 ,2
,2Advances on the roles of m 6A in tumorigenesis
Tiangong Wang1, Meng Ye,2通讯作者:
编委: 宋旭
收稿日期:2018-05-18修回日期:2018-07-31网络出版日期:2018-12-20
| 基金资助: |
Received:2018-05-18Revised:2018-07-31Online:2018-12-20
| Fund supported: |
作者简介 About authors
王天工,硕士研究生,专业方向:肿瘤学E-mail:tonywoolf21@gmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (447KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王天工, 叶孟. m 6A甲基化与肿瘤研究进展 [J]. 遗传, 2018, 40(12): 1055-1065 doi:10.16288/j.yczz.18-098
Tiangong Wang, Meng Ye.
肿瘤是一种基因组疾病,具有可遗传性,也可能由于各种理化因素和(或)生物因素等所导致,通常表现为正常细胞出现异常分化与增殖,不受正常的细胞周期、凋亡等因素调控,甚至具有侵袭性、转移性和促血管生成的能力。m6A甲基化是在RNA分子上的一种表观遗传修饰。早在20世纪70年代,遗传学家们就在真核生物的信使RNA (message RNA, mRNA)上发现了这一甲基化修饰[1]。由于检测技术手段的局限,起初科学家们认为m6A甲基化位点只存在于mRNA中,但近年来的研究发现多种类型RNA中均存在m6A甲基化的身影[2]。随着高通量测序技术的快速发展以及表观遗传学研究领域的逐步深入,m6A甲基化在不同生物学过程中的功能和作用再次受到人们的关注。目前,已有成熟的单一位点甚至高通量单一位点检测手段可对m6A位点进行准确而有效地检测。但是,由于全转录组检测通常需要花费大量的时间和资金,因此人们也开发了多种预测m6A甲基化位点的生物信息学平台[3]。这些工具为人们对m6A甲基化的研究提供了极大的帮助。
DNA上携带的遗传信息首先转录为mRNA,然后才被进一步翻译为有功能的蛋白质。在细胞癌变的过程中,m6A甲基化这一表观遗传修饰能通过调控癌基因、抑癌基因的mRNA分子的表达水平来影响肿瘤的发生发展。与DNA甲基化相似,m6A甲基化受甲基转移酶和去甲基酶的调控,且在不改变碱基序列的情况下调控基因的转录后表达水平,但其调控机制却远比DNA甲基化复杂得多。结合近年来发表的与肿瘤相关的m6A甲基化的研究,本文对m6A甲基化的相关概念、检测和预测手段以及与肿瘤的关系等几方面进行阐述,以期能更深入地探寻m6A甲基化与肿瘤发生发展的关联,并希望能够为肿瘤的分子病理诊断和分子靶向治疗寻找新的标志物和潜在靶点。
1 RNA的m6A甲基化
1.1 m6A甲基化的相关概念
RNA的m6A甲基化是腺嘌呤(A)第6位N通过甲基转移酶催化形成的一种甲基化修饰[4,5]。m6A甲基化在mRNA上最为常见,同时也可以出现在转运RNA (transfer RNA, tRNA)、核糖体RNA (ribosome RNA, rRNA)甚至非编码RNA (non-coding RNA, ncRNA)中[6]。也有研究发现m6A甲基化可出现在多个常见的长链非编码RNA (long non-coding RNA, LncRNA)中,如MALAT1、HOTAIR、XIST和SRA1[7]。近期,有文献报道了在微小RNA (microRNA和miRNA)中也发现了m6A甲基化[8]。值得关注的是,m6A甲基化不但可以通过影响微小RNA前体的生成来改变微小RNA的表达,m6A甲基化同样可以出现在成熟的微小RNA中[9]。大量的研究证明,m6A甲基化主要与3类蛋白质相互作用。第一类是m6A甲基转移酶,它会促进RNA中的m6A甲基化修饰,其编码基因被称为写入基因(Writers),最早发现的写入基因有METTL3、METTL14和WATP等,它们形成的复合物共同促使m6A甲基化基团写入RNA中[10,11,12]。随后,又有文献报道发现METTL16、KIAA1429和RBM15等更多新的写入基因[13,14,15,16]。第二类蛋白质是m6A去甲基酶,它可以去除RNA中的m6A甲基化基团,其编码基因为擦除基因(Erasers),常见的擦除基因有FTO和ALKBH5[17]。由此可见,m6A甲基化其实是一个动态、可逆的过程(图1)。最后一类蛋白则能够与RNA中的m6A甲基化位点结合进而发挥特定作用,它们的编码基因被称为读取基因(Readers)。最早发现的读取基因是YTH结构域家族蛋白的编码基因,包括YTHDFs和YTHDCs两个亚型。