 ,2, 吴元奇,1
,2, 吴元奇,1Comparation and utilization of crop-omics databases
Jie Song1,2, Yongbo Wu2, Yueheng Zhou2, Bojuan Liu2, Nan Wang2, Zhuanfang Hao,2, Yuanqi Wu,1第一联系人:
编委: 赵方庆
收稿日期:2018-03-5修回日期:2018-06-4网络出版日期:2018-07-20
| 基金资助: |
Received:2018-03-5Revised:2018-06-4Online:2018-07-20
| Fund supported: |
作者简介 About authors
作者简介:宋洁,硕士研究生,专业方向:玉米遗传育种E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (484KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
宋洁, 吴永波, 周跃恒, 柳波娟, 王楠, 郝转芳, 吴元奇. 作物组学数据库的比较和应用. 遗传[J], 2018, 40(7): 534-545 doi:10.16288/j.yczz.18-004
Jie Song, Yongbo Wu, Yueheng Zhou, Bojuan Liu, Nan Wang, Zhuanfang Hao, Yuanqi Wu.
作物生命科学研究是我国农业科技可持续发展的强大支撑[1],基于高通量数据发展起来的基因组学、转录组学、蛋白质组学、代谢组学及表观组学研究是作物生命科学研究的核心领域。利用组学研究高通量信息化的特点,可有效解决阻碍农业科学发展中复杂性状的遗传及相关学科交叉问题,包括种质资源利用和创新,重大应用价值基因的克隆、功能注释及解析,全基因组选择以及基因预测等。因此,必须加速推进作物组学研究的进程,发掘更多的优异种质资源,解析重要基因遗传机制和培育突破性的新品种,从而满足农业科技发展对组学研究的需求。
21世纪以来,水稻(Oryza sativa L.)[2,3]、玉米(Zea mays L.)[4]、大豆(Glycine max L.)[5]和小麦(Triticum aestivum L.)[6,7]等主要农作物的基因组相继被破译。其中,我国牵头完成了水稻和小麦等重要农作物以及黄瓜(Cucumis sativus L.)和甘薯(Dioscorea esculenta (Lour.) Burkill)等重要经济作物的基因组测序,其重要性状的遗传机制及相应基因功能得到了解析,基因组学的研究水平已走在国际前列。随着高通量测序技术的发展以及科学研究需求的不断提高,目前全球各数据库中的高通量测序数据呈指数级增长,生物信息学成为整合并有效提取高通量数据信息的主要方法。数据库是生物信息学研究的主要载体,各种数据库几乎覆盖了生命科学的全部领域。其中,组学数据库不仅为各组学的研究和发展提供了丰富的信息来源,同时强化了不同组学之间及与其他系统生物学分支之间的联系,为科学研究提供了诸多便利。
根据对数据处理分析程度的不同,组学数据库可分为一级数据库(基础数据库)和二级数据库。其中,一级数据库只收集大量的数据,进行数据的存储和呈现;二级数据库在存储数据的基础上还对数据进行处理和加工[8]。根据数据的存储内容,组学数据库又可分为基因组学数据库、转录组学数据库、蛋白质组学数据库、代谢组学数据库和表型组学数据库。目前,这5大组学数据库是生物信息学研究的热点,充分了解各组学数据库的信息内容并对其进行深度的挖掘和利用,也是建立作物组学研究系统的前提和条件。
1 作物组学及相关数据库
1.1 作物基因组学及相关数据库
1.1.1 基因组学在作物科学研究中的应用基因组学(genomics)是对生物体的所有基因组信息及相关数据进行分析利用的科学,包括对基因的序列、位置、结构、功能及相互作用等的研究。研究内容可分为两部分:一是以全基因组测序为目标的结构基因组学(structural genomics);二是以基因功能鉴定为目标的功能基因组学(functional genomics),又称为后基因组学(post-genomics)[9]。
基因组学是组学中最早提出的概念,起源于物种高通量的基因组序列,目的是为了构建和完善基因连锁图谱、物理图谱和转录图谱,以及对基因功能进行探索。目前,作物基因组学研究主要涉及两个方面:一是基因组测序,对物种的不同个体进行基因组de novo测序或重测序,从而对个体或群体进行差异性分析;二是通过大量遗传标记分析相关的目标性状,进而采用全基因组关联分析等方法,发掘影响该性状的基因变异以及对目标候选基因进行功能注释等。例如在水稻中,Huang等[10]通过对950个国内外水稻品种进行基因组重测序,利用全基因组关联分析,分别在粳稻(Japonica)、籼稻(Indica)群体中找到与开花时间相关的32个新的遗传位点。在玉米中,Chia等[11]通过对105个野生和栽培玉米品种进行测序和分析,成功构建了第二代玉米单体型图谱(Maize HapMap 2),可以进一步对玉米遗传多样性进行更全面更系统地分析。在小麦中,凌宏清等[12]基于A基因组测序结果,发现TuGASR7和TaGASR7是控制水稻粒长基因OsGASR7的同源基因,并在乌拉尔图小麦(Triticum urartu)和普通小麦中获得该基因的单倍型,关联分析进一步证明TuGASR7和TaGASR7基因参与调控小麦粒长和千粒重的基因表达;另外,李爱丽等[13]基于D基因组测序结果,发现NBS-LRRl等基因与小麦抗病能力相关,为研究小麦的抗逆性及适应性奠定了基础。
1.1.2 基因组学相关数据库
NCBI (National Center for Biotechnology Information) GenBank、EMBL (The European Molecular Biology Laboratory) EBI和DDBJ (DNA Data Bank of Japan)共同组成了世界3大基因组学综合数据库[14]。这3大基因组学综合数据库又分别包含许多子数据库。GenBank收集了各种基因组序列,包括高通量基因组序列(high throughput genomic sequences, HTGS)、表达序列标记(expressed sequence tags, EST)、序列标记位点(sequence tagged sites, STS)和基因组概览序列(genome survey sequences, GSS);EBI可进行核苷酸序列检索及序列相似性查询;DDBJ主要搜集DNA序列信息并赋予每个数据不同的存取号。为保证数据完整性,这3大数据库之间又建立了相互交换数据的合作关系。
此外,与基因组有关的数据库还有很多。如植物基因组和基因家族的数据分析中心(Phytozome),该数据库涵盖了多种植物的基因信息,包括基因序列、基因结构、基因家族信息,以及基因相关的功能注释;美国基因组资源中心(NCGR)的基因组序列数据库(genome sequence database, GSDB)主要搜集、管理和发布完整的DNA序列及其相关信息,以满足基因组测序中心的需要;作物基因组信息数据库(Gramene)具备对各基因组进行高级分析的功能;与Gramene有共建合作关系的植物物种科学基因组中心(Ensembl Plants),包含参考序列、基因注释、RNA和蛋白质排列等与全基因组相关的所有数据;植物基因组序列数据库(PlantGDB)主要包含ESTs数据以及对这些数据的注释、定位和与其他数据库的链接,满足基础的数据查询功能;植物基础功能信息的数据库(planteome)主要涉及到植物的器官结构、不同发育时期的基因组信息;另外,还有用于提供一系列未知基因的功能注释及微阵列探针信息的数据库(EasyGO),它包含15个物种(主要是植物)40多个数据类型。以上这些基因组数据库基本可以满足作物大多数基因组信息的查询。
1998年,国际水稻基因组测序计划正式启动;2002年,籼稻和粳稻两个亚种全基因组框架图以及全长序列的测定相继完成,形成了水稻基因组精确序列数据[15]。随后,我国启动了中国水稻功能基因组计划(China rice functional genomics program, CRFGP),该计划根据测序得到的精确序列,鉴定了水稻中控制产量、口感、香味、抗病虫害等重要农艺性状的基因,进而提高了水稻产量并改善了水稻品质[16]。2005年,中国农业科学院作物科学研究所和中国水稻研究所提出并主持了水稻基因数据库(Rice Data)的构建工作[17],该工作一方面收集整理国内外发现的水稻基因(含数量性状位点信息),包括基因名称、功能、定位等信息及参考文献等,另一方面,为了加强水稻基因数据库与水稻分子育种信息平台内其他相关数据库的关联,重新构建了ONTOLOGY系统、水稻分子标记数据库、遗传图谱数据库及相关的参考文献数据库。另外,还有一些针对水稻的专用数据库,如美国基因组研究所数据库(the institute of genomic research database-TDB, TIGR)中的水稻基因组数据库(The TIGR Rice Database)、水稻基因组注释数据库(national center of plant gene research, NCPGR)、水稻信息共享数据库(information commons for rice, IC4R)、水稻EST数据库(REDB)、水稻突变体数据库(RMD)、水稻基因组的基因预测工具(BGF)、水稻信息系统(BGI-RIS)以及韩国的水稻基因组数据库(KOME),密歇根州立大学的水稻基因组注释数据库(RGAP)等。
2009年,美国宣布以高产玉米自交系B73为对象的测序工作完成[18],这项成果为玉米基因组学的研究提供了强大的数据支撑。目前,玉米相关数据库大多来自美国,包括TIGR研究所维护的玉米基因组数据库(The TIGR Maize Database);玉米遗传学和基因组学数据库(maize genetics and genomics database, MaizeGDB),该数据库主要包含玉米序列、表型、基因型和核型变异以及染色体图谱数据等,涵盖了所有遗传学、基因产物、功能分析以及相关文献查阅等的玉米基因组学研究信息;另外,还有可用于玉米基因组中分子和功能多样性分析的玉米基因组工程数据库(Panzea)、储存玉米基因组研究进展的玉米基因组进展信息数据库(maize genetics cooperation newsletter, MGCN)、收集显著突变体的玉米定点诱导突变体数据库(maize targeted mutagenesis database, MTM)以及德国玉米基因组数据库(plant genome and systems biology, PGSB)等。
2013年,中国科学院遗传与发育生物学研究所联合我国多家单位,首次测序完成了普通小麦A基因组序列草图[19]。同年,中国农业科学院作物科学研究所联合多家单位对粗山羊草(Aegilops tauschii,含DD基因组)基因组进行测序,完成了小麦D基因组供体种—粗山羊草基因组草图的绘制[20];随后对D基因组重新测序和组装,完成了染色体级别的D基因组精细图谱的绘制[21]。由于小麦基因组的多倍性,其基因和功能分析极具挑战,相关研究也相对滞后,而小麦A基因组和D基因组序列信息为研究和改良小麦提供了一个宝贵的平台。基于小麦基因组相关数据库的建立,科学家们通过基因同源性比对分析,鉴定出一批控制小麦籽粒长度、千粒重、株高、落粒性等重要农艺性状的基因,极大推动了小麦基因组数据库的发展。由TIGR研究所维护的小麦基因组数据库(The TIGR Wheat Database),它主要包含小麦的基因组及基因注释信息;谷类作物信息数据库(Grain Genes)也包含小麦等麦类品种的遗传信息和遗传图谱。另外,还有一些针对小麦的专用数据库,如由捷克共和国作物种植研究所维护的小麦数据库(european cooperative programme for plant genetic resources, ECPGR)、由北海道大学等日本高校联合维护的日本小麦网(shared information of genetic resources, SHIGEN)和德国植物基因组计划中心数据库包含的EST数据库(the crop EST database, CR-EST)等。
1.2 作物转录组学及相关数据库
1.2.1 转录组学在作物科学研究中的应用转录组(transcriptome)是活细胞所能转录出来的所有RNA的总和,是研究细胞表型和基因功能的一个重要方法[22]。转录组学(tanscriptomics)从RNA水平研究基因的表达情况,是一门在整体水平上研究基因转录及转录调控规律的学科。目前,转录组学主要基于RNA测序技术(RNA-seq)对器官发育、杂种优势、生物或非生物胁迫的应答、表观遗传学的应用以及分子标记的筛选等进行研究。在水稻中,Zhai等[23,24]分别对超级杂交稻协优9308及其亲本R9308、协青早B的分蘖期和抽穗期的根部RNA进行测序,研究了水稻杂种优势的分子机制,并阐述了RNA-seq在器官发育和杂种优势中的应用。在玉米中,Trevisan等[25]对硝酸盐胁迫处理的玉米根系中的RNA进行测序,筛选差异表达基因,从而证明了根表皮细胞中存在一氧化氮(NO)合成/清除基因的共转录模式。在小麦中,傅艳萍等[26]对小麦苗期和成株期叶片进行RNA-seq,分别获得苗期和成株期差异表达基因以及大量的抗病相关基因,解析了器官发育机制以及后续功能标记的应用。
1.2.2 转录组学相关数据库
转录组学研究建立的数据库大多是综合数据库,目前最有影响的3个转录学研究平台是NCBI、EBI和SMD。NCBI中的基因表达数据库(gene expression omnibus, GEO)是最大、最全面的基因表达和微阵列数据库;EBI中的基因表达数据的微阵列数据库(ArrayExpress),不仅存储基因注释数据,还包含多个基因表达数据集以及与实验相关的原始图像集;另外,Oracle (甲骨文股份有限公司,全球大型数据库软件公司)作为数据库管理软件的微阵列数据库(stanford microarray database, SMD),存储了微阵列实验的原始数据、归一化数据和对应的图像文件。
Genevestigator和我国的植物转录因子数据库(plant transcription factor database, PlantTFDB)也可用于转录组学研究。Genevestigator可查询基因表达和进行元分析(Meta-analysis),而且提供了许多植物基因注释信息。PlantTFDB收录了包括拟南芥(Arabidopsis thaliana L.)、水稻等20个种植物84个转录因子家族,还包含28 193个蛋白模型和26 184条特异蛋白序列。另外,还有一些与表达谱数据相关的数据库,如德国植物基因组计划的中心数据库(genomanalyse im biologischen system pflanze, GABI),收集了实验中获得的一些2-D蛋白胶图,以及基因表达谱数据、代谢途径等数据信息;美国公共的植物和其病原体的基因表达数据库(PLEXdb),用于整合迅速扩展的基因表达谱数据和传统的基因组结构数据以及表型数据;美国的表达谱数据库(HarcEST)也收集了大豆、水稻、小麦等作物大量的表达谱数据。此外,还有专门为数量性状研究建立的数据库(Plant QTL-GE),主要收集水稻、拟南芥等植物的芯片数据、表达谱数据和基因组标签序列,同时也提供基因注释及顺式调控元件注释信息。
除了以上这些综合数据库,一些特定物种的数据库中也包含转录组学的数据信息。如我国的水稻转录因子数据库(a database of rice transcription factors, DRTF)、IC4R的水稻表达数据库(rice expression database, RED)、美国的水稻功能基因组表达数据库(rice functional genomic express database, RiceGE)和日本的水稻表达谱数据库(rice expression profile database, RiceXPro)等。
1.3 作物蛋白质组学及相关数据库
1.3.1 蛋白质组学在作物科学研究中的应用蛋白质组学(proteomics)是研究细胞、组织或生物体中蛋白质组成及其变化规律的科学[27]。
蛋白质组学主要根据基因组表达的变化,获得不同条件下的蛋白质差异图谱,并对差异蛋白质和不同时期、不同器官或组织、不同器官间相互关系、细胞器及胁迫相关蛋白质组学进行研究。在水稻中,Uchiumi等[28]采用单向SDS-PAGE电泳结合LC-MS质谱分析技术研究水稻花粉母细胞的主要蛋白质组成;邵彩虹等[29]应用蛋白质组学分析技术,对养分胁迫下抗早衰的水稻生育后期叶片蛋白质组差异表达进行了研究,揭示了抗早衰水稻耐养分胁迫机理。在玉米中,王秀玲等[30]采用双向电泳技术(2-DE)研究玉米苗期受淹后叶片的蛋白质组差异,进一步阐明玉米对淹水胁迫的响应机制;于涛等[31]从蛋白质组学的角度找到玉米早期发育阶段上、中部籽粒差异表达的蛋白质并分析其功能,探明玉米不同部位籽粒发育差异的分子机理。在小麦中,张戴静等[32]采用水培方法,在0~60 mg/L铜浓度范围内,测定不同浓度对小麦幼苗生长发育、转录组表达和蛋白质组表达的差异,探索外源铜胁迫对小麦幼根的影响及其抵御重金属毒害的分子机制;王志军等[33]以小麦单子房品系77 (2)及其多子房近等基因系Mu77 (2)为材料,采用TCA-丙酮法提取穗分化至四分体时期的幼穗总蛋白,并通过IEF/SDS-PAGE双向凝胶电泳分离,获得分辨率和重复性较好的蛋白质组差异图谱。
