 ,中国科学院北京基因组研究所,中国科学院精准基因组医学重点实验室,北京 100101
,中国科学院北京基因组研究所,中国科学院精准基因组医学重点实验室,北京 100101Precision genomic and translational medicine for acute myeloid leukemia
Xuexin Yu, Aili Chen, Yueying Li, Dan Liu, Qianfei Wang,CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Bejijng 100101, China通讯作者:
编委: 刘峰
收稿日期:2018-07-3修回日期:2018-09-12网络出版日期:2018-11-20
Received:2018-07-3Revised:2018-09-12Online:2018-11-20
作者简介 About authors
于雪新,博士,助理研究员,研究方向:白血病生物信息学E-mail:yuxx@big.ac.cn。

摘要
关键词:
Abstract
Keywords:
PDF (3050KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
于雪新, 陈艾莉, 李玥莹, 刘丹, 王前飞. 白血病的精准基因组医学研究与转化应用[J]. 遗传, 2018, 40(11): 988-997 doi:10.16288/j.yczz.18-188
Xuexin Yu, Aili Chen, Yueying Li, Dan Liu, Qianfei Wang.
自20世纪60年代末,随着肿瘤治疗新药的不断发现和支持治疗的加强,白血病的疗效有了逐步提高。急性白血病(acute leukemia)是常见的血液系统恶性肿瘤,发病率大约为2.76/100000人。急性白血病的治疗主要以化疗为主,但是该病的总体治疗效果欠佳,5年无病生存率只有30%~40%[1]。因此,急性白血病的研究急需新的突破口。国际肿瘤基因组计划的完成为我们打开了白血病研究的新大门,白血病基因组、表观组以及转录组的图谱已被初步绘制出来,上述发现可能对白血病的诊断、治疗以及预后有指导性意义。因此深入挖掘转录组、基因组以及表观修饰改变的临床意义,有助于制定针对特定患者的个体化治疗策略。
近年来,中国科学院北京基因组研究所精准基因组医学重点实验室在白血病研究中取得了一系列重要进展。同时,精准基因组医学重点实验室与多家医院开展了密切的临床合作,利用高通量测序技术、病人大规模临床数据分析以及细胞和动物模型的集成体系,系统解析白血病基因组和表观组特征并揭示其在肿瘤发生发展过程中和药物作用下的异质性及克隆演化规律,发现白血病难治性亚型的发病机制和药物靶点,上述成果推动了基因组研究在白血病精准治疗中的应用。结合本研究所精准基因组医学重点实验室多年来的工作,本文对白血病的发病机制、新抑癌基因的挖掘、白血病克隆演化以及低剂量化疗方案临床疗效等方面的研究进行了综述,旨在为白血病诊断及治疗方案的改进提供新的契机。
1 白血病发病机制
急性白血病是一种常见的、具有高度异质性的血液系统恶性肿瘤。造血系统在分化的不同阶段发生恶变而引发血细胞分化的阻滞,导致急性白血病的发生。深入探究急性白血病的分子发病机制,有助于发现诊断及治疗的靶点,实现个性化精准诊疗,最终达到改善急性白血病治疗效果的目的。1.1 白血病转录调控机制
混合谱系白血病(mixed lineage leukemia, MLL)是一类恶性程度高且预后差的难治性急性白血病,因染色体易位形成MLL融合蛋白而得名。MLL融合蛋白激活下游一系列在血细胞分化和发育过程中起关键作用的基因表达,继而引发造血细胞的发育紊乱和分化停滞,最终导致白血病[2]。此外,转录因子MEIS1的持续过度表达是混合谱系白血病的特征之一。小鼠实验证实MEIS1过度表达能够缩短小鼠MLL相关白血病的潜伏期并加速其发展,但分子机理尚不明确[3]。精准基因组医学重点实验室与美国芝加哥大学合作开展了探究MLL中MEIS1过表达机制的研究[4],研究发现位于近3°端内含子区内的远端增强子E9能上调MEIS1的表达且带有很强的组蛋白H3K27ac和H3K4me1修饰。E9增强子所在的基因组区域是小鼠白血病模型中的“热点”攻击位点,反转录病毒通过整合插入到E9位点直接诱发白血病的发生。该研究揭示了转录因子MEIS1通过远端增强子调控自身表达水平的机制,为深入开展白血病的转录调控机理研究奠定了良好基础。虽然MLL融合蛋白在MLL中非常重要,但是只有MLL融合蛋白却不足以引起MLL白血病。是否存在其他关键的独立致病因子,一直是白血病领域中广受关注的问题。