YTHDCs亚型中的读取基因为YTHDC1和YTHDC2[14,18]。而YTHDFs亚型的读取基因分别为 YTHDF1、YTHDF2和YTHDF3[19]。除了YTH结构域家族蛋白基因,随后又有多个读取基因被发现,分别为eIF3、hnRNP C和hnRNP A2/B1基因[20,21,22]。读取蛋白会与RNA上的m6A位点结合,进而产生各种生物学功能。目前对于m6A甲基化的生物学功能尚未完全了解,但已有研究发现YTHDFs亚型的蛋白质主要定位在细胞质中,YTHDF1和YTHDF3蛋白会提高mRNA的翻译效率,而YTHDF2蛋白与m6A位点结合则与缩短mRNA的半衰期有关[23,24]。但是,也有研究发现YTHDF2蛋白会在核内通过与m6A位点结合以阻止FTO蛋白去除5°UTR区的m6A甲基化进而促进RNA的帽非依赖性翻译[25]。而YTHDCs亚型的蛋白质则主要在核内发挥功能。有报道显示,YTHDC1蛋白对编码和非编码基因的转录都具有调控作用,且可通过影响mRNA前体的可变剪接来调控mRNA的表达水平[18,26]。目前对于YTHDC2与m6A甲基化的功能了解甚少,有文献报道YTHDC2蛋白在核内核外均可存在,且会选择性地与非编码RNA上的m6A位点结合[14],但所产生的生物学功能依然是一个谜。另外,在hnRNP家族中发现hnRNP A2/B1蛋白可能参与了前体miRNA转录的调控过程[20],而hnRNP C蛋白则会影响mRNA和LncRNA的局部二级结构[21]。由此推测,m6A甲基化很可能参与了更为广泛的生物学调控过程。目前,与m6A甲基化有关的新的写入蛋白和读取蛋白依然在源源不断地被人们鉴别出来,可见人们对RNA上m6A甲基化水平的动态调控以及潜在的生物学调控功能依然有非常广阔的探索空间。期望未来,人们可以更全面地了解m6A甲基化的生物学功能,使表观遗传在对于转录后层面的基因表达有更多的认识和补充。图1
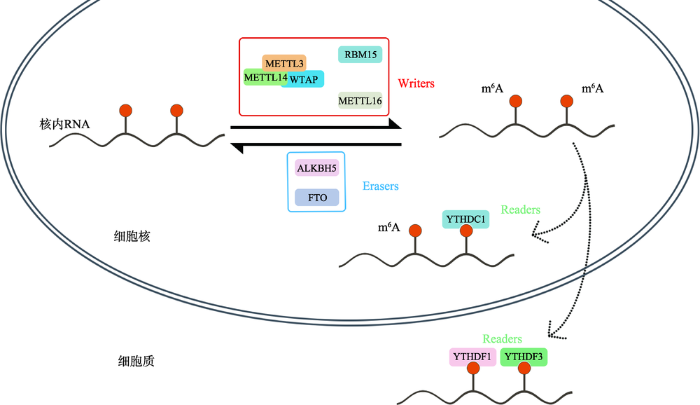 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1m6A甲基化的分子机制
METTL3、METTL14和WTAP形成的复合物,RBM15和METTL16为常见的Writers(写入蛋白)。 ALKBH5、FTO为常见的Erasers(擦除蛋白)。 YTHDC1、YTHDF1和YTHDF3为常见的Readers(读取蛋白)。核内RNA在Writers的作用下,出现了m6A甲基化位点。核内RNA的m6A甲基化位点也可在Erasers的作用下被擦除。随后,在核内RNA的进一步加工中,核内的Readers(读取蛋白)会与m6A甲基化位点结合;当成熟的RNA出核后,依然会有一些存在与核外的Readers会与其m6A位点结合。值得注意的是,不同的Reader结合到m6A位点上会产生不同的生物学作用。
Fig. 1The molecule mechanism of m6A methylation
1.2 m6A甲基化的检测方法
早期由于检测技术落后,科研工作者们对于m6A甲基化位点知之甚少。由于RNA的m6A甲基化并不影响其逆转录,也不能像m7G甲基化那样可以被特异性地切割,以至于m6A位点的鉴别在初期研究中显得非常困难[27,28]。但随着二代测序的出现,两种相似的m6A甲基化位点检测技术应运而生,这就是m6A-seq[29]和MeRIP-seq[30]技术。这两种检测方法其实是应用免疫共沉淀技术将具有m6A甲基化的RNA片段捕捉后再使用二代测序来鉴定其序列。随后,大量的m6A甲基化位点被发现,科研工作者分别在人和小鼠的7000余个基因中发现了多达12 000个m6A信号峰,这些信号峰都富集在靠近3'端的终止密码子附近,且发现这些位点在人和小鼠中都具有高度保守性。这一发现为m6A甲基化在转录后对与基因表达水平的调控提供了有力证据,且可能与各类遗传疾病有关。这种检测手段的缺陷是该技术所捕获的RNA片段被限制在100~200nt左右,并且该技术无法识别两个非常接近的m6A位点,所以这种方法不能对全转录组的m6A甲基化位点作出精准的鉴别。另外有文献报道,在mRNA中的5'端含有m6Am修饰[31],因为m6Am修饰同m6A修饰一样具有第六位甲基,因而m6A-seq和MeRIP-seq都可能将这种修饰误读为m6A修饰。由于以上种种原因,人们对检测技术做出了改进。有3个实验室在2015年均发文报道了使用紫外交联免疫共沉淀技术可以更为精确地在RNA的单个碱基上去捕捉m6A甲基化位点。这3种相似的技术分别为miCLIP[32]、PA-m6A-seq[33]和m6A-CLIP(或称为UV-CLIP)[34]。