1.3.2 蛋白质组学相关数据库
蛋白质组学技术将基因组序列信息和在特定组织、细胞或细胞器中执行生命功能的蛋白质有机地联系起来,并能全面检测不同组织器官、不同发育阶段、不同条件下蛋白质表达的差异[34]。常用的数据库包含二维凝胶电泳图谱库(GELBANK)、细胞通讯网络数据库(EndoNet)、蛋白质组分析数据库(proteome analysis database)和核仁蛋白组数据库(nucleolar proteome database, NOPdb)等。在作物科学研究中,蛋白质功能注释是蛋白质组学研究的核心对象,其相关数据库也被广泛应用。例如,GO (gene ontology)具有三级结构的标准语言(Ontologies),分别根据基因产物的分子功能、生物过程、细胞组分进行注释。COG (cluster of orthologous groups of proteins)和KOG (clusters of orthologous groups for eukaryotic complete genomes)这两个数据库对同源的蛋白质进行注释;蛋白质序列数据库(SWISS- PROT, TrEMBL)和蛋白质信息资源数据库(protein information resource, PIR),这两个数据库利用GOA (gene ontology annotation)按照标准化的GO词汇表对UniProt (The Universal Protein Resource)基因进行分子注释。蛋白质家族集合数据库(Pfam)也被广泛使用,它又分为高质量、人工管理的Pfam-A和低质量、未经注释的Pfam-B两个数据库。除了以上提到的蛋白质数据库外,还有一些其他数据库,如蛋白质综合数据库(InterPro)和蛋白质互作及网络的数据库,如解析已知蛋白质和预测蛋白质之间相互作用关系的数据库(STRING)、结构域互作数据库(DOMINE)、蛋白质磷酸化位点数据库(Phospho.ELM)和蛋白质水解路径数据库(PMAP)等,以及其他转录因子结构域预测数据库(transcription regulatory regions database, TRRD)、植物蛋白簇预测数据库(PHYTOPROT)等。
此外,还有一些特定物种的蛋白质组学专用数据库,如日本的水稻蛋白质组学数据库(RPD)和水稻蛋白结构数据库(RPSD)、美国的水稻蛋白激酶数据库(RKD)等。
1.4 作物代谢组学及相关数据库
1.4.1 代谢组学在作物科学研究中的应用代谢组学(metabolomics)是对生物体内所有代谢物进行定量分析,并寻找代谢物与生理、病理变化相互关系的学科,其研究对象大多是相对分子质量在1000以内的小分子物质[35]。
目前,代谢组学研究已广泛应用于作物的生物和非生物胁迫、基因功能、辅助育种及转基因安全等方面。例如,Shu等[36]运用气相色谱-质谱联用技术(GC-MC)研究水稻种子萌发过程中代谢物的变化;周艳明等[37]利用GC-MC技术对转植酸酶基因玉米代谢物指纹图谱进行研究,结果发现转基因与其对照之间存在显著的差异性;包雨卓等[38]对不同温度条件下的东农冬麦1号幼苗分蘖节进行GC- TOF/MS代谢组学检测,并对获得的数据进行多元变量模式识别分析,明确了东农冬麦1号耐寒越冬的生理机制。
1.4.2 代谢组学相关数据库
代谢组学发展相对较晚,目前有关植物代谢组学的数据库主要有:综合数据库KEGG (kyoto encyclopedia of genes and genomes),该数据库包含代谢通路和互作网络信息;针对拟南芥、水稻等12种植物建立的PlantCyc数据库,该数据库又包括几个子数据库,其中MetaCyc收集了这些物种大量的代谢通路以及酶的信息;代谢物鉴定数据库(the golm metabolome database, GMD)主要包含GC-MS、GC- TOF-MS质谱图库;代谢质谱数据库(scripps center for metabolomics, Metlin)可免费在线查询各个化合物的LC-MS、MS/MS、FTMS质谱数据;Madison代谢组学联合数据库(madison metabolomics consortium database, MMCD)收集了超过10 000种代谢物(拟南芥代谢物为主)的信息以及它们的质谱和核磁共振谱数据;日本多所大学及研究机构共同建立的质谱图数据库MassBank,支持用户通过输入文本格式的质谱进行搜索,并实现与三维可视化质谱的比较;由美国加州大学圣地亚哥分校(University of California, San Diego)创立的基于代谢组学的系统生物学整合数据库BiGG Models,含有各类模式生物的代谢谱图模型,用户可以直观的调取各种生物的整体代谢通路,查看某个具体的生化反应,同时也可以进行代谢产物搜索。
1.5 作物表型组学及相关数据库
1.5.1 表型组学在作物科学研究中的应用表型组学(phenomics)是系统研究某一生物或细胞在不同环境条件下所有表型的学科[39]。
21世纪以来,随着传统表型观测手段瓶颈的突破,自动化、智能化农业装备得到重视,使得表型组学研究逐渐兴起,国际上尤其是欧美发达国家植物表型组学研究迅速发展。我国在表型组学方面的研究虽然起步较晚,但部分研究所和农业院校近几年开始重视表型组的平台和团队建设,取得了一定的阶段性进展。如Yang等[40]和Duan等[41]研究发现,作物高通量植株表型测量平台可以自动提取水稻株高、叶面积、分蘖数、穗数等表型数据,24小时可测量1920株水稻。Yang等[42]研究证明数字化水稻考种机可测量总粒数、实粒数、结实率、单株产量、粒长、粒宽、粒面积等,效率为1分钟/株,将获取的表型数据与全基因组关联分析相结合,不仅可以鉴定传统表型观测手段已鉴定的遗传位点,还能鉴定新的遗传位点。
1.5.2 表型组学相关数据库
表型组学数据形式丰富多样,其数据库大多以物种进行分类。如作物数据库(Gramene)中只包含玉米、水稻、小麦等大田作物的表型数据;谷类作物信息数据库(Grain Genes)中只包含小麦等麦类的分子和表型信息数据;棉花表型数据库CottonQTLd和CottonGEN中也只包含不同棉花品种的表型数 据;大豆诱变突变体数据库(TILLING, Forrest and Williams82)中仅有约3000个M2代(第2代突变体)材料表型及分类;BASC系统是芸苔属(Brassica campestris L.)遗传、基因组和表型数据的整合挖掘和浏览工具。
2 作物组学数据库比较及利用
近年来,随着数据内容或形式的多样化,数据库也变得更加丰富。按照研究对象,作物组学数据库可分为综合数据库和专用数据库,其中综合数据库包含了针对多种组学、多个物种、多种类型的数据(表1,相应网址信息见附表1)。如NCBI包含了基因组学方面的子数据库GenBank和转录组学的子数据库GEO等数据信息;PlantGDB包含了多种植物的序列及注释的转录本装配数据等。此外,还有大量专用数据库,如特定物种的专用数据库。如Rice Data收集了与水稻相关的各种数据,MaizeGDB只包含了与玉米相关的各种数据。另外,还有一些特定研究内容的专用数据库,如REDB包含水稻EST数据,RMD包含水稻突变体数据等。Table 1
表1
表1 5大组学常用数据库
Table 1
| 分类 | 综合数据库 | 特定数据库 | |||
|---|---|---|---|---|---|
| 水稻 | 玉米 | 小麦 | 其他作物 | ||
| 基因组学 | GenBank (NCBI)、EBI (EMBL)、DDBJ (DNA Data Bank of Japan)、Phytozome、GSDB、Gramene、Ensembl Plants、PlantGDB、planteome、EasyGO | Rice Data、The TIGR Rice Database、NCPGR、IC4R、REDB、RMD、BGF、BGI-RIS | The TIGR Maize Database、 MaizeGDB、Panzea、MGCN、MTM、PGSB | The TIGR Wheat Database、GrainGenes、ECPGR、SHIGEN、CR-EST | |
| 转录组学 | GEO(NCBI)、ArrayExpress、SMD、PlantTFDB、Genevestigator、GABI、PLEXdb、PlantQTL-GE、HarcEST | DRTF、RED、RiceGE、RiceXPro | |||
| 蛋白质组学 | GO、COG/KOG、GOA、UniProt (SWISS-PROT, TrEMBL, PIR)、Pfam、InterPro、STRING、DOMINE、Phospho.ELM、PMAP、TRRD、PHYTOPROT | RPD、RPSD、RKD | |||
| 代谢组学 | KEGG、MetaCyc (PlantCyc)、GMD (GC-MS、GC-TOF-MS)、Metlin (LC-MS、MS/MS、FTMS)、MMCD、MassBank、BiGG Models | ||||
| 表型组学 | Gramene、GrainGenes | CottonQTLdb、CottonGEN、TILLING、BASC | |||
新窗口打开|下载CSV
各个组学和各个数据库之间都存在着千丝万缕的交互联系。整合从综合或者专用数据库中提取的数据信息,挖掘物种在某个组学上的生物过程,可阐明不同生物体、不同环境条件、不同时期以及不同组织或器官等对代谢的影响,揭示出更多的生命奥秘。例如,以具有地下茎的长雄野生稻和拟高粱,以及耐冷水稻品种(丽江新团黑谷)和冷敏感水稻品种(IR29)为材料,采用基因芯片及转录组测序等技术,对地下茎发育及耐冷性进行系统的功能基因组学和比较转录组学分析,发掘与地下茎发生发育相关功能的候选基因,并解析水稻耐冷胁迫调控的分子遗传机制[43]。利用转录组学和蛋白质组学有机结合,在不同小麦品种和小麦野生近缘种中鉴定出大量参与小麦抗旱分子调控网络的基因和蛋白质[44],实现对分子调控网络进行分析并相互验证。在高通量技术支持的基础上,多学科、多层次、多角度、多时空(发育和进化)的组学整体研究格局正在逐渐形成。水稻、玉米、小麦等主要作物各相关领域的研究也在不断拓展,更加趋于系统化和规模化。
为进一步了解近年来我国作物科学研究对数据库的利用情况,对常用的中文文献数据库进行检索,以水稻、玉米、小麦3大作物及相关数据库为关键词,筛选出近10年(2007~2017年)发表的130篇中文文献,对文献中利用的数据库进行了不完全统计和分类。在所检测的文献中,研究对象是水稻、玉米、小麦的分别为49、39、42篇(附表2),分别占文献总数的38%、30%、32% (图1A)。从图1可以看出,3大主要作物在数据库的利用上比较均等。但是,按年代来分,3大作物在每年中利用数据库进行研究的比例不完全一致,如利用数据库研究水稻的相关文献在2012和2013年数量较多,利用数据库研究玉米的相关文献在2008、2010、2012年较少,而小麦的相关文献在2013和2015年相对较少(图1B)。对所检测的文献中涉及的数据库按组学研究内容进行分类,结果发现,涉及基因组学相关的数据库31个,其中包含15个综合数据库(不含3个专用的子数据库)和13个专用数据库;涉及转录组学相关的数据库5个,包含4个综合数据库和1个专用数据库;涉及蛋白质组学相关的数据库11个,包含10个综合数据库1和1个专用数据库;涉及代谢组学相关的数据库只有1个常用的综合数据库;在所有文献中没有涉及与表型组学相关的单一数据库。由此可以推断,基因组学的相关研究仍然是组学研究的主要内容,转录组学和蛋白质组学的研究还有很大的挖掘空间,而对于起步较晚的代谢组学和表型组学的研究应当提出更多创新的思维,以促进组学研究的整体发展。进一步对这些中文文献中涉及到的数据库按基因组学数据库进行分类,发现利用综合数据库NCBI的文献数量最多,而其中GenBank又是文献使用量较多的子数据库(图1C);此外,分析文献中对GenBank数据库的使用情况,发现10年间其使用量逐步减少(图1D),而更多的专用数据库被应用在这些文献中。可以看出,随着研究的进一步发展,数据库的利用已经从早年间的综合数据库单一利用转变成了专用数据库或综合数据库结合利用。
图1
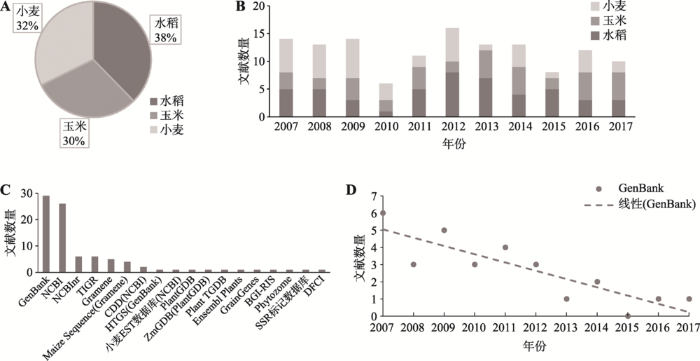 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1近10年(2007~2017年)有关水稻、玉米、小麦中文文献统计分析
A:水稻、玉米、小麦中文文献数量及比例;B:3大作物中文文献年发表数量;C:文献中基因组学相关数据库利用情况分布;D:文献中GenBank数据库的利用情况。
Fig. 1Statistic analysis of the literatures (in Chinese) on rice, maize, and wheat in the last decade (2007-2017)
通过分析这些数据库的应用情况,可以看出,五大组学数据库在作物科学研究中的发展是不均衡的。其中,基因组学和转录组学相关数据库内容和形式都相对丰富,尤其基因组学相关数据库,不仅有大量的综合数据库,还有各种专用数据库;蛋白质组学相关数据库常用的是一些功能注释数据库;而目前对代谢组学和表型组学相关数据库的利用都相对较少。
作物科学和数据库的发展是相辅相成的,在以后的科学研究中,我们要充分利用各种综合和专用数据库,促进作物科学的发展,从而也会促进这些数据库的发展和利用。
3 结 语
在高通量测序技术快速发展的背景下,作物科学研究从传统的常规育种逐渐演化为多种组学技术相结合的方式。结合多组学多数据库中的数据信息对重要性状、基因等的解析和注释,有助于准确和全面地揭示其形成机制,从而为育种工作提供更有效的方案[45]。同时,作物科学研究和数据库的发展也相互影响。随着研究的不断进步,单一数据库的利用已经不能满足研究现状,各组学数据库的利用愈加综合化,多组学数据库的交叉整合利用也必将成为高通量数据时代研究的重要方向。尽管作物科学研究已经取得了长足的进展,但目前的研究成果也只是冰山一角。随着测序技术的不断发展,需要多层次、多手段全面地解析作物的各项研究,并利用新技术聚焦研究目的,开辟研究新思路,在传统方法的基础上不断开拓创新,使作物科学研究领域迎来新的突破。附录:
附表1和附表2见文章电子版www.chinagene.cn。Supplementary Table 1
附表1
附表1 文章中列举的数据库网址
Supplementary Table 1
| 数据库名称及缩写 | 网址链接 |
|---|---|
| GenBank(NCBI) | https://www.ncbi.nlm.nih.gov/genbank/ |
| EBI(EMBL) | https://www.ebi.ac.uk/ |
| DDBJ(DNA Data Bank of Japan) | http://www.ddbj.nig.ac.jp/ |
| Phytozome | https://phytozome.jgi.doe.gov/pz/portal.html |
| GSDB | http://www.ncgr.org/research/sequence/ |
| Gramene | http://www.gramene.org/ |
| Ensembl Plants | http://plants.ensembl.org/index.html |
| PlantGDB | http://www.plantgdb.org/ |
| planteome | http://planteome.org/ |
| EasyGO | http://bioinformatics.cau.edu.cn/easygo/ |
| Rice Data | http://www.ricedata.cn/ |
| The TIGR Rice Database | http://www.tigr.org/tdb/rice/ |
| NCPGR | http://www.ncpgr.cn/web/ |
| IC4R | http://www.ic4r.org/ |
| REDB | http://redb.ncpgr.cn/ |
| RMD | http://rmd.ncpgr.cn/ |
| BGF | http://tlife.fudan.edu.cn/bgf/ |
| BGI-RIS | http://rice.genomics.org.cn/ |
| The TIGR Maize Database | http://maize.jcvi.org/ |
| MaizeGDB | https://www.maizegdb.org/ |
| Panzea | https://www.panzea.org/ |
| MGCN | http://www.agron.missouri.edu/index.html |
| MTM | http://mtm.cshl.org/ |
| PGSB | http://mips.helmholtz-muenchen.de/plant/barley/ |
