 ,1,2
,1,2Application of BIG-Annotator in the genome sequencing data functional annotation and genetic diagnosis
Ying Huang1,2, Qi Liu1, Lianjiang Chi1, Chengmin Shi1, Zhen Wu1, Min Hu3, Hong Shi4, Hua Chen,1,2通讯作者:
编委: 杨昭庆
收稿日期:2018-09-29修回日期:2018-11-2网络出版日期:2018-11-20
| 基金资助: |
Received:2018-09-29Revised:2018-11-2Online:2018-11-20
| Fund supported: |
作者简介 About authors
黄莹,硕士研究生,专业方向:群体与计算遗传学E-mail:huangying@big.ac.cn。

摘要
关键词:
Abstract
Keywords:
PDF (550KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄莹, 刘琪, 池连江, 石承民, 吴祯, 胡敏, 石宏, 陈华. BIG-Annotator:基因组测序数据高效功能注释及其在遗传诊断中的应用[J]. 遗传, 2018, 40(11): 1015-1023 doi:10.16288/j.yczz.18-274
Ying Huang, Qi Liu, Lianjiang Chi, Chengmin Shi, Zhen Wu, Min Hu, Hong Shi, Hua Chen.
精准医疗是随着基因组测序技术的进步和生物信息与大数据科学的交叉应用而发展起来的、以个体化医疗为目的的新型医学理念与医疗模式[1]。进行精准医疗的诊疗依据来自于高通量二代测序(next generation sequencing, NGS)数据。随着测序成本的大幅度降低以及测序通量的不断提升,全基因组测序已经成为研究人类疾病最为快速有效的方法之一。然而,二代测序技术产生的原始数据并不能直接应用于遗传病诊断和临床治疗,还需要一系列后续的生物信息学分析,包括NGS数据基本分析,如碱基和序列质控、序列比对和变异识别,以及基于数据库或算法的变异注释和对注释结果的验证与解读 等[2]。对于精准医疗和遗传诊断来说,在遗传变异注释的基础上还需要进一步给出临床诊断指导和可能的用药指南。
目前常用的遗传变异注释的软件有两类:一类基于功能数据库完成各种变异注释,如ANNOVAR[3];另一类更侧重于对遗传变异的临床意义给出判断,如InterVar[4]。ANNOVAR软件基于Perl语言开发,能够确定SNP或CNV是否导致蛋白质编码区变化以及受影响的氨基酸,并且能够由用户自主选择不同类型的基因定义系统(如RefSeq、UCSC、Ensemble等)。ANNOVAR能独立识别非编码区的特定功能区域的变异,如保守性区域、转录因子结合位点、DNase I高度敏感的活性染色质区域、组蛋白结合修饰区域;或关联其他数据库,如dbSNP (https://www.ncbi. nlm.nih.gov/SNP)和1000 Genome Project[5]等,对遗传变异进行注释。此外,ANNOVAR还能检索用户自定义的基因组位置,识别与孟德尔遗传性疾病相关的可能的基因列表。InterVar是一个基于Python语言开发的生物信息分析软件,应用ACMG-AMP 《解读报告标准指南》,实现对ACMG 28条判读标准中的18条进行自动化评分(其余10条由于需要后续证据输入或者参数调整,如Sanger测序验证结果),将变异分类为“良性”,“可能良性”,“不确定重要性”,“可能致病性”和“致病性”。
虽然这些遗传变异注释软件在NGS数据分析和遗传诊断中成为极重要的分析工具,但是在性能上仍存在不足。一方面,ANNOVAR和InterVar在使用上不够便捷,计算速度慢,对单个个体全基因组突变的分析动辄耗时10多个小时。另一方面,各类对遗传变异的功能注释和临床作用的注释分散在不同软件中,而且缺少复杂疾病,如肿瘤的信息。鉴于此,本研究基于Java语言编写了分析软件BIG- Annotator。该软件运行更高效,集成了对二代测序数据的变异注释的方法与流程,整合了包括肿瘤在内的多种遗传疾病变异数据库,以及2015年美国分子遗传协会(ACMG)协同全美分子病理协会(AMP)共同出台的临床数据解读与报告的标准指南[6]和2017年中国遗传协会遗传咨询分会拟定的《ACMG遗传变异分类标准中文版专家共识指南》,临床可靠性更强。
本文首先介绍了BIG-Annotator的分析流程,并将BIG-Annotator和常用的注释软件进行了性能上的比较,进而以一个成骨发育不全症的家系数据和一例癌症组织全基因组测序的遗传变异数据为例,应用于实际数据的注释分析,鉴定出可信度较高的可疑致病突变,为临床确诊和治疗提供依据。
