 ,华南农业大学动物科学学院,国家生猪种业工程技术研究中心,广州 510642
,华南农业大学动物科学学院,国家生猪种业工程技术研究中心,广州 510642Progress and application of genome-edited pigs in biomedical research
Yaoqiang Huang, Guoling Li, Huaqiang Yang, Zhenfang Wu,National Engineering Research Center for Breeding Swine Industry, College of Animal Science, South China Agricultural University, Guangzhou 510642, China通讯作者:
编委: 任军
收稿日期:2018-01-6修回日期:2018-05-18网络出版日期:2018-08-16
| 基金资助: |
Received:2018-01-6Revised:2018-05-18Online:2018-08-16
| Fund supported: |
作者简介 About authors
黄耀强,硕士研究生,专业方向:分子遗传与动物育种E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (545KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄耀强, 李国玲, 杨化强, 吴珍芳. 基因编辑猪在生物医学研究中的应用[J]. 遗传, 2018, 40(8): 632-646 doi:10.16288/j.yczz.18-026
Yaoqiang Huang, Guoling Li, Huaqiang Yang, Zhenfang Wu.
基因组编辑技术(genome editing technologies, GETs)能够精确靶向修饰生物体基因组特定位点、人为改造生物体遗传信息。以锌指核酸酶(zinc finger nucleases, ZFN)、类转录激活因子效应物核酸酶(transcription activator-like effector nucleases, TALEN)和成簇的规律间隔短回文重复序列及其相关系统(clustered regularly interspaced short palindromic repeats/Cas endonucleases, CRISPR/Cas)为代表的新型基因组编辑技术已经掀起了生命科学研究的热潮,在生物学、医学和农业等各领域被广泛应用。与产生基因随机位点整合的传统转基因修饰技术不同,人工构建的基因组编辑系统可以实现基因组靶序列的精确切割,激发细胞内源性DNA损伤修复继而实现基因组定点修饰,成为研究基因功能、修改遗传信息的重要手段,在动物模型建立、异种器官移植等生物医学领域具有重要作用。尽管啮齿类动物的生物学基础研究价值无法替代,但由于其与人类的生理特征相距较远等因素,在模拟人类疾病病症病程与器官移植等方面的实际应用受到限制。猪的心血管系统和脏器尺寸等生理解剖特征与人体高度相似,免疫特征与免疫反应也与人体比较接近,这就决定了其可作为人类疾病动物模型和异种器官移植研究的理想对象。然而在相当长的一段时间里,猪的定点基因修饰操作是非常困难的。由于研究人员至今尚未建立猪的胚胎干细胞(embryonic stem cells, ESC)或诱导多能干细胞(induced pluripotent stem cells, IPSC)等具有生殖系嵌合能力的细胞系,基因修饰猪只能通过体细胞核移植技术(somatic cell nuclear transfer, SCNT)进行克隆制备。而传统的同源重组技术对体细胞进行基因修饰的能力极低,极大地限制了基因修饰猪的制作和研究。具有较高打靶效率的基因组编辑技术的出现,大大提升了基因修饰动物的制备效率。SCNT结合基因组编辑技术,推动了猪在生物医学领域的应用。本文结合ZFN、TALEN和CRISPR/Cas9等新型基因组编辑技术的发展概况和作用机理,对基因编辑猪在模拟人类疾病、异种器官移植等生物医学方面具有重要意义的最新研究进展进行了综述。
1 基因组编辑技术的发展
传统的转基因技术是指将已知外源基因整合进细胞基因组并使之表达的技术,如原核显微注射法、慢病毒介导法等。传统转基因技术产生的基因整合是随机的,其诱导效率与适用性低,同时转基因动物受内源、外源基因的双重影响而表现出遗传性状的不确定性。自20世纪70年代报道了基于酵母细胞进行外源基因的同源重组(homologous recombination)和内源基因敲除(knock-out)后,基因打靶技术迅猛发展[1,2,3]。1986年,Thomas等[4]利用同源重组恢复了抗新霉素基因缺陷细胞的抗药性,这是首次基于哺乳动物细胞开展的基因编辑。由于猪的ES细胞系尚未稳定建立,因此基因编辑猪的制备主要依靠体细胞同源重组结合SCNT技术。然而体细胞体外增殖能力有限,同源重组效率极其低下,因此利用传统基因工程技术制备转基因猪的效率极低。随着多种生物的基因组测序相继完成,生物遗传信息与生命体间的遗传差异成为重要研究资源,加上对细胞基因组同源重组机制的认识不断加深[5,6],以ZFN、TALEN和CRISPR/Cas9系统为主的新型基因组编辑技术应运而生,为解析基因功能及其调控、精确改造生物遗传信息的研究带来了突破性进展。1996年,Kim等[7]将锌指蛋白(zinc finger protein, ZFP)与FokⅠ融合,首次构建出具有靶向切割DNA功能的ZFN。在ZFN诞生15年后,第二代基因组编辑技术TALEN开始发展。TALEN技术最初由Christian等[8]建立,此后经历了逐步完善的技术改进过程。ZFN与TALEN技术的发展成熟,重新定义了遗传学研究的界限和范畴。1987年,Ishino等[9]在大肠杆菌的碱性磷酸酶基因中首次发现规律间 隔性成簇短回文重复序列的特殊结构,此后Mojica 等[10]和Jasen等[11]在其他细菌和古菌基因组也发现此类结构,并将其命名为CRISPR,这一结构与病毒等外源遗传物质之间存在的同源性以及二者间的免疫调控关系也逐渐明确。CRISPR/Cas9技术作为第三代基因组编辑系统,正是基于细菌等微生物抵御外界DNA的获得性免疫系统发展起来的,2012年被报道后,便迅速突破了以往的基因组编辑技术在实际应用上所面临的多种限制,成为基因组定点编辑的主要工具。2 ZFN、TALEN与CRISPR/Cas9基因组编辑技术的作用机制
基因组编辑技术通过限制性核酸酶对基因组特定位点进行切割,产生DNA双链断裂(double strand breaks, DSB)。DSB经细胞内DNA的主要修复机制非同源末端连接(non-homologous end joining, NHEJ)或同源重组(homologous directed recombination, HDR)后实现靶基因的遗传学修饰。NHEJ是真核生物基因组的主要修复机制[12],不需要或仅需有限的同源序列即可将断开的DNA游离末端重新缝合,常伴随着核苷酸的插入或缺失(insertions and deletions,indels)并形成编码区移码突变,继而敲除内源基因[13,14]。在同源片段存在的情况下,DSB处发生HDR的概率大幅提高,通过将同源DNA 置换或重组至DSB以恢复或修改遗传信息,实现基因定点编辑或敲入(knock-in)[15,16](图1)。无论NHEJ还是HDR都依赖于DSB的产生,然而基因组中自然发生DSB的频率极低,因此如何诱导特定基因位点产生DSB成为对动物进行基因编辑所面临的主要问题。ZFN、TALEN和CRISPR/Cas9系统等新型基因组编辑技术可以靶向切割基因组,产生DSB,继而激发细胞的NHEJ或HDR修复,实现多种生物基因组的高效而精确的编辑。图1
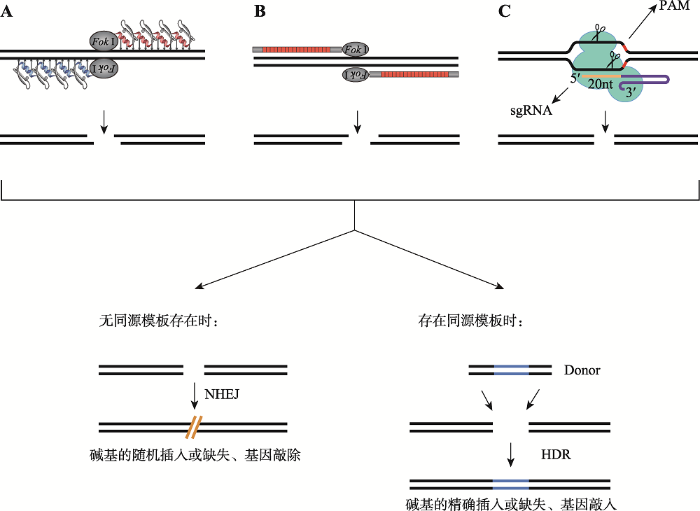 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1ZFN、TALEN和CRISPR/Cas9系统介导的基因组编辑原理
A:ZFN介导的基因组编辑原理;B:TALEN介导的基因组编辑原理;C:CRISPR/Cas9介导的基因组编辑原理。ZFN、TALEN和CRISPR/Cas9 诱导基因组靶位点形成DSB,DSB被细胞内源性修复机制NHEJ和HDR等修复,实现靶基因的敲除或精确编辑。根据文献[21,37,53]等修改绘制。
Fig. 1Schematic illustration of the principles of ZFN-, TALEN- and CRISPR/Cas9-mediated genome-editing technologies
ZFN由负责特异性识别DNA序列的Cys2 His2锌指蛋白(Cys2 His2 ZFP)与非特异性的ⅡS型限制性核酸内切酶FokⅠ经连接区(linker)融合而成[7,17]。Cys2 His2锌指最初在非洲爪蟾(Xenopus laevis)的转录因子TFIIIA中被发现[18],是由约30个氨基酸组成的肽链。锌指的Cys、His氨基酸残基与Zn+螯合,使得肽链折叠成β-β-α的结构。α-螺旋的-1、+3、+6位氨基酸直接同靶序列的三联碱基相互作用,介导Cys2 His2 ZF基序嵌合至DNA链,因此若干与密码子对应的ZF模块微组(modular assembly)后可特异性结合相应DNA片段[19,20,21],使用者只需根据靶基因设计8~10个锌指结构域,并与核酸内切酶的催化域结合,可制备出靶向特异性的ZFN。FokⅠ属Ⅱ型限制性内切酶,通过识别非回文序列5°-GGATG- 3°,在此序列的下游处切割DNA[22, 23]。ZFNs对DNA的切割需要两个ZFN单体以精确的方向分别结合至靶序列两侧,一个FokⅠ先与DNA反应,激活切割域,再与另一个FokⅠ反应形成二聚物并切割DNA (图1A)。FokⅠ酶切结构域二聚化是ZFN切割DNA所需的条件,也利于减少对DNA的非特异剪切,因为激活FokⅠ催化域并切割DNA磷酸键需要二者形成二聚体[7, 21, 23, 24]。此外,ZFP与FokⅠ之间的连接序列及其长度同样会影响ZFN的活性及靶向特异 性[25]。2001年,Bikikova等[26]将特制的ZFN注射于非洲爪蟾卵母细胞核后,发现ZFN可靶向切割DNA的特定序列并激发同源重组。随后,该技术在黑腹果蝇(Drosophila melanogaster)基因研究上得到应用[17, 27]。基于模块微组装策略构建的ZFN在斑马鱼(Danio rerio)、兔(Oryctolagus cuniculus)、小鼠(Mus musculus)和猪(Sus scrofa)等物种中也均实现了基因敲除或敲入[28,29,30,31,32]。在TALEN基因打靶技术出现之前,ZFN为实现诸多物种的靶向基因修饰提供了可能[33, 34],然而由于ZFN存在作用靶点受限、打靶效率不稳定、载体构建困难等弊端,限制了ZFN系统的实际应用。
