 ,1,2
,1,2The inheritance and reprogramming of chromatin structure in early animal embryos
Yuwen Ke1,2, Jiang Liu,1,2通讯作者:
编委: 高绍荣
收稿日期:2018-07-4修回日期:2018-10-8网络出版日期:2018-11-20
| 基金资助: |
Received:2018-07-4Revised:2018-10-8Online:2018-11-20
| Fund supported: |
作者简介 About authors
柯玉文,博士,副研究员,研究方向:表观遗传E-mail:keyw@big.ac.cn。

摘要
关键词:
Abstract
Keywords:
PDF (443KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
柯玉文, 刘江. 动物早期胚胎发育中染色质结构的继承和重编程[J]. 遗传, 2018, 40(11): 977-987 doi:10.16288/j.yczz.18-189
Yuwen Ke, Jiang Liu.
多细胞生物个体里的绝大部分细胞都携带同样的遗传物质DNA,然而不同类型细胞的形态、功能却千差万别。这种异质性是由各种细胞不同的基因表达模式造成的,不同的基因表达模式由表观遗传所调控从而决定细胞命运。表观遗传是指由非DNA序列改变引起的、可遗传的基因表达调控方式,主要包括DNA修饰、组蛋白修饰(histone modification)、非编码RNA (no-coding RNA)、染色质开放性(chromatin accessibility)及染色质三维高级结构(3D chromatin organization)等[1,2,3]。表观遗传广泛参与分化、发育、肿瘤发生等细胞生理过程。
两性生殖动物生命起始于卵子和精子的结合,经过细胞分裂、基因正确有序激活表达等过程形成胚胎,并进一步分化出不同的细胞类型和器官发育成新的个体。终端分化的精子和卵子的表观遗传状态有着巨大的差别,融合后形成全能性的受精卵(zygote),来自父源和母源的基因组在表观遗传状态逐步趋向一致,最终会重编程为相同的状态。这种重编程对胚胎的早期发育起着至关重要的作用,一旦出现差错会导致胚胎致死或者疾病、畸形[4, 5]。
由于早期胚胎材料的稀缺及研究手段有限,长期以来人们对早期胚胎表观遗传的继承和重编程规律不甚了解。近几年来基因组测序技术高速发展,以及各种微量高效DNA分析新技术不断涌现,例如甲基化测序(whole-genome bisulfite sequencing methylC- seq or BS-seq)[6,7,8]、染色质免疫共沉淀测序(chromatin immunoprecipitation sequencing, ChIP-seq)[9,10,11,12]、转座酶可接近性核染色质区域测序(assay for transposase accessible chromatin, ATAC-seq)[13,14,15,16]、DNA核酸酶超敏感位点测序(DNase I hypersensitive sites sequencing, DNase-seq)[17,18,19,20]、高通量染色质构象捕获测序(high input chromosome conformation capture, Hi-C)[21,22,23,24,25]等。这些技术使得研究早期胚胎表观遗传动态变化成为可能。鉴于脊椎动物早期胚胎的甲基化、组蛋白修饰的重编程规律和调控作用已有多篇优秀文章研究讨论[3, 26~28],本文重点介绍近年来动物早期胚胎发育中染色质开放性及染色质三维高级结构方面重编程规律的研究进展。
1 染色质开放性重编程
在真核生物中,顺式调控元件(cis-regulatory elements, 包括启动子promoter、增强子enhancer、沉默子silencer、隔离子insulator等)与细胞类型特异的转录因子(transcription factor)共同指导了细胞的命运决定和个体的发育。转录因子通过motif识别或其他蛋白质介导结合到特定的DNA片段上,发挥调控基因表达的重要作用。基因组DNA某个特定区域上的核小体包装不紧密甚至缺乏核小体,DNA暴露出来,可被核酸酶Ⅰ(DNase Ⅰ)识别并特异酶切,称之为染色质开放性或可接近性区域[29,30,31]。诸多研究表明转录因子往往结合在开放的染色质片段上。因此定位染色质的开放区域能够鉴定细胞或组织特异的调控元件和重要的转录因子,并可研究染色质的开放状态改变与基因转录的关系。目前DNase-seq或ATAC-seq方法是定位染色质开放区域的两种常用方法。DNase-seq利用核酸酶Ⅰ特异酶切无核小体包装的裸露DNA,对酶切下来的DNA小片段建库测序,得到核酸酶Ⅰ超敏感位点DHS (DNase Ⅰ hypersensitive sites)[18, 19];而ATAC-seq同样是利用转座酶Transposase不能进入核小体连接致密的地方,但能进入松散的区域并切割下暴露的DNA并同时连接上特异性的接头adapters,连接上adapters的DNA片段被分离出来用于二代测序[16]。因此,DNase-seq和ATAC-seq得到的都是全基因组尺度上处于开放状态的染色质区域。在分化的细胞或组织中,DNase- seq和ATAC-seq得到的结果基本一致。在哺乳动物早期胚胎发育过程中,染色质开放性经历了剧烈的变化,并且染色质的开放决定了受精卵全基因组转录激活(zygotic genome activation, ZGA)事件。Lu等[32]用少量细胞DNase-seq方法生成小鼠(Mus musculus)受精卵至桑椹胚(morula)时期的染色质开放性图谱发现:受精卵时期开放区域较少,仅有844个;随着胚胎发育进行,DHS数目逐步增加,尤其是8细胞时期增长显著,达到12 000多个;并且发现早期建立的DHS会一直维持至后面的发育阶段。各个时期DHS的分布情况显示:受精卵时期DHS有87.2%位于启动子区,8细胞胚胎时期DHS将近一半位于远端调控区(distal region),该结果说明基因表达随着发育进行而趋向受到多种调控元件的特异性调控。通过分析精子和卵子的DHS信号发现:两者的染色质开放程度比较低,但卵子比精子更开放一些。手动分离受精卵期间的雌雄原核分别分析父源和母源的染色质开放状态发现:在受精后7.5 h的PN3阶段受精卵中,父源基因组的开放状态已经与母源基本一致。而在后续的胚胎发育时期,两个亲本的基因组开放状态高度相似,仅仅发现一些区域的开放状态呈现亲本偏好形式,其中部分是印记基因(imprinting gene)。这结果说明受精后,父源和母源的染色质开放性便迅速重编程为相似状态。
同样使用DNase-seq方法研究人早期胚胎,结果发现DHS数量也是随着胚胎发育逐渐增加,总的趋势和小鼠高度相似,说明人和小鼠早期胚胎的染色质开放性建立模式比较保守[33]。小鼠ZGA发生在2细胞胚胎时期而人发生在8细胞胚胎时期[34],有意思的是DHS与ZGA密切相关。结合DHS和ZGA激活的基因分析发现:小鼠2细胞胚胎时期的DHS富集于启动子区域并且将近32%的ZGA激活基因的启动子具有DHS;但人4细胞时期胚胎才仅有8% ZGA基因的启动子具有DHS,而8细胞阶段达到了81%[33]。这一结果说明小鼠和人启动子区DHS的建立与ZGA密切相关。
在大部分分化组织和细胞中,使用DNase-seq和ATAC-seq得到的染色质开放性图谱基本一致[16],但在精子和早期胚胎中却有较大的差别。使用ATAC-seq方法研究小鼠精子时发现,精子里大部分启动子和增强子区处于开放状态,并与胚胎干细胞或其他成体分化细胞高度相似[35],但精子的DHS信号不强[32]。另外与DNase-seq结果不同,ATAC-seq信号在小鼠和人早期胚胎尤其是ZGA前的染色质上广泛存在[36, 37]。在小鼠早2细胞(early 2-cell) ZGA前ATAC-seq信号会发现它们富集于重复元件(repeat elements)上,并且这些信号里较强的一部分位于MERVL上或附近,与此一致的是RNA分析结果显示MERVL在早2细胞ZGA前活跃表达。总体来说,小鼠早2细胞胚胎染色质的ATAC-seq信号暗示其可能处于一种更松散的状态。而在2细胞至内细胞团(inner cell mass, ICM)时期,早期胚胎与胚胎干细胞相似,大部分ATAC-seq信号位于启动子区和远端元件区。另一方面,2细胞时期在转录终止位点下游(transcription end site, TES)有非同寻常的强烈ATAC-seq信号,这种富集在后面时期减弱,在体细胞里已经非常微弱。与ATAC-seq小鼠早期胚胎结论一致,人在ZGA前的1~4细胞时期染色质上也广泛存在ATAC peaks信号。这些1~4细胞时期的ATAC peaks很多集中在CpG含量较高的启动子区,并且大部分在8细胞时期维持,说明启动子区的提前开放与基因的未来激活相关;同时很多远端非启动子区也有很强的ATAC peaks信号,并且这些区域富集于转录因子的结合位点,然而这些ATAC peaks信号随着ZGA反而大量消失,随后胚胎细胞在很多新的调控元件位置建立起开放染色质区域。在ZGA后胚胎时期,ATAC-seq得到的染色质开放性图谱与DNase-seq比较类似,例如在人胚胎中,2细胞时期ATAC peaks有22 977个,但DHS仅有729个,8细胞时期两者的差异就小,分别为40 426个和39 813个[33, 37]。这说明早期胚胎ZGA前的染色质结构非常独特,不同的检测方法得到的信号不同,哪种方法得到的信号更接近真实的染色质开放状态有待进一步的验证。