| The TIGR Wheat Database | http://www.tigr.org/tdb/e2k1/tae1/ |
| GrainGenes | https://wheat.pw.usda.gov/GG3/ |
| ECPGR | https://www.ecpgr.cgiar.org/ |
| SHIGEN | http://www.shigen.nig.ac.jp/wheat/top.html |
| CR-EST | http://pgrc.ipk-gatersleben.de/cr-est/ |
| GEO(NCBI) | https://www.ncbi.nlm.nih.gov/geo/ |
| ArrayExpress | https://www.ebi.ac.uk/arrayexpress/ |
| SMD | http://smd.stanford.edu/ |
| PlantTFDB | http://planttfdb.cbi.pku.edu.cn/ |
| Genevestigator | https://genevestigator.com/gv/ |
| GABI | http://gabi.rzpd.de |
| PLEXdb | http://www.barleybase.org/ |
| PlantQTL-GE | http://www.scbit.org/qtl2gene/new/ |
| HarcEST | http://harvest.ucr.edu/ |
| DRTF | http://drtf.cbi.pku.edu.cn/ |
| RED | http://red.dna.affrc.go.jp/RED/ |
| RiceGE | http://signal.salk.edu/cgi-bin/RiceGE |
| RiceXPro | http://ricexpro.dna.affrc.go.jp/ |
| GELBANK | http://gelbank.anl.gov/ |
| EndoNet | http://endonet.bioinf.med.uni-goettingen.de/ |
| 数据库名称及缩写 | 网址链接 |
| Proteome Analysis Database | http://www.ebiac.uk.proteome/ |
| Nucleolar Proteome Database, NOPdb | http://www.lamondlab.com/NOPdb3.0/ |
| GO | http://www.geneontology.org/ |
| COG/KOG | http://www.ncbi.nlm.nih.gov/COG/ |
| GOA | http://www.ebi.ac.uk/GOA |
| UniProt(SWISS-PROT, TrEMBL, PIR) | http://www.uniprot.org/ |
| Pfam | http://pfam.xfam.org/ |
| InterPro | http://www.ebi.ac.uk/interpro/ |
| STRING | https://string-db.org/cgi/input.pl |
| DOMINE | http://domine.utdallas.edu/ |
| Phospho.ELM | http://phospho.elm.eu.org/ |
| PMAP | http://www.proteolysis.org |
| TRRD | http://wwwmgs.bionet.nsc.ru/mgs/gnw/trrd/ |
| PHYTOPROT | http://urgi.versailles.inra.fr/phytoprot/ |
| RPD | http://gene64.dna.affrc.go.jp/RPD/ |
| RPSD | http://structure.rice.dna.affrc.go.jp/ |
| RKD | http://rkd.ucdavis.edu/ |
| KEGG | http://www.kegg.jp/kegg/ |
| MetaCyc(PlantCyc) | https://www.plantcyc.org/databases/plantcyc/ |
| GMD(GC-MS、GC-TOF-MS) | http://gmd.mpimp-golm.mpg.de/ |
| Metlin(LC-MS、MS/MS、FTMS) | https://metlin.scripps.edu/landing_page.php?pgcontent=mainPage |
| MMCD | http://mmcd.nmrfam.wisc.edu/ |
| MassBank | http://www.massbank.jp/en/database.html |
| BiGG Models | http://bigg.ucsd.edu/ |
| CottonQTLdb | http://www2.cottonqtldb.org:8081/ |
| CottonGEN | https://www.cottongen.org/ |
| TILLING | http://www.soybeantilling.org/ |
| BASC | http://hornbill.cspp.latrobe.edu.au/cgi-binpub/brassica/index.pl |
新窗口打开|下载CSV
Supplementary Table 2
附表2
附表2 130篇统计文献相关信息
Supplementary Table 2
| 作物名称 | 发表年份 | 利用数据库 | 文献名称 |
|---|---|---|---|
| 水稻 | 2007 | NCBI | 一个低氮诱导表达的水稻Dof转录因子OsDof-13的分离和转化 |
| NCBI、Pfam | 水稻Ds插入双分蘖突变体形成机理的分析 | ||
| GenBank | 水稻谷蛋白基因 GluB-6的cDNA克隆及表达 | ||
| 水稻基因芯片注释整合数据库 | RiceDB: 基于Web界面的水稻基因芯片注释整合数据库 | ||
| MSDB (MySQL) | 红莲型水稻细胞质雄性不育花粉总蛋白质初步比较分析 | ||
| 2008 | NCBI | 水稻 SLR1 基因的克隆及其植物表达载体的构建 | |
| GenBank | 水稻胚胎发育过程相关基因的鉴定(简报) | ||
| GenBank | 一个新的水稻逆境响应基因OsMsr1的表达与克隆 | ||
| TIGR | 利用阵列分析水稻AP2/EREBP家族基因 的表达特性 | ||
| TIGR | 水稻单侧卷叶突变体B157遗传分析及基因初步定位 | ||
| 作物名称 | 发表年份 | 利用数据库 | 文献名称 |
| 水稻 | 2009 | TIGR | 水稻U-Box 蛋白质在不同发育时期的表达分析 |
| SWISS-PROT MSDB (MySQL) | 水稻灌浆期叶片蛋白质差异表达及其作用机理分析 | ||
| SSR标记数据库、 水稻基因组数据库 | 一个水稻开颖不育突变体ohs1 (t)的遗传分析及基因定位 | ||
| 2010 | GenBank | 一个新的水稻低温应答类糖基转移酶基因(OsCrGtl)的表达分析与克隆 | |
| 2011 | NCBI | 水稻维生素C合成相关基因的表达分析 | |
| GenBank | 水稻多逆境响应基因OsMsr13的克隆与功能分析 | ||
| TIGR、Pfam | 水稻油体钙蛋白家族的进化及对干旱胁迫的响应性分析 | ||
| Plant MetGenMAP (MySQL) | Plant MetGenMAP数据库分析水稻表达谱芯片数据的方法初探 | ||
| Rice Data | 中国水稻品种及其系谱数据库 | ||
| 2012 | NCBI | 水稻中介体亚基的表达谱分析及亚细胞定位 | |
| GenBank | 水稻OsWRKY17基因定位表达载体的构建 | ||
| GEO | GEO水稻相关基因表达数据研究现状 | ||
| TIGR | 一个水稻叶片衰老上调表达基因的初步生物学功能分析 | ||
| 水稻基因组数据库 | 2个水稻PLT基因启动子的克隆及其表达分析 | ||
| GO、UniProtKB | 外源茉莉酸甲酯诱导的水稻叶片蛋白质差异表达分析 | ||
| RiceXPro | 水稻OsSsr1基因的生物信息学分析、亚细胞定位及其表达模式 | ||
| SCI-EXPANDED | 基于Web of Science的水稻研究态势分析 | ||
| 2013 | NCBI、Pfam | 水稻Ds插入稃片失绿突变体的初步鉴定 | |
| NCBI | 水稻磷转运蛋白OsPHT2-1在提高磷素利用率方面的作用 | ||
| GenBank | OsPIN1a基因在水稻根负向光性中的作用初探 | ||
| 水稻基因组数据库 | 几条水稻MAR序列的克隆及其功能分析 | ||
| 水稻基因组数据库 | 水稻WRKY80转录调节蛋白基因的分离与表达模式 | ||
| Gramene | 水稻茸毛基因GL6的遗传学分析与精细定位 | ||
| GO、KEGG | 水稻CYP81A6基因干扰对其他基因表达的影响 | ||
| 2014 | Gramene | 水稻窄卷叶突变体nrl(t)的遗传分析与基因定位 | |
| 蛋白质数据库 | 耐热和热敏感水稻应答灌浆初期高温胁迫过程中的差异表达蛋白质鉴定 | ||
| Plant TGDB | 水稻抗逆相关基因的分子进化分析 | ||
| SOGO顺式作用元件数据库 | 水稻胚乳特异表达启动子DXCP35的克隆及功能鉴定 | ||
| 2015 | NCBI | 基于EST数据的水稻基因表达大规模初步分析 | |
| Rice Data | 不同环境下水稻株高和穗长的QTL分析 | ||
| Rice Data | 中国两系杂交水稻光温敏核不育基因的鉴定与演化分析 | ||
| TFDB、ExPASy、RGAP、KOME | 水稻Trihelix转录因子家族全基因组分析及功能预测 | ||
| MySQL | 控制水稻抽穗延迟基因OsSET34的鉴定和图位克隆 | ||
| 2016 | NCBInr、GO、KEGG | 水稻颖花开放前浆片转录组变化 | |
| 水稻基因组注释数据库(RGAP) | 水稻OsABC1K3突变体鉴定及其对强光胁迫的响应 | ||
| 水稻基因组注释计划数据库 | 水稻中过敏原蛋白全基因组预测及生物学功能分析 | ||
| 作物名称 | 发表年份 | 利用数据库 | 文献名称 |
| 水稻 | 2017 | BGI-RIS | 基于比较转录组的水稻苗期耐冷相关基因BGIOSGA032296 TALEN基因组编辑技术构建 |
| MySQL | 小RNA高通量测序数据分析方法 | ||
| MySQL | 基于PHP技术的玉米大豆水稻农艺性状专网的设计与建立 | ||
| 玉米 | 2007 | GenBank | 基于同源克隆技术分离玉米漆酶基因片段 |
| MaizeGDB | 玉米抗病基因一致性图谱的构建 | ||
| 玉米的核酸数据库 | 应用抑制差减杂交法分离玉米幼苗期叶片土壤干旱诱导的基因 | ||
| 2008 | NCBI | 玉米C型细胞质雄性不育系C48-2及其保持系线粒体差异蛋白分析 | |
| MaizeGDB、Gramene | 玉米产量相关性状QTL通用图谱的构建及功能候选基因的开发 | ||
| 2009 | HTGS (GenBank) | 玉米CACTA型转座子携带宿主基因片段转移新功能的发现与特性分析 | |
| GenBank | 玉米ZmCBL6基因的克隆及其植物表达载体构建 | ||
| GenBank | 玉米对二氧化硫胁迫响应的相关cDNA克隆与分析 | ||
| NCBI、MaizeGDB、 Maize Sequence (Gramene) | 玉米蔗糖转运蛋白基因ZmERD6 cDNAs的克隆与逆境条件下的表达 | ||
| 2010 | PlantGDB、 Maize Sequence (Gramene) | 利用生物信息学方法进行基于表达序列标签的玉米单核苷酸多态性标记的开发 | |
| MaizeGDB | 玉米Mu转座子及其在反向遗传研究中的应用 | ||
| 2011 | GenBank | 干旱和盐碱共胁迫玉米EST数据库分析 | |
| GenBank、miRBase | 基于EST和GSS序列的玉米未知微RNA的数据挖掘 | ||
| PDB | Cd胁迫对发芽玉米抗氧化酶体系的影响分析 | ||
| NCBInr、SWISS-PROT、 MSDB (MySQL) | 不同抗性玉米自交系接种纹枯病菌后蛋白表达的差异分析 | ||
| 2012 | NCBI | 玉米AFB2基因的克隆、表达及功能分析 | |
| NCBI | 玉米温光敏雄性不育系9417叶片叶绿体蛋白质差异分析 | ||
| 2013 | NCBInr | 盐胁迫下玉米叶片差异蛋白的双向电泳分析 | |
| Plant TFDB | 玉米转录因子结构与功能研究进展 | ||
| MaizeGDB、Plant GDB (ZmGDB) | 玉米D亚族bZIP转录因子基因的数据库挖掘、分析、克隆与表达 | ||
| Maize Sequence (Gramene) | 玉米ZmCIPK42克隆及逆境胁迫后表达特异性分析 | ||
| Panzea | 利用核心SNP位点鉴别玉米自交系的研究 | ||
| 2014 | NCBInr | 砷胁迫对丛枝菌根接种玉米叶片蛋白表达谱的影响 | |
| MaizeGDB | 玉米穗行数性状QTL的元分析 | ||
| 玉米EST数据库 | 干旱和盐碱共胁迫下玉米EST文库的构建与分析 | ||
| 玉米基因组数据库 | 甜玉米自交系含糖量的测定及甜质 等位基因的分子标记检测 | ||
| TIGR、TAIR、 Maize Sequence(Gramene) | 玉米BURP家族基因的鉴定和分析 | ||
| 2015 | Gramene | 基于热胁迫甜玉米雌穗发育差异表达基因的SSR标记开发 | |
| PLEXdb | 玉米1,3,4-三磷酸肌醇5/6激酶ITPK 家族基因的鉴定和分析 | ||
| 2016 | GenBank | 耐盐碱转基因玉米的获得及其抗性分析 | |
| GEO、MaizeGDB | 玉米新型氧甲基转移酶基因的全基因组鉴定、进化与表达研究 | ||
| 作物名称 | 发表年份 | 利用数据库 | 文献名称 |
| 玉米 | 2016 | CDD (NCBI)、MaizeGDB、Pfam | 玉米Glyco-hydro-16糖苷酶家族全基因组的 鉴定及其遗传分化 |
| CDD (NCBI)、Gramene、Pfam | 玉米 HSP70 基因家族的全基因组鉴定与分析 | ||
| MaizeGDB | 玉米Zmcen基因的克隆、表达与生物信息学分析 | ||
| 2017 | NCBI | 2 个玉米光敏色素 C 基因的转录丰度对多种光质处理的响应 | |
| NCBI、Phytozome | 玉米ZmNAC6基因克隆及盐碱逆境胁迫下的表达分析 | ||
| GenBank | 转cspB基因玉米获得及其耐盐碱性分析 | ||
| MaizeGDB、Pfam、Interpro | 玉米FLA蛋白家族的生物信息学分析 | ||
| 玉米蛋白数据库 | 蛋白质组学分析揭示玉米籽粒发育过程中胁迫相关蛋白的表达特性 | ||
| 小麦 | 2007 | NCBI | 利用cDNA-RAPD方法筛选与V型小麦细胞质雄性不育育性相关的线粒体转录产物的研究 |
| GenBank | 小麦高分子量谷蛋白亚基一级结构分析 | ||
| GenBank | 小麦耐盐相关基因TaSTK的克隆 | ||
| GenBank | 小麦脂质转运蛋白cDNA结构及相应肽链构造特征的分析 | ||
| GenBank | 小麦种子萌发期水分胁迫诱导表达差异cDNA的研究 | ||
| ACCESS | 基于VB的小麦性状数据记录查询系统的设计 | ||
| 2008 | NCBI、 SOPMA数据库 | 小麦液泡膜反转运蛋白基因TaNHX1的生物信息学分析 | |
| NCBI | 抑制差减杂交分离赤霉病菌诱导的小麦特异表达基因 | ||
| GenBank | 小麦拔节过程中基部茎节的基因差异表达 | ||
| GrainGenes | 小麦AFLP_SCAR标记的开发及应用 | ||
| MIPS | 水分胁迫条件下“洛旱2号”小麦根系的基因表达谱 | ||
| MSDB (MySQL) | 小麦灌浆期籽粒粒重差异的代谢蛋白质组学初步研究 | ||
| 2009 | NCBI、GenBank、GO | 小麦温敏雄性不育系YM3314育性转换相关基因分析 | |
| 小麦EST数据库(NCBI) | 一个小麦AP2-ERF转录因子家族单独亚族基因的克隆及分析 | ||
| GenBank | 小麦和水稻auxin基因家族的生物信息学比较分析 | ||
| GenBank | 小麦品种“中国春”主要过敏原CM16基因的克隆及序列分析 | ||
| NCBI、MSDB (MySQL) | 不同抗寒性冬小麦品种分蘖节低温诱导蛋白比较 | ||
| NCBInr、MSDB (MySQL) | 小麦抗白粉病基因Pm 21抗病差异的蛋白质组学研究 | ||