TALEN由特异性识别蛋白与FokⅠ内切酶结合而成的嵌合体,其DNA识别结构域由自然界的效应蛋白—TALEs (transcription activator like effectors)衍生构建所得。TAL效应蛋白模块串联后识别DNA的特定碱基序列[35,36,37],直到2009年两个研究团队分别报道了植物病原菌黄单胞杆菌(Xanthomona)的TALE可结合DNA特定序列[38, 39],这种特性才开始被用于基因打靶载体的构建。TALE整体结构包括由约33~35个氨基酸残基(典型长度为34个氨基酸)的模块串联成的中间重复结构域、N端分泌转运信号结构域以及C端核定位信号与转录激活结构域。中间串联重复序列是TALE对DNA的识别模块及结合域,重复片段间的氨基酸序列高度保守,彼此间差异在于第12、13位的重复可变残基(repeat variable di-residues , RVDs)。RVDs与DNA碱基之间的识别密码为:组氨酸-天冬氨酸(HD)识别C,天冬氨酸-异亮氨酸(NI)识别A,天冬氨酸-甘氨酸(NG)识别T,天冬氨酸-天冬氨酸(NN)或天冬氨酸-赖氨酸(NK)识别G,同时DNA甲基化也会影响RVD与碱基对结合的特异性[38,39,40]。成对的TALEN分子结合到DNA靶序列之后,其碳末端的FokⅠ酶切结构域会发生二聚化,并切割DNA双链(图1B)。另有研究表明,直接与碱基结合的是RVD的第二个氨基酸残基,第一个氨基酸残基主要稳定RVD环状构型[41]。自2010年TALEN技术建立后,陆续有实验室尝试改进相关的合成技术,包括Zhang等[42]基于分层连接(hierarchical ligation)策略构建出TALE工具盒,有效解决了TALE串联重复片段的构建难题。Miller等[37]利用TALE特定截断变异型,将TALEN系统改造为更适合哺乳动物细胞的基因组编辑系统。随着对TALEN靶向机制的了解以及产物合成技术的改善[42,43,44],TALEN技术的应用范围迅速扩展,相继被应用于人类细胞和斑马鱼、小鼠、猴(Macaca fascicularis)、猪等动物的基因组编辑[45]。与ZFN比较,TALEN对细胞的毒性较小,基因靶向特异性更强,且TALEN的构建也较为方便,因此得到了广泛应用。
CRISPR/Cas系统是迄今为止最新的RNA导向式基因组编辑系统,来源于细菌和古菌等原核生物识别和摧毁外源入侵的病毒或质粒等遗传物质时获得的免疫防御系统[46, 47]。化脓链球菌(Streptococcus pyogenes SF370)获得性免疫系统的DNA序列主要包括以下部分:CRISPR序列,分别编码CRISPR序列相关核酸内切酶Cas9、Cas1、Cas2和Cns2的基因片段,以及反式激活RNA (trans-activating RNA,tracrRNA)的编码序列等,其中,CRISPR序列的高度重复片段之间存在非重复的间隔序列。当细菌首次遭到某种病毒或外源质粒入侵时,可将病毒基因的特定序列整合到自身基因组的CRISPR区;当相应病毒再次入侵时,CRISPR区的特定序列可转录生成crRNA前体(pre-crRNA),pre-crRNA经内源RNase III和Cas9等蛋白的催化剪接后,形成crRNA[48,49,50,51,52],引导CRISPR/Cas9复合物寻找病毒同源序列,由Cas9的HNH与RuvC两个酶切域分别切割DNA双链[53,54,55]。基于II型CRISPR/Cas构建的基因打靶系统已经被广泛应用,其系统包括具有核酸内切酶活性的Cas9、靶向特异性的crRNA以及tracrRNA。将crRNA和tracrRNA组合为一条单链的向导RNA (single-strand RNA, sgRNA),极大推动了CRISPR/ Cas9系统的应用[53] (图1C)。2013年,Hwang等[56]率先应用CRISPR/Cas9系统在斑马鱼胚胎内进行高效的基因打靶。此后,CRISPR/Cas9技术被广泛应用在对多种动物基因组靶向修饰的研究。与此同时,研究者利用晶体学等多种技术手段,深入解析了CRISPR/Cas9对DNA靶序列的识别与切割等机理。Jinek等[57]率先解析了Cas9的二叶状晶体结构,电镜显示Cas9分子内沟槽在gRNA诱导下可发生结构重取向反应并形成容纳RNA/DNA的中心通道(central channel),表明RNA的载入可激发Cas9对DNA的捕获并激活CRISPR/Cas9。Sternberg等[58]借助晶体分析与分子内荧光共振能量转移(intramolecular Förster resonance energy transfer)等方法对Cas9 的催化域进行相对定位,认为Cas9切割DNA靶位点的效率与HNH活性构象的状态有关,并揭示了一种确保HNH与RuvC切割DNA的变构通讯模式,即HNH通过动态变构对RuvC的别构调控施加影响,活性状态的HNH可通过二者之间的信号传感器激活RuvC的酶切功能。这些结果支持了Cas9精确切割DNA的多重调控模式,如gRNA载入与DNA定向解螺旋之后,对靶向DNA的正确识别促使HNH变构并触发RuvC催化活性等“校勘”机制。这些分子机制的解析有助于科研人员对Ca9进行遗传改造以适应不同需求的应用。2016年,Komor等[59]基于CRISPR/Cas9与胞嘧啶脱氨酶的融合,构建出可将胞嘧啶转换为胸腺嘧啶的单碱基编辑系统(base editor, BE)。2017年,Gaudelli等[60]将RNA腺苷酸脱氨酶与Cas9突变体融合,经多轮选择与蛋白修饰后,建立了腺嘌呤单碱基转换系统(adenine base editor, ABE),在基因组上成功实现了腺嘌呤到鸟嘌呤的精确转换,再次显示了CRISPR/Cas9系统的基因编辑能力[61]。基于噬菌体辅助持续演化系统(Phage assistant continuous evolution, PACE)得到的xCas9可识别NG等更多类型的PAM序列,在更精确的基因组操作方面具有优势。同时,xCas9靶向基因组特定序列的特异性有所改善,表明CRISPR/ Cas9技术可能会在基因组编辑效率、特异性与PAM可容性等方面实现兼容[62]。CRISPR/Cas9具备了ZFN和TALEN等基因打靶技术未能达到的特异性和精确性,摆脱了合成DNA特异识别模块的繁琐操作,仅需sgRNA引导即可对基因组任意位点进行打靶,切割能力不受基因组甲基化影响[63]。但是,在应用上也面临着脱靶效应[64,65]等困扰。
基因组编辑技术导致的脱靶效应是指由其导致的基因组非预期、不可控的意外断裂。由于基因组中同源序列及高度相似序列的存在,因此ZFN、TALEN与CRISPR 3种基因组编辑技术均存在着潜在脱靶效应,这是基因组编辑技术应用于生物医学研究与临床治疗时所面临的主要瓶颈。如何降低脱靶效应是该领域研究的焦点之一。DNA结合域靶向DNA的特异性不足与酶切域发生非特异性切割,是导致脱靶效应的两大因素。目前,科研人员已找到了针对性的方法来降低脱靶效应,如优化DNA结合域长度,对其进行改善以量化评估并增强DNA结合特异性,对FokⅠ酶改造以及通过选择进化以筛选更具靶向特异性的ZFN/TALEN二聚体等手段,均可弱化由于对基因组特定位点识别能力有限而导致的脱靶。Pattanayak等[66]依据能量补偿模式,解释了ZFN结合能(binding energy)过剩对其靶位点识别影响,认为避免使用DNA结合能过剩的ZFN可减少脱靶。也有研究表明,当构建的ZF数量为4~6时,ZFN对DNA的靶向特异性更强[67]。对核酸内切酶结构域进行改造是增强基因组编辑技术打靶特异性的另一策略。FokⅠ具有随机同源二聚化的性质,因此通过对FokⅠ进行异质化改造,使其转变为必须依赖ZF或TALE等靶向定位结构才能形成稳态二聚化的性质,能在一定程度上避免非特异性酶切的发生[68]。单切口(single-strand break,SSB)趋向于高保真修复且不能诱导NHEJ,因此构建切口酶(nickase)也可降低非特异性酶切的几率,如通过ZFNickase定点诱导DNA形成单链断裂,能有效改进基因组编辑的靶向性[69]。Cas9的D10A突变型Cas9-nickase (Cas9n)具有切口酶性质,结合1对sgRNAs以诱导DNA相应范围内的单链断裂后即可形成DNA双切口,能明显降低CRISPR技术导致的脱靶效应[70,71]。Slaymaker等[72]筛选出具备更强打靶特异性的eSpCas9,并验证了Cas9结合DNA以及DNA重杂化等反应过程对脱靶效应的影响。由于CRISPR/Cas9系统的特异性主要取决于sgRNA的定点靶向能力,考虑到基因组潜在脱靶位点的复杂性,以及gRNA与DNA靶序列之间错配碱基的具体数量与定位对CRISPR/Cas9打靶特异性的影响,因此设计sgRNA时应保证其足够的特异性[73],通过对基因组潜在脱靶位点进行高通量分析并量化评估Cas9对特定位点的潜在脱靶效应,可为sgRNA的选择与相关的脱靶检测提供依据[74]。
3 基因编辑猪在医学领域中的应用
基于ES和同源重组的传统基因打靶技术在猪等缺乏体外稳定培养的ES细胞系的大动物上的应用效率极低,大大限制了基因打靶大动物的制备。ZFN、TALEN和CRISPR/Cas9等基因组编辑技术可定点切割基因组并激发细胞DNA损伤修复,高效介导基因定点修饰,提高基因编辑大动物的制备效率。猪具有与人类相似的解剖学、生理学等特征,遗传亲缘关系和营养代谢生理与人类接近,寿命较长且具有繁殖周期短、产仔量高等特征,允许研究者对疾病模型猪的病程发展进行长期研究,并可通过遗传改造获得移植器官,可用于人类疾病动物模型和器官移植等生物医学领域的研究。基因组编辑技术结合SCNT制备基因编辑猪,可为生物医学研究提供遗传稳定的材料。3.1 利用基因组编辑技术建立人类疾病模型猪
基于基因操作手段获得的人类疾病动物模型对于研究疾病的发病机理、开发医疗诊断新技术具有重要意义。长期以来,人类疾病动物模型以基因修饰小鼠为主,然而小鼠体型较小、寿命较短,生理功能等各方面与人类存在较大差异,与人类具有同样遗传缺陷的小鼠可能无法完全模拟人类疾病特征,同时小鼠模型无法对疾病进程和药物疗效进行长期跟踪,因此限制了其在人类医学上的转化应用,如小鼠模型获得的抗肿瘤药物临床前研究成果仅有约5%可在临床Ⅲ期试验中表现出期望的药效[75]。非人灵长类动物与人类遗传亲缘最为接近,其生理解剖特征与代谢反应等特征与人类相似度最高,基于灵长类动物取得的实验结果能客观地反映人体内的各种相应机制。然而,非人灵长类动物不仅资源匮乏,对其进行基因修饰与胚胎操作也较难实施,而且对非人灵长类动物开展实验也受到伦理限制。以资源丰富的猪作为实验动物,既弥补了非人灵长类动物所面临的资源匮乏,也克服了小鼠寿命短、亲缘关系远等方面的不足,且猪实验成本较低、遗传修饰与胚胎操作技术成熟,作为实验模型受到的伦理争议较小,是理想的大动物模型。具有特定遗传缺陷基因编辑猪可模拟多种人类疾病,以此为平台进行肿瘤、心血管系统疾病、代谢性疾病和神经退行性疾病等的发病机理研究、药物评估等临床前试验,有利于加速推动实验动物研究成果的临床转化。3.1.1 心血管系统疾病模型