1 软件设计
1.1 软件分析流程
BIG-Annotator软件的流程和运行过程如图1所示。首先,软件对变异遗传位点进行功能注释,该步骤和ANNOVAR功能一致。软件允许输入未注释的VCF格式文件,或者经过预注释的ANNOVAR输出文件。在功能注释部分,BIG-Annotator采用3种不同的注释方式:基于基因的注释、基于区域的注释和基于过滤的注释。软件提供6种主要的功能:数据库下载、蛋白质序列推断、多种文件格式转换、物种转录本自建、不同类型的注释以及灵活注释流程定制。在实际使用中,BIG-Annotator软件已包含人类基因组已建好的转录本和各种人类疾病数据库。对于其他物种的变异注释,使用者可以根据基因组定义文件或者GFF3文件、GTF文件及序列的FASTA文件构建数据库。图1
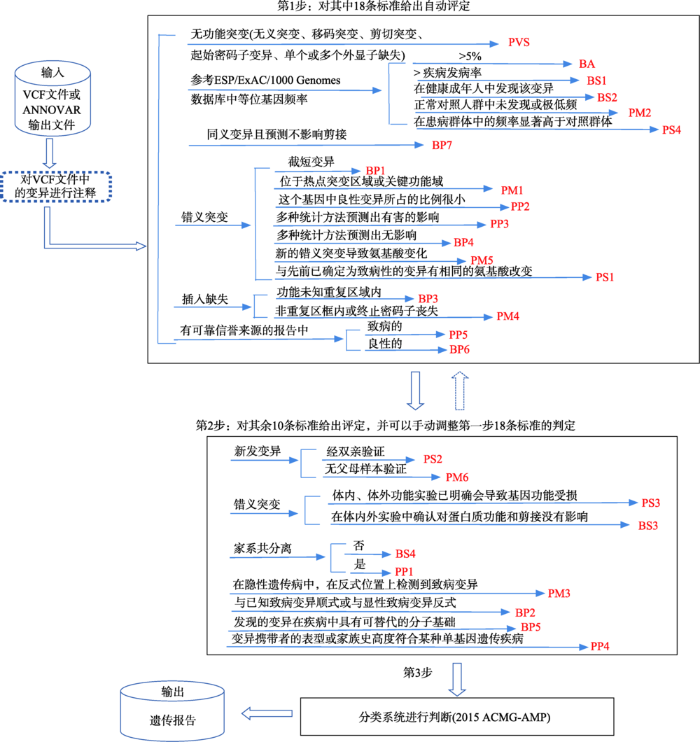 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1BIG-Annotator分析流程
PVS:当一个疾病的致病机制为功能丧失(LOF)时的无功能变异(无义突变、移码突变、经典±1或2的剪接突变、起始密码子变异、单个或多个外显子缺失)。PM1:位于热点突变区域,和/或位于已知无良性变异的关键功能域(如酶的活性位点)。PM4:非重复区框内插入/缺失或终止密码子丧失导致的蛋白质长度变化。PM5:新的错义突变到氨基酸变化,此变异之前未曾报道,但是在同一位点,导致另外一种氨基酸的变异已经确认是致病性的。PP2:对某个基因来说,如果这个基因的错义变异是造成某种疾病的原因,并且这个基因中良性变异所占的比例很小,在这样的基因中所发现的新的错义变异。PP3:多种统计方法预测出该变异会对基因或基因产物造成有害的影响,包括保守性预测、进化预测、剪接位点影响等。PP5:有可靠信誉来源的报告认为该变异为致病的,但证据尚不足以支持进行实验室独立评估。BS2:对于早期完全外显的疾病,在健康成年人中发现该变异(隐性遗传病发现纯合、显性遗传病发现杂合,或者X连锁半合子)。BP1:已知一个疾病的致病原因是由于某基因的截短变异,在此基因中所发现的错义变异。BP2:在显性遗传病中又发现了另一条染色体上同一基因的一个已知致病变异,或者是任意遗传模式遗传病中又发现了同一条染色体上同一基因的一个已知致病变异。BP3:功能未知重复区域内的缺失/插入,同时没有导致基因编码框改变。BP4:多种统计方法预测出该变异会对基因或基因产物无影响,包括保守性预测、进化预测、剪接位点影响等。注意事项:由于做预测时许多生物信息算法使用相同或非常相似的输入,每个算法不应该算作一个独立的标准。BP4在一个任何变异的评估中只能使用一次。BP6:有可靠信誉来源的报告认为该变异为良性的,但证据尚不足以支持进行实验室独立评估。
Fig. 1Pipeline of BIG-Annotator
在功能注释的基础上,BIG-Annotator执行以下步骤:第一步,依据ACMG给出的28条标准对SNP做判定,软件根据功能注释对其中18条标准给出自动评定;第二步,根据用户提供的资料和证据,对其余10条标准给出评定,并可以手动调整第一步18条标准的判定;第三步,根据指南给出的分类联合标准规则,对SNP生成最终分类结果,最后输出报告文件。
根据ACMG 2015年发布的变异位点临床意义判读指南中的28条评估标准,可以将变异位点分为5级:致病变异、疑似致病变异、临床意义不明确变异、疑似良性变异和良性变异。在指南中这28条评估标准具体可以分为7类证据:(1) 1条极强的致病证据PVS;(2) 4条强的致病证据PS1~4;(3) 6条中等的致病证据PM1~6;(4) 5条支持的致病证据PP1~5;(5) 1条极强的良性证据BA,(6) 4条强烈的良性证据BS1~4;(7) 7条支持的良性证据BP1~7。对每个变异根据遗传变异分类联合标准规则进而从5级系统中选择一个分类(例如满足2个以上的PS证据,或者满足1个PS证据和3个以上PM证据,则会判定为致病变异)。
此外,重要的遗传突变往往影响核苷酸的变化和蛋白质的结构功能,因此软件集合了一些常用的疾病遗传变异数据库信息和一些突变功能预测工具。
1.2 数据库和分析工具集成