| MySQL | 基于 JSP 技术的四川小麦种质资源信息系统的设计和实现 | ||
| 2010 | GenBank | 小麦大粒突变体的SSH文库构建及分析 | |
| GenBank | 小麦铝胁迫相关基因的研究进展 | ||
| MySQL | 基于.NET的小麦性状数据查询分析系统的构建 | ||
| 2011 | GenBank | 小麦 RPL21 基因同源克隆与表达分析 | |
| SWISS-PROT | 小麦生理型和遗传型雄性不育系及其保持系小花完整叶绿体蛋白质组分比较研究 | ||
| 2012 | NCBI | 利用基因芯片分析小麦茎秆伸长过程中的基因表达谱 | |
| NCBI | 小麦TaPHYA基因亚家族的克隆及表达分析 | ||
| GenBank | 小麦SKC1-like基因的克隆及多样性研究 | ||
| 作物名称 | 发表年份 | 利用数据库 | 文献名称 |
| 小麦 | 2012 | GenBank | 小麦生长素结合基因 TaABP1-D的克隆、功能标记开发及其与株高的关联 |
| NCBInr | 小麦籽粒清蛋白和球蛋白表达对干旱胁迫的响应 | ||
| MySQL | 基于Web的小麦品种布局优化决策支持系统 | ||
| 2013 | NCBI | 小麦逆境胁迫相关基因TaCBL7的克隆、生物信息学及表达特性分析 | |
| 2014 | NCBI | 小麦SOD基因的电子克隆及生物信息学分析 | |
| NCBI | 小麦胁迫相关基因TaLEA2的克隆、生物信息学及表达特性分析 | ||
| GenBank | 普通小麦祖先种类TaNAC2a基因的鉴定分析和表达模式研究 | ||
| GenBank | 小麦高分子量麦谷蛋白亚基基因Dx5的克隆与生物信息学分析 | ||
| 2015 | 小麦基因组测序数据库 | 小麦R2R3-MYB转录因子TaMYB3-4D的克隆及功能分析 | |
| 2016 | NCBI | 小麦胁迫相关基因TaSF3B2的克隆及表达特性分析 | |
| NCBI、DFCI | 小麦F-box家族基因的分类及克隆分析 | ||
| COG | 低温胁迫下小麦幼穗中差异表达蛋白质的鉴定 | ||
| ExPASy | 小麦脱水素基因WDHN1-2的克隆及其表达分析 | ||
| 2017 | EnsemblPlants | 小麦锌指转录因子TaDi19A对低温的响应及其互作蛋白的筛选 | |
| 突变品种数据库(MVD) | 小麦诱发突变技术育种研究进展 |
新窗口打开|下载CSV
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:27535938 [本文引用: 1]

Abstract Asian cultivated rice consists of two subspecies: Oryza sativa subsp. indica and O. sativa subsp. japonica Despite the fact that indica rice accounts for over 70% of total rice production worldwide and is genetically much more diverse, a high-quality reference genome for indica rice has yet to be published. We conducted map-based sequencing of two indica rice lines, Zhenshan 97 (ZS97) and Minghui 63 (MH63), which represent the two major varietal groups of the indica subspecies and are the parents of an elite Chinese hybrid. The genome sequences were assembled into 237 (ZS97) and 181 (MH63) contigs, with an accuracy >99.99%, and covered 90.6% and 93.2% of their estimated genome sizes. Comparative analyses of these two indica genomes uncovered surprising structural differences, especially with respect to inversions, translocations, presence/absence variations, and segmental duplications. Approximately 42% of nontransposable element related genes were identical between the two genomes. Transcriptome analysis of three tissues showed that 1,059-2,217 more genes were expressed in the hybrid than in the parents and that the expressed genes in the hybrid were much more diverse due to their divergence between the parental genomes. The public availability of two high-quality reference genomes for the indica subspecies of rice will have large-ranging implications for plant biology and crop genetic improvement.
URLPMID:19965430 [本文引用: 1]
We report an improved draft nucleotide sequence of the 2.3-gigabase genome of maize, an important crop plant and model for biological research. Over 32,000 genes were predicted, of which 99.8% were placed on reference chromosomes. Nearly 85% of the genome is composed of hundreds of families of transposable elements, dispersed nonuniformly across the genome. These were responsible for the capture and amplification of numerous gene fragments and affect the composition, sizes, and positions of centromeres. We also report on the correlation of methylation-poor regions with Mu transposon insertions and recombination, and copy number variants with insertions and/or deletions, as well as how uneven gene losses between duplicated regions were involved in returning an ancient allotetraploid to a genetically diploid state. These analyses inform and set the stage for further investigations to improve our understanding of the domestication and agricultural improvements of maize.
URL [本文引用: 1]
[本文引用: 1]
URLPMID:23535592 [本文引用: 1]
About 8,000 years ago in the Fertile Crescent, a spontaneous hybridization of the wild diploid grass Aegilops tauschii (2n = 14; DD) with the cultivated tetraploid wheat Triticum turgidum (2n = 4x = 28; AABB) resulted in hexaploid wheat (T. aestivum; 2n = 6x = 42; AABBDD)(1,2). Wheat has since become a primary staple crop worldwide as a result of its enhanced adaptability to a wide range of climates and improved grain quality for the production of baker's flour(2). Here we describe sequencing the Ae. tauschii genome and obtaining a roughly 90-fold depth of short reads from libraries with various insert sizes, to gain a better understanding of this genetically complex plant. The assembled scaffolds represented 83.4% of the genome, of which 65.9% comprised transposable elements. We generated comprehensive RNA-Seq data and used it to identify 43,150 protein-coding genes, of which 30,697 (71.1%) were uniquely anchored to chromosomes with an integrated high-density genetic map. Whole-genome analysis revealed gene family expansion in Ae. tauschii of agronomically relevant gene families that were associated with disease resistance, abiotic stress tolerance and grain quality. This draft genome sequence provides insight into the environmental adaptation of bread wheat and can aid in defining the large and complicated genomes of wheat species.
URL [本文引用: 1]
近20年来,转基因植物在全球范围内发展迅速。转基因作物因其抗虫、抗病、抗逆、高产、优质等优良性状,得到了很好的推广与应用。商业化的转基因植物以大豆、棉花、玉米、油菜这四种作物为主,此外,有20多种植物的转基因事件已被批准商业化,或仍在商业化进程中。转基因植物的推广与人类健康和环境安全息息相关,转基因育种的安全性问题一直是困扰该技术广泛应用的关键问题。随着转基因植物的迅速发展,如何对转基因植物进行合理的安全性评价日益体现出它的重要性。本文围绕转基因安全性评价的实质等同性原则,利用代谢组学手段对多个自然栽培大豆品种的代谢谱进行了分析,对转基因安全性评价进行了一系列的研究,并获得了重要的研究成果。此外,本研究还提出建立一个植物种子分子特征参考数据库来为安全性评价提供参考。 为了探索利用组学方法研究转基因植物安全性的新思路,本研究以第一代抗草甘膦转基因大豆GTS40-3-2(抗农达)为例,从代谢组学层面综合分析了具有代表性的中国大豆品种的代谢谱差异,以及相对于自然品种的代谢物组分,转基因大豆在代谢物种类与含量上的差异。在代谢组水平上,本研究以29种来自全国不同生态区的主要大豆品种作为自然变异的研究对象,以转EPSPS大豆GTS40-3-2为代表,采取了整体的无偏向性的代谢轮廓分析方法,用GC-MS,HPLC-MS手段,共测得大豆种子中的代谢物169种。对这些代谢物在不同品种间的差异性分析表明,代谢物含量差异广泛存在于不同的自然品种之中,转基因大豆与非转基因大豆之间代谢物含量的差异都基本上在自然差异范围之内。此外对大豆种子代谢组学的研究还发现了大豆特有的代谢物、对大豆生态区划分有重要作用的代谢物、代谢物和代谢物之间的关系、代谢物和表型之间的关系。这种全局的无偏向性的代谢组学研究方法比靶标性的研究方法更能够全面体现大豆种子中的代谢物成分,应用于转基因大豆的安全性评价时也更有可能发现非预期效应。 为了使转基因植物的安全性评价有一个客观的量化的参考,本研究设计了一个植物种子分子特征数据库。这个数据库包含了四种作物(水稻、大豆、玉米、小麦)的栽培品种的种子在四个组学水平(基因组、转录组、蛋白组、代谢组)上的实验数据。通过综合不同栽培品种间的数据,找到一个不同栽培品种各个组学表达量的安全变化范围,为这四种作物转基因的安全性评价提供依据。用户可以在本数据库中提交这四种作物栽培品种的实验数据,帮助我们拥有更广的数据来源,使计算出的安全变化范围更加客观,更能代表整体栽培品种品种间的变化范围;用户还可以提交转基因品种标准化后的实验数据,通过本数据库与其他栽培品种比较,观察其表达量是否与栽培品种显著不同,从而找到转基因和栽培品种的差异,为预测转基因品种安全性提供量化的证据。
URL [本文引用: 1]
近20年来,转基因植物在全球范围内发展迅速。转基因作物因其抗虫、抗病、抗逆、高产、优质等优良性状,得到了很好的推广与应用。商业化的转基因植物以大豆、棉花、玉米、油菜这四种作物为主,此外,有20多种植物的转基因事件已被批准商业化,或仍在商业化进程中。转基因植物的推广与人类健康和环境安全息息相关,转基因育种的安全性问题一直是困扰该技术广泛应用的关键问题。随着转基因植物的迅速发展,如何对转基因植物进行合理的安全性评价日益体现出它的重要性。本文围绕转基因安全性评价的实质等同性原则,利用代谢组学手段对多个自然栽培大豆品种的代谢谱进行了分析,对转基因安全性评价进行了一系列的研究,并获得了重要的研究成果。此外,本研究还提出建立一个植物种子分子特征参考数据库来为安全性评价提供参考。 为了探索利用组学方法研究转基因植物安全性的新思路,本研究以第一代抗草甘膦转基因大豆GTS40-3-2(抗农达)为例,从代谢组学层面综合分析了具有代表性的中国大豆品种的代谢谱差异,以及相对于自然品种的代谢物组分,转基因大豆在代谢物种类与含量上的差异。在代谢组水平上,本研究以29种来自全国不同生态区的主要大豆品种作为自然变异的研究对象,以转EPSPS大豆GTS40-3-2为代表,采取了整体的无偏向性的代谢轮廓分析方法,用GC-MS,HPLC-MS手段,共测得大豆种子中的代谢物169种。对这些代谢物在不同品种间的差异性分析表明,代谢物含量差异广泛存在于不同的自然品种之中,转基因大豆与非转基因大豆之间代谢物含量的差异都基本上在自然差异范围之内。此外对大豆种子代谢组学的研究还发现了大豆特有的代谢物、对大豆生态区划分有重要作用的代谢物、代谢物和代谢物之间的关系、代谢物和表型之间的关系。这种全局的无偏向性的代谢组学研究方法比靶标性的研究方法更能够全面体现大豆种子中的代谢物成分,应用于转基因大豆的安全性评价时也更有可能发现非预期效应。 为了使转基因植物的安全性评价有一个客观的量化的参考,本研究设计了一个植物种子分子特征数据库。这个数据库包含了四种作物(水稻、大豆、玉米、小麦)的栽培品种的种子在四个组学水平(基因组、转录组、蛋白组、代谢组)上的实验数据。通过综合不同栽培品种间的数据,找到一个不同栽培品种各个组学表达量的安全变化范围,为这四种作物转基因的安全性评价提供依据。用户可以在本数据库中提交这四种作物栽培品种的实验数据,帮助我们拥有更广的数据来源,使计算出的安全变化范围更加客观,更能代表整体栽培品种品种间的变化范围;用户还可以提交转基因品种标准化后的实验数据,通过本数据库与其他栽培品种比较,观察其表达量是否与栽培品种显著不同,从而找到转基因和栽培品种的差异,为预测转基因品种安全性提供量化的证据。
URL [本文引用: 1]
基因组学是研究生物体的整个基因组结构、功能及进化的一门学科,内容包括基因组作图、序列分析、基因定位、基因功能分析、基因组的进化分析等,内容多且与其他课程内容相互渗透,紧跟时代发展前沿,发展变化较快.本研究从教学内容选择、不同教学方法的运用、网络课堂的构建及考核方式的改革等方面进行了探讨,以期提高教学质量,培养具有创新和实践能力的高素质生物技术专业人才.