精准基因组医学重点实验室和美国辛辛那提儿童医院合作,发现与以往研究中转录因子PU.1在t (8;21)和t (15;17)急性白血病中“抑癌基因”的作用相反,PU.1在MLL白血病中高表达且可诱发和维持肿瘤细胞生长的作用,且PU.1不是MLL融合蛋白的下游靶基因[5]。研究人员鉴定出15个只接受PU.1调控的靶基因,其与细胞生长、造血分化及炎性反应等功能有关,包括CSF1R、PU.1、PBX3、MEIS1、KIT、FLT3、TPM4、LY86、CD180、CCL3、CTSH、TBXAS1、AIF1、NFKB1以及SKY。此外,上述基因中包含了MLL融合蛋白下游的MEIS-HOX通路上的关键调控因子。研究者们利用大规模急性白血病病人的临床生存数据证实PU.1调控的靶基因与病人的预后显著相关。该研究发现转录因子PU.1具有维持MLL白血病的作用,并证实PU.1是独立于MLL融合蛋白的关键调控因子,可与MEIS1协同作用,是调控血细胞分化发育的重要基因(图1)。该研究为MLL白血病的临床诊断、预后评估及靶向治疗提供了新的线索。
图1
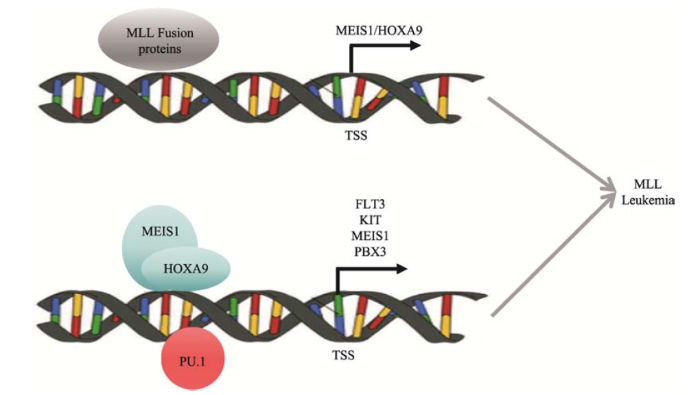 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1转录因子PU.1在MLL白血病中的调控模式图
Fig. 1Regulated patterns of transcriptional factors PU.1 in MLL leukemia
1.2 白血病表观遗传调控机制
在MLL的研究中,除转录调控外,表观组的变异也是研究的热点之一。精准基因组医学重点实验室与美国迈阿密大学米勒医学院合作,揭示急性髓系白血病(acute myeloid leukemia,AML)中重现性突变基因ASXL2在正常造血干细胞功能维持中的抑癌作用[6]。ASXL2是一种表观调控因子,其突变主要出现于伴t (8;21)染色体异常的AML中(~23%),此类患者的复发率可高至36%。研究人员发现Asxl2基因缺失可导致小鼠的造血干细胞(hematopoietic stem cell, HSC)向髓系谱系分化倾斜,并使其出现骨髓增生异常综合征样疾病;Asxl2敲除后,会使得小鼠长期造血干细胞频率的增加且自我更新潜能提高;Asxl2敲除小鼠的骨髓细胞能引起白血病发生。Asxl2敲除引起了与HSC功能、细胞凋亡和髓系发育相关的基因表达改变以及转录激活相关的组蛋白修饰H3K27ac和H3K4me1/2的特异性改变。这些数据表明,ASXL2通过组蛋白修饰调控造血转录程序,维持正常HSC的功能和抑制髓系肿瘤的发生(图2)。该研究在机制上证明了ASXL2通过维持正常造血干细胞功能而抑制髓系肿瘤的发生。图2
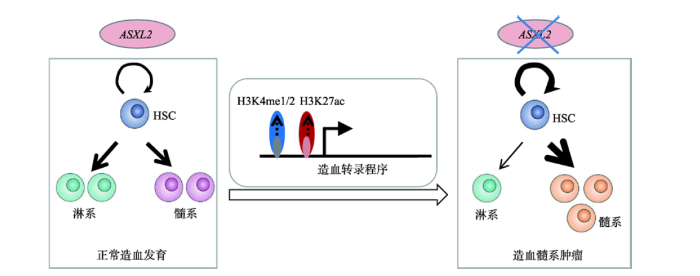 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2表观调控因子ASXL2通过组蛋白修饰在造血干细胞功能维持中起抑癌作用
Fig. 2The epigenetic regulator ASXL2 plays a critical role in maintenance of normal HSC functions and suppression of myeloid malignancies via histone modification
同时,精准基因组医学重点实验室联合武汉大学,发现了AML表观靶向治疗新靶点,有望通过靶向酸性核磷蛋白ANP32A调节表观遗传修饰来治疗AML[7]。研究人员发现ANP32A通过调节表观组蛋白H3乙酰化(acetyl-H3)修饰,促进白血病的发生发展。在AML病人细胞中,ANP32A异常高表达,对白血病细胞增殖、生存和克隆形成具有促进作用。而在ANP32A缺失的AML细胞中,acetyl-H3富集变化与基因表达变化显著正相关,其中包括与脂代谢通路相关的基因如APOC1。进一步功能实验证明,ANP32A缺失降低了acetyl-H3在基因APOC1启动子区的富集水平,下调APOC1基因的表达,而过表达APOC1能够恢复因ANP32A缺失引起的生长抑制(图3)。该研究首次揭示了ANP32A在白血病中作为致癌因子发挥功能;ANP32A蛋白只有249个氨基酸,适合利用小分子抑制剂或多肽有效干扰其功能,有望通过靶向ANP32A调节异常升高的acetyl- H3修饰来治疗白血病。
图3
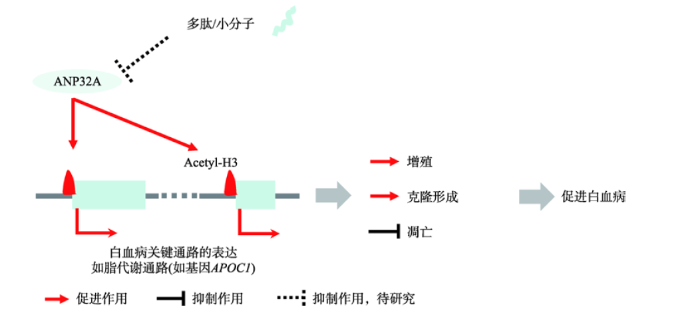 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3表观调控新因子ANP32A通过调节表观acetyl-H3促进白血病
