR-301b-3p[15]和miR-3584-5p[15]促进支持细胞的增殖而抑制其凋亡,miR-638促进支持细胞凋亡而抑制其增殖[16]。尽管如此,仍有许多miRNA调控猪支持细胞增殖和凋亡的机制尚不清楚。
研究表明,miR-362与miR-500在基因组中成簇位于X染色体上,形成X-linked miRNA,而X-linked miRNA的快速进化及表达特异性与多种哺乳动物雄性生殖功能密切相关[17]。miR-362-5p通过靶向TUBB (tubulin beta)基因在非梗阻性无精子症过程中具有重要的调控作用[18]。此外,RNA-seq结果显示miR-362在60胎龄至30日龄的猪睾丸组织中的表达水平明显高于60~180日龄[11],结合支持细胞的增殖特点,推测miR-362在猪未成熟支持细胞的增殖过程中具有调控作用。因此,本研究利用双荧光素酶报告基因检测技术、实时荧光定量PCR (quantitativereal time PCR, qRT-PCR)技术、流式细胞术、CCK8 (cell counting kit-8)和EdU (5-ethynyl-2°- deoxyuridine)试剂盒研究了miR-362调控猪未成熟支持细胞增殖和凋亡的作用机制,为进一步揭示miR- 362调节猪睾丸发育或精子生成的机制奠定基础。
1 材料和方法
1.1 实验动物及样品采集
沙子岭公猪(Sus scrofa)由湖南省湘潭市沙子岭猪资源场提供,包括1日龄(D1)、30日龄(D30)、60日龄(D60)、90日龄(D90)、120日龄(D120)、150日龄(D150)、180日龄(D180)各3头。以人工阉割法取其两侧睾丸组织,经预冷的磷酸盐缓冲液(0.01 mol/L)快速漂洗并沥去溶液后转入1.5 mL RNase-free离心管,置于液氮中速冷5 h,样品置于-80℃保存。1.2 细胞系
猪睾丸细胞系(swine testicular, ST) ATCC? CRL-1746?购自武汉博士德生物科技有限公司。该细胞系已被证实是猪睾丸组织中未成熟支持细胞,并广泛应用于相关研究[13, 16, 19]。1.3 靶基因预测
利用miRWalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html)、RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/submission.html)、TargetScan (http://www.targetscan.org/)和miRanda (http://www.microrna.org/)网站分别预测人(Homo sapiens)和小鼠(Mus musculus)的ZNF644基因3′UTR与miRNA-362的结合位点。随后,利用NCBI中BLAST功能分析此结合位点在猪、人、黑猩猩(Pan troglodytes)、恒河猴(Macacamulatta)、小鼠、牛(Bostraurus)、绵羊(Ovisaries)、狗(Canis lupus familiari)、猫(Felis catus)和大象(Elephantide)10种哺乳类动物中的保守性。1.4 细胞培养
细胞培养液按胎牛血清(PAN-Biotech,德国)∶DMEM high glucose培养基(Hyclone,美国)为1∶9比例配置,并将ST细胞置于37℃、5% CO2的培养箱中培养。1.5 双荧光素酶基因报告载体构建和酶活性检测
参照GenBank中猪ZNF644基因3′UTR序列(NC_010446.5),分别合成正常型和突变型3′UTR序列,其中突变型序列在miR-362预测结合位点中突变2个碱基(CAAGGAUA→CAAGCUUA),两条序列的5′和3′端均分别加上CTCGAG(XhoⅠ内切酶位点)和GCGGCCGC (NotⅠ内切酶位点)。序列由上海铂尚生物股份有限公司合成。将上述两条序列分别克隆至psi-CHECK-2载体(Promega,美国),构建野生型psiCHECK2-ZNF644-WT 3′UTR和突变型psiCHECK2-ZNF644-MT 3′UTR双荧光素酶报告质粒。将ST细胞接种至12孔细胞培养板,当细胞融合度达70%时使用lipofectamine 2000试剂盒(Invitrogen,美国)进行转染。转染分组为:(1) psiCHECK2- ZNF644-WT 3′UTR + miR-362 mimic;(2) psiCHECK2- ZNF644-WT 3′UTR + mimic NC;(3) psiCHECK2- ZNF644-MT 3′UTR + miR-362 mimic;(4) psiCHECK2- ZNF644-MT 3′UTR + mimic NC。每组设置3个平行孔,转染48 h后收集细胞,利用多功能酶标仪(Molecular Devices, spectra max m5e,美国)检测双荧光素酶活性。
1.6 细胞转染
将ST细胞接种至6孔细胞培养板,在融合度达70%时进行转染。为过表达或抑制表达miR-362,采用Lipofectamine 2000试剂分别将miR-362 mimic/ mimic NC (negative control)/miR-362 inhibitor/inhibitor NC (300 ng/μL,苏州吉玛基因股份有限公司)转染细胞。为抑制表达ZNF644基因,设计合成3条ZNF644 siRNA序列,转染入ST细胞,采用qRT- PCR技术检测抑制效率,挑选出最佳ZNF644 siRNA序列,采用Lipofectamine2000试剂分别将ZNF644 siRNA/siRNA NC转染入ST细胞(300 ng/μL,广州锐博生物科技有限公司)。以上各组细胞转染实验,设置空白对照组,各组设置3个平行孔。将6孔细胞培养板置于37℃、5% CO2培养箱(ThermoFisher, Forma3111,美国)中,5 h后换成不含Penicillin- Streptomycin的培养基,继续培养24 h,收集细胞。1.7 总RNA提取和qRT-PCR
采用TRIzol试剂盒提取不同发育时期的猪睾丸组织样品和ST细胞样品总RNA。采用核酸/蛋白质浓度测定仪检查总RNA质量,要求A260/280介于1.8~2.1之间,rRNA Ratio (28S:18S)≥1.8:1,浓度≥200 ng/μL。采用qRT-PCR技术定量分析miR-362、ZNF644、Bcl2 (B-cell lymphoma-2)、BAX(BCL2 associated X protein)和Caspase-3基因在猪7个不同发育时期的睾丸组织和ST细胞中的表达水平。采用Oligo 6.0软件设计qRT-PCR引物,引物信息见表1,由生工生物工程(上海)股份有限公司合成。分别采用PrimeScriptTM RT试剂盒(TaKaRa公司)和SYBR Premix Ex TaqTM试剂盒(TaKaRa公司)进行cDNA逆转录和qRT-PCR反应,所有操作严格按照试剂盒说明书于IQ-5荧光定量PCR仪(Bio-Rad,美国)上进行。qRT-PCR反应体系为25 μL,包括12.5 μL SYBR Premix Ex TaqTM(2×)、1 μL上游引物、1 μL下游引物、2.0 μL cDNA模板和8.5 μL ddH2O。反应程序:95℃ 30 s;95℃ 5 s,60℃ 30 s,40循环;55~95℃溶解30 s,81个循环。采用GAPDH和U6基因分别作为蛋白编码基因和miR-362的内参基因,每组设置3个生物学重复。
Table 1
表1
表1 引物序列信息
Table 1
| 名称 | 引物序列(5′→3′) |
|---|---|
| miR-362 | RT:GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCACTCA |
| F:TCG GAATCC TTGGAACCTAGGTG | |
| R:ATCCAGTGCAGGGTCCGAGG | |
| ZNF644 | F:CATCCTGGAAGAGCCGATAG |
| R:GAGGATGTTCCGAATGTTGG | |
| Bcl2 | F:GAGGATTGTGGCCTTCTTTG |