URL [本文引用: 1]
基因组学是研究生物体的整个基因组结构、功能及进化的一门学科,内容包括基因组作图、序列分析、基因定位、基因功能分析、基因组的进化分析等,内容多且与其他课程内容相互渗透,紧跟时代发展前沿,发展变化较快.本研究从教学内容选择、不同教学方法的运用、网络课堂的构建及考核方式的改革等方面进行了探讨,以期提高教学质量,培养具有创新和实践能力的高素质生物技术专业人才.
URLPMID:22138690 [本文引用: 1]
A high-density haplotype map recently enabled a genome-wide association study (GWAS) in a population of indica subspecies of Chinese rice landraces. Here we extend this methodology to a larger and more diverse sample of 950 worldwide rice varieties, including the Oryza sativa indica and Oryza sativa japonica subspecies, to perform an additional GWAS. We identified a total of 32 new loci associated with flowering time and with ten grain-related traits, indicating that the larger sample increased the power to detect trait-associated variants using GWAS. To characterize various alleles and complex genetic variation, we developed an analytical framework for haplotype-based de novo assembly of the low-coverage sequencing data in rice. We identified candidate genes for 18 associated loci through detailed annotation. This study shows that the integrated approach of sequence-based GWAS and functional genome annotation has the potential to match complex traits to their causal polymorphisms in rice.
URLPMID:2 [本文引用: 1]
Whereas breeders have exploited diversity in maize for yield improvements, there has been limited progress in using beneficial alleles in undomesticated varieties. Characterizing standing variation in this complex genome has been challenging, with only a small fraction of it described to date. Using a population genetics scoring model, we identified 55 million SNPs in 103 lines across pre-domestication and domesticated Zea mays varieties, including a representative from the sister genus Tripsacum. We find that structural variations are pervasive in the Z. mays genome and are enriched at loci associated with important traits. By investigating the drivers of genome size variation, we find that the larger Tripsacum genome can be explained by transposable element abundance rather than an allopolyploid origin. In contrast, intraspecies genome size variation seems to be controlled by chromosomal knob content. There is tremendous overlap in key gene content in maize and Tripsacum, suggesting that adaptations from Tripsacum (for example, perennialism and frost and drought tolerance) can likely be integrated into maize.
[本文引用: 1]
URL [本文引用: 1]
Based on both cDNA sequence of barley powdery mildew resistance control element Mlo and DNA sequence of the known putative disease resistance gene from Triticum monococcum L., we designed some primers to amplify resistant homologous sequences in the near isogenic lines (NILs) of powdery mildew resistance using RT-PCR method. Two expressed cDNA fragments were isolated from wheat genome. One showed 83% homology to the Mlo gene of barley. The other contained two possible open reading frames (ORFs). NBS conservative domains 2, 3 of disease resistance gene and 13 LRR structures similar to rice Pib protein terminal were found respectively in the two ORFs. It indicated that the latter fragment belongs to NBS-LRR-like genes. The obvious difference of RT-PCR products was observed between the before challenged and the challenged for 72 h by Blumeria graminis f. sp. tritici, which implied that this sequence could be associated with disease resistance of wheat. Using nulli-tetrasomic lines of hinese Spring", the NBS-LRR-like gene had been located on chromosome 1D.
URL [本文引用: 1]
The GenBank sequence database is an annotated collection of all publicly available nucleotidesequences and their protein translations. This database is produced at National Center forBiotechnology Information (NCBI) as part of an international collaboration with the EuropeanMolecular Biology Laboratory (EMBL) Data Library from the European Bioinformatics Institute (EBI) and the DNA Data Bank of Japan (DDBJ). GenBank and its collaborators receive sequencesproduced in laboratories throughout the world from more than 100,000 distinct organisms. GenBank continues to grow at an exponential rate, doubling every 10 months. Release 134, produced inFebruary 2003, contained over 29.3 billion nucleotide bases in more than 23.0 million sequences.GenBank is built by direct submissions from individual laboratories, as well as from bulksubmissions from large-scale sequencing centers.Direct submissions are made to GenBank using BankIt [http://www.ncbi.nlm.nih.gov/BankIt/],which is a Web-based form, or the stand-alone submission program, Sequin [http://www.ncbi.nlm. nih.gov/Sequin/index.html]. Upon receipt of a sequence submission, the GenBank staff assigns an Accession number to the sequence and performs quality assurance checks. The submissions arethen released to the public database, where the entries are retrievable by Entrez or downloadable by FTP. Bulk submissions of Expressed Sequence Tag (EST), Sequence Tagged Site (STS),Genome Survey Sequence (GSS), and High-Throughput Genome Sequence (HTGS) data are most often submitted by large-scale sequencing centers. The GenBank direct submissions group alsoprocesses complete microbial genome sequences.
Magsci [本文引用: 1]
综述了水稻基因组测序的工作进展和水稻基因组信息。至2002年,籼、粳稻两个亚种基因组工作“框架图”的测定及粳稻基因组全长序列的精确测定已相继完成。初步得到了水稻非冗余序列389.81~409.76 Mb;预测水稻基因总数达32 000~56 000个,少于30%的基因被功能分类;基因内GC含量梯度明显;外显子变异少、内含子变化大;籼稻和粳稻的序列差异达22%以上;水稻与其他禾谷类基因之间有广泛的共线性,但与拟南芥的共线性是有限的。基因组测序不仅开辟了基因组学这一生命科学的新兴学科,而且带动了生物信息学、蛋白质组学等一批新兴学科和技术的发展。
Magsci [本文引用: 1]
综述了水稻基因组测序的工作进展和水稻基因组信息。至2002年,籼、粳稻两个亚种基因组工作“框架图”的测定及粳稻基因组全长序列的精确测定已相继完成。初步得到了水稻非冗余序列389.81~409.76 Mb;预测水稻基因总数达32 000~56 000个,少于30%的基因被功能分类;基因内GC含量梯度明显;外显子变异少、内含子变化大;籼稻和粳稻的序列差异达22%以上;水稻与其他禾谷类基因之间有广泛的共线性,但与拟南芥的共线性是有限的。基因组测序不仅开辟了基因组学这一生命科学的新兴学科,而且带动了生物信息学、蛋白质组学等一批新兴学科和技术的发展。
URLPMID:12850444 [本文引用: 1]
To discover genes essential for rice genetic improvement, the Ministry of Science and Technology of China has started the China Rice Functional Genomics Program (CRFGP). During the past three years, the CRFGP has focused on developing rice functional genomics tools and resources to identify genes of agronomic significance. Here, we highlight recent progress made by the CRFGP and discuss the possible integration of global resources for rice functional genomics.
Magsci [本文引用: 1]
水稻基因数据库系统是在建的国家作物科学数据共享中心的一个重要模块,也是国家水稻数据中心的一个重要子系统。功能上,该子系统提供了有关水稻基因各方面的数据信息,包括基因序列、基因产物、基因功能、参考文献等;系统设计上,基于ASP.NET的程序骨架使得系统在网络上运行时更快捷、更安全、更易维护。用户可通过因特网(http://gene.ricedata.cn/)对数据库进行检索操作。
Magsci [本文引用: 1]
水稻基因数据库系统是在建的国家作物科学数据共享中心的一个重要模块,也是国家水稻数据中心的一个重要子系统。功能上,该子系统提供了有关水稻基因各方面的数据信息,包括基因序列、基因产物、基因功能、参考文献等;系统设计上,基于ASP.NET的程序骨架使得系统在网络上运行时更快捷、更安全、更易维护。用户可通过因特网(http://gene.ricedata.cn/)对数据库进行检索操作。
URL [本文引用: 1]
URLMagsci [本文引用: 1]
选取已定位的大麦lH染色体的STS标记MWG913为引物,在普通小麦(TritiumaestivumL.)及其4个可能的起源种乌拉尔图小麦(T.urartuT.)、栽培一粒小麦(T.monococcumL.)栽培二粒小麦(T.dicoccumS.)、方穗山羊草(Ae.squarrosaL.)上特异性扩增。扩增产物克隆测序后对其进行序列分析,由序列差异的程度来确定这几个物种之间的亲缘关系.实验结果表明,普通小麦(TritiumaestivumL.)的A基因组此段序列和乌拉尔图小麦(T.urartuT.)、栽培一粒小麦(T.monococcumL.)、栽培二粒小麦(T.dicoccumS.)A基因组此段序列完全相同;普通小麦的D基因组此段序列与方穗山羊草(Ae.squarrosaL.)也完全相同;普通小麦的B基因组此段序列和栽培二粒小麦B基因组此段序列有0.61%的差异。研究结果一方面对现有的普通小麦A、B、D基因组起源和进化理论给予了分子水平上的证明,同时也揭示了同一物种不同的基因组进化速度存在差异。
URLMagsci [本文引用: 1]
选取已定位的大麦lH染色体的STS标记MWG913为引物,在普通小麦(TritiumaestivumL.)及其4个可能的起源种乌拉尔图小麦(T.urartuT.)、栽培一粒小麦(T.monococcumL.)栽培二粒小麦(T.dicoccumS.)、方穗山羊草(Ae.squarrosaL.)上特异性扩增。扩增产物克隆测序后对其进行序列分析,由序列差异的程度来确定这几个物种之间的亲缘关系.实验结果表明,普通小麦(TritiumaestivumL.)的A基因组此段序列和乌拉尔图小麦(T.urartuT.)、栽培一粒小麦(T.monococcumL.)、栽培二粒小麦(T.dicoccumS.)A基因组此段序列完全相同;普通小麦的D基因组此段序列与方穗山羊草(Ae.squarrosaL.)也完全相同;普通小麦的B基因组此段序列和栽培二粒小麦B基因组此段序列有0.61%的差异。研究结果一方面对现有的普通小麦A、B、D基因组起源和进化理论给予了分子水平上的证明,同时也揭示了同一物种不同的基因组进化速度存在差异。
URLPMID:23535592 [本文引用: 1]
About 8,000 years ago in the Fertile Crescent, a spontaneous hybridization of the wild diploid grass Aegilops tauschii (2n = 14; DD) with the cultivated tetraploid wheat Triticum turgidum (2n = 4x = 28; AABB) resulted in hexaploid wheat (T. aestivum; 2n = 6x = 42; AABBDD)(1,2). Wheat has since become a primary staple crop worldwide as a result of its enhanced adaptability to a wide range of climates and improved grain quality for the production of baker's flour(2). Here we describe sequencing the Ae. tauschii genome and obtaining a roughly 90-fold depth of short reads from libraries with various insert sizes, to gain a better understanding of this genetically complex plant. The assembled scaffolds represented 83.4% of the genome, of which 65.9% comprised transposable elements. We generated comprehensive RNA-Seq data and used it to identify 43,150 protein-coding genes, of which 30,697 (71.1%) were uniquely anchored to chromosomes with an integrated high-density genetic map. Whole-genome analysis revealed gene family expansion in Ae. tauschii of agronomically relevant gene families that were associated with disease resistance, abiotic stress tolerance and grain quality. This draft genome sequence provides insight into the environmental adaptation of bread wheat and can aid in defining the large and complicated genomes of wheat species.
[本文引用: 1]
[本文引用: 1]
URLPMID:2332425710001000 [本文引用: 1]
pAbstract/p pBackground/p pHeterosis is a phenomenon in which hybrids exhibit superior performance relative to parental phenotypes. In addition to the heterosis of above-ground agronomic traits on which most existing studies have focused, root heterosis is also an indispensable component of heterosis in the entire plant and of major importance to plant breeding. Consequently, systematic investigations of root heterosis, particularly in reproductive-stage rice, are needed. The recent advent of RNA sequencing technology (RNA-Seq) provides an opportunity to conduct in-depth transcript profiling for heterosis studies./p pResults/p pUsing the Illumina HiSeq 2000 platform, the root transcriptomes of the super-hybrid rice variety Xieyou 9308 and its parents were analyzed at tillering and heading stages. Approximately 391 million high-quality paired-end reads (100-bp in size) were generated and aligned against the Nipponbare reference genome. We found that 38,872 of 42,081 (92.4%) annotated transcripts were represented by at least one sequence read. A total of 829 and 4186 transcripts that were differentially expressed between the hybrid and its parents (DGsubHP/sub) were identified at tillering and heading stages, respectively. Out of the DGsubHP/sub, 66.59% were down-regulated at the tillering stage and 64.41% were up-regulated at the heading stage. At the heading stage, the DGsubHP/sub were significantly enriched in pathways related to processes such as carbohydrate metabolism and plant hormone signal transduction, with most of the key genes that are involved in the two pathways being up-regulated in the hybrid. Several significant DGsubHP/sub that could be mapped to quantitative trait loci (QTLs) for yield and root traits are also involved in carbohydrate metabolism and plant hormone signal transduction pathways./p pConclusions/p pAn extensive transcriptome dataset was obtained by RNA-Seq, giving a comprehensive overview of the root transcriptomes at tillering and heading stages in a heterotic rice cross and providing a useful resource for the rice research community. Using comparative transcriptome analysis, we detected DGsubHP/sub and identified a group of potential candidate transcripts. The changes in the expression of the candidate transcripts may lay a foundation for future studies on molecular mechanisms underlying root heterosis./p
URLPMID:23613738 [本文引用: 1]
Hybridization, a common process in nature, can give rise to a vast reservoir of allelic variants. Combination of these allelic variants may result in novel patterns of gene action and is thought to contribute to heterosis. In this study, we analyzed genome-wide allele-specific gene expression (ASGE) in the super-hybrid rice variety Xieyou9308 using RNA sequencing technology (RNA-Seq). We identified 9325 reliable single nucleotide polymorphisms (SNPs) distributed throughout the genome. Nearly 68% of the identified polymorphisms were CT and GA SNPs between R9308 and Xieqingzao B, suggesting the existence of DNA methylation, a heritable epigenetic mark, in the parents and their F1 hybrid. Of 2793 identified transcripts with consistent allelic biases, only 480 (17%) showed significant allelic biases during tillering and/or heading stages, implying that trans effects may mediate most transcriptional differences in hybrid offspring. Approximately 67% and 62% of the 480 transcripts showed R9308 allelic expression biases at tillering and heading stages, respectively. Transcripts with higher levels of gene expression in R9308 also exhibited R9308 allelic biases in the hybrid. In addition, 125 transcripts were identified with significant allelic expression biases at both stages, of which 74% showed R9308 allelic expression biases. R9308 alleles may tend to preserve their characteristic states of activity in the hybrid and may play important roles in hybrid vigor at both stages. The allelic expression of 355 transcripts was highly stage-specific, with divergent allelic expression patterns observed at different developmental stages. Many transcripts associated with stress resistance were differently regulated in the F1 hybrid. The results of this study may provide valuable insights into molecular mechanisms of heterosis.