建立心血管疾病动物模型对于研究心血管疾病机理与药物研发具有重要意义。小型啮齿类动物与人类的心血管系统差异明显,如小鼠心跳频率每分钟可达500次以上,与人类有较大差距。猪的心血管系统,特别在解剖结构、运动机能等方面,与人类相似度高,是人类血管疾病模型制备的理想对象。胰岛素增敏剂噻唑烷二酮类药物(troglitazone, TZD)是2型糖尿病的治疗药物,可作为配体激活过氧化物酶体增殖物激活受体-γ (peroxisome proliferators- activated receptors-γ, Ppar-γ),对小鼠心血管疾病模型表现出一定的疗效,但在病患的临床使用上却面临着不良心血管事件的限制[76]。PPAR-γ与人类心血管疾病、机体免疫及胰岛素敏感的关系密切。为探究其在人类心血管疾病中的作用,2011年Yang等[77]应用ZFN技术,靶向敲除了猪成纤维细胞中的Ppar-γ基因,对获得单碱基突变细胞系进行SCNT,成功制备出单等位Ppar-γ基因敲除猪。这是首次将锌指核酸酶基因打靶技术应用于猪的内源基因敲除,并结合SCNT技术获得基因敲除猪,该模型可作为心血管疾病发病机理、Ppar-γ的体内精确调控机制以及Ppar-γ靶标药物与治疗新方法开发等研究的对象,在基因治疗和转化医学研究上具有重要意义。家族性肥厚型心脏病(familial hypertrophic cardiomyopathy, HCM) 是主要的遗传性心脏病,HCM的病理机制尚未明确,病因包括编码β-心肌球蛋白重链(β-myosin heavy chain, MyHC)的MYH7基因发生突变。啮齿类等动物的心肌球蛋白亚型异于人类,因此MYH7突变的小鼠模型并不能有效模拟人类的HCM。猪的电生理学等与人类类似,具备作为HCM模型动物的可能。2017年,Montag等[78]利用TALEN技术对猪MYH7位点进行R723G碱基突变并获得单碱基突变的克隆猪,该模型表现出明显的由R723G型MYH7突变导致的HCM早期症状,如心肌排列紊乱、核畸形以及MHY7过表达导致引起的α/β- MyHC失调,但是该克隆猪存活时间极短(产前死亡、产后24 h内死亡),这可能与MYH表达紊乱有关。该研究是首次基于Selection-free SCNT通过诱导点突变构建出的心血管疾病模型猪。由于在医学研究上,病变心脏组织较难获得,因此对HCM的长期研究需要动物模型加以辅助。以猪为代表的大动物模型实验成果有利于研究HCM病人早期心脏衰竭等的遗传机制。
3.1.2 免疫缺陷模型
X染色体上的白细胞介素-2受体共同γ链基因(IL-2RG)编码的受体蛋白对动物体自然杀伤细胞(natural killer cell,NK cell)的发育起着重要作用。IL-2RG缺陷会导致NK细胞活性丧失,机体免疫功能严重受损。2013年,Watanabe等[79]向猪胎儿成纤维细胞转入ZFN表达载体后,靶向敲除了细胞的IL2RG基因,经SCNT得到的IL2RG-KO猪表现出与人类X染色体连锁重症联合免疫缺陷(X-severe combined immunodeficiency disease, X-SCID)相似的症状,包括一个胸腺完全缺失、T与NT细胞量不足等。在成熟前T细胞发育为成熟T细胞以及成熟前B细胞发育为成熟B细胞的免疫组库分化过程中,发生V(D)J基因片段的重排,形成淋巴细胞表面的高度多样性抗体可变区的编码基因。V(D)J重排的一个早期步骤是B、T细胞内IG或TCR基因SRR双链断裂,该过程由重组激活基因(Recombination Activating gene 1/2, RAG1/2)的蛋白产物催化,RAG1/2中任意一者缺失都会导致T、B淋巴细胞分化阻滞、外周血不具成熟淋巴细胞而引发SCID[80,81]。猪与人类免疫系统具有相似性,RAG突变猪可模拟人类SCID,为进一步解析SCID机制和探究治疗方法提供帮助。2014年,Lee等[82]利用TALEN系统对猪体细胞的RAG2进行靶向修饰,经SCNT后获得的RAG2突变纯合子胸腺异常,并有脾脏内炎症与细胞凋亡等发育停滞现象。在接受人类诱导多能干细胞移植后,RAG2基因修饰猪发育为成熟的三胚层畸胎瘤,说明该RAG2-KO猪具有免疫缺陷的表 型,是成功的SCID大动物模型。同年,Huang等[83]根据巴马猪的RAG1、RAG2基因,分别设计了TALEN表达载体,对成纤维细胞打靶后建立RAG1、RAG2基因双敲除的细胞系,并克隆出RAG1/2双敲猪,RAG1/2功能障碍导致胸腺胰腺发育出现缺陷,免疫器官内成熟T、B淋巴细胞大幅减少且淋巴细胞V(D)J基因片段重排消失,表现出典型的SCID特征。由于小鼠的生理指标以及炎症免疫反应等机制与人类差距较大,因此基于SCID小鼠模型进行的药物筛选与临床评价、干细胞治疗长期跟踪等方面的研究,获得的成果难以有效地转化为临床应用,而SCID模型猪的应用有望填补这方面的空白。同时,免疫缺陷疾病的致病因素复杂,制备免疫缺陷模型猪有利于研究者对相关功能障碍基因与环境因素进行筛查,推动人体免疫细胞的分化以及机体免疫系统的建立或损伤等研究。
3.1.3 脂代谢异常疾病模型
家族性高胆固醇血症(familial hypercholesterolemia, FH)作为一种导致人体脂代谢紊乱的常染色体单基因显性遗传病,其发病源于低密度脂蛋白受体(low density lipoprotein receptor,LDLR)基因缺陷,致使细胞表面LDLR蛋白功能障碍,LDLR无法介导血液中携带胆固醇的LDL、β-VLDL进入细胞进行清除代谢,导致二者血浆浓度增高继而引发动脉粥样硬化(atherosclerosis)等临床症状。2012年,Carlson等[84]利用TALEN打靶技术敲除猪成纤维细胞基因组的LDLR,经SCNT后获得LDLR-/- 克隆猪,对模拟人类FH等脂代谢综合征具有重要生物医学价值。2017年,Huang等[85]将靶向ApoE (apolipoprotein E)及LDLR基因的sgRNA与Cas9载体同时转入巴马小型猪的胚胎成纤维细胞后,获得11个ApoE与LDLR基因双敲的细胞克隆;经SCNT后获得6头ApoE-/-/LDLR-/-后代,其血清低密度脂蛋白胆固醇(LDL-C)、总胆固醇(TC)和LDL的主要载脂蛋白APOB浓度较阴性猪显著增高,表现出动脉粥样硬化相关的脂代谢异常。该研究首次获得脂代谢紊乱及血管粥样硬化的基因修饰猪。猪的消化系统和新陈代谢接近人类,ApoE/LDLR双基因修饰猪等代谢性疾病猪模型为研究人类代谢性疾病分子机理、开发相关药物和治疗手段提供重要动物平台。
3.1.4 癌症模型
世界首例癌症相关基因打靶猪——重组腺相关病毒介导的乳腺癌相关基因1 (breast cancer associated gene 1, BRCA1)敲除猪于2010年被报道[86]。随着CRISPR/Cas9等新型基因组编辑技术的出现,构建可模拟人类癌症的克隆猪发展也逐步加快。2017年,Wang等[87]利用TALEN 技术,在猪基因组上插入Cas9基因,获得可在Cre酶诱导下表达Cas9核酸酶分子的基因编辑猪。研究人员利用包含重组酶与靶向肿瘤相关基因的gRNAs慢病毒感染猪的肺脏后,成功诱导了猪肺癌相关抑癌基因以及原癌基因的突变,率先在体内利用基因组编辑技术建立原发性肺癌大动物模型。该研究成果的技术突破之一在于Cas9条件性表达体系的建立,达到了跳过胚胎注射和SCNT等阶段,仅需使用重组酶即可启动sgRNA引导的体内基因组编辑的目的。该基因编辑猪的建立,将推动猪基因功能研究,加快生物医药研究与重要基因修饰模型的制备,人类癌症模型猪将在癌症治疗策略与诊断技术研发、药物评估中具有重要作用。
3.1.5 神经退行性疾病模型
亨廷顿舞蹈症(Huntington disease, HTT)、脊髓侧缩硬化症(Amyotrophic Lateral Sclerosis, ALS)和帕金森病(Parkinson’s disease, PD)等神经退行性疾病的特征之一是伴随年龄增长逐渐加重的神经病理学症状与选择性神经元退化。神经退行性疾病模型猪可模拟病患脑组织致病蛋白沉淀聚集、神经元死亡等特征病变,显示出比转基因小鼠模型更为优越的临床前应用前景。2015年,Zhou等[88]应用CRISPR/ Cas9技术获得了PARK2与PINK1基因突变的猪细胞系,进行核移植克隆后得到基因编辑猪,经检测,克隆猪的神经元无法表达PINK 1与Parkin蛋白,而且7月龄猪未表现出明显的帕金森综合征,与人类神经退行性疾病随着年龄增长愈渐明显的病程相符。该研究首次实现了一个世代内大动物双基因的等位敲除,加速了大动物疾病模型的建立和异种器官移植研究进展。亨廷顿舞蹈症是常染色体显性遗传病,以特定神经元退化为特征,由亨廷顿基因上CAG密码子重复突变所致。CAG三碱基重复过多会导致毒性蛋白产生,聚集于神经元并引起神经元死亡,最终出现亨廷顿舞蹈症。2018年,Yan等[89]应用CRISPR/Cas9技术精准地将包含150CAG重复的人源亨廷顿突变基因插入猪HTT内源基因位点,经SCNT建立了表达人源突变型HTT的基因编辑猪,这是国际上首次建立的模拟神经退行病人基因突变的大动物模型。克隆猪表现出HTT患者神经元选择性退行病变的典型病理特征以及亨廷顿舞蹈样、呼吸衰竭等疾病表型,且这些疾病特征均可遗传至克隆动物后代。作为神经退行性疾病研究领域的重大突破,亨廷顿基因敲入猪的建立既可促进神经退行性疾病的新药研发筛选、干细胞治疗手段的临床前评价,也为更深入了解神经细胞死亡的机制及寻找有效的治疗方法提供支持。
3.1.6 其他疾病模型
血管性血友病因子(von Willebrand factor, vWF)作为重要的血浆成分,在血凝过程中具有重要作用。vWF基因突变可导致遗传性血管性血友病(von Willebrand disease, vWD)的发生,病人表现为出血时间延长等凝血障碍。Hai等[90]通过将靶向切割vWF的Cas9与sgRNA注射受精卵胞浆后,利用胚胎移植技术高效获得存活的vWF双等位基因敲除猪。猪的心血管与循环系统与人体具有可比性,vWD-KO猪的肺部vWF表达基本被阻断,凝血因子FVⅢ降解失活且机体出血时间延长,与vWD患者的凝血功能障碍等临床表型相似。这是首次利用CRISPR/ Cas9技术构建的具有特定疾病表型的哺乳动物疾病模型,胚胎注射结合胚胎移植的发育方式,为基因修饰大动物模型的快速建立提供了一种可行方法,对人类vWF等遗传性疾病的发病机理与治疗方法的研究起到推动作用。2015年,Zhou等[88]利用Cas9- nickase载体对猪胎儿成纤维细胞进行转染,获得纯合的酪氨酸酶基因(Tyrosinase, TYR)敲除细胞系,经体细胞克隆后建立TYR基因敲除的克隆猪。由于酪氨酸酶的失活,克隆猪体内黑色素等酪氨酸酶反应产物缺失,其皮肤、虹膜异常白化,表现为典型的白化病特征。该研究应用Cas9-nickase,避免了因脱靶效应而对克隆胚胎的发育过程造成过大影响。猪皮肤的结构、厚度、色素沉着及血液供应方面都与人类更为相似,因此该克隆猪可在白化病发病机制方面的研究上发挥作用。纯发-甲型外胚叶发育不良症(ectodermal dysplasia-9, ED-9)患者毛发稀少且甲营养不良。Han等[91]通过单链寡核苷酸结合CRISPR/ Cas9和SCNT的技术路线,成功在猪体内引入导致ED-9的Hoxc13基因无义突变,制备出毛发缺失、毛囊与蹄发育异常的ED-9克隆猪,可应用于ED-9与皮肤癌等疾病的发病机制、动物体毛发再生以及伤口愈合等需要无毛动物模型的研究。
3.2 新型基因组编辑技术在异种器官移植和人源化器官研究上的应用