Fig. 3ANP32A as a novel epigenetic regulator of histone H3 acetylation promotes leukemogenesis in AML
2 白血病新抑癌基因的发现
精准基因组医学重点实验室与中国医学科学院血液学研究所和美国辛辛那提儿童医院等多个团队合作,首次发现并证实组蛋白修饰酶SETD2在白血病中的抑癌基因功能,表明SETD2突变引起的组蛋白修饰紊乱是白血病发生中新的关键通路[8]。该研究发现在241例急性白血病病人中,6.2%的病人含有SETD2突变并携带MLL异位或其他常见染色体异常;同时发现SETD2在人类急性白血病中具有抑癌基因的突变失活特征,但突变谱显著不同于实体瘤,点突变和表达下调是急性白血病中SETD2功能破坏的主要机制,而非大的删除和表观沉默;SETD2突变在人类急性白血病中造成基因功能失活,致使白血病细胞的染色质修饰H3K36me3的整体水平降低。研究人员进一步证实,在含有其他常见染色体异常时敲低SETD2能增强白血病干细胞的自我更新促进白血病的起始和进展,抑制mTOR信号通路能阻碍SETD2敲低的急性白血病细胞的增殖(图4)。该研究揭示了SETD2是新的白血病抑制基因,而且SETD2-H3K36me3通路的功能破坏是白血病发生发展的一种表观遗传机制。SETD2是一个新的分子治疗靶点,为急性白血病的临床预测、诊断和治疗提供了新的机遇。针对此项研究,Nature Reviews Cancer发表专文点评,指出“近年肿瘤测序发现的染色质调控因子突变的生物学功能不清”,而此项研究“揭示了SETD2突变在白血病中的重要作用”。Cancer Discovery杂志在“研究观察”专栏中点评“SETD2是协同血液肿瘤发生和维持中的抑癌基因”的重要影响。中国中央电视台、新华社、人民日报以及国际肿瘤干细胞新闻等24家国内外媒体也进行了专题采访或报道,指出这一发现作为急性白血病表观发病机制的重要科学意义。图4
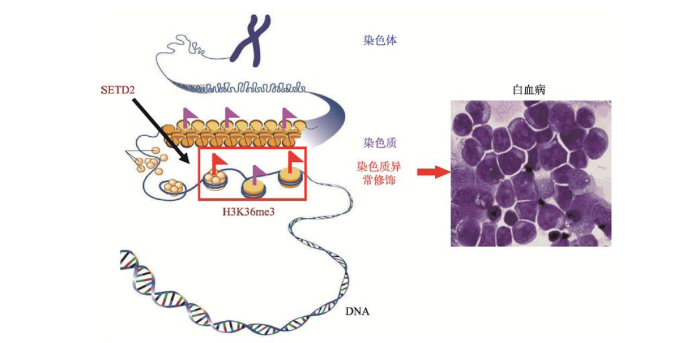 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4SETD2突变可导致染色质的异常修饰促进白血病发生
Fig. 4SETD2 mutations lead to deregulated histone modification and promote leukemogenesis in AML
基于上述发现,精准基因组医学重点实验室与美国辛辛那提儿童医院合作进一步探索了携带SETD2突变的MLL白血病中不同组蛋白表观修饰间的相互作用。发现在SETD2野生型的MLL白血病中不仅H3K79me2异常高水平修饰,H3K36me3水平也显著升高,并且两种组蛋白修饰显著富集于同一个基因集;而在SETD2功能缺失的MLL白血病中,H3K36me3修饰降低,H3K79me2修饰水平则升高;该动态的组蛋白修饰并没有进一步激活传统认识的MLL靶基因,而是调控了一组新的基因集。其中抑癌基因ASXL1等被抑制,癌基因ERG等被激活,进而促进MLL白血病的发生。 该研究首次揭示了表观调控通路的功能协同,表明MLL白血病中致癌通路DOT1L-H3K79me2和抑癌通路SETD2-H3K36me3在基因调控过程中相互作用(图5)[9]。该项研究的成果为临床提供了新的治疗靶点,有望逆转SETD2突变介导的化疗耐药。
图5
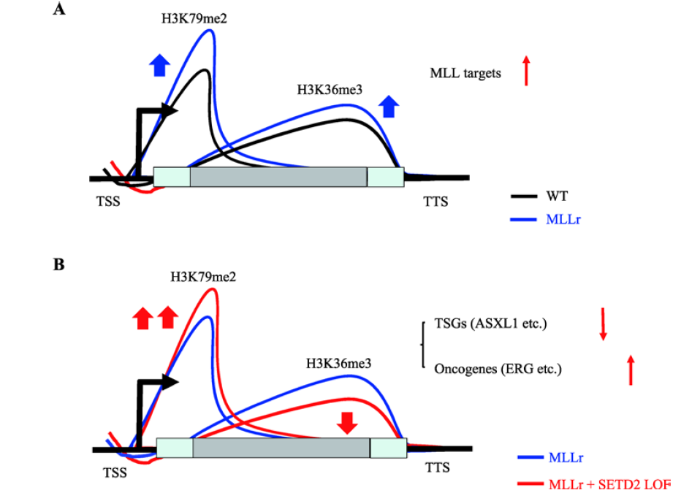 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5携带SETD2突变的MLL白血病中不同组蛋白表观修饰间的相互作用
A:SETD2野生型的MLL白血病中(MLLr)高水平H3K79me2修饰促进H3K36me3修饰升高,表达水平发生变化的主要是MLL靶基因被激活;B:SETD2突变后(SETD2 LOF)MLL白血病中H3K36me3修饰降低,但H3K79me2修饰进一步升高,传统的MLL靶基因表达水平并无进一步改变,而新的抑癌基因被抑制,癌基因被激活。
Fig. 5SETD2-mediated crosstalk between H3K36me3 and H3K79me2 in MLL-rearranged leukemia
3 白血病的精准诊治与临床转化
