 ,中国科学院北京基因组研究所,中国科学院基因组科学与信息重点实验室,北京 100101
,中国科学院北京基因组研究所,中国科学院基因组科学与信息重点实验室,北京 100101Advances of sequencing and assembling technologies for complex genomes
Shenghan Gao, Haiying Yu, Shuangyang Wu, Sen Wang, Jianing Geng, Yingfeng Luo, Songnian Hu,CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China通讯作者:
编委: 赵方庆
收稿日期:2018-09-10修回日期:2018-10-29网络出版日期:2018-11-20
Received:2018-09-10Revised:2018-10-29Online:2018-11-20
作者简介 About authors
高胜寒,博士,助理研究员,研究方向:基因组结构与稳定性E-mail:gaoshh@big.ac.cn。

摘要
关键词:
Abstract
Keywords:
PDF (1120KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
高胜寒, 禹海英, 吴双阳, 王森, 耿佳宁, 骆迎峰, 胡松年. 复杂基因组测序技术研究进展[J]. 遗传, 2018, 40(11): 944-963 doi:10.16288/j.yczz.18-255
Shenghan Gao, Haiying Yu, Shuangyang Wu, Sen Wang, Jianing Geng, Yingfeng Luo, Songnian Hu.
基因组是所有生命遗传物质的集合,为生命行使生物学功能提供指导,基因组中的碱基序列信息记录着生命进化的历史。因而,基因组序列的完整解析可极大促进基因功能研究,更为物种相互作用和基因组比较等生命科学研究提供基础信息。大多数生物的基因组均由A、T、G、C 4种碱基组成,其组合顺序和总长度各不相同,如何快速和低成本地获取基因组序列一直是基因组学领域的重心。由于测序技术或测序仪器的内在缺陷,测序读长仍小于基因组长度,所以除少数基因组较小的DNA病毒外,绝大多数基因组仍无法通过一次测序直接获得全部的序列信息,需要通过高覆盖度测序和序列组装获得完整的基因组信息。而复杂基因组指的是无法使用常规测序和组装手段直接解析的一类基因组,通常是指包含高比例重复序列、高杂合度、存在难以消除的异源DNA污染的基因组。本文从复杂基因组的特点和来源入手,结合实例介绍了复杂基因组测序的技术解决途径和发展历程,为制订合适的复杂基因组测序策略提供参考。
1 复杂基因组概念
复杂基因组是根据重复序列比例和杂合度高低来定义的。通常杂合率大于0.8%、重复序列比例大于60%就称为复杂基因组。基因组的复杂度主要来源于下面几个方面。1.1 重复序列
复杂基因组组装一直是一个难题,很大原因是由于重复序列含量高并且分布在基因组的不同位置,往往造成组装的基因组偏小于实际的基因组大小。重复序列是基因组中重复出现的序列。重复序列在各物种中的比例从病毒(小于1%)、细菌(啤酒酵母:3.4%)到真核生物(人:47%;玉米:77%)逐步升高。在对44种植物和68种脊椎动物分析其全基因组重复水平和基因组大小关系时发现,重复序列与脊椎动物的基因组大小关联性更高,植物基因组的重复序列通常比脊椎动物的更高(图1)[1]。图1
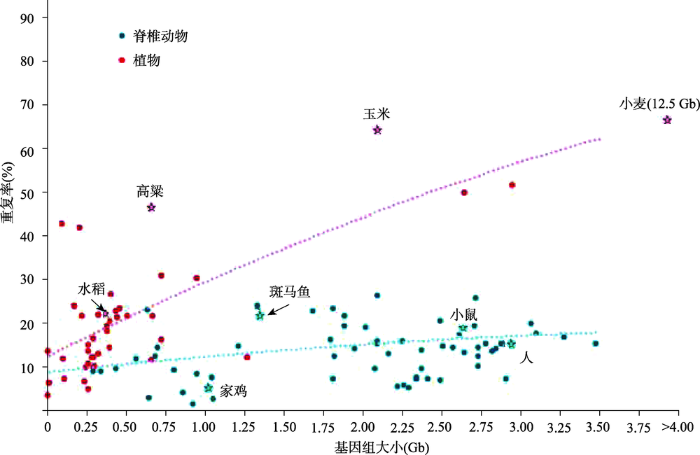 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因组大小和重复序列的相关性
根据文献[1]修改绘制。
Fig. 1The correlation between repetitive sequences and genome size
根据重复序列结构、位置及功能方面的差别,可分为散在重复序列(interspersed repeat)、串联重复序列(tandem repeat)和片段重复序列(segmental duplication)。散在重复序列比较均匀地分布在基因组中,包含长散在重复(long interspersed nuclear elements, LINE)、短散在重复(short interspersed nuclear elements, SINE)、类反转录病毒转座子(long terminal repeat- retrotransposon, LTR-RT)和DNA转座子(DNA transposon)。其中,LTR-RT是植物中分布最为广泛的一类转座子,是基因组重复区域的主要成分,如橡胶(Hevea brasiliensis)基因组中71.2%为重复序列,其中LTR-RT占主要部分,它们的大规模复制插入是橡胶基因组明显大于其他近缘物种如木薯(Manihot esculenta)、杨树(Populus trichocarpa)、蓖麻(Ricinus communis)等的主要原因[2];墨西哥蝾螈(Ambystoma mexicanum)基因组的重复序列为65.6%,LTR-RT是主要成分,且几乎都分布在Contig序列的末端,给组装带来巨大挑战[3]。
串联重复是由1~500个碱基的重复单元构成,一般在基因组中重复几十到几百万次,包含简单重复(simple sequence repeat)和卫星DNA。如人类基因组的着丝粒周边区域以及染色体近端短臂含有卫星DNA和串联重复序列;一些遗传调控区域序列如核小体结合单元、甲基化位点等都与串联重复有关;产生茶叶风味的次生代谢产物合成酶基因在基因组上发生拷贝数扩增主要是由串联重复产生[4]。
1.2 杂合度
杂合度对基因组组装产生很大影响,以二倍体基因组为例,通常只组装出一套染色体,对于杂合度高的区域,会将两条染色单体都组装出来,从而造成组装的基因组偏大于实际的基因组大小。对秀丽线虫(Caenorhabditis elegans)模拟不同杂合度的数据进行组装,当杂合度升高时,各组装软件的Contig N50指标都明显下降(图2)[5]。相比于动物基因组而言,植物基因组更加复杂,很多植物因远源杂交、自交不亲和等因素,具有基因组杂合高、倍性高等特征,加上基因组本身比较大,这些都增加了基因组组装的难度。如自交不亲和的茶树(Camellia sinensis)基因组由于种间频繁杂交导致杂合度高达2.8%[4];异源多倍体的陆地棉(Gossypium hirsutum)[6]、油菜(Brassica napus)[7]等物种需要借助二代和三代测序结合进行组装。图2
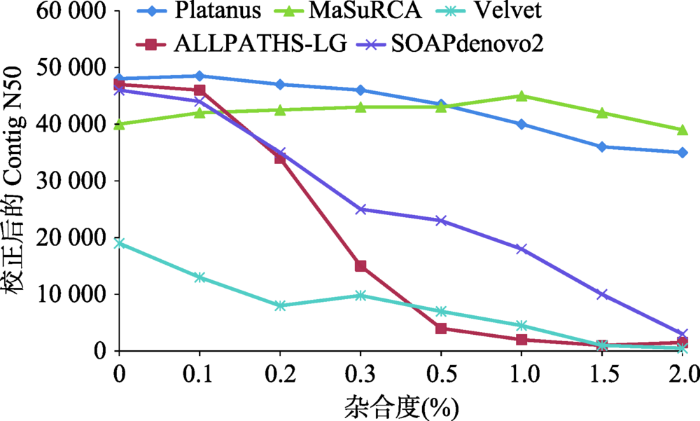 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2杂合度对不同算法组装指标的影响
根据文献[5]修改绘制。
Fig. 2The impact of heterozygosity on genome assembly using different algorithms
1.3 极端GC含量
尽管二代测序(next-generation sequencing, NGS)具有很大优势,但是极端碱基组成一直是造成NGS数据组装具有挑战性的因素之一。在PCR扩增、桥式簇扩增以及测序等NGS数据产生过程中(主要是Illumina测序平台),由于GC偏好性使得基因组中低GC或高GC区域的测序读长(Reads)覆盖度不均一。因为Reads覆盖度是许多组装软件的关键参数,这种极端的GC区域会导致基因组组装碎片化,使其完整性降低[8]。如恶性疟原虫(Plasmodium falciparum)的基因组平均GC含量低于25%,导致许多低GC区域的Reads覆盖度很少甚至没有Reads覆盖[9];脐形紫菜(Porphyra umbilicalis)基因组中GC含量高达65.8%,利用二代测序几乎无法进行组装[10]。1.4 基因组污染
基因组中存在污染也是造成其复杂性的一个因素。基因组污染一方面可能来源于DNA提取或扩增过程,如DNA提取试剂盒、化学试剂和实验室环境中的杂菌很容易造成污染;另一方面可能来源于物种间相互作用/共生/共栖的生活环境。对于藻类来说,共生微生物较多,如紫菜基因组,即使经过抗生素处理,提取出的DNA中依然有50%的测序数据来自共生微生物和其他真核生物污染[10]。最近,水熊虫(Hypsibius dujardini)基因组就因为污染序列事件成为各方争论的热点,最终Koutsovoulos等[11]根据 测序Reads的覆盖度和GC含量不均一性证明了Boothby 等[12]发表的版本组装结果中存在大量的(30%)来自细菌的污染序列。2 高复杂基因组测序技术解决途径
2.1 针对高复杂基因组测序的实验方法
复杂基因组测序领域一直是基因组学的重要关注点。在第二代高通量测序之前,测序费用高且通量低,对获取高重复序列物种的全基因组序列难度太大或费用太高的物种,科研人员主要尝试提高“有用”基因组区域的比例,如利用杂交退火法提高重复序列较少的基因区域的相对丰度[13],采用甲基化碱基致死大肠杆菌突变体提高甲基化程度较低基因区及其邻近调控区在质粒克隆中的比例[14],或通过外显子芯片杂交获取近缘物种基因区域[15]。实验手段“简化”复杂基因组是目前一个重要的研究方向,成功应用案例包括构建单倍体品系[16]、染色体分离[17, 18]以及低识别位点限制性内切酶完全酶切[19]等。但以上方法都有其技术局限性:并不是每个物种都可以获得稳定的单倍体品系;染色体分离技术要求染色体完整且各条染色体具有不同长度或标记信息,目标样品较少,而微量样品扩增可能引入偏差;酶切法无法有效分离不同来源的同源染色体。
2.2 现有高通量测序技术的特点
目前,使用最多的高通量测序手段分为两种主要类型:(1) 短读长高通量测序,主要包括Illumina的HiSeq和10X Genomics测序平台;(2) 长读长单分子测序,主要包括PacBio的SMRT平台和Oxford Nanopore Technologies的MinION平台。此外,还包括一系列辅助分子标记测序系统,如BioNano的Saphyr光学酶切图谱系统等,可用于辅助复杂基因组的组装,并对组装的准确性进行评估。从头测序组装(de novo assembly)复杂基因组的关键之一是获得较长的读长序列,以降低重复序列或高相似基因组片段对基因组组装的影响。在第二代高通量测序仪问世之初,Illumina/Solexa读长不足35 bp,基本难以实现对基因组进行有效的组装,对于复杂基因组更是“束手无策”。为了克服这一困难,当时发展起来的酶切“延伸”[20]和局部组装[21]技术对基因组的组装具有明显的促进作用。经过多年发展,Illumina的HiSeq平台目前的常见读长为2×100 bp (Illumina HiSeq 2000)至2×250 bp (Illumina MiSeq),虽然较早期有很大提升,但仍然难以满足复杂基因组组装的需求。尽管Illumina测序平台读长较短,但与长读长技术相比,其序列的准确性具有明显的优势。因此,目前针对复杂基因组组装,需要依靠长读长数据与二代Illumina短读长数据相结合的方法来实现,以充分利用两者之间的互补优势。