转录因子在早期胚胎染色质开放性建立及启动ZGA发挥着至关重要的作用。OCT4是一种干细胞多能性维持的重要转录因子,在胚胎发育、胚层分化中起着重要的调控作用,但它在小鼠和人中的作用规律不完全相同[38]。分析早期胚胎OCT4基因上的DHS位点发现,在人中DHSs位于OCT4基因内区域,而在小鼠中位于启动子区域[33]。通过转录因子结合片段富集分析(TF-binding motif enrichment analysis)发现OCT4在人ZGA期(8细胞期)高度富集在DHS上,相比之下,OCT4在小鼠ZGA(2细胞期)期是不富集在DHSs上或者富集很少,这个结果暗示OCT4可能在人ZGA中具有重要作用[33]。敲低OCT4 (siOCT4)实验证明了这一点:OCT4表达下降后,人8细胞期近25%的ZGA基因表达随之下调,其中85%下调基因的启动子区在8细胞期已建立DHS;但小鼠4细胞期只有1.4%的ZGA基因表达随之下调,说明OCT4及其密切相关的DHS在人ZGA事件中发挥了重要的作用而对小鼠ZGA影响较小[33]。另外,研究还发现在小鼠中OCT4表达下降后,部分8细胞时期获得的DHS消失或减弱,并且这些DHS较多富集于远端调控区,暗示OCT4在小鼠胚胎中参与基因的远端调控作用[32]。这些结果说明OCT4在小鼠和人ZGA事件和胚胎发育中发挥不同的调控作用。同样通过富集分析发现:小鼠2细胞启动子DHS富集于Nfya结合片段。Nfya是Nfy复合物的一个亚基,该复合物能结合到基因组上并促进染色质开放[39]。在小鼠胚胎中敲低Nfya后,约28.4%的2细胞DHS减弱并且减弱的DHS富集于启动子区,同时发现15.1%的ZGA基因表达下调,说明Nfya在小鼠中参与部分启动子DHS的形成及ZGA的激活[32]。总之,转录因子广泛参与哺乳动物早期胚胎DHS的建立,调控着ZGA事件和胚胎发育。还有哪些转录因子在其中发挥作用有待于进一步研究。
胚胎发育早期的染色质开放性与重复元件表达活跃密切相关。小鼠和人基因组包含着大量的重复元件,重复元件在物种进化中具有非常重要的作用,部分重复元件在早期胚胎中转录活跃但在终端分化的细胞、组织中不表达[40,41,42]。与此对应的是无论是ATAC-seq还是DNase-seq信号在早期胚胎中均发现有富集于重复元件上[33, 36, 37]。例如在小鼠早2细胞时期,富集于重复元件特别是MERVL上;小鼠2细胞时期基因启动子和远端调控区的染色质开放信号特异地集中在基因组的重复序列上,尤其是SINE、ERVL最为富集,并与此时期重复序列表达较活跃相关联。在人中部分转座元件例如:HERV-K、HERV-H、SVA、AluS,在早期胚胎中高表达而在分化组织中没有表达。分析人早期胚胎DNase-seq信号发现相较于其他分化组织,这些转座子区域的DHS在早期胚胎更加富集,说明转座子活化与染色质开放性相关联[33]。同样在人8细胞时期,远端调控区的ATAC- seq信号特异地集中在Alu、SINE、LTR,而ERVK,SVA和ERV1同时富集于8细胞时期和ICM[37]。这些结果说明早期胚胎重复元件的转录活跃与其区域上染色质的开放性密切相关。
在哺乳动物早期胚胎发育过程中,DNA甲基化会经历一个重编程的过程:精子和卵子中的DNA甲基化程度都很高并且精子的甲基化水平显著高于卵子;受精后父源和母源基因组上甲基化均被大规模擦除,到植入前的囊胚阶段,胚胎的DNA甲基化水平降到最低点;当胚胎着床以后,通过从头甲基化(de novo methylation)的方式,胚胎的DNA甲基化水平渐渐升高[43,44,45]。分析DHSs与DNA甲基化的相关性发现,开放区域富集于低甲基化区域,具有DHS信号的区域的甲基化水平比没有DHS信号区域的低[33];无论DNase-seq还是ATAC-seq都发现人早期胚胎中高开放性的启动子一般都是高CpG密度,而低开放性的启动子一般都是低CpG密度[33, 37];同时发现高CpG的启动子区域趋向于在早期发育阶段建立DHSs,而低CpG的启动子区域趋向则相反[33]。ZGA前检测到的ATAC-seq信号很大程度上与卵子的部分甲基化区域(partially methylated domains, PMDs)重合,例如人2细胞胚胎有73%的ATAC peaks与PMD重合[37]。另外通过小鼠实验发现,雌原核的远端ATAC peaks与卵子PMD区域及受精卵母源非经典H3K3me3宽峰有极大程度的重合,而雄原核则没有。ZGA后,PMD区域上的ATAC开放信号和非经典H3K3me3均消失。父源基因组上ZGA前建立的H3K3me3也与父源的ATAC开放信号相重叠。这些结果说明染色质开放性与DNA甲基化及组蛋白修饰有一定程度上的相关性。
综上所述,小鼠和人在早期胚胎发育过程中的染色质开放性总体建立模式非常相似,都经历了逐步建立的过程,并且与ZGA密切相关。
2 染色质三维高级结构重编程
在高等真核生物细胞里,例如人线性非组装DNA长度可达2 m,而细胞核直径仅有10 μm。为适应有限的细胞核空间,在组蛋白及其他蛋白介导下,染色质经过非常复杂有序的折叠形成多层次立体空间结构,同时正确地按照时空顺序执行功能以确保生物机体正常生活及生长发育。染色质三维高级结构是表观遗传非常重要的部分,在基因组调控中发挥着非常重要的作用,如基因表达、DNA复制、分裂期基因组从母代传递到子代、维持基因组稳定性等[46,47,48]。目前一般认为染色质按不同长度尺度分层次组装:染色质疆域(chromosome territories),即不同的染色质在细胞核内折叠、占据不同的特定的区域;活性或惰性区室(A/B-compartment)结构,与基因表达活性密切相关,活性的开放的A-compartment的基因往往表达水平较高,而惰性的封闭的B-compartment基因表达水平较低;拓扑相关区域(topologically associating domains, TADs)结构,染色质上一段连续的区域,此区域上的所有位点(loci)互相之间相互作用频率高,但与区域外上的位点相互作用极少,TADs普遍存在,目前普遍认为TAD结构是基本的结构和功能单元;亚TAD结构及环状(loop)结构,具有组织特异性,亚TAD结构与功能元件间的特殊loop相互作用直接相关,loop相互作用很普遍,常发生在活跃基因启动子、活跃增强子及CTCF结合位点上。TAD和compartment是染色质三维高级结构的基础组成部分,存在于所有的体细胞、组织和体外培养细胞株中。TAD结构在种属间相当保守[49],胚胎干细胞(ESC)分化或诱导多能干细胞(iPSC)转化过程中,TAD结构基本不变,仅有小部分compartment发生活性(A)/惰性(B)状态改变[50, 51]。在细胞有丝分裂期,基本检测不到染色体的TAD和compartment结构[52]。传统上主要借助于显微镜研究染色质三维高级结构,但显微镜在分辨率及高通量产出方面有很大的局限性。2002年以来,发展起一类基于分子生化的染色质构象捕获技术(3C、4C、5C、Hi-C、ChIA-PET等),这些方法利用的是近距离连接反应原理,即低浓度下分子内的连接反应优先发生。通过检测基因组上任意一对位点的相对相互作用频率(relative frequency of interaction),而描绘出染色质的精细分子构象。它们超越了显微镜的分辨率局限,是一种侧面反映染色质构象的方法[53]。其中高通量全基因组染色质构象捕获技术Hi-C以能检测全基因组的相互作用图谱及其高通量特性,得到较为广泛的应用。通过改进Hi-C方法适用于少量细胞,能帮助研究人员了解动物早期胚胎发育中的染色质三维高级结构动态变化规律。图1
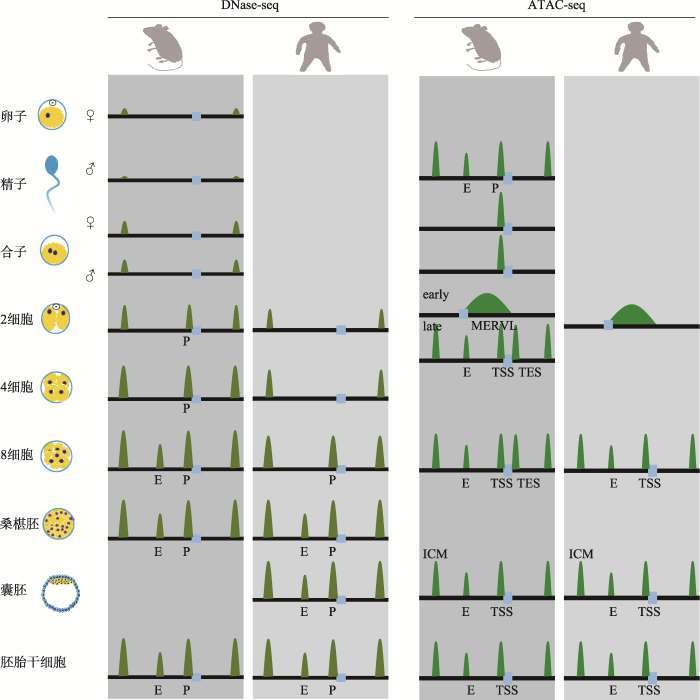 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1染色质开放性在小鼠和人早期胚胎发育的重编程
左侧部分显示DNase-seq方法检测到小鼠和人早期胚胎中的染色质开放状态模式,DNase-seq信号在ZGA前基本检测不到;右侧部分显示ATAC-seq方法检测到小鼠和人早期胚胎中的染色质开放状态模式,ATAC-seq信号在ZGA前的染色质上广泛存在。在ZGA后,DNase-seq与ATAC-seq得到的染色质开放性图谱比较类似。P代表启动子(promoter);E代表增强子(enhancer);TSS代表转录起始位点(transcription start site);TES代表转录终止位点(transcription end site)。
Fig. 1The reprogramming of chromatin accessibility during early embryo development in the mouse and human
小鼠成熟精子里染色质上大部分组蛋白被精蛋白取代,仅保留约1%的组蛋白。小鼠成熟卵子停滞于第二次减数分裂中期(MⅡ),受精后才完成分裂排出第二极体。显微镜形态学观察结果显示:小鼠配子和体细胞的染色质结构具有显著性差异,精子、卵子染色质形态高度浓缩紧致,而体细胞间期的染色质比较松散。然而通过Hi-C测序方法得到的小鼠精子高分辨率染色质三维构象图谱结果却出乎意料,结果显示:小鼠精子中存在典型的TAD和compartment结构,无论是TAD数目、TAD结构模式还是compartment模式与小鼠体细胞的高度相似[35, 54, 55]。对精子染色质结构进行进一步的分析发现:精子同一条染色质内普遍存在着超远程(大于4 Mb)的相互作用,这种超远程的相互作用往往以TAD为基本单元,发生在TAD-TAD间;同时还发现精子不同染色质间的相互作用频率相对其他胚胎时期也高得多[54],这些结果非常符合精子染色质高度紧缩的生理特点。小鼠未成熟卵细胞(non-surrounded nucleolus, NSN)的染色质结构与体细胞的很相似,具有典型的loop、TAD、compartment结构[56];成熟为卵子(surrounded- nucleolus, SN)过程中,逐渐关闭转录,染色质与核膜连接消失,loop、TAD、compartment强度逐渐降低;最终成熟卵子染色质呈现出一种均一性结构,并且缺乏TADs和compartment结构[54, 55],卵子的这种均一性结构与细胞分裂期染色质结构高度相似,这非常符合成熟卵子处于MⅡ期的生理特点。
