 ,2, 曹新2
,2, 曹新2Roles of the FGF signaling pathway in regulating inner ear development and hair cell regeneration
Zhi Yang1, Jun Yao,2, Xin Cao2第一联系人:
编委: 袁慧军
收稿日期:2018-03-21修回日期:2018-05-8网络出版日期:2018-07-20
| 基金资助: |
Received:2018-03-21Revised:2018-05-8Online:2018-07-20
| Fund supported: |
作者简介 About authors
杨志,本硕连读生,专业方向:临床医学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (451KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨志, 姚俊, 曹新. FGF信号通路在内耳发育调控和毛细胞再生中的作用. 遗传[J], 2018, 40(7): 515-524 doi:10.16288/j.yczz.17-407
Zhi Yang, Jun Yao, Xin Cao.
成熟脊椎动物的内耳是一种高度精密且复杂的结构,由前庭器和耳蜗组成,分别行使着维持平衡和探测声音的功能。内耳起源于胚胎期增厚的外胚层,在神经胚期,第5菱脑节两侧对称形成清晰可辨的听基板(otic placode, OP),OP内陷形成听窝(otic cup),然后与外胚层演变成听囊(otocyst)[1]。听囊形成并封闭后,一部分成神经细胞从听囊底部分离,向腹内侧移行一小段距离后融合形成螺旋神经节(statoacoustic ganglion, SAG)[2]。此后,球状听囊在经历一系列复杂的变化后,形成背侧的前庭区和腹侧的耳蜗区。内耳发育的过程中需要多种信号 通路的参与,包括FGF、Notch、Wnt、BMP、Shh等[3,4,5]。其中,成纤维生长因子(fibroblast growth factors, FGFs)是一类由Fgf基因家族编码的分泌信号蛋白,FGF信号通路参与了OP的诱导、SAG的形成和内耳感觉上皮细胞的分化等过程,FGF信号通路的异常可以引起包括综合征型耳聋在内的多种遗传病[6]。本文将对FGF信号通路对内耳发育过程的调控及其在毛细胞再生中的作用进行综述,旨在为毛细胞再生过程中FGF信号通路的调控机制及其关键信号因子的研究奠定理论基础。
1 FGF信号通路与内耳发育调控
FGFs家族成员包括FGF1-23,根据FGFs的作用机制可将其分为3类:FGF11-14为非分泌信号且不依赖于FGF受体(fibroblast growth factor receptors, FGFRs)而发挥作用,可以影响神经元的电兴奋性,被称为胞内FGFs;其余两类FGFs皆依赖于FGFRs,通过局部信号的扩散而作用于附近的靶细胞,主要作为胚胎发育中组织形态和器官形成的旁分泌因子(包括FGF1/2/5, FGF3/4/6, FGF7/10/22, FGF8/17/18和FGF9/16/20亚家族)和多种代谢过程中的内分泌信号的内分泌因子(FGF15/19/21/23亚家族)[6]。在FGFs信号通路中,FGFs与细胞膜表面FGFRs结合标志着FGF信号通路的激活。目前有4种包含细胞内酪氨酸激酶结构域的FGFRs (FGFR1~4)。许多研究发现FGFRs细胞内区域的酪氨酸磷酸化可激活细胞内关键的信号通路,包括促分裂素原活化蛋白激酶(mitogenactivated protein kinase, MAPK)、磷脂酰肌醇3-激酶-Akt(phosphoinositide 3-kinase-AKT, PI3K-Akt)、磷脂酶Cγ/蛋白激酶Cα(phospholipase Cγ/proteinkinase Cα, PLCγ/PKCα)和信号转导子与转录激活子(signal transducer and activator of transcription, STAT)信号通路[7]。FGF信号通路在肝脏、肾脏、脑部和骨骼等组织器官的发育以及生理和病理过程中均发挥重要作用,内耳的发育也与FGF信号通路密切相关。1.1 FGF信号通路与OP的诱导
外胚层的前基板区(pre-placodal region, PPR)被认为是内耳及其他颅面部感觉器官发育的起源区域。神经板和中胚层分泌的FGF信号分子可诱导PPR的形成,这一过程由多种转录因子共同决定:GBX2将OTX2的表达限制在PPR前部,可能与维持PPR前部特性相关;而FOXI1/3和SIX1/EYA2复合体以一个正反馈环的形式相互调节,可能与维持PPR后部特性有关(图1)。在原肠胚后期至神经胚早期,来自心和脑中胚层的FGFs可上调这些PPR相关基因在PPR形成区的表达(如鸡中胚层分泌的FGF4和FGF8,蟾蜍神经板分泌的FGF8)[8],而分别来自外胚层和神经板的BMP和Wnt信号可抑制PPR基因的表达[9]。FGF信号可直接促进PPR相关基因的表达,或通过抑制BMP和Wnt信号间接上调PPR相关基因的表达,最终使PPR得以诱导[10]。在内耳发育初期,PPR先分化形成表达PAX2的细胞区域,这一区域既可以作为听祖细胞(otic progenitor cell)前体分化形成内耳结构,也可以分化形成上皮板结构或鳃上板结构,故PAX2区域也被称为耳前体区域或内耳-上鳃基板域(otic-epibranchial progenitor domain, OEPD),而OP由OEPD分化而来。图1
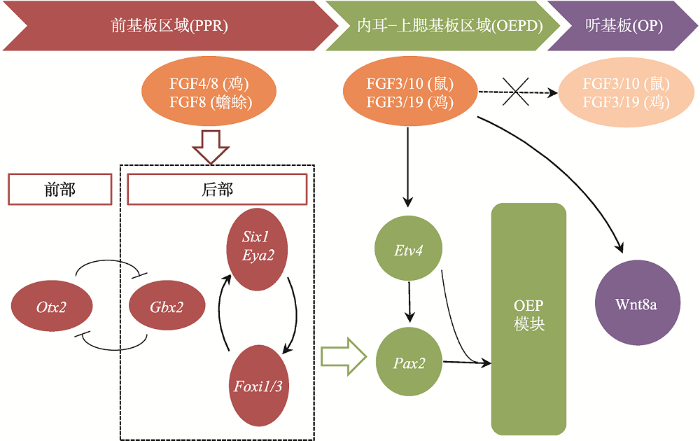 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1OP诱导的基因调控网络
FGF信号通过调控后部PPR区域标志基因(Six1、Eya2、Foxi1/3、Gbx2)的表达诱导PPR形成,随后在FGF信号靶基因Etv4的共同参与下诱导转录因子Pax2的表达;在ETV4和PAX2的协同作用下激活OEP模块向OEPD的诱导;最后FGF信号关闭并激活Wnt信号诱导OP从OEPD中分离。
Fig. 1The regulatory network mediated by the otic placode gene
FGF信号通路在PPR向OEPD转化的过程中起关键的调控作用:FGF3在鸡胚胎的中胚层和后脑 的表达和OEPD的诱导过程相一致,并发挥重要作用[11];但在小鼠Fgf3突变体中,后期内耳发育虽然受损,内耳仍然能被诱导。而Fgf10(在鸡胚胎中为Fgf19)有与Fgf3相似的表达模式,只有当Fgf3与Fgf10同时突变时,才无法诱导OEPD[12]。Chen等[13]建立了第一个内耳早期发育的基因网络,表明PPR后部的Six1、Eya2、Foxi1/3、Gbx2和FGF的目标基因Etv4共同诱导了转录因子Pax2的表达(图1)。而ETV4和PAX2协同作用激活了下游的OEP模块,包括Sox8、Lmx1a、Zbtb16、Sox13、Prdm1、Sall4、Znf385c和Irx5,并最终诱导出OEPD。Anwar等[14]的研究也表明,FGF信号通过激活下游的转录因子(如Etv5, Foxi3和Gbx2)诱导相关基因的转录,其中,Etv5是FGF信号的唯一靶基因。一旦FGF信号激活了其下游基因网络,靶细胞不需要FGF信号也能维持其特性。
多种信号调节机制参与决定了OEPD的区域分化。在OP区域分化过程中,因FGF信号通路负调控基因(如Sprouty和MKP3)的表达上调,FGF信号通路受到抑制;而Wnt信号通路被激活并上调Notch配体Jag1的表达,Jag1与Notch受体结合后进一步增强Wnt信号通路的强度;此外,在鳃上板区域未检测到Sprouty基因的表达[15,16,17]。上述结果说明, FGF信号通路的抑制和Wnt/Notch信号通路的高表达可促进OP方向的分化,而FGF信号通路的高表达和Wnt/Notch信号通路的抑制则可促进鳃上基板的分化[18]。FGF信号通路与Wnt信号通路可通过共同的网络节点发生交互和协同作用,如Wnt信号可通过磷酸化GSK3β使其失活,以允许β-catenin进入细胞核促进目标基因转录,而FGF信号也可通过激活PI3K-AKT级联通路磷酸化GSK3β使得β-catenin转入细胞核发挥作用[19]。有研究发现FGF信号可以诱导一组转录抑制因子(Foxg1、Hesx1、Sall1和Znf217)时空表达,形成一个抑制环以抑制FGF信号效应(如Hesx1可抑制Eya2、Foxi3和Etv5等转录因子),这即可能是FGF信号调控与反馈抑制的分子基础[14]。随后表达于后脑尾部的WNT8a参与诱导OP的形成并与FGF信号密切相关。而在鸡和小鼠中,WNT8a都是由FGF信号诱导的[20]。但是小鼠Wnt8a突变体依然可以诱导出OP,说明可能有其他WNTs弥补了WNT8a的缺失[21]。
1.2 FGF信号通路与SAG发育
FGF的差异性表达调控了SAG发育的不同阶段。SAG负责支配内耳的位听觉,它起源于听囊腹侧的SAG祖细胞,即成神经细胞。这种新特化的成神经细胞迅速与听囊分离一小段距离后经历短暂的增殖以扩大祖细胞数量,这一过程称作转移放大(transit- amplification, TA)效应。这些成神经细胞最终分化成熟为SAG神经元,FGF信号促进了SAG神经元的发育成熟。正常表达的FGF促使听囊腹侧的神经感觉区域快速表达高迁移率族蛋白组(high mobility group, HMG)录因子SOX2,从而诱导神经元发育所必需的神经营养因子NEUROG1 (NGN1)的表达,并促进了内耳的神经发生[22,23,24]。此外,Wang等[25]的研究表明,FGFR-PI3K/Akt通路被激活后,其下游分子SOX9a和ATOH1a结合到它们的目标基因(Eya2和Tlx2)也促进了内耳的神经发生。新生的成神经细胞将表达Notch的配体DELTA1以激活邻近细胞的配体Notch1,通过HES调节的侧抑制途径抑制NGN1表达,从而抑制神经发生。成神经细胞转移之后,神经感觉区域的细胞将发育为感觉祖细 胞[23]。NGN1虽然参与诱导了成神经细胞的形成,但不足以诱导成神经细胞的分层。最近的研究表明,一种组织者基因Goosecoid (Gsc)在FGF信号的调节下引发了成神经细胞的分层[26]。随后,离开听囊的成神经细胞因缺失NGN1引起相关促神经因子NEUROD的表达,这些表达NEUROD的细胞形成了一组增殖和转移的祖细胞,即所谓的转移放大效应。随着这些细胞分化为成熟的SAG神经元,NEUROD停止表达,同时,一些成熟神经元的标志基因被激活。另一方面,FGF信号分子对SAG的发育成熟也有抑制作用,从而形成一个负反馈环路,FGF5即为一个重要的负反馈调控因子。由成熟SAG神经元特异性表达的FGF5不断在听囊外积累,终止了听囊神经发生,从而维持了转移放大过程,同时抑制了处于转移放大状态细胞的成熟。这一机制使得成神经细胞的特化与神经元的分化成熟相适应,因而SAG可在稳定状态下发育(图2)。在斑马鱼Fgf5突变体中可以见到听囊腹侧区NGN1呈高水平持续表达,成神经细胞的形成和分离的阶段也得以延长,且Fgf5基因的敲除只会高度特异地影响SAG的发育,而对听囊其他区域标记物的表达和感觉毛细胞的初期发育无显著影响[2]。
图2
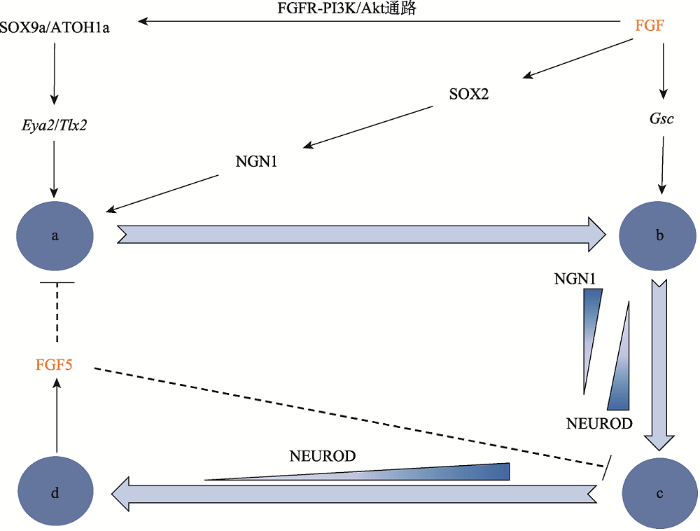 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2FGF信号因子在SAG发育调控机制中的作用
→为正调控作用,---|为负调控作用;a:成神经细胞的特化;b:分层;c:转移放大效应;d:成熟神经元。
Fig. 2Roles of FGF signaling factors in the regulatory network of the SAG development
1.3 FGF信号通路与内耳感觉上皮的发育
FGF信号通路也参与调节了蜗管的长度及Corti器感觉上皮的分化。感觉祖细胞的数量决定了蜗管长度,分泌于感觉上皮的FGF20信号和非感觉上皮的FGF9信号可通过间充质细胞中FGFR1和FGFR2的表达促进感觉祖细胞的增殖并延长蜗管[27]。FGF20及其受体FGFR1调控了感觉祖细胞向毛细胞和支持细胞的分化。碱性螺旋-环-螺旋(basic helix-loop-helix, bHLH)转录因子ATOH1是启动祖细胞向毛细胞分化程序的关键因子,而FGF20/FGFR1可能通过激活MEKK4-ERK通路上调ATOH1的表达以调节毛细胞的发育[28]。此外,SOX2转录因子能够激活ATOH1而诱导鸡听囊中异位毛细胞的形成[29],而FGFR1对MAPK的激活可以稳定SOX2在感觉祖细胞中的表达并促进它们向感觉细胞分 化[30]。在内耳发育过程中,NOTCH的配体JAG1表达早于FGF20,在小鼠Jag1突变体中观察不到FGF20的表达。在毛细胞发育之前,向人工培养的蜗管中加入DAPT(Notch抑制剂)后,FGF20以及毛细胞和支持细胞的发生都受到抑制,而加入FGF20可以逆转抑制效应[31],这些发现表明FGF20在Notch信号通路的调控下参与了前感觉区域的维持和特化。FGF通路也参与了毛细胞和支持细胞的进一步分化。毛细胞在耳蜗形成之后,内耳毛细胞表达FGF8,而外耳毛细胞和支持细胞表达FGFR3[32]。缺乏FGF8/FGFR3信号的小鼠突变体会出现柱细胞受损甚至听力障碍,说明内毛细胞分泌的FGF8可以控制局部的支持细胞分化为柱细胞[33]。在携带Fgfr3突变[p.(Pro244Arg)]的Muenke综合征小鼠模型中可以观察到低频听力损失,Corti器含有过量的柱细胞和少量的Deiters细胞,以及蜗管顶端出现过量的外毛细胞[34],而下调FGF10的表达可以逆转这一模型中耳蜗组织形态结构的紊乱[35]。有趣的是,小鼠Map3k1 (又称MEK激酶, MKKKs)突变体可同时引起FGFR3、FGF8和FGF10表达下调,并出现过量的外毛细胞和Deiters细胞,进而导致听力障碍[36],其机制有待探明。