Pacific Biosciences (PacBio)公司于2010年发布的基于单分子实时技术(single molecule real time, SMRT)的测序仪PacBio RS,目前已经更新到RSII和Sequel版本。PacBio测序反应是在SMRT Cell反应管中进行,每个测序芯片(Cell)都有一个厚度为100 nm的金属小芯片,其上面固定着大约15万个零模波导孔(zero-mode wave guide, ZMW)。ZMW是测序技术的核心[22]。DNA 聚合酶以共价结合的方式锚定在ZMW底部,用来结合单链DNA分子模板。PacBio测序得到的序列是真实的单分子DNA序列,且其读长较长,典型情况下可达到平均20~40 kb,与Illumina的最长250 bp相比具有明显的优势,但测序的准确度相对较低,平均准确度约为80%左右。PacBio产生的数据更适用于复杂基因组的组装,但需要先进行复杂的校正工作,才能达到组装要求。目前,PacBio是用于复杂基因组组装的主流方法。
Oxford Nanopore Technologies Limited公司在2012 年推出第一款基于纳米孔测序技术的测序仪。目前测序平台包括MinION、GridION X5、PromethION和SmidgION。其中SmidgION是迄今为止体积最小的测序设备,可在任何地点与智能手机配套使用。纳米孔测序技术的测序原理是:在纳米孔两边加上一定的电压,在电势的作用下,DNA电泳通过纳米孔,由于4种核苷酸的电离水平和空间结构不同,通过纳米孔时电流强度不同,根据电流强度准确判断碱基种类[23]。纳米孔测序的读长可达数百kb,在解决复杂基因组组装时,与PacBio相比具有更大的优势。
10X Genomics平台本质上是一种改进的二代Illumina测序技术,其核心是一种条码标记(barcoding)技术,根据Barcode信息组装短Reads从而获得跨度为几十kb到几百kb的连锁读长(linked reads),进而将基因组组装划分成数万乃至数百万个局部组装,再将局部组装进一步组装到全基因组。该技术可显著降低复杂度,获得更完整的组装结果,因此也十分适用于复杂基因组的组装。
对于植物等复杂物种基因组的组装项目,现有的二代和三代测序仍然难以准确跨过重复序列区域,而光学图谱技术的出现可以有效克服这一基因组组装难题[24, 25]。基因组光学图谱是指利用荧光标记酶切技术在全基因水平上构建限制性内切酶酶切图谱。BioNano Genomics公司分别在2014年和2017年推出了Irys分析平台和Saphyr分析平台。该平台利用限制性内切酶对DNA分子进行酶切,并利用DNA聚合酶和不同荧光标记的核苷酸合成带有荧光标记的核酸链;再利用微流控装置的毛细管电泳将DNA分子线性化;当DNA分子通过纳米孔的时候进行高分辨率荧光成像,从而生成酶切图谱。利用BioNano技术和三代PacBio/Nanopore相结合,可有效进行基因组从头测序组装,解决复杂基因组的组装难题。
Hi-C (high-throughput chromosome conformation capture)测序是一种以生物细胞核(动物/植物)为研究对象,研究染色质之间相互作用的技术。该技术可有效进行染色体构象捕获,从而获得基因组序列信息及其在基因组中的位置信息[26]。其处理过程:首先利用染色质与甲醛等交联剂进行交联反应;再利用HindⅢ、MboⅠ等限制性内切酶进行酶切反应而获得粘性末端,并加入生物素标记;最后进行解交联反应,利用带有标记的产物进行建库测序。由于Hi-C数据可以准确区分细胞核中的不同染色体,因此对于基因组组装来说,该技术和三代测序技术结合可以高效进行Scaffold乃至染色体级别基因组的构建[27]。
2.3 复杂基因组组装难点与解决方案
高重复和高杂合对于基因组组装的影响,在组装结果中表现为两个相反的特性:由于在组装过程中会将相似的重复序列组装到一起,因此重复序列会导致基因组组装大小的收缩(小于预估的实际基因组大小);对于高杂合来说,染色体组的杂合序列之间存在一定的序列差异,因此在组装的时候会被分别独立组装,从而导致基因组组装大小的扩张(大于预估的实际基因组大小)。因此,对于具有高重复和高杂合成分的基因组来说,对其进行正确组装具有较大的挑战性。目前常用于基因组组装的两种算法DBG (De brujin Graph)[28]和OLC (overlap-layout-consensus)[29],虽然在原理和速度上具有较大的差别,但其本质都是寻找特定序列的最佳连续匹配,因此在处理高重复和高杂合时,都存在上述的弱点。相对而言,由于DBG算法是通过K-mer的精确匹配进行组装,可以区分细微的序列差别,因此在一定程度上可以区分不同的重复序列;但对于本身具有较大差异的杂合区段来说,DBG会将其组装成独立的序列,因此DBG对于高重复组装具有相对的优势,但对于高杂合表现则不佳。而OLC算法在寻找最佳比对时,允许一定的错配,因此在一定程度上可将杂合区段合并组装,但对于重复序列来说,由于大部分重复序列上的差别小于OLC允许的错配,重复序列可能被错误合并组装在一起,因此OLC算法对于高杂合具有相对的优势。如果将两种算法适当地结合在一起,则可以在一定程度上解决由于高重复和高杂合引起的组装难题。
无论使用哪一种组装方法,高重复与高杂合在本质上是无法被完美解决的,只存在解决这两种问题的相对方法。如前所述,基因组的组装是通过寻找测序数据之间的最佳比对来实现的,但重复序列是高度相似的,杂合区段是存在差异的,因此总会发生将序列错误合并组装,将本应合并的序列错误分离。为了尽可能降低发生错误的可能性,就需要在组装时寻找特异性的最佳比对(unique alignment)。由于比对结果的可信度(得分)与比对的长度成正比,越长的序列,得到的比对越长,得到最佳比对的可能性也越高,因此就要求用于组装的测序数据尽可能长。对于高重复基因组组装来说,最理想的情况就是测序数据将高度重复序列完全包含在读长中间,即形成特异-重复-特异(unique-repeat-unique)序列结构,方能保证重复序列被放置到正确的位置[30];对于高杂合基因组来说情况类似,最理想的情况就是将杂合区段完全包含在读长中间,形成特异-杂合-特异(unique-hetero-unique)序列结构,才能将杂合区段正确识别出来,避免杂合区段被重复组装的问题[5]。
然而,在实际的测序和组装过程中,最理想的情况是不易获得的。在测序平台方面,目前用于基因组组装的测序平台主要有以Illumina HiSeq为代表的二代平台和以PacBio为代表的三代平台。这两种平台具有各自不同的特点,前者测序的精度较高(错误率<1%),但测序较短(100~300 bp),而后者测序片段很长(平均可达20 kb),但错误率很高(一般>15%)。由于基因组中STR序列和LTR序列的存在,二代测序平台得到的数据无法将重复序列和高杂合区域跨过,导致组装时出现大量的分支(branch)和环(loop),使组装结果碎片化;而三代测序平台得到的数据虽然有助于跨过这些区域,但因其错误率较高,不仅会引入组装错误,同时在一些极端情况下,由于无法和其他序列正确比对,导致组装无法进行(事实上等同于人为引入了杂合)。此外,尽管长片段测序能够帮助解决一部分高重复和高杂合组装的问题,但并不是全部。根据基因组本身特性的差异,即使在PacBio平台上得到长度分布完全一样的测序数据,能够跨过重复或者杂合区域的比例也仅在30%~60%之间浮动。因此,根据基因组本身的特征对测序策略进行优化是十分必要的过程。
针对于不同测序数据的特点,目前产生了多种不同的组装方法,以利用测序数据的特点来尽可能降低高重复和高杂合对于基因组组装的影响,得到尽可能连续的基因组序列。
在产生较为有效的三代数据组装的算法之前,已经有研究对于仅使用二代数据进行高重复和高杂合基因组组装进行了尝试,结果比传统方法具有十分显著的提升。以Platanus为例,其组装策略分为Contig生成、Scaffold构建和空洞填补(Gapfill) 3个部分(图3)[5]。该软件主要基于DBG算法,并根据DBG算法的特点,重复与杂合成分会在DBG中形成“接合”(Junction)与“鼓泡”(Bubble),而测序的错误会形成“断头”(Tip)。进行组装优化的目的就是将Junction、Bubble和Tip尽可能去除,形成线性图(straight)。在Contig构建时,Platanus采取了3种创新的策略:(1) 通过拟合泊松分布,将低频“拐点”之下的K-mer全部去除,从而有效减少了Tips的数量;(2) 将Reads直接比对定位到Junction节点,通过定位的质量确定Junction的走向,而不是使用K-mer深度,可以解决连续Junction的组合问题;(3) 使用多K-mer延伸策略,从较小的K-mer向较大的K-mer进行延伸,既可以在初始构建DBG时避免杂合造成较低的K-mer深度,又可以有效利用测序数据的长度。通过这一策略,杂合形成的Bubble被有效地鉴定并被分离出来(注意不是去除)。而在Scaffold阶段,Platanus将大片段Mate-pair文库统一定位到Contig和去掉的Bubble上,将带有杂合的序列作为整体考虑,从而有效利用构建形成Scaffold时所包含的Contig两端的连接数。在后续过程中,Platanus同样会识别Scaffold图中可能存在的杂合,将覆盖度较低且内部不含Bubble的Scaffold分支识别为杂合从而去除,带有Bubble的进行保留(默认二倍体基因组中不应存在多重杂合)。最后,在空洞填补阶段,将Reads重新定位到组装成的Scaffold上,将在空洞(Gap)附近的Reads筛选出来,进行局部重新组装,将空洞进行填补。综上所述,Platanus进行高杂合基因组的核心策略是将含有杂合的序列鉴定出来并进行合并去除,相对于传统的方法,可以有效避免在分支处将序列切断,因此可以得到更加连续的组装结果。
图3
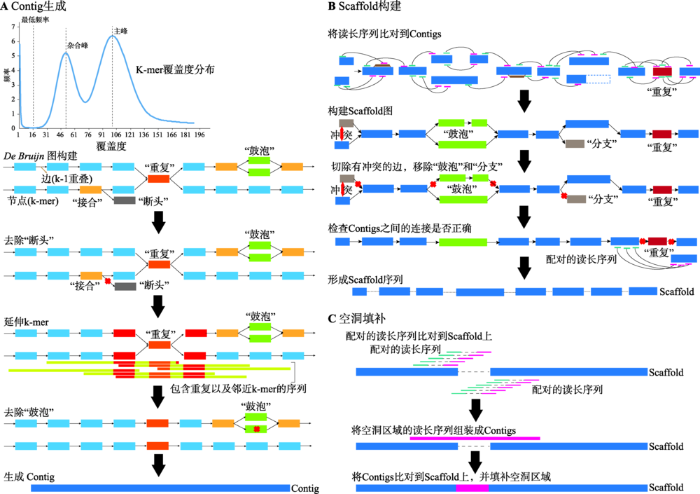 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Platanus组装策略