在精子和卵子结合形成受精卵的过程中,父源染色质上的精蛋白替换回组蛋白,母源染色质解压缩。利用电子光谱成像研究小鼠早期胚胎发现:受精卵时期染色质呈现普遍消散(dispersed)状态,在核膜附近没有明显的聚集;2细胞时期开始出现离散凝集区块,在核膜附近聚集开始增多;在8细胞时期和多能外胚层细胞里,染色质呈现纤维网状结构,与胚胎干细胞的极度相似[57]。小鼠早期胚胎高分辨率染色质三维构象图谱与此成像表型相一致,三维构象图谱分析结果显示[54, 55]:受精后,染色质的三维结构迅速呈现为一种极其松散的状态;受精卵和2细胞时期胚胎的TAD和compartment结构不清晰;清晰的TAD和compartment结构随着胚胎发育的进行逐步建立起来;8细胞时期胚胎具有了比较经典的TAD和compartment结构。值得注意的是,分析受精后处于细胞间期的受精卵和2细胞时期胚胎发现:这两个时期胚胎细胞里的染色质竟然没有明显的TAD和compartment结构,这是一种非常独特的结构。进一步分析发现,这种结构与其他处于细胞间期的体细胞或体外培养细胞株的染色质结构都不同,与有丝分裂期染色体结构也显著不同。仔细比对分析受精卵时期和胚胎发育后面时期的染色质相互作用图谱发现:在后面时期的典型TAD的位置上,受精卵在该位置有较高频率的相互作用(相对于背景而言),可以称之为TAD雏形或者类TAD结构,同样受精卵时期存在着compartment结构雏形[55, 56]。随着胚胎?发育的进行,TAD雏形内部的相互作用频率增高,与TAD外部的相互作用减少,边界隔绝(insulation of boundary)效应增强,compartment 强度增加,直至8细胞时期胚胎形成经典的TAD和compartment结构。
精子和卵子的染色质三维高级结构差异显著,受精后父源和母源的染色质结构会发生怎样的变化呢?利用品系间的单核苷酸多态性(single nucleotide polymorphisms, SNPs)分别对来源于父源和母源的基因组进行研究发现,两者染色质结构建立模式高度相似:同样在受精卵和2细胞时期胚胎中没有清晰的TAD和compartment结构,随着发育的进行逐渐地建立起来经典的TAD和compartment结构[54, 55]。在PN3、PN5阶段的受精卵中,发现父源染色质的远距离相互作用比母源的少一些,意味着受精卵中父源染色质在组蛋白替换后迅速呈现更为松散的状态。而在compartment结构上,受精卵中父源和母源的染色质也有细微差别,父源染色质compartment强度要高一些,这种差别一直持续到8细胞[56, 58, 59]。总的来说,受精后父源和母源染色质迅速呈现相似而又有微小差别的结构,发育至胚胎后期已基本一致,仅在一些等位特异(allele-specific)区域例如印记基因[54]具有结构微小差异。
小鼠ZGA发生在晚2细胞期(late 2-cell),TAD和compartment结构伴随着ZGA的发生而逐步建立。在果蝇(Drosophila melanogaster)胚胎发育中也同样发现TAD和compartment结构伴随着ZGA的发生而逐步建立。果蝇受精后仅细胞核进行分裂,并没有细胞质的分裂,当细胞核分裂至第13个周期(nuclear cycle, nc13)时才形成细胞膜。在nc14发生ZGA,同时果蝇胚胎里染色质的TAD和compartment结构开始逐步建立[60]。使用药物α-amanitin处理小鼠受精卵,抑制ZGA的发生,发现染色质仍然能够建立TAD和compartment结构,说明染色质三维高级结构的建立不依赖于ZGA[54, 55]。同样,果蝇早期胚胎染色质三维高级结构的建立不依赖于ZGA[60]。染色质三维高级结构的逐步建立伴随着ZGA发生
但又同时不依赖于ZGA这一规律特点是否在其他物种中也存在还有待于进一步验证,各物种早期胚胎染色质三维高级结构的建立机制还知之甚少,需要更多的研究探索。另一方面通过药物aphidicolin抑制小鼠受精卵DNA复制,发现不能建立TAD和compartment结构,说明染色质三维高级结构的建立依赖于基因组的复制[54]。小鼠受精卵复制分裂至2细胞时期而果蝇复制分裂至nc14开始建立染色质三维高级结构,这其中蕴含着怎样的调节机制还有待进一步探索。
胚胎干细胞的分化过程和体细胞重编程为iPSCs过程都会发生部分compartment状态转换,及伴随基因表达的协同变化。与此现象相一致,在胚胎发育过程中同样有部分compartment发生了A/B状态的转换。使用基因本体(Gene Ontology, GO)方法分析时期特异(stage-specific) compartment上的基因发现:这些基因富集于生殖系统发育或胚胎发育过程,这说明染色质三维高级结构参与调控生殖发育及胚胎发育的进行[54]。分析染色质三维高级结构与其他表观遗传信息发现有着密切的关联[54]:例如非甲基化CpG相对比较富集于A-compartment;A-compartment上的启动子(promoter)有60%处于非甲基化状态,而B-compartment上的启动子仅有20%处于非甲基化状态。总之,非甲基化启动子往往调控基因更好的表达,位于A-compartment的基因一般表达水平更高。早期胚胎发育会发生全基因组的去甲基化,分析发现A-compartment上的CpG甲基化的下降水平比B-compartment上的CpG甲基化的下降水平要高,暗示A-compartment更利于DNA甲基转移酶接近执行去甲基化功能[54, 58]。通过整合小鼠早期胚胎染色质开放性数据发现,各时期DHSs富集于A-compartment,同时某个特定胚胎时期新获得的DHS位点也是富集于A-compartment。总而言之,A-compartment甲基化程度更低,开放性更好,转录更活跃。
综上所述,早期胚胎染色质具有独特的结构,伴随着ZGA发生逐步建立起经典的高级结构。这种建立模式在其他种属生物是否一样,以及何种机制指导胚胎细胞建立正确的染色质结构,都是亟待解决的问题。
图2
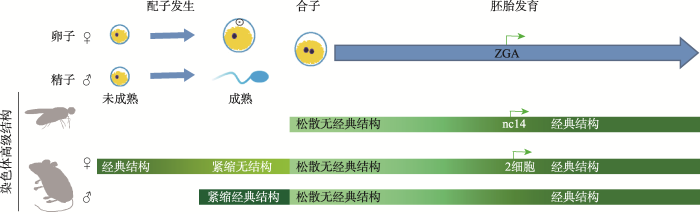 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2染色质三维高级结构在早期胚胎的重编程
果蝇和小鼠早期胚胎发育过程中,伴随着ZGA发生逐步建立起经典的染色质三维高级结构。ZGA代表受精卵基因组转录激活。nc14代表细胞核分裂第14个周期。
Fig. 2The reprogramming of chromatin high-order structures during embryogenesis
3 结语与展望
动物早期胚胎发育是一个非常特异的过程,终端分化的配子融合形成全能性的胚胎,最终分化形成各种细胞、组织和器官,除了DNA序列,表观遗传信息、RNA、蛋白质、信号通路网络、代谢甚至细胞形态都发生了剧烈的变化。微量DNA分析技术的不断发展使得早期胚胎的表观遗传图谱初步浮现,然而还有太多的谜题等待解答:表观遗传信息有些能遗传到下一代而有些不能,是什么在其中发挥了选择作用?早期胚胎中在没有模板的情况下,某些表观遗传信息是如何建立起新的模式?表观遗传信息之间更深层次的关联以及如何协调指导早期胚胎发育等等,这些都是仍然有待阐明的热点问题。另一方面,近年来蓬勃发展的单细胞转录组测序技术显示即使同类型细胞也存在细胞-细胞之间的异质性,这也暗示着细胞-细胞之间表观遗传调控的异质性。单细胞转录组技术显示高度对称相似的早期胚胎4细胞时期胚胎已呈现细胞-细胞之间表达谱的异质性[61]。单细胞转录组和表观遗传信息整合分析将帮助我们研究何种因素影响细胞个体基因表达模式,每个细胞将要分化形成的细胞类型,哪些关键基因和表观遗传信息驱动发育阶段前进,帮助我们全面了解绘制早期胚胎发育的表观遗传图谱及细胞命运决定的调控机理。因此单细胞表观遗传测序分析技术将是未来研究热点之一。早期胚胎表观遗传的研究有助于人们全面认识人早期胚胎发育机制,为解决临床生殖医学的各种问题提供线索,从而提高人口质量。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URL [本文引用: 1]
.
URL [本文引用: 1]
.
URL [本文引用: 2]