临床器官移植是治疗终末期器官衰竭的重要方法,器官移植医学领域所面临的一大困境就是器官资源匮乏。异种器官移植和人源化器官培育等方法有望解决这个难题,而猪器官被视为合适的器官移植供体。虽然猪的心脏、肾脏等器官在规模尺寸、生理机能上与特定病理反应上跟人类器官接近,但种属间器官不相容性和猪内源逆转录病毒(porcine endogenous retrovirus, PERV)跨物种感染的隐患仍然限制异种器官移植的临床应用[92, 93]。通过基因编辑技术对器官移植过程中与免疫排斥反应相关的基因加以修饰,有望使猪源器官更加接近人类免疫系统可接受的范围;嵌合体大动物体内培育人源化器官同样是解决移植器官供源匮乏的可能途径。基因编辑猪的制备,有望加速猪-人种间器官移植的研究,推动人源化器官的培育,缓解移植器官供源不足的紧张局面。3.2.1 猪-灵长类器官移植过程的免疫不相容
外源器官植入后与人类免疫系统的不相容阻碍了异种器官移植的临床应用进展。特异性敲除猪体内抗原分子或其合成酶基因可缓解异种器官移植出现的免疫排斥反应。导致宿主对猪器官发生超急性排斥反应(hyperacute rejection)的表面抗原主要包括由GGTA1 (α-1,3-galactosyltransferase)基因编码的α-1,3-galactosyltransferase、CMAH (cytidine monophosphate-N-acetylneuraminic acid hydroxylase)基因编码的Meu5Gc,以及B4GALNT2 (beta-1,4-N-acetyl- galactosa-minyl transferase 2)基因编码的Sda blood group等。α-1,3-galactose (α-gal)表面抗原作为在猪细胞表面广泛表达的多聚糖分子,是猪-灵长类动物器官移植进程中导致超急性排斥反应的最主要抗原分子,因此敲除α-gal的合成酶基因GGTA1,对于解决免疫排斥反应、延长移植器官存活具有重要意义。2002年,Lai等[94]基于传统基因打靶载体介导的同源重组和SCNT技术对猪细胞GGA1基因进行同源置换后,制备出首例敲除半乳糖苷转移酶的克隆猪,对异种器官移植研究的具有重大意义。2011年,Hauschild等[95]应用ZFN技术对GGTA1基因进行高效双敲除,获得细胞表面完全无Gal表达的转基因猪,其抗补体介导溶解(complement-mediated lysis)能力与利用传统同源重组技术获得的转基因猪相似,很大程度上抑制了猪源器官移植人体后超急排斥反应的发生。CMAH编码的N-羟乙酰神经氨酸(N-g1yco1ylneuraminicacid, Neu5Gc)是除Gal之外的主要免疫抗原,Neu5Gc可在GGTA1-KO猪上皮细胞广泛表达[96]。Lutz等[97]结合ZFN技术和SCNT构建出GGTA1/CMAH双基因敲除猪,并证明人体血清对GGTA1/CMAH-KO猪细胞的免疫排斥性较GGTA1- KO猪更小。2017年,Martens等[98]利用CRISPR/Cas9敲除猪胚胎内上述3种糖类抗原的编码基因GGTA1、CMAH和B4GALNT2后克隆出三基因敲除猪,检测发现人类血清对三基因敲除细胞的异种免疫抗性结合显著减少。这些特异性抗原敲除猪将在抗免疫排斥药物的筛选、建立更安全器官供体、临床转化等研究上发挥重要作用,一系列技术路线的建立也为最终培育出适用于临床应用的猪器官奠定基础。尽管目前应用基因修饰技术降低猪异种器官移植中的免疫排斥反应已取得了显著进展,然而在实际应用中仍有诸多问题需要解决,猪器官的异种移植仍然任重而道远。
3.2.2 PERV的跨物种感染
PERV属γ-逆转录病毒(γ-retroviruses)。目前尽管尚未有PERV感染人体的报道,但PERV感染体外培养条件下的人类细胞并整合入细胞基因组仍然是猪器官移植必须解决的隐患。为克服猪-人器官移植过程中PERV传播至宿主这一隐患,在实现了对PK15细胞内62拷贝的PERV pol序列的靶向敲除 后[99],2017年,Niu等[100]基于CRISPR/Cas9系统联合细胞凋亡抑制因子等策略,培育出全部PERV pol拷贝突变、PERV失活的克隆猪。检测发现,克隆猪不仅生理表现正常,而且其PERV对人类细胞的感染率大幅降低,从而消除了器官移植中PERV感染的潜在隐患。这一研究成果通过基因组修饰解决了异种器官移植中潜在的内源病毒感染问题,为移植医学研究提供相对稳定安全的研究材料,再次展示了基因组编辑技术对推动解决供体器官紧缺局面的意义,“定制”异种器官的医学应用前景光明。
3.2.3 培育人源化器官
人类多能干细胞(human pluripotent stem cells, hPSCs)具备多潜能分化能力,可在动物胚胎内整合发育,通过囊胚间的互补,可形成异种嵌合体并获得再生器官。以小鼠为宿主培育hPSC衍生的人-小鼠异种嵌合体的效率低,以猪为活体系统培育人-猪嵌合体则有望培育出正常的人源器官。2013年,Matsunaria等[101]利用异体猪的卵裂球胚胎对因Hes1基因表达异常而导致胰腺发育缺陷的克隆猪胚进行补偿,成功制备出健康可育的嵌合体猪,胰腺器官由异体胚胎发育而来,证实了同种异源间囊胚互补与再生器官的可行性。2017年,Wu等[102]应用CRISPR/Cas9系统对猪胚胎中胰腺发育相关基因PDX1进行敲除,为结合hPSC移植建立大动物体内再生人类胰腺的平台奠定基础。随后,Wu等[103]利用Cas9/gRNA获得小鼠基因修饰胚,建立了多能囊胚补偿平台,研究人员通过将大鼠PSC注射进器官发育缺陷小鼠的胚胎内,验证了大鼠PSC对嵌合体的生成以及再生器官发育的贡献作用。该研究还首次评估了基于naive、intermediated以及primed等不同分化状态的hiPSCs (human induced pluripotent stem cells)对人-猪嵌合胚胎发育的作用,不同状态的hiPSCs在猪胚胎中形成不等程度的嵌合,为在动物体内再生人类器官打下了理论基础。这项研究有力地推动基于基因组编辑技术制备人-猪嵌合胚胎以及人类器官再生的研究进程,同时也必须认识到,以基因编辑猪为载体再生人体器官的科学进程仍受多方面条件的制约,既包括对胚胎微环境调控尚缺精确认识等技术短板,也包括研究过程中必须面临道德与监管等问题。
4 结语与展望
基因组编辑技术已经成为生命科学发展进程的里程碑式的突破,是生命科学研究方法的革新。多种生物基因组测序的相继完成,为解析具体基因功能、创造符合研究目的遗传变异提供了资源。CRISPR/Cas9等基因组编辑技术可对动物体基因进行高效精确的修饰,同时可以避免外源遗传物质整合入宿主基因组,体现出传统转基因技术无可比拟的优势。基于基因组编辑技术与体细胞克隆技术的策略制备基因编辑猪,在遗传病病理研究、医疗技术开发等医学研究的应用上具有独特优势,并已在模拟人类疾病和异种器官移植研究上获得突破。此外,基因组编辑技术还可以应用于基因治疗,为遗传性的机体代谢障碍、器官衰竭病变的医治提供有别于药物治疗的新思路。利用基因组编辑操作对疾病模型动物开展疾病相关突变基因位点的碱基编辑、基因替换或敲除等基因修复治疗,可为多种遗传病开发有效的治疗手段,并提供重要的临床前安全与策略指导,其医学应用前景巨大。然而,基因组编辑技术的应用也面临着体内导入效率不高、潜在脱靶效应和基因编辑效率不足等缺点,且目前制备的疾病模型猪种类偏少,仍然需要大量工作以建立更多能精确模拟人类疾病表型的疾病模型猪。此外,如何最大限度地解决异种器官移植过程中宿主对外源器官的免疫排斥、彻底清除外源器官携带的内源性病毒仍需继续研究;动物体内培育人源器官的研究应用更是处于初级阶段,相关法律条例的评估与颁布也迫在眉睫。尽管如此,基因编辑大动物的研究无疑将为生物医学的发展注入更大的动力。随着人们对生物遗传机制认识的深入,对猪等大动物进行精确的基因编辑操作将会更为简易和高效,基因编辑猪也会在生物医学研究中得到更为广泛的应用并使得相关领域受益。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:6273866 [本文引用: 1]

DNA molecules that integrate into yeast chromosomes during yeast transformation do so by homologous recombination. We have studied the way in which circular and linear molecules recombine with homologous chromosomal sequences. We show that DNA ends are highly recombinogenic and interact directly with homologous sequences. Circular hybrid plasmids can integrate by a single reciprocal crossover, but only at a low frequency. Restriction enzyme digestion within a region homologous to yeast chromosomal DNA greatly enhances the efficiency of integration. Furthermore, if two restriction cuts are made within the same homologous sequence, thereby removing an internal segment of DNA, the resulting deleted-linear molecules are still able to transform at a high frequency. Surprisingly, the integration of these gapped-linear molecules results in replacement of the missing segment using chromosomal information. The final structure is identical to that obtained from integration of a circular molecule. The integration of linear and gapped-linear molecules, but not of circular molecules, is blocked by the rad52-1 mutation. Consideration of models for plasmid integration and gene conversion suggests that RAD52 may be involved in the DNA repair synthesis required for these processes. Implications of this work for the isolation of integrative transformants, fine-structure mapping, and the cloning of mutations are discussed.