A:Contig生成阶段:首先由原始数据生成k-mer分布并拟合泊松分布,确定最低频率、杂合峰和主峰,并去除最低频率下的所有k-mer。然后,依次通过De brujin图构建、去除“断头”、延伸k-mer和去除“鼓泡”4个步骤,生成尽可能连续的Contig序列。B:Scaffold构建阶段:将原始数据比对到生成的Contig序列上,通过配对关系,首先构建Scaffold图,然后通过切除有冲突的边,移除“鼓泡”和“分支”,生成尽可能连续的Scaffold序列。C:空洞填补阶段:通过比对获取空洞内及其临近的配对序列,对这些序列进行局部组装,再通过比对将空洞进行填补。根据文献[5]修改绘制。
Fig. 3The assembling workflow of Platanus
与Platanus有所不同,另一个组装软件ALLPATHS-LG[31]则更适用于处理高重复基因组。与一般的组装程序不同,ALLPATHS-LG要求在DNA文库构建时同时构建小片段(Fragment)文库和长跨度(Jumping)文库。其中,小片段文库在构建时,要求插入片段长度小于Reads读长两倍的文库,例如插入长度为180 bp的2×100 bp Pair-end文库。在组装时,ALLPATHS-LG会将小片段文库两端的重叠区域结合起来,形成平均长度近似于插入长度的接合片段(end-overlap fragments)。然后,ALLPATHS-LG使用大小K-mer组合的策略,使用较大K-mer将结合片段组装成不含任何分支的独立路径(unipath)片段,通过长跨度文库,将路径片段连接成组装图(assembly graph),并通过覆盖度等信息将包含重复序列的分支“扁平化”(flatten),从而形成包含尽可能少的分支或者环路的线性组装序列。ALLPATHS-LG的组装方法具有以下优势:(1) 基于180 bp的3′末端对接片段,使用较大的K-mer (默认为K=96)进行初始组装,可以有效地避免重复序列区域产生过多的分支;(2) 在组装前,使用24-mer进行测序数据的矫正,可以有效降低由于测序错误或者低频SNP造成的复杂度;(3) 在处理长跨度文库(包括Mate-pair文库和BAC-end文库)时,首先进行嵌合和非环化Reads的检查,消除构建Scaffold时的负面影响;(4) 对于由PCR Bias或者极端GC造成的低覆盖度(<10×)的基因组区域,由于低覆盖度区域的Reads之间的重叠长度可能非常短,因此ALLPATHS-LG尝试采用极低的K-mer (默认为K=15)对初始组装中的空洞(可能来自极低覆盖度区域)进行填补。得益于这些特性,ALLPATHS-LG可以在仅使用Illumina测序数据的情况下,获得连续性较好的组装结果。以人和小鼠的数据为例,其N50分别可以达到11.5 Mb和7.2 Mb。
在以PacBio为代表的三代测序技术逐渐兴起之后,由于三代测序数据的读长更长,因此能够有效地解决高重复和高杂合的问题。但受限于三代数据的通量和成本,在早期,利用三代数据的方法主要为三代和二代混合组装,因此也产生了众多的二代、三代数据混合组装的算法,包括PBcR[32]、SPAdes[33]、DBG2OLC[34]和MaSuRCA[35]等。其中,MaSuRCA对于高杂合和高重复基因组的混合组装表现较好。MaSuRCA是基于OLC算法诞生的组装程序,其最早主要利用二代Illumina数据进行组装,并比较成功地组装了基因组大小约22 Gb的火炬松(Pinus taeda)为代表的超大植物基因组[36]。MaSuRCA在3.2.0版本之后开始支持二、三代数据混合组装,并已经完成了粗山羊草(Aegilops tauschii)基因组(古小麦D基因组,4.25 Gb)的组装[37]。与使用纯二代数据的SOAPdenovo[38]和DenovoMagic方法相比,MaSuRCA可以将N50提升30倍以上(N50分别为2.1 kb、16.4 kb和486.8 kb)[35]。MaSuRCA的混合组装策略如图4所示。首先,对二代Illumina数据进行质控,去掉低质量和接头序列,并使用Quorum算法[39]进行错误矫正;然后,对矫正后的Reads进行预组装,形成称为超级读长(Super- reads)的长片段;将Super-reads比对到三代PacBio数据上,根据比对的顺序和重叠,将Super-reads进一步融合形成更长的巨型读长(Mega-reads);最后,使用Celera Assembler中的OLC算法,将Mega-reads组装成基因组序列。与只使用二代数据进行组装的算法相比,得益于PacBio数据的引导作用,MaSuRCA可以更好地组装包含重复序列的基因组片段,同时得益于OLC算法允许一定SNP差异的特性,MaSuRCA对于高杂合基因组也有较好的适用性。为了进一步降低高杂合成分对于组装的影响,在组装最后阶段,MaSuRCA会使用MUMMer[40]将基因组中较长的冗余片段合并去除,从而有效降低高杂合造成的冗余组装问题。但同时也需要注意,由于MaSuRCA的组装策略是使用三代长序列作为“骨架”引导二代数据组装,三代数据并不构成序列的主要部分,因此仍然会受限于Illumina测序平台对GC含量和扩增偏好性(PCR Bias)的敏感性,理论上仍会存在基因组组装的覆盖度盲区。此外,由于Super-reads的产生仍然依赖于较短的测序长度,因此对于较长的重复序列,如LTR等,仍然会存在较大的困难。
图4
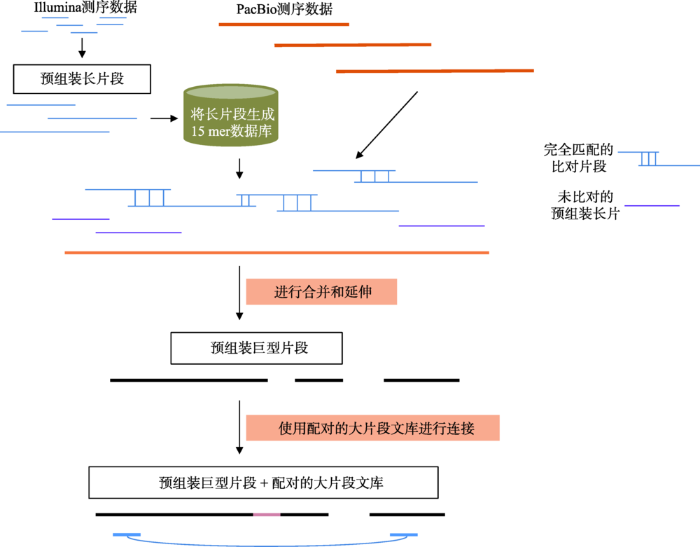 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4MaSuRCA混合组装策略
根据文献[35]修改绘制。
Fig. 4The hybrid assembling strategy of MaSuRCA
随着三代测序技术的进步,且三代测序在偏好性方面有二代技术无法比拟的优势,新的策略开始着眼于仅使用三代数据来进行基因组的组装。除了PacBio官方SMRT Pipeline中的PBcR和Falcon之外,仅使用三代数据的组装方法还包括Canu[30]、miniasm[41]、MECAT[42]、SMARTdenovo和WTDBG 等。在这些软件中,Canu具有较好的组装准确性,但是由于其基于完全的OLC算法,对于资源消耗十分巨大;而以miniasm为代表的加速算法,虽然在准确性上欠佳,但是其资源消耗小,更适用于深度测序或对超大基因组进行组装[43]。以目前使用较多且组装准确性较高的Canu为例,该方法是CABOG组装程序和PBcR自我矫正程序的延伸版本,其基本组装策略同样为OLC算法,但其核心的优势在于整合了高效的比对算法MHAP[32]和矫正算法Falcon[44],从而在连续性和准确性方面具有较好的平衡性。如图5所示,Canu的组装流程分为3个阶段:矫正(correction)、修剪(trim)和组装(assemble)。在矫正阶段,Canu首先将原始数据使用MHAP算法进行比对,根据比对结果将Reads进行聚类,然后根据聚类结果生成一致性(consensus)序列,从而对测序数据进行自我矫正。在修剪阶段,Canu采用CABOG中的重叠修剪(overlap-based trim)方法,将测序数据中不产生重叠的部分切除。最后,Canu使用矫正与修剪后的Reads进行基于OLC算法的组装,生成Contig,从而完成组装。目前,根据Canu官方的测试结果,Canu在完整程度和速度上,均优于Falcon (纯三代数据组装)和SPAdes (二代三代混合组装)。但是,对于高杂合的基因组,Canu目前仍然将差异较大(超过1.5%以上的差异)的单倍型(Haplotypes)序列分开组装,且目前尚无较好的方法来继续分辨与处理这些序列。目前,使用Canu进行高杂合或多倍基因组组装,建议的解决思路有两个:(1) 尽可能保留单倍型组装,然后再去掉或合并同源区段:该方法通过设置严格的比对错误率,尽可能将杂合部分单独组装,产生多倍于预期基因组的组装,然后通过“Purge Haplotigs”方法[45]去除杂合或者多倍部分,但有可能去除掉部分基因组重复;(2) 尽可能在组装时合并杂合或多倍部分:该方法通过设置较宽松的错误率,将杂合或多倍部分合并到一起,但有可能造成组装错误。同时,Canu计划在未来与10X Genomics或者Hi-C等数据结合,综合解决该问题。
图5
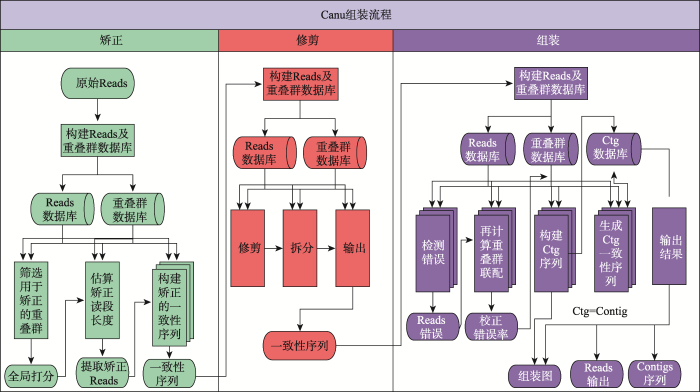 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5Canu组装流程
根据文献[30]修改绘制。
Fig. 5The workflow of Canu pipeline
随着以Oxford Nanopore Technology (ONT)为代表的纳米孔测序技术的发展,其测序的读长和通量相对于PacBio测序更胜一筹,因此基于ONT数据完成的组装实例也日渐增多。与PacBio相比,ONT 在数据上的特点表现为平均读长更长,但是错误率相对来说较高。目前,可应用于PacBio数据的组装方法,包括Canu[30]、miniasm[41]和TULIP等,同样适用于ONT数据,但需要对参数进行调整[46]。以Canu为例,根据软件作者的测试,在处理ONT数据时,需要将矫正预期的错误率(corrected error rate)适当调高,比如在组装Nanopore R7 2D和R9 1D数据时,建议将预期错误率从8.5%调整到14.4%,以适应ONT数据的特点。
正如前面所述,解决复杂基因组组装难题的关键,在于获得尽可能长的测序数据。但受限于现有的DNA实验技术与测序技术,可获得的测序长度是有限的。即便是宣称最长读长可超过1 Mb的ONT技术,在实际的复杂基因组组装中,其一般建库测序方法得到的文库平均长度与PacBio相比并无显著优势[43]。因此,目前尚不能从根本上解决复杂基因组的精准组装问题。但近年来,一些特殊的技术如BioNano、10X Genomics和Hi-C,虽然无法直接获得完整的DNA长片段,但可以在现有测序技术的基础上获得更大的跨度,在一定程度上解决了复杂基因组组装的难题,为复杂基因组结构分析提供帮助。