2 FGF信号通路与毛细胞再生
声创伤、耳毒性药物的使用以及衰老等有害因素都可导致毛细胞受损或缺失[37],从而引起听觉和平衡觉的紊乱。对于哺乳动物,内耳感觉上皮一旦成熟即失去了再生能力,一旦受损将导致不可逆的听力损失。但是,低等脊椎动物的内耳感觉上皮取却始终拥有强大的再生能力,与毛耳蜗细胞相间排列的支持细胞是其再生的主要来源:(1)通过有丝分裂再生,即一个支持细胞可增殖分裂成两个子细胞,其中的一个或两个可以分化成为毛细胞;(2)支持细胞直接向毛细胞转分化[38]。一些关键的因子(ATOH1 和p27Kip1)和细胞信号通路在耳蜗毛细胞再生中发挥重要作用[39],FGF信号通路也是其调控路径之一。2.1 FGF信号通路与毛细胞的自发再生
在毛细胞自发再生过程中,需要先下调FGF信号以促进支持细胞增殖分裂,随后上调FGF信号以促进毛细胞再生。Ku等[40]发现当鸡椭圆囊受耳毒性损伤后支持细胞开始大量增殖,FGF20、FGF3以及FGFR3的表达水平下降,而支持细胞增殖终止时可以观察到FGF20的表达水平上升。此外,加入外源性FGF20亦可以抑制支持细胞增殖,说明FGF信号具有抗增殖效应。Jiang等[41]发现,在支持细胞增殖过程FGF和Notch信号的下调与细胞周期依赖性蛋白激酶(cyclin-dependent kinase, CDK)抑制因子CDKN1b (p27/kip1)的下调相一致,可能FGF信号与Notch信号都可以激活CDKN1b使得细胞退出细胞周期,因此FGF与Notch信号的下调成为支持细胞增殖的触发事件。然而,Lee等[42]在新霉素处理过的斑马鱼侧线中未观察到FGF3和FGF10a的表达,但随着毛细胞的修复最终回复至正常表达水平;而通过药物和基因阻断FGF信号发现,抑制FGF信号可明显抑制神经丘毛细胞的再生,说明毛细胞的再生需要FGF信号的上调;分布于腹背轴的FGFR1a阳性支持细胞可能是产生新毛细胞的前体,而分布于前后轴的FGFR1a阴性支持细胞有显著的增殖能力以补充消耗的毛细胞前体。FGF信号也参与了毛细胞再生的表观遗传调控过程。赖氨酸特异性去甲基化酶1 (lysine-specific demethylase 1, LSD1)在毛细胞再生过程中表达水平显著上调,对LSD1的抑制可导致新霉素处理的斑马鱼神经丘再生毛细胞数量显著降低。推测可能的机制是,毛细胞再生过程需要Wnt信号和FGF信号的激活,包括FGF3、FGF10a以及其下游分子EVT4,对LSD1的抑制导致这些信号的显著下调从而抑制了毛细胞的再生[43]。组蛋白H3赖氨酸9 (H3K9me2)与LSD1发挥类似作用,对H3K9me2的抑制同样可以降低Wnt信号和FGF信号水平以抑制毛细胞再生,此外,在使用H3K9me2抑制剂的同时加入Wnt信号和FGF信号效应物可以逆转这一过程[44]。
低等脊椎动物毛细胞的再生需要包括FGF信号在内的多种信号通路的共同参与,深入探索这些信号通路网络在毛细胞再生过程中的交互作用将为实现人类毛细胞损伤修复提供新的思路。
2.2 FGF信号与干细胞的定向分化
干细胞移植和诱导是实现毛细胞损伤修复(再生)的另一途径[45]。类似于内耳的发育过程,FGF信号在干细胞向毛细胞诱导分化中也发挥了关键作用。FGF3和FGF10参与了OP的诱导,有研究证实FGF3和FGF10可以诱导人类胚胎干细胞(human embryonic stem cells, hESCs)在培养过程中表达内耳发育早期的标志基因产物,如SIX1, EYA1, DLX5,PAX2和PAX8[46,47]。而在hESCs诱导过程中同时使用SU5402抑制FGF信号可导致PAX2、PAX8以及背侧耳囊标记物Oc90分别下降96%、93%和100%[48]。以上说明内耳早期细胞的诱导与FGF信号调控密切相关。此外,由于bFGF(basic FGF, 即FGF2)可以激活不同的FGFR亚型,Oshima等[49]用bFGF替代FGF3和FGF10发现bFGF的诱导效率更高。Ohnishi等[50]还单独用bFGF对人类诱导多能干细胞(induced pluripotent stem cells, iPSCs)向OP细胞诱导,结果表明仅仅使用FGF信号诱导听觉祖细胞的效率还很低,干细胞向内耳毛细胞的诱导还需其他信号通路的共同参与。Ealy等[51]在hESCs由非神经外胚层向OEPDs的诱导过程中使用bFGF同时激活Wnt信号,结果仅诱导出前部基板标记物PAX6而检测不到后部基板标记物PAX2和PAX8。而当先后激活FGF、Wnt、Bmp和维甲酸通路可以使得后部基板标记物强烈表达。Koehler等[52,53,54]先对胚胎干细胞以BMP和TGFβ抑制剂培养诱导出非神经外胚层,4~5天后加以bFGF和BMP信号抑制剂培养,随后可以观察到PAX8、SOX2、TFAP2A、ECAD和NCAD等标记物的表达,表明将出现类似OEPD的表型,8天后将培养的细胞团块转移到无血清的悬浮培养系统,让其自行引导分化并最终形成内耳类器官,这说明FGF信号通路及其相关信号通路的时空表达在干细胞向内耳毛细胞诱导的过程中发挥重要作用,这一过程更可为研究毛细胞发育的生理和病理机制以及毛细胞再生提供新平台,因而如何调节FGF信号的时空表达以达到最佳的诱导效率具有重要的研究 价值。
3 FGF通路异常与遗传性听力损失相关综合征
FGF信号通路参与了内耳发育的多个过程,其异常可导致内耳发育组织结构异常并导致听觉功能障碍。目前,已经发现多种由FGF通路异常引起的耳聋相关遗传病。如颅缝早闭综合征(craniosynostosis syndromes)合并其他综合征(包括Muenke、Apert、Pfeiffer、Crouzon、Beare-Stevenson和Jackson- Weiss综合征等)、LADD综合征(lacrimo-auriculo- dento-digital syndrome)、LAMM综合征(labyrinthine aplasia, microtia and microdontia syndrome, LAMM syndrome)和Kallmann综合征等[55]。其中最具代表性的是Muenke综合征和LADD综合征。Muenke综合征是一种常染色体显性遗传病,以耳聋、冠状缝颅缝早闭、发育迟缓、腕骨或跟骨的融合和行为异常为特征[56],在新生儿中的发病率为1/10000,占颅缝早闭综合征的8%~10%。FGFR3基因(4p16.3)功能获得性突变(c.749 C>G)导致第250位密码子编码的脯氨酸被精氨酸取代[p.(Pro250Arg)],引发了Muenke综合征[57]。Kruszka等[58]对来自71个家庭的106位携带该突变的患者进行了研究,在51位先证者中有33例(64.7%)是遗传性的,70.8%的患者有不同程度的耳聋。这一突变会影响FGFR3b和FGFR3c这两个亚型并增加其与FGFs的亲和力(如FGF9和FGF10)[35],而FGF8/FGFR3信号对于柱细胞的发育至关重要。过表达的FGF信号分子可导致过多的柱细胞发育分化,而柱细胞对基底膜的运动和毛细胞顶端纤毛的偏移运动起联动作用,其数量异常可导致传动异常,从而阻断机械能向电信号的转变,最终导致耳聋[59]。在小鼠Muenke综合征模型中(包括携带Fgfr3杂合与纯合突变的小鼠),可以观察到异常分化的Corti器,其中柱细胞数量增多,Deiter细胞减少,还出现额外的外毛细胞,仅内毛细胞数量显示正常,这种异常现象在耳蜗低频区(顶回)比在高频区(底回)更加明显。此外,携带Fgfr3纯合突变的小鼠耳聋程度更加严重。事实上,在Muenke综合征患者和小鼠Muenke综合征模型中都有程度不同的低频神经性耳聋,人类可能丧失对20~40 dB声音的听觉,小鼠则可能损失对60 dB以下声音的听觉[35]。
LADD综合征也被称作Levy-Hollister综合征,是一种罕见的多发性先天异常,为常染色体显性遗传病,其特征为耳聋、小而低位的杯状耳、泪腺和唾液腺发育不良以及牙齿和手指或足趾的异常。LADD综合征可以由编码FGF10、FGFR2、FGFR3以及TP63(tumor protein 63)的基因突变导致[60]。FGF10中发现3种错义突变[p.(Ile156Arg)、p.(Cys106Phe)、p.(Lys137stop)],其中,p.(Ile156Arg)突变体无法结合FGFR2b;p.(Cys106Phe)突变体在生理温度下缺乏稳定性,其合成后迅速被降解而无法转运至靶细胞;p.(Lys137stop)突变体缺失FGF10分子的C-端,导致FGF10无表达或表达无活性。此外,FGFR2中发现3种错义突变[p.(Ala628Thr),p.(Ala648Thr),p.(Arg649Ser)]和FGFR3中发现两种错义突变[p.(Asp513Asn),p.(Asp628Asn)]也将使FGF信号表达水平不足以维持细胞正常的生理功能,并导致LADD综合征[61, 62]。
FGF信号通路参与了多器官的发育,其异常可引起包括内耳在内的多种器官的病理变化,阐明导致这些异常表型的基因突变及其致病机制可以为遗传性听力损失相关综合征的临床诊断与治疗提供方向。但FGF信号通路异常对人类内耳结构和功能的影响不仅源于其相关基因的致病性突变,可能还与环境因素、环境-遗传因素的相互作用等密切相关,其具体机制还待进一步阐明。
4 结语与展望
近年来,随着内耳发育生物学研究的深入,内耳发育过程及相关信号调控途径逐步被阐明,多个信号通路的众多信号因子和调控基因通过有序的识别、传递和双向作用,在时空上精确表达并相互协调,从而形成调控网络机制,最终诱导复杂内耳结构和功能的成熟。理解并明确这些信号通路及其关键因子在内耳发育过程中的作用可能会为内耳的组织修复与再生提供新途径。大量动物模型实验已揭示Notch、Wnt、BMP、Shh和FGF等信号通路的网络效应不仅共同调控内耳发育的进程,在内耳毛细胞增殖、分化和再生等方面同样具有重要作用。其中,FGF信号通路在OP的诱导、SAG的形成和内耳感觉上皮的发育过程的调控网络机制中发挥着关键作用,FGF信号还参与了毛细胞再生的调控与hESCs的定向分化过程。由于FGF信号通路在进化上高度保守,因此推测人的内耳发育过程也存在类似的调控作用。然而,基于内耳发育过程调控机制的复杂性,FGF信号通路的调控机制仍未完全明确:为何不同的FGF信号水平可以诱导出不同的细胞命运以及FGF信号如何在多变的发育条件下稳定地诱导出成熟内耳仍需要进一步的研究;FGF信号通路与其他信号通路的交互作用也有待进一步阐明;此外,如何对FGF信号以及其他信号通路关键因子进行精确的时空调控以稳定诱导出具有正常功能和存活能力的毛细胞将是未来的重要研究方向。随着FGF等信号通路在内耳发育过程中调控机制的阐 明,不仅将对听力发生(损失)的生理(病理)过程有更加深入的理解,而且可为靶向调控FGF及其他信号通路关键因子的内耳修复与毛细胞再生奠定理论 基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:4322616 [本文引用: 1]

Various cellular replacement therapies using in vitro generated cells to replace damaged tissue have been proposed as strategies to alleviate hearing loss. All such therapies must involve a complete understanding of the earliest steps in inner ear development; its induction as a thickened plate of cells in the non-neural, surface ectoderm of the embryo, to its internalization as an otocyst embedded in the head mesenchyme of the embryo. Such knowledge informs researchers addressing the feasibility of the proposed strategy and present alternatives if needed. In this review we describe the mechanisms of inner ear induction, concentrating on the factors that steer the fate of ectoderm into precursors of the inner ear. Induction then leads to inner ear morphogenesis and we describe the cellular changes that occur as the inner ear is converted from a superficial placode to an internalized otocyst, and how they are coordinated with a particular emphasis on how the signalling environment surrounding the inner ear influences these processes.