作为白血病治疗的最主要手段,常规化疗取得了显著临床效果并成为骨髓移植前的必需桥接疗法。然而,常规化疗方案也存在着较为突出的问题: ①传统化疗方案对白血病难治亚型以及复发难治的白血病患者的治疗效果欠佳,且医学界也没有统一的治疗标准和指南。②严重毒副作用导致的早期死亡与长期并发症现象普遍。这一问题在发展中国家由于支持治疗条件的不完善而更加突出,我国单中心报道长期无病生存率比发达国家低20%~40%。 ③高额治疗费用造成因经济原因放弃治疗的普遍现象在我国部分医院高达 30%。针对上述存在的临床问题,以临床病例为研究对象所开展的白血病的精准基因组医学研究能够为临床提供新的治疗思路。近年来,中科院基因组所精准基因组医学重点实验室与多家医院和医药研发单位合作,开展了多项与临床相关的研究,在侵袭性NK细胞白血病(aggressive natural killer leukemia, ANKL)、急性淋巴细胞白血病(acute lymphoblastic leukemia, ALL)以及急性髓系白血病(AML)中取得了一定的研究成果。3.1 白血病难治亚型及耐药的研究
ANKL起源于NK细胞异常增殖,病情进展迅速,多数患者在极短的时间内发生多器官衰竭,部分患者还会出现吞噬血细胞的现象,病情十分凶险。ANKL患者即使积极接受正规治疗,平均生存时间也仅仅只有几个月。目前临床上ANKL的治疗面临两大主要问题:一个是患者对传统的化疗方案不敏感,且无统一的治疗标准和指南;另一个是NK细胞白血病发病具有明显的地域差异,在亚洲(报道病例多以中国、日本和韩国为主)和中南美洲更为常见。NK细胞白血病发病机理不明,也严重制约着临床医生选择和制定有效的治疗方案[10]。精准基因组医学重点实验室联合华中科技大学附属武汉同济医院,对近50例中国ANKL患病人群进行基因组、转录组以及代谢组的整合分析,结果显示JAK/STAT信号转导通路的基因在NK细胞白血病中频繁发生突变[11]。突变增强了JAK/STAT的信号传递功能,促使下游能够控制细胞代谢水平的MYC基因活化,进而一批参与代谢功能的基因过量的表达,NK白血病细胞呈现了代谢极其旺盛的特点(核苷酸和糖的代谢最突出)。此外,研究者还发现了在NK细胞白血病中携带能够修改遗传物质的表观修饰基因的突变,如TET2等基因。本研究揭示,NK细胞白血病存在代谢活跃的特征,这提示传统化疗方案联合抗代谢药物如左旋门冬酰胺酶可以有效缓解疾病进展;同时研究发现的JAK/STAT通路以及高度活化的MYC基因,将是开展新型治疗的靶点(图6)。图6
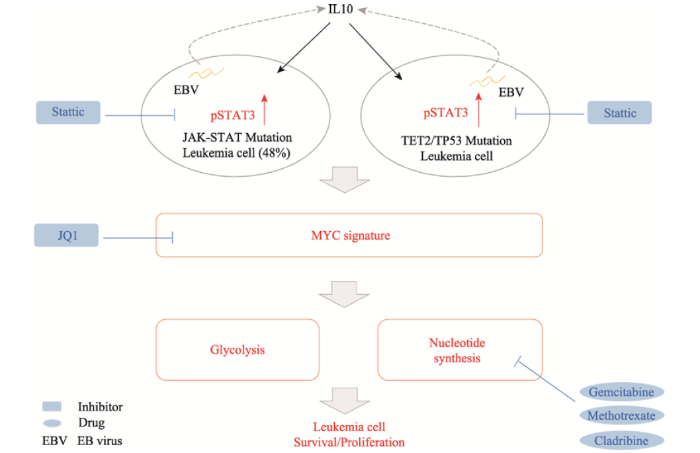 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6ANKL发病机理模型及潜在的治疗新靶标
Fig. 6The pathogenesis model of ANKL and novel potential therapeutic targets
儿童ALL是一种以癌变的、不成熟的淋巴母细胞在骨髓中异常增生为特点的急性白血病,具有起病急、贫血、感染、出血、器官侵润等临床特征。在中国,每年的ALL患儿大概有1.5万人,约占儿童白血病的70%。近年来临床上发现酪氨酸激酶抑制剂(tyrosine-kinase inhibitor, TKI)可用于儿童ALL的治疗,但该抑制剂的耐药性已成为临床上亟待解决的问题[12]。精准基因组医学重点实验室联合中国医学科学院血液病医院,首次系统的揭示了TKI临床治疗ALL的耐药机制,并提出新的治疗策略[13]。研究团队发现使用TKI治疗缓解后又复发的儿童ALL病人携带了一种新的致病融合基因(AGGF1- PDGFRB),其致病性在BaF3/Arf-/-细胞系中得到验证。另外,研究团队还刻画了该病人疾病进程中的克隆演化模式,发现PDGFRB点突变在TKI治疗过程中特异出现,并使白血病细胞获得生长优势,成长为TKI耐药复发时的主克隆;而体外细胞系功能实验也证实了PDGFRB突变可直接导致TKI耐药这一论断。此外,由于PDGFRB主要下游通路为JAK- STAT通路,研究人员使用广谱JAK抑制剂CHZ868进一步处理携带PDGFRB突变的白血病细胞,发现其对该抑制剂高度敏感,提示CHZ868可能是一种治疗该类耐药病人的潜在治疗手段。本研究揭示了靶向药物的耐药和治疗过程中的基因组演化有关,融合基因或点突变的出现都可能影响药物的治疗效果;而针对不同的基因组异常,可给予针对性的治疗措施以逆转耐药。
3.2 儿童白血病的低剂量化疗方案研究