BIG-Annotator集成了ClinVAR、HGMD和OMIM 3个常用的疾病遗传变异数据库相关信息。ClinVAR (https://www.ncbi.nlm.nih.gov/clinvar/)是一个将遗传变异、临床表型、实证数据以及功能注解与分析4个方面的信息通过专家评审,逐步形成一个遗传变异-临床表型关联的数据库[7]。该数据库整合了NCBI下的dbSNP、dbVar、gene、GTR、MedGen和ACMG等10多个数据库的信息,截止目前收集的条目超过67万,分布在30 181个基因中。数据库每一个条目的信息包括基因、变异、发生频率、表型、临床意义、评审状态及染色体上的位置等。ClinVAR的注释在PVS、PS1、PM5、PM1、PP5和BP6的判定中提供证据。HGMD (http://www.hgmd.cf. ac.uk/ac/index.php)数据库是由英国卡尔地夫医学遗传研究所构建的专门用于收录整理已发表文献中与人类遗传病密切相关的致病位点数据库,每个位点都有参考文献的PMID[8]。HGMD从大约250种期刊中收集突变信息,截至目前收录了9073个基因中的127 200个突变。查询条目详细列出了变异的染色体定位,突变类型列表和相关的表型列表,并将基因内的所有突变定位到HGMD的参考序列上。HGMD对变异的类型做了10多种划分,包括编码区、调控区和剪切区域的点突变,大片段或小片段的插入和缺失,基因重组、序列重复,致病性点突变及移码突变,影响可变剪切和疾病相关的多态性位点等。HGMD根据与疾病的相关程度将变异位点分为6个等级:致病突变、疑似致病突变、有功能证据支持的疾病相关多态性变异、实验证明有功能的多态性变异、疾病相关的多态性变异移码或截短突变。HGMD的注释在PP5和BP6的判定中提供证据。OMIM (http://www.omim.org/)数据库是一个保持更新的免费的人类孟德尔遗传性疾病数据库。该数据库的信息由世界各地的研究者上传,并提供相关文献,由约翰霍普斯金大学医学院的研究人员进行维护。截止2018年6月,数据库共收集了24 576个条目,记录了15 909个基因位点和8973种表型信息,包含有3917个致病性基因变异。现在OMIM关注的表型主要分为单个基因的孟德尔遗传性疾病,具有显著性的单基因致病的复杂疾病,以及染色体缺失和重复综合症。OMIM的每个条目还包括相关染色体基因位置、临床简介、参考文献的摘要、表型描述、相似表型的遗传异质性和疾病的分类等。OMIM的注释在BA、BS1、BS2、PS4和PM2的判定中提供证据。BIG-Annotator中同时还集成了SIFT数据分析工具的功能。SIFT (http://sift.jcvi.org/)是一个突变功能预测工具,基于同源蛋白每一个位点上的氨基酸保守性的评估,给出所提交蛋白质每个氨基酸位点发生突变后的评估分数,分数小于0.05被认为可能影响到蛋白质功能。
BIG-Annotator还集成了本课题组开发的一个肿瘤组织遗传变异的数据库。在对肿瘤组织的遗传变异报告中,除了上述的注释,还增加了响应药物的突变、耐药突变、驱动型突变和继发性突变来描述突变的意义。
2 软件应用实例
2.1 成骨不全症致病性突变研究
运用BIG-Annotator对来自云南的成骨不全症(osteogenesis imperfect, OI)家系进行全基因组遗传变异的注释。该家系样本包括5名OI患者和2名正常个体。对原始数据的测序质量评估后,以GRCh37 (hg19)为参考基因组,使用Samtools v1.0识别所有的单核苷酸多态性位点(SNPs),获得7例样本的原始SNP变异信息对应的VCF文件,每例样包含约 309万个SNP位点。以此VCF文件作为输入文件,运用BIG-Annotator,并结合全基因组多态性分析,进一步确定致病变异位点。首先,从5名患者的SNP数据中提取出共有的等位基因位点(shared allele),约79万个候选SNP变异,所有患者在这些位点携带至少一个相同的等位基因。其次,对这79万个SNPs需要进一步过滤和筛选,下载了103个中国汉族(CHB, 1000 genome project phase III)样本约8000万SNP的VCF数据。从79万个候选SNPs中选取那些在这8000万位点中不存在的,或者最小等位基因频率小于0.01的等位基因位点,作为候选的疾病突变位点。然后,将候选突变位点集合与测序的2个非患者亲属个体的SNPs进行比对,滤除掉在非患者中具有相同等位基因的位点,进一步缩小候选致病位点的范围。最后,通过分析剩下的SNP的注释信息,找到OI的致病突变位点。2.2 癌症组织变异注释
运用BIG-Annotator软件对一例肺鳞癌症患者组织的全基因测序数据进行遗传变异的注释。首先,对样本的全基因组的变异位点进行了注释;然后,对引起蛋白变化的变异进行筛选,包括非同义突变、移码突变等;最后,结合肿瘤注释数据库,挑选出可能致病的位点。3 实例分析结果
3.1 BIG-Annotator运行性能
BIG-Annotator集成了目前常用的二代测序遗传变异注释数据库,以及临床数据解读与报告的标准指南(2015 ACMG-AMP《解读报告标准指南》),并且增加了针对肿瘤组织遗传变异注释的信息。因此,对于遗传疾病的致病性变异分析,BIG-Annotator软件最后会生成各个变异位点的详细风险报告,报告中包含的具体信息如表1所示。Table 1