URLPMID:21762167 [本文引用: 1]
090004Given the importance of nitrogen for plant growth and the environmental costs of intense fertilization, an understanding of the molecular mechanisms underlying the root adaptation to nitrogen fluctuations is a primary goal for the development of biotechnological tools for sustainable agriculture. This research aimed to identify the molecular factors involved in the response of maize roots to nitrate.090004cDNA-amplified fragment length polymorphism was exploited for comprehensive transcript profiling of maize (Zea mays) seedling roots grown with varied nitrate availabilities; 336 primer combinations were tested and 661 differentially regulated transcripts were identified. The expression of selected genes was studied in depth through quantitative real-time polymerase chain reaction and in situ hybridization.090004Over 50% of the genes identified responded to prolonged nitrate starvation and a few were identified as putatively involved in the early nitrate signaling mechanisms. Real-time results and in situ localization analyses demonstrated co-regulated transcriptional patterns in root epidermal cells for genes putatively involved in nitric oxide synthesis/scavenging.090004Our findings, in addition to strengthening already known mechanisms, revealed the existence of a new complex signaling framework in which brassinosteroids (BRI1), the module MKK2090009MAPK6 and the fine regulation of nitric oxide homeostasis via the co-expression of synthetic (nitrate reductase) and scavenging (hemoglobin) components may play key functions in maize responses to nitrate.
URL [本文引用: 1]
条锈病是我国小麦生产上的重要病害,利用抗病性是控制小麦条锈病最经济有效的措施。由于条锈菌的毒性变异频繁,常常导致全生育期抗病品种丧失抗锈性而失去利用价值。小麦成株抗条锈性具有抗性持久的特征,目前在抗病品种选育与应用中受到广泛重视。然而,成株抗性品种其抗病性由苗期的感病转变为成株期抗病的内在调控机理尚未清楚。本研究应用新一代高通量测序技术Illumina SolexaⅡ对小麦兴资9104苗期和成株期叶片分别进行转录组测序,并通过生物信息学分析成株期与苗期差异表达基因,挖掘其中参与成株抗性的小麦基因。测序结果显示,兴资9104转录组测序共获得157689条Unigene,其中苗期转录组测序获得109606条Unigene,成株期获得159931条Unigene。通过FDR≤0.001且(log2Ratio)≥1参数进行筛选,共获得27204个在苗期和成株期差异表达基因,其中苗期上调的为13976个,成株期上调的是13228个。另外,本研究还获得了大量抗病相关基因,其中在苗期特异表达表达的抗性基因为221个,在成株期特异表达表达为107个。这些基因的获得为后续基因的功能分析,及其小麦抗条锈性的遗传改良奠定了重要基础。
URL [本文引用: 1]
蛋白质是生命功能的执行体,蛋白质组学是研究细胞、组织或生物体中蛋白质组成及其变化规律的科学,目前蛋白质组学在植物上的研究已经陆续展开,但在棉花上还没有被广泛应用。双向电泳作为蛋白质组研究的三大核心技术之一,是目前常用的唯一一种能够连续在一块胶上分离数千种蛋白质的方法,广泛应用于生物学研究的各个方面。 利用双向电泳技术(two-dimensional electrophoresis,2-DE)对植物发育过程中基因表达进行分析,能够从蛋白质水平揭示植物发育的内在机制。在植物雄性不育分子机制的研究中,全蛋白双向电泳技术正得到越来越多的应用。但是迄今为止,关于棉花雄性不育蛋白质组学的研究,国内外未见报道。利用蛋白质组学技术分析棉花花药总蛋白,旨在找到花粉发育过程中与败育相关的蛋白质,为更进一步研究棉花及其他植物败育发生的机理提供依据。 本实验陆地棉花药为材料,通过对蛋白质提取方法、等电聚焦方法、样品上样量等因素进行比较和优化,以期提高双向电泳的分辨率和重复性,试图建立一套适用于棉花花药蛋白质组学分析的双向电泳方法,为使蛋白质组学更好的应用于棉花提供一些借鉴。将棉花花药用液氮研磨,然后用TCA/丙酮沉淀法和苯酚抽提结合甲醇/醋酸铵沉淀法提取蛋白。采取载体两性电解质PH梯度等电聚焦/SDS-PAGE和固相PH梯度等电聚焦/SDS-PAGE双向凝胶电泳,对陆地棉花药总蛋白质进行了分离。结果表明,苯酚抽提结合甲醇/醋酸铵沉淀的方法较适合棉花花药蛋白质的提取,采用载体两性电解质PH梯度聚焦方法较PH梯度等电聚焦方法的效果好,凝胶用硝酸银染色,上样量为500ug时,得到的电泳图谱分辨率高、重复性好,经ImageMaster2D platinum 5.5软件分析后,大约可识别1300-1400个蛋白质点。 采用载体两性电解质pH梯度-SDS-PAGE双向电泳对陆地棉核雄性不育株和可育株双核期花药总蛋白质进行了分离,通过银染显色,获得了分辨率和重复性较好的双向电泳图谱。ImageMaster2D platinum 5.5软件可识别约1300个蛋白质点,其中差异表达的蛋白质数为79个.将其中61个差异点采用基质辅助激光解析电离飞行时间质谱(matrix a ssisted laser de sorption/ionizaton time of flight mass spectrometry, MALDI-TOF-MS)进行了肽指纹图谱分析,通过Mascot软件利用MSDB数据库进行检索,得到了部分差异蛋白的相关信息,并对这些差异蛋白的表达情况、功能等做了比较分析,以期为探讨棉花核雄性不育过程中的基因表达调控机制奠定基础。 质谱分析鉴定出来的蛋白质按功能主要有以下几类:与信号转导相关的蛋白、与基因调控相关的蛋白、与糖代谢及能量代谢有关的酶、与光合作用有关的酶等。推测这些在陆地棉核雄性不育花药中出现的表达量上调或下调的蛋白质或酶类,在棉花败育过程中具有重要的生物学功能。
URL [本文引用: 1]
蛋白质是生命功能的执行体,蛋白质组学是研究细胞、组织或生物体中蛋白质组成及其变化规律的科学,目前蛋白质组学在植物上的研究已经陆续展开,但在棉花上还没有被广泛应用。双向电泳作为蛋白质组研究的三大核心技术之一,是目前常用的唯一一种能够连续在一块胶上分离数千种蛋白质的方法,广泛应用于生物学研究的各个方面。 利用双向电泳技术(two-dimensional electrophoresis,2-DE)对植物发育过程中基因表达进行分析,能够从蛋白质水平揭示植物发育的内在机制。在植物雄性不育分子机制的研究中,全蛋白双向电泳技术正得到越来越多的应用。但是迄今为止,关于棉花雄性不育蛋白质组学的研究,国内外未见报道。利用蛋白质组学技术分析棉花花药总蛋白,旨在找到花粉发育过程中与败育相关的蛋白质,为更进一步研究棉花及其他植物败育发生的机理提供依据。 本实验陆地棉花药为材料,通过对蛋白质提取方法、等电聚焦方法、样品上样量等因素进行比较和优化,以期提高双向电泳的分辨率和重复性,试图建立一套适用于棉花花药蛋白质组学分析的双向电泳方法,为使蛋白质组学更好的应用于棉花提供一些借鉴。将棉花花药用液氮研磨,然后用TCA/丙酮沉淀法和苯酚抽提结合甲醇/醋酸铵沉淀法提取蛋白。采取载体两性电解质PH梯度等电聚焦/SDS-PAGE和固相PH梯度等电聚焦/SDS-PAGE双向凝胶电泳,对陆地棉花药总蛋白质进行了分离。结果表明,苯酚抽提结合甲醇/醋酸铵沉淀的方法较适合棉花花药蛋白质的提取,采用载体两性电解质PH梯度聚焦方法较PH梯度等电聚焦方法的效果好,凝胶用硝酸银染色,上样量为500ug时,得到的电泳图谱分辨率高、重复性好,经ImageMaster2D platinum 5.5软件分析后,大约可识别1300-1400个蛋白质点。 采用载体两性电解质pH梯度-SDS-PAGE双向电泳对陆地棉核雄性不育株和可育株双核期花药总蛋白质进行了分离,通过银染显色,获得了分辨率和重复性较好的双向电泳图谱。ImageMaster2D platinum 5.5软件可识别约1300个蛋白质点,其中差异表达的蛋白质数为79个.将其中61个差异点采用基质辅助激光解析电离飞行时间质谱(matrix a ssisted laser de sorption/ionizaton time of flight mass spectrometry, MALDI-TOF-MS)进行了肽指纹图谱分析,通过Mascot软件利用MSDB数据库进行检索,得到了部分差异蛋白的相关信息,并对这些差异蛋白的表达情况、功能等做了比较分析,以期为探讨棉花核雄性不育过程中的基因表达调控机制奠定基础。 质谱分析鉴定出来的蛋白质按功能主要有以下几类:与信号转导相关的蛋白、与基因调控相关的蛋白、与糖代谢及能量代谢有关的酶、与光合作用有关的酶等。推测这些在陆地棉核雄性不育花药中出现的表达量上调或下调的蛋白质或酶类,在棉花败育过程中具有重要的生物学功能。
URLPMID:17558543 [本文引用: 1]
The female gamete, the egg cell, is a specially differentiated haploid cell that develops into an embryo following fertilization. In the present study, we analyzed egg cell lysates by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequent mass spectrometry-based proteomics technology and identified the major proteins expressed in rice egg cells. The proteins identified included glyceraldehyde-3-phosphate dehydrogenase, ascorbate peroxidase and heat shock protein 90. The abundant existence of chaperons and antioxidant enzymes in plant egg cells indicates that the major protein components of plant egg cells are partly analogous to those of mammalian eggs and oocytes.