URLPMID:23166517 [本文引用: 2]
Neuroblasts of the statoacoustic ganglion (SAG) initially form in the floor of the otic vesicle during a relatively brief developmental window. They soon delaminate and undergo a protracted phase of proliferation and migration (transit-amplification). Neuroblasts eventually differentiate and extend processes bi-directionally to synapse with hair cells in the inner ear and various targets in the hindbrain. Our studies in zebrafish have shown that Fgf signaling controls multiple phases of this complex developmental process. Moderate levels of Fgf in a gradient emanating from the nascent utricular macula specify SAG neuroblasts in laterally adjacent otic epithelium. At a later stage, differentiating SAG neurons express Fgf5, which serves two functions: First, as SAG neurons accumulate, increasing levels of Fgf exceed an upper threshold that terminates the initial phase of neuroblast specification. Second, elevated Fgf delays differentiation of transit-amplifying cells, balancing the rate of progenitor renewal with neuronal differentiation. Laser-ablation of mature SAG neurons abolishes feedback-inhibition and causes precocious neuronal differentiation. Similar effects are obtained by Fgf5-knockdown or global impairment of Fgf signaling, whereas Fgf misexpression has the opposite effect. Thus Fgf signaling renders SAG development self-regulating, ensuring steady production of an appropriate number of neurons as the larva grows.
URLPMID:25040109 [本文引用: 1]
Abstract The membranous labyrinth of the inner ear is a highly complex organ that detects sound and balance. Developmental defects in the inner ear cause congenital hearing loss and balance disorders. The membranous labyrinth consists of three semicircular ducts, the utricle, saccule, and endolymphatic ducts, and the cochlear duct. These complex structures develop from the simple otic placode, which is established in the cranial ectoderm adjacent to the neural crest at the level of the hindbrain at the early neurula stage. During development, the otic placode invaginates to form the otic vesicle, which subsequently gives rise to neurons for the vestibulocochlear ganglion, the non-sensory and sensory epithelia of the membranous labyrinth that includes three ampullary crests, two maculae, and the organ of Corti. Combined paracrine and autocrine signals including fibroblast growth factor, Wnt, retinoic acid, hedgehog, and bone morphogenetic protein regulate fate determination, axis formation, and morphogenesis in the developing inner ear. Juxtacrine signals mediated by Notch pathways play a role in establishing the sensory epithelium, which consists of mechanosensory hair cells and supporting cells. The highly differentiated organ of Corti, which consists of uniformly oriented inner/outer hair cells and specific supporting cells, develops during fetal development. Developmental alterations/arrest causes congenital malformations in the inner ear in a spatiotemporal-restricted manner. A clearer understanding of the mechanisms underlying inner ear development is important not only for the management of patients with congenital inner ear malformations, but also for the development of regenerative therapy for impaired function.
URLMagsci [本文引用: 1]
<p>在内耳发育过程中, Sonic Hedgehog(Shh)信号通路参与确定了内耳的腹侧极性、螺旋神经元的诱导及毛细胞发育。Shh由菱脑底端分泌, 与顶端产生的Wnt相互拮抗, 共同调节内耳的背腹轴形成。Shh作为神经元细胞的促分裂因子, 能够直接促进螺旋神经元细胞的发育。Shh信号的激活可导致Tbx1对Ngn1的抑制减弱, 间接上调了Ngn1的表达, 调控内耳的神经形成过程。通过调节耳蜗前体细胞的细胞周期, Shh通路参与了内耳毛细胞的分化过程。Shh从蜗管底部至顶部的浓度逐渐降低保证了毛细胞的正常发育顺序。动物实验及对听力障碍患者的研究均表明, Shh通路的传导缺陷将影响靶基因的转录, 进而干扰内耳的正常发育, 引起听力障碍。人类异常Shh信号所致听力障碍的疾病包括Greig cephalopolysyndactyly syndrome (GCPS)、Pallister-Hall syndrome (PHS)、Waardenburg syndrome (WS)及髓母细胞瘤等。文章总结了Shh信号通路在内耳发育调控领域的最新研究进展, 为内耳发育的分子生物学机制及临床应用奠定了理论基础。</p>
URLMagsci [本文引用: 1]
<p>在内耳发育过程中, Sonic Hedgehog(Shh)信号通路参与确定了内耳的腹侧极性、螺旋神经元的诱导及毛细胞发育。Shh由菱脑底端分泌, 与顶端产生的Wnt相互拮抗, 共同调节内耳的背腹轴形成。Shh作为神经元细胞的促分裂因子, 能够直接促进螺旋神经元细胞的发育。Shh信号的激活可导致Tbx1对Ngn1的抑制减弱, 间接上调了Ngn1的表达, 调控内耳的神经形成过程。通过调节耳蜗前体细胞的细胞周期, Shh通路参与了内耳毛细胞的分化过程。Shh从蜗管底部至顶部的浓度逐渐降低保证了毛细胞的正常发育顺序。动物实验及对听力障碍患者的研究均表明, Shh通路的传导缺陷将影响靶基因的转录, 进而干扰内耳的正常发育, 引起听力障碍。人类异常Shh信号所致听力障碍的疾病包括Greig cephalopolysyndactyly syndrome (GCPS)、Pallister-Hall syndrome (PHS)、Waardenburg syndrome (WS)及髓母细胞瘤等。文章总结了Shh信号通路在内耳发育调控领域的最新研究进展, 为内耳发育的分子生物学机制及临床应用奠定了理论基础。</p>
URL [本文引用: 1]
Wnt信号通路在生物发育和维持内环境稳态过程中起着重要作用。Wnt配体通过与Frizzle受体结合参与体轴的形成、细胞分化和细胞命运决定等生命活动。在小鼠内耳发育过程中,Wnt信号通路扮演了重要角色:在内耳发育早期阶段,参与听基板的特化和听泡的形成;在内耳发育后期阶段,调控毛细胞分化及毛细胞纤毛束的定向。本文综述了Wnt信号通路在内耳毛细胞发育分化及再生过程中的研究进展,以期为从事相关领域的科研人员提供参考。
URL [本文引用: 1]
Wnt信号通路在生物发育和维持内环境稳态过程中起着重要作用。Wnt配体通过与Frizzle受体结合参与体轴的形成、细胞分化和细胞命运决定等生命活动。在小鼠内耳发育过程中,Wnt信号通路扮演了重要角色:在内耳发育早期阶段,参与听基板的特化和听泡的形成;在内耳发育后期阶段,调控毛细胞分化及毛细胞纤毛束的定向。本文综述了Wnt信号通路在内耳毛细胞发育分化及再生过程中的研究进展,以期为从事相关领域的科研人员提供参考。
URLPMID:19247306 [本文引用: 2]
Abstract The family of fibroblast growth factors (FGFs) regulates a plethora of developmental processes, including brain patterning, branching morphogenesis and limb development. Several mitogenic, cytoprotective and angiogenic therapeutic applications of FGFs are already being explored, and the recent discovery of the crucial roles of the endocrine-acting FGF19 subfamily in bile acid, glucose and phosphate homeostasis has sparked renewed interest in the pharmacological potential of this family. This Review discusses traditional applications of recombinant FGFs and small-molecule FGF receptor kinase inhibitors in the treatment of cancer and cardiovascular disease and their emerging potential in the treatment of metabolic syndrome and hypophosphataemic diseases.
URLPMID:25772309 [本文引用: 1]
Abstract The signaling component of the mammalian Fibroblast Growth Factor (FGF) family is comprised of eighteen secreted proteins that interact with four signaling tyrosine kinase FGF receptors (FGFRs). Interaction of FGF ligands with their signaling receptors is regulated by protein or proteoglycan cofactors and by extracellular binding proteins. Activated FGFRs phosphorylate specific tyrosine residues that mediate interaction with cytosolic adaptor proteins and the RAS-MAPK, PI3K-AKT, PLC纬, and STAT intracellular signaling pathways. Four structurally related intracellular non-signaling FGFs interact with and regulate the family of voltage gated sodium channels. Members of the FGF family function in the earliest stages of embryonic development and during organogenesis to maintain progenitor cells and mediate their growth, differentiation, survival, and patterning. FGFs also have roles in adult tissues where they mediate metabolic functions, tissue repair, and regeneration, often by reactivating developmental signaling pathways. Consistent with the presence of FGFs in almost all tissues and organs, aberrant activity of the pathway is associated with developmental defects that disrupt organogenesis, impair the response to injury, and result in metabolic disorders, and cancer. For further resources related to this article, please visit the WIREs website.
URLPMID:22327005 [本文引用: 1]
Despite the vital importance of Fgf for otic induction, previous attempts to study otic induction through Fgf misexpression have yielded widely varying and contradictory results. There are also discrepancies regarding the ability of Fgf to induce otic tissue in ectopic locations, raising questions about the sufficiency of Fgf and the degree to which other local factors enhance or restrict otic potential. Using heat shock-inducible transgenes to misexpress Fgf3 or Fgf8 in zebrafish, we found that the stage, distribution and level of misexpression strongly influence the response to Fgf. Fgf misexpression during gastrulation can inhibit or promote otic development, depending on context, whereas misexpression after gastrulation leads to expansion of otic markers throughout preplacodal ectoderm surrounding the head. Elevated Fgf also expands expression of the putative competence factor Foxi1, which is required for Fgf to expand other otic markers. Misexpression of downstream factors Pax2a or Pax8 also expands otic markers but cannot bypass the requirement for Fgf or Foxi1. Co-misexpression of Pax2/8 with Fgf8 potentiates formation of ectopic otic vesicles expressing a full range of otic markers. These findings document the variables critically affecting the response to Fgf and clarify the roles of foxi1 and pax2/8 in the otic response.
