 ,1,2,3
,1,2,3Database resources of the reference genome and genetic variation maps for the Chinese population
Shuhui Song1,2,3, Xufei Teng1,3, Jingfa Xiao,1,2,3通讯作者:
编委: 赖江华
收稿日期:2018-05-24修回日期:2018-09-10网络出版日期:2018-11-20
| 基金资助: |
Editorial board:
Received:2018-05-24Revised:2018-09-10Online:2018-11-20
| Fund supported: |
作者简介 About authors
宋述慧,博士,副研究员,研究方向:生物信息学E-mail:songshh@big.ac.cn。
滕徐菲,在读硕士研究生,专业方向:生物信息学E-mail:tengxufei@big.ac.cn,宋述慧和滕徐菲并列第一作者。

摘要
关键词:
Abstract
Keywords:
PDF (465KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
宋述慧, 滕徐菲, 肖景发. 中国人群参考基因组及基因组变异图谱资源库[J]. 遗传, 2018, 40(11): 1048-1054 doi:10.16288/j.yczz.18-177
Shuhui Song, Xufei Teng, Jingfa Xiao.
自1990年10月1日启动“人类基因组计划”,到2003年4月15日,国际人类基因组组织正式宣布全部完成,历时10多年的国际人类基因组计划绘制了物理、遗传、序列和基因4张图谱[1, 2],开启了人类对自身(包括癌症在内的人类疾病的发生)的深入认识和研究,推动了测序技术、基因组学和生物信息学等的发展,并相继启动了国际单倍体型计划(HapMap计划)[3, 4]、“国际千人基因组计划”[5]、“肿瘤基因组解剖计划”[6,7,8]和“环境基因组学计划”[9]等一系列与健康相关的研究计划。其中,2008年1月22日启动的“国际千人基因组计划”[5]是举世闻名的人类基因组计划的延续和发展,该计划于2012年3月29日完成,是基因组科学研究向临床医学迈进的重要转折点,不仅绘制了迄今为止最详尽的、最有医学应用价值的人类基因组遗传多态性图谱,还贡献了海量的源于不同国家和不同人群的、包含着大量遗传变异信息的个人基因组数据[10, 11]。该计划产生的392个中国人(283个汉族和109个少数民族)样本的全基因组测序数据,为中国人群特异性的遗传特征和相关医学分析研究提供了宝贵的数据资源。科学家们通过分析,发现不同人种之间的基因组单核苷酸多态性位点及频率存在明显的差异,因此许多国家纷纷启动了面向本国或本地区的基因组测序计划,目标是建立更加精细的参考基因组及变异组。例如,英国于2010年和2012年分别启动了英国万人基因组计划和10万人基因组计划[12, 13],旨在通过大规模的基因组测序寻找英国人群特有的基因组变异,挖掘与健康和疾病相关联的遗传风险因素。2016年,日本人参考基因组计划(Japanese Reference Genome)通过新一代DNA测序技术构建了日本人参考基因组序列[14]。此外,澳大利亚、冰岛、加拿大、新加坡、韩国、荷兰、丹麦、沙特阿拉伯等国家和地区都纷纷启动了相应的基因组计划。中国人要有自己的基因组数据和参考基因组序列,才能解决中国人特有的疾病遗传问题。2007年10月,第一个黄种人个人基因序列“炎黄一号”完成[15],是首例基于二代测序技术完成的参考基因组序列。2016年6月,中国人个体基因组“华夏一号”公布,该个体基因组采用三代单分子测序和二代测序技术相结合,大幅度提高了基因组组装的完整性和准确性[16]。