表1
表1 BIG-Annotator软件生成报告解析
Table 1
| 属性 | 描述 |
|---|---|
| Chr. | 染色体号 |
| Position | 变异位点在染色体上的绝对位置 |
| Ref | 参考基因组碱基型 |
| Alt | 样本基因组碱基型 |
| Gene (refGene) | 基于refGene注释的基因名称,列出该变异所在的基因 |
| Type | 软件关于ACMG的分类判定 |
| Clinvar | ClinVAR注释 |
| ExonicFunc (refGene) | 外显子区的SNV或InDel变异类型 |
| Gene (ensembl) | 基于Ensembl注释的基因号 |
| SNP | dbSNP数据库关于该位点的描述 |
| Transcripts (ensembl) | 基于Ensembl的转录本和变异注释 |
| MAF in ExAC_ALL | ExAC_ALL数据库中的次等位基因频率 |
| MAF in 1000g2014oct | 千人数据库中的次等位基因频率 |
| SIFT_score | SIFT分值,表示该变异对蛋白序列的影响 |
| GERP++_RS | 注释变异位点的保守性的GERP++_RS分值 |
| dbscSNV | 关于splicing区的变异注释数据库,基于不同算法给出Ada和RF分值 |
| OMIM | OMIM数据库注释的疾病号 |
| Interpro_domain | 蛋白序列和蛋白分类数据库InterPro中关于结构域的注释 |
新窗口打开|下载CSV
BIG-Annotator提供多线程运行模式,运行高效,适用于人群队列研究和大样本全基因组关联分析等数据量庞大、时效性要求高的分析需求。在计算性能的比较中,用InterVar软件和BIG-Annotator软件分别对一个30X的全基因组测序数据进行了遗传变异注释。两种软件都是基于以下数据库做出注释:refGene、refGeneMrna、esp6500siv2_all、ALL.sites. 2015_08、avsnp147、dbnsfp33a、clinvar_20170905、exac03、dbscsnv11、dbnsfp31a_interpro、rmsk、ensGene、ensGeneMrna、kgXref、knownGene和knownGeneMrna。两个软件的运行都在一个主频为1009.734 MHz,64核的服务器上进行。二者都注释了全基因组共4 451 891个变异位点,InterVar耗时930 min,而BIG-Annotator运行耗时56 min (表2)。InterVar每秒钟可注释近80个位点,而BIG-Annotator每秒钟可注释1325个位点(表2)。BIG-Annotator的运行时间为InterVar的6%,而注释速度超过后者的16倍,更为高效,能更好的满足大样本数据分析的需求。
Table 2
表2
表2 BIG-Annotator和InterVar运行性能比较
Table 2
| 软件 | 运行耗时(min) | 注释速率(variants/s) |
|---|---|---|
| InterVar | 930 | 79.78 |
| BIG-Annotator | 56 | 1324.97 |
新窗口打开|下载CSV
3.2 成骨不全症致病性突变的鉴定
成骨不全症是一组罕见的伴有全身性结缔组织异常的遗传性疾病[10],在临床上OI亚型多达15种,不同亚型患者可能是由不同的基因突变导致,但在表型上是呈连续型变化,典型表现为身材矮小,骨骼发育异常,关节松弛,有多发性骨折[11],因此仅从临床症状和体征上很难对患者进行疾病亚型的精确诊断与治疗。本研究对来自云南地区的一个成骨不全症家系的全基因组测序数据进行了分析和注释。经过过滤和筛选,找到了33个可能的候选致病位点。这些候选位点都满足以下特点:在患者样本中具有一致性,在患者和非患者样本的对比中具有特异性,在正常人群中最小等位基因频率具有极小性。通过分析候选致病位点SNP注释,挑选出所有的位于基因外显子区段上的非同义突变,最终找到23个致病变异的候选SNP位点(表3)。运用BIG-Annotator对这23个SNP位点的注释和判定以及用DAVID工具对这些候选基因功能注释的结果表明致病变异基因功能和OI相关。在该家系测序的16名成员中,最终确定SNP位点rs66612022 (COL1A2: p.Gly328Ser)上AG的基因型在患者中是稳定发生的致病性突变(表4),该位点位于常染色体上且表现显性遗传模式(图2)。BIG-Annotator对该SNP位点生成的报告如表5所示。这一发现为该家系中OI患者的确诊和个性化治疗具有指导意义。