| R:GCCGGTTCAGGTACTCAGTC | |
| BAX | F:TCCGGGGAGCAACCCA |
| R:AAACCCTGAAGCAAAAGGGC | |
| Caspase-3 | F:TGGATGCTGCAAATCTCA |
| R:TCCCACTGTCCGTCTCAA | |
| GAPDH | F:GTTTGTGATGGGCGTGAAC |
| R:ATGGACCTGGGTCATGAGT | |
| U6 | F:AACGCTTCACGAATTTGCGT |
| R:CGCTTCACGAATTTGCGTGTCAT |
新窗口打开|下载CSV
1.8 细胞增殖检测
为检测过表达miR-362或抑制表达ZNF644基因对猪未成熟支持细胞增殖的影响,采用流式细胞术检测细胞周期,CCK8 (上海碧云天生物技术有限公司)和EdU试剂盒(广州锐博生物科技有限公司)检测细胞增殖情况。将miR-362 mimic、mimic NC、ZNF644 siRNA或siRNA NC分别转染ST细胞,24 h后收集各组细胞,PBS洗涤2次,使用70%乙醇于4℃过夜,1000 r/min离心3 min后收集细胞,使用流式细胞仪(BD公司,FACSCalibur)检测细胞周期。细胞于96孔板转染24 h、48 h、72 h后,每孔加入10 μL CCK8试剂,置于37℃、5% CO2的培养箱中继续培养1 h,随后使用酶标仪于450 nm波长下检测吸光度(OD)值。细胞于6孔板转染24 h后,每孔加入100 μL EdU试剂,置于37℃、5% CO2的培养箱中孵育2 h,严格按照EdU试剂盒说明书对细胞进行染色,置于荧光显微镜(Nikon,Ti-E,日本)下拍照,并使用Image J软件对图片中的细胞进行计数。
1.9 细胞凋亡检测
为检测过表达miR-362或抑制表达ZNF644基因对猪未成熟支持细胞凋亡的影响,采用Annexin V-FITC/PI方法检测细胞的凋亡率,利用qRT-PCR技术检测细胞凋亡相关基因的表达水平。将miR-362 mimic、mimic NC、ZNF644 siRNA和siRNA NC分别转染ST细胞,24 h后收集细胞于1.5 mL离心管,PBS洗涤2次,2000 r/min离心5 min。每管细胞中分别加入500 μL染色缓冲液和5 μL Annexin V-FITC (江苏凯基生物股份有限公司),混匀后再加入5 μL PI染色液,室温避光静止20 min,使用流式细胞仪检测细胞凋亡情况。利用qRT-PCR技术检测各组细胞中Bcl2、BAX和Caspase-3基因(细胞凋亡相关基因)的表达水平。
1.10 统计分析
采用2-△△Ct公式计算qRT-PCR结果,并用IBM SPSS 22.0软件进行数据统计分析,采用One-Way ANOVA方差进行显著性分析,并采用Duncan氏多重比较法以评估各组之间的差异。数据以平均值±标准差表示。2 结果与分析
2.1 靶基因预测及初步验证
采用qRT-PCR技术检测miR-362和ZNF644基因在沙子岭猪7个不同发育时期睾丸组织中的表达水平。结果显示,miR-362在D1、D30和D180发育时期的表达水平较高,而在D60、D90、D120、D150发育时期的表达水平较低(图1A);而ZNF644基因在7个发育时期呈现出先升高后降低的表达模式(图1B)。采用SPSS 22.0将miR-362和ZNF644基因的表达量进行相关性分析,发现二者呈负相关(r=-0.723,P<0.01)。图1
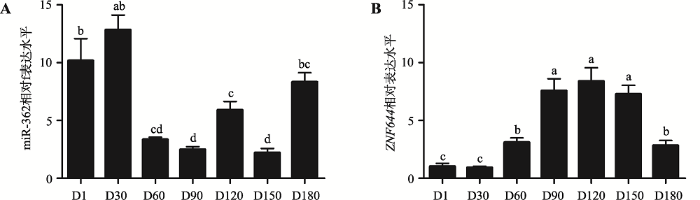 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1miR-362和ZNF644基因在猪不同发育时期睾丸组织中的mRNA表达水平
A:miR-362在猪不同发育时期睾丸组织中的表达水平(U6为内参基因);B:ZNF644基因在猪不同发育时期睾丸组织中的表达水平(GAPDH为内参)。D1~D180表示1日龄、30日龄、60日龄、90日龄、120日龄、150日龄、180日龄的猪睾丸组织。肩标不同字母表示差异显著。
Fig. 1Relative expression levels of miR-362 and ZNF644 gene during the development of porcine testes
采用miRWalk、RNAhybrid、TargetScan和miRanda网站预测miR-362的靶基因,发现ZNF644基因为miR-362的潜在靶基因之一,ZNF644基因3′UTR存在一个与miR-362相结合的潜在位点,且该位点在10种哺乳动物中完全保守(图2),因此推测miR-362靶向ZNF644基因3′UTR。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2miR-362靶基因预测结果和结合位点保守性分析
黑色方框中的序列表示miR-362与ZNF644基因3′UTR的潜在结合位点在物种间完全保守。
Fig. 2The predicted results of miR-362 target genes and conservation analysis of the target site
2.2 双荧光素酶报告基因载体构建和酶活性检测结果
将合成的ZNF644基因3′UTR-WT、3′UTR-MT序列与psiCHECK2载体连接,提取psiCHECK2- ZNF644-WT 3′UTR和psiCHECK2-ZNF644-MT 3′UTR双荧光素酶报告质粒,经NotⅠ和XhoⅠ双酶切,进行凝胶电泳检测(图3,A和B)。结果显示,两个双荧光素酶报告质粒均含有插入的ZNF644基因3′UTR序列片段,表明已成功构建ZNF644基因3′UTR双荧光素酶报告质粒。图3
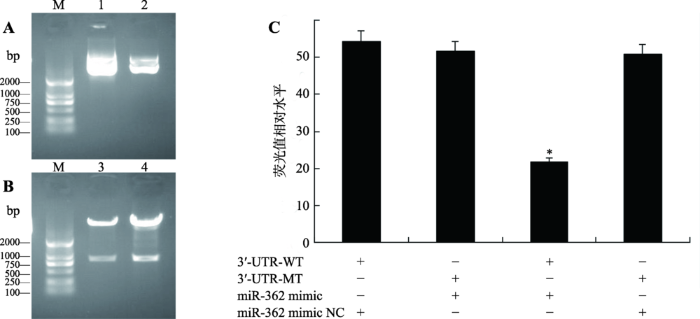 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3psiCHECK2-ZNF644-3′UTR-WT和psiCHECK2-ZNF644-3′UTR-MT质粒验证及双荧光素酶检测结果
A:psiCHECK2-ZNF644-3′UTR-WT和psiCHECK2-ZNF644-3′UTR-MT质粒鉴定;B:采用XhoⅠ和NotⅠ内切酶对psiCHECK2-ZNF644- 3′UTR-WT和psiCHECK2-ZNF644-3′UTR-MT质粒进行双酶切鉴定;C:ST细胞中荧光素酶活性检测miR-362对ZNF644 3′UTR的调控。M:DL2000 marker;1:psiCHECK2-ZNF644-3′UTR-WT重组质粒;2:psiCHECK2-ZNF644-3′UTR-MT重组质粒;3:siCHECK2- ZNF644-3′UTR-MT重组质粒双酶切结果;4:psiCHECK2-ZNF644-3′UTR-WT重组质粒双酶切结果;*P<0.05表示差异显著。
Fig. 3Characterization of psiCHECK2-ZNF644-3′UTR-WT and psiCHECK2-ZNF644-3′UTR-MT plasmid and results of dual-luciferase assays
将psiCHECK2-ZNF644-WT 3′UTR和psiCHECK2-ZNF644-MT 3′UTR与miR-362 mimic和miR- 362 mimic NC两两组合共转染至融合度达70%的ST细胞中,48 h后收集细胞并检测双荧光素酶活性。结果显示,miR-362 mimic + psiCHECK2-ZNF644-WT3′UTR组的荧光素酶活性显著低于其余3组(P<0.05) (图3C),表明miR-362靶向ZNF644基因3′UTR。
2.3 miR-362对猪未成熟支持细胞中ZNF644基因mRNA表达水平的影响
将miR-362 mimic、mimic NC、miR-362 inhibitor、inhibitor NC转染ST细胞中。qRT-PCR检测以上各组细胞中miR-362和ZNF644基因的表达水平。结果显示:miR-362 mimic显著提高miR-362的表达水平(P<0.05)而抑制ZNF644基因的表达水平(P<0.05),miR-362 inhibitor显著抑制miR-362的表达水平(P<0.05)而提高ZNF644基因的表达水平(P<0.05) (图4,A和B),表明miR-362显著抑制ZNF644基因mRNA的表达。图4