基因组数据的测定为鉴定和研究遗传变异及多态特点提供了基础。国际人类基因组单体型图谱计划(HapMap计划)测定了全球11个人群,获得约500万单核苷酸多态性位点(single nucleotide polymerphisms, SNPs)。国际千人基因组计划对全球不同人类种群的2500人进行了全基因组测序,获得了8470万SNPs、360万序列插入删除(insertion or deletion)和6万结构变异。20世纪90年代以来,我国也先后启动和实施了“中华民族基因组SNP研究”、“中华民族基因组中若干位点基因结构的研究”和“中国人群若干群体的基因组多态性研究”等重大项目。这些项目的开展都为创建我国人群遗传资源库打下了重要的基础,如:通过对20 635例中国人群样本的主要组织相容性复合体(major histocompatibility complex, MHC)目标区域进行高深度测序和分析,建立了世界上最大样本量的中国人群MHC全区域完整遗传变异数据库[17],展示了中国人群MHC区域突变位点和HLA基因的多态性图谱,为开展中国人群复杂疾病与MHC区域的相关性研究奠定了坚实的基础。
尽管在人类(尤其是中国人)基因组的解析和发展中取得了长足的进步,但在基因组学研究中广泛用于序列比对分析的人类基因组参考序列,仅是基于有限的人类个体全基因组测序后的结果,这个不包含任何遗传变异信息的静态基因组显然不足以支持高度复杂的基因组学、转录组学、表观基因组学以及全基因组关联分析等研究;此外,目前国际上公开的人类基因组变异数据也主要来源于西方白种人,利用这些变异数据作为参比数据,常造成我国基因组研究和临床应用结果的不准确。面向未来中国精准医学研究的新需求,中科院北京基因组研究所发展并建立了基于中国人群全基因组测序数据的虚拟中国人基因组数据库(Virtual Chinese Genome Data Base, VCGDB)[18, 19]和基因组变异数据库(Genome Variation Map, GVM)[20]资源(图1),有效并全面展示了中国人群的遗传变异特征,更好服务于中国的人类遗传学、基因组学和生物医学的研究和应用。
图1
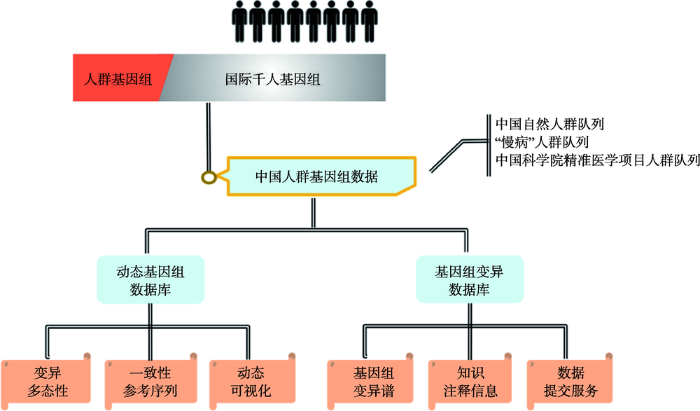 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1中国人群参考基因组和变异组数据库建立示意及主要特征
Fig. 1Schematic for Chinese reference genome and variome databases and their main characteristics
1 虚拟中国人基因组数据库
国际千人基因组计划提供了丰富的全基因组测序数据资源,其中包含中国南方汉族人群数据(Southern Han Chinese, CHS)、北方汉族人群数据(Han Chinese in Beijing, CHB)以及中国西双版纳傣族的中国人基因组数据(Dai Chinese in Xishuangbanna, CDX)。为了充分利用这些信息,选取该计划中包含中国南方人群和北方人群数据共计194个高覆盖度个体的全基因组序列数据,通过标准化数据分析和处理流程[18],构建了虚拟中国人基因组数据库(VCGDB, http://bigd.big.ac.cn/vcg/) (图2)。VCGDB提供了中国人群基因组多态性信息,共包括3500万个单核苷酸变异位点信息(SNPs)、50万个基因组插入删除片段信息、2900万个罕见变异位点信息,及其对应的基因组注释信息[18]。同时VCGDB还分别提供了中国人群体、南方人群体和北方人群体的一致性基因组参考序列。此外,通过真实的基因组测序数据序列比对分析,将其与已有的人类基因组参考序列以及“炎黄一号”进行比较,表明基于中国人群体高频遗传变异位点构建的中国人基因组一致性参考序列更能体现中国人群体的基因组特征。图2