URL [本文引用: 1]
URLPMID:26952139 [本文引用: 1]
The sensory organs of the vertebrate head originate from simple ectodermal structures known as cranial placodes. All cranial placodes derive from a common domain adjacent to the neural plate, the preplacodal region, which is induced at the border of neural and non-neural ectoderm during gastrulation. Induction and specification of the preplacodal region is regulated by the fibroblast growth factor, bone morphogenetic protein, WNT, and retinoic acid signaling pathways, and characterized by expression of the EYA and SIX family of transcriptional regulators. Once the preplacodal region is specified, different combinations of local signaling molecules and placode-specific transcription factors, including competence factors, promote the induction of individual cranial placodes along the neural axis of the head region. In this review, we summarize the steps of cranial placode development and discuss the roles of the main signaling molecules and transcription factors that regulate these steps during placode induction, specification, and development. For further resources related to this article, please visit the WIREs website.
URLPMID:7789270 [本文引用: 1]
FGF-3 has been implicated in the development of the hindbrain and otocyst in vertebrate embryos. Since the chicken embryo offers a favourable system in which to study the development of these structures, we have isolated and characterised cDNAs for chicken Fgf-3 and determined its pattern of expression in chick embryos from stage 3 (primitive streak) to stage 25 (early organogenesis). Within the developing cranial neural tube, Fgf-3 exhibits dynamic spatial and temporal expression. During extension of the head process, RNA is detected in the midline of the developing neural plate. In neurulating embryos, transcripts are observed initially in rhombomeres 4 and 5 of the hindbrain and later, in rhombomere 6. During hindbrain development, expression is lost from these rhombomeres, but becomes restricted to rhombomere boundaries, providing an intracellular marker which distinguishes a population of cells within boundary regions. Fgf-3 expression is elevated in ventral and medial boundary regions and is greatly reduced in dorsal parts. Studies of regenerating rhombomere boundaries show that Fgf-3 expression is induced in reforming boundaries when even-numbered rhombomere tissue is grafted next to odd, but not when like is juxtaposed to like. Fgf-3 disappears from boundary regions just prior to the loss of the morphological boundaries suggesting a boundary-associated function. Other sites of expression have also been identified. At early stages of development Fgf-3 is expressed in the epiblast and mesendoderm of the primitive streak, in mesoderm lateral to the streak and in Hensen's node. In older embryos transcripts are detected in the endoderm of the pharyngeal pouches, the ectoderm of the second and third pharyngeal arches and the stomodeum. Expression was also detected in the segmental plate and in the posterior half of the three most-recently generated somites.
URLPMID:12810586 [本文引用: 1]
The inner ear, which contains the sensory organs specialised for audition and balance, develops from an ectodermal placode adjacent to the developing hindbrain. Tissue grafting and recombination experiments suggest that placodal development is directed by signals arising from the underlying mesoderm and adjacent neurectoderm. In mice, Fgf3 is expressed in the neurectoderm prior to and concomitant with placode induction and otic vesicle formation, but its absence affects only the later stages of otic vesicle morphogenesis. We show here that mouse Fgf10 is expressed in the mesenchyme underlying the prospective otic placode. Embryos lacking both Fgf3 and Fgf10 fail to form otic vesicles and have aberrant patterns of otic marker gene expression, suggesting that FGF signals are required for otic placode induction and that these signals emanate from both the hindbrain and mesenchyme. These signals are likely to act directly on the ectoderm, as double mutant embryos showed normal patterns of gene expression in the hindbrain. Cell proliferation and survival were not markedly affected in double mutant embryos, suggesting that the major role of FGF signals in otic induction is to establish normal patterns of gene expression in the prospective placode. Finally, examination of embryos carrying three out of the four mutant Fgf alleles revealed intermediate phenotypes, suggesting a quantitative requirement for FGF signalling in otic vesicle formation.
URLPMID:28264836 [本文引用: 1]
The inner ear is a complex vertebrate sense organ, yet it arises from a simple epithelium, the otic placode. Specification towards otic fate requires diverse signals and transcriptional inputs that act sequentially and/or in parallel. Using the chick embryo, we uncover novel genes in the gene regulatory network underlying otic commitment and reveal dynamic changes in gene expression. Functional analysis of selected transcription factors reveals the genetic hierarchy underlying the transition from progenitor to committed precursor, integrating known and novel molecular players. Our results not only characterize the otic transcriptome in unprecedented detail, but also identify new gene interactions responsible for inner ear development and for the segregation of the otic lineage from epibranchial progenitors. By recapitulating the embryonic programme, the genes and genetic sub-circuits discovered here might be useful for reprogramming na ve cells towards otic identity to restore hearing loss. Summary:Transcriptome analysis and knock down of select transcription factors reveals a genetic hierarchy as cells become committed to inner ear fate.
URLPMID:5522468 [本文引用: 2]
Abstract During development cell commitment is regulated by inductive signals that are tightly controlled in time and space. In response, cells activate specific programmes, but the transcriptional circuits that maintain cell identity in a changing signalling environment are often poorly understood. Specification of inner ear progenitors is initiated by FGF signalling. Here, we establish the genetic hierarchy downstream of FGF by systematic analysis of many ear factors combined with a network inference approach. We show that FGF rapidly activates a small circuit of transcription factors forming positive feedback loops to stabilise otic progenitor identity. Our predictive network suggests that subsequently, transcriptional repressors ensure the transition of progenitors to mature otic cells, while simultaneously repressing alternative fates. Thus, we reveal the regulatory logic that initiates ear formation and highlight the hierarchical organisation of the otic gene network.
URLPMID:16452098 [本文引用: 1]
The otic placode, the anlagen of the inner ear, develops from an ectodermal field characterized by expression of the transcription factor Pax2. Previous fate mapping studies suggest that these Pax2(+) cells will give rise to both otic placode tissue and epidermis, but the signals that divide the Pax2(+) field into placodal and epidermal territories are unknown. We report that Wnt signaling is normally activated in a subset of Pax2(+) cells, and that conditional inactivation of beta-catenin in these cells causes an expansion of epidermal markers at the expense of the otic placode. Conversely, conditional activation of beta-catenin in Pax2(+) cells causes an expansion of the otic placode at the expense of epidermis, and the resulting otic tissue expresses exclusively dorsal otocyst markers. Together, these results suggest that Wnt signaling acts instructively to direct Pax2(+) cells to an otic placodal, rather than an epidermal, fate and promotes dorsal cell identities in the otocyst.
URLPMID:22562792 [本文引用: 1]
Inner ear hair cells are specialized sensory cells essential for auditory function. Previous studies have shown that the sensory epithelium is postmitotic, but it harbors cells that can behave as progenitor cells in vitro, including the ability to form new hair cells. Lgr5, a Wnt target gene, marks distinct supporting cell types in the neonatal cochlea. Here, we tested the hypothesis that Lgr562 cells are Wnt-responsive sensory precursor cells. In contrast to their quiescent in vivo behavior, Lgr562 cells isolated by flow cytometry from neonatal Lgr5EGFPCreERT2/+ mice proliferated and formed clonal colonies. After 10 d in culture, new sensory cells formed and displayed specific hair cell markers (myo7a, calretinin, parvalbumin, myo6) and stereocilia-like structures expressing F-actin and espin. In comparison with other supporting cells, Lgr562 cells were enriched precursors to myo7a62 cells, most of which formed without mitotic division. Treatment with Wnt agonists increased proliferation and colony-formation capacity. Conversely, small-molecule inhibitors of Wnt signaling suppressed proliferation without compromising the myo7a62 cells formed by direct differentiation. In vivo lineage tracing supported the idea that Lgr562 cells give rise to myo7a62 hair cells in the neonatal Lgr5EGFP-CreERT2/+ cochlea. In addition, overexpression of β-catenin initiated proliferation and led to transient expansion of Lgr562 cells within the cochlear sensory epithelium. These results suggest that Lgr5 marks sensory precursors and that Wnt signaling can promote their proliferation and provide mechanistic insights into Wnt-responsive progenitor cells during sensory organ development.
URLPMID:25535395 [本文引用: 1]
正With the support of the National Natural Science Foundation of China and the Ministry of Science and Technology of China,Prof.Li Huawei's laboratory at the EyeEnt Hospital of Fudan University discovered a new function of Notch and a new route to regenerate hair cells from inner ear progenitor cells,which was published in PNAS(2015,112(1):166—171).Notch signaling pathway is known as a fundamental pathway that regulates the cell-fate determination in
URLPMID:18799542 [本文引用: 1]
The development of the vertebrate inner ear is an emergent process. Its progression from a relatively simple disk of thickened epithelium within head ectoderm into a complex organ capable of sensing sound and balance is controlled by sequential molecular and cellular interactions. Fibroblast growth factor (FGF) and Wnt signals emanating from mesoderm and neural ectoderm have been shown to direct inner ear fate. However, the role of these multiple signals during inner ear induction is unclear. We demonstrate that the action of the FGFs and Wnts is sequential, and that their roles support a model of hierarchical fate decisions that progressively restrict the developmental potential of the ectoderm until otic commitment. We show that signalling by Fgf3 and Fgf19 is required to initiate a proliferative progenitor region that is a precursor to both the inner ear and the neurogenic epibranchial placodes. Significantly, we find that only after FGF action is attenuated can the subsequent action of Wnt signalling allow otic differentiation to proceed. In addition, gain and loss of function of Wnt-signalling components show a role for this signalling in repressing epibranchial fate. This interplay of signalling factors ensures the correct and ordered differentiation of both inner ear and epibranchial systems.
URLPMID:4594887 [本文引用: 1]
Background In multiple vertebrate organisms, including chick, Xenopus, and zebrafish, Fibroblast Growth Factor (FGF) and Wnt signaling cooperate during formation of the otic placode. However, in the mouse, although FGF signaling induces Wnt8a expression during induction of the otic placode, it is unclear whether these two signaling pathways functionally cooperate. Sprouty (Spry) genes encode intracellular antagonists of receptor tyrosine kinase signaling, including FGF signaling. We previously demonstrated that the Sprouty1 (Spry1) and Sprouty2 (Spry2) genes antagonize FGF signaling during induction of the otic placode. Here, we investigate cross talk between FGF/SPRY and Wnt signaling during otic placode induction and assess whether these two signaling pathways functionally cooperate during early inner ear development in the mouse. Methods Embryos were generated carrying combinations of a Spry1 null allele, Spry2 null allele, ??-catenin null allele, or a Wnt reporter transgene. Otic phenotypes were assessed by in situ hybridization, semi-quantitative reverse transcriptase PCR, immunohistochemistry, and morphometric analysis of sectioned tissue. Results Comparison of Spry1, Spry2, and Wnt reporter expression in pre-otic and otic placode cells indicates that FGF signaling precedes and is active in more cells than Wnt signaling. We provide in vivo evidence that FGF signaling activates the Wnt signaling pathway upstream of TCF/Lef transcriptional activation. FGF regulation of Wnt signaling is functional, since early inner ear defects in Spry1 and Spry2 compound mutant embryos can be genetically rescued by reducing the activity of the Wnt signaling pathway. Interestingly, we find that although the entire otic placode increases in size in Spry1 and Spry2 compound mutant embryos, the size of the Wnt-reporter-positive domain does not increase to the same extent as the Wnt-reporter-negative domain. Conclusions This study provides genetic evidence that FGF and Wnt signaling cooperate during early inner ear development in the mouse. Furthermore, our data suggest that although specification of the otic placode may be globally regulated by FGF signaling, otic specification of cells in which both FGF and Wnt signaling are active may be more tightly regulated.
URLPMID:20171206 [本文引用: 1]
The inner ear epithelium, with its complex array of sensory, non-sensory, and neuronal cell types necessary for hearing and balance, is derived from a thickened patch of head ectoderm called the otic placode. Mouse embryos lacking both Fgf3 and Fgf10 fail to initiate inner ear development because appropriate patterns of gene expression fail to be specified within the pre-otic field. To understand the transcriptional “blueprint” initiating inner ear development, we used microarray analysis to identify prospective placode genes that were differentially expressed in control and Fgf361/61;Fgf1061/61 embryos. Several genes in the down-regulated class, including Hmx3, Hmx2, Foxg1, Sox9, Has2, and Slc26a9 were validated by in situ hybridization. We also assayed candidate target genes suggested by other studies of otic induction. Two placode markers, Fgf4 and Foxi3, were down-regulated in Fgf361/61;Fgf1061/61 embryos, whereas Foxi2, a cranial epidermis marker, was expanded in double mutants, similar to its behavior when WNT responses are blocked in the otic placode. Assays of hindbrain Wnt genes revealed that only Wnt8a was reduced or absent in FGF-deficient embryos, and that even some Fgf361/61;Fgf1061/+ and Fgf361/61 embryos failed to express Wnt8a, suggesting a key role for Fgf3, and a secondary role for Fgf10, in Wnt8a expression. Chick explant assays showed that FGF3 or FGF4, but not FGF10, were sufficient to induce Wnt8a. Collectively, our results suggest that Wnt8a provides the link between FGF-induced formation of the pre-otic field and restriction of the otic placode to ectoderm adjacent to the hindbrain.