Abstract Drastic epigenetic reprogramming takes place during preimplantation development, leading to the conversion of terminally differentiated gametes to a totipotent embryo. Deficiencies in remodeling of the epigenomes can cause severe developmental defects, including embryonic lethality. However, how chromatin modifications and chromatin organization are reprogrammed upon fertilization in mammals has long remained elusive. Here, we review recent progress in understanding how the epigenome is dynamically regulated during early mammalian development. The latest studies, including many from genome-wide perspectives, have revealed unusual principles of reprogramming for histone modifications, chromatin accessibility, and 3D chromatin architecture. These advances have shed light on the regulatory network controlling the earliest development and maternal-zygotic transition.
.
URL [本文引用: 1]
.
URL [本文引用: 1]
.
URL [本文引用: 1]
.
URLPMID:23828890 [本文引用: 1]
DNA methylation is implicated in mammalian brain development and plasticity underlying learning and memory. We report the genome-wide composition, patterning, cell specificity, and dynamics of DNA methylation at single-base resolution in human and mouse frontal cortex throughout their lifespan. Widespread methylome reconfiguration occurs during fetal to young adult development, coincident with synaptogenesis. During this period, highly conserved non-CG methylation (mCH) accumulates in neurons, but not glia, to become the dominant form of methylation in the human neuronal genome. Moreover, we found an mCH signature that identifies genes escaping X-chromosome inactivation. Last, whole-genome single-base resolution 5-hydroxymethylcytosine (hmC) maps revealed that hmC marks fetal brain cell genomes at putative regulatory regions that are CG-demethylated and activated in the adult brain and that CG demethylation at these hmC-poised loci depends on Tet2 activity.
.
URLPMID:334363962285043916846 [本文引用: 1]
A high-resolution, genome-wide view of methylation in the mouse brain reveals sequence signatures for methylation at canonical CG sites, methylation at noncanonical sequences, and new information about imprinted alleles that are differentially methylated.
.
URLPMID:4636926 [本文引用: 1]
Chromatin profiling provides a versatile means to investigate functional genomic elements and their regulation. However, current methods yield ensemble profiles that are insensitive to cell-to-cell variation. Here we combine microfluidics, DNA barcoding and sequencing to collect chromatin data at single-cell resolution. We demonstrate the utility of the technology by assaying thousands of individual cells, and using the data to deconvolute a mixture of ES cells, fibroblasts and hematopoietic progenitors into high-quality chromatin state maps for each cell type. The data from each single cell is sparse, comprising on the order of 1000 unique reads. However, by assaying thousands of ES cells, we identify a spectrum of sub-populations defined by differences in chromatin signatures of pluripotency and differentiation priming. We corroborate these findings by comparison to orthogonal single-cell gene expression data. Our method for single-cell analysis reveals aspects of epigenetic heterogeneity not captured by transcriptional analysis alone.
.
URLPMID:25607992 [本文引用: 1]
Combined chromatin immunoprecipitation and next-generation sequencing (ChIP-seq) has enabled genome-wide epigenetic profiling of numerous cell lines and tissue types. A major limitation of ChIP-seq, however, is the large number of cells required to generate high-quality data sets, precluding the study of rare cell populations. Here, we present an ultra-low-input micrococcal nuclease-based native ChIP (ULI-NChIP) and sequencing method to generate genome-wide histone mark profiles with high resolution from as few as 10cells. We demonstrate that ULI-NChIP-seq generates high-quality maps of covalent histone marks from 10to 10embryonic stem cells. Subsequently, we show that ULI-NChIP-seq H3K27me3 profiles generated from E13.5 primordial germ cells isolated from single male and female embryos show high similarity to recent data sets generated using 50 180 more material. Finally, we identify sexually dimorphic H3K27me3 enrichment at specific genic promoters, thereby illustrating the utility of this method for generating high-quality and -complexity libraries from rare cell populations.