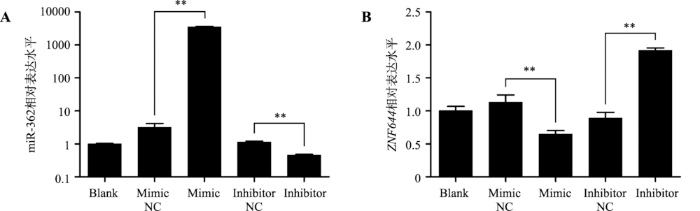 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4miR-362调节ZNF644基因在猪未成熟支持细胞中的表达
A:qRT-PCR技术检测各组细胞中miR-362的表达水平(U6为内参基因);B:qRT-PCR技术检测各组细胞中ZNF644基因的表达水平(GAPDH为内参基因)。** P<0.01表示差异极显著。
Fig. 4miR-362 regulates the expression levels of ZNF644 gene in porcine immature Sertoli cells
2.4 过表达miR-362抑制猪未成熟支持细胞的增殖而促进其凋亡
采用流式细胞术检测转染miR-362 mimic或NC后ST细胞周期的分布情况,结果显示转染miR-222 mimic后,处于G1期的细胞比例显著增加(P<0.05),而处于S期的细胞比例显著减少(P<0.05),表明细胞周期被阻滞在了G1期(图5A)。利用CCK8试剂盒检测转染miR-362 mimic或NC后,ST细胞的增殖情况,结果显示过表达miR-362后,0~72 h内细胞增殖能力极显著减弱(P<0.01) (图5B)。此外,利用EdU试剂盒检测转染miR-362 mimic或NC后,ST细胞的增殖情况,结果表明转染miR-362 mimic 24 h后,处于增殖期的细胞数量显著低于NC转染组(P<0.05) (图5,C和D)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5miR-362抑制猪未成熟支持细胞增殖
A:miR-362对猪未成熟支持细胞周期的影响;B:CCK8检测miR-362对猪未成熟支持细胞增殖的影响;C:EdU检测miR-362对猪未成支持细胞增殖影响的统计结果;D:EdU检测miR-362对猪未成熟支持细胞增殖影响的代表图。*P<0.05表示差异显著,** P<0.01表示差异极显著;蓝色细胞为Hoechst染色,代表所有的活细胞;红色细胞为EdU染色,代表正在增殖的细胞;Overlay为Hoechst染色图和EdU染色图重叠;标尺:50 μm。
Fig. 5miR-362 inhibited porcine immature Sertoli cell proliferation
采用Annexin V-FITC/PI方法检测过表达miR- 362对细胞凋亡的影响(图6,A和B),相比于NC,转染miR-362 mimic后处于凋亡早期和凋亡晚期的细胞比例明显增加,统计结果显示miR-362 mimic细胞转染组的凋亡率显著高于NC组(P<0.05) (图6C)。qRT-PCR检测两组细胞中Bcl2、BAX和Caspase-3基因的表达水平,结果显示过表达miR-362显著增加BAX和Caspase-3基因(细胞凋亡促进基因[20, 21])的表达水平(P<0.05),而对Bcl2基因(细胞凋亡抑制基因[22])的表达水平未见明显影响(图6D)。综合以上实验结果,过表达miR-362抑制猪未成熟支持细胞增殖而促进其凋亡。
图6
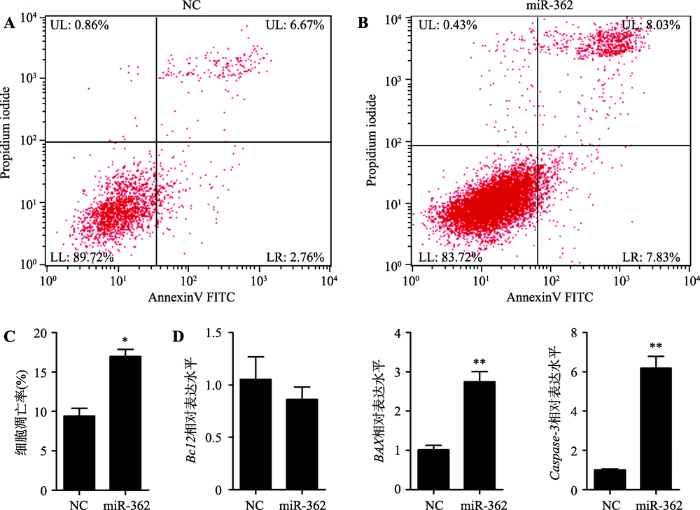 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6miR-362促进猪未成熟支持细胞凋亡
A,B:采用Annexin V-FITC/PI法于流式细胞仪上检测各组细胞凋亡情况;C:流式细胞术检测细胞凋亡情况的统计结果;D:采用qRT-PCR技术检测两组细胞中Bcl2、BAX和Caspase-3基因的表达水平(GAPDH为内参基因)。UL:upper left,为碎片及损失细胞;UR:upper right,为晚期凋亡细胞和死亡细胞;LL:lower left,为正常细胞;LR:lower right,为早期凋亡细胞;*P<0.05表示差异显著,** P<0.01表示差异极显著。
Fig. 6miR-362 promoted porcine immature Sertoli cell apoptosis
2.5 抑制表达ZNF644基因抑制猪未成熟支持细胞增殖而促进其凋亡
为抑制ZNF644基因在猪未成熟支持细胞中的表达,设计合成了3条ZNF644 siRNA序列,并分别转染至猪未成熟支持细胞中。qRT-PCR检测结果显示,3条ZNF644 siRNA序列均能极显著抑制ZNF644基因的表达水平(P<0.01) (图7)。因此,本文将3条ZNF644 siRNA序列等量混合转染至猪未成熟支持细胞以抑制ZNF644基因的表达。流式细胞术检测结果显示,转染ZNF644 siRNA后,处于G2期的细胞比例显著增加(P<0.05),处于S期的细胞比例明显降低,表明细胞被阻滞在了G2期(图8A)。CCK8结果显示,转染ZNF644 siRNA后,0~72 h内细胞增殖能力显著降低(图8B)。此外,EdU染色结果显示,抑制表达ZNF644基因同样显著抑制细胞的增殖能力(图8,C和D)。图7
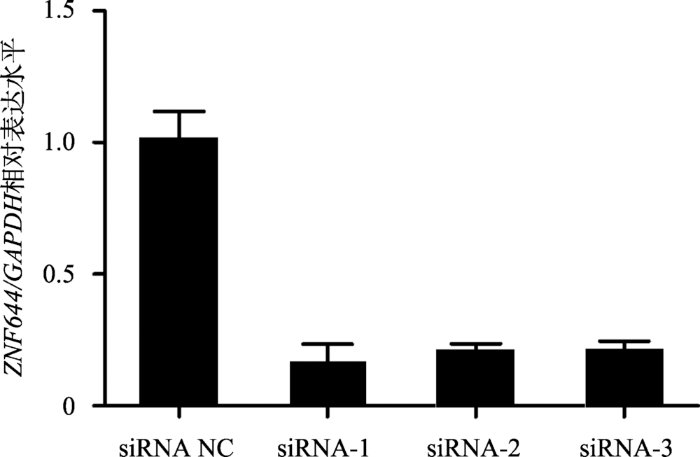 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7ZNF644 siRNA有效抑制ZNF644基因表达水平
设计并合成3条ZNF644 siRNA和1条siRNA NC,分别转染至ST细胞,24 h后收集细胞,采用qRT-PCR技术检测各组细胞中ZNF644基因的表达水平。GAPDH为内参基因,** P<0.01表示差异极显著。
Fig. 7ZNF644 siRNAs effectively suppressed the ZNF644 gene expression
图8
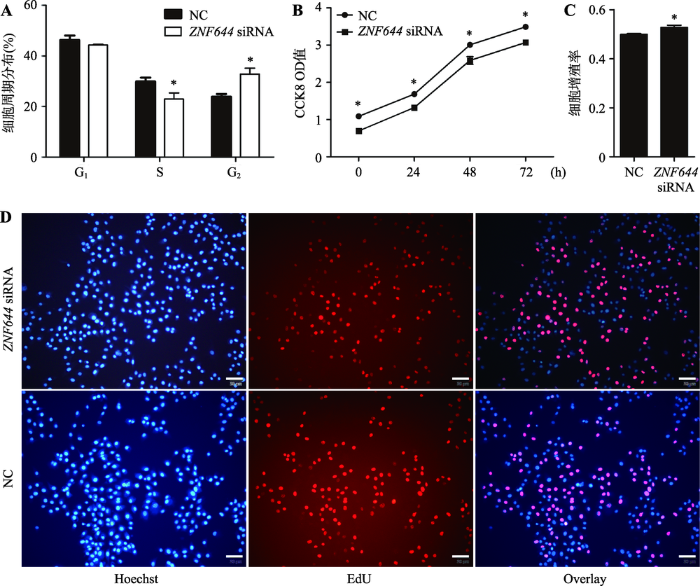 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8ZNF644 siRNA抑制猪未成熟支持细胞增殖
A:利用流式细胞术检测两组细胞中ZNF644基因对猪未成熟支持细胞周期的影响;B:CCK8检测ZNF644基因对猪未成熟支持细胞增殖的影响;C:EdU检测ZNF644基因对猪未成支持细胞增殖影响的统计结果;D:EdU检测ZNF644基因对猪未成熟支持细胞增殖情况。*P<0.05表示差异显著;蓝色细胞为Hoechst染色,代表所有的活细胞;红色细胞为EdU染色,代表正在增殖的细胞;Overlay为Hoechst染色图和EdU染色图重叠;标尺:50 μm。
Fig. 8ZNF644 siRNA inhibited porcine immature Sertoli cell proliferation
采用Annexin V-FITC/PI方法检测抑制表达ZNF644基因对细胞凋亡的影响(图9,A和B),相比于NC,转染ZNF644 siRNA后处于凋亡早期和凋亡晚期的细胞比例明显增加,统计结果显示ZNF644 siRNA细胞转染组的凋亡率显著高于NC组(P<0.05) (图9C)。qRT-PCR检测两组细胞中Bcl2、BAX和Caspase-3基因的表达水平,结果显示抑制表达ZNF644基因显著增加BAX和Caspase-3基因的表达水平(P<0.05),而对Bcl2基因的表达水平显著降低(图9D)。综合以上实验结果,抑制表达ZNF644基因抑制猪未成熟支持细胞增殖而促进其凋亡。
图9
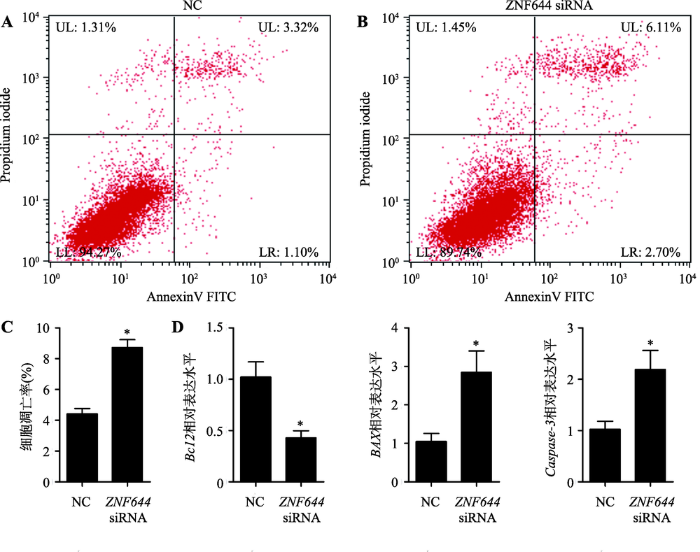 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9ZNF644 siRNA促进猪未成熟支持细胞凋亡