Table 3
表3
表3 成骨不全症相关的23个候选SNP位点及其对应基因
Table 3
| 基因 | SNP | |||
|---|---|---|---|---|
| 错义突变 | 终止密码突变 | 未知 | ||
| AGAP3 | CHR7_150783917_T2G_L2R | - | - | |
| CD22 | rs182604067 | - | - | |
| COL1A2 | rs66612022 | - | - | |
| DNAJC21 | rs77129269 | - | - | |
| DNASE1L3 | rs12491947 | - | - | |
| DNTT | CHR10_98079087_G2T_K2N | - | - | |
| HMMR | CHR5_162898459_G2C_E2Q | - | - | |
| HSPA6 | rs41297718 | - | - | |
| IL17RD | rs140930246 | - | - | |
| KCNJ12 | rs4985866 | - | - | |
| KCNJ18 | rs4985866 | - | - | |
| MCM7 | CHR7_99693641_C2T_A2T | - | - | |
| PDPR | rs10852462 | - | - | |
| PRIM2 | - | - | rs927192 | |
| PTPRG | rs372086949 | - | - | |
| RNF19B | rs113840389 | - | - | |
| SORBS1 | - | CHR10_97096994_G2A_R2X | - | |
| TACC2 | rs140280635 | - | - | |
| TRPS1 | CHR8_116427025_T2G_Q2H | - | - | |
| XKR9 | - | rs115507207 | - | |
| ZAN | - | - | rs191137 | |
| - | - | rs80067406 | ||
| ZHX2 | CHR8_123965019_C2G_I2M | - | - | |
新窗口打开|下载CSV
Table 4
表4
表4 COL1A2基因外显子片段上发现的变异列表
Table 4
| ID | SNP编号及其最小等位基因频率 | |||||
|---|---|---|---|---|---|---|
| rs1801182 (C/0.3398) | rs1800222 (C/0.3835) | rs66612022 (A/na) | rs42524 (G/0.0680) | rs1800238 (T/0.3738) | rs1800248 (T/0.1165) | |
| Ind19 | CT | - | - | GG | GT | - |
| Ind20 | CC | - | GA | GG | TT | - |
| Ind21 | CC | CC | GA | GG | TT | - |
| Ind22 | - | - | - | CG | GT | - |
| Ind23 | - | CT | GA | GG | GT | - |
| Ind24 | CT | CT | GA | GG | GT | CT |
| Ind25 | CT | CT | GA | CG | GT | - |
新窗口打开|下载CSV
图2
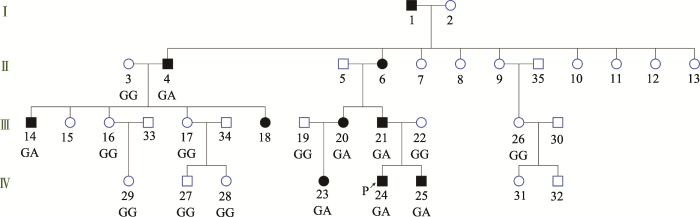 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2COL1A2基因rs66612022位点基因型在家系上的分布
I~IV表示家系中的代数,圆形表示女性,正方形表示男性,加黑个体表示为患者,P表示该个体为家系中的先证者。
Fig. 2Genotypes of rs66612022 of COL1A2 in the studied pedigree
Table 5
表5
表5 BIG-Annotator生成的有关rs66612022的报告
Table 5
| 属性 | 描述 | 属性 | 描述 |
|---|---|---|---|
| Chr. | 7 | SNP | rs66612022 |
| Position | 94039080 | Transcripts (ensembl) | ENST00000297268 c.982G>A p.G328S |
| Ref | G | MAF in ExAC_ALL | . (. means absent) |
| Alt | A | MAF in 1000g2014oct | .(. means absent) |