.
URLPMID:21642965 [本文引用: 1]
Abstract Genome-wide profiling of transcription factors based on massive parallel sequencing of immunoprecipitated chromatin (ChIP-seq) requires nanogram amounts of DNA. Here we describe a high-fidelity, single-tube linear DNA amplification method (LinDA) for ChIP-seq and reChIP-seq with picogram DNA amounts obtained from a few thousand cells. This amplification technology will facilitate global analyses of transcription-factor binding and chromatin with very small cell populations, such as stem or cancer-initiating cells.
.
URL [本文引用: 1]
.
URLPMID:4685948 [本文引用: 1]
Cell-to-cell variation is a universal feature of life that impacts a wide range of biological phenomena, from developmental plasticity1,2to tumor heterogeneity3. While recent advances have improved our ability to document cellular phenotypic variation4 8the fundamental mechanisms that generate variability from identical DNA sequences remain elusive. Here we reveal the landscape and principles of cellular DNA regulatory variation by developing a robust method for mapping the accessible genome of individual cells via assay for transposase-accessible chromatin using sequencing (ATAC-seq). Single-cell ATAC-seq (scATAC-seq) maps from hundreds of single-cells in aggregate closely resemble accessibility profiles from tens of millions of cells and provides insights into cell-to-cell variation. Accessibility variance is systematically associated with specifictrans-factors andcis-elements, and we discover combinations oftrans-factors associated with either induction or suppression of cell-to-cell variability. We further identify sets oftrans-factors associated with cell-type specific accessibility variance across 8 cell types. Targeted perturbations of cell cycle or transcription factor signaling evoke stimulus-specific changes in this observed variability. The pattern of accessibility variation incisacross the genome recapitulates chromosome topological domains9de novo,linking single-cell accessibility variation to three-dimensional genome organization. All together, single-cell analysis of DNA accessibility provides new insight into cellular variation of the egulome.
URLPMID:4374986 [本文引用: 1]
Abstract This unit describes Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq), a method for mapping chromatin accessibility genome-wide. This method probes DNA accessibility with hyperactive Tn5 transposase, which inserts sequencing adapters into accessible regions of chromatin. Sequencing reads can then be used to infer regions of increased accessibility, as well as to map regions of transcription-factor binding and nucleosome position. The method is a fast and sensitive alternative to DNase-seq for assaying chromatin accessibility genome-wide, or to MNase-seq for assaying nucleosome positions in accessible regions of the genome. Copyright 2015 John Wiley & Sons, Inc.
.
URLPMID:4836442 [本文引用: 1]
Technical advances have enabled the collection of genome and transcriptome data sets with single-cell resolution. However, single-cell characterization of the epigenome has remained challenging. Furthermore, because cells must be physically separated before biochemical processing, conventional single-cell preparatory methods scale linearly. We applied combinatorial cellular indexing to measure chromatin accessibility in thousands of single cells per assay, circumventing the need for compartmentalization of individual cells. We report chromatin accessibility profiles from more than 15,000 single cells and use these data to cluster cells on the basis of chromatin accessibility landscapes. We identify modules of coordinately regulated chromatin accessibility at the level of single cells both between and within cell types, with a scalable method that may accelerate progress toward a human cell atlas.
.
URLPMID:24097267 [本文引用: 3]
We describe an assay for transposase-accessible chromatin using sequencing (ATAC-seq), based on direct in vitro transposition of sequencing adaptors into native chromatin, as a rapid and sensitive method for integrative epigenomic analysis. ATAC-seq captures open chromatin sites using a simple two-step protocol with 500-50,000 cells and reveals the interplay between genomic locations of open chromatin, DNA-binding proteins, individual nucleosomes and chromatin compaction at nucleotide resolution. We discovered classes of DNA-binding factors that strictly avoided, could tolerate or tended to overlap with nucleosomes. Using ATAC-seq maps of human CD4(+) T cells from a proband obtained on consecutive days, we demonstrated the feasibility of analyzing an individual's epigenome on a timescale compatible with clinical decision-making.
.
URLPMID:4697938 [本文引用: 1]
DNase I hypersensitive sites (DHSs) provide important information on the presence of transcriptional regulatory elements and the state of chromatin in mammalian cells1 3. Conventional DNase-Seq for genome-wide DHSs profiling is limited by the requirement of millions of cells4,5. Here we report an ultrasensitive strategy, called Pico-Seq, for detection of genome-wide DHSs in single cells. We show that DHS patterns at the single cell level are highly reproducible among individual cells. Among different single cells, highly expressed gene promoters and the enhancers associated with multiple active histone modifications display constitutive DHS while chromatin regions with fewer histone modifications exhibit high variation of DHS. Furthermore, the single-cell DHSs predict enhancers that regulate cell-specific gene expression programs and the cell-to-cell variations of DHS are predictive of gene expression. Finally, we apply Pico-Seq to pools of tumor cells and pools of normal cells, dissected from formalin-fixed paraffin-embedded (FFPE) tissue slides from thyroid cancer patients, and detect thousands of tumor-specific DHSs. Many of these DHSs are associated with promoters and enhancers critically involved in cancer development. Analysis of the DHS sequences uncovers one single-nucleotide variant (chr18:52417839 G>C) in the tumor cells of a follicular thyroid carcinoma patient, which affects the binding of the tumor suppressor protein p53 and correlates with decreased expression of its target geneTXNL1. In conclusion, Pico-Seq can reliably detect DHSs in single cells, greatly extending the range of applications of DHS analysis for both basic and translational research and may provide critical information for personalized medicine.
URLPMID:20150147 [本文引用: 2]
INTRODUCTIONIdentification of active gene regulatory elements is a key to understanding transcriptional control governing biological processes such as cell-type specificity, differentiation, dev ...
.
URLPMID:23821440 [本文引用: 2]
Abstract DNase I-seq is a global and high-resolution method that uses the nonspecific endonuclease DNase I to map chromatin accessibility. These accessible regions, designated as DNase I hypersensitive sites (DHSs), define the regulatory features, (e.g., promoters, enhancers, insulators, and locus control regions) of complex genomes. In this unit, methods are described for nuclei isolation, digestion of nuclei with limiting concentrations of DNase I, and the biochemical fractionation of DNase I hypersensitive sites in preparation for high-throughput sequencing. DNase I-seq is an unbiased and robust method that is not predicated on an a priori understanding of regulatory patterns or chromatin features. 2013 by John Wiley & Sons, Inc.
.
URL [本文引用: 1]
.
URL [本文引用: 1]
.
URLPMID:25497547 [本文引用: 1]
Using 3D genome sequencing, we find 6510,000 DNA loops across the human genome. Loop anchors typically occur at domain boundaries and bind CTCF in a convergent orientation, with the asymmetric motifs “facing” one another. On the inactive X chromosome, large loops are anchored at CTCF-binding repeats. Loops are conserved across cell types and species.
.
URLPMID:24067610 [本文引用: 1]
Large-scale chromosome structure and spatial nuclear arrangement have been linked to control of gene expression and DNA replication and repair. Genomic techniques based on chromosome conformation capture (3C) assess contacts for millions of loci simultaneously, but do so by averaging chromosome conformations from millions of nuclei. Here we introduce single-cell Hi-C, combined with genome-wide statistical analysis and structural modelling of single-copy X chromosomes, to show that individual chromosomes maintain domain organization at the megabase scale, but show variable cell-to-cell chromosome structures at larger scales. Despite this structural stochasticity, localization of active gene domains to boundaries of chromosome territories is a hallmark of chromosomal conformation. Single-cell Hi-C data bridge current gaps between genomics and microscopy studies of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and how this structure is probabilistically linked with genome activity patterns.
.
URLPMID:28289288 [本文引用: 1]
The folding of genomic DNA from the beads-on-a-string-like structure of nucleosomes into higher-order assemblies is crucially linked to nuclear processes. Here we calculate 3D structures of entire mammalian genomes using data from a new chromosome conformation capture procedure that allows us to first image and then process single cells. The technique enables genome folding to be examined at a scale of less than 100 b, and chromosome structures to be validated. The structures of individual topological-associated domains and loops vary substantially from cell to cell. By contrast, A and B compartments, lamina-associated domains and active enhancers and promoters are organized in a consistent way on a genome-wide basis in every cell, suggesting that they could drive chromosome and genome folding. By studying genes regulated by pluripotency factor and nucleosome remodelling deacetylase (NuRD), we illustrate how the determination of single-cell genome structure provides a new approach for investigating biological processes.