URLMagsci [本文引用: 1]
为揭示不易早衰水稻耐养分胁迫机理,应用蛋白质组学分析技术,对养分胁迫下不易早衰水稻隆平001生育后期叶片蛋白质组差异表达进行了研究。叶片蛋白质图谱经软件Imagemaster 2D Elite 5.0分析,有26个蛋白质发生差异表达,其中17个蛋白质功能经质谱分析(ESI-Q MS/MS)得到鉴定。差异表达蛋白质功能分析显示,养分胁迫下,隆平001叶片中光合及养分再利用相关蛋白质表达量增强,从而满足养分胁迫下植株生长发育的需要,是其具有较强抗养分胁迫的内在机理,而参与活性氧清除及逆境保护相关蛋白质表达量增强,可以保护光合机构免受胁迫伤害,从而对养分胁迫有更强的耐性、不易发生早衰。
URLMagsci [本文引用: 1]
为揭示不易早衰水稻耐养分胁迫机理,应用蛋白质组学分析技术,对养分胁迫下不易早衰水稻隆平001生育后期叶片蛋白质组差异表达进行了研究。叶片蛋白质图谱经软件Imagemaster 2D Elite 5.0分析,有26个蛋白质发生差异表达,其中17个蛋白质功能经质谱分析(ESI-Q MS/MS)得到鉴定。差异表达蛋白质功能分析显示,养分胁迫下,隆平001叶片中光合及养分再利用相关蛋白质表达量增强,从而满足养分胁迫下植株生长发育的需要,是其具有较强抗养分胁迫的内在机理,而参与活性氧清除及逆境保护相关蛋白质表达量增强,可以保护光合机构免受胁迫伤害,从而对养分胁迫有更强的耐性、不易发生早衰。
URL [本文引用: 1]
采用双向电泳技术(2-DE)研究玉米苗期受淹后叶片的蛋白质组学差异,以进一步阐明玉米对淹水胁迫的响应机制。玉米种植10 d后进行72 h淹水处理,提取叶片总蛋白质、采用2-DE,利用图像分析,获得12个差异蛋白点。经鉴定得到9种蛋白质,这些蛋白质分别参与光合作用、氨基酸代谢、防御与胁迫和碳水化合物代谢等多个代谢过程。其中,参与光合作用、蛋白质折叠和氨基酸代谢的蛋白质表达基本被负调,而参与淀粉生物合成的2个蛋白质均上调。这说明淹水胁迫抑制了玉米的光合作用,而玉米植株则可能通过提高淀粉生物合成增加有机物质储备,从而有利于植株恢复。玉米对淹水胁迫的适应是一个复杂的生物过程,涉及到多种蛋白质的相互作用,构成了一个复杂的调控网络。
URL [本文引用: 1]
采用双向电泳技术(2-DE)研究玉米苗期受淹后叶片的蛋白质组学差异,以进一步阐明玉米对淹水胁迫的响应机制。玉米种植10 d后进行72 h淹水处理,提取叶片总蛋白质、采用2-DE,利用图像分析,获得12个差异蛋白点。经鉴定得到9种蛋白质,这些蛋白质分别参与光合作用、氨基酸代谢、防御与胁迫和碳水化合物代谢等多个代谢过程。其中,参与光合作用、蛋白质折叠和氨基酸代谢的蛋白质表达基本被负调,而参与淀粉生物合成的2个蛋白质均上调。这说明淹水胁迫抑制了玉米的光合作用,而玉米植株则可能通过提高淀粉生物合成增加有机物质储备,从而有利于植株恢复。玉米对淹水胁迫的适应是一个复杂的生物过程,涉及到多种蛋白质的相互作用,构成了一个复杂的调控网络。
URL [本文引用: 1]
【目的】从蛋白质组学的角度研究早期发育阶段玉米上、中部籽粒差异表达的蛋白质,分析其功能,探明玉米粒位发育差异的分子机理。【方法】在大田条件下,以粒位效应显著的玉米品种登海661(DH661)为供试材料,90 000株/hm~2密度下种植,在开花期人工饱和授粉后0、3、6、12 d取果穗上部与中部籽粒。采用TCA-丙酮沉淀法提取籽粒总蛋白,双向电泳分离后获得蛋白质图谱。分别以0、3、6、12 d中部籽粒的凝胶作为参考胶,将上部籽粒凝胶与其进行比对,利用Image master 2D 7.0软件分析籽粒早期发育不同阶段上、中粒位蛋白质表达的差异。通过MALDI-TOF/TOF MS质谱分析及NCBI数据库搜索,对差异表达蛋白质进行鉴定并分析其涉及的生物学功能。【结果】较高密度种植后,果穗籽粒早期发育阶段共检测到超过1000个清晰蛋白质点。通过图像处理软件成对匹配分析,果穗上、中部籽粒早期发育阶段差异蛋白质点为66个,其中52个蛋白质点与NCBI数据库匹配,鉴定率为78.8%。差异蛋白质涉及籽粒呼吸与能量代谢(10个蛋白质点,19%)、胁迫与防御(9个蛋白质点,17%)、蛋白质代谢(9个蛋白质点,17%)、氮代谢(6个蛋白质点,11%)、细胞分化与增殖(5个蛋白质点,10%)、转录与翻译(5个蛋白质点,10%)、次生物质代谢(3个蛋白质点,6%)等功能范畴。对相关的差异蛋白质表达丰度分析,与中部籽粒相比,上部籽粒涉及细胞分化与增殖、呼吸与能量代谢的大部分蛋白质在一个或多个时间段内均显著下调,说明上部籽粒胚乳细胞增殖及呼吸能量代谢能力显著降低。同时,上部籽粒涉及胁迫与防御的多种抗氧化酶系、乙二醛酶1以及涉及蛋白质代谢的4个分子伴侣蛋白质在发育早期也处于低水平表达,说明上部籽粒应对逆境条件防御能力较弱且蛋白质结构不稳定。另外,与中部籽粒相比,上部籽粒氮代谢中丙氨酸转氨酶以及S-腺苷甲硫氨酸合成酶1在授粉后6—12 d内均下调表达,说明上部籽粒氮同化能力较弱,影响后续的氨基酸合成与蛋白质代谢过程。【结论】与果穗中部籽粒相比,上部籽粒细胞分化与增殖相关蛋白质的表达水平较低,呼吸与能量代谢能力较弱,导致上部籽粒库容与库活性降低。另外,面对氧化应激等逆境时,上部籽粒相关的抗氧化酶以及分子伴侣蛋白表达水平较低,致使其防御能力低于中部籽粒。丙氨酸转氨酶、S-腺苷甲硫氨酸合成酶1(SAMS)的差异表达也可能是导致粒位效应的重要原因。
URL [本文引用: 1]
【目的】从蛋白质组学的角度研究早期发育阶段玉米上、中部籽粒差异表达的蛋白质,分析其功能,探明玉米粒位发育差异的分子机理。【方法】在大田条件下,以粒位效应显著的玉米品种登海661(DH661)为供试材料,90 000株/hm~2密度下种植,在开花期人工饱和授粉后0、3、6、12 d取果穗上部与中部籽粒。采用TCA-丙酮沉淀法提取籽粒总蛋白,双向电泳分离后获得蛋白质图谱。分别以0、3、6、12 d中部籽粒的凝胶作为参考胶,将上部籽粒凝胶与其进行比对,利用Image master 2D 7.0软件分析籽粒早期发育不同阶段上、中粒位蛋白质表达的差异。通过MALDI-TOF/TOF MS质谱分析及NCBI数据库搜索,对差异表达蛋白质进行鉴定并分析其涉及的生物学功能。【结果】较高密度种植后,果穗籽粒早期发育阶段共检测到超过1000个清晰蛋白质点。通过图像处理软件成对匹配分析,果穗上、中部籽粒早期发育阶段差异蛋白质点为66个,其中52个蛋白质点与NCBI数据库匹配,鉴定率为78.8%。差异蛋白质涉及籽粒呼吸与能量代谢(10个蛋白质点,19%)、胁迫与防御(9个蛋白质点,17%)、蛋白质代谢(9个蛋白质点,17%)、氮代谢(6个蛋白质点,11%)、细胞分化与增殖(5个蛋白质点,10%)、转录与翻译(5个蛋白质点,10%)、次生物质代谢(3个蛋白质点,6%)等功能范畴。对相关的差异蛋白质表达丰度分析,与中部籽粒相比,上部籽粒涉及细胞分化与增殖、呼吸与能量代谢的大部分蛋白质在一个或多个时间段内均显著下调,说明上部籽粒胚乳细胞增殖及呼吸能量代谢能力显著降低。同时,上部籽粒涉及胁迫与防御的多种抗氧化酶系、乙二醛酶1以及涉及蛋白质代谢的4个分子伴侣蛋白质在发育早期也处于低水平表达,说明上部籽粒应对逆境条件防御能力较弱且蛋白质结构不稳定。另外,与中部籽粒相比,上部籽粒氮代谢中丙氨酸转氨酶以及S-腺苷甲硫氨酸合成酶1在授粉后6—12 d内均下调表达,说明上部籽粒氮同化能力较弱,影响后续的氨基酸合成与蛋白质代谢过程。【结论】与果穗中部籽粒相比,上部籽粒细胞分化与增殖相关蛋白质的表达水平较低,呼吸与能量代谢能力较弱,导致上部籽粒库容与库活性降低。另外,面对氧化应激等逆境时,上部籽粒相关的抗氧化酶以及分子伴侣蛋白表达水平较低,致使其防御能力低于中部籽粒。丙氨酸转氨酶、S-腺苷甲硫氨酸合成酶1(SAMS)的差异表达也可能是导致粒位效应的重要原因。
URLMagsci [本文引用: 1]
为探讨外源铜胁迫对小麦幼根的影响,采用水培方法,测定0~60 mg/L不同质量浓度铜处理下小麦幼苗生长发育、转录组表达和蛋白质组表达的差异,探索其抵御重金属毒害的分子机制。结果表明:高质量浓度的铜影响小麦生长,随胁迫时间的延长,小麦幼苗萎蔫发黄,幼根生长受抑。小麦幼根转录组测序检测,共得到2 283个差异表达基因(DEGs),其中826个DEGs表达上调,1 457个DEGs表达下调;将DEGs进行KEGG pathway注释,被注释到31个代谢途径中。双向电泳试验检测,小麦幼根中有2 049个蛋白质点,30 mg/L铜处理96 h后,与对照(0 mg/L处理)相比,约130个蛋白质点表达出现显著差异,其中56个丰度增加,74个丰度降低;质谱鉴定部分差异表达蛋白,抗性蛋白如谷胱甘肽转移酶、27K蛋白等在胁迫下表达上升,而生理代谢相关蛋白表达降低。
URLMagsci [本文引用: 1]
为探讨外源铜胁迫对小麦幼根的影响,采用水培方法,测定0~60 mg/L不同质量浓度铜处理下小麦幼苗生长发育、转录组表达和蛋白质组表达的差异,探索其抵御重金属毒害的分子机制。结果表明:高质量浓度的铜影响小麦生长,随胁迫时间的延长,小麦幼苗萎蔫发黄,幼根生长受抑。小麦幼根转录组测序检测,共得到2 283个差异表达基因(DEGs),其中826个DEGs表达上调,1 457个DEGs表达下调;将DEGs进行KEGG pathway注释,被注释到31个代谢途径中。双向电泳试验检测,小麦幼根中有2 049个蛋白质点,30 mg/L铜处理96 h后,与对照(0 mg/L处理)相比,约130个蛋白质点表达出现显著差异,其中56个丰度增加,74个丰度降低;质谱鉴定部分差异表达蛋白,抗性蛋白如谷胱甘肽转移酶、27K蛋白等在胁迫下表达上升,而生理代谢相关蛋白表达降低。
URLMagsci [本文引用: 1]
小麦多子房性状的蛋白质组学特性和形成机制及其在小麦杂种优势中的应用可能提供理论依据。为揭示以小麦单子房品系77(2)及其多子房近等基因系Mu77(2)为材料, 采用TCA-丙酮法提取穗分化至四分体时期的幼穗总蛋白, 并通过IEF/SDS-PAGE双向凝胶电泳分离, 获得了分辨率和重复性较好的蛋白质组差异图谱。在等电点4~7、分子量14.4~97.4 kD之间发现约450个肉眼可辨的蛋白点, 其中上调2倍以上且达到99%统计学显著水平的差异表达蛋白点30个。对6个特异性差异蛋白点作二级质谱(LC-MS/MS)分析, 结果表明它们是富含甘氨酸RNA结合蛋白(S1)、SGT1-1(S2)、HMG-I/Y(S3)、谷胱甘肽转移酶(S4)、果糖1,6-二磷酸醛缩酶(S5)和未知蛋白(S6)。这些蛋白质对DNA转录、蛋白质翻译、能量转换及代谢、抗逆防卫等生理生化过程起调控作用, 可能与小麦多子房性状的形成有关。
URLMagsci [本文引用: 1]
小麦多子房性状的蛋白质组学特性和形成机制及其在小麦杂种优势中的应用可能提供理论依据。为揭示以小麦单子房品系77(2)及其多子房近等基因系Mu77(2)为材料, 采用TCA-丙酮法提取穗分化至四分体时期的幼穗总蛋白, 并通过IEF/SDS-PAGE双向凝胶电泳分离, 获得了分辨率和重复性较好的蛋白质组差异图谱。在等电点4~7、分子量14.4~97.4 kD之间发现约450个肉眼可辨的蛋白点, 其中上调2倍以上且达到99%统计学显著水平的差异表达蛋白点30个。对6个特异性差异蛋白点作二级质谱(LC-MS/MS)分析, 结果表明它们是富含甘氨酸RNA结合蛋白(S1)、SGT1-1(S2)、HMG-I/Y(S3)、谷胱甘肽转移酶(S4)、果糖1,6-二磷酸醛缩酶(S5)和未知蛋白(S6)。这些蛋白质对DNA转录、蛋白质翻译、能量转换及代谢、抗逆防卫等生理生化过程起调控作用, 可能与小麦多子房性状的形成有关。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:19053355 [本文引用: 1]
A metabolite profiling approach based on gas chromatography-mass spectrometry (GC-MS) was used to investigate time-dependent metabolic changes in the course of the germination of rice. Brown rice kernels were soaked and incubated for a total of 96 h under ambient conditions. Samples taken during the germination process were subjected to an extraction and fractionation procedure covering a broad spectrum of lipophilic (e.g., fatty acid methyl esters, hydrocarbons, fatty alcohols, sterols) and hydrophilic (e.g., sugars, acids, amino acids, amines) low molecular weight rice constituents. Investigation of the obtained fractions by GC resulted in the detection of 615 distinct peaks, of which 174 were identified by means of MS. Statistical assessment of the data via principal component analysis demonstrated that the metabolic changes during the germination process are reflected by time-dependent shifts of the scores, which were similar for the three rice materials investigated. Analysis of the corresponding loadings showed that polar metabolites were major contributors to the separation along the first principal component. Relative quantifications based on standardized peak heights revealed dynamic changes of the metabolites in the course of the germination.
URL [本文引用: 1]
目的:建立气相色谱-质谱联用技术(GC-MS)的代谢组学方法,初步研究转基因株系与对照株系之间代谢物指纹图谱的差异性,为转基因作物安全的评价提供参考。方法:优化提取条件,考察色谱条件,并采用主成分分析(PCA)数据处理方法对转基因株系及对照进行模式识别。结果:优化了提取条件及色谱条件,建立了GC-MS的代谢组学方法,获得了小分子的代谢产物的表达谱,发现转基因与其对照之间呈现出显著性差异。结论:优化的GC-MS的代谢组学方法可以从代谢水平检测转基因作物,找出差异性,为转基因作物的检测与评价提供技术支持。
URL [本文引用: 1]
目的:建立气相色谱-质谱联用技术(GC-MS)的代谢组学方法,初步研究转基因株系与对照株系之间代谢物指纹图谱的差异性,为转基因作物安全的评价提供参考。方法:优化提取条件,考察色谱条件,并采用主成分分析(PCA)数据处理方法对转基因株系及对照进行模式识别。结果:优化了提取条件及色谱条件,建立了GC-MS的代谢组学方法,获得了小分子的代谢产物的表达谱,发现转基因与其对照之间呈现出显著性差异。结论:优化的GC-MS的代谢组学方法可以从代谢水平检测转基因作物,找出差异性,为转基因作物的检测与评价提供技术支持。
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
Plant phenomics has the potential to accelerate progress in understanding gene functions and environmental responses. Progress has been made in automating high-throughput plant phenotyping. However, few studies have investigated automated rice panicle counting. This paper describes a novel method for automatically and nonintrusively determining rice panicle numbers during the full heading stage by analyzing color images of rice plants taken from multiple angles. Pot-grown rice plants were transferred via an industrial conveyer to an imaging chamber. Color images from different angles were automatically acquired as a turntable rotated the plant. The images were then analyzed and the panicle number of each plant was determined. The image analysis pipeline consisted of extracting the i2 plane from the original color image, segmenting the image, discriminating the panicles from the rest of the plant using an artificial neural network, and calculating the panicle number in the current image. The panicle number of the plant was taken as the maximum of the panicle numbers extracted from all 12 multi-angle images. A total of 105 rice plants during the full heading stage were examined to test the performance of the method. The mean absolute error of the manual and automatic count was 0.5, with 95.3% of the plants yielding absolute errors within 1. The method will be useful for evaluating rice panicles and will serve as an important supplementary method for high-throughput rice phenotyping.