| Gene (refGene) | COL1A2 | SIFT_score | 0 |
| Type | Pathogenic | GERP++_RS | 5.58 |
| Clinvar | UNK | dbscSNV | . (. means absent) |
| ExonicFunc (refGene) | Nonsynonymous SNV | OMIM | 120160 |
| Gene (ensembl) | ENSG00000164692 | Interpro_domain | . (. means absent) |
新窗口打开|下载CSV
3.3 癌症组织变异注释分析结果
在对一例肺鳞癌症患者组织的全基因测序数据遗传变异的注释中,BIG-Annotator分析确定了2个可能致病的位点(表6)。这2个位点分别位于2号染色体79523268碱基位点和X染色体123885987碱基位点上,在分析个体中均以杂合状态存在,都是由于碱基插入导致移码突变,并分别引起了CTNNA2基因和XIAP基因的编码蛋白质的变化,从而影响代谢通路。在数据库的注释中,CTNNA2基因上的该位点突变与胃癌,子宫内膜癌相关,XIAP基因上位点的突变与小细胞肺癌相关。目前,尚无对这2个位点及其相关基因的靶向药物(表6)。Table 6
表6
表6 肿瘤组织全基因组测序变异注释报告
Table 6
| 染色体 | 碱基位置 | 突变 | 突变种类 | 影响基因 | 变异类型 | 蛋白变化 | 临床意义 | 相关疾病 | 影响方式 | 靶向药物 |
|---|---|---|---|---|---|---|---|---|---|---|
| Chr.2 | 79523268 | T→TAG TTAAAA ATAATA TACAAA TTTATA | 杂合 | CTNNA2 | Frame shift Insertion | CTNNA2:NM_001282 598:exon1:c.56_57 insAGTTAAAAATAATA TACAAATTTATA:p.F 19fs | 可能致病 | 胃癌 子宫内膜癌 | 癌症上的 通路 | 无 |
| Chr.X | 123885987 | G→GAT | 杂合 | XIAP | Frame shift Insertion | XIAP:NM_001167:exo n2:c.325_326insAT:p. G109fs,XIAP:NM_001 204401:exon2:c.325_ 326insAT:p.G109fs | 可能致病 | 小细胞 肺癌 | 癌症上的 通路 | 无 |
新窗口打开|下载CSV
4 讨 论
本研究基于JAVA语言开发编写了软件BIG- Annotator,且提供了多线程的选项。与现有的同类软件工具相比,BIG-Annotator在运行性能上有很大提高,时间缩短了10多倍,因此能满足大样本二代测序数据注释分析的需求。BIG-Annotator既可作为一个二代测序变异注释工具,也可作为一个临床诊断辅助工具,不仅可以对遗传变异给出注释,也提供遗传变异的临床意义和解释,以及对肿瘤的遗传变异给出初步的功能性判断。目前,还鲜有专门针对肿瘤组织的遗传变异的注释解析工具。由于肿瘤细胞突变多为体细胞突变,其意义与遗传性突变的意义(如致病性突变)不同[9],因此在常见遗传病领域的规范化指导和报告难以直接使用。BIG-Annotator集成了课题组归纳收集的肿瘤变异数据库,可以对肿瘤组织变异给出初步的遗传注释。当然,BIG-Annotator输出结果的可靠性依赖于现有的多个公开的变异位点数据库,在临床实践应用上也受其局限。目前还缺少非常全面的信息库资源,能够保证对每个遗传变异的28条标准评估实现自动化判定。因此,在BIG-Anonotaor分析流程中允许用户根据自己的主观判断修改某些标准的评估结果。此外,一个可以改进的地方在于,现在只是按照2015 ACMG-AMP的28条指南来给出临床意义,没有借助一些统计学方法对这28条标准做任何的综合性分析,这可能还需要未来能收集到大量实例的软件分析结果和相应的临床验证的比较。最后,对于肿瘤组织的遗传变异的临床解析,目前各种肿瘤组织的变异的临床意义的信息很少,尤其是对耐药性和驱动性突变的鉴定,这方面的数据库信息还有待补充。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
[Dissertation].
URL [本文引用: 1]
URLPMID:2938201 [本文引用: 1]

Abstract. High-throughput sequencing platforms are generating massive amounts of genetic variation data for diverse genomes, but it remains a challenge to pinp