A,B:采用Annexin V-FITC/PI法于流式细胞仪上检测各组细胞的凋亡情况;C:流式细胞术检测细胞凋亡情况的统计结果;D:采用qRT-PCR技术检测两组细胞中ZNF644基因对猪未成熟支持细胞中凋亡相关基因表达水平的影响(GAPDH为内参基因)。UL:upper left,为碎片及损失细胞;UR:upper right,为晚期凋亡细胞和死亡细胞;LL:lower left,为正常细胞;LR:lower right,为早期凋亡细胞;*P<0.05表示差异显著,** P<0.01表示差异极显著。
Fig. 9 ZNF644 siRNA promoted porcine immature Sertoli cell apoptosis
3 讨 论
支持细胞作为雄性动物精子生成过程中的“保姆”细胞,在精子生长发育过程中发挥着重要的作用。近年来,随着RNA-seq技术的发展,在猪睾丸组织中鉴定出大量miRNA[11, 23~25],对miRNA功能研究的不断深入,发现miRNA对猪未成熟支持细胞的增殖和凋亡具有重要的调控作用,如miR-762[13]、miR- 1285[14]和miR-638[16]等。另外,本课题组也证实miR- 26a、miR-10b、miR-34c和miR-222对猪未成熟支持的增殖、凋亡或自噬具有调控作用(结果尚未发表)。在前期研究中,本课题组利用miRNA-mRNA关联分析从不同发育阶段的猪睾丸组织中鉴定出557个miR-362的靶基因[11]。但是,目前关于miR-362的研究多集中于癌细胞。例如,miR-362-5p通过靶向PI3K-C2β (Phosphatidylinositol 3-kinase-C2β)基因抑制神经母细胞瘤细胞的增殖、迁移和侵袭[26];miR- 362-5p通过靶向CYLD基因抑制人乳腺癌MCF7细胞的增殖、迁移和侵袭[27]。此外,CD82[28]、GADD45α[29]和CYLD[30]等基因与miR-362-5p的靶向关系也得到了确定,表明miR-362广泛参与调控细胞生物学过程。为揭示miR-362是否参与调控猪未成熟支持细胞增殖和凋亡,本研究在猪未成熟支持细胞中过表达miR-362后,经流式细胞术、CCK8和EdU试剂盒检测,结果表明miR-362抑制猪未成熟支持细胞增殖。为进一步验证miR-362对猪未成熟支持细胞凋亡的调控作用,本研究采用流式细胞术检测了过表达miR-362后细胞的凋亡情况,结果表明miR-362促进猪未成熟支持细胞凋亡。同时,采用qRT-PCR技术检测了细胞凋亡相关基因的表达水平,包括BCL2、BAX和Caspase-3基因。BCL2和BAX基因作为BCL2基因家族的成员,广泛参与调控细胞凋亡,其中BCL2蛋白位于细胞质与线粒体外膜连接从而发挥抗凋亡作用,而BAX蛋白通过调节线粒体外膜通透性而发挥促凋亡作用[22, 31]。Caspase-3蛋白作为Caspase级联反应中的一个重要的公共凋亡效应因子,通过切割多种蛋白质底物,引起瀑布式的级联反应,进而导致细胞死亡,在细胞凋亡程序中发挥着最后的枢纽作用[21]。本研究结果显示,过表达miR-362后,BCL2基因的表达水平显著降低,而BAX和Caspase-3基因的表达水平显著增加,结合流式细胞术结果证明miR-362促进猪未成熟支持细胞凋亡。
为进一步确定miR-362抑制猪未成熟支持细胞增殖而促进其凋亡的靶基因,本文通过对前期miRNA-mRNA关联分析结果的筛选,并结合多款miRNA靶基因预测软件对miR-362靶基因进行预测的结果,分析预测出ZNF644基因可能为miR-362的靶基因。本研究在验证了miR-362和ZNF644基因在7个发育阶段的猪睾丸组织中的表达水平呈显著负相关的基础上,采用双荧光素酶报告基因系统验证了miR-362在猪支持细胞中靶向ZNF644基因3′UTR,且miR-362抑制ZNF644基因在猪支持细胞中的mRNA表达水平。以上结果表明,miR-362调控猪支持细胞增殖和凋亡可能通过抑制ZNF644基因的表达而得以实现。
锌指蛋白是指含有通过结合Zn2+稳定的、短的、可以自我折叠形成“手指”结构的一类蛋白质,通过与靶分子DNA、RNA、DNA-RNA序列特异性结合,以及与自身或其它锌指蛋白的结合,在转录和翻译水平上调控基因的表达。研究表明,锌指蛋白基因家族成员广泛的表达于动物不同发育阶段的睾丸组织以及精子细胞、间质细胞和支持细胞[32,33,34],提示其在动物睾丸发育和精子生成过程中发挥着重要作用。ZNF644(Zinc finger protein 644)蛋白结构含有多个C2H2结构,为C2H2型锌指蛋白,研究显示ZNF644能与常染色质组蛋白甲基转移酶中的G9a和GLP形成稳定的复合物并共同调控组蛋白H3K9二甲基化,从而改变染色质结构及影响基因转录,且该生物学过程与胚胎形成中细胞分化和增殖相关[35, 36]。尽管如此,目前尚未有关于ZNF644基因在猪未成熟支持细胞中的功能研究报道。本研究不仅将ZNF644基因确定为miR-362的一个靶基因,且证明其表达水平受miR-362的负向调控。在此基础上,本研究设计合成ZNF644 siRNA并转染至猪未成熟支持细胞,实验结果表明抑制表达ZNF644基因可以抑制猪支持细胞增殖而促进其凋亡,该结果与过表达miR-362一致。综上所述,miR-362可能通过靶向ZNF146基因抑制猪未成熟支持细胞增殖而促进其凋亡。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:26732153 [本文引用: 1]

Spermatogenesis is contingent upon hormones and growth factors acting through endocrine and paracrine pathways either in vivo or in vitro. Sertoli cells (SCs) furnish essential factors for the successful advancement of spermatogenesis and spermiogenesis. Moreover, receptors for follicle stimulating hormone (FSH) and testosterone, which are the main hormonal regulators of spermatogenesis, are identified on SCs. Testosterone, FSH and luteinizing hormone are known to determine the destiny of germ cells and in their absence germ cells undergo apoptosis. Bcl-2 family proteins determine one signaling pathway which seems to be crucial for the homeostasis of male gametes. In addition to paracrine signals, germ cell development also relies on signals generated by SCs via direct membrane contact. The regulatory peptide somatostatin has an important role in the regulation of the proliferation of the male germ cells. Activin A, follistatin and FSH control germ cell development. In vitro culture systems have provided initial evidence supporting the achievement of the completion of the first and second male meiotic division in vitro. This review article provides an overview of the literature regarding the hormonal pathways governing spermatogenesis and spermiogenesis.