URL [本文引用: 1]
URLPMID:3002636 [本文引用: 1]
We corrected a defective gene residing in the chromosome of a mammalian cell by injecting into the nucleus copies of the same gene carrying a different mutation. We determined how the number, the arrangement, and the chromosomal position of the integrated gene, as well as the number of injected molecules influence the gene-targeting frequency. Recombination between the newly introduced DNA and its chromosomal homolog occurred at a frequency of 1 in 10 3 cells receiving DNA. Correction events were mediated by either double reciprocal recombination or gene conversion. This resulted in sequences in the genome being replaced by sequences of the introduced DNA or, in separate experiments, sequences in the incoming DNA being replaced by chromosomal sequences. Both point mutations and deletion mutations were corrected; however, the nature of the mutation carried by the respective sequence influenced whether the integrated or injected sequence was corrected.
URL [本文引用: 1]
URLPMID:73116 [本文引用: 1]
Abstract Double-strand breaks (DSBs) are recombinogenic lesions in chromosomal DNA in yeast, Drosophila and Caenorhabditis elegans. Recent studies in mammalian cells utilizing the I-Scel endonuclease have demonstrated that in some immortalized cell lines DSBs in chromosomal DNA are also recombinogenic. We have now tested embryonic stem (ES) cells, a non-transformed mouse cell line frequently used in gene targeting studies. We find that a DSB introduced by I-Scel stimulates gene targeting at a selectable neo locus at least 50-fold. The enhanced level of targeting is achieved by transient expression of the I-Scel endonuclease. In 97% of targeted clones a single base pair polymorphism in the transfected homologous fragment was incorporated into the target locus. Analysis of the targeted locus demonstrated that most of the homologous recombination events were 'two-sided', in contrast to previous studies in 3T3 cells in which 'one-sided' homologous events predominated. Thus ES cells may be more faithful in incorporating homologous fragments into their genome than other cells in culture.
URLPMID:8577732 [本文引用: 3]
A long-term goal in the field of restriction-modification enzymes has been to generate restriction endonucleases with novel sequence specificities by mutating or engineering existing enzymes. This will avoid the increasingly arduous task of extensive screening of bacteria and other microorganisms for new enzymes. Here, we report the deliberate creation of novel site-specific endonucleases by linking two different zinc finger proteins to the cleavage domain of Fok I endonuclease. Both fusion proteins are active and under optimal conditions cleave DNA in a sequence-specific manner. Thus, the modular structure of Fok I endonuclease and the zinc finger motifs makes it possible to create ``artificial'' nucleases that will cut DNA near a predetermined site. This opens the way to generate many new enzymes with tailor-made sequence specificities desirable for various applications.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
Genetic variation within and between species is based on recombination of DNA molecules. Recombination also plays a very important role in the repair of damaged DNA. Clarity about the mechanism by which recombination occurs is of profound interest not only to understand how this process assures the maintenance of genome integrity and at the same time is the driving force of evolution, but also for its application in biotechnology. The isolation of genes involved in recombination and the elucidation of the role of many of the corresponding gene products in Escherichia coli and Saccharomyces cerevisiae has formed the basis for comparative analysis in other, more complex eukaryotic systems. The identification of homologous genes from different organisms, including plants, suggests a conservation of the general mechanisms of recombination. Transgenes introduced in an organism may be incorporated in the genome by either homologous or nonhomologous recombination (end joining). The preferred pathway differs strongly between organisms. In plants there is a preference for random integration of the introduced DNA by nonhomologous recombination, which might lead to the accidental inactivation of important genes and to variable and unpredictable expression of the transgene itself. Therefore, there is an urgent need for the development and improvement of techniques for the directed integration of transgenes at specific locations in the genome. The integration of transgenes by homologous recombination would allow specific modification or disruption of endogenous genes, providing a tool for more detailed analysis of gene function. In combination with the recent introduction of site-specific recombination systems from E. coli or yeast into plants, this may lead to the development of versatile systems for modification of the plant genome.
URLPMID:10494824 [本文引用: 1]
The targeting of chromosomal genes via homologous recombination (HR) is an essential tool of reverse genetics as applied for the functional assay of genes within complex genomes. However, in higher plants, foreign DNA integrates almost exclusively at random, non-homologous sites. A variety of environmental parameters known to influence levels of HR do not increase targeting frequencies when combined in gene-targeting experiments. The identification of cellular factors that may control the level of chromosomal HR in plant somatic cells is required. Plant genes encoding proteins similar to those involved in HR in other organisms can be found in the expanding sequence databases. Evidence for evolutionary conservation should help to decipher mechanisms of plant HR and possibly detect limiting factors. At present, however, only one genetic locus influencing levels of chromosomal recombination in plants has been well defined. Here we summarise current knowledge of HR and the status of gene targeting (GT) in plants, focusing on genetic approaches to molecular factors regulating HR levels.
URLPMID:20192759 [本文引用: 1]
Abstract Double-strand DNA breaks are common events in eukaryotic cells, and there are two major pathways for repairing them: homologous recombination (HR) and nonhomologous DNA end joining (NHEJ). The various causes of double-strand breaks (DSBs) result in a diverse chemistry of DNA ends that must be repaired. Across NHEJ evolution, the enzymes of the NHEJ pathway exhibit a remarkable degree of structural tolerance in the range of DNA end substrate configurations upon which they can act. In vertebrate cells, the nuclease, DNA polymerases, and ligase of NHEJ are the most mechanistically flexible and multifunctional enzymes in each of their classes. Unlike repair pathways for more defined lesions, NHEJ repair enzymes act iteratively, act in any order, and can function independently of one another at each of the two DNA ends being joined. NHEJ is critical not only for the repair of pathologic DSBs as in chromosomal translocations, but also for the repair of physiologic DSBs created during variable (diversity) joining [V(D)J] recombination and class switch recombination (CSR). Therefore, patients lacking normal NHEJ are not only sensitive to ionizing radiation (IR), but also severely immunodeficient.
URLPMID:3401455 [本文引用: 1]
Homologous recombination (HR) is critical both for repairing DNA lesions in mitosis and for chromosomal pairing and exchange during meiosis. However, some forms of HR can also lead to undesirable DNA rearrangements. Multiple regulatory mechanisms have evolved to ensure that HR takes place at the right time, place and manner. Several of these impinge on the control of Rad51 nucleofilaments that play a central role in HR. Some factors promote the formation of these structures while others lead to their disassembly or the use of alternative repair pathways. In this article, we review these mechanisms in both mitotic and meiotic environments and in different eukaryotic taxa, with an emphasis on yeast and mammal systems. Since mutations in several proteins that regulate Rad51 nucleofilaments are associated with cancer and cancer-prone syndromes, we discuss how understanding their functions can lead to the development of better tools for cancer diagnosis and therapy.
URL [本文引用: 1]
URLPMID:12730594 [本文引用: 2]
Author information: (1)Department of Biochemistry, University of Utah School of Medicine, 20 North 1900 East, Salt Lake City, UT 84132-3201, USA.
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:20717154 [本文引用: 2]
Abstract Reverse genetics in model organisms such as Drosophila melanogaster, Arabidopsis thaliana, zebrafish and rats, efficient genome engineering in human embryonic stem and induced pluripotent stem cells, targeted integration in crop plants, and HIV resistance in immune cells - this broad range of outcomes has resulted from the application of the same core technology: targeted genome cleavage by engineered, sequence-specific zinc finger nucleases followed by gene modification during subsequent repair. Such 'genome editing' is now established in human cells and a number of model organisms, thus opening the door to a range of new experimental and therapeutic possibilities.
[本文引用: 1]
URLPMID:11491302 [本文引用: 2]
FokI is a bipartite restriction endonuclease that recognizes a non-palindromic DNA sequence, and then makes double-stranded cuts outside of that sequence to leave a 5 overhang. Earlier kinetic and crystallographic studies suggested that FokI might function as a dimer. Here, we show, using dynamic light-scattering, gel-filtration and analytical ultracentrifugation, that FokI dimerizes only in the presence of divalent metal ions. Furthermore, analysis of the DNA-bound complex reveals that two copies of the recognition sequence are incorporated into the dimeric complex and that formation of this complex is essential for full activation of cleavage. These results have broad implications for the mechanism by which monomeric type II endonucleases achieve high fidelity.
URL [本文引用: 1]
FokI is a type IIs restriction endonuclease comprised of a DNA recognition domain and a catalytic domain. The structural similarity of the FokI catalytic domain to the type II restriction endonuclease BamHI monomer suggested that the FokI catalytic domains may dimerize. In addition, the FokI structure, presented in an accompanying paper in this issue of Proceedings, reveals a dimerization interface between catalytic domains. We provide evidence here that FokI catalytic domain must dimerize for DNA cleavage to occur. First, we show that the rate of DNA cleavage catalyzed by various concentrations of FokI are not directly proportional to the protein concentration, suggesting a cooperative effect for DNA cleavage. Second, we constructed a FokI variant, FokN13Y, which is unable to bind the FokI recognition sequence but when mixed with wild-type FokI increases the rate of DNA cleavage. Additionally, the FokI catalytic domain that lacks the DNA binding domain was shown to increase the rate of wild-type FokI cleavage of DNA. We also constructed an FokI variant, FokD483A, R487A, which should be defective for dimerization because the altered residues reside at the putative dimerization interface. Consistent with the FokI dimerization model, the variant FokD483A, R487A revealed greatly impaired DNA cleavage. Based on our work and previous reports, we discuss a pathway of DNA binding, dimerization, and cleavage by FokI endonuclease.
URL [本文引用: 1]
URLPMID:88802 [本文引用: 1]
Chimeric nucleases that are hybrids between a nonspecific DNA cleavage domain and a zinc finger DNA recognition domain were tested for their ability to find and cleave their target sites in living cells. Both engineered DNA substrates and the nucleases were injected into Xenopus laevis oocyte nuclei, in which DNA cleavage and subsequent homologous recombination were observed. Specific cleavage required two inverted copies of the zinc finger recognition site in close proximity, reflecting the need for dimerization of the cleavage domain. Cleaved DNA molecules were activated for homologous recombination; in optimum conditions, essentially 100% of the substrate recombined, even though the DNA was assembled into chromatin. The original nuclease has an 18-amino-acid linker between the zinc finger and cleavage domains, and this enzyme cleaved in oocytes at paired sites separated by spacers in the range of 6 to 18 bp, with a rather sharp optimum at 8 bp. By shortening the linker, we found that the range of effective site separations could be narrowed significantly. With no intentional linker between the binding and cleavage domains, only binding sites exactly 6 bp apart supported efficient cleavage in oocytes. We also showed that two chimeric enzymes with different binding specificities could collaborate to stimulate recombination when their individual sites were appropriately placed. Because the recognition specificity of zinc fingers can be altered experimentally, this approach holds great promise for inducing targeted recombination in a variety of organisms.