URLPMID:28132688 [本文引用: 1]
In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published updated standards and guidelines for the clinical interpretation of sequence variants with respect to human diseases on the basis of 28 criteria. However, variability between individual interpreters can be extensive because of reasons such as the different understandings of these guidelines and the lack of standard algorithms for implementing them, yet computational tools for semi-automated variant interpretation are not available. To address these problems, we propose a suite of methods for implementing these criteria and have developed a tool called InterVar to help human reviewers interpret the clinical significance of variants. InterVar can take a pre-annotated or VCF file as input and generate automated interpretation on 18 criteria. Furthermore, we have developed a companion web server, wInterVar, to enable user-friendly variant interpretation with an automated interpretation step and a manual adjustment step. These tools are especially useful for addressing severe congenital or very early-onset developmental disorders with high penetrance. Using results from a few published sequencing studies, we demonstrate the utility of InterVar in significantly reducing the time to interpret the clinical significance of sequence variants.
URL [本文引用: 1]
URLPMID:25356965 [本文引用: 1]
These recommendations are designed primarily as an educational resource for medical geneticists and other health-care providers to help them provide quality medical genetics services. Adherence to these recommendations does not necessarily ensure a successful medical outcome. These recommendations should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, geneticists and other clinicians should apply their own professional judgment to the specific clinical circumstances presented by the individual patient or specimen. It may be prudent, however, to document in the patient's record the rationale for any significant deviation from these recommendations.