.
URLPMID:5330809 [本文引用: 1]
Abstract We present single-cell combinatorial indexed Hi-C (sciHi-C), a method that applies combinatorial cellular indexing to chromosome conformation capture. In this proof of concept, we generate and sequence six sciHi-C libraries comprising a total of 10,696 single cells. We use sciHi-C data to separate cells by karyotypic and cell-cycle state differences and identify cell-to-cell heterogeneity in mammalian chromosomal conformation. Our results demonstrate that combinatorial indexing is a generalizable strategy for single-cell genomics.
.
URLPMID:25559156 [本文引用: 1]
5-Methylcytosine (5mC) is a major epigenetic modification in . The programming and inheritance of parental DNA methylomes ensures the compatibility for totipotency and . In , the DNA methylomes of sperm and oocyte are significantly different. During early , the paternal and maternal methylomes will reset to the same state. Herein, we focus on recent advances in how offspring obtain the information from parents in .
.
Magsci [本文引用: 1]
<p>DNA甲基化通过调节基因转录、印记、X染色体灭活和防御外源性遗传物质入侵等, 在细胞分化、胚胎发育、环境适应和疾病发生发展上发挥重要作用, 是当前表观遗传学研究的热点领域之一。文章介绍了在过去几年中TET介导的DNA羟甲基化及其在早期胚胎发育中的作用, DNA主动去甲基化及其与被动去甲基化的关系, DNA甲基化建立及其与组蛋白修饰、染色质构象、多梳蛋白和非编码RNA结合等关系方面的重要研究进展和存在的问题以及DNA甲基化的转化应用前景。</p>
Magsci [本文引用: 1]
<p>DNA甲基化通过调节基因转录、印记、X染色体灭活和防御外源性遗传物质入侵等, 在细胞分化、胚胎发育、环境适应和疾病发生发展上发挥重要作用, 是当前表观遗传学研究的热点领域之一。文章介绍了在过去几年中TET介导的DNA羟甲基化及其在早期胚胎发育中的作用, DNA主动去甲基化及其与被动去甲基化的关系, DNA甲基化建立及其与组蛋白修饰、染色质构象、多梳蛋白和非编码RNA结合等关系方面的重要研究进展和存在的问题以及DNA甲基化的转化应用前景。</p>
.
URL [本文引用: 1]
.
URLPMID:18059368 [本文引用: 1]
Abstract Assembly, mobilization and disassembly of nucleosomes can influence the regulation of gene expression and other processes that act on eukaryotic DNA. Distinct nucleosome-assembly pathways deposit dimeric subunits behind the replication fork or at sites of active processes that mobilize pre-existing nucleosomes. Replication-coupled nucleosome assembly appears to be the default process that maintains silent chromatin, counteracted by active processes that destabilize nucleosomes. Nucleosome stability is regulated by the combined effects of nucleosome-positioning sequences, histone chaperones, ATP-dependent nucleosome remodellers, post-translational modifications and histone variants. Recent studies suggest that histone turnover helps to maintain continuous access to sequence-specific DNA-binding proteins that regulate epigenetic inheritance, providing a dynamic alternative to histone-marking models for the propagation of active chromatin.
.
URLPMID:25473421 [本文引用: 1]
Transcriptional activation throughout the eukaryotic lineage has been tightly linked with disruption of nucleosome organization at promoters, enhancers, silencers, insulators and locus control regions due to transcription factor binding. Regulatory DNA thus coincides with open or accessible genomic sites of remodeled chromatin. Current chromatin accessibility assays are used to separate the genome by enzymatic or chemical means and isolate either the accessible or protected locations. The isolated DNA is then quantified using a next-generation sequencing platform. Wide application of these assays has recently focused on the identification of the instrumental epigenetic changes responsible for differential gene expression, cell proliferation, functional diversification and disease development. Here we discuss the limitations and advantages of current genome-wide chromatin accessibility assays with especial attention on experimental precautions and sequence data analysis. We conclude with our perspective on future improvements necessary for moving the field of chromatin profiling forward.
.
URLPMID:27259149 [本文引用: 4]
The DNase I-hypersensitive site mapping of mouse preimplantation embryos reveals how the chromatin regulatory landscape in the mouse embryos is established from differentially packaged sperm and egg genomes and identifies key transcription factors crucial for this process.
.
URLPMID:29526463 [本文引用: 11]
SummaryThe dynamics of the chromatin regulatory landscape during human early embryogenesis remains unknown. Using DNase I hypersensitive site (DHS) sequencing, we report that the chromatin accessibility landscape is gradually established during human early embryogenesis. Interestingly, the DHSs with OCT4 binding motifs are enriched at the timing of zygotic genome activation (ZGA) in humans, but not in mice. Consistently, OCT4 contributes to ZGA in humans, but not in mice. We further find that lower CpG promoters usually establish DHSs at later stages. Similarly, younger genes tend to establish promoter DHSs and are expressed at later embryonic stages, while older genes exhibit these features at earlier stages. Moreover, our data show that human active transposons SVA and HERV-K harbor DHSs and are highly expressed in early embryos, but not in differentiated tissues. In summary, our data provide an evolutionary developmental view for understanding the regulation of gene and transposon expression.
.
URL [本文引用: 1]
.
URLPMID:28178516 [本文引用: 2]
The sperm nucleus appears to be transcriptionally inert. Jung et al. find that mouse sperm contain promoters and regulatory sequences in an active chromatin state. Many transcription factors remain on sperm chromatin, including CTCF and cohesin. These proteins fold the sperm genome into a 3D architecture as in somatic cells.
.
URLPMID:27309802 [本文引用: 2]
In mammals, extensive chromatin reorganization is essential for reprogramming terminally committed gametes to a totipotent state during preimplantation development. However, the global chromatin landscape and its dynamics in this period remain unexplored. Here we report a genome-wide map of accessible chromatin in mouse preimplantation embryos using an improved assay for transposase-accessible chromatin with high throughput sequencing (ATAC-seq) approach with CRISPR/Cas9-assisted mitochondrial DNA depletion. We show that despite extensive parental asymmetry in DNA methylomes, the chromatin accessibility between the parental genomes is globally comparable after major zygotic genome activation (ZGA). Accessible chromatin in early embryos is widely shaped by transposable elements and overlaps extensively with putative cis-regulatory sequences. Unexpectedly, accessible chromatin is also found near the transcription end sites of active genes. By integrating the maps of cis-regulatory elements and single-cell transcriptomes, we construct the regulatory network of early development, which helps to identify the key modulators for lineage specification. Finally, we find that the activities of cis-regulatory elements and their associated open chromatin diminished before major ZGA. Surprisingly, we observed many loci showing non-canonical, large open chromatin domains over the entire transcribed units in minor ZGA, supporting the presence of an unusually permissive chromatin state. Together, these data reveal a unique spatiotemporal chromatin configuration that accompanies early mammalian development.
.
URLPMID:29720659 [本文引用: 6]
Abstract Upon fertilization, drastic chromatin reorganization occurs during preimplantation development 1 . However, the global chromatin landscape and its molecular dynamics in this period remain largely unexplored in humans. Here we investigate chromatin states in human preimplantation development using an improved assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) 2 . We find widespread accessible chromatin regions in early human embryos that overlap extensively with putative cis-regulatory sequences and transposable elements. Integrative analyses show both conservation and divergence in regulatory circuitry between human and mouse early development, and between human pluripotency in vivo and human embryonic stem cells. In addition, we find widespread open chromatin regions before zygotic genome activation (ZGA). The accessible chromatin loci are readily found at CpG-rich promoters. Unexpectedly, many others reside in distal regions that overlap with DNA hypomethylated domains in human oocytes and are enriched for transcription factor-binding sites. A large portion of these regions then become inaccessible after ZGA in a transcription-dependent manner. Notably, such extensive chromatin reorganization during ZGA is conserved in mice and correlates with the reprogramming of the non-canonical histone mark H3K4me3, which is uniquely linked to genome silencing 3-5 . Taken together, these data not only reveal a conserved principle that underlies the chromatin transition during mammalian ZGA, but also help to advance our understanding of epigenetic reprogramming during human early development and in vitro fertilization.
.