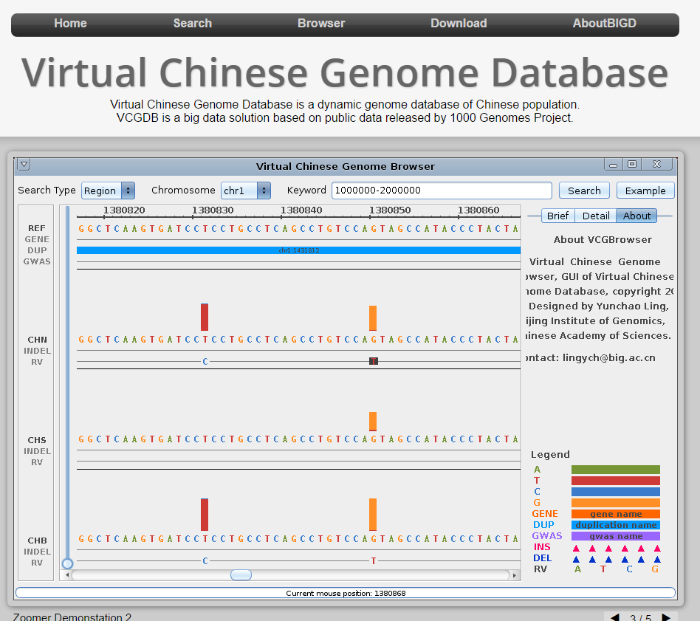 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2虚拟中国人基因组数据库主页
Fig. 2A screen shot of the home page of VCGDB
虚拟中国人基因组数据库具有以下特点:(1) VCGDB是一个“动态”的数据库,通过信息熵等方法来计算中国人群体之间各个位点遗传变异的动态变化水平和发生率,能够展示基因组中不同位点的遗传变异多态性信息和各位点不同基因型的发生频率信息;(2) VCGDB是一个“虚拟”的数据库,通过整合中国人群体高频遗传变异位点信息,以标准参考基因组为参照,分别构建了中国人群体、南方人群体和北方人群体的一致性基因组参考序列。构建的一致性基因组参考序列并不属于和代表任何一个真实存在的个体,而是源于对200多个个体TB级大规模数据进行综合分析的结果,也因此可以更好地描述中国人群体的遗传变异特征;(3) VCGDB提供高度交互的、友善的、融合多种全新功能的中国人动态基因组浏览器(VCGBrowser),相较于传统的基因组浏览器如UCSC Genome Browser和JBrowse Genome Browser并不能显示群体的基因组动态信息,VCGBrowser可根据用户的不同需求,从染色体、固定片段、指定基因和指定位点等多层次展示所有位点在不同群体的位点动态信息以及相关的基因组注释信息。总体上,虚拟中国人基因组数据库实现了对国际千人基因组计划中中国人群基因组测序数据的精细整合分析,并提供了中国人群体基因组变异的动态信息,为今后开展大规模人群基因组测序数据的分析和展示提供了参照[18]。
2 中国人群基因组变异数据库
虚拟中国人基因组数据库的建设为中国人基因组数据比对分析提供了较为精准的参考基因组,为进一步满足基于变异位点基因型和表型的关联分析及知识发现的研究需求,在VCGDB的基础上又发展和构建了中国人群基因组变异数据库(Genome Variation Map, GVM)(http://bigd.big.ac.cn/gvm, 图3)[20]。利用国际千人基因组计划中的215个(测序覆盖度>5)全基因组序列数据,采用统一的变异位点鉴定和注释分析流程[20],提供了截至日前最全的中国人群变异位点、人群频率和位点知识的注释信息,共包括13327822个单核苷酸变异位点信息(SNPs)、3 019 815个基因组插入删除片段信息、16 739 583个低发生概率(minor allele frequency, MAF <0.05)的变异位点信息,5 343 882个罕见发生概率(MAF <0.005)的变异位点信息,以及与这些位点和序列片段相关的基因组注释信息,包括位点突变效应、临床表型效应、人类孟德尔遗传疾病效应等。图3
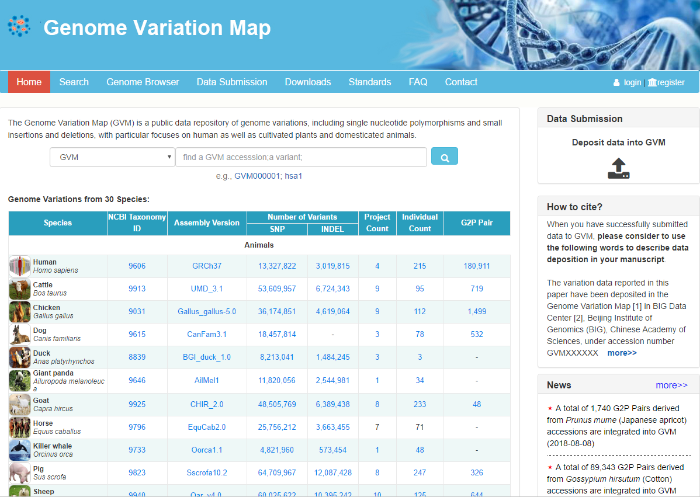 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基因组变异数据库主页
Fig. 3A screen shot of the home page of GVM