BioNano(以Irys版本为例)是利用光学标记构建基因组酶切图谱的技术。在进行复杂基因组组装时,BioNano技术的作用体现在两方面:(1) 由于BioNano读取的分子长度最高可以达到1 Mb以上,平均长度也可以达到200 kb,因此在构建Scaffold时可以有效跨过较长的重复序列区域,获得较为连续的Scaffold序列;(2) BioNano可以用于鉴定单倍型基因组上的酶切位点差异,从而将单倍型基因组序列分离开,因此有助于将高杂合基因组中的杂合区域完全分离组装。同时,BioNano读取的光学酶切图谱可以有效纠正组装中的结构错误。对于复杂基因组组装来说,使用BioNano官方的BioNano Sovle软件进行辅助组装的基本流程包括:(1) 生成基因组草图的BioNano酶切图谱;(2) 将特定酶切的BioNano Molecule进行自身一致性组装,生成光学图谱(optical map);(3) 将光学图谱和基因组序列的酶切图谱进行共线性比对,以光学图谱为骨架生成Scaffold序列;(4) 如果使用双酶切文库,可进一步使用TGH Pipeline生成双酶切一致的Scaffold,实现最佳组装效果。
10X Genomics提供的技术(以Chromium为例)类似于早期的BAC混合池(BAC pooling)技术,该技术通过油包水微体系,为DNA片段加入条码标记(barcode)并测序,之后可通过条码标记将原本属于同一DNA片段的测序数据进行聚类。10X Genomics技术可以有效降低组装的复杂度,通过局部的Contig组装和不同条码标记聚类之间的重叠,可以有效地构建出连续的基因组组装。与BioNano技术类似,10X Genomics技术对复杂基因组组装的作用也体现在两个方面:(1) 目前其条码标记序列的平均长度可达到50 kb左右,理论上可以跨过大部分的转座子重复,且限制在局部片段上,其复杂度相对于全基因组来说要低很多,因此该技术可以有效减少由于重复序列造成的组装碎片化问题;(2) 由于10X Genomics技术可以在50 kb的分辨率上对单倍型差异通过条码标记进行分离,因此同样可以对高杂合基因组中的每一个单倍型基因组进行独立组装,在一定程度上解决杂合造成的组装难题。目前,已经有多个利用10X Genomics技术和其官方提供的Supernova组装方法完成的基因组组装实例,其Scaffold N50基本都可以达到Mb级别[47, 48]。
Hi-C技术是通过固定染色体在三维空间上的交联,对染色体的空间结构进行概率性的推断。由于相邻序列之间发生交联的概率要大于相距较远的序列,因此,同一染色体内的空间交联要高于不同染色体之间的交联,因此利用Hi-C技术,可以在染色体级别的长跨度上对染色体进行组装和分离。同时,由于单倍型基因组分属于不同的染色体组,因此利用Hi-C技术,同样可以实现对异源二倍体基因组中的单倍型基因组进行分离。对于多倍体超大基因组(如六倍体小麦基因组),Hi-C同样可以将多倍体基因组中不同来源的基因组成分的分离,从而在一定程度上解决多倍超大基因组的组装难题。相对于早期利用遗传图谱进行染色体构建的方法,Hi-C技术优势主要表现在:(1) 不需要构建大量的F1群体,只需个体即可完成;(2) 不需要对亲本进行纯化,就可以对单倍型基因组进行分离,因此适用于不易纯化的高杂合基因组组装。以具有代表性的使用Juicer方法组装的埃及伊蚊(Aedes aegypti)基因组[49]为例,使用Juicer进行辅助组装的步骤包括:生成与Hi-C文库对应的全基因组电子酶切图谱;将Hi-C文库比对定位到基因组上,筛选酶切位点附近的测序数据,并将完全重复的位点去除;(3) 构建基于Hi-C文库的全基因组交联图(contact map);(4) 根据交联图中Contig之间的频率,将属于同一个拓扑结构域(TAD)的序列进行聚类,并对Contig进行重排;(5) 构建出Hi-C Scaffolds,并通过多次迭代纠正组装的错误,将无法正确放置的序列碎片剔除。
综上所述,利用BioNano、10X Genomics或Hi-C技术可以有效弥补现有测序技术在测序长度上的不足,辅助构建出更加连续完整的基因组序列。同时,从3种技术对于杂合处理的趋势来看,进行多倍型基因组完整组装是复杂基因组组装未来发展的趋势。
3 复杂基因组测序经典案例
3.1 梨基因组
梨(Pyrus bretschneideri)由于其自交不亲和、排斥近交等特点使其基因组杂合度达到1%,是高度杂合的物种。因此,不适合采用全基因组鸟枪法(whole genome shotgun, WGS)测序组装策略。研究人员在梨基因组测序中采用新一代Illumina测序平台并结合BAC-to-BAC策略,其中BAC-to-BAC策略是成功的关键[50]。该方法首先构建插入片段长度为80~180 kb的BAC文库,文库构建按照Agilent公司标准流程,在文库构建过程中加入了不同接头,从而实现将2208个样品混合进行1个测序泳道(lane)的测序,最终得到38 304个BAC的测序结果,相当于10倍基因组的长度。同时,进行了全基因组的Pair-end测序(插入片段长度180~800 bp)和Mate-pair测序(插入片段长度大于2000 bp)。在组装方面,首先利用SOAPdenovo软件和Pair-end的数据进行单个BAC的测序组装,然后利用SSPACE软件[51]和全基因组的Mate-pair数据进行Scaffolds构建。利用几轮的相互叠加的测序验证将单个BAC的组装结果进行混合组装,同时将Scaffold末端3 kb的序列和Scaffold进行比对,相同序列进行组装,同时去除冗余序列,最终得到可信的Scaffolds,进一步利用SSPACE软件、Mate-pair的全基因组测序数据和Pair-end的测序数据将Scaffold组装成Super-scaffold。最后利用NUCmer[52]和BLAST[53]将BAC的组装结果和Scaffold进行组装。梨基因组研究在没有物理图谱辅助的情况下,完成了高杂合、高重复序列的二倍体果树基因组组装,积累的组装经验对于其他高度杂合的基因组研究具有很好的借鉴价值。3.2 太平洋牡蛎基因组
太平洋牡蛎(Crassostrea gigas)与大多数海洋无脊椎动物类似,具有高度杂合的基因组,其杂合度高达2.3%,基于常规的策略,几乎不可能对其进行从头组装。研究人员采用Fosmid克隆混合池(Fosmid pooling)、辅助短序列以及逐级组装策略,比较成功地解决了牡蛎基因组高杂合造成的组装难题[54]。首先将145 170个Fosmid克隆混合成1613个混合池进行测序,每个混合池测序数据量达到60×覆盖度。将得到的序列按照每个混合池单独组装,得到Contig序列,这些序列通过Contig之间的重叠序列进行混合组装得到Super-contig。利用LASTZ和测序程度信息对Super-contig进行自我全基因组比对,移除组装过程中产生的冗余序列,将得到的序列进一步用Pair-end数据矫正组装得到最终的Scaffold序列。最后利用短序列对Scaffold序列进行矫正,最终得到的牡蛎基因组大约为559 Mb,总共约有28 000个基因。牡蛎基因组的这种Fosmid混合池结合短序列的测序和组装方法,为研究人员更好地破译具有高度杂合性和高度多态性的基因组开辟了一条新途径。3.3 硬橡胶树杜仲基因组
杜仲(Eucommia ulmoides)基因组杂合率约为0.9%~1%,重复序列在66%以上,属于高杂合、高重复的复杂基因组。在杜仲基因组测序策略上,采用了全基因组鸟枪法和第二代(Illumina HiSeq 2000和MiSeq)、第三代测序技术(PacBio)和BioNano光学图谱技术的有机结合[55]。利用Platanus组装软件将高质量数据组装成Contigs和Scaffolds,然后用SSPACE软件,利用Pair-end和Mate-pair数据将Scaffolds组装成更长的Super-scaffolds。使用PBJelly[56]和PacBio数据完成上述Scaffold的补洞工作。最后利用BioNano数据完成Scaffold的定位。在组装过程中,采用了适用于杂合物种的软件Platanus,同时利用PBJelly软件进一步通过三代数据完善组装结果。3.4 六倍体小麦基因组
六倍体小麦(Triticum aestivum)基因组庞大,是典型的异源多倍体基因组,由3套相似而又不同的基因组整合形成一个极为复杂的六倍体基因组。其中3套不同的基因组分别是A、B、D基因组,每个基因组含有7条染色体,共有21条染色体组成,并且每套基因组上都有一套同源性比较高的相关基因,而且这些基因在每个同源染色体上的排列顺序也都不相同,这就使基因组序列的组装工作变的非常复杂。由于其高倍性,加之高度重复序列,给小麦基因组的研究带来巨大的挑战,对六倍体小麦的基因组研究更是延续了13年之久。对于这种基因组体量大、重复序列含量高的复杂基因组,研究者采取“分而治之”的组装策略:首先利用NRGene的DeNovoMagic2软件对小麦基因组进行组装,然后通过BAC克隆文库,对分属于3套不同基因组的序列进行分离。然后,在分离组装的基础上,结合三维基因组Hi-C技术和高密度遗传图谱(population sequencing, POPSEQ)对基因组序列进行染色体挂载,重新修正了基因组,大小为15.4~15.8 Gb。在组装过程中,研究者还参照已经发表的乌拉尔图小麦(Triticum urartu) (A基因组)、粗山羊草(Aegilops tauschii) (D基因组)、拟斯卑尔脱山羊草(Aegilops speltoides) (B基因组)和硬粒小麦(Triticum durum) (B基因组)基因组。最终,组装得到1601条Scaffold序列,总大小为14.5 Gb,是迄今为止完成度最高、质量值最好的小麦基因组序列。同时,为了验证组装的准确性,研究者还构建了BioNano光学图谱、辐射杂交物理图谱等,用于检查和修正基因组Scaffolds中存在的组装错误[57]。综上所述,对于高倍体基因组,对其中的单倍型基因组进行分离是完成该类型复杂基因组组装的关键。3.5 野生番茄基因组更新
野生番茄(Solanum pennellii)基因组大小约为1.2 Gb,属于较大的复杂基因组。之前已有研究报道过基于二代数据组装的野生番茄基因组,但受限于技术水平,其组装的Scaffold N50为1.7 Mb,组装大小为942 Mb,但序列中存在较多的空洞,其Contig N50仅为2.18 kb[58]。随着Nanopore技术的进步,近期有研究团队对野生番茄基因组进行了基于纯Nanopore测序数据的更新。Nanopore单分子实时测序技术,其一般建库方法的平均读长为10 kb,超长建库方法平均读长可达到100 kb,因此可以用来测序、组装Gb级别的高质量基因组,尤其是可以用于高杂合、高重复等复杂基因组。在该研究中,通过Nanopore测序得到了约110.96 Gb的有效数据,并使用Canu和SMARTdenovo两个软件联合进行组装,最终得到的更新版本基因组组装大小为915 Mb,Contig数目为889,Contig N50达到了2.52 Mb,与之前发表的2.18 kb相比有了质的飞跃[59]。该研究充分表明了使用Nanopore进行复杂大型基因组进行组装的可行性。同时,该研究还对各种Nanopore数据组装方法的效果进行了对比,具有一定的参考价值。4 本课题组研究领域及成果