[本文引用: 1]
[本文引用: 1]
URLPMID:19628861 [本文引用: 1]
Abstract The toolbox of rat genetics currently lacks the ability to introduce site-directed, heritable mutations into the genome to create knockout animals. By using engineered zinc-finger nucleases (ZFNs) designed to target an integrated reporter and two endogenous rat genes, Immunoglobulin M (IgM) and Rab38, we demonstrate that a single injection of DNA or messenger RNA encoding ZFNs into the one-cell rat embryo leads to a high frequency of animals carrying 25 to 100% disruption at the target locus. These mutations are faithfully and efficiently transmitted through the germline. Our data demonstrate the feasibility of targeted gene disruption in multiple rat strains within 4 months time, paving the way to a humanized monoclonal antibody platform and additional human disease models.
URLPMID:18500334 [本文引用: 1]
We describe the use of zinc-finger nucleases (ZFNs) for somatic and germline disruption of genes in zebrafish (Danio rerio), in which targeted mutagenesis was previously intractable. ZFNs induce a targeted double-strand break in the genome that is repaired to generate small insertions and deletions. We designed ZFNs targeting the zebrafish golden and no tail/Brachyury (ntl) genes and developed a budding yeast-based assay to identify the most active ZFNs for use in vivo. Injection of ZFN-encoding mRNA into one-cell embryos yielded a high percentage of animals carrying distinct mutations at the ZFN-specified position and exhibiting expected loss-of-function phenotypes. Over half the ZFN mRNA-injected founder animals transmitted disrupted ntl alleles at frequencies averaging 20%. The frequency and precision of gene-disruption events observed suggest that this approach should be applicable to any loci in zebrafish or in other organisms that allow mRNA delivery into the fertilized egg.
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URLPMID:21631241 [本文引用: 1]
Zinc-finger nucleases (ZFNs) are a powerful tool that can be used to edit the human genome ad libitum. The technology has experienced remarkable development in the last few years with regard to both the target site specificity and the engineering platforms used to generate zinc-finger proteins. As a result, two phase I clinical trials aimed at knocking out the CCR5 receptor in T cells isolated from HIV patients to protect these lymphocytes from infection with the virus have been initiated. Moreover, ZFNs have been successfully employed to knockout or correct disease-related genes in human stem cells, including hematopoietic precursor cells and induced pluripotent stem cells. Targeted genome engineering approaches in multipotent and pluripotent stem cells hold great promise for future strategies geared toward correcting inborn mutations for personalized cell replacement therapies. This review describes how ZFNs have been applied to models of gene therapy, discusses the opportunities and the risks associated with this novel technology, and suggests future directions for their safe application in therapeutic genome engineering.
URL [本文引用: 1]
URLPMID:3381595 [本文引用: 1]
Molecular Therapy — Nucleic Acids is a new international all open-access journal publishing top-quality basic, translational and clinical research in the broad fields of nucleic acid-based therapeutics to treat and/or correct genetic and acquired disease.
URLPMID:21179091 [本文引用: 2]
Nucleases that cleave unique genomic sequences in living cells can be used for targeted gene editing and mutagenesis. Here we develop a strategy for generating such reagents based on transcription activator-like effector (TALE) proteins from Xanthomonas. We identify TALE truncation variants that efficiently cleave DNA when linked to the catalytic domain of FokI and use these nucleases to generate discrete edits or small deletions within endogenous human NTF3 and CCR5 genes at efficiencies of up to 25%. We further show that designed TALEs can regulate endogenous mammalian genes. These studies demonstrate the effective application of designed TALE transcription factors and nucleases for the targeted regulation and modification of endogenous genes.
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:21248753 [本文引用: 2]
Abstract The ability to direct functional proteins to specific DNA sequences is a long-sought goal in the study and engineering of biological processes. Transcription activator-like effectors (TALEs) from Xanthomonas sp. are site-specific DNA-binding proteins that can be readily designed to target new sequences. Because TALEs contain a large number of repeat domains, it can be difficult to synthesize new variants. Here we describe a method that overcomes this problem. We leverage codon degeneracy and type IIs restriction enzymes to generate orthogonal ligation linkers between individual repeat monomers, thus allowing full-length, customized, repeat domains to be constructed by hierarchical ligation. We synthesized 17 TALEs that are customized to recognize specific DNA-binding sites, and demonstrate that they can specifically modulate transcription of endogenous genes (SOX2 and KLF4) in human cells.
URL [本文引用: 1]
URLPMID:3141260 [本文引用: 1]
Abstract Transcription activator-like effector (TALE) DNA binding proteins show tremendous potential as molecular tools for targeted binding to any desired DNA sequence. Their DNA binding domain consists of tandem arranged repeats, and due to this repetitive structure it is challenging to generate designer TALEs (dTALEs) with user-defined specificity. We present a cloning approach that facilitates the assembly of multiple repeat-encoding DNA fragments that translate into dTALEs with pre-defined DNA binding specificity. This method makes use of type IIS restriction enzymes in two sequential cut-ligase reactions to build dTALE repeat arrays. We employed this modular approach for generation of a dTALE that differentiates between two highly similar DNA sequences that are both targeted by the Xanthomonas TALE, AvrBs3. These data show that this modular assembly system allows rapid generation of highly specific TALE-type DNA binding domains that target binding sites of predefined length and sequence. This approach enables the rapid and flexible production of dTALEs for gene regulation and genome editing in routine and high-throughput applications.
URL [本文引用: 1]
URLPMID:20528693 [本文引用: 1]
Clustered regularly interspaced short palindromic repeats (CRISPRs) along with Cas proteins is a widespread system across bacteria and archaea that causes interference against foreign nucleic acids. The CRISPR/Cas system acts in at least two general stages: the adaptation stage, where the cell acquires new spacer sequences derived from foreign DNA, and the interference stage, which uses the recently acquired spacers to target and cleave invasive nucleic acid. The CRISPR/Cas system participates in a constant evolutionary battle between phages and bacteria through addition or deletion of spacers in host cells and mutations or deletion in phage genomes. This review describes the recent progress made in this fast-expanding field.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:19141480 [本文引用: 1]
An RNA-based gene silencing pathway that protects bacteria and archaea from viruses and other genome invaders is hypothesized to arise from guide RNAs encoded by CRISPR loci and proteins encoded by the cas genes. CRISPR loci contain multiple short invader-derived sequences separated by short repeats. The presence of virus-specific sequences within CRISPR loci of prokaryotic genomes confers resistance against corresponding viruses. The CRISPR loci are transcribed as long RNAs that must be processed to smaller guide RNAs. Here we identified Pyrococcus furiosus Cas6 as a novel endoribonuclease that cleaves CRISPR RNAs within the repeat sequences to release individual invader targeting RNAs. Cas6 interacts with a specific sequence motif in the 5' region of the CRISPR repeat element and cleaves at a defined site within the 3' region of the repeat. The 1.8 angstrom crystal structure of the enzyme reveals two ferredoxin-like folds that are also found in other RNA-binding proteins. The predicted active site of the enzyme is similar to that of tRNA splicing endonucleases, and concordantly, Cas6 activity is metal-independent. cas6 is one of the most widely distributed CRISPR-associated genes. Our findings indicate that Cas6 functions in the generation of CRISPR-derived guide RNAs in numerous bacteria and archaea.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:22745249 [本文引用: 2]
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
URL [本文引用: 1]
URLPMID:21048762 [本文引用: 1]
Bacteria and Archaea have developed several defence strategies against foreign nucleic acids such as viral genomes and plasmids. Among them, clustered regularly interspaced short palindromic repeats (CRISPR) loci together with cas (CRISPR-associated) genes form the CRISPR/Cas immune system, which involves partially palindromic repeats separated by short stretches of DNA called spacers, acquired from extrachromosomal elements. It was recently demonstrated that these variable loci can incorporate spacers from infecting bacteriophages and then provide immunity against subsequent bacteriophage infections in a sequence-specific manner. Here we show that the Streptococcus thermophilus CRISPR1/Cas system can also naturally acquire spacers from a self-replicating plasmid containing an antibiotic-resistance gene, leading to plasmid loss. Acquired spacers that match antibiotic-resistance genes provide a novel means to naturally select bacteria that cannot uptake and disseminate such genes. We also provide in vivo evidence that the CRISPR1/Cas system specifically cleaves plasmid and bacteriophage double-stranded DNA within the proto-spacer, at specific sites. Our data show that the CRISPR/Cas immune system is remarkably adapted to cleave invading DNA rapidly and has the potential for exploitation to generate safer microbial strains.
URLPMID:23360964 [本文引用: 1]
In bacteria, foreign nucleic acids are silenced by clustered, regularly interspaced, short palindromic repeats (CRISPR) CRISPR-associated (Gas) systems. Bacterial type II CRISPR systems have been adapted to create guide RNAs that direct site-specific DNA cleavage by the Cas9 endonuclease in cultured cells. Here we show that the CRISPR-Cas system functions in vivo to induce targeted genetic modifications in zebrafish embryos with efficiencies similar to those obtained using zinc finger nucleases and transcription activator like effector nucleases.
URLPMID:24505130 [本文引用: 1]
Abstract Type II CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated) systems use an RNA-guided DNA endonuclease, Cas9, to generate double-strand breaks in invasive DNA during an adaptive bacterial immune response. Cas9 has been harnessed as a powerful tool for genome editing and gene regulation in many eukaryotic organisms. We report 2.6 and 2.2 angstrom resolution crystal structures of two major Cas9 enzyme subtypes, revealing the structural core shared by all Cas9 family members. The architectures of Cas9 enzymes define nucleic acid binding clefts, and single-particle electron microscopy reconstructions show that the two structural lobes harboring these clefts undergo guide RNA-induced reorientation to form a central channel where DNA substrates are bound. The observation that extensive structural rearrangements occur before target DNA duplex binding implicates guide RNA loading as a key step in Cas9 activation.
URLPMID:26524520 [本文引用: 1]
Cas9 is an RNA-guided DNA endonuclease that targets foreign DNA for destruction as part of a bacterial adaptive immune system mediated by CRISPR (clustered regularly interspaced short palindromic repeats)1,2. Together with single-guide RNAs (sgRNA)3, Cas9 also functions as a powerful genome engineering tool in plants and animals4–6, and efforts are underway to increase the efficiency and specificity of DNA targeting for potential therapeutic applications7,8. Studies of off-target effects have shown that DNA binding is far more promiscuous than DNA cleavage9–11, yet the molecular cues that govern strand scission have not been elucidated. Here we show that the conformational state of the HNH nuclease domain directly controls DNA cleavage activity. Using intramolecular F02rster resonance energy transfer (FRET) experiments to detect relative orientations of the Cas9 catalytic domains when associated with on- and off-target DNA, we find that DNA cleavage efficiencies scale with the extent to which the HNH domain samples an activated conformation. We furthermore uncover a surprising mode of allosteric communication that ensures concerted firing of both Cas9 nuclease domains. Our results highlight a proofreading mechanism beyond initial PAM recognition12and RNA–DNA base-pairing3that serves as a final specificity checkpoint before DNA double-strand break formation.