白血病在儿童恶性肿瘤中发病率和死亡率均居首位,患儿死亡率高达40%。我国目前至少有400万白血病患者,新增病例中有一半是儿童。其中有超过75%的患儿来自农村,家庭年收入不足3万元。平均30万元的治疗费用给患儿家庭带来了沉重负担,许多患儿家庭只能放弃治疗。然而,半个多世纪以来主流医学界在临床实践和治疗理念上秉承的剂量“越高越好”的最大耐受剂量化疗也存在着较为突出问题:毒副作用大以及高额的治疗费用[14]。因此,革新儿童白血病的治疗方案,既能最大限度的清除肿瘤细胞、减少复发风险并大幅度降低治疗费用,是临床上亟待解决的问题。精准基因组医学重点实验室与苏州大学附属儿童医院合作,对140例儿童AML患者分别在诱导缓解疗程使用低剂量化疗联合G-CSF的方案(简称低剂量方案)或标准剂量诱导缓解方案(简称标准方案)治疗并评估疗效。临床数据表明低剂量方案与标准方案相比具有相似的诱导缓解率和4年总体生存率,同时低剂量组的患者粒缺时间和血小板恢复时间较标准剂量组明显缩短,感染程度大大降低,患者在前两个诱导缓解疗程的治疗中平均可节省花费约5万元。通过高通量靶向测序检测和追踪9例低剂量组和11例标准剂量组取得完全缓解的患者在初诊、诱导缓解疗程、巩固强化疗程和疗程结束后的随访期的突变清除情况,全面精确的证实低剂量方案能够取得与标准方案相似的分子疗效并提示两者具有相似的长期复发风险。这些结果表明低剂量化疗联合G-CSF的方案在保持与标准剂量化疗相当临床疗效的同时能够显著降低毒副作用和治疗费用,对于感染较重的不耐受患者及经济困难放弃治疗者有十分重大的意义。
为了进一步推广低剂量化疗方案在儿童白血病治疗中的应用,中科院基因组研究所已联合多家医院进行以下研究:从基因组水平分析确立常规高剂量化疗方案和低剂量化疗方案的敏感人群,实现急性髓系白血病的精准分层治疗;明确低剂量方案清除肿瘤突变的分子疗效和起效机制,在全国范围内推广低剂量化疗方案并使该方案成为国际认可的儿童急性髓系白血病的一线临床治疗方案。
3.3 药物治疗下白血病患者基因组克隆演化的研究
化疗作为一种主流的治疗白血病的手段,可以由不同的药物种类、用药剂量以及用药密度而形成不同的化疗方案。然而近年来的多个研究表明,化疗是一把“双刃剑”,它既能杀死肿瘤细胞,也能筛选、诱导耐药突变,驱动肿瘤基因组克隆演化。即便是在相同的化疗药物作用下,不同的用药剂量或用药密度也会对肿瘤内部产生不同的选择压力,而导致肿瘤产生不同的克隆演化过程。在治疗白血病时,化疗药物驱动着白血病细胞产生不同的基因突变、结构及拷贝数变异、表观修饰的改变及基因表达异常等诸多改变,从而导致了同一白血病细胞群体内存在遗传背景和表型功能显著不同的多个细胞克隆。而白血病不同细胞克隆的异质性也是造成白血病化疗耐药的根本原因之一。针对基因组克隆演化介导的白血病化疗耐药,我们开展了以下3个方面的工作:(1) 明确化疗耐药患者的基因组克隆演化模式;(2) 探索化疗耐药的分子机制;(3) 寻找可以逆转化疗耐药的手段。目前,精准基因组医学重点实验室与苏州大学附属儿童医院合作,运用全基因组高通量深度测序研究不同化疗方案下患者的突变清除以及基因组克隆演化模式。初步结果显示,低剂量化疗联合G-CSF (预激方案)对特定患者可以全面清除主要的肿瘤克隆,达到分子水平的缓解。而未来对于难治型白血病的研究,将侧重于低剂量化疗是否能通过减缓肿瘤耐药克隆演化来延长某些患者的生存期。该研究有望建立基于肿瘤基因组演化规律推广肿瘤新疗法的典范,并拓宽和修正医学界对于首选大剂量化疗治疗肿瘤策略的传统认识。
同时,精准基因组医学重点实验室与河南省肿瘤医院和南京医科大学第一附属医院合作对初诊的老年AML患者(50~75岁)开展了低剂量化疗联合G-CSF的临床试验。将2013~2017年间入院就诊的154例老年AML患者按照治疗方案进行分组,包括低剂量化疗联合G-CSF组和常规剂量化疗组。初步研究发现,低剂量化疗联合G-CSF能够更有效的清除表观因子突变,且接受常规剂量化疗的老年AML患者在达到完全缓解后,残留的基因突变主要为表观因子突变,因此我们推断表观因子突变可能是导致白血病化疗耐药的原因之一。此外,我们还将进一步探究上述表观因子突变是否在造血干细胞时期就已经存在,这些携带表观因子突变的前白血病克隆又是如何一步一步演化为白血病克隆的,通过药物抑制上述克隆的膨胀是否可以有效的改善白血病化疗耐药。此外,为了进一步探究化疗药物驱动下复发难治成人AML患者的克隆演化模式,精准基因组医学重点实验室也与北京大学人民医院展开了合作,对复发难治的成人AML患者进行回顾性研究。研究者们探究了化疗联合G-CSF方案对表观因子突变的清除作用并刻画出患者在不同方案治疗下基因组克隆演化的模式,期望可以准确的刻画出不同化疗方案下敏感和耐药克隆的基因组特征,据此筛选出化疗联合G-CSF方案敏感人群的分子特征并在患者治疗过程中动态的调整用药方案和剂量。我们希望通过该研究进一步完善临床上对于复发难治AML患者治疗方案的选择依据并将化疗联合G-CSF方案推广为部分复发难治成人AML患者的一线治疗方案。