URL [本文引用: 1]
Results and Problems in Cell Differentiation is an up-to-date book series that presents and explores selected questions of cell and developmental biology. Each volume focuses on a single, well-defined topic. Reviews address basic questions and phenomena, but also provide concise information on the most recent advances. Together, the volumes provide a valuable overview of this exciting and dynamically expanding field. This series is indexed in PubMed.
URLPMID:26556893 [本文引用: 1]
The gap junction protein CONNEXIN43 (CX43) plays a vital role in mammalian spermatogenesis by allowing for direct cytoplasmic communication between neighboring testicular cells. In addition, different publications suggest that CX43 in Sertoli cells (SC) might be important for blood-testis barrier (BTB) formation and BTB homeostasis. Thus, through the use of the Cre-LoxP recombination system, a transgenic mouse line was developed in which only SC are deficient of the Gja1 (gap junction protein, alpha 1) gene. Gja1 codes for the protein CX43. This transgenic mouse line has been commonly defined as the SC specific connexin43 knockout (SCCx43KO) mouse line. Within the seminiferous tubule, SC aid in spermatogenesis by nurturing germ cells (GC) and help them to proliferate and mature. Due to the absence of CX43 within the SC, homozygous KO mice are infertile, have reduced testis size, and mainly exhibit spermatogenesis arrest at the level of spermatogonia, seminiferous tubules containing only SC (SC-only syndrome, SCO) and intratubular SC-clusters. Although the SC specific knockout of CX43 does not seem to have an adverse effect on BTB integrity, CX43 influences BTB composition as the expression pattern of different BTB proteins (like OCCLUDIN, 尾-CATENIN, N-CADHERIN and CLAUDIN11) is altered in mutant males. The supposed roles of CX43 in dynamic BTB regulation, BTB assembly and/or dissassembly and its possible interaction with other junctional proteins composing this unique barrier are discussed. Data collectively indicate that CX43 might represent an important regulator for dynamic BTB formation, composition and function.
URLPMID:18242891 [本文引用: 1]
Abstract Testicular function is under the control of expression and repression of several genes and gene products, and many of these works through Sertoli cells. The capability of Sertoli cells to regulate spermatogenesis is dependent on Sertoli cell functions and Sertoli cell number. Sertoli cell number has long been thought to be stable in adults with no proliferation of Sertoli cells once adult numbers have been reached. However, adult horses do not have stable Sertoli cell numbers, and new studies indicate that adult Sertoli cells can be made to re-enter mitotic phase under certain experimental conditions. This review discusses roles of Sertoli cells in regulation of spermatogenesis and methods for estimating the number of Sertoli cells, in a testis, that overcome the problems (assumptions) associated with the indented, pear-shaped of Sertoli cell nuclei which make it difficult to estimate the volume of individual nuclei. Using several approaches to overcome the problems associated with any one method, the horse is identified as a species in which Sertoli cell number is not fixed, but it fluctuates with season. In addition to Sertoli cell numbers, the functions of Sertoli cells that are very important in signaling and controlling spermatogenesis are discussed. Recent studies have shown that "post-mitotic terminally differentiated Sertoli cells" from adult animals could, under certain conditions, re-enter the cell division cycle. Can seasonal influences be a natural set of conditions to induce the Sertoli cells of the horse testis to seasonally re-enter the cell division cycle and explain the seasonal differences in Sertoli cell number as summarized in this review? Alternatively, can seasonal differences in Sertoli cell number reflect, in the horse to a greater extent, but in adults of most species, the presence of some mitotic-capable Sertoli cells in adults? In any case, both Sertoli cell number and function are important in regulation of spermatogenesis.
URLPMID:26846984 [本文引用: 2]
Summary It has been one and a half centuries since Enrico Sertoli published the seminal discovery of the testicular ‘nurse cell’, not only a key cell in the testis, but indeed one of the most amazing cells in the vertebrate body. In this review, we begin by examining the three phases of morphological research that have occurred in the study of Sertoli cells, because microscopic anatomy was essentially the only scientific discipline available for about the first 7502years after the discovery. Biochemistry and molecular biology then changed all of biological sciences, including our understanding of the functions of Sertoli cells. Immunology and stem cell biology were not even topics of science in 1865, but they have now become major issues in our appreciation of Sertoli cell's role in spermatogenesis. We end with the universal importance and plasticity of function by comparing Sertoli cells in fish, amphibians, and mammals. In these various classes of vertebrates, Sertoli cells have quite different modes of proliferation and epithelial maintenance, cystic vs. tubular formation, yet accomplish essentially the same function but in strikingly different ways.
URLPMID:25986756 [本文引用: 1]
61miRNAs were altered in Sertoli cells exposed to MC-LR.61Alerted genes were involved in different cell functions including the cell morphology.61MC-LR adversely affected Sertoli cell junction formation through the regulating miRNAs.
URLMagsci [本文引用: 1]
MicroRNA(miRNA)是一类长约22 nt的非编码小RNA,广泛存在于各种生物中,调节生物体生长、发育和凋亡等过程。研究表明,miRNA在猪肌肉、脂肪、生殖系统以及免疫系统等的发育过程中发挥着重要的调控作用。此外,高通量的新一代测序技术在猪miRNA的挖掘和差异表达研究中发挥着巨大的作用。文章综述了高通量的新一代测序技术在挖掘猪miRNA中的应用以及一些miRNA在猪脂肪代谢、肌肉发育、卵母细胞成熟和B、T淋巴细胞发育中的调控作用,旨在为猪miRNA的研究提供参考,为利用miRNA调控和改善猪肉品质、生长性能、繁殖性能以及免疫机能提供理论基础和研究思路。
URLMagsci [本文引用: 1]
MicroRNA(miRNA)是一类长约22 nt的非编码小RNA,广泛存在于各种生物中,调节生物体生长、发育和凋亡等过程。研究表明,miRNA在猪肌肉、脂肪、生殖系统以及免疫系统等的发育过程中发挥着重要的调控作用。此外,高通量的新一代测序技术在猪miRNA的挖掘和差异表达研究中发挥着巨大的作用。文章综述了高通量的新一代测序技术在挖掘猪miRNA中的应用以及一些miRNA在猪脂肪代谢、肌肉发育、卵母细胞成熟和B、T淋巴细胞发育中的调控作用,旨在为猪miRNA的研究提供参考,为利用miRNA调控和改善猪肉品质、生长性能、繁殖性能以及免疫机能提供理论基础和研究思路。
Magsci [本文引用: 1]
MicroRNA(miRNA)是一类长约22nt的非编码小RNA, 广泛存在于各种生物中, 调节生物体生长、发育和凋亡等过程。研究表明, miRNA在人和动物睾丸发育及精子生成等过程也起着重要的调控作用。但miRNA在不同种属的睾丸组织及其不同发育时间段均存在特异性表达。此外, miRNA在动物精子生成过程中也存在时空特异性。文章综述了睾丸发育和精子生成过程中miRNA的差异性表达、表达调控以及一些miRNA对精子生成的调节作用, 旨在为睾丸miRNA的进一步研究提供参考, 为利用miRNA调控和促进种公畜精液品质提供研究思路。
Magsci [本文引用: 1]
MicroRNA(miRNA)是一类长约22nt的非编码小RNA, 广泛存在于各种生物中, 调节生物体生长、发育和凋亡等过程。研究表明, miRNA在人和动物睾丸发育及精子生成等过程也起着重要的调控作用。但miRNA在不同种属的睾丸组织及其不同发育时间段均存在特异性表达。此外, miRNA在动物精子生成过程中也存在时空特异性。文章综述了睾丸发育和精子生成过程中miRNA的差异性表达、表达调控以及一些miRNA对精子生成的调节作用, 旨在为睾丸miRNA的进一步研究提供参考, 为利用miRNA调控和促进种公畜精液品质提供研究思路。
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 4]
To understand the complex physiological process underlying pig testis development and spermatogenesis, this study aims to characterize the change in miRNA and mRNA profiles at four developmental stages of embryonic and postnatal testes, including 60 dpc (days post coitus, E60), 90 dpc (E90), 30-day-old (D30) and 180-day-old (D180). A total of 304 mature, 50 novel miRNAs, and 8343 differentially-expressed genes were identified. 93 (48 up and 45 down), 104 (49 up and 55 down), 122 (49 up and 73 down) differentially-expressed miRNAs, as well as 1007 (646 up and 361 down), 1929 (911 up and 1018 down), 7420 (3998 up and 3422 down) differentially-expressed genes were identified in E90vs.E60, D30vs.E90 and D180vs.D30, respectively. Integrating analysis of miRNA and mRNA expression profiles predicted more than 50 000 miRNA RNA interaction sites. GO and KEGG pathway analysis of the predicted target genes illustrated the likely roles of differentially expressed miRNAs in testis development and spermatogenesis. For example, PI3K-Akt signaling pathway and Hippo signaling pathway related development, and carbon metabolism, fatty acid metabolism, protein digestion and absorption, were involved in metabolite synthesis. These integrated high-throughput expression data show that miRNA is a critical factor in porcine testis development, providing a useful resource to understand global genome expression change in porcine testis development and spermatogenesis.