URLPMID:28953884 [本文引用: 1]
Abstract Despite their fundamental biological and clinical importance, the molecular mechanisms that regulate the first cell fate decisions in the human embryo are not well understood. Here we use CRISPR-Cas9-mediated genome editing to investigate the function of the pluripotency transcription factor OCT4 during human embryogenesis. We identified an efficient OCT4-targeting guide RNA using an inducible human embryonic stem cell-based system and microinjection of mouse zygotes. Using these refined methods, we efficiently and specifically targeted the gene encoding OCT4 (POU5F1) in diploid human zygotes and found that blastocyst development was compromised. Transcriptomics analysis revealed that, in POU5F1-null cells, gene expression was downregulated not only for extra-embryonic trophectoderm genes, such as CDX2, but also for regulators of the pluripotent epiblast, including NANOG. By contrast, Pou5f1-null mouse embryos maintained the expression of orthologous genes, and blastocyst development was established, but maintenance was compromised. We conclude that CRISPR-Cas9-mediated genome editing is a powerful method for investigating gene function in the context of human development.
.
URLPMID:23332751 [本文引用: 1]
The sequence-specific transcription factor NF-Y binds the CCAAT box, one of the sequence elements most frequently found in eukaryotic promoters. NF-Y is composed of the NF-YA and NF-YB/NF-YC subunits, the latter two hosting histone-fold domains (HFDs). The crystal structure of NF-Y bound to a 25 bp CCAAT oligonucleotide shows that the HFD dimer binds to the DNA sugar-phosphate backbone, mimicking the nucleosome H2A/H2B-DNA assembly. NF-YA both binds to NF-YB/NF-YC and inserts an a helix deeply into the DNA minor groove, providing sequence-specific contacts to the CCAAT box. Structural considerations and mutational data indicate that NF-YB ubiquitination at Lys138 precedes and is equivalent to H2B Lys120 monoubiquitination, important in transcriptional activation. Thus, NF-Y is a sequence-specific transcription factor with nucleosome-like properties of nonspecific DNA binding and helps establish permissive chromatin modifications at CCAAT promoters. Our findings suggest that other HFD-containing proteins may function in similar ways.
.
URLPMID:19763152 [本文引用: 1]
Their ability to move within genomes gives transposable elements an intrinsic propensity to affect genome evolution. Non-long terminal repeat (LTR) retrotransposons--including LINE-1, Alu and SVA elements--have proliferated over the past 80 million years of primate evolution and now account for approximately one-third of the human genome. In this Review, we focus on this major class of elements and discuss the many ways that they affect the human genome: from generating insertion mutations and genomic instability to altering gene expression and contributing to genetic innovation. Increasingly detailed analyses of human and other primate genomes are revealing the scale and complexity of the past and current contributions of non-LTR retrotransposons to genomic change in the human lineage.
.
URLPMID:25896322 [本文引用: 1]
Endogenous retroviruses (ERVs) are remnants of ancient retroviral infections, which comprise nearly 8% of the human genome1. The most recently acquired human ERV is HERV-K (HML-2), which repeatedly infected the primate lineage both before and after the divergence of humans and chimpanzees2,3. Unlike most other human ERVs, HERV-K retained multiple copies of intact open reading frames (ORFs) encoding retroviral proteins4. However, HERV-K is transcriptionally silenced by the host with exception of certain pathological contexts, such as germ cell tumors, melanoma, or HIV infection5 7. Here we demonstrate that DNA hypomethylation at LTR elements representing the most recent genomic integrations, together with transactivation by OCT4, synergistically facilitate HERV-K expression. Consequently, HERV-K is transcribed during normal human embryogenesis beginning with embryonic genome activation (EGA) at the 8-cell stage, continuing through the emergence of epiblast cells in pre-implantation blastocysts, and ceasing during hESC derivation from blastocyst outgrowths. Remarkably, HERV-K viral-like particles and Gag proteins are detected in human blastocysts, indicating that early human development proceeds in the presence of retroviral products. We further show that overexpression of one such product, HERV-K accessory protein Rec, in a pluripotent cell line is sufficient to increase IFITM1 levels on the cell surface and inhibit viral infection, suggesting at least one mechanism through which HERV-K can induce viral restriction pathways in early embryonic cells. Moreover, Rec directly binds a subset of cellular RNAs and modulates their ribosome occupancy, arguing that complex interactions between retroviral proteins and host factors can fine-tune regulatory properties of early human development.
.
URLPMID:15469847 [本文引用: 1]
A comprehensive analysis of transposable element (TE) expression in mammalian full-grown oocytes reveals that LTR class III retrotransposons make an unexpectedly high contribution to the maternal mRNA pool, which persists in cleavage stage embryos. The most abundant transcripts in the mouse oocyte are from the mouse transcript (MT) retrotransposon family, and expression of this and other TE families is developmentally regulated. Furthermore, TEs act as alternative promoters and first exons for a subset of host genes, regulating their expression in full-grown oocytes and cleavage stage embryos. To our knowledge, this is the first example of TEs initiating synchronous, developmentally regulated expression of multiple genes in mammals. We propose that differential TE expression triggers sequential reprogramming of the embryonic genome during the oocyte to embryo transition and in preimplantation embryos.
.
URLPMID:24813617 [本文引用: 1]
Allele-specific, single-base-resolution sequencing of mouse early embryos shows that mammalian paternal methylome and a significant proportion of maternal methylome undergo active demethylation during early embryonic development.
.
URL [本文引用: 1]
.
URLPMID:25079557 [本文引用: 1]
Abstract DNA methylation is a crucial element in the epigenetic regulation of mammalian embryonic development. However, its dynamic patterns have not been analysed at the genome scale in human pre-implantation embryos due to technical difficulties and the scarcity of required materials. Here we systematically profile the methylome of human early embryos from the zygotic stage through to post-implantation by reduced representation bisulphite sequencing and whole-genome bisulphite sequencing. We show that the major wave of genome-wide demethylation is complete at the 2-cell stage, contrary to previous observations in mice. Moreover, the demethylation of the paternal genome is much faster than that of the maternal genome, and by the end of the zygotic stage the genome-wide methylation level in male pronuclei is already lower than that in female pronuclei. The inverse correlation between promoter methylation and gene expression gradually strengthens during early embryonic development, reaching its peak at the post-implantation stage. Furthermore, we show that active genes, with the trimethylation of histone H3 at lysine 4 (H3K4me3) mark at the promoter regions in pluripotent human embryonic stem cells, are essentially devoid of DNA methylation in both mature gametes and throughout pre-implantation development. Finally, we also show that long interspersed nuclear elements or short interspersed nuclear elements that are evolutionarily young are demethylated to a milder extent compared to older elements in the same family and have higher abundance of transcripts, indicating that early embryos tend to retain higher residual methylation at the evolutionarily younger and more active transposable elements. Our work provides insights into the critical features of the methylome of human early embryos, as well as its functional relation to the regulation of gene expression and the repression of transposable elements.
.
URLPMID:25966907 [本文引用: 1]
Dominated by microscopy for decades the nuclear genome organization field has recently undergone a dramatic transition fuelled by new next generation sequencing technologies that are beginning to bridge the gap between microscopic observations and molecular scale studies. It is no longer in doubt that the nucleus is spatially compartmentalized and that the genome organization with respect to these compartments is cell type specific. However, it is still unclear if and how this organization contributes to genome function, or whether it is simply a consequence of it. This uncertainty is partly due to the cell-to-cell variability of genome organization, but also due to limitations of the measurement techniques and the scale of the problem at hand. Here we discuss some of the exciting recent progress made towards understanding three-dimensional genome architecture and function.
.
URLPMID:4517094 [本文引用: 1]
In humans, nearly two meters of genomic material must be folded to fit inside each micrometer-scale cell nucleus while remaining accessible for gene transcription, DNA replication, and DNA repair. This fact highlights the need for mechanisms governing genome organization during any activity and to maintain the physical organization of chromosomes at all times. Insight into the functions and three-dimensional structures of genomes comes mostly from the application of visual techniques such as fluorescence in situ hybridization (FISH) and molecular approaches including chromosome conformation capture (3C) technologies. Recent developments in both types of approaches now offer the possibility of exploring the folded state of an entire genome and maybe even the identification of how complex molecular machines govern its shape. In this review, we present key methodologies used to study genome organization and discuss what they reveal about chromosome conformation as it relates to transcription regulation across genomic scales in mammals.
.