4 巨核细胞和血小板体外发育及再生
巨核细胞(MK)是骨髓中高度多倍体化且高效生成血小板的重要细胞,而血小板在止血、伤口愈合以及维持机体稳态等过程中具有重要作用。临床上输注血小板和(或)巨核细胞可用于多种具有凝血功能障碍的疾病,包括:血小板减少症、肿瘤化疗后、手术、HIV感染以及外伤出血等,其临床需求量大。据统计,血小板的民间临床应用在美国每年有大于10亿美元的市场需求。然而,因为捐赠者非常有限且血小板成分保存困难,人体捐赠作为目前最主要的供血来源,远不能满足临床的需求。利用多能干细胞以及造血干细胞体外诱导生成功能血细胞是国际科学界及生物科技公司的关注热点和前沿。由于对造血干细胞自我更新和下游功能分化的关键调控靶点和机制了解不清,目前体外再生获得的血细胞的研发面临极大的技术挑战,如体外产生巨核细胞或血小板具有效率低下和功能有限的特点,效率比体内低将近1000倍。因此,亟需不断探索巨核细胞发育的过程和机制,开发新的可促进体外人巨核细胞生成的技术手段,进而满足日益增长的临床需要。已有研究发现转录因子RUNX1单等位基因胚系突变会引起MK发育障碍和血小板减少的家族性血小板疾病(familial platelet disorders, FPD),但具体机制尚不清楚。合作方美国约翰霍普金斯大学医学院的程临钊教授前期工作发现利用FPD病人来源的多能干细胞(hiPSCs)体外模型可重现病人表型,而通过基因打靶修复RUNX1 突变后可以恢复其巨核细胞的生成能力。为了揭示FPD巨核发育障碍和血小板减少的发病机制,促进体外巨核细胞或血小板再生,精准基因组医学重点实验室利用疾病干细胞体外模型结合功能基因组学,找到了一系列受RUNX1调控并对巨核细胞生成具有重要作用的靶基因和通路[15]。其中,首次揭示NOTCH4作为转录因子RUNX1的直接靶基因,负向调控体外巨核细胞发育;并筛选出Notch通路的小分子抑制剂,使得从多能干细胞和脐血来源的造血干祖细胞生成巨核细胞的产量提高近十倍。该成果有望应用于临床输注,治疗包括血小板减少症、肿瘤化疗后和手术外伤出血等在内的多种凝血功能障碍疾病,具有较高的科学意义和临床价值。此外,新靶点NOTCH4及其抑制剂的医药用途已申请国内及国际专利。
5 未来研究展望
白血病发病机制的研究不仅可以帮助研究者们进一步了解白血病的致病机理,也促使更多的可用于诊断及治疗的新靶点得以发现。此外,以临床问题为切入点并结合高通量测序技术所开展的白血病的精准基因组医学研究也为白血病的临床治疗提供了崭新的治疗思路。随着研究的不断深入以及测序技术的日益发展,研究者们已经意识到白血病具有高度异质性,肿瘤细胞的恶性程度、耐药性、复发能力和转移能力均不同,在疾病进展和药物治疗下不断演化,最终引起难治和复发。而肿瘤异质性的演化是白血病难治和复发的重要原因,也是实现白血病精准治疗所面临的巨大挑战。因此,探究不同化疗方案以及相同方案的不同用药剂量的驱动下的患者基因组特征以及克隆演化模式,筛选出化疗方案在不同治疗阶段适用人群的分子标记,最终实现在治疗过程中动态调整化疗药物使用剂量已成为临床上急需解决的问题。我们认为基于白血病基因组克隆演化而展开的适应性治疗,不仅可以改善白血病患者的化疗耐药,也将提高患者的完全缓解率并延长其整体生存期,最终实现改善白血病临床治疗效果的目的。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:17957188 [本文引用: 1]

Translocations that involve the mixed lineage () gene identify a unique group of , and often predict a poor prognosis. The gene encodes a that methylates lysine 4 (H3K4), and positively regulates gene expression including multiple genes. Leukaemogenic translocations encode fusion proteins that have lost H3K4 activity. A key feature of fusion proteins is their ability to efficiently transform haematopoietic cells into . The link between a modulator and provides support for epigenetic landscapes as an important part of and normal stem-.