URLPMID:26755652 [本文引用: 1]
Sertoli cells play critical roles in regulating spermatogenesis and they can be reprogrammed to the cells of other lineages, highlighting that they have significant applications in reproductive and regenerative medicine. The fate determinations of Sertoli cells are regulated precisely by epigenetic factors. However, the expression, roles, and targets of microRNA (miRNA) in human Sertoli cells remain unknown. Here we have for the first time revealed that 174 miRNAs were distinctly expressed in human Sertoli cells between Sertoli-cell-only syndrome (SCOS) patients and obstructive azoospermia (OA) patients with normal spermatogenesis using miRNA microarrays and real time PCR, suggesting that these miRNAs may be associated with the pathogenesis of SCOS. MiR-133b is upregulated in Sertoli cells of SCOS patients compared to OA patients. Proliferation assays with miRNA mimics and inhibitors showed that miR-133b enhanced the proliferation of human Sertoli cells. Moreover, we demonstrated that GLI3 was a direct target of miR-133b and the expression of Cyclin B1 and Cyclin D1 was enhanced by miR-133b mimics but decreased by its inhibitors. Gene silencing of GLI3 using RNA inference stimulated the growth of human Sertoli cells. Collectively, miR-133b promoted the proliferation of human Sertoli cells by targeting GLI3. This study thus sheds novel insights into epigenetic regulation of human Sertoli cells and the etiology of azoospermia and offers new targets for treating male infertility
URLPMID:5011707 [本文引用: 3]
A growing number of reports have revealed that microRNAs (miRNAs) play critical roles in spermatogenesis. Our previous study showed that miR-762 is differentially expressed in immature and mature testes of Large White boars. Our present data shows that miR-762 directly binds the 3′ untranslated region (3′UTR) of ring finger protein 4 (RNF4) and down-regulates RNF4 expression. A single nucleotide polymorphism (SNP) in theRNF43′UTR that is significantly associated with porcine sperm quality traits leads to a change in the miR-762 binding ability. Moreover, miR-762 promotes the proliferation of and inhibits apoptosis in porcine immature Sertoli cells, partly by accelerating DNA damage repair and by reducing androgen receptor (AR) expression. Taken together, these findings suggest that miR-762 may play a role in pig spermatogenesis by regulating immature Sertoli cell growth.
URLPMID:26287402 [本文引用: 2]
Abstract This study investigated the capacity of 10 0204M 170205-estradiol to inhibit immature boar Sertoli cell (SC) proliferation and the involvement of microRNA (miR)-1285 in this process. SC viability and cell cycle progression were investigated using a cell counting kit-8 and flow cytometry, respectively. Expression of AMP-activated protein kinase (AMPK), S phase kinase-associated protein 2 (Skp2), and miR-1285 was analyzed by real-time RT-PCR and Western blotting. 170205-Estradiol (10 0204M) reduced SC viability and miR-1285 expression and promoted AMPK phosphorylation. A double-stranded synthetic miR-1285 mimic promoted SC viability, increased levels of ATP, and phosphorylated mammalian target of rapamycin (mTOR) and Skp2 mRNA and protein, whereas p53 and p27 expression decreased, and 170205-estradiol-mediated effects on SCs were significantly attenuated. A single-stranded synthetic miR-1285 inhibitor produced the opposite effects on these measures. Activation of AMPK inhibited SC viability, reduced levels of ATP, phosphorylated mTOR and Skp2 mRNA and protein, and increased p53 and p27 expression. An AMPK inhibitor (compound C) attenuated the effects of 170205-estradiol on SCs. This indicated that 170205-estradiol (10 0204M) reduced SC proliferation by inhibiting miR-1285 and thus activating AMPK. Phosphorylated AMPK is involved in the regulation of 170205-estradiol-mediated inhibition of SC viability through increasing p53 and p27 expression and inhibiting mTOR and Skp2 expression. Our findings also implicated Skp2 as the downstream integration point of p53 and mTOR. These findings indicated that miR-1285 may represent a target for the manipulation of boar sperm production.
URLPMID:29162477 [本文引用: 2]
To investigate the possible molecular mechanism of low concentration plasticizer mono-n-butyl phthalate (MBP) -induced juvenile Sertoli cells (SCs) proliferation, we evaluated global alterations of miRNA and mRNA expression in rat SCs treated with 0.1 mM MBP. Microarray analysis revealed that miR-3584-5p and miR-301b-3p were up-regulated and their common target gene Dexamethasone-induced Ras-related protein 1 (Rasd1) was down-regulated. Further work suggested that SCs proliferation induced by low concentration MBP in vitro might be mediated by Rasd1 regulating ERK1/2 signaling pathway. The present study is first to investigate the effect of low-dose MBP on SCs proliferation and may enhance our understanding on the modes of action of low concentration MBP on male reproductive system. We hope the results will contribute to explain the causes of precocious puberty and testicular tumors induced by exogenous chemicals.
URLPMID:29119857 [本文引用: 3]
react-text: 413 Various factors influencing toxicity of 4-nitroquinoline 1-oxide (4-NQO) in Chinese hamster ovary cells were determined. Cell density during 4-NQO treatment and volume of treatment medium had a great effect on cell survival indicating that not the 4-NQO concentration per se, but the amount of 4-NQO per cell determines the toxic effect. When the cell-cycle response for 4-NQO-induced cell... /react-text react-text: 414 /react-text [Show full abstract]
URLPMID:2660371 [本文引用: 1]
MicroRNAs (miRNAs), which are small, non-coding RNAs approximately 21-nucleotides in length, have become a major focus of research in molecular biology. Mammalian miRNAs are proposed to regulate approximately 30% of all protein-coding genes. Previous studies have focused on highly conserved miRNAs, but nonconserved miRNAs represent a potentially important source of novel functionalities during evolution.An analysis of the chromosome distribution of miRNAs showed higher densities of miRNAs on the X chromosome compared to the average densities on autosomes in all eight mammalian species analyzed. The distribution pattern did not, however, apply well to species beyond mammals. In addition, by comparing orthologous human and mouse miRNAs, we found that X-linked miRNAs had higher substitution rates than autosomal miRNAs. Since the highest proportion of X-linked miRNAs were found in mouse testis, we tested the hypothesis that testis miRNAs are evolving faster on the X chromosome than on autosomes. Mature X-linked testis miRNAs had an average substitution rate between mouse and human that was almost 25-fold higher than mature testis miRNAs on autosomes. In contrast, for mature miRNAs with precursors not expressed in testis, no significant difference in the substitution rate between the X chromosome and autosomes was found. Among mammals, the rapid evolution of X-linked testis miRNAs was also observed in rodents and primates.The rapid evolution of X-linked testis miRNAs implies possible important male reproductive functions and may contribute to speciation in mammals.
URLPMID:27976743 [本文引用: 1]
Abstract Microcystin-leucine arginine (MC-LR) is a potent toxin for Sertoli cells. However, the specific molecular mechanisms of MC-induced cytotoxicity still remain unclear. In this study, we performed a comprehensive analyses of changes of miRNAs and mRNAs in Sertoli cells treated with MC-LR. Through computational approaches, we showed the pivotal roles of differentially expressed miRNAs that were associated with cell metabolism, cellular growth and proliferation, cell-to-cell signaling and interaction and cellular movement. Ingenuity Pathway Analyses (IPA) revealed some differentially expressed miRNAs and mRNAs that may cause reproductive system diseases. Target gene analyses suggested that destruction in tight junctions (TJ) and adherens junctions (AJ) in testes may be mediated by miRNAs. Consistent with a significant enrichment of chemokine signaling pathways, we observed numerous macrophages in the testes of mice following treatment with MC-LR, which may cause testicular inflammation. Moreover, miR-98-5p and miR-758 were predicted to bind the 3'-UTR region of the mitogen-activated protein kinase 11 (MAPK11, p38 0205 isoform) gene which stimulates tumor necrosis factor-02± (TNF-02±) expression in Sertoli cells. TNF-02± could interact with the tumor necrosis factor receptor 1 (TNFR1) on germ cells leading to induction of germ cell apoptosis. Collectively, our integrated miRNA/mRNA analyses provided a molecular paradigm, which was experimentally validated, for understanding MC-LR-induced cytotoxicity.