URLPMID:27096365 [本文引用: 1]
Current genome-editing technologies introduce double-stranded (ds) DNA breaks at a target locus as the first step to gene correction.1,2Although most genetic diseases arise from point mutations, current approaches to point mutation correction are inefficient and typically induce an abundance of random insertions and deletions (indels) at the target locus from the cellular response to dsDNA breaks.1,2Here we report the development of base editing, a new approach to genome editing that enables the direct, irreversible conversion of one target DNA base into another in a programmable manner, without requiring dsDNA backbone cleavage or a donor template. We engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks, and mediate the direct conversion of cytidine to uridine, thereby effecting a C→T (or G→A) substitution. The resulting “base editors” convert cytidines within a window of approximately five nucleotides (nt), and can efficiently correct a variety of point mutations relevant to human disease. In four transformed human and murine cell lines, second- and third-generation base editors that fuse uracil glycosylase inhibitor (UGI), and that use a Cas9 nickase targeting the non-edited strand, manipulate the cellular DNA repair response to favor desired base-editing outcomes, resulting in permanent correction of 6515-75% of total cellular DNA with minimal (typically ≤ 1%) indel formation. Base editing expands the scope and efficiency of genome editing of point mutations.
URLPMID:29720650 [本文引用: 1]
Abstract The spontaneous deamination of cytosine is a major source of transitions from C090004G to T090004A base pairs, which account for half of known pathogenic point mutations in humans. The ability to efficiently convert targeted A090004T base pairs to G090004C could therefore advance the study and treatment of genetic diseases. The deamination of adenine yields inosine, which is treated as guanine by polymerases, but no enzymes are known to deaminate adenine in DNA. Here we describe adenine base editors (ABEs) that mediate the conversion of A090004T to G090004C in genomic DNA. We evolved a transfer RNA adenosine deaminase to operate on DNA when fused to a catalytically impaired CRISPR-Cas9 mutant. Extensive directed evolution and protein engineering resulted in seventh-generation ABEs that convert targeted A090004T base pairs efficiently to G090004C (approximately 50% efficiency in human cells) with high product purity (typically at least 99.9%) and low rates of indels (typically no more than 0.1%). ABEs introduce point mutations more efficiently and cleanly, and with less off-target genome modification, than a current Cas9 nuclease-based method, and can install disease-correcting or disease-suppressing mutations in human cells. Together with previous base editors, ABEs enable the direct, programmable introduction of all four transition mutations without double-stranded DNA cleavage.
URL [本文引用: 1]
近年发展起来的人工核酸酶可通过引起特定位点的DNA双链断裂实现对目的片段的有效编辑。为进一步提高碱基修改的效率和精确度,2016年研究者们利用CRISPR/Cas9识别特定DNA序列的功能,结合胞嘧啶脱氨酶的生化活性发明了将胞嘧啶高效转换为胸腺嘧啶(CT)的嘧啶单碱基编辑系统(base editor)。这一系统虽然能精准实现嘧啶直接转换,大大提高精确基因编辑效率,但美中不足的是无法对嘌呤进行修改。近期,Nature报道了将细菌中的t RNA腺嘌呤脱氨酶定向进化形成具有催化DNA腺嘌呤底物的脱氨酶,将其与Cas9系统融合发明了具有高效催化腺嘌呤转换为鸟嘌呤的新工具—腺嘌呤单碱基编辑系统(ABEs,adenine base editors)。本文总结了单碱基编辑工具的发展历程和最新研究进展,着重介绍ABEs的研发过程,并对单碱基编辑工具今后的应用方向和研发方向进行展望。
URL [本文引用: 1]
近年发展起来的人工核酸酶可通过引起特定位点的DNA双链断裂实现对目的片段的有效编辑。为进一步提高碱基修改的效率和精确度,2016年研究者们利用CRISPR/Cas9识别特定DNA序列的功能,结合胞嘧啶脱氨酶的生化活性发明了将胞嘧啶高效转换为胸腺嘧啶(CT)的嘧啶单碱基编辑系统(base editor)。这一系统虽然能精准实现嘧啶直接转换,大大提高精确基因编辑效率,但美中不足的是无法对嘌呤进行修改。近期,Nature报道了将细菌中的t RNA腺嘌呤脱氨酶定向进化形成具有催化DNA腺嘌呤底物的脱氨酶,将其与Cas9系统融合发明了具有高效催化腺嘌呤转换为鸟嘌呤的新工具—腺嘌呤单碱基编辑系统(ABEs,adenine base editors)。本文总结了单碱基编辑工具的发展历程和最新研究进展,着重介绍ABEs的研发过程,并对单碱基编辑工具今后的应用方向和研发方向进行展望。
URLPMID:29512652 [本文引用: 1]
Programmable DNA nucleases have provided scientists with the unprecedented ability to probe, regulate, and manipulate the human genome. Zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeat-Cas9 system (CRISPR-Cas9) represent a powerful array of tools that can bind to and cleave a specified DNA... [Show full abstract]
URLPMID:23873081 [本文引用: 1]
The Streptococcus pyogenes Cas9 (SpCas9) nuclease can be efficiently targeted to genomic loci by means of single-guide RNAs (sgRNAs) to enable genome editing(1-10). Here, we characterize SpCas9 targeting specificity in human cells to inform the selection of target sites and avoid off-target effects. Our study evaluates >700 guide RNA variants and SpCas9-induced indel mutation levels at >100 predicted genomic off-target loci in 293T and 293FT cells. We find that SpCas9 tolerates mismatches between guide RNA and target DNA at different positions in a sequence-dependent manner, sensitive to the number, position and distribution of mismatches. We also show that SpCas9-mediated cleavage is unaffected by DNA methylation and that the dosage of SpCas9 and sgRNA can be titrated to minimize off-target modification. To facilitate mammalian genome engineering applications, we provide a web-based software tool to guide the selection and validation of target sequences as well as off-target analyses.
URLPMID:23792628 [本文引用: 1]
Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) have rapidly emerged as a facile and efficient platform for genome editing. Here, we use a human cell-based reporter assay to characterize off-target cleavage of CRISPR-associated (Cas) 9-based RGNs. We find that single and double mismatches are tolerated to varying degrees depending on their position along the guide RNA (gRNA)-DNA interface. We also readily detected off-target alterations induced by four out of six RGNs targeted to endogenous loci in human cells by examination of partially mismatched sites. The off-target sites we identified harbored up to five mismatches and many were mutagenized with frequencies comparable to (or higher than) those observed at the intended on-target site. Our work demonstrates that RGNs can be highly active even with imperfectly matched RNA-DNA interfaces in human cells, a finding that might confound their use in research and therapeutic applications.
.
[本文引用: 1]
[本文引用: 1]
URLPMID:21822273 [本文引用: 1]
Abstract Engineered zinc-finger nucleases (ZFNs) are promising tools for genome manipulation, and determining off-target cleavage sites of these enzymes is of great interest. We developed an in vitro selection method that interrogates 10(11) DNA sequences for cleavage by active, dimeric ZFNs. The method revealed hundreds of thousands of DNA sequences, some present in the human genome, that can be cleaved in vitro by two ZFNs: CCR5-224 and VF2468, which target the endogenous human CCR5 and VEGFA genes, respectively. Analysis of identified sites in one cultured human cell line revealed CCR5-224-induced changes at nine off-target loci, though this remains to be tested in other relevant cell types. Similarly, we observed 31 off-target sites cleaved by VF2468 in cultured human cells. Our findings establish an energy compensation model of ZFN specificity in which excess binding energy contributes to off-target ZFN cleavage and suggest strategies for the improvement of future ZFN design.
URLPMID:23222846 [本文引用: 1]
Zinc-finger nucleases (ZFNs) are important tools for genome engineering. Despite intense interest by many academic groups, the lack of robust noncommercial methods has hindered their widespread use. The modular assembly (MA) of ZFNs from publicly available one-finger archives provides a rapid method to create proteins that can recognize a very broad spectrum of DNA sequences. However, three-and four-finger arrays often fail to produce active nucleases. Efforts to improve the specificity of the one-finger archives have not increased the success rate above 25%, suggesting that the MA method might be inherently inefficient due to its insensitivity to context-dependent effects. Here we present the first systematic study on the effect of array length on ZFN activity. ZFNs composed of six-finger MA arrays produced mutations at 15 of 21 (71%) targeted loci in human and mouse cells. A novel drop-out linker scheme was used to rapidly assess three-to six-finger combinations, demonstrating that shorter arrays could improve activity in some cases. Analysis of 268 array variants revealed that half of MA ZFNs of any array composition that exceed an ab initio B-score cutoff of 15 were active. These results suggest that, when used appropriately, MA ZFNs are able to target more DNA sequences with higher success rates than other current methods.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:23992846 [本文引用: 1]
Targeted genome editing technologies have enabled a broad range of research and medical applications. The Cas9 nuclease from the microbial CRISPR-Cas system is targeted to specific genomic loci by a 20 nt guide sequence, which can tolerate certain mismatches to the DNA target and thereby promote undesired off-target mutagenesis. Here, we describe an approach that combines a Cas9 nickase mutant with paired guide RNAs to introduce targeted double-strand breaks. Because individual nicks in the genome are repaired with high fidelity, simultaneous nicking via appropriately offset guide RNAs is required for double-stranded breaks and extends the number of specifically recognized bases for target cleavage. We demonstrate that using paired nicking can reduce off-target activity by 50- to 1,500-fold in cell lines and to facilitate gene knockout in mouse zygotes without sacrificing on-target cleavage efficiency. This versatile strategy enables a wide variety of genome editing applications that require high specificity.
.
URLPMID:24253446 [本文引用: 1]
Abstract RNA-guided endonucleases (RGENs), derived from the prokaryotic adaptive immune system known as CRISPR/Cas, enable targeted genome engineering in cells and organisms. RGENs are ribonucleoproteins that consist of guide RNA and Cas9, a protein component originated from Streptococcus pyogenes. These enzymes cleave chromosomal DNA, whose sequence is complementary, to guide RNA in a targeted manner, producing site-specific DNA double-strand breaks (DSBs), the repair of which gives rise to targeted genome modifications. Despite broad interest in RGEN-mediated genome editing, these nucleases are limited by off-target mutations and unwanted chromosomal translocations associated with off-target DNA cleavages. Here, we show that off-target effects of RGENs can be reduced below the detection limits of deep sequencing by choosing unique target sequences in the genome and modifying both guide RNA and Cas9. We found that both the composition and structure of guide RNA can affect RGEN activities in cells to reduce off-target effects. RGENs efficiently discriminated on-target sites from off-target sites that differ by two bases. Furthermore, exome sequencing analysis showed that no off-target mutations were induced by two RGENs in four clonal populations of mutant cells. In addition, paired Cas9 nickases, composed of D10A Cas9 and guide RNA, which generate two single-strand breaks (SSBs) or nicks on different DNA strands, were highly specific in human cells, avoiding off-target mutations without sacrificing genome-editing efficiency. Interestingly, paired nickases induced chromosomal deletions in a targeted manner without causing unwanted translocations. Our results highlight the importance of choosing unique target sequences and optimizing guide RNA and Cas9 to avoid or reduce RGEN-induced off-target mutations.
URLPMID:26628643 [本文引用: 1]
The RNA-guided endonuclease Cas9 is a versatile genome-editing tool with a broad range of applications from therapeutics to functional annotation of genes. Cas9 creates double-strand breaks (DSBs) at targeted genomic loci complementary to a short RNA guide. However, Cas9 can cleave off-target sites that are not fully complementary to the guide, which poses a major challenge for genome editing. Here, we use structure-guided protein engineering to improve the specificity of Streptococcus pyogenes Cas9 (SpCas9). Using targeted deep sequencing and unbiased whole-genome off-target analysis to assess Cas9-mediated DNA cleavage in human cells, we demonstrate that “enhanced specificity” SpCas9 (eSpCas9) variants reduce off-target effects and maintain robust on-target cleavage. Thus, eSpCas9 could be broadly useful for genome-editing applications requiring a high level of specificity.