[本文引用: 1]
URLPMID:5481345 [本文引用: 1]
Neurons of the cochleovestibular ganglion (CVG) transmit hearing and balance information to the brain. During development, a select population of early otic progenitors express NEUROG1, delaminate from the otocyst, and coalesce to form the neurons that innervate all inner ear sensory regions. At present, the selection process that determines which otic progenitors activate NEUROG1 and adopt a neuroblast fate is incompletely understood. The transcription factor SOX2 has been implicated in otic neurogenesis, but its requirement in the specification of the CVG neurons has not been established. Here we tested SOX2 requirement during inner ear neuronal specification using a conditional deletion paradigm in the mouse. SOX2 deficiency at otocyst stages caused a near-absence of NEUROG1-expressing neuroblasts, increased cell death in the neurosensory epithelium, and significantly reduced the CVG volume. Interestingly, a milder decrease in neurogenesis was observed in heterozygotes, indicating SOX2 levels are important. Moreover, fate-mapping experiments revealed that the timing of SOX2 expression did not parallel the established vestibular-then-auditory sequence. These results demonstrate that SOX2 is required for the initial events in otic neuronal specification including expression of NEUROG1, although fate-mapping results suggest SOX2 may be required as a competence factor rather than a direct initiator of the neural fate.
URLPMID:5364141 [本文引用: 2]
Abstract Integration between cell signals and bHLH transcription factors plays a prominent role during the development of hair cells of the inner ear. Hair cells are the sensory receptors of the inner ear, responsible for the mechano-transduction of sound waves into electrical signals. They derive from multipotent progenitors that reside in the otic placode. Progenitor commitment is the result of cell signaling from the surrounding tissues that result in the restricted expression of SoxB1 transcription factors, Sox2 and Sox3. In turn, they induce the expression of Neurog1 and Atoh1, two bHLH factors that specify neuronal and hair cell fates, respectively. Neuronal and hair cell development, however, do not occur simultaneously. Hair cell development is prevented during neurogenesis and prosensory stages, resulting in the delay of hair cell development with respect to neuron production. Negative interactions between Neurog1 and Atoh1, and of Atoh1 with other bHLH factors driven by Notch signaling, like Hey1 and Hes5, account for this delay. In summary, the regulation of Atoh1 and hair cell development relies on interactions between cell signaling and bHLH transcription factors that dictate cell fate and timing decisions during development. Interestingly, these mechanisms operate as well during hair cell regeneration after damage and during stem cell directed differentiation, making developmental studies instrumental for improving therapies for hearing impairment.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:25568117 [本文引用: 1]
Fibroblast growth factors (Fgfs) play important roles in developmental processes of the inner ear, including the ontogeny of the statoacoustic ganglia (SAG) and hair cells. However, the detailed genetic mechanism(s) underlying Fgf/Fgfr-dependent otic neural development remains elusive. Using conditional genetic approaches and inhibitory small molecules, we have revealed that Fgfr-PI3K/Akt signaling is mainly responsible for zebrafish SAG development and have determined that Sox9a and Atoh1a act downstream of Fgfr-Akt signaling to specify and/or maintain the otic neuron fate during the early segmentation stage. Sox9a and Atoh1a coregulate numerous downstream factors identified through our ChIP-seq analyses, including Tlx2 and Eya2. Fgfr-Erk1/2 signaling contributes to ultricular hair cell development during a critical period between 9 and 15 hours postfertilization. Our work reveals that a genetic network of the previously known sensory determinant Atoh1 and the neural crest determinant Sox9 plays critical roles in SAG development. These newly uncovered roles for Atoh1and Sox9 in zebrafish otic development may be relevant to study in other species.
URLPMID:27791112 [本文引用: 1]
Neurons of the Statoacoustic Ganglion (SAG), which innervate the inner ear, originate as neuroblasts in the floor of the otic vesicle and subsequently delaminate and migrate toward the hindbrain before completing differentiation. In all vertebrates, locally expressed Fgf initiates SAG development by inducing expression of () in the floor of the otic vesicle. However, not all Ngn1-positive cells undergo delamination, nor has the mechanism controlling SAG delamination been elucidated. Here we report that (), best known for regulating cellular dynamics in the Spemann organizer, regulates delamination of neuroblasts in the otic vesicle. In zebrafish, Fgf coregulates expression of and Ngn1 in partially overlapping domains, with delamination occurring primarily in the zone of overlap. Loss of Gsc severely inhibits delamination, whereas overexpression of Gsc greatly increases delamination. Comisexpression of Ngn1 and induces ectopic delamination of some cells from the medial wall of the otic vesicle but with a low incidence, suggesting the action of a local inhibitor. The medial marker Pax2a is required to restrict the domain of expression, and misexpression of Pax2a is sufficient to block delamination and fully suppress the effects of . The opposing activities of and Pax2a correlate with repression or up-regulation, respectively, of E-cadherin (). These data resolve a genetic mechanism controlling delamination of otic neuroblasts. The data also elucidate a developmental role for consistent with a general function in promoting epithelial-to-mesenchymal transition (EMT).
URLPMID:4434254 [本文引用: 1]
The sensory and supporting cells (SCs) of the organ of Corti are derived from a limited number of progenitors. The mechanisms that regulate the number of sensory progenitors are not known. Here, we show that Fibroblast Growth Factors (FGF) 9 and 20, which are expressed in the non-sensory (Fgf9) and sensory (Fgf20) epithelium during otic development, regulate the number of cochlear progenitors. We further demonstrate that Fgf receptor (Fgfr) 1 signaling within the developing sensory epithelium is required for the differentiation of outer hair cells and SCs, while mesenchymal FGFRs regulate the size of the sensory progenitor population and the overall cochlear length. In addition, ectopic FGFR activation in mesenchyme was sufficient to increase sensory progenitor proliferation and cochlear length. These data define a feedback mechanism, originating from epithelial FGF ligands and mediated through periotic mesenchyme that controls the number of sensory progenitors and the length of the cochlea. DOI: http://dx.doi.org/10.7554/eLife.05921.001 Mammalian ears contain several structures that are involved in hearing. Within the inner ear is a spiral-shaped structure called the cochlea. This contains an array of cells called sensory hair cells that convert sound vibrations into electrical signals, which are then conveyed to the brain. Sounds of differing pitch are detected at different points along the cochlea, so its overall length helps to determine the range of sounds that an individual can hear. In the embryo, sensory hair cells and their associated supporting cells develop from ochlear progenitor cells. The final length of the cochlea is determined by the numbers of progenitor cells that commit to becoming either sensory hair cells or supporting cells. Two proteins called FGF9 and FGF20 are involved in the formation of the cochlea. FGF20 promotes the formation of the hair cells and supporting cells, but the precise roles of both proteins are not clear. Here, Huh et al. studied FGF9 and FGF20 in the inner ear of mice at an early stage of development. The experiments show that these proteins work together to control the number of progenitor cells and the length of the cochlea. FGF20 is produced by the same tissue structure (called an pithelium ) that gives rise to the hair cells and supporting cells. In contrast, FGF9 is produced in another epithelium tissue that produces the cells that line the fluid-filled tubes of the inner ear. The experiments also show that both FGF9 and FGF20 act as signals to cells in an adjacent tissue called the mesenchyme, where they activate other proteins known as FGF receptors. These receptors, in turn, regulate an unknown molecule in the mesenchyme that influences the growth of progenitor cells and the length of the cochlea. Sensory hair cells can be injured or lost by excessive sound exposure, some medications and as part of normal aging. These cells are not replaced, and so their loss is a major cause of permanent hearing loss. Understanding the signals that produce the progenitor cells will take us one step closer to being able to grow these cells in the laboratory for use in therapies to replace or repair damaged sensory hair cells. DOI: http://dx.doi.org/10.7554/eLife.05921.002
URLPMID:26818521 [本文引用: 1]
Mechanosensory hair cells (HCs) residing in the inner ear are critical for hearing and balance. Precise coordination of proliferation, sensory specification, and differentiation during development is essential to ensure the correct patterning of HCs in the cochlear and vestibular epithelium. Recent studies have revealed that FGF20 signaling is vital for proper HC differentiation. However, the mechanisms by which FGF20 signaling promotes HC differentiation remain unknown. Here, we show that mitogen-activated protein 3 kinase 4 (MEKK4) expression is highly regulated during inner ear development and is critical to normal cytoarchitecture and function. Mice homozygous for a kinase-inactive MEKK4 mutation exhibit significant hearing loss. Lack of MEKK4 activity in vivo also leads to a significant reduction in the number of cochlear and vestibular HCs, suggesting that MEKK4 activity is essential for overall development of HCs within the inner ear. Furthermore, we show that loss of FGF20 signaling in vivo inhibits MEKK4 activity, whereas gain of Fgf20 function stimulates MEKK4 expression, suggesting that Fgf20 modulates MEKK4 activity to regulate cellular differentiation. Finally, we demonstrate, for the first time, that MEKK4 acts as a critical node to integrate FGF20-FGFR1 signaling responses to specifically influence HC development and that FGFR1 signaling through activation of MEKK4 is necessary for outer hair cell differentiation. Collectively, this study provides compelling evidence of an essential role for MEKK4 in inner ear morphogenesis and identifies the requirement of MEKK4 expression in regulating the specific response of FGFR1 during HC development and FGF20/FGFR1 signaling activated MEKK4 for normal sensory cell differentiation. Sensory hair cells (HCs) are the mechanoreceptors within the inner ear responsible for our sense of hearing. HCs are formed before birth, and mammals lack the ability to restore the sensory deficits associated with their loss. In this study, we show, for the first time, that MEKK4 signaling is essential for the development of normal cytoarchitecture and hearing function as MEKK4 signaling-deficient mice exhibit a significant reduction of HCs and a hearing loss. We also identify MEKK4 as a critical hub kinase for FGF20-FGFR1 signaling to induce HC differentiation in the mammalian cochlea. These results reveal a new paradigm in the regulation of HC differentiation and provide significant new insights into the mechanism of Fgf signaling governing HC formation.
URLPMID:22292066 [本文引用: 1]
The proneural geneAtoh1is crucial for the development of inner ear hair cells and it requires the function of the transcription factor Sox2 through yet unknown mechanisms. In the present work, we used the chicken embryo and HEK293T cells to explore the regulation ofAtoh1by Sox2. The results show that hair cells derive from Sox2-positive otic progenitors and that Sox2 directly activatesAtoh1through a transcriptional activator function that requires the integrity of Sox2 DNA binding domain.Atoh1activation depends on Sox transcription factor binding sites (SoxTFBS) present in theAtoh13 enhancer where Sox2 directly binds, as shown by site directed mutagenesis and chromatin immunoprecipitation (ChIP). In the inner ear,Atoh1enhancer activity is detected in the neurosensory domain and it depends on Sox2. Dominant negative competition (Sox2HMG-Engrailed) and mutation of the SoxTFBS abolish the reporter activity in vivo. Moreover, ChIP assay in isolated otic vesicles shows that Sox2 is bound to theAtoh1enhancer in vivo. However, besides activatingAtoh1, Sox2 also promotes the expression ofAtoh1negative regulators and the temporal profile ofAtoh1activation by Sox2 is transient suggesting that Sox2 triggers an incoherent feed-forward loop. These results provide a mechanism for the prosensory function of Sox2 in the inner ear. We suggest that sensory competence is established early in otic development through the activation ofAtoh1by Sox2, however, hair cell differentiation is prevented until later stages by the parallel activation of negative regulators ofAtoh1function.
URLPMID:3900395 [本文引用: 1]
Inner ear mechanosensory hair cells transduce sound and balance information. Auditory hair cells emerge from a Sox2-positive sensory patch in the inner ear epithelium, which is progressively restricted during development. This restriction depends on the action of signaling molecules. Fibroblast growth factor (FGF) signalling is important during sensory specification: attenuation of Fgfr1 disrupts cochlear hair cell formation; however, the underlying mechanisms remain unknown. Here we report that in the absence of FGFR1 signaling, the expression of Sox2 within the sensory patch is not maintained. Despite the down-regulation of the prosensory domain markers, p27Kip1, Hey2, and Hes5, progenitors can still exit the cell cycle to form the zone of non-proliferating cells (ZNPC), however the number of cells that form sensory cells is reduced. Analysis of a mutant Fgfr1 allele, unable to bind to the adaptor protein, Frs2/3, indicates that Sox2 maintenance can be regulated by MAP kinase. We suggest that FGF signaling, through the activation of MAP kinase, is necessary for the maintenance of sensory progenitors and commits precursors to sensory cell differentiation in the mammalian cochlea.