URLPMID:24234437 [本文引用: 1]
ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) provides a freely available archive of reports of relationships among medically important variants and phenotypes. ClinVar accessions submissions reporting human variation, interpretations of the relationship of that variation to human health and the evidence supporting each interpretation. The database is tightly coupled with dbSNP and dbVar, which maintain information about the location of variation on human assemblies. ClinVar is also based on the phenotypic descriptions maintained in MedGen (http://www.ncbi.nlm.nih.gov/medgen). Each ClinVar record represents the submitter, the variation and the phenotype, i.e. the unit that is assigned an accession of the format SCV000000000.0. The submitter can update the submission at any time, in which case a new version is assigned. To facilitate evaluation of the medical importance of each variant, ClinVar aggregates submissions with the same variation/phenotype combination, adds value from other NCBI databases, assigns a distinct accession of the format RCV000000000.0 and reports if there are conflicting clinical interpretations. Data in ClinVar are available in multiple formats, including html, download as XML, VCF or tab-delimited subsets. Data from ClinVar are provided as annotation tracks on genomic RefSeqs and are used in tools such as Variation Reporter (http://www.ncbi.nlm.nih.gov/variation/tools/reporter), which reports what is known about variation based on user-supplied locations.
[本文引用: 1]
URLPMID:29618041 [本文引用: 1]
Abstract Background: In germline genetic testing, variants from understudied ancestries have been disproportionately classified as being of uncertain significance. We hypothesized that the rate of variant reclassification likewise differs by ancestry. Methods: Nonbenign variants in actionable genes were collected from consenting subjects undergoing genetic testing at two Southern California sites from September 1996 through December 2016. Variant reclassifications were recorded as they were received, until February 2017 or reclassification to benign. Excluding duplicate variants (same ancestry, laboratory, classification), generalized linear models for the hereditary breast cancer genes (BRCA1/2) and other variants investigated whether rate of reclassification differed for seven categories of ancestry compared with non-Hispanic European. Models took into account laboratory, year, gene, sex, and current classification (handled as a time-dependent covariate) and were adjusted for multiple hypothesis testing. Results: Among 1483 nonbenign variants, 693 (46.7%) involved BRCA1/2. Overall, 268 (18.1%) variants were reclassified at least once. Few (9.7%) reclassified variants underwent a net upgrade in pathogenicity. For BRCA1/2 variants, reclassification rates varied by ancestry and increased over time, more steeply for ancestries with lower initial rates (African, Ashkenazi, Chinese) than for ancestries whose initial rates were high (Middle Eastern) or similar to non-Hispanic European (non-Chinese Asian, Native American, Hispanic). In contrast, reclassification rates of non-BRCA1/2 variants did not vary over time but were elevated for most minority ancestries except non-Chinese Asian and Native American. Conclusions: For nonbenign variants in cancer-related genes, the rates at which reclassifications are issued vary by ancestry in ways that differ between BRCA1/2 and other genes.
URLPMID:2940745 [本文引用: 1]
Osteogenesis imperfecta (OI) is the most common bone genetic disorder and it is characterized by bone brittleness and various degrees of growth disorder. Clinical severity varies widely; nowadays eight types are distinguished and two new forms have been recently described although not yet classified. The approach to such a variable and heterogeneous disease should be global and therefore multidisciplinary. For simplicity, the objectives of treatment can be reduced to three typical situations: the lethal perinatal form (type II), in which the problem is survival at birth; the severe and moderate forms (types III–IX), in which the objective is ‘autonomy’; and the mild form (type I), in which the aim is to reach ‘normal life’. Three types of treatment are available: non-surgical management (physical therapy, rehabilitation, bracing and splinting), surgical management (intramedullary rod positioning, spinal and basilar impression surgery) and medical-pharmacological management (drugs to increase the strength of bone and decrease the number of fractures as bisphosphonates or growth hormone, depending on the type of OI). Suggestions and guidelines for a therapeutic approach are indicated and updated with the most recent findings in OI diagnosis and treatment.
URLPMID:17875077 [本文引用: 1]
Background: Osteogenesis imperfecta (OI) is an autosomal dominant disorder of connective tissue characterized by bone fragility and low bone mass. COL1A1 and COL1A2 genes are very large and have been rarely analyzed systematically in Japan. The aim of this project was to develop an effective and convenient method of finding mutations in the COL1A1 and COL1A2 gene by using denaturing high-performance liquid chromatography (DHPLC).

{kind=link}
{kind=link}
{kind=link}
{kind=link}