本课题组主要从事基因组结构和功能解析,从病毒、细菌和原生生物等简单生物到高等生物(橡胶、木薯等)皆有涉及。本课题组根据物种特点结合当时测序技术分别制定测序策略,以获得较好的基因组组装结果。在二代测序技术发展早期,细菌等基因组主要采用一代测序技术,该技术具有准确度高、测序读长长等优点,是简单基因组测序的优先选择,如本课题组已测序完成的副血链球菌(Streptococcus parasanguinis)[60]等。但是由于一代测序技术的花费高、通量低,耗时长等缺点,导致该技术在复杂基因组上的应用具有很大的局限性。随着二代测序技术的应用以及相应软件的开发,大量物种采用该技术进行测序。本课题组开展了多个物种的测序与分析工作,包括枣椰树(Phoenixdactylifera)[61]、牛带绦虫(Taenia saginata)[62]、橡胶树(Hevea brasiliensis)[2]、茶薪菇(Agrocybe chaxingu)等。
以橡胶树基因组的测序与组装策略为例(图6),主要过程包括:(1) 构建不同片段插入长度的文库,包括Pair-end和Mate-pair文库;(2) 利用二代组装软件将测序数据搭建成Scaffolds;(3) 使用cDNA 进一步延伸Scaffolds;(4) 筛选到5912个BAC克隆,将这些BAC分到143个384板中,构建BAC Pooling文库,利用Illumina测序平台获得数据,基于序列质量,将其中124个384板的数据单独组装;(5) 基于BAC组装数据,模拟10 kb、20 kb、30 kb、40 kb、50 kb、70 kb和100 kb插入长度片段的测序数据;(6) 将原有Scaffolds从空洞区域断开,拆分成Contigs,然后利用模拟数据将Contigs重新搭建成Scaffolds。橡胶树基因组采用BAC数据模拟成不同插入长度片段文库测序数据来辅助基因组组装,这与之前已发表的牡蛎基因组[54]所采用Fosmid混合池辅助组装的策略不同。在牡蛎基因组测序与组装中,每个Fosmid混合池数据单独组装并矫正组装数据;然后,利用OLC方法将所有混合池组装互相连接成Super-contigs;最后,利用二代Mate-pair大片段数据将Super-contigs进一步组装成Scaffold,并对空洞区域进行填补。由于在橡胶树基因组中BAC测序数据大约为4×,理论上只覆盖基因组98%的区域,并且只有78%的转录本比对到BAC数据上,因此本实验室采用不同的方法处理BAC数据,包括BAC-Scaffold与WGS-Scaffold合并,BAC-Scaffold比对到WGS-Scaffold上以延长WGS-Scaffold以及BAC数据模拟同插入长度片段文库测序数据等。综合比较这些策略,BAC数据模拟是最优策略。该策略的优点主要体现在:(1) 降低Mate-pair文库的测序量;(2) 模拟产生更大的Mate-pair片段,提高Scaffold完整性;(3) 相比Mate-pair文库,通过BAC克隆得到的数据可避开环化失败、PCR重复扩增导致的偏好性,产生更多有效数据。
图6
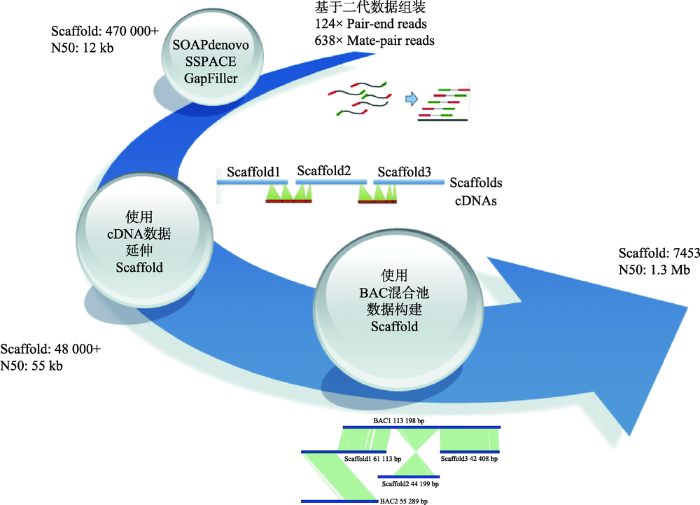 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6橡胶树基因组的组装流程
根据文献[2]修改绘制。
Fig. 6The workflow of para rubber tree genome assembly
枣椰树基因组采用454、SOLiD和3730XL测序平台相结合的策略,具体过程包括:(1) 利用454数据,使用Newbler程序构建Contigs;(2) 使用SOLiD的不同插入片段长度的LMP (long mate-pair)文库搭建Scaffolds;(3) 利用高质量测序数据填补Scaffolds中的空洞区域;(4) 采用BAC末端测序数据进一步延伸Scaffolds。为了降低高重复引起的复杂度问题,本实验室采用BAC混合池测序来辅助枣椰树基因组的组装(图7)。
图7
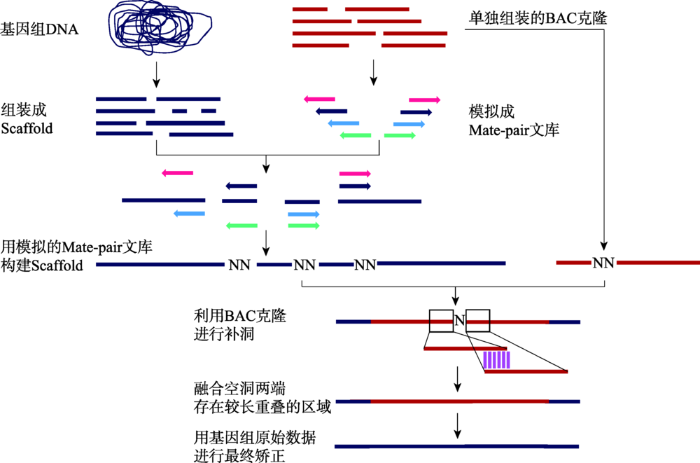 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7BAC混合池辅助基因组组装流程
根据文献[61]修改绘制。
Fig. 7The workflow of BAC pooling assisted genome assembly
二代测序数据读长较短,快速获得高重复高杂合基因组的高质量序列图谱仍面临着极大困难,而三代测序数据读长较长,可更好地处理重复和杂合问题。本实验室已将PacBio等三代测序技术作为主流策略,现已利用PacBio技术对水稻(Oryza sativa ssp. indica cv. 93-11)进行测序,并使用BioNano光学图谱和Hi-C三维基因组技术,对基因组进行染色体级别的Scaffold组装。在此基础上,进一步循环使用Hi-C和BioNano光学图谱数据,对组装中存在的错误进行多轮校正,优化提升组装质量(图8)。
图8
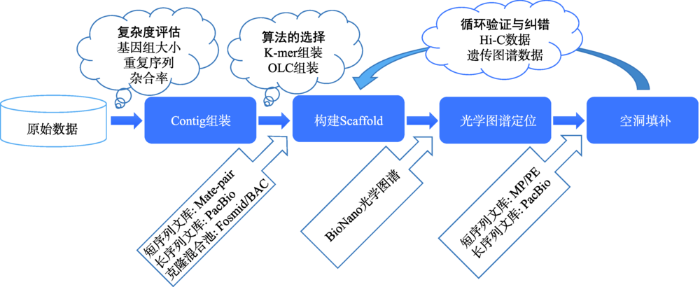 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8使用Hi-C和BioNano技术进行辅助优化组装流程
Fig. 8The workflow of genome assembly improvement using Hi-C and BioNano data
综合考虑当前的测序技术特点,针对如何挑选进行基因组测序的物种或样品,我们建议应充分考
虑基因组测序难度,相近物种间或品种间可能存在的异质性,优先选择容易测序的,如杂合度较低、重复比例较低、易获得高质量基因组DNA、易获得单倍体材料的物种或品种;同时也要考虑到同一物种不同组织间也可能存在差异,优先选择方便提取高质量基因组DNA,且外源DNA污染可能性小的组织。而如何进行复杂基因组测序,总原则是先短序列评估,再进行长短结合深度测序,具体可参考如下流程:(1) 首先采用二代测序数据进行基因组复杂度评估,估计基因组大小,杂合度和重复序列比例,完成GC含量分布图,比较公共微生物基因组数据库,评估是否存在基因组污染,可对多个近缘物种或品种平行测序评估,选择复杂度最低的物种或品种进行深度测序,并测试能否提取高质量长片段基因组DNA;(2) 考虑到测序费用仍然较高,进行深度测序前,可以使用已完成全测序的基因组大小和重复序列比例最接近的近缘基因组序列,参考前期评估的杂合度,进行基于10X Genomics或PacBio/ Nanopore测序的技术的数据模拟,评估方案效果和费用;(3) 进行高通量测序,获得二代和三代测序数据,三代序列质量矫正和组装,整合BioNano光学图谱,参考Hi-C结果和遗传图谱评估和反馈优化组装结果,使用长短序列进行组装结果矫正,最终获得高质量的高杂合基因组序列图谱。
5 结语与展望
总结近10年来基因组测序技术领域的发展脉络,不难看出新老技术不断融合与发展的力量:BAC混合池测序实质是BAC-to-BAC测序策略与二代段片段测序技术的结合,PacBio和Nanopore测序虽然是基于实时测序的新技术,测序组装策略也采用直接对全基因组DNA进行测序,但其数据处理的算法则可以追溯到Sanger测序时代。此外,二代和三代测序技术流程基本已经成熟,组装成功与否和质量的好坏已不再受制于测序技术本身,而是取决于是否能够获得高质量长片段基因组DNA。尤其对于植物而言,如何降低多样化次生代谢物质的影响,以及如何从包含共生微生物的藻类中提取高质量DNA,仍然充满挑战。而目前最有效的高质量基因组DNA提取流程的源头就是Sanger测序时代的BAC文库构建技术。从这个层面上看,充分认识和尊重基因组学技术的继承性和发展连续性特点弥足珍贵。随着技术的发展,基因组测序难易的概念也随之发展和变化,早期认为难以测序的复杂基因组将成为容易测序的“简单”基因组。当然,基因组完成图的标准也将随之不断提升,即使是通常认为的高质量基因组完成图,如人类基因组和水稻基因组,实际上仍然存在着大量未知区域。未来,使用最新技术更新重要物种基因组,解析未知区域的序列信息也将是未来数年基因组研究领域的重点之一。
毫无疑问,随着测序技术的进步,获取高质量基因组序列的时间和费用成本将变得越来越低,更多物种都将有高质量的基因组序列以及其它多组学数据信息。因此,基因组学专家的工作重点应逐渐转为利用比较基因组学技术解决生物学问题,或通过整合多组学数据进行信息挖掘,指导和推动以生物学问题为中心的科学研究。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:28231512 [本文引用: 2]

Since the introduction of next generation sequencing, plant genome assembly projects do not need to rely on dedicated research facilities or community-wide consortia anymore, even individual research groups can sequence and assemble the genomes they are interested in. However, such assemblies are typically not based on the entire breadth of genomic technologies including genetic and physical maps and their contiguities tend to be low compared to the full-length gold standard reference sequences. Recently emerging third generation genomic technologies like long-read sequencing or optical mapping promise to bridge this quality gap and enable simple and cost-effective solutions for chromosomal-level assemblies.