GVM数据库中的中国人群变异模块具有以下特点:(1)提供了强大实用的变异数据检索服务,支持用户根据变异位点编号、变异类型、变异效应、基因名称、基因功能、已知临床效应、遗传病和其他表型等条件进行组合检索,初步检索结果还可以进一步根据上述条件组合进行二次过滤。检索结果以表格形式返回,并提供了变异位点的详细注释信息、各个体的基因型在线浏览、全部检索结果条目的下载服务等;(2)提供了高度交互的、友善的、融合多种功能和信息的基于GBrowser技术的在线浏览功能,支持用户自主选择感兴趣的个体和区间,提供统一的坐标系直接展示和比较全基因组水平的所有动态变异信息,可以根据用户需求缩放至单碱基水平,从基因组的水平展示某个变异位点的频率信息和其他详细细节;(3)提供了数据递交服务,支持用户选择在线或离线两种不同的方式递交数据,用户通过创建BioProjct填写数据元信息,系统对所提交变异数据自动分配唯一编号(GVMXXXXXX),该编号可以直接应用于科技论文的数据获取。总体上,中国人群基因组变异数据库实现了对国际千人基因组计划海量中国人数据的完整、全面的整合和高效展示,体现了中国人群的变异特征。是未来精准医学研究表型与基因型关联分析的重要基础,为未来我国精准医学队列人群计划大数据的处理和分析、数据管理等提供了示范指导,也为基于基因组序列变异的遗传检测、药物研发等提供数据支持。
3 未来展望
国际大型基因组研究计划如HapMap和千人基因组计划等虽已将汉族样本作为主要亚洲人群进行了研究。然而,这些重要参考数据却存在着很大的局限:一方面样本量较少,因而对于低频基因组多态性的代表性差;另一方面对应样本没有表型信息,无法将遗传多态性与表型进行有效关联。中国科学院于2015年率先启动了“中国人群精准医学研究计划(中科院)”重点部署项目,已产出上千人的高覆盖度的全基因组测序数据,并已经提交到中国生命与健康大数据中心[21]的组学原始数据库GSA中管理[22]。2016年,我国“十三五”重点研发计划中已设置了为实施精准医学研究而构建百万人以上的自然人群国家大型健康队列和重大疾病专病队列的项目,随着国家“慢病”、“精准医学”大型人群队列项目的启动,未来将产生百万人群的多达EB级组学数据。这些人群队列的基因组数据将进一步丰富和完善中国人群的参考基因组和基因组变异数据。此外,VCGDB和GVM数据库将为未来大规模人群队列基因组数据的汇交、存储、管理、共享与分析提供支持和指导。国家“慢病”、“精准医学”大型人群队列项目,未来不仅产出多层次的组学分子数据,还将产出海量多维的生物医学数据,包括基线数据、随访和临床表型组数据等。未来将建立包含多层次多维度信息数据的VCGDB和GVM数据库,开展大数据驱动的创新应用研究和疾病防诊治方案的精准化研究,助力于发现疾病或表型、用药等遗传关联位点,为生物医学数据转换为能够支撑临床决策的辅助诊疗信息,服务大众实现疾病预测和预警提供重要基础性保障,真正实现数据到信息和知识的转化。
发展可用于精准医学研究组学数据分析的大数据系统的应用体系和解析体系,开发及完善基因组分析算法及软硬件,开发基于通路分析和网络模块的基因型-表型深度解析方法,形成可用于海量基因型-表型数据解析流程、汇交管理、数据挖掘整合的算法体系,并在VCGDB和GVM中为临床和其他科研人员提供完善的在线分析和研究系统,助力我国的精准医学研究与应用。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:12690187 [本文引用: 1]

The Human Genome Project has been the first major foray of the biological and medical research communities into ``big science.'' In this Viewpoint, we present some of our experiences in organizing and managing such a complicated, publicly funded, international effort. We believe that many of the lessons we learned will be applicable to future large-scale projects in biology.