URLPMID:22973011 [本文引用: 1]
Hearing loss is becoming an increasingly prevalent problem affecting more than 250 million people worldwide. During development, fibroblast growth factors (FGFs) are required for inner ear development as well as hair cell formation in the mammalian cochlea and thus make attractive therapeutic candidates for the regeneration of sensory cells. Previous findings showed that Fgfr1 conditional knock out mice exhibited hair cell and support cell formation defects. Immunoblocking with Fgf20 antibody in vitro produced a similar phenotype. While hair cell differentiation in mice starts at embryonic day (E)14.5, beginning with the inner hair cells, Fgf20 expression precedes hair cell differentiation at E13.5 in the cochlea. This suggests a potential role for Fgf20 in priming the sensory epithelium for hair cell formation. Treatment of explants with a gamma-secretase inhibitor, DAPT, decreased Fgf20 mRNA, suggesting that Notch is upstream of Fgf20. Notch signaling also plays an early role in prosensory formation during cochlear development. In this report we show that during development, Notch-mediated regulation of prosensory formation in the cochlea occurs via Fgf20. Addition of exogenous FGF20 compensated for the block in Notch signaling and rescued Sox2, a prosensory marker, and Gfi1, an early hair cell marker in explant cultures. We hypothesized that Fgf20 plays a role in specification, amplification, or maintenance of Sox2 expression in prosensory progenitors of the developing mammalian cochlea.
URLPMID:17634195 [本文引用: 1]
The mammalian auditory sensory epithelium (the organ of Corti) contains a number of unique cell types that are arranged in ordered rows. Two of these cell types, inner and outer pillar cells (PCs), are arranged in adjacent rows that form a boundary between a single row of inner hair cells and three rows of outer hair cells (OHCs). PCs are required for auditory function, as mice lacking PCs owing to a mutation in Fgfr3 are deaf. Here, using in vitro and in vivo techniques, we demonstrate that an Fgf8 signal arising from the inner hair cells is the key component in an inductive pathway that regulates the number, position and rate of development of PCs. Deletion of Fgf8 or inhibition of binding between Fgf8 and Fgfr3 leads to defects in PC development, whereas overexpression of Fgf8 or exogenous Fgfr3 activation induces ectopic PC formation and inhibits OHC development. These results suggest that Fgf8-Fgfr3 interactions regulate cellular patterning within the organ of Corti through the induction of one cell fate (PC) and simultaneous inhibition of an alternate fate (OHC) in separate progenitor cells. Some of the effects of both inhibition and overactivation of the Fgf8-Fgfr3 signaling pathway are reversible, suggesting that PC differentiation is dependent upon constant activation of Fgfr3 by Fgf8. These results suggest that PCs might exist in a transient state of differentiation that makes them potential targets for regenerative therapies.
URL [本文引用: 1]
URLPMID:18818193 [本文引用: 1]
The heterozygous Pro250Arg substitution mutation in fibroblast growth factor receptor 3 (FGFR3), which increases ligand-dependent signalling, is the most common genetic cause of craniosynostosis in humans and defines Muenke syndrome. Since FGF signalling plays dosage-sensitive roles in the differentiation of the auditory sensory epithelium, we evaluated hearing in a large group of Muenke syndrome subjects, as well as in the corresponding mouse model (Fgfr3(P244R)). The Muenke syndrome cohort showed significant, but incompletely penetrant, predominantly low-frequency sensorineural hearing loss, and the Fgfr3(P244R) mice showed dominant, fully penetrant hearing loss that was more severe than that in Muenke syndrome individuals, but had the same pattern of relative high-frequency sparing. The mouse hearing loss correlated with an alteration in the fate of supporting cells (Deiters'-to-pillar cells) along the entire length of the cochlear duct, with the most extreme abnormalities found at the apical or low-frequency end. In addition, there was excess outer hair cell development in the apical region. We conclude that low-frequency sensorineural hearing loss is a characteristic feature of Muenke syndrome and that the genetically equivalent mouse provides an excellent model that could be useful in testing hearing loss therapies aimed at manipulating the levels of FGF signalling in the inner ear.
URL [本文引用: 3]
The stereotyped arrangement of cochlear sensory and supporting cells is critical for auditory function. Our previous studies showed that Muenke syndrome model mice (Fgfr3(P244R/+)) have hearing loss associated with a supporting cell fate transformation of two Deiters' cells to two pillar cells. We investigated the developmental origins of this transformation and found that two prospective Deiters' cells switch to an outer pillar cell-like fate sequentially between embryonic day 17.5 (E17.5) and postnatal day 3 (P3). Unexpectedly, the Fgfr3(P244R/+) hearing loss and supporting cell fate transformation are not rescued by genetically reducing fibroblast growth factor 8 (FGF8), the FGF receptor 3c (FGFR3c) ligand required for pillar cell differentiation. Rather, reducing FGF10, which normally activates FGFR2b or FGFR1b, is sufficient for rescue of cochlear form and function. Accordingly, we found that the P244R mutation changes the specificity of FGFR3b and FGFR3c such that both acquire responsiveness to FGF10. Moreover, Fgf10 heterozygosity does not block the Fgfr3(P244R/+) supporting cell fate transformation but instead allows a gradual reversion of fate-switched cells toward the normal phenotype between P5 and at least P14. This study indicates that Deiters' and pillar cells can reversibly switch fates in an FGF-dependent manner over a prolonged period of time. This property might be exploited for the regulation of sensory cell regeneration from support cells.
URLPMID:4728323 [本文引用: 1]
MAP3K1 is a serine/threonine kinase that is activated by a diverse set of stimuli and exerts its effect through various downstream effecter molecules, including JNK, ERK1/2 and p38. In humans, mutant alleles ofMAP3K1are associated with 46,XY sex reversal. Until recently, the only phenotype observed inMap3k1tm1Yxiamutant mice was open eyelids at birth. Here, we report that homozygousMap3k1tm1Yxiamice have early-onset profound hearing loss accompanied by the progressive degeneration of cochlear outer hair cells. In the mouse inner ear, MAP3K1 has punctate localization at the apical surface of the supporting cells in close proximity to basal bodies. Although the cytoarchitecture, neuronal wiring and synaptic junctions in the organ of Corti are grossly preserved,Map3k1tm1Yxiamutant mice have supernumerary functional outer hair cells (OHCs) and Deiters' cells. Loss of MAP3K1 function resulted in the downregulation ofFgfr3,Fgf8,Fgf10andAtf3expression in the inner ear. Fgfr3, Fgf8 and Fgf10 have a role in induction of the otic placode or in otic epithelium development in mice, and their functional deficits cause defects in cochlear morphogenesis and hearing loss. Our studies suggest that MAP3K1 has an essential role in the regulation of these key cochlear morphogenesis genes. Collectively, our data highlight the crucial role of MAP3K1 in the development and function of the mouse inner ear and hearing. Summary:Map3k1mutant mice exhibit early-onset profound hearing loss and supernumerary outer hair cells, along with dysregulation of the FGF signaling pathway, accentuating its function in otic epithelium development and morphogenesis.
URLPMID:25676005 [本文引用: 1]
Abstract Mechanisms that lead to the death of hair cells are reviewed. Exposure to noise, the use of ototoxic drugs that damage the cochlea and old age are accompanied by hair cell death. Outer hair cells are often more susceptible than inner hair cells, partly because of an intrinsically greater susceptibility; high frequency cells are also more vulnerable. A common factor in hair cell loss following age-related changes and exposure to ototoxic drugs or high noise levels is the generation of reactive oxygen species, which can trigger intrinsic apoptosis (the mitochondrial pathway). However, hair cell death is sometimes produced via an extracellular signal pathway triggering extrinsic apoptosis. Necrosis and necroptosis also play a role and, in various situations in which cochlear damage occurs, a balance exists between these possible routes of cell death, with no one mechanism being exclusively activated. Finally, the numerous studies on these mechanisms of hair cell death have led to the identification of many potential therapeutic agents, some of which have been used to attempt to treat people exposed to damaging events, although clinical trials are not yet conclusive. Continued work in this area is likely to lead to clinical treatments that could be used to prevent or ameliorate hearing loss.
URLPMID:17891722 [本文引用: 1]
Abstract Regeneration of sensory hair cells in the mature avian inner ear was first described just over 20 years ago. Since then, it has been shown that many other non-mammalian species either continually produce new hair cells or regenerate them in response to trauma. However, mammals exhibit limited hair cell regeneration, particularly in the auditory epithelium. In birds and other non-mammals, regenerated hair cells arise from adjacent non-sensory (supporting) cells. Hair cell regeneration was initially described as a proliferative response whereby supporting cells re-enter the mitotic cycle, forming daughter cells that differentiate into either hair cells or supporting cells and thereby restore cytoarchitecture and function in the sensory epithelium. However, further analyses of the avian auditory epithelium (and amphibian vestibular epithelium) revealed a second regenerative mechanism, direct transdifferentiation, during which supporting cells change their gene expression and convert into hair cells without dividing. In the chicken auditory epithelium, these two distinct mechanisms show unique spatial and temporal patterns, suggesting they are differentially regulated. Current efforts are aimed at identifying signals that maintain supporting cells in a quiescent state or direct them to undergo direct transdifferentiation or cell division. Here, we review current knowledge about supporting cell properties and discuss candidate signaling molecules for regulating supporting cell behavior, in quiescence and after damage. While significant advances have been made in understanding regeneration in non-mammals over the last 20 years, we have yet to determine why the mammalian auditory epithelium lacks the ability to regenerate hair cells spontaneously and whether it is even capable of significant regeneration under additional circumstances. The continued study of mechanisms controlling regeneration in the avian auditory epithelium may lead to strategies for inducing significant and functional regeneration in mammals.
URLPMID:27527363 [本文引用: 1]
Abstract Sensory hair cells in the inner ear are responsible for sound recognition. Damage to hair cells in adult mammals causes permanent hearing impairment because these cells cannot regenerate. By contrast, newborn mammals possess limited regenerative capacity because of the active participation of various signaling pathways, including Wnt and Notch signaling. The Wnt and Notch pathways are highly sophisticated and conserved signaling pathways that control multiple cellular events necessary for the formation of sensory hair cells. Both signaling pathways allow resident supporting cells to regenerate hair cells in the neonatal cochlea. In this regard, Wnt and Notch signaling has gained increased research attention in hair cell regeneration. This review presents the current understanding of the Wnt and Notch signaling pathways in the auditory portion of the inner ear and discusses the possibilities of controlling these pathways with the hair cell fate determiner Atoh1 to regulate hair cell regeneration in the mammalian cochlea.
URLPMID:3942572 [本文引用: 1]
Sensory hair cell loss is the major cause of hearing and balance disorders. Mammals are incapable of sustained hair cell regeneration, but lower vertebrates can regenerate these mechano-electrical transducers. We present the first comprehensive transcriptome (by mRNA-Seq) of hair cell regeneration in the chick utricle. We provide pathway and pattern annotations and correlate these with the phenotypic events that occur during regeneration. These patterns are surprisingly synchronous and highly punctuated. We show how these patterns are a new resource for identifying components of the hair cell transcriptome and identify 494 new putative hair-cell-specific genes and validate three of these (of three tested) by immunohistochemical staining. We describe many surprising new components and dynamic expression patterns, particularly within NOTCH signaling. For example, we show that HES7 is specifically expressed during utricle hair cell regeneration and closely parallels the expression of HES5. Likewise, the expression of ATOH1 is closely correlated with HEYL and the HLH inhibitory transcription factors ID1, ID2, and ID4. We investigate the correlation between fibroblast growth factor signaling and supporting cell proliferation and show that FGF20 inhibits supporting cell proliferation. We also present an analysis of 212 differentially expressed transcription factor genes in the regenerative time course that fall into nine distinct gene expression patterns, many of which correlate with phenotypic events during regeneration and represent attractive candidates for future analysis and manipulation of the regenerative program in sensory epithelia and other vertebrate neuroepithelia.
URLPMID:24706903 [本文引用: 1]
Deafness caused by the terminal loss of inner ear hair cells is one of the most common sensory diseases. However, nonmammalian animals (e.g., birds, amphibians, and fish) regenerate damaged hair cells. To understand better the reasons underpinning such disparities in regeneration among vertebrates, we set out to define at high resolution the changes in gene expression associated with the regeneration of hair cells in the zebrafish lateral line. We performed RNA-Seq analyses on regenerating support cells purified by FACS. The resulting expression data were subjected to pathway enrichment analyses, and the differentially expressed genes were validated in vivo via whole-mount in situ hybridizations. We discovered that cell cycle regulators are expressed hours before the activation of Wnt/ -catenin signaling following hair cell death. We propose that Wnt/ -catenin signaling is not involved in regulating the onset of proliferation but governs proliferation at later stages of regeneration. In addition, and in marked contrast to mammals, our data clearly indicate that the Notch pathway is significantly down-regulated shortly after injury, thus uncovering a key difference between the zebrafish and mammalian responses to hair cell injury. Taken together, our findings lay the foundation for identifying differences in signaling pathway regulation that could be exploited as potential therapeutic targets to promote either sensory epithelium or hair cell regeneration in mammals.