URL [本文引用: 1]
Oncogenic mutations of the MLL histone methyltransferase confer an unusual ability to transform non-self-renewing myeloid progenitors into leukemia stem cells (LSCs) by mechanisms that remain poorly defined. Misregulation of Hox genes is likely to be critical for LSC induction and maintenance but alone it does not recapitulate the phenotype and biology of MLL leukemias, which are clinically heterogeneous--presumably reflecting differences in LSC biology and/or frequency. TALE (three-amino-acid loop extension) class homeodomain proteins of the Pbx and Meis families are also misexpressed in this context, and we thus employed knockout, knockdown, and dominant-negative genetic techniques to investigate the requirements and contributions of these factors in MLL oncoprotein-induced acute myeloid leukemia. Our studies show that induction and maintenance of MLL transformation requires Meis1 and is codependent on the redundant contributions of Pbx2 and Pbx3. Meis1 in particular serves a major role in establishing LSC potential, and determines LSC frequency by quantitatively regulating the extent of self-renewal, differentiation arrest, and cycling, as well as the rate of in vivo LSC generation from myeloid progenitors. Thus, TALE proteins are critical downstream effectors within an essential homeoprotein network that serves a rate-limiting regulatory role in MLL leukemogenesis.
URLPMID:5774621 [本文引用: 1]
Abstract Aberrant activation of the three-amino-acid-loop extension homeobox gene MEIS1 shortens the latency and accelerates the onset and progression of acute leukemia, yet the molecular mechanism underlying persistent activation of the MEIS1 gene in leukemia remains poorly understood. Here we used a combined comparative genomics analysis and an in vivo transgenic zebrafish assay to identify six regulatory DNA elements that are able to direct green fluorescent protein expression in a spatiotemporal manner during zebrafish embryonic hematopoiesis. Analysis of chromatin characteristics and regulatory signatures suggests that many of these predicted elements are potential enhancers in mammalian hematopoiesis. Strikingly, one of the enhancer elements (E9) is a frequent integration site in retroviral-induced mouse acute leukemia. The genomic region corresponding to enhancer E9 is differentially marked by H3K4 monomethylation and H3K27 acetylation, hallmarks of active enhancers, in multiple leukemia cell lines. Decreased enrichment of these histone marks is associated with downregulation of MEIS1 expression during hematopoietic differentiation. Further, MEIS1/HOXA9 transactivate this enhancer via a conserved binding motif in vitro, and participate in an autoregulatory loop that modulates MEIS1 expression in vivo. Our results suggest that an intronic enhancer regulates the expression of MEIS1 in hematopoiesis and contributes to its aberrant expression in acute leukemia.
URLPMID:24445817 [本文引用: 1]
Mixed lineage leukemia (MLL) fusion proteins directly activate the expression of key downstream genes such as MEIS1, HOXA9 to drive an aggressive form of human leukemia. However, it is still poorly understood what additional transcriptional regulators, independent of the MLL fusion pathway, contribute to the development of MLL leukemia. Here we show that the transcription factor PU.1 is essential for MLL leukemia and is required for the growth of MLL leukemic cells via the promotion of cell-cycle progression and inhibition of apoptosis. Importantly, PU.1 expression is not under the control of MLL fusion proteins. We further identified a PU.1-governed 15-gene signature, which contains key regulators in the MEIS-HOX program (MEIS1, PBX3, FLT3, and c-KIT). PU.1 directly binds to the genomic loci of its target genes in vivo, and is required to maintain active expression of those genes in both normal hematopoietic stem and progenitor cells and in MLL leukemia. Finally, the clinical significance of the identified PU.1 signature was indicated by its ability to predict survival in acute myelogenous leukemia patients. Together, our findings demonstrate that PU.1 contributes to the development of MLL leukemia, partially via crosstalk with the MEIS/HOX pathway.