URL [本文引用: 1]
URLPMID:14685227 [本文引用: 1]
The goal of the International HapMap Project is to determine the common patterns of DNA sequence variation in the human genome and to make this information freely available in the public domain. An international consortium is developing a map of these patterns across the genome by determining the genotypes of one million or more sequence variants, their frequencies and the degree of association between them, in DNA samples from populations with ancestry from parts of Africa, Asia and Europe. The HapMap will allow the discovery of sequence variants that affect common disease, will facilitate development of diagnostic tools, and will enhance our ability to choose targets for therapeutic intervention. 2003 Nature Publishing Group.
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
URLPMID:21720365 [本文引用: 1]
A catalogue of molecular aberrations that cause ovarian cancer is critical for developing and deploying therapies that will improve patients' lives. The Cancer Genome Atlas project has analysed messenger RNA expression, microRNA expression, promoter methylation and DNA copy number in 489 high-grade serous ovarian adenocarcinomas and the DNA sequences of exons from coding genes in 316 of these tumours. Here we report that high-grade serous ovarian cancer is characterized by TP53 mutations in almost all tumours (96%); low prevalence but statistically recurrent somatic mutations in nine further genes including NF1, BRCA1, BRCA2, RB1 and CDK12; 113 significant focal DNA copy number aberrations; and promoter methylation events involving 168 genes. Analyses delineated four ovarian cancer transcriptional subtypes, three microRNA subtypes, four promoter methylation subtypes and a transcriptional signature associated with survival duration, and shed new light on the impact that tumours with BRCA1/2 (BRCA1 or BRCA2) and CCNE1 aberrations have on survival. Pathway analyses suggested that homologous recombination is defective in about half of the tumours analysed, and that NOTCH and FOXM1 signalling are involved in serous ovarian cancer pathophysiology.
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:28796226 [本文引用: 1]
Gene-edited embryos are exciting, but the truly urgent conversations concern genomic medicine, says Vivienne Parry.
URLPMID:29691228 [本文引用: 1]
In partnership with NHS England, Genomics England’s ambitious plans to embed genomic medicine into routine patient care are well underway. Clare Turnbull and colleagues discuss its progressMany disorders we encounter in clinical medicine have a genomic basis, from rare “single gene” disorders such as cystic fibrosis, to complex, polygenic disorders such as ischaemic heart disease, drug toxicity, and tumour evolution driven by serial somatic mutations. Next generation technology has transformed the capacity, speed, and cost of genomic sequencing. This has provided important advances and new opportunities for the clinical application of genomics (fig 1). However, radical expansion of genomic medicine within clinical care requires new infrastructure, extended skills, education of the workforce, and diligent engagement with the public. The Genomics England 10065000 Genomes Project was initiated in 2013 to establish the use of whole genome sequencing in the NHS and drive change within NHS services to adopt this technology.Fig 1 Potential applications of genomics in medicineThe UK has long been at the forefront of discovery in human genomics and is recognised for its world leading genetic research studies, such as UK Biobank and Deciphering Developmental Disorders (fig 2).123 In parallel the UK has evolved a mature network of NHS funded regional genetics laboratories and