URLPMID:27255837 [本文引用: 3]
Abstract The Para rubber tree (Hevea brasiliensis) is an economically important tropical tree species that produces natural rubber, an essential industrial raw material. Here we present a high-quality genome assembly of this species (1.37090005Gb, scaffold N50090005=0900051.28 Mb) that covers 93.8% of the genome (1.47090005Gb) and harbours 43,792 predicted protein-coding genes. A striking expansion of the REF/SRPP (rubber elongation factor/small rubber particle protein) gene family and its divergence into several laticifer-specific isoforms seem crucial for rubber biosynthesis. The REF/SRPP family has isoforms with sizes similar to or larger than SRPP1 (204 amino acids) in 17 other plants examined, but no isoforms with similar sizes to REF1 (138 amino acids), the predominant molecular variant. A pivotal point in Hevea evolution was the emergence of REF1, which is located on the surface of large rubber particles that account for 93% of rubber in the latex (despite constituting only 6% of total rubber particles, large and small). The stringent control of ethylene synthesis under active ethylene signalling and response in laticifers resolves a longstanding mystery of ethylene stimulation in rubber production. Our study, which includes the re-sequencing of five other Hevea cultivars and extensive RNA-seq data, provides a valuable resource for functional genomics and tools for breeding elite Hevea cultivars.
URL [本文引用: 1]
Salamanders serve as important tetrapod models for developmental, regeneration and evolutionary studies. An extensive molecular toolkit makes the Mexican axolotl (Ambystoma mexicanum) a key representative salamander for molecular investigations. Here we report the sequencing and assembly of the 32-gigabase-pair axolotl genome using an approach that combined long-read sequencing, optical mapping and development of a new genome assembler (MARVEL). We observed a size expansion of introns and intergenic regions, largely attributable to multiplication of long terminal repeat retroelements. We provide evidence that intron size in developmental genes is under constraint and that species-restricted genes may contribute to limb regeneration. The axolotl genome assembly does not contain the essential developmental gene Pax3. However, mutation of the axolotl Pax3 paralogue Pax7 resulted in an axolotl phenotype that was similar to those seen in Pax3and Pax7mutant mice. The axolotl genome provides a rich biological resource for developmental and evolutionary studies.
URLPMID:29678829 [本文引用: 2]
Abstract Tea, one of the world's most important beverage crops, provides numerous secondary metabolites that account for its rich taste and health benefits. Here we present a high-quality sequence of the genome of tea, Camellia sinensis var. sinensis (CSS), using both Illumina and PacBio sequencing technologies. At least 64% of the 3.1-Gb genome assembly consists of repetitive sequences, and the rest yields 33,932 high-confidence predictions of encoded proteins. Divergence between two major lineages, CSS and Camellia sinensis var. assamica (CSA), is calculated to 0908040.38 to 1.54 million years ago (Mya). Analysis of genic collinearity reveals that the tea genome is the product of two rounds of whole-genome duplications (WGDs) that occurred 09080430 to 40 and 09080490 to 100 Mya. We provide evidence that these WGD events, and subsequent paralogous duplications, had major impacts on the copy numbers of secondary metabolite genes, particularly genes critical to producing three key quality compounds: catechins, theanine, and caffeine. Analyses of transcriptome and phytochemistry data show that amplification and transcriptional divergence of genes encoding a large acyltransferase family and leucoanthocyanidin reductases are associated with the characteristic young leaf accumulation of monomeric galloylated catechins in tea, while functional divergence of a single member of the glutamine synthetase gene family yielded theanine synthetase. This genome sequence will facilitate understanding of tea genome evolution and tea metabolite pathways, and will promote germplasm utilization for breeding improved tea varieties.
[本文引用: 5]
[本文引用: 1]
URLPMID:28849613 [本文引用: 1]
react-text: 64 Improving the trait based the results from GWAS /react-text react-text: 65 /react-text
URLPMID:3639258 [本文引用: 1]
Next-generation-sequencing (NGS) has revolutionized the field of genome assembly because of its much higher data throughput and much lower cost compared with traditional Sanger sequencing. However, NGS poses new computational challenges tode novogenome assembly. Among the challenges, GC bias in NGS data is known to aggravate genome assembly. However, it is not clear to what extent GC bias affects genome assembly in general. In this work, we conduct a systematic analysis on the effects of GC bias on genome assembly. Our analyses reveal that GC bias only lowers assembly completeness when the degree of GC bias is above a threshold. At a strong GC bias, the assembly fragmentation due to GC bias can be explained by the low coverage of reads in the GC-poor or GC-rich regions of a genome. This effect is observed for all the assemblers under study. Increasing the total amount of NGS data thus rescues the assembly fragmentation because of GC bias. However, the amount of data needed for a full rescue depends on the distribution of GC contents. Both low and high coverage depths due to GC bias lower the accuracy of assembly. These pieces of information provide guidance toward a betterde novogenome assembly in the presence of GC bias.
URLPMID:19287394 [本文引用: 1]
Abstract Amplification artifacts introduced during library preparation for the Illumina Genome Analyzer increase the likelihood that an appreciable proportion of these sequences will be duplicates and cause an uneven distribution of read coverage across the targeted sequencing regions. As a consequence, these unfavorable features result in difficulties in genome assembly and variation analysis from the short reads, particularly when the sequences are from genomes with base compositions at the extremes of high or low G+C content. Here we present an amplification-free method of library preparation, in which the cluster amplification step, rather than the PCR, enriches for fully ligated template strands, reducing the incidence of duplicate sequences, improving read mapping and single nucleotide polymorphism calling and aiding de novo assembly. We illustrate this by generating and analyzing DNA sequences from extremely (G+C)-poor (Plasmodium falciparum), (G+C)-neutral (Escherichia coli) and (G+C)-rich (Bordetella pertussis) genomes.
URLPMID:28716924 [本文引用: 2]
Author(s): Brawley, SH; Blouin, NA; Ficko-Blean, E; Wheeler, GL; Lohr, M; Goodson, HV; Jenkins, JW; Blaby-Haas, CE; Helliwell, KE; Chan, CX; Marriage, TN; Bhattacharya, D; Klein, AS; Badis, Y; Brodie, J; Cao, Y; Coll n, J; Dittami, SM; Gachon, CMM; Green, BR; Karpowicz, SJ; Kim, JW; Kudahl, UJ; Lin, S; Michel, G; Mittag, M; Olson, BJSC; Pangilinan, JL; Peng, Y; Qiu, H; Shu, S; Singer, JT; Smith, AG; Sprecher, BN; Wagner, V; Wang, W; Wang, ZY; Yan, J; Yarish, C; Z uner-Riek, S; Zhuang, Y; Zou, Y; Lindquist, EA; Grimwood, J; Barry, KW; Rokhsar, DS; Schmutz, J; Stiller, JW; Grossman, AR; Prochnik, SE | Abstract: Porphyra umbilicalis (laver) belongs to an ancient group of red algae (Bangiophyceae), is harvested for human food, and thrives in the harsh conditions of the upper intertidal zone. Here we present the 87.7-Mbp haploid Porphyra genome (65.8% G + C content, 13,125 gene loci) and elucidate traits that inform our understanding of the biology of red algae as one of the few multicellular eukaryotic lineages. Novel features of the Porphyra genome shared by other red algae relate to the cytoskeleton, calcium signaling, the cell cycle, and stress-Tolerance mechanisms including photoprotection. Cytoskeletal motor proteins in Porphyra are restricted to a small set of kinesins that appear to be the only universal cytoskeletal motors within the red algae. Dynein motors are absent, and most red algae, including Porphyra, lack myosin. This surprisingly minimal cytoskeleton offers a potential explanation for why red algal cells and multicellular structures are more limited in size than in most multicellular lineages. Additional discoveries further relating to the stress tolerance of bangiophytes include ancestral enzymes for sulfation of the hydrophilic galactan-rich cell wall, evidence for mannan synthesis that originated before the divergence of green and red algae, and a high capacity for nutrient uptake. Our analyses provide a comprehensive understanding of the red algae, which are both commercially important and have played a major role in the evolution of other algal groups through secondary endosymbioses.
URLPMID:27035985 [本文引用: 1]
National Academy of Sciences
URL [本文引用: 1]
URLPMID:14684821 [本文引用: 1]
Approximately 80% of the maize genome comprises highly repetitive sequences interspersed with single-copy, gene-rich sequences, and standard genome sequencing strategies are not readily adaptable to this type of genome. Methodologies that enrich for genic sequences might more rapidly generate useful results from complex genomes. Equivalent numbers of clones from maize selected by techniques called methylation filtering and High$C_{0}t$selection were sequenced to generate ~200,000 reads (approximately 132 megabases), which were assembled into contigs. Combination of the two techniques resulted in a sixfold reduction in the effective genome size and a fourfold increase in the gene identification rate in comparison to a nonenriched library.