URL [本文引用: 1]
水稻是我国乃至世界最重要的粮食作物之一,如何挖掘水稻育种的最大潜力,培育高产且环境适应力强(抗旱、抗虫、抗盐碱、营养利用率高)的优良水稻品种一直是水稻育种热门研究领域。水稻产量相关性状参数,包括谷粒性状参数,由于其和产量以及品质直接相关,是水稻表型参数中最重要也是最直接反映水稻育种水平的指标,因此水稻育种的各个筛选和鉴定时期都需要测量大量水稻产量相关性状参数,主要包括每株总粒数、实粒数、结实率、千粒重、粒长、粒宽等。 传统谷粒表型测量方法主要依靠人工测量,存在低效、测量精度依赖于操作人员的主观因素、可重复性差等特点。而目前现有水稻产量相关参数测量仪器和测量技术,比如数粒仪,存在自动化程度低、测量参数单一、效率低、对测量物要求清洁度较高等问题。随着育种技术的快速发展,一天可产生成百上千种新育种材料。因此需要一种自动化程度高、精度高、多参数的数字化考种技术,以突破传统谷粒表型测量方法所带来的瓶颈。 本文针对上述需求,对水稻表型参数中的产量相关性状参数数字化提取技术展开研究,并在此基础上研发全自动数字化考种机样机。主要研究内容包括(1)用于单株水稻谷粒高精度考种的全自动谷粒分离系统的设计与实现,同时自动清除杂质,以解决谷物自动高精度分离和单株之间杂质自动清除的难题。(2)产量相关性状参数自动测量系统的设计与实现。应用机器视觉技术和自动输送技术对脱粒后的谷粒进行自动测量,以解决谷粒性状高通量、自动、高精度和多参数测量的难题;同时设计高精度风选装置实现饱粒和空瘪粒的自动筛分,解决谷粒高精度自动风选的难题。(3)产量相关性状参数数字化管理系统的设计与实现。采用物联技术和数据库管理技术,实现将测量完毕的单株谷粒自动包装入库,同时将该株水稻对应产量相关表型数据自动存储至数据库,二者以统一的种子编号进行索引实现智能化识别、定位和追溯,解决了单株种子性状参数数字化管理,以及和对应库存种子智能化识别、定位和追溯的难题。最终将上述技术进行转化,形成全自动数字化考种机样机,系统样机性状参数提取相对误差均值均在5%以内,系统整体运行稳定,测量效率达到720株/天,约为人工测量效率的70倍,和传统测量方法相比,具有客观性、自动化、高通量、多参数等优点。 此外,对核心技术加以改进,易于推广于小麦、大麦、玉米等作物育种研究应用,有利的突破了国外表型测量技术封锁,顺应我国现代农业工程技术和精准农业技术发展潮流和趋势。本系统的研发和建立不仅将直接推动表型组学的快速发展,而且会带动和促进功能基因组学发展,以其产量相关性状特性高通量鉴定特点,成为植物生理学家和育种学家快速解码大量未知基因功能的重要科学工具之一
URL [本文引用: 1]
野生稻为改良栽培稻重要农艺性状提供了宝贵遗传资源。长雄野生稻具有和亚洲栽培稻相同的AA基因组,其地下茎无性繁殖特性是多年生性的理想供体。同时,长雄野生稻对低温等非生物胁迫的抗性也为采用遗传工程手段培育抗逆水稻新品种提供了基因资源。 本研究论文以具地下茎的长雄野生稻和拟高粱、耐冷及冷敏感水稻品种丽江新团黑谷和IR29为材料,采用基因芯片及转录组测序等技术平台,对地下茎发育及耐冷性进行系统的功能基因组学和比较转录组学分析,发掘地下茎发生发育相关功能候选基因及解析水稻耐冷胁迫调控的分子遗传机制,为进一步克隆地下茎发育及耐冷基因打下基础。具体研究结果如下: 1)构建了长雄野生稻地下茎茎尖均一化cDNA文库,并随机挑选10,283个克隆进行测序分析,获得原始ESTs序列10,136条,经高质量ESTs拼接得到4,419条非重复序列。在≥80%序列一致性情况下,分别有4,285(96.97%)和4,151(93.94%)条非重复序列定位到日本晴和9311基因组上。41条非重复序列表现为特异的可变剪接形式,516条非重复序列中共检测到666个简单序列重复(SSR).地下茎茎尖中表达的178条非重复序列共定位到10个地下茎相关QTLs区间。此外,还对OLRR1在五个组织中进行实时荧光定量PCR分析,组织原位杂交进一步验证其在地下茎茎尖的顶端分生组织中高表达。 2)采用异源芯片杂交策略,利用Agilent水稻寡核苷酸长探针芯片对拟高粱五个组织进行全基因组表达谱比较分析,总共检测到548个组织高水平表达基因,其中有31和114个基因在地下茎茎尖和节间中高水平表达。在地下茎茎尖特异高表达基因中发现三个顺式调控元件可能在地下茎发生发育过程中起重要作用,即ABA响应的RY重复序列CATGCA.蔗糖抑制蛋白相关元件TTATCC和GA响应元件TAACAA.对比分析前人报道的长雄野生稻和拟高粱地下茎特异表达基因,发现包括脱落酸、生长素、赤霉素和水杨酸等植物激素在地下茎发生发育中起重要调控作用。 3)鉴于异源芯片表达谱分析的局限性,本研究采用新一代RNA测序技术对拟高粱地下茎和地上茎表达谱进行比较分析。结果显示拟高粱基因组中超过70%的基因在地上茎和地下茎中检测到表达,同时发现1,963和599个基因在地上茎和地下茎特异或高表达,其中分别包括122和55个转录因子,功能聚类分析表明它们在地上或地下茎组织生长发育过程中起重要作用。进一步分析发现ACGT box、GCCAC、GATC和TGACG box等顺式调控元件在地上茎特异表达基因中显著富集;而MYB和ROOTMOTIFTAPOX1顺式元件、10-promoter element TATTCT及响应细胞分裂素元件TATTAG在地下茎特异表达基因中显著富集,表明组织特异及复杂的分子调控网络参与调控地上茎和地下茎的生长发育。此外,27.9%的拟高粱基因检测到可变剪接,其中60%的可变剪接具有组织特异性,表明可变剪接可能在组织特异细胞功能决定中起重要作用。 对本研究中鉴定的地下茎高水平表达基因和已报道的地下茎高水平表达基因进行比较分析,共鉴定了111个基因在至少两个不同平台中的地下茎高水平表达。这些在至少两个不同平台中尤其是在三个不同平台中都是地下茎高水平表达的基因,不仅验证了其表达水平的可靠性,还表明这些基因在拟高粱和长雄野生稻中的功能可能是保守的,为地下茎发生发育相关基因克隆奠定基础。 4)采用Affymetrix水稻全基因组芯片对水稻耐冷品种丽江新团黑谷(LTH)和冷敏感品种IR29进行冷胁迫转录组比较分析。结果表明,部分胁迫反应及信号传导相关基因的组成性高表达与LTH耐冷性相关:同时LTH和IR29在连续时间冷胁迫条件下,基因表达水平呈现基本相同的早期反应和品种特异的后期反应,早期反应主要表现为转录因子和信号传导相关基因上调表达;而在冷胁迫处理后期,不同品种由于对持续冷胁迫的功能性适应而表现为不同的差异表达,包括ROS相关基因在LTH特异高表达,而在IR29中抑制表达。在冷胁迫终止后的恢复过程中,LTH冷胁迫差异表达基因大部分都迅速而有效地回复到正常水平,而IR29冷胁迫差异表达基因则恢复缓慢或不能恢复。进一步分析表明包括CBF和MYBS3调控元在内的许多调控途径参与冷胁迫反应。 5)采用RNA-seq分析长雄野生稻地上茎及地下茎冷胁迫全基因组表达谱。总测序数据量25gigabases (Gb),约覆盖长野基因组58倍。约10%的转录本不能定位于已测序水稻基因组或基因区域,暗示长雄野生稻与水稻基因组序列间存在差异。冷胁迫共导致913和884个基因在地上茎和地下茎中差异表达,包括共同上调表达的33个转录因子。在两组织中发现大量冷胁迫特异可变剪接事件,且这些可变剪接基因广泛参与信号转导、生物学调控、定位和细胞组分生物合成途径,表明可变剪接在冷胁迫信号转导与基因调控网络中起重要作用。此外,在地上茎和地下茎冷处理及对照中共鉴定8,005个新转录本和3,916个融合基因。 进一步对长雄野生稻、LTH和IR29冷胁迫全基因组表达谱进行比较分析,鉴定了154个三个基因型共同冷胁迫诱导上调基因,其中包括34个转录因子,功能注释揭示这些转录因子在栽培稻和野生稻冷胁迫应答中的功能是保守的。另外56个基因(占总数36.4%)的启动子区域至少含有一个CRT/DRE核心元件A/GCCGAC,表明这些COR基因在栽培稻和野生稻中共同参与冷胁迫分子调控网络。
URL [本文引用: 1]
野生稻为改良栽培稻重要农艺性状提供了宝贵遗传资源。长雄野生稻具有和亚洲栽培稻相同的AA基因组,其地下茎无性繁殖特性是多年生性的理想供体。同时,长雄野生稻对低温等非生物胁迫的抗性也为采用遗传工程手段培育抗逆水稻新品种提供了基因资源。 本研究论文以具地下茎的长雄野生稻和拟高粱、耐冷及冷敏感水稻品种丽江新团黑谷和IR29为材料,采用基因芯片及转录组测序等技术平台,对地下茎发育及耐冷性进行系统的功能基因组学和比较转录组学分析,发掘地下茎发生发育相关功能候选基因及解析水稻耐冷胁迫调控的分子遗传机制,为进一步克隆地下茎发育及耐冷基因打下基础。具体研究结果如下: 1)构建了长雄野生稻地下茎茎尖均一化cDNA文库,并随机挑选10,283个克隆进行测序分析,获得原始ESTs序列10,136条,经高质量ESTs拼接得到4,419条非重复序列。在≥80%序列一致性情况下,分别有4,285(96.97%)和4,151(93.94%)条非重复序列定位到日本晴和9311基因组上。41条非重复序列表现为特异的可变剪接形式,516条非重复序列中共检测到666个简单序列重复(SSR).地下茎茎尖中表达的178条非重复序列共定位到10个地下茎相关QTLs区间。此外,还对OLRR1在五个组织中进行实时荧光定量PCR分析,组织原位杂交进一步验证其在地下茎茎尖的顶端分生组织中高表达。 2)采用异源芯片杂交策略,利用Agilent水稻寡核苷酸长探针芯片对拟高粱五个组织进行全基因组表达谱比较分析,总共检测到548个组织高水平表达基因,其中有31和114个基因在地下茎茎尖和节间中高水平表达。在地下茎茎尖特异高表达基因中发现三个顺式调控元件可能在地下茎发生发育过程中起重要作用,即ABA响应的RY重复序列CATGCA.蔗糖抑制蛋白相关元件TTATCC和GA响应元件TAACAA.对比分析前人报道的长雄野生稻和拟高粱地下茎特异表达基因,发现包括脱落酸、生长素、赤霉素和水杨酸等植物激素在地下茎发生发育中起重要调控作用。 3)鉴于异源芯片表达谱分析的局限性,本研究采用新一代RNA测序技术对拟高粱地下茎和地上茎表达谱进行比较分析。结果显示拟高粱基因组中超过70%的基因在地上茎和地下茎中检测到表达,同时发现1,963和599个基因在地上茎和地下茎特异或高表达,其中分别包括122和55个转录因子,功能聚类分析表明它们在地上或地下茎组织生长发育过程中起重要作用。进一步分析发现ACGT box、GCCAC、GATC和TGACG box等顺式调控元件在地上茎特异表达基因中显著富集;而MYB和ROOTMOTIFTAPOX1顺式元件、10-promoter element TATTCT及响应细胞分裂素元件TATTAG在地下茎特异表达基因中显著富集,表明组织特异及复杂的分子调控网络参与调控地上茎和地下茎的生长发育。此外,27.9%的拟高粱基因检测到可变剪接,其中60%的可变剪接具有组织特异性,表明可变剪接可能在组织特异细胞功能决定中起重要作用。 对本研究中鉴定的地下茎高水平表达基因和已报道的地下茎高水平表达基因进行比较分析,共鉴定了111个基因在至少两个不同平台中的地下茎高水平表达。这些在至少两个不同平台中尤其是在三个不同平台中都是地下茎高水平表达的基因,不仅验证了其表达水平的可靠性,还表明这些基因在拟高粱和长雄野生稻中的功能可能是保守的,为地下茎发生发育相关基因克隆奠定基础。 4)采用Affymetrix水稻全基因组芯片对水稻耐冷品种丽江新团黑谷(LTH)和冷敏感品种IR29进行冷胁迫转录组比较分析。结果表明,部分胁迫反应及信号传导相关基因的组成性高表达与LTH耐冷性相关:同时LTH和IR29在连续时间冷胁迫条件下,基因表达水平呈现基本相同的早期反应和品种特异的后期反应,早期反应主要表现为转录因子和信号传导相关基因上调表达;而在冷胁迫处理后期,不同品种由于对持续冷胁迫的功能性适应而表现为不同的差异表达,包括ROS相关基因在LTH特异高表达,而在IR29中抑制表达。在冷胁迫终止后的恢复过程中,LTH冷胁迫差异表达基因大部分都迅速而有效地回复到正常水平,而IR29冷胁迫差异表达基因则恢复缓慢或不能恢复。进一步分析表明包括CBF和MYBS3调控元在内的许多调控途径参与冷胁迫反应。 5)采用RNA-seq分析长雄野生稻地上茎及地下茎冷胁迫全基因组表达谱。总测序数据量25gigabases (Gb),约覆盖长野基因组58倍。约10%的转录本不能定位于已测序水稻基因组或基因区域,暗示长雄野生稻与水稻基因组序列间存在差异。冷胁迫共导致913和884个基因在地上茎和地下茎中差异表达,包括共同上调表达的33个转录因子。在两组织中发现大量冷胁迫特异可变剪接事件,且这些可变剪接基因广泛参与信号转导、生物学调控、定位和细胞组分生物合成途径,表明可变剪接在冷胁迫信号转导与基因调控网络中起重要作用。此外,在地上茎和地下茎冷处理及对照中共鉴定8,005个新转录本和3,916个融合基因。 进一步对长雄野生稻、LTH和IR29冷胁迫全基因组表达谱进行比较分析,鉴定了154个三个基因型共同冷胁迫诱导上调基因,其中包括34个转录因子,功能注释揭示这些转录因子在栽培稻和野生稻冷胁迫应答中的功能是保守的。另外56个基因(占总数36.4%)的启动子区域至少含有一个CRT/DRE核心元件A/GCCGAC,表明这些COR基因在栽培稻和野生稻中共同参与冷胁迫分子调控网络。
URL [本文引用: 1]
干旱是影响小麦种植区域分布、产量和品质的最严重的非生物胁迫因素之一。随着气候变暖、淡水资源日益短缺,干旱对小麦的影响呈现加重趋势。当前,转录组和蛋白质组技术已经成为研究小麦抗旱分子机制的常规和可靠工具,利用这两种技术已在不同小麦品种和小麦野生近缘种中鉴定出了大量参与小麦抗旱分子调控网络的基因和蛋白质。本文简要介绍了近年来利用转录组和蛋白质组学技术获得的对小麦响应干旱分子机制的认识,指出了存在的主要问题,并展望了未来发展趋势,对应用已有的研究成果改良小麦抗旱性及进一步应用转录组和蛋白质组学技术更好地揭示小麦抗旱分子机理具有参考意义。
URL [本文引用: 1]
干旱是影响小麦种植区域分布、产量和品质的最严重的非生物胁迫因素之一。随着气候变暖、淡水资源日益短缺,干旱对小麦的影响呈现加重趋势。当前,转录组和蛋白质组技术已经成为研究小麦抗旱分子机制的常规和可靠工具,利用这两种技术已在不同小麦品种和小麦野生近缘种中鉴定出了大量参与小麦抗旱分子调控网络的基因和蛋白质。本文简要介绍了近年来利用转录组和蛋白质组学技术获得的对小麦响应干旱分子机制的认识,指出了存在的主要问题,并展望了未来发展趋势,对应用已有的研究成果改良小麦抗旱性及进一步应用转录组和蛋白质组学技术更好地揭示小麦抗旱分子机理具有参考意义。
URL [本文引用: 1]

{kind=link}
{kind=link}