URLPMID:27351484 [本文引用: 1]
Unlike mammals, the non-mammalian vertebrate inner ear can regenerate the sensory cells, hair cells, either spontaneously or through induction after hair cell loss, leading to hearing recovery. The mechanisms underlying the regeneration are poorly understood. By microarray analysis on a chick model, we show that chick hair cell regeneration involves the activation of proliferation genes and downregulation of differentiation genes. BothMYCandFGFare activated in chick hair cell regeneration. Using a zebrafish lateral line neuromast hair cell regeneration model, we show that the specific inhibition of Myc or Fgf suppresses hair cell regeneration, demonstrating that both pathways are essential to the process. Rapid upregulation of Myc and delayed Fgf activation during regeneration suggest a role of Myc in proliferation and Fgf in differentiation. The dorsal-ventral pattern offgfr1ain the neuromasts overlaps with the distribution of hair cell precursors. By laser ablation, we show that thefgfr1a-positive supporting cells are likely the hair cell precursors that directly give rise to new hair cells; whereas the anterior-posteriorfgfr1a-negative supporting cells have heightened proliferation capacity, likely to serve as more primitive progenitor cells to replenish lost precursors after hair cell loss. Thusfgfr1ais likely to mark compartmentalized supporting cell subtypes with different capacities in renewal proliferation and hair cell regeneration. Manipulation of c-MYC and FGF pathways could be explored for mammalian hair cell regeneration.
URLPMID:26008620 [本文引用: 1]
Lysine-specific demethylase 1 (LSD1/KDM1A) plays an important role in complex cellular processes such as differentiation, proliferation, apoptosis, and cell cycle progression. It has recently been demonstrated that during development, downregulation of LSD1 inhibits cell proliferation, modulates the expression of cell cycle regulators, and reduces hair cell formation in the zebrafish lateral line, which suggests that LSD1-mediated epigenetic regulation plays a key role in the development of hair cells. However, the role of LSD1 in hair cell regeneration after hair cell loss remains poorly understood. Here, we demonstrate the effect of LSD1 on hair cell regeneration following neomycin-induced hair cell loss. We show that the LSD1 inhibitor trans-2-phenylcyclopropylamine (2-PCPA) significantly decreases the regeneration of hair cells in zebrafish after neomycin damage. In addition, immunofluorescent staining demonstrates that 2-PCPA administration suppresses supporting cell proliferation and alters cell cycle progression. Finally, in situ hybridization shows that 2-PCPA significantly downregulates the expression of genes related to Wnt/ -catenin and Fgf activation. Altogether, our data suggest that downregulation of LSD1 significantly decreases hair cell regeneration after neomycin-induced hair cell loss through inactivation of the Wnt/ -catenin and Fgf signaling pathways. Thus, LSD1 plays a critical role in hair cell regeneration and might represent a novel biomarker and potential therapeutic approach for the treatment of hearing loss.
URLPMID:4880589 [本文引用: 1]
The activation of neuromast (NM) supporting cell (SC) proliferation leads to hair cell (HC) regeneration in the zebrafish lateral line. Epigenetic mechanisms have been reported that regulate HC regeneration in the zebrafish lateral line, but the role of H3K9me2 in HC regeneration after HC loss remains poorly understood. In this study, we focused on the role of H3K9me2 in HC regeneration following neomycin-induced HC loss. To investigate the effects of H3K9me2 in HC regeneration, we took advantage of the G9a/GLP-specific inhibitor BIX01294 that significantly reduces the dimethylation of H3K9. We found that BIX01294 significantly reduced HC regeneration after neomycin-induced HC loss in the zebrafish lateral line. BIX01294 also significantly reduced the proliferation of NM cells and led to fewer SCs in the lateral line.In situhybridization showed that BIX01294 significantly down-regulated the Wnt and Fgf signaling pathways, which resulted in reduced SC proliferation and HC regeneration in the NMs of the lateral line. Altogether, our results suggest that down-regulation of H3K9me2 significantly decreases HC regeneration after neomycin-induced HC loss through inactivation of the Wnt/ -catenin and Fgf signaling pathways. Thus H3K9me2 plays a critical role in HC regeneration.
URL [本文引用: 1]
干细胞治疗耳聋是近些年耳科的研究热点之一,本文通过对干细胞耳聋治疗相关的文献进行综述,从干细胞用于内耳结构的替代或修复的基础、干细胞植入的方式、干细胞移植安全性方面,对干细胞治疗耳聋的可行性进行论述,认为干细胞的内耳移植治疗已具备一定的基础,在移植前需对细胞的分化进行更精准的控制,并且可以与传统的治疗方式(如人工耳蜗植入术)结合使用。
URL [本文引用: 1]
干细胞治疗耳聋是近些年耳科的研究热点之一,本文通过对干细胞耳聋治疗相关的文献进行综述,从干细胞用于内耳结构的替代或修复的基础、干细胞植入的方式、干细胞移植安全性方面,对干细胞治疗耳聋的可行性进行论述,认为干细胞的内耳移植治疗已具备一定的基础,在移植前需对细胞的分化进行更精准的控制,并且可以与传统的治疗方式(如人工耳蜗植入术)结合使用。
URLPMID:22972191 [本文引用: 1]
Abstract Deafness is a condition with a high prevalence worldwide, produced primarily by the loss of the sensory hair cells and their associated spiral ganglion neurons (SGNs). Of all the forms of deafness, auditory neuropathy is of particular concern. This condition, defined primarily by damage to the SGNs with relative preservation of the hair cells, is responsible for a substantial proportion of patients with hearing impairment. Although the loss of hair cells can be circumvented partially by a cochlear implant, no routine treatment is available for sensory neuron loss, as poor innervation limits the prospective performance of an implant. Using stem cells to recover the damaged sensory circuitry is a potential therapeutic strategy. Here we present a protocol to induce differentiation from human embryonic stem cells (hESCs) using signals involved in the initial specification of the otic placode. We obtained two types of otic progenitors able to differentiate in vitro into hair-cell-like cells and auditory neurons that display expected electrophysiological properties. Moreover, when transplanted into an auditory neuropathy model, otic neuroprogenitors engraft, differentiate and significantly improve auditory-evoked response thresholds. These results should stimulate further research into the development of a cell-based therapy for deafness.
URLPMID:26615761 [本文引用: 1]
Sensorineural hearing loss and vestibular dysfunction have become the most common forms of sensory defects. Stem cell-based therapeutic strategies for curing hearing loss are being developed. Several attempts to develop hair cells by using chicken utricle stromal cells as feeder cells have resulted in phenotypic conversion of stem cells into inner ear hair-cell-like cells. Here, we induced the differentiation of human embryonic stem cells (hESCs) into otic epithelial progenitors (OEPs), and further induced the differentiation of OEPs into hair-cell-like cells using different substrates. Our results showed that OEPs cultured on the chicken utricle stromal cells with the induction medium could differentiate into hair-cell-like cells with stereociliary bundles. Co-culture with stromal cells, however, may be problematic for subsequent examination of the induced hair-cell-like cells. In order to avoid the interference from stromal cells, we cultured OEPs on laminin with different induction media and examined the effects of the induction medium on the differentiation potentials of OEPs into hair-cell-like cells. The results revealed that the culture of OEPs on laminin with the conditioned medium from chicken utricle stromal cells supplemented with EGF and all-trans retinoic acid (RA) could promote the organization of cells into epithelial clusters displaying hair-cell-like cells with stereociliary bundles. These cells also displayed the expected electrophysiological properties.
URLPMID:8980132210002224512547 [本文引用: 1]
Abstract In mammals, the permanence of many forms of hearing loss is the result of the inner ear's inability to replace lost sensory hair cells. Here, we apply a differentiation strategy to guide human embryonic stem cells (hESCs) into cells of the otic lineage using chemically defined attached-substrate conditions. The generation of human otic progenitor cells was dependent on fibroblast growth factor (FGF) signaling, and protracted culture led to the upregulation of markers indicative of differentiated inner ear sensory epithelia. Using a transgenic ESC reporter line based on a murine Atoh1 enhancer, we show that differentiated hair cell-like cells express multiple hair cell markers simultaneously. Hair cell-like cells displayed protrusions reminiscent of stereociliary bundles, but failed to fully mature into cells with typical hair cell cytoarchitecture. We conclude that optimized defined conditions can be used in vitro to attain otic progenitor specification and sensory cell differentiation.
URLPMID:20478259 [本文引用: 1]
【Key Words】:
URLPMID:26003451 [本文引用: 1]
Disease-specific induced pluripotent stem cells (iPS) cells are expected to contribute to exploring useful tools for studying the pathophysiology of inner ear diseases and to drug discovery for treating inner ear diseases. For this purpose, stable induction methods for the differentiation of human iPS cells into inner ear hair cells are required. In the present study, we examined the efficacy of a simple induction method for inducing the differentiation of human iPS cells into hair cells. The induction of inner ear hair cell-like cells was performed using a stepwise method mimicking inner ear development. Human iPS cells were sequentially transformed into the preplacodal ectoderm, otic placode, and hair cell-like cells. As a first step, preplacodal ectoderm induction, human iPS cells were seeded on a Matrigel-coated plate and cultured in a serum free N2/B27 medium for 8 days according to a previous study that demonstrated spontaneous differentiation of human ES cells into the preplacodal ectoderm. As the second step, the cells after preplacodal ectoderm induction were treated with basic fibroblast growth factor (bFGF) for induction of differentiation into otic-placode-like cells for 15 days. As the final step, cultured cells were incubated in a serum free medium containing Matrigel for 48 days. After preplacodal ectoderm induction, over 90% of cultured cells expressed the genes that express in preplacodal ectoderm. By culture with bFGF, otic placode marker-positive cells were obtained, although their number was limited. Further 48-day culture in serum free media resulted in the induction of hair cell-like cells, which expressed a hair cell marker and had stereocilia bundle-like constructions on their apical surface. Our results indicate that hair cell-like cells are induced from human iPS cells using a simple stepwise method with only bFGF, without the use of xenogeneic cells.
URLPMID:27402757 [本文引用: 1]
Efficient pluripotent stem cell guidance protocols for the production of human posterior cranial placodes such as the otic placode that gives rise to the inner ear do not exist. Here we use a systematic approach including defined monolayer culture, signaling modulation, and single-cell gene expression analysis to delineate a developmental trajectory for human otic lineage specification in vitro. We found that modulation of bone morphogenetic protein (BMP) and WNT signaling combined with FGF and retinoic acid treatments over the course of 18 days generates cell populations that develop chronological expression of marker genes of non-neural ectoderm, preplacodal ectoderm, and early otic lineage. Gene expression along this differentiation path is distinct from other lineages such as endoderm, mesendoderm, and neural ectoderm. Single-cell analysis exposed the heterogeneity of differentiating cells and allowed discrimination of non-neural ectoderm and otic lineage cells from off-target populations. Pseudotemporal ordering of human embryonic stem cell and induced pluripotent stem cell-derived single-cell gene expression profiles revealed an initially synchronous guidance toward non-neural ectoderm, followed by comparatively asynchronous occurrences of preplacodal and otic marker genes. Positive correlation of marker gene expression between both cell lines and resemblance to mouse embryonic day 10.5 otocyst cells implied reasonable robustness of the guidance protocol. Single-cell trajectory analysis further revealed that otic progenitor cell types are induced in monolayer cultures, but further development appears impeded, likely because of lack of a lineage-stabilizing microenvironment. Our results provide a framework for future exploration of stabilizing microenvironments for efficient differentiation of stem cell-generated human otic cell types.
URLPMID:23842490 [本文引用: 1]
The inner ear contains sensory epithelia that detect head movements, gravity and sound. It is unclear how to derive these sensory epithelia from pluripotent stem cells, a process which will be critical for modeling inner ear disorders or developing cell-based therapies for profound hearing loss and balance disorders1,2. To date, attempts to derive inner ear mechanosensitive hair cells and sensory neurons have resulted in inefficient or incomplete phenotypic conversion of stem cells into inner ear-like cells3 7. A key insight lacking from these previous studies is the importance of the non-neural and pre-placodal ectoderm, two critical precursors during inner ear development8 11. Here we report the step-wise differentiation of inner ear sensory epithelia from mouse embryonic stem cells (ESCs) in three-dimensional culture12,13. We show that by recapitulatingin vivodevelopment with precise temporal control of BMP, TGF and FGF signaling, ESC aggregates transform sequentially into non-neural, pre-placodal and otic placode-like epithelia. Remarkably, in a self-organized process that mimics normal development, vesicles containing prosensory cells emerge from the presumptive otic placodes and give rise to hair cells bearing stereocilia bundles and a kinocilium. Moreover, these stem cell-derived hair cells exhibit functional properties of native mechanosensitive hair cells and form specialized synapses with sensory neurons that have also arisen from ESCs in the culture. Finally, we demonstrate how these vesicles are structurally and biochemically comparable to developing vestibular end organs. Our data thus establish a novelin vitromodel of inner ear differentiation that can be used to gain deeper insight into inner ear development and disorder.