URLPMID:5472177 [本文引用: 1]
react-text: 170 In Reply.— We appreciate the comments of Dr Goldstein and Drs Mazur and Herbert regarding our recent report in JAMA. We have data available on changes in low-density lipoprotein and high-density lipoprotein (HDL) cholesterol concentrations in five of the eight patients reported (Table). In three of these patients HDL-cholesterol decreased slightly, but in all instances the decrease in... /react-text react-text: 171 /react-text [Show full abstract]
URLPMID:29467488 [本文引用: 1]
To investigate the role of SH-2-containing protein tyrosine phosphatase 1, SHP-1, in IL-4-induced IL-4 receptor (IL-4R) expression, we examined IL-4 receptor α-chain (IL-4Rα) mRNA expression in Na3VO4-treated wild type (WT) spleen cells and measured IL-4R mRNA in IL-4-stimulated spleen cells of viable motheaten mice (me v/mev). It is found that IL-4-induced IL-4R mRNA expression was impaired... [Show full abstract]
URLPMID:24509477 [本文引用: 1]
Acute leukemia characterized by chromosomal rearrangements requires additional molecular disruptions to develop into full-blown malignancy, yet the cooperative mechanisms remain elusive. Using whole-genome sequencing of a pair of monozygotic twins discordant for MLL (also called KMT2A) gene-rearranged leukemia, we identified a transforming MLL-NRIP3 fusion gene and biallelic mutations in SETD2 (encoding a histone H3K36 methyltransferase). Moreover, loss-of-function point mutations in SETD2 were recurrent (6.2%) in 241 patients with acute leukemia and were associated with multiple major chromosomal aberrations. We observed a global loss of H3K36 trimethylation (H3K36me3) in leukemic blasts with mutations in SETD2. In the presence of a genetic lesion, downregulation of SETD2 contributed to both initiation and progression during leukemia development by promoting the self-renewal potential of leukemia stem cells. Therefore, our study provides compelling evidence for SETD2 as a new tumor suppressor. Disruption of the SETD2-H3K36me3 pathway is a distinct epigenetic mechanism for leukemia development.
URLPMID:29249820 [本文引用: 1]
Hemophagocytic lymphohistiocytosis is a life-threatening syndrome characterized by overwhelming immune activation. A steroid and chemotherapy-based regimen remains as the first-line of therapy but it has substantial morbidity. Thus, novel, less toxic therapy for hemophagocytic lymphohistiocytosis is urgently needed. Although differences exist between familial hemophagocytic lymphohistiocytosis... [Show full abstract]
URLPMID:21116747 [本文引用: 1]
Extranodal NK/T cell lymphoma, nasal type (ENKL) with advanced stage and aggressive NK-cell leukemia (ANKL) are highly aggressive neoplasms with a dismal clinical outcome. It is well known that P-glycoprotein, which is a product of MDR1 gene and related to multi-drug resistance, is expressed on tumor cells of ENKL or ANKL. This is a major reason for the refractoriness to conventional chemotherapeutic regimens for malignant lymphoma containing anthracycline. However, recent studies have identified that several drugs including l -asparaginase, methotrexate and alkylators show excellent effect for these tumors. The SMILE (steroid, methotrexate, ifosfamide, l -asparaginase and etoposide) regimen is one of the promising regimens for advanced or relapsed/refractory ENKL, but its myelotoxicity is strong. ANKL needs another treatment strategy because of a systemic disease progression and extensive organ insufficiency. Optimal treatment scheme using such effective agents for these unfavorable NK-cell tumors should further be explored.
URLPMID:29148541 [本文引用: 1]
react-text: 293 When oxygen is limiting, cells adjust their metabolism through the transcription factors hypoxia-inducible factors (HIFs). Loss of function of VHL, a protein necessary to maintain the low abundance of HIFs under normoxic conditions, results in constitutively active HIFs and contributes to some forms of cancer, in particular clear cell renal carcinoma (ccRCC). Li et al. found through... /react-text react-text: 294 /react-text [Show full abstract]
URL [本文引用: 1]
URLPMID:20929322 [本文引用: 1]
Data on childhood acute myeloid leukemia (AML) in developing countries are limited. Herein we report the outcome of childhood AML treated with modified NPCLC-AML97 in our institution from 1997 to 2005. One hundred and eighty-five children with newly diagnosed AML were admitted. The 7-year overall survival (OS) and event free survival (EFS) rates for the whole cohort were 33.1 00± 4.1% and 31.2 00± 3.7%, respectively. Sixty patients (32.4%) refused chemotherapy and 123 were eligible for protocol evaluation. Among eligible patients, 111 (90.2%) achieved complete remission (CR). The estimated 7-year OS and EFS rates were 50.2 00± 5.5% and 46.9 00± 5.1%, respectively. APL was more curable than non-APL (7-year EFS: 63.5 00± 7.9% vs. 35.9 00± 6.3%, p = 0.005). Thirty-one patients (25.2%) relapsed, but no central nervous system leukemia was observed. Although the cure rate of childhood AML in China was low, the treatment outcome for patients who could adhere to the treatment protocol was satisfactory.
URL [本文引用: 1]
Philadelphia chromosome–like acute lymphoblastic leukemia (Ph-like ALL) is a recently described B-cell precursor ALL with a gene expression profile and a high frequency of IKZF1 gene alteration similar to that of Ph-positive ALL. Its prevalence is approximately 12% in children, 21% in adolescents (16-20 years of age), and 20% to 24% in adults older than 40 years, with a peak (27%) in young... [Show full abstract]
URLPMID:29101237 [本文引用: 1]
Publisher's Note: There is a [ Blood Commentary][1] on this article in this issue. [1]: /lookup/doi/10.1182/blood-2017-11-815464

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}