clinical genetics departments.Fig 2 Genomics in the UK: timelines of clinical testing and research achievementsUntil recently, genomic technologies available in the clinic have enabled us to look for the “causative mutation” just one segment of a gene at a time, limiting both the speed and volume of clinical testing. Over the past decade, next generation sequencing has made it possible to sequence millions of fragments of DNA simultaneously. This step change in scale enables us to offer genetic testing to many more people and test one person for hundreds or thousands …
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:4931320 [本文引用: 1]
Short-read sequencing has enabled thede novoassembly of several individual human genomes, but with inherent limitations in characterizing repeat elements. Here we sequence a Chinese individual HX1 by single-molecule real-time (SMRT) long-read sequencing, construct a physical map by NanoChannel arrays and generate ade novoassembly of 2.9365Gb (contig N50: 8.365Mb, scaffold N50: 22.065Mb, including 39.365Mb N-bases), together with 20665Mb of alternative haplotypes. The assembly fully or partially fills 274 (28.4%) N-gaps in the reference genome GRCh38. Comparison to GRCh38 reveals 12.865Mb of HX1-specific sequences, including 4.165Mb that are not present in previously reported Asian genomes. Furthermore, long-read sequencing of the transcriptome reveals novel spliced genes that are not annotated in GENCODE and are missed by short-read RNA-Seq. Our results imply that improved characterization of genome functional variation may require the use of a range of genomic technologies on diverse human populations. Short-read sequencing has inherent limitations in the characterisation of long repeat elements. Shi and Guoet al.combine single-molecule real-time sequencing and IrysChip to construct a Chinese reference genome that fills many gaps in the reference genome, and identify novel spliced genes.
URL [本文引用: 1]
URLPMID:4028056 [本文引用: 4]
Background The data released by the 1000 Genomes Project contain an increasing number of genome sequences from different nations and populations with a large number of genetic variations. As a result, the focus of human genome studies is changing from single and static to complex and dynamic. The currently available human reference genome (GRCh37) is based on sequencing data from 13 anonymous Caucasian volunteers, which might limit the scope of genomics, transcriptomics, epigenetics, and genome wide association studies. Description We used the massive amount of sequencing data published by the 1000 Genomes Project Consortium to construct the Virtual Chinese Genome Database (VCGDB), a dynamic genome database of the Chinese population based on the whole genome sequencing data of 194 individuals. VCGDB provides dynamic genomic information, which contains 35 million single nucleotide variations (SNVs), 0.5 million insertions/deletions (indels), and 29 million rare variations, together with genomic annotation information. VCGDB also provides a highly interactive user-friendly virtual Chinese genome browser (VCGBrowser) with functions like seamless zooming and real-time searching. In addition, we have established three population-specific consensus Chinese reference genomes that are compatible with mainstream alignment software. Conclusions VCGDB offers a feasible strategy for processing big data to keep pace with the biological data explosion by providing a robust resource for genomics studies; in particular, studies aimed at finding regions of the genome associated with diseases.
[学位论文].
URL [本文引用: 1]