URLPMID:26967279 [本文引用: 1]
Proper expression of genes requires communication with their regulatory elements that can be located elsewhere along the chromosome. The physics of chromatin fibers imposes a range of constraints on such communication. The molecular and biophysical mechanisms by which chromosomal communication is established, or prevented, have become a topic of intense study, and important roles for the spatial organization of chromosomes are being discovered. Here we present a view of the interphase 3D genome characterized by extensive physical compartmentalization and insulation on the one hand and facilitated long-range interactions on the other. We propose the existence of topological machines dedicated to set up and to exploit a 3D genome organization to both promote and censor communication along and between chromosomes.
.
URL [本文引用: 1]
.
URL [本文引用: 1]
.
URLPMID:26971819 [本文引用: 1]
Krijger et al. report that the reprogramming of four somatic cell types with highly distinct 3D genomes results in pluripotent cells with largely identical, ESC-like, genome conformations carrying founder-dependent topological hallmarks. The latter are not remnants of somatic chromosome topologies but are acquired during reprogramming in a cell-of-origin-dependent manner.
.
URLPMID:4040465 [本文引用: 1]
Mitotic chromosomes are among the most recognizable structures in the cell, yet for over a century their internal organization remains largely unsolved. We applied chromosome conformation capture methods, 5C and Hi-C, across the cell cycle and revealed two distinct three-dimensional folding states of the human genome. We show that the highly compartmentalized and cell type-specific organization described previously for nonsynchronous cells is restricted to interphase. In metaphase, we identified a homogenous folding state that is locus-independent, common to all chromosomes, and consistent among cell types, suggesting a general principle of metaphase chromosome organization. Using polymer simulations, we found that metaphase Hi-C data are inconsistent with classic hierarchical models and are instead best described by a linearly organized longitudinally compressed array of consecutive chromatin loops.
.
URL [本文引用: 1]
URLPMID:28709003 [本文引用: 11]
Abstract High-order chromatin structure plays important roles in gene expression regulation. Knowledge of the dynamics of 3D chromatin structures during mammalian embryo development remains limited. We report the 3D chromatin architecture of mouse gametes and early embryos using an optimized Hi-C method with low-cell samples. We find that mature oocytes at the metaphase II stage do not have topologically associated domains (TADs). In sperm, extra-long-range interactions (>4 Mb) and interchromosomal interactions occur frequently. The high-order structures of both the paternal and maternal genomes in zygotes and two-cell embryos are obscure but are gradually re-established through development. The establishment of the TAD structure requires DNA replication but not zygotic genome activation. Furthermore, unmethylated CpGs are enriched in A compartment, and methylation levels are decreased to a greater extent in A compartment than in B compartment in embryos. In summary, the global reprogramming of chromatin architecture occurs during early mammalian development. Copyright 2017 Elsevier Inc. All rights reserved.
.
URLPMID:28703188 [本文引用: 6]
Abstract In mammals, chromatin organization undergoes drastic reprogramming after fertilization. However, the three-dimensional structure of chromatin and its reprogramming in preimplantation development remain poorly understood. Here, by developing a low-input Hi-C (genome-wide chromosome conformation capture) approach, we examined the reprogramming of chromatin organization during early development in mice. We found that oocytes in metaphase II show homogeneous chromatin folding that lacks detectable topologically associating domains (TADs) and chromatin compartments. Strikingly, chromatin shows greatly diminished higher-order structure after fertilization. Unexpectedly, the subsequent establishment of chromatin organization is a prolonged process that extends through preimplantation development, as characterized by slow consolidation of TADs and segregation of chromatin compartments. The two sets of parental chromosomes are spatially separated from each other and display distinct compartmentalization in zygotes. Such allele separation and allelic compartmentalization can be found as late as the 8-cell stage. Finally, we show that chromatin compaction in preimplantation embryos can partially proceed in the absence of zygotic transcription and is a multi-level hierarchical process. Taken together, our data suggest that chromatin may exist in a markedly relaxed state after fertilization, followed by progressive maturation of higher-order chromatin architecture during early development.
.
URLPMID:28355183 [本文引用: 3]
Chromatin is reprogrammed after fertilization to produce a totipotent zygote with the potential to generate a new organism1. The maternal genome inherited through the oocyte and the paternal genome provided by sperm coexist as separate haploid nuclei in the zygote. How these two epigenetically distinct genomes are spatially organized is poorly understood. Existing chromosome conformation capture-based methods2–5are inapplicable to oocytes and zygotes due to a paucity of material. To study the 3D chromatin organization in rare cell types, we developed a single-nucleus Hi-C (snHi-C) protocol that provides >10-fold more contacts per cell than the previous method2. Here we show that chromatin architecture is uniquely reorganized during the mouse oocyte-to-zygote transition and is distinct in paternal and maternal nuclei within single-cell zygotes. Features of genomic organization including compartments, topologically associating domains (TADs) and loops are present in individual oocytes when averaged over the genome; each feature at a locus is variable between cells. At the sub-megabase level, we observe stochastic clusters of contacts that violate TAD boundaries but average into TADs. Strikingly, we found that TADs and loops but not compartments are present in zygotic maternal chromatin, suggesting that these are generated by different mechanisms. Our results demonstrate that the global chromatin organization of zygote nuclei is fundamentally different from other interphase cells. An understanding of this zygotic chromatin “ground state” has the potential to provide insights into reprogramming to totipotency.
.
URL [本文引用: 1]
.
URLPMID:29203909 [本文引用: 2]
Abstract In mammals, all somatic development originates from lineage segregation in early embryos. However, the dynamics of transcriptomes and epigenomes acting in concert with initial cell fate commitment remains poorly characterized. Here we report a comprehensive investigation of transcriptomes and base-resolution methylomes for early lineages in peri- and postimplantation mouse embryos. We found allele-specific and lineage-specific de novo methylation at CG and CH sites that led to differential methylation between embryonic and extraembryonic lineages at promoters of lineage regulators, gene bodies, and DNA-methylation valleys. By using Hi-C experiments to define chromatin architecture across the same developmental period, we demonstrated that both global demethylation and remethylation in early development correlate with chromatin compartments. Dynamic local methylation was evident during gastrulation, which enabled the identification of putative regulatory elements. Finally, we found that de novo methylation patterning does not strictly require implantation. These data reveal dynamic transcriptomes, DNA methylomes, and 3D chromatin landscapes during the earliest stages of mammalian lineage specification.
.
URL [本文引用: 1]
Fertilization triggers assembly of higher‐order chromatin structure from a condensed maternal and a na07ve paternal genome to generate a totipotent embryo. Chromatin loops and domains have been detected in mouse zygotes by single‐nucleus Hi‐C (snHi‐C), but not bulk Hi‐C. It is therefore unclear when and how embryonic chromatin conformations are assembled. Here, we investigated whether a mechanism of cohesin‐dependent loop extrusion generates higher‐order chromatin structures within the one‐cell embryo. Using snHi‐C of mouse knockout embryos, we demonstrate that the zygotic genome folds into loops and domains that critically depend on Scc1‐cohesin and that are regulated in size and linear density by Wapl. Remarkably, we discovered distinct effects on maternal and paternal chromatin loop sizes, likely reflecting differences in loop extrusion dynamics and epigenetic reprogramming. Dynamic polymer models of chromosomes reproduce changes in snHi‐C, suggesting a mechanism where cohesin locally compacts chromatin by active loop extrusion, whose processivity is controlled by Wapl. Our simulations and experimental data provide evidence that cohesin‐dependent loop extrusion organizes mammalian genomes over multiple scales from the one‐cell embryo onward.
URLPMID:28388407 [本文引用: 2]
Chromatin architecture is fundamental in regulatinggene expression. To investigate when spatial genome organization is first established during development, we examined chromatin conformation during Drosophila embryogenesis and observed the emergence of chromatin architecture within a tight time window that coincides with the onset of transcription activation in the zygote. Prior to zygotic genome activation, the genome is mostly unstructured. Early expressed genes serve as nucleation sites for topologically associating domain (TAD) boundaries. Activation of gene expression coincides with the establishment of TADs throughout the genome and co-localization of housekeeping gene clusters, which remain stable in subsequent stages of development. However, the appearance of TAD boundaries is independent of transcription and requires the transcription factor Zelda for locus-specific TAD boundary insulation. These results offer insight into when spatial organization of the genome emerges and identify a key factor that helps trigger this architecture.
.
URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}