URLPMID:24784820 [本文引用: 1]
Abstract This protocol describes a culture system in which inner-ear sensory tissue is produced from mouse embryonic stem (ES) cells under chemically defined conditions. This model is amenable to basic and translational investigations into inner ear biology and regeneration. In this protocol, mouse ES cells are aggregated in 96-well plates in medium containing extracellular matrix proteins to promote epithelialization. During the first 14 d, a series of precisely timed protein and small-molecule treatments sequentially induce epithelia that represent the mouse embryonic non-neural ectoderm, preplacodal ectoderm and otic vesicle epithelia. Ultimately, these tissues develop into cysts with a pseudostratified epithelium containing inner ear hair cells and supporting cells after 16-20 d. Concurrently, sensory-like neurons generate synapse-like structures with the derived hair cells. We have designated the stem cell-derived epithelia harboring hair cells, supporting cells and sensory-like neurons as inner ear organoids. This method provides a reproducible and scalable means to generate inner ear sensory tissue in vitro.
URLPMID:28459451 [本文引用: 1]
Abstract The derivation of human inner ear tissue from pluripotent stem cells would enable in vitro screening of drug candidates for the treatment of hearing and balance dysfunction and may provide a source of cells for cell-based therapies of the inner ear. Here we report a method for differentiating human pluripotent stem cells to inner ear organoids that harbor functional hair cells. Using a three-dimensional culture system, we modulate TGF, BMP, FGF, and WNT signaling to generate multiple otic-vesicle-like structures from a single stem-cell aggregate. Over 2 months, the vesicles develop into inner ear organoids with sensory epithelia that are innervated by sensory neurons. Additionally, using CRISPR-Cas9, we generate an ATOH1-2A-eGFP cell line to detect hair cell induction and demonstrate that derived hair cells exhibit electrophysiological properties similar to those of native sensory hair cells. Our culture system should facilitate the study of human inner ear development and research on therapies for diseases of the inner ear.
[本文引用: 1]
URL [本文引用: 1]
To investigate executive function and adaptive behavior in individuals with Muenke syndrome using validated instruments with a normative population and unaffected siblings as controls. Participants in this cross-sectional study included individuals with Muenke syndrome (P250R mutation in FGFR3) and their mutation-negative siblings. Participants completed validated assessments of executive functioning (Behavior Rating Inventory of Executive Function [BRIEF]) and adaptive behavior skills (Adaptive Behavior Assessment System, Second Edition [ABAS-II]). Forty-four with a positive FGFR3 mutation, median age 902years, range 702months to 5202years were enrolled. In addition, 10 unaffected siblings served as controls (5 males, 5 females; median age, 1302years; range, 3-1802years). For the General Executive Composite scale of the BRIEF, 32.1% of the cohort had scores greater than +1.5 SD, signifying potential clinical significance. For the General Adaptive Composite of the ABAS-II, 28.2% of affected individuals scored in the 3rd-8th percentile of the normative population, and 56.4% were below the average category (<25th percentile). Multiple regression analysis did not identify craniosynostosis as a predictor of BRIEF (P02=02.70) or ABAS-II scores (P02=02.70). In the sibling pair analysis, affected siblings performed significantly poorer on the BRIEF General Executive Composite and the ABAS-II General Adaptive Composite. Individuals with Muenke syndrome are at an increased risk for developing adaptive and executive function behavioral changes compared with a normative population and unaffected siblings.
URLPMID:9042914 [本文引用: 1]
Abstract The underlying basis of many forms of syndromic craniosynostosis has been defined on a molecular level. However, many patients with familial or sporadic craniosynostosis do not have the classical findings of those craniosynostosis syndromes. Here we present 61 individuals from 20 unrelated families where coronal synostosis is due to an amino acid substitution (Pro250Arg) that results from a single point mutation in the fibroblast growth factor receptor 3 gene on chromosome 4p. In this instance, a new clinical syndrome is being defined on the basis of the molecular finding. In addition to the skull findings, some patients had abnormalities on radiographs of hands and feet, including thimble-like middle phalanges, coned epiphyses, and carpal and tarsal fusions. Brachydactyly was seen in some cases; none had clinically significant syndactyly or deviation of the great toe. Sensorineural hearing loss was present in some, and developmental delay was seen in a minority. While the radiological findings of hands and feet can be very helpful in diagnosing this syndrome, it is not in all cases clearly distinguishable on a clinical basis from other craniosynostosis syndromes. Therefore, this mutation should be tested for in patients with coronal synostosis.
URLPMID:26740388 [本文引用: 1]
Muenke syndrome is an autosomal dominant disorder characterized by coronal suture craniosynostosis, hearing loss, developmental delay, carpal, and calcaneal fusions, and behavioral differences. Reduced penetrance and variable expressivity contribute to the wide spectrum of clinical findings. Muenke syndrome constitutes the most common syndromic form of craniosynostosis, with an incidence of 1 in 30,000 births and is defined by the presence of the p.Pro250Arg mutation in FGFR3 . Participants were recruited from international craniofacial surgery and genetic clinics. Affected individuals, parents, and their siblings, if available, were enrolled in the study if they had a p.Pro250Arg mutation in FGFR3 . One hundred and six patients from 71 families participated in this study. In 51 informative probands, 33 cases (64.7%) were inherited. Eighty-five percent of the participants had craniosynostosis (16 of 103 did not have craniosynostosis), with 47.5% having bilateral and 28.2% with unilateral synostosis. Females and males were similarly affected with bicoronal craniosynostosis, 50% versus 44.4% ( P = 0.84), respectively. Clefting was rare (1.1%). Hearing loss was identified in 70.8%, developmental delay in 66.3%, intellectual disability in 35.6%, attention deficit/hyperactivity disorder in 23.7%, and seizures in 20.2%. In patients with complete skeletal surveys (upper and lower extremity x-rays), 75% of individuals were found to have at least a single abnormal radiographical finding in addition to skull findings. This is the largest study of the natural history of Muenke syndrome, adding valuable clinical information to the care of these individuals including behavioral and cognitive impairment data, vision changes, and hearing loss. 2016 Wiley Periodicals, Inc.
URLPMID:24686979 [本文引用: 1]
There are a number of craniosynostosis syndromes with hearing loss-including Muenke, Apert, Pfeiffer, Crouzon, Beare-Stevenson, Crouzon with acanthosis nigricans, and Jackson-Weiss syndromes-that result from mutations in the fibroblast growth factor receptor (FGFR) genes. Studies of FGFRs and their ligands, fibroblast growth factors (FGFs), have revealed clues to the precise contribution of aberrant FGFR signaling to inner ear morphogenesis and the hearing loss encountered in craniosynostoses. The purpose of this article is to review basic studies of FGFRs with emphasis on their function and expression in the inner ear and surrounding structures. A Medline search was performed to find basic science articles regarding FGFR, their ligands, and their expression and relevant mouse models. Additional items searched included clinical descriptions and studies of individuals with FGFR-related craniosynostosis syndromes. The FGF signaling pathway is essential for the morphogensis and proper function of the inner ear and auditory sensory epithelium. The variable auditory phenotypes seen in individuals with Muenke syndrome may have a genetic basis, likely due to multiple interacting factors in the genetic environment or modifying factors. Further analysis and studies of mouse models of Muenke syndrome, in particular, may provide clues to the specific effects of the defining mutation in FGFR3 in the inner ear not only at birth but also into adulthood. In particular, investigations into these models may give insight into the variable expression and incomplete penetrance of this phenotype.
URLPMID:5373035 [本文引用: 1]
Lacrimo-auriculo-dento-digital (LADD) syndrome is an autosomal dominant disorder displaying variable expression of multiple congenital anomalies including hypoplasia or aplasia of the lacrimal and salivary systems causing abnormal tearing and dry mouth. Mutations in theFGF10,FGFR2, andFGFR3genes were found to cause some cases of LADD syndrome in prior genetic studies. The goal of this study is to identify the genetic basis of a case of LADD syndrome with glaucoma and thin central corneal thickness (CCT). Whole exome sequencing was performed, and previously described disease-causing genes (FGF10, FGFR2,andFGFR3) were first evaluated for mutations. Fifty-eight additional prioritized candidate genes were identified by searching gene annotations for features of LADD syndrome. The potential pathogenicity of the identified mutations was assessed by determining their frequency in large public exome databases; through sequence analysis using the Blosum62 matrix, PolyPhen2, and SIFT algorithms; and through homology analyses. A structural analysis of the effects of the top candidate mutation in tumor protein 63 (TP63) was also conducted by superimposing the mutation over the solved crystal structure. No mutations were detected inFGF10,FGFR2, orFGFR3. The LADD syndrome patient exome data was searched for mutations in the 58 candidate genes and only one mutation was detected, an Arg343Trp mutation in the tumor protein 63 (TP63) gene. ThisTP63mutation is absent from the gnomAD sequence database. Analysis of the Arg343Trp mutation with Blosum62, PolyPhen2, and SIFT all suggest it is pathogenic. This arginine residue is highly conserved in orthologous genes. Finally, crystal structure analysis showed that the Arg343Trp mutation causes a significant alteration in the structure of TP63 DNA binding domain. We report a patient with no mutations in known LADD syndrome genes (FGF10,FGFR2, andFGFR3). Our analysis provides strong evidence that the Arg343Trp mutation inTP63caused LADD syndrome in our patient and thatTP63is a fourth gene contributing to this condition.TP63encodes a transcription factor involved in the development and differentiation of tissues affected by LADD syndrome. These data suggest thatTP63is a novel LADD syndrome gene and may also influence corneal thickness and risk for open-angle glaucoma.
URLPMID:28483234 [本文引用: 1]
Abstract Lacrimo-auriculo-dento-digital syndrome (LADD) is a multiple congenital anomaly and a genetically heterogeneous disorder. The aim of this study was to identify the pathogenic gene in an Iranian family with LADD syndrome and review the literature on reported mutations that involved in pathogenesis of LADD syndrome. One novel variant, c.1882 G>A, in fibroblast growth factor receptor 3 (FGFR3) was identified by next generation sequencing and Sanger sequencing. The heterozygous FGFR3 c.1882 G>A variant results in substitution of aspartic acid with asparagine at amino acid 628 (p.D628N) and co-segregated with the phenotype in the LADD family. Our findings suggest that the heterozygous FGFR3 c.1882 G>A variant might be the pathogenic mutation, because this amino acid is conserved in several species. Our data extend the mutation spectrum of the FGFR3 gene and have important implications for genetic counseling for the families. This is the second report of FGFR3 involvement in syndromic deafness in humans, and confirms the gene positive role in inner ear development. In addition, this is the first FGFR3 mutation recognized in the Iranian LADD family.
URLPMID:2099236 [本文引用: 1]
Lacrimo-auriculo-dento-digital (LADD) syndrome is characterized by abnormalities in lacrimal and salivary glands, in teeth, and in the distal limbs. Genetic studies have implicated heterozygous mutations in fibroblast growth factor 10 (FGF10) and in FGF receptor 2 (FGFR2) in LADD syndrome. However, it is not clear whether LADD syndrome mutations (LADD mutations) are gain- or loss-of-function mutations. In order to reveal the molecular mechanism underlying LADD syndrome, we have compared the biological properties of FGF10 LADD and FGFR2 LADD mutants to the activities of their normal counterparts. These experiments show that the biological activities of three different FGF10 LADD mutants are severely impaired by different mechanisms. Moreover, haploinsufficiency caused by defective FGF10 mutants leads to LADD syndrome. We also demonstrate that the tyrosine kinase activities of FGFR2 LADD mutants expressed in transfected cells are strongly compromised. Since tyrosine kinase activity is stimulated by ligand-induced receptor dimerization, FGFR2 LADD mutants may also exert a dominant inhibitory effect on signaling via wild-type FGFR2 expressed in the same cell. These experiments underscore the importance of signal strength in mediating biological responses and that relatively small changes in receptor signaling may influence the outcome of developmental processes in cells or organs that do not possess redundant signaling pathway.

{kind=link}
{kind=link}
{kind=link}
{kind=link}