URLPMID:26744029 [本文引用: 1]
Swine testicular (ST) cell line is isolated from swine fetal testes and has been widely used in biomedical research fields related to pig virus infection. However, the potential benefit and...
URLPMID:4047880 [本文引用: 1]
Lipids are key regulators of cell physiology through the control of many aspects of cellular life and survival. In particular, lipids have been implicated at different levels and through many different mechanisms in the cell death program called apoptosis. Here, we discuss the action of lipids in the regulation of the activation and the integration of Bax into the mitochondrial outer membrane, a key pro-apoptotic member of the BCL-2 family. We describe how, during apoptosis, lipids can act simultaneously or in parallel as receptors or ligands for Bax to stimulate or inhibit its pro-death activity.
URL [本文引用: 2]
URLPMID:24162659 [本文引用: 2]
Abstract Apoptosis, a mechanism for programmed cell death, has key roles in human health and disease. Many signals for cellular life and death are regulated by the BCL-2 family proteins and converge at mitochondria, where cell fate is ultimately decided. The BCL-2 family includes both pro-life (e.g. BCL-XL) and pro-death (e.g. BAX, BAK) proteins. Previously, it was thought that a balance between these opposing proteins, like a simple 'rheostat', could control the sensitivity of cells to apoptotic stresses. Later, this rheostat concept had to be extended, when it became clear that BCL-2 family proteins regulate each other through a complex network of bimolecular interactions, some transient and some relatively stable. Now, studies have shown that the apoptotic circuitry is even more sophisticated, in that BCL-2 family interactions are spatially dynamic, even in nonapoptotic cells. For example, BAX and BCL-XL can shuttle between the cytoplasm and the mitochondrial outer membrane (MOM). Upstream signaling pathways can regulate the cytoplasmic-MOM equilibrium of BAX and thereby adjust the sensitivity of cells to apoptotic stimuli. Thus, we can view the MOM as the central locale of a dynamic life-death rheostat. BAX invariably forms extensive homo-oligomers after activation in membranes. However, recent studies, showing that activated BAX monomers determine the kinetics of MOM permeabilization (MOMP), perturb the lipid bilayer and form nanometer size pores, pose questions about the role of the oligomerization. Other lingering questions concern the molecular mechanisms of BAX redistribution between MOM and cytoplasm and the details of BAX/BAK-membrane assemblies. Future studies need to delineate how BCL-2 family proteins regulate MOMP, in concert with auxiliary MOM proteins, in a dynamic membrane environment. Technologies aimed at elucidating the structure and function of the full-length proteins in membranes are needed to illuminate some of these critical issues.
URLPMID:22240065 [本文引用: 1]
MicroRNAs (miRNAs) are small noncoding regulatory RNAs that play key roles in many diverse biological processes such as spermatogenesis. However, no study has been performed on the miRNA transcriptome of developing porcine testes. Here, we employed Solexa deep sequencing technology to extend the repertoire of porcine testis miRNAs and extensively compare the expression patterns of sexually immature and mature porcine testes. Solexa sequencing of two small RNA libraries derived from immature (30 days) and mature (180 days) pig testis samples yielded over 25 million high-quality reads. Overall, the two developmental stages had significantly different small RNA compositions. A custom data analysis pipeline identified 398 known and/or homologous conserved porcine miRNAs, 15 novel pig-specific miRNAs and 56 novel candidate miRNAs. We further observed multiple mature miRNA variants and identified a new bidirectional transcribed miRNA locus, ssc-mir-181a. A total of 122 miRNAs were differentially expressed in the immature and mature testes, and 10 were validated using quantitative RT-PCR. Furthermore, GO and KEGG pathway analyses of the predicted miRNA targets further illustrate the likely roles for these differentially expressed miRNAs in spermatogenesis. This study is the first comparative profile of the miRNA transcriptome in immature and mature porcine testes using a deep sequencing approach, and it provides a useful resource for future studies on the role of miRNAs in spermatogenesis and male infertility treatment.
URL
URLPMID:25563581 [本文引用: 1]
Background Spermatogenesis is an intricate biological event wherein an undifferentiated spermatogonium develops into mature sperms. MicroRNAs are a type of...
URLPMID:26073258 [本文引用: 1]
Abstract miR-362-5p is down-regulated in high-risk neuroblastoma and can function as a tumor suppressor. However, its role remains poorly understood. We show that miR-362-5p is down-regulated in metastatic neuroblastoma compared with primary neuroblastoma. Overexpression of miR-362-5p inhibits cell proliferation, migration and invasion of neuroblastoma cells in vitro and suppresses tumor growth of neuroblastoma in vivo. Phosphatidylinositol 3-kinase (PI3K)-C20205 is a target of miR-362-5p. Knockdown of PI3K-C20205 by siRNA had a similar effect to overexpression of miR-362-5p on SH-SY5Y cells. Overexpression of PI3K-C20205 partially reversed tumor-suppressive effects of miR-362-5p. We suggest that miR-362-5p suppresses neuroblastoma cell growth and motility, partially by targeting PI3K-C20205. Copyright 0008 2015 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
URLPMID:26893711 [本文引用: 1]
An increasing number of studies have indicated that the deregulation of microRNAs (miRNAs) contributes to tumorigenesis and metastasis. In the present study, significant upregulation of miR62362625p was identified in the breast cancer MDA62MB6223102and MCF702cell lines compared with the control CCD621095Sk cell line. The inhibition of miR62362625p was demonstrated to significantly inhibit the cell proliferation, migration and invasion of human breast cancer MCF702cells. In addition, the knockdown of miR62362625p induced G102arrest and promoted apoptosis in the breast cancer cells. Mechanistic investigations confirmed that the tumor suppressor gene CYLD is a direct target of miR62362625p. The ectopic expression of miR62362625p represses CYLD expression, whereas miR62362625p inhibitor treatment induces CYLD protein expression and decreases NF62κB expression in the downstream signaling pathway. Thus, these findings may provide novel insights into the molecular mechanisms through which miR62362625p regulates breast cancer cell proliferation, migration and invasion. This study also suggests that miR62362625p may act as a novel potential therapeutic target for the treatment of breast cancer.
URLPMID:25652145362382 [本文引用: 1]
Aim This study was to investigate the effects and mechanisms of miR-362-3p on regulation of gastric cancer (GC) cell metastasis potential. Methods We detected miR-362-3p level in GC and adjacent normal tissues and investigated the relationship with clinicopathological factors. Next, we analyzed the level of miR-362-3p expression and CD82 in different differentiated GC cells compared with a normal gastric mucosa cell by RT-PCR and Western blot. Dual-luciferase reporter assay and Western blot confirmed a direct interaction between miR-362-3p and CD82 3 TR. After miR-362-3p and CD82 were silenced in GC cells, we compared the transfected GC cells migration and invasion capacity by transwell assay. In addition, we detected the effects on cells angiogenesis by tube formation assay. Western blot was used to detect the impact of CD82 and miR-362-3p on epithelial-to-mesenchymal transition markers in treated GC cells. Results Level of miR-362-3p expression was much higher in GC cells than in normal gastric mucosa cell, and miR-362-3p expression negatively correlated with CD82 mRNA expression in these cell lines. Furthermore, miR-362-3p expression inhibited GC cell metastasis capacity by suppression of CD82 expression. Level of miR-362-3p may mediate E-cadherin, N-cadherin, and vimentin expression in GC cells. Conclusion This study illuminated that downregulation of miR-362-3p along with the upregulation of CD82 in GC cells resulted in the inhibition of GC migration and invasion. Thus, our results suggested that miR-362-3p or CD82 can be exploited as a new potential target for control of GC in the future.