URLPMID:14684820 [本文引用: 1]
Gene enrichment strategies offer an alternative to sequencing large and repetitive genomes such as that of maize. We report the generation and analysis of nearly 100,000 undermethylated (or methylation filtration) maize sequences. Comparison with the rice genome reveals that methylation filtration results in a more comprehensive representation of maize genes than those that result from expressed sequence tags or transposon insertion sites sequences. About 7% of the repetitive DNA is unmethylated and thus selected in our libraries, but potentially active transposons and unmethylated organelle genomes can be identified. Reverse transcription polymerase chain reaction can be used to finish the maize transcriptome.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:21169219 [本文引用: 1]
Abstract The two haploid genome sequences that a person inherits from the two parents represent the most fundamentally useful type of genetic information for the study of heritable diseases and the development of personalized medicine. Because of the difficulty in obtaining long-range phase information, current sequencing methods are unable to provide this information. Here, we introduce and show feasibility of a scalable approach capable of generating genomic sequences completely phased across the entire chromosome.
URLPMID:21170043 [本文引用: 1]
Abstract Conventional experimental methods of studying the human genome are limited by the inability to independently study the combination of alleles, or haplotype, on each of the homologous copies of the chromosomes. We developed a microfluidic device capable of separating and amplifying homologous copies of each chromosome from a single human metaphase cell. Single-nucleotide polymorphism (SNP) array analysis of amplified DNA enabled us to achieve completely deterministic, whole-genome, personal haplotypes of four individuals, including a HapMap trio with European ancestry (CEU) and an unrelated European individual. The phases of alleles were determined at 09080499.8% accuracy for up to 09080496% of all assayed SNPs. We demonstrate several practical applications, including direct observation of recombination events in a family trio, deterministic phasing of deletions in individuals and direct measurement of the human leukocyte antigen haplotypes of an individual. Our approach has potential applications in personal genomics, single-cell genomics and statistical genetics.
URLPMID:20123915 [本文引用: 1]
ABSTRACT We have developed a novel approach for using massively parallel short-read sequencing to generate fast and inexpensive de novo genomic assemblies comparable to those generated by capillary-based methods. The ultrashort (<100 base) sequences generated by this technology pose specific biological and computational challenges for de novo assembly of large genomes. To account for this, we devised a method for experimentally partitioning the genome using reduced representation (RR) libraries prior to assembly. We use two restriction enzymes independently to create a series of overlapping fragment libraries, each containing a tractable subset of the genome. Together, these libraries allow us to reassemble the entire genome without the need of a reference sequence. As proof of concept, we applied this approach to sequence and assembled the majority of the 125-Mb Drosophila melanogaster genome. We subsequently demonstrate the accuracy of our assembly method with meaningful comparisons against the current available D. melanogaster reference genome (dm3). The ease of assembly and accuracy for comparative genomics suggest that our approach will scale to future mammalian genome-sequencing efforts, saving both time and money without sacrificing quality.
URL [本文引用: 1]
URLPMID:20081835 [本文引用: 1]
Abstract We demonstrate subassembly, an in vitro library construction method that extends the utility of short-read sequencing platforms to applications requiring long, accurate reads. A long DNA fragment library is converted to a population of nested sublibraries, and a tag sequence directs grouping of short reads derived from the same long fragment, enabling localized assembly of long fragment sequences. Subassembly may facilitate accurate de novo genome assembly and metagenome sequencing.
URL [本文引用: 1]
URLPMID:5124260 [本文引用: 1]
Nanopore DNA strand sequencing has emerged as a competitive, portable technology. Reads exceeding 150 kilobases have been achieved, as have in-field detection and analysis of clinical pathogens. We summarize key technical features of the Oxford Nanopore MinION, the dominant platform currently available. We then discuss pioneering applications executed by the genomics community.
URLPMID:5066648 [本文引用: 1]
The assembly of a reference genome sequence of bread wheat is challenging due to its specific features such as the genome size of 1702Gbp, polyploid nature and prevalence of repetitive sequences.BAC‐by‐BACsequencing based on chromosomal physical maps, adopted by the International Wheat Genome Sequencing Consortium as the key strategy, reduces problems caused by the genome complexity and polyploidy, but the repeat content still hampers the sequence assembly. Availability of a high‐resolution genomic map to guide sequence scaffolding and validate physical map and sequence assemblies would be highly beneficial to obtaining an accurate and complete genome sequence. Here, we chose the short arm of chromosome 7D (7DS) as a model to demonstrate for the first time that it is possible to couple chromosome flow sorting with genome mapping in nanochannel arrays and create ade novogenome map of a wheat chromosome. We constructed a high‐resolution chromosome map composed of 371 contigs with an N50 of 1.302Mb. LongDNAmolecules achieved by our approach facilitated chromosome‐scale analysis of repetitive sequences and revealed a ~800‐kb array of tandem repeats intractable to currentDNAsequencing technologies. Anchoring 7DSsequence assemblies obtained by clone‐by‐clone sequencing to the 7DSgenome map provided a valuable tool to improve theBAC‐contig physical map and validate sequence assembly on a chromosome‐arm scale. Our results indicate that creating genome maps for the whole wheat genome in a chromosome‐by‐chromosome manner is feasible and that they will be an affordable tool to support the production of improved pseudomolecules.
URLPMID:28212831 [本文引用: 1]
@@
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URLPMID:18952627 [本文引用: 1]
Motivation: DNA sequence reads from Sanger and pyrosequencing platforms differ in cost, accuracy, typical coverage, average read length and the variety of available paired-end protocols. Both read types can complement one another in a 'hybrid' approach to whole-genome shotgun sequencing projects, but assembly software must be modified to accommodate their different characteristics. This is true even of pyrosequencing mated and unmated read combinations. Without special modifications, assemblers tuned for homogeneous sequence data may perform poorly on hybrid data.Results: Celera Assembler was modified for combinations of ABI 3730 and 454 FLX reads. The revised pipeline called CABOG (Celera Assembler with the Best Overlap Graph) is robust to homopolymer run length uncertainty, high read coverage and heterogeneous read lengths. In tests on four genomes, it generated the longest contigs among all assemblers tested. It exploited the mate constraints provided by paired-end reads from either platform to build larger contigs and scaffolds, which were validated by comparison to a finished reference sequence. A low rate of contig mis-assembly was detected in some CABOG assemblies, but this was reduced in the presence of sufficient mate pair data.Availability: The software is freely available as open-source from http://wgs-assembler.sf.net under the GNU Public License.Contact: jmillercvi.orgSupplementary information: Supplementary data are available at Bioinformatics online.
URLPMID:28298431 [本文引用: 4]
Abstract Long-read single-molecule sequencing has revolutionized de novo genome assembly and enabled the automated reconstruction of reference-quality genomes. However, given the relatively high error rates of such technologies, efficient and accurate assembly of large repeats and closely related haplotypes remains challenging. We address these issues with Canu, a successor of Celera Assembler that is specifically designed for noisy single-molecule sequences. Canu introduces support for nanopore sequencing, halves depth-of-coverage requirements, and improves assembly continuity while simultaneously reducing runtime by an order of magnitude on large genomes versus Celera Assembler 8.2. These advances result from new overlapping and assembly algorithms, including an adaptive overlapping strategy based on tf-idf weighted MinHash and a sparse assembly graph construction that avoids collapsing diverged repeats and haplotypes. We demonstrate that Canu can reliably assemble complete microbial genomes and near-complete eukaryotic chromosomes using either PacBio or Oxford Nanopore technologies, and achieves a contig NG50 of greater than 21 Mbp on both human and Drosophila melanogaster PacBio datasets. For assembly structures that cannot be linearly represented, Canu provides graph-based assembly outputs in graphical fragment assembly (GFA) format for analysis or integration with complementary phasing and scaffolding techniques. The combination of such highly resolved assembly graphs with long-range scaffolding information promises the complete and automated assembly of complex genomes. Published by Cold Spring Harbor Laboratory Press.
URLPMID:21187386 [本文引用: 1]
Massively parallel DNA sequencing technologies are revolutionizing genomics by making it possible to generate billions of relatively short (~100-base) sequence reads at very low cost. Whereas such data can be readily used for a wide range of biomedical applications, it has proven difficult to use them to generate high-quality de novo genome assemblies of large, repeat-rich vertebrate genomes. To date, the genome assemblies generated from such data have fallen far short of those obtained with the older (but much more expensive) capillary-based sequencing approach. Here, we report the development of an algorithm for genome assembly, ALLPATHS-LG, and its application to massively parallel DNA sequence data from the human and mouse genomes, generated on the Illumina platform. The resulting draft genome assemblies have good accuracy, short-range contiguity, long-range connectivity, and coverage of the genome. In particular, the base accuracy is high (鈮99.95%) and the scaffold sizes (N50 size = 11.5 Mb for human and 7.2 Mb for mouse) approach those obtained with capillary-based sequencing. The combination of improved sequencing technology and improved computational methods should now make it possible to increase dramatically the de novo sequencing of large genomes. The ALLPATHS-LG program is available at http://www.broadinstitute. org/science/programs/genome-biology/crd.
URLPMID:26006009 [本文引用: 2]
Abstract Long-read, single-molecule real-time (SMRT) sequencing is routinely used to finish microbial genomes, but available assembly methods have not scaled well to larger genomes. We introduce the MinHash Alignment Process (MHAP) for overlapping noisy, long reads using probabilistic, locality-sensitive hashing. Integrating MHAP with the Celera Assembler enabled reference-grade de novo assemblies of Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster and a human hydatidiform mole cell line (CHM1) from SMRT sequencing. The resulting assemblies are highly continuous, include fully resolved chromosome arms and close persistent gaps in these reference genomes. Our assembly of D. melanogaster revealed previously unknown heterochromatic and telomeric transition sequences, and we assembled low-complexity sequences from CHM1 that fill gaps in the human GRCh38 reference. Using MHAP and the Celera Assembler, single-molecule sequencing can produce de novo near-complete eukaryotic assemblies that are 99.99% accurate when compared with available reference genomes.
URLPMID:22506599 [本文引用: 1]
Abstract The lion's share of bacteria in various environments cannot be cloned in the laboratory and thus cannot be sequenced using existing technologies. A major goal of single-cell genomics is to complement gene-centric metagenomic data with whole-genome assemblies of uncultivated organisms. Assembly of single-cell data is challenging because of highly non-uniform read coverage as well as elevated levels of sequencing errors and chimeric reads. We describe SPAdes, a new assembler for both single-cell and standard (multicell) assembly, and demonstrate that it improves on the recently released E+V-SC assembler (specialized for single-cell data) and on popular assemblers Velvet and SoapDeNovo (for multicell data). SPAdes generates single-cell assemblies, providing information about genomes of uncultivatable bacteria that vastly exceeds what may be obtained via traditional metagenomics studies. SPAdes is available online ( http://bioinf.spbau.ru/spades ). It is distributed as open source software.
URL [本文引用: 1]
The highly anticipated transition from next generation sequencing (NGS) to third generation sequencing (3GS) has been difficult primarily due to high error rates and excessive sequencing cost. The high error rates make the assembly of long erroneous reads of large genomes challenging because existing software solutions are often overwhelmed by error correction tasks. Here we report a hybrid assembly approach that simultaneously utilizes NGS and 3GS data to address both issues. We gain advantages from three general and basic design principles: (i) Compact representation of the long reads leads to efficient alignments. (ii) Base-level errors can be skipped; structural errors need to be detected and corrected. (iii) Structurally correct 3GS reads are assembled and polished. In our implementation, preassembled NGS contigs are used to derive the compact representation of the long reads, motivating an algorithmic conversion from ade Bruijngraph to an overlap graph, the two major assembly paradigms. Moreover, since NGS and 3GS data can compensate for each other, our hybrid assembly approach reduces both of their sequencing requirements. Experiments show that our software is able to assemble mammalian-sized genomes orders of magnitude more quickly than existing methods without consuming a lot of memory, while saving about half of the sequencing cost.