URL [本文引用: 1]
URLPMID:25513782 [本文引用: 1]
Abstract CRISPR RNA-guided nucleases (RGNs) are widely used genome-editing reagents, but methods to delineate their genome-wide, off-target cleavage activities have been lacking. Here we describe an approach for global detection of DNA double-stranded breaks (DSBs) introduced by RGNs and potentially other nucleases. This method, called genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq), relies on capture of double-stranded oligodeoxynucleotides into DSBs. Application of GUIDE-seq to 13 RGNs in two human cell lines revealed wide variability in RGN off-target activities and unappreciated characteristics of off-target sequences. The majority of identified sites were not detected by existing computational methods or chromatin immunoprecipitation sequencing (ChIP-seq). GUIDE-seq also identified RGN-independent genomic breakpoint 'hotspots'. Finally, GUIDE-seq revealed that truncated guide RNAs exhibit substantially reduced RGN-induced, off-target DSBs. Our experiments define the most rigorous framework for genome-wide identification of RGN off-target effects to date and provide a method for evaluating the safety of these nucleases before clinical use.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:4690471 [本文引用: 1]
Cryo-EM structures of synaptic RAG complexes reveal a recombination signal sequence (RSS)-induced closed conformation that enables catalytic activation and explain the molecular basis for the 12/23 rule, a dogma in which 12-RSS and 23-RSS flanking the V, D, and J segments are synapsed.
URL [本文引用: 1]
URLPMID:24973446 [本文引用: 1]
Pigs share many physiological, biochemical, and anatomical similarities with humans and have emerged as valuable large animal models for biomedical research. Considering the advantages in immune system resemblance, suitable size, and longevity for clinical practical and monitoring purpose, SCID pigs bearing dysfunctional RAG could serve as important experimental tools for regenerative medicine, allograft and xenograft transplantation, and reconstitution experiments related to the immune system. In this study, we report the generation and phenotypic characterization of RAG1 and RAG2 knockout pigs using transcription activator-like effector nucleases. Porcine fetal fibroblasts were genetically engineered using transcription activator-like effector nucleases and then used to provide donor nuclei for somatic cell nuclear transfer. We obtained 27 live cloned piglets; among these piglets, 9 were targeted with biallelic mutations in RAG1, 3 were targeted with biallelic mutations in RAG2, and 10 were targeted with a monoallelic mutation in RAG2. Piglets with biallelic mutations in either RAG1 or RAG2 exhibited hypoplasia of immune organs, failed to perform V(D)J rearrangement, and lost mature B and T cells. These immunodeficient RAG1/2 knockout pigs are promising tools for biomedical and translational research.
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URLPMID:29146772 [本文引用: 1]
Abstract Despite being time-consuming and costly, generating genome-edited pigs holds great promise for agricultural, biomedical, and pharmaceutical applications. To further facilitate genome editing in pigs, we report here establishment of a pig line with Cre-inducible Cas9 expression that allows a variety of ex vivo genome editing in fibroblast cells including single- and multigene modifications, chromosome rearrangements, and efficient in vivo genetic modifications. As a proof of principle, we were able to simultaneously inactivate five tumor suppressor genes ( TP53 , PTEN , APC , BRCA1 , and BRCA2 ) and activate one oncogene ( KRAS ), achieved by delivering Cre recombinase and sgRNAs, which caused rapid lung tumor development. The efficient genome editing shown here demonstrates that these pigs can serve as a powerful tool for dissecting in vivo gene functions and biological processes in a temporal manner and for streamlining the production of genome-edited pigs for disease modeling. 2017 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
URLPMID:25274063 [本文引用: 2]
The domestic pig has been widely used as an important large animal model. Precise and efficient genetic modification in pig provides a great promise in biomedical research. Recently, clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system has been successfully used to produce many gene-targeted animals. However, these animals have been generated by co-injection of Cas9 mRNA and single-guide RNA (sgRNA) into one-cell stage embryos, which mostly resulted in mosaicism of the modification. One or two rounds of further breeding should be performed to obtain homozygotes with identical genotype and phenotype. To address this issue, gene-targeted somatic cells can be used as donor for somatic cell nuclear transfer (SCNT) to produce gene-targeted animals with single and identical mutations. In this study, we applied Cas9/sgRNAs to effectively direct gene editing in porcine fetal fibroblasts and then mutant cell colonies were used as donor to generate homozygous gene-targeted pigs through single round of SCNT. As a result, we successfully obtained 15 tyrosinase ( TYR ) biallelic mutant pigs and 20 PARK2 and PINK1 double-gene knockout (KO) pigs. They were all homozygous and no off-target mutagenesis was detected by comprehensive analysis. TYR 61/61 pigs showed typical albinism and the expression of parkin and PINK1 were depleted in PARK2 61/61 / PINK1 61/61 pigs. The results demonstrated that single- or double-gene targeted pigs can be effectively achieved by using the CRISPR/Cas9 system combined with SCNT without mosaic mutation and detectable off-target effects. This gene-editing system provides an efficient, rapid, and less costly manner to generate genetically modified pigs or other large animals.
[本文引用: 1]
URLPMID:24481528 [本文引用: 1]
One-step generation of knockout pigs by zygote injection of CRISPR/Cas systemCell Research advance online publication, January 31 2014. doi:10.1038/cr.2014.11Authors: Tang Hai, Fei ...
URLPMID:28011715 [本文引用: 1]
Abstract Atrichia and sparse hair phenotype cause distress to many patients. Ectodermal dysplasia-9 (ED-9) is a congenital condition characterized by hypotrichosis and nail dystrophy without other disorders, and Hoxc13 is a pathogenic gene for ED-9. However, mice carrying Hoxc13 mutation present several other serious disorders, such as skeletal defects, progressive weight loss and low viability. Mouse models cannot faithfully mimic human ED-9. In this study, we generated an ED-9 pig model via Hoxc13 gene knockout through single-stranded oligonucleotides (c.396C090009>090009A) combined with CRISPR/Cas9 and somatic cell nuclear transfer. Eight cloned piglets with three types of biallelic mutations (five piglets with Hoxc13c.396C090009>090009A/c.396C090009>090009A, two piglets with Hoxc13c.396C090009>090009A/c.396C090009>090009A090009+0900091 and one piglet with Hoxc13020040/020040) were obtained. Hoxc13 was not expressed in pigs with all three mutation types, and the expression levels of Hoxc13-regulated genes, namely, Foxn1, Krt85 and Krt35, were decreased. The hair follicles displayed various abnormal phenotypes, such as reduced number of follicles and disarrayed hair follicle cable without normal hair all over the body. By contrast, the skin structure, skeleton phenotype, body weight gain and growth of Hoxc13 knockout pigs were apparently normal. The phenotypes of Hoxc13 mutation in pigs were similar to those in ED-9 patients. Therefore, Hoxc13 knockout pigs could be utilized as a model for ED-9 pathogenesis and as a hairless model for hair regeneration research. Moreover, the hairless pigs without other major abnormal phenotypes generated in this study could be effective models for other dermatological research because of the similarity between pig and human skins. 0008 The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
URL [本文引用: 1]
URLPMID:9338777 [本文引用: 1]
Le Tissier P, Stoye JP, Takeuchi Y, Patience C, Weiss RA.
URLPMID:11778012 [本文引用: 1]
The presence of galactose α-1,3-galactose residues on the surface of pig cells is a major obstacle to successful xenotransplantation. Here, we report the production of four live pigs in which one allele of the α-1,3-galactosyltransferase locus has been knocked out. These pigs were produced by nuclear transfer technology; clonal fetal fibroblast cell lines were used as nuclear donors for embryos reconstructed with enucleated pig oocytes.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:26456528 [本文引用: 1]
Abstract The shortage of organs for transplantation is a major barrier to the treatment of organ failure. Although porcine organs are considered promising, their use has been checked by concerns about the transmission of porcine endogenous retroviruses (PERVs) to humans. Here we describe the eradication of all PERVs in a porcine kidney epithelial cell line (PK15). We first determined the PK15 PERV copy number to be 62. Using CRISPR-Cas9, we disrupted all copies of the PERV pol gene and demonstrated a >1000-fold reduction in PERV transmission to human cells, using our engineered cells. Our study shows that CRISPR-Cas9 multiplexability can be as high as 62 and demonstrates the possibility that PERVs can be inactivated for clinical application of porcine-to-human xenotransplantation. Copyright 2015, American Association for the Advancement of Science.
URLPMID:28798043 [本文引用: 1]
Xenotransplantation is a promising strategy to alleviate the shortage of organs for human transplantation. In addition to the concerns about pig-to-human immunological compatibility, the risk of cross-species transmission of porcine endogenous retroviruses (PERVs) has impeded the clinical application of this approach. We previously demonstrated the feasibility of inactivating PERV activity in an immortalized pig cell line. We now confirm that PERVs infect human cells, and we observe the horizontal transfer of PERVs among human cells. Using CRISPR-Cas9, we inactivated all of the PERVs in a porcine primary cell line and generated PERV-inactivated pigs via somatic cell nuclear transfer. Our study highlights the value of PERV inactivation to prevent cross-species viral transmission and demonstrates the successful production of PERV-inactivated animals to address the safety concern in clinical xenotransplantation.
[本文引用: 1]
URLPMID:28874671 [本文引用: 1]
Abstract Genome editing using programmable nucleases has revolutionized biomedical research. CRISPR-Cas9 mediated zygote genome editing enables high efficient production of knockout animals suitable for studying development and relevant human diseases. Here we report efficient disabling pancreatogenesis in pig embryos via zygotic co-delivery of Cas9 mRNA and dual sgRNAs targeting the PDX1 gene, which when combined with chimeric-competent human pluriopotent stem cells may serve as a suitable platform for the xeno-generation of human tissues and organs in pigs.
URLPMID:28129541 [本文引用: 1]
Abstract Interspecies blastocyst complementation enables organ-specific enrichment of xenogenic pluripotent stem cell (PSC) derivatives. Here, we establish a versatile blastocyst complementation platform based on CRISPR-Cas9-mediated zygote genome editing and show enrichment of rat PSC-derivatives in several tissues of gene-edited organogenesis-disabled mice. Besides gaining insights into species evolution, embryogenesis, and human disease, interspecies blastocyst complementation might allow human organ generation in animals whose organ size, anatomy, and physiology are closer to humans. To date, however, whether human PSCs (hPSCs) can contribute to chimera formation in non-rodent species remains unknown. We systematically evaluate the chimeric competency of several types of hPSCs using a more diversified clade of mammals, the ungulates. We find that na ve hPSCs robustly engraft in both pig and cattle pre-implantation blastocysts but show limited contribution to post-implantation pig embryos. Instead, an intermediate hPSC type exhibits higher degree of chimerism and is able to generate differentiated progenies in post-implantation pig embryos. Copyright 2017 Elsevier Inc. All rights reserved.

{kind=link}
{kind=link}