URLPMID:4636774 [本文引用: 1]
MicroRNAs (miR, miRNAs) play pivotal roles in numerous physiological and pathophysiological contexts. We investigated whether miR-362-5p act as an oncogene in chronic myeloid leukaemia (CML) and aimed to understand its potential underlying mechanisms. We compared the miR-362-5p expression levels between CML and non-CML cell lines, and between fresh blood samples from CML patients and normal healthy controls using quantitative real-time PCR (qPCR). Cell counting kit-8 (CCK-8) and Annexin V-FITC/PI analyses were used to measure the effects of miR-362-5p on proliferation and apoptosis, and Transwell assays were used to evaluate migration and invasion. A xenograft model was used to examine in vivo tumourigenicity. The potential target of miR-362-5p was confirmed by a luciferase reporter assay, qPCR and western blotting. Involvement of the JNK1/2and P38 pathways was investigated by western blotting. miR-362-5p was up-regulated in CML cell lines and fresh blood samples from CML patients, and was associated with Growth arrest and DNA damage-inducible (GADD)45 down-regulation. Inhibition of miR-362-5p simultaneously repressed tumour growth and up-regulatedGADD45 expression in a xenograft model. Consistently, the knockdown ofGADD45 expression partially neutralized the effects of miR-362-5p inhibition. Furthermore study suggested thatGADD45 mediated downstream the effects of miR-362-5p, which might indirectly regulates the activation of the JNK1/2and P38 signalling pathways. miR-362-5p acts as an oncomiR that down-regulatesGADD45 , which consequently activates the JNK1/2and P38 signalling. This finding provides novel insights into CML leukaemogenesis and may help identify new diagnostic and therapeutic targets. The online version of this article (doi:10.1186/s12943-015-0465-3) contains supplementary material, which is available to authorized users.
URLPMID:25909817 [本文引用: 1]
Background: Natural killer(NK) cells are critical effectors in the immune response against malignancy and infection, and micro RNAs(mi RNAs) play important roles in NK cell biology. However, the micro RNA transcriptomes of the different phenotypes and functions of human NK cell subsets are incompletely understood. Objective: To better understand the physiologic significance of mi RNAs in the regulation of NK cell function. Methods: We examined mi RNA profiles of human NK cells from different cell compartments(peripheral blood, cord blood, and uterine deciduas) and of NKT and T cells from peripheral blood, and highly expressed mi RNAs in human NK cells were identified preferentially. Results: We identified a novel mi RNA, mi R-362-5p, that is highly expressed in human peripheral blood NK(p NK) cells. We also demonstrated that CYLD, a negative regulator of NF- B signaling, was a target of mi R-362-5p in NK cells. Furthermore, we showed that the over-expression of mi R-362-5p enhanced the expression of IFN- , perforin, granzyme-B, and CD107 a in human primary NK cells, and we found that silencing CYLD with a small interfering RNA(si RNA) mirrored the effect of mi R-362-5p over-expression. In contrast, the inhibition of mi R-362-5p had the opposite effect in NK cells, which was abrogated by CYLD si RNA, suggesting that mi R-362-5p promotes NK-cell function, at least in part, by the down-regulation of CYLD. Conclusion: Our results provide a resource for studying the roles of mi RNAs in human NK cell biology and suggest that the modulation of mi R-362-5p may be a useful therapeutic strategy to enhance NK-cell effector function against viral infections and malignancy.
URLPMID:258279522 [本文引用: 1]
61Here we review recent advances in understanding Bcl-2 family members.61Recent X-ray crystal structures have provided insight into Bax and Bak activation.61Some lymphoma-associated BCL2 mutations were shown to enhance Bcl-2 function.61The Bcl-2-selective BH3 mimetic ABT-199 has shown promising clinical activity.
URLPMID:18823591 [本文引用: 1]
Znf230, the mouse homologue of the human spermatogenesis-related gene, ZNF230, has been cloned by rapid amplification of cDNA ends (RACE). This gene is expressed predominantly in testis, but its expression in different testicular cells and spermatogenic stages has not been previously analyzed in detail. In the present study, the cellular localization of the Znf230 protein in mouse testis and epididymal spermatozoa was determined by RT-PCR, immunoblotting, immunohistochemistry and immunofluorescence. It is primarily expressed in the nuclei of spermatogonia and subsequently in the acrosome system and the entire tail of developing spermatids and spermatozoa. The results indicate that Znf230 may play an important role in mouse spermatogenesis, including spermatogenic cell proliferation and sperm maturation, as well as motility and fertilization.
URLPMID:17394778 [本文引用: 1]
We have cloned a novel mouse zinc finger protein gene Znf313 by rapid amplification of cDNA ends (RACE) according to the homologue of human ZNF313 gene. The cDNA is 2,163 base pairs (bp) in length and encodes a 229 amino acids (aa) protein with a C(3)HC(4) ring finger domain and three C(2)H(2) domains. 89% and 93% nucleotide (nt) and aa sequence identity is observed with its human homologue. Revealed by Northern blot and RT-PCR, full mRNA consists of 2.16 kb and widely expresses in tissues as a single transcript, most abundantly in heart, liver, kidney and testis. The expression of Znf313 in testis is detected in all development stages. Western blot analysis also reveals that Znf313 is expressed in the tissues. Immunohistochemical staining and subcellular localization demonstrate that Znf313 is expressed both in the cytoplasm and nucleus whereas predominantly localized in the nucleus. Present data suggests that Znf313 gene might play a fundamental role in gene transcription and regulation in organism and relates to spermatogenesis.
URLPMID:18584306 [本文引用: 1]
The ZNF230 gene is a recently cloned gene which is transcribed only in fertile male testes and may be related to human spermatogenesis. To characterize the multiple stage-specific transcription elements necessary for ZNF230 expression, we cloned ZNF230 promoter and constructed chimeric luciferase reporter Plasmids. Overexpression and site-directed mutation test were used to characterize the cis -element. The results showed ZNF230 gene promoter to be GC rich and not contain a TATA box. Deletion analysis of the 5 -flanking region of ZNF230 in HEK293 cells indicated that the sequence encompassing from nt 131 to +152 has a basal transcriptional activity. Site-directed mutation test and mithramycin A treatment demonstrated that the ZNF230 promoter contained a functional Sp1 site. Overexpression of the Sox5 protein activated the promoter activity. A 312-bp fragment surrounding the transcription start site exhibits a characteristic CpG island which overlaps with the promoter region. We also provided evidence that both the human and mouse znf230 promoter consist of Sp1 binding site and GC-rich sequences, suggesting Sp1 is required for the transcription of human and mouse ZNF230 genes. In conclusion, these findings suggest that ZNF230 is tightly controlled at transcriptional level and a common mechanism controls the basal transcription of ZNF230 gene.
URLPMID:27546533 [本文引用: 1]
Proliferating progenitor cells undergo changes in competence to give rise to post-mitotic progeny of specialized function. These cell-fate transitions typically involve dynamic regulation of gene expression by histone methyltransferase (HMT) complexes. However, the composition, roles, and regulation of these assemblies in regulating cell-fate decisions invivo are poorly understood. Using unbiased affinity purification and mass spectrometry, we identified the uncharacterized C2H2-like zinc finger protein ZNF644 as a G9a/GLP-interacting protein and co-regulator of histone methylation. In zebrafish, functional characterization of ZNF644 orthologs, znf644a and znf644b , revealed complementary roles in regulating G9a/H3K9me2-mediated gene silencing during neurogenesis. The non-overlapping requirements for znf644a and znf644b during retinal differentiation demarcate critical aspects of retinal differentiation programs regulated by differential G9a-ZNF644 associations, such as transitioning proliferating progenitor cells toward differentiation. Collectively, our data point to ZNF644 as a critical co-regulator of G9a/H3K9me2-mediated gene silencing during neuronal differentiation.
URLPMID:27034204 [本文引用: 1]
AimsHigh myopia is a common visual disorder in the world. The ZNF644, GRM6, and CTNND2 genes are expressed in the retina. This study aims to investigate the associations of these genes with high myopia in Han Chinese population.MethodsThe case-control association included high myopia cases (n=430) and controls (n=430) recruited from a population-based study, 'Jiangsu Eye Study'. Fourteen single-nucleotide polymorphisms (SNPs) in three genes were genotyped by the TaqMan method using the real-time PCR system.ResultsThree SNPs GRM6-rs11746675, GRM6-rs2067011, and GRM6-rs2645339 were associated with high myopia (odds ratio (OR)=0.74, P=0.003; OR=0.78, P=0.018; and OR=0.78, P=0.023; respectively). The significances of rs2067011 and rs2645339 disappeared after multiple testing corrections. Rs11746675 remained significant after correction for multiple testing. The genetic model analysis found that GRM6-rs11746675 and GRM6-rs2067011 were suggestively associated with high myopia in the recessive model (OR=0.54, P=0.004; OR=0.52, P=0.003; respectively). Haplotype GAT for GRM6 markers rs2067011-rs2645339-rs762724 showed significance (P=0.0239), but such association did not remain significant after multiple testing corrections.ConclusionsOur data suggested that genetic variants in GRM6 are associated with high myopia. The mechanism of GRM6 in the development of high myopia need to be further investigated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}