URL [本文引用: 3]
URLPMID:24653210 [本文引用: 1]
Conifers are the predominant gymnosperm. The size and complexity of their genomes has presented formidable technical challenges for whole-genome shotgun sequencing and assembly. We employed novel strategies that allowed us to determine the loblolly pine (Pinus taeda) reference genome sequence, the largest genome assembled to date. Most of the sequence data were derived from whole-genome shotgun sequencing of a single megagametophyte, the haploid tissue of a single pine seed. Although that constrained the quantity of available DNA, the resulting haploid sequence data were well-suited for assembly. The haploid sequence was augmented with multiple linking long-fragment mate pair libraries from the parental diploid DNA. For the longest fragments, we used novel fosmid DiTag libraries. Sequences from the linking libraries that did not match the megagametophyte were identified and removed. Assembly of the sequence data were aided by condensing the enormous number of paired-end reads into a much smaller set of longer uper-reads, rendering subsequent assembly with an overlap-based assembly algorithm computationally feasible. To further improve the contiguity and biological utility of the genome sequence, additional scaffolding methods utilizing independent genome and transcriptome assemblies were implemented. The combination of these strategies resulted in a draft genome sequence of 20.15 billion bases, with an N50 scaffold size of 66.9 kbp.
[本文引用: 1]
[本文引用: 1]
URLPMID:4471408 [本文引用: 1]
Illumina Sequencing data can provide high coverage of a genome by relatively short (most often 100 bp to 150 bp) reads at a low cost. Even with low (advertised 1%) error rate, 100 coverage Illumina data on average has an error in some read at every base in the genome. These errors make handling the data more complicated because they result in a large number of low-count erroneousk-mers in the reads. However, there is enough information in the reads to correct most of the sequencing errors, thus making subsequent use of the data (e.g. for mapping or assembly) easier. Here we use the term rror correction to denote the reduction in errors due to both changes in individual bases and trimming of unusable sequence. We developed an error correction software called QuorUM. QuorUM is mainly aimed at error correcting Illumina reads for subsequent assembly. It is designed around the novel idea of minimizing the number of distinct erroneousk-mers in the output reads and preserving the most truek-mers, and we introduce a composite statistic that measures how successful we are at achieving this dual goal. We evaluate the performance of QuorUM by correcting actual Illumina reads from genomes for which a reference assembly is available. We produce trimmed and error-corrected reads that result in assemblies with longer contigs and fewer errors. We compared QuorUM against several published error correctors and found that it is the best performer in most metrics we use. QuorUM is efficiently implemented making use of current multi-core computing architectures and it is suitable for large data sets (1 billion bases checked and corrected per day per core). We also demonstrate that a third-party assembler (SOAPdenovo) benefits significantly from using QuorUM error-corrected reads. QuorUM error corrected reads result in a factor of 1.1 to 4 improvement in N50 contig size compared to using the original reads with SOAPdenovo for the data sets investigated. QuorUM is distributed as an independent software package and as a module of the MaSuRCA assembly software. Both are available under the GPL open source license athttp://www.genome.umd.edu. gmarcais@umd.edu.
URLPMID:395750 [本文引用: 1]
The newest version of MUMmer easily handles comparisons of large eukaryotic genomes at varying evolutionary distances, as demonstrated by applications to multiple genomes. Two new graphical viewing tools provide alternative ways to analyze genome alignments. The new system is the first version of MUMmer to be released as open-source software. This allows other developers to contribute to the code base and freely redistribute the code. The MUMmer sources are available at http://www.tigr.org/software/mummer .
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
URLPMID:27749838 [本文引用: 1]
Abstract While genome assembly projects have been successful in many haploid and inbred species, the assembly of noninbred or rearranged heterozygous genomes remains a major challenge. To address this challenge, we introduce the open-source FALCON and FALCON-Unzip algorithms (https://github.com/PacificBiosciences/FALCON/) to assemble long-read sequencing data into highly accurate, contiguous, and correctly phased diploid genomes. We generate new reference sequences for heterozygous samples including an F1 hybrid of Arabidopsis thaliana, the widely cultivated Vitis vinifera cv. Cabernet Sauvignon, and the coral fungus Clavicorona pyxidata, samples that have challenged short-read assembly approaches. The FALCON-based assemblies are substantially more contiguous and complete than alternate short- or long-read approaches. The phased diploid assembly enabled the study of haplotype structure and heterozygosities between homologous chromosomes, including the identification of widespread heterozygous structural variation within coding sequences.
URL [本文引用: 1]
Recent developments in third-gen long read sequencing and diploid-aware assemblers have resulted in the rapid release of numerous reference-quality assemblies for diploid genomes. However, assembling highly heterozygous genomes is still facing a major problem where the two haplotypes for a region are highly polymorphic and the synteny is not recognised during assembly. This causes issues with downstream analysis, for example variant discovery using the haploid assembly, or haplotype reconstruction using the diploid assembly. A new pipeline--Purge Haplotigs--was developed specifically for third-gen assemblies to identify and reassign the duplicate contigs. The pipeline takes a draft haplotype-fused assembly or a diploid assembly, and read alignments to produce an improved assembly. The pipeline was tested on a simulated dataset and on four recent diploid (phased) de novo assemblies from third-generation long-read sequencing. All assemblies after processing with Purge Haplotigs were less duplicated with minimal impact on genome completeness. The software is available at https://bitbucket.org/mroachawri/purge_haplotigs under a permissive MIT licence.
[本文引用: 1]
.
[本文引用: 1]
URLPMID:28902843 [本文引用: 1]
Constituting approximately 10% of flowering plant species, orchids (Orchidaceae) display unique flower morphologies, possess an extraordinary diversity in lifestyle, and have successfully colonized almost every habitat on Earth. Here we report the draft genome sequence of Apostasia shenzhenica, a representative of one of two genera that form a sister lineage to the rest of the Orchidaceae, providing a reference for inferring the genome content and structure of the most recent common ancestor of all extant orchids and improving our understanding of their origins and evolution. In addition, we present transcriptome data for representatives of Vanilloideae, Cypripedioideae and Orchidoideae, and novel third-generation genome data for two species of Epidendroideae, covering all five orchid subfamilies. A. shenzhenica shows clear evidence of a whole-genome duplication, which is shared by all orchids and occurred shortly before their divergence. Comparisons between A. shenzhenica and other orchids and angiosperms also permitted the reconstruction of an ancestral orchid gene toolkit. We identify new gene families, gene family expansions and contractions, and changes within MADS-box gene classes, which control a diverse suite of developmental processes, during orchid evolution. This study sheds new light on the genetic mechanisms underpinning key orchid innovations, including the development of the labellum and gynostemium, pollinia, and seeds without endosperm, as well as the evolution of epiphytism; reveals relationships between the Orchidaceae subfamilies; and helps clarify the evolutionary history of orchids within the angiosperms.
[本文引用: 1]
URL [本文引用: 1]
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
URL [本文引用: 1]
URLPMID:12034836 [本文引用: 1]
We describe a suffix-tree algorithm that can align the entire genome sequences of eukaryotic and prokaryotic organisms with minimal use of computer time and memory. The new system, MUMmer 2, runs three times faster while using one-third as much memory as the original MUMmer system. It has been used successfully to align the entire human and mouse genomes to each other, and to align numerous smaller eukaryotic and prokaryotic genomes. A new module permits the alignment of multiple DNA sequence fragments, which has proven valuable in the comparison of incomplete genome sequences. We also describe a method to align more distantly related genomes by detecting protein sequence homology. This extension to MUMmer aligns two genomes after translating the sequence in all six reading frames, extracts all matching protein sequences and then clusters together matches. This method has been applied to both incomplete and complete genome sequences in order to detect regions of conserved synteny, in which multiple proteins from one organism are found in the same order and orientation in another. The system code is being made freely available by the authors.
URLPMID:2231712 [本文引用: 1]
Tire discovery of sequence Immology to a known protein or family of proteins often provides tire first clues about, the fimction of a newly sequenced gene. As tire DNA and amino acid sequence databases continue to grow in size they become increasingly nsefld in tim analysis of newly sequenced genes and proteins because of the greater chance of finding such homologies. There are a number of software tools for searching sequence databases but all use some measure of similarity between sequences to distinguish biologically significant relationships from chance similarities. Perhai)s the best studied measures are those used in conjunction with varia- tions of the dynamic programming algorithm (Needleman & Wunsch, 1970; Sellers, 1974; Sankoff & Kruskal, 1983; Waterman, 1984). These methods assign scores to insertions, deletions and replace- ments, and compute an aligmnent of two" sequences that corresI)onds to the least costly set of such mutations. Such an alignment may be thought of as minimizing the evolutionary distance or maximizing the similarity between tire two sequences compared. In either case, the cost of this alignment is a measure of similarity; tim algorithm guarantees' it is
URL [本文引用: 2]
URL [本文引用: 1]
URLPMID:23185243 [本文引用: 1]
Abstract Many genomes have been sequenced to high-quality draft status using Sanger capillary electrophoresis and/or newer short-read sequence data and whole genome assembly techniques. However, even the best draft genomes contain gaps and other imperfections due to limitations in the input data and the techniques used to build draft assemblies. Sequencing biases, repetitive genomic features, genomic polymorphism, and other complicating factors all come together to make some regions difficult or impossible to assemble. Traditionally, draft genomes were upgraded to "phase 3 finished" status using time-consuming and expensive Sanger-based manual finishing processes. For more facile assembly and automated finishing of draft genomes, we present here an automated approach to finishing using long-reads from the Pacific Biosciences RS (PacBio) platform. Our algorithm and associated software tool, PBJelly, (publicly available at https://sourceforge.net/projects/pb-jelly/) automates the finishing process using long sequence reads in a reference-guided assembly process. PBJelly also provides "lift-over" co-ordinate tables to easily port existing annotations to the upgraded assembly. Using PBJelly and long PacBio reads, we upgraded the draft genome sequences of a simulated Drosophila melanogaster, the version 2 draft Drosophila pseudoobscura, an assembly of the Assemblathon 2.0 budgerigar dataset, and a preliminary assembly of the Sooty mangabey. With 24 mapped coverage of PacBio long-reads, we addressed 99% of gaps and were able to close 69% and improve 12% of all gaps in D. pseudoobscura. With 4 mapped coverage of PacBio long-reads we saw reads address 63% of gaps in our budgerigar assembly, of which 32% were closed and 63% improved. With 6.8 mapped coverage of mangabey PacBio long-reads we addressed 97% of gaps and closed 66% of addressed gaps and improved 19%. The accuracy of gap closure was validated by comparison to Sanger sequencing on gaps from the original D. pseudoobscura draft assembly and shown to be dependent on initial reference quality.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}