国际千人基因组计划贡献了海量的源于不同国家和不同人群的、包含着大量遗传变异信息的个人基因组数据。生物大数据的产生对科学家提出了新的问题和挑战,即如何有效地利用如此大规模的数据,合理规划数据的传输、分析和存储流程,最终发现隐藏在数据中的知识和规律,已经成为非常紧迫的问题。目前基因组学研究已经从原有单一的、静态的人类基因组向更加复杂的、动态的个体化基因组转型。然而,基因组学研究中作为指导标准存在的,广泛用于比对过程中的人类基因组参考序列,仅是基于有限的人类个体全基因组测序后的结果。这个不包含任何遗传变异信息的静态基因组显然不足以用于高度复杂的基因组学、转录组学、表观基因组学以及全基因组关联分析等研究。 本课题基于国际千人基因组计划中来自于两个中国人群体的194个全基因组序列数据,构建了虚拟中国人动态基因组数据库(VCGDB)。VCGDB提供了一系列动态基因组学信息,共包括3500万个单核苷酸变异位点信息(SNVs)、50万个基因组插入删除片段信息(InDels)、2900万个罕见发生概率的变异位点信息,以及与这些位点和序列片段相关的基因组注释信息。综合这些基因组变异信息,我们构建了一条中国人群体的基因组一致性参考序列,并使用真实的基因组测序数据进行比对,将其与已有的人类基因组参考序列进行比较,结果表明基于动态基因组构建的中国人基因组参考序列更能体现中国人群体的基因组特征。 VCGDB是“虚拟”的数据库。因为虚拟中国人基因组并不属于和代表任何一个真实存在的中国人个体,而是源于对几百个中国人个体的TB级大规模数据进行综合分析的结果,也因此可以描述中国人群体的遗传变异特性和各个位点上的碱基偏好性。VCGDB同时又是“动态”的数据库。我们从样本和人群等多个水平,使用信息熵等方法来分析和评估中国人个体之间以及人群之间各个单核苷酸变异位点、插入删除信息、结构变异信息的动态变化水平和发生率。VCGDB将动态变异与个体特征以及基因组注释信息,比如相关的基因信息、基因组重复片段信息和全基因组关联临床特征信息等进行了有机地整合,汇总得到与中国人群体相关的所有动态信息。 VCGDB同时提供高度交互的、友善的、融合多种全新功能的虚拟中国人基因组浏览器(VCGBrowser)。该浏览器支持从网页直接使用或以客户端形式使用,也支持本地跨平台使用,具有高兼容性特性。不论是在单个群体内或是多个群体之间,它提供了一个全方位的视角和一个统一的坐标系,来直接地展示和比较全基因组水平的所有动态变异信息。VCGBrowser具有高度灵活特性,支持对动态基因组进行实时、无极缩放到任意分辨率,从基因组的水平展示某个基因组区域的动态变异分布信息,到位点水平展示各位点的动态变异细节信息。得益于高度结构化和索引优化的虚拟中国人基因组数据库,VCGBrowser支持由浏览器点击触发实时搜索,并返回细节信息。 总体上,虚拟中国人基因组数据库实现了对国际千人基因组计划海量数据的高效利用和成功展示,为生物大数据的处理和分析提供了成功案例,并且将在数据持续增长的情况下提供稳定、有效的资源,以求对基因组学以及其他与疾病相关领域,特别是个体化基因组方面的研究有所帮助。
URL [本文引用: 3]
react-text: 179 Background/Question/Methods General theories for macroecological patterns have become increasingly prevalent in the last decade. These theories potentially allow predictions to be made in the absence of detailed understanding of the processes structuring an ecosystem. We discuss research testing one of these general theories, the Maximum Entropy Theory of Ecology, which posits that many... /react-text react-text: 180 /react-text [Show full abstract]
URLPMID:29140455 [本文引用: 1]
Abstract Cell types in cell populations change as the condition changes: some cell types die out, new cell types may emerge and surviving cell types evolve to adapt to the new condition. Using single-cell RNA-sequencing data that measure the gene expression of cells before and after the condition change, we propose an algorithm, SparseDC, which identifies cell types, traces their changes across conditions and identifies genes which are marker genes for these changes. By solving a unified optimization problem, SparseDC completes all three tasks simultaneously. SparseDC is highly computationally efficient and demonstrates its accuracy on both simulated and real data. The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
URLPMID:5339404 [本文引用: 1]
With the rapid development of sequencing technologies towards higher throughput and lower cost, sequence data are generated at an unprecedentedly explosive rate. To provide an efficient and easy-to-use platform for managing huge sequence data, here we presentGenome Sequence Archive(GSA;http://bigd.big.ac.cn/gsaorhttp://gsa.big.ac.cn), a data repository for archivingraw sequence data. In compliance with data standards and structures of the International Nucleotide Sequence Database Collaboration (INSDC), GSA adopts four data objects (BioProject, BioSample, Experiment, and Run) for data organization, accepts raw sequence reads produced by a variety of sequencing platforms, stores both sequence reads and metadata submitted from all over the world, and makes all these data publicly available to worldwide scientific communities. In the era ofbig data, GSA is not only an important complement to existing INSDC members by alleviating the increasing burdens of handling sequence data deluge, but also takes the significant responsibility for global big data archive and provides free unrestricted access to all publicly available data in support of research activities throughout the world.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}