另有一项检测m6A甲基化水平的技术为m6A-LAIC- seq[35],这项技术在m6A-seq的基础上引入了spike-in RNAs作为内参,从而计算出全转录组中每段基因上的m6A甲基化水平,其缺点是无法检测出单个m6A甲基化位点。除了高通测序外,单基因的m6A甲基化位点检测手段也非常重要。其中最有名的当属SCARLET[36]检测法。这种方法可以在mRNA和LncRNA中精确地检测单个m6A甲基化位点,并由此计算出整段RNA的m6A甲基化水平。虽然SCARLET属于低通量检测且费用较为昂贵,但极高的精准性使其成为用于检验高通量检测m6A甲基化位点的准确性的一种常用方法。也有文献报道,SCARLET同样可以用于检测其他类型的RNA表观遗传修饰,例如RNA的m5C修饰和Ψ修饰[37]。荧光定量PCR同样也可以作为检测m6A甲基化水平的方法。Golovina等[38]研究了多种RNA,发现在同一种RNA中不同的m6A甲基化水平在荧光定量PCR检测下会产生不同的熔解曲线,这是由于m6A甲基化水平的不同使RNA-DNA复合物的熔解温度发生改变。由此作者提出了HRM检测方法,该技术可以在RNA中检测已知的m6A甲基化水平是否发生变化。但是,Golovina等只在rRNA、tRNA以及snRNA中进行了实验,所以该技术能否推广到其他RNA中还有待验证。时至今日,高通量测序技术已经非常成熟,但荧光定量检测依然是目前最为经济和便捷的分子检测手段之一,开发荧光定量PCR用于m6A的检测仍然非常重要。随着m6A甲基化位点的检测手段的逐步提高,人们对其认识日渐深入,从而为研究m6A甲基化与各类疾病特别是与肿瘤的关系打下坚实的基础。1.3 m6A甲基化位点的预测方法
由于m6A甲基化位点的检测需要花费大量的人力物力,而通过生物信息学预测则可以极大地提高研究效率。近年来,生物信息学发展迅速,在各类分子生物学的研究中应用广泛,以下几种方法可以帮助人们更有效地预测m6A的甲基化位点。Zhang等[39]最早提出了使用隐藏马尔科夫模型(HIDDEN MARKOV MODEL, HMM)来预测已知位点周边的残余位点。随后,Liu等[40]又开发了更快速更稳定的pRNAm-PC方法来预测位点。同时,Chen等[41]也开发了iRNA-Methyl方法。两者的共同特点是都应用了支持向量机(support vector machine, SVM)模型。在此基础上,Jia等[42]开发了RNA-methylPred方法,这要比前两者更为稳定和高效。随后Li等[43]提出了更好的TargetM6A方法,但是这种方法只能在初级RNA序列中预测m6A的甲基化位点。而Zhou等[44]则综合了多种数学模型,提出了SRAMP方法。这种方法可以更有效地在哺乳动物的RNA中预测m6A甲基化位点。近期有文献报道,名为RMBaseV2.0[45]数据库网站已经建立(网址:http://rna.sysu. edu.cn/rmbase/),其收录了13个物种的多个RNA表观遗传修饰的测序数据,其中包括了大量关于m6A的甲基化位点的数据。此数据库可对需要研究的RNA的m6A甲基化信息加以预测(表1)。Table 1
表1
表1 m6A甲基化的检测和预测手段
Table 1
| 方法 | 特点 | 参考文献 |
|---|---|---|
| 检测方法 | ||
| m6A-Seq | 高通量 | [29] |
| MeRIP-Seq | 高通量 | [30] |
| miCLIP | 高通量、单位点 | [32] |
| PA-m6A-Seq | 高通量、单位点 | [33] |
| m6A-CLIP | 高通量、单位点 | [34] |
| m6A-LAIC-Seq | 高通量、精确 | [35] |
| SCARLET | 低通量、精确 | [36] |
| HRM | 低通量 | [37] |
| 预测方法 | ||
| HMM | 数据库 | [39] |
| pRNAm-PC | 数据库 | [40] |
| iRNA-Methyl | 数据库 | [41] |
| RNA-methylPred | 数据库 | [42] |
| TargetM6A | 数据库 | [43] |
| SRAMP | 数据库 | [44] |
| RMBaseV2.0 | 网站 | [45] |
新窗口打开|下载CSV
2 m6A甲基化与肿瘤
2.1 m6A高甲基化与肿瘤
在早期的基础研究中,已有国内****发现m6A甲基化参与了脊椎动物的造血干细胞的分化过程[46],这提示m6A甲基化很可能与血液系统肿瘤的发生发展有着密切联系。Barbieri等[47]研究发现,METTL3基因对小鼠和人的多种急性髓细胞白血病(acute myelocytic leukemia,AML)细胞系的增值和生长均有不同程度的影响,其中对小鼠KMT2A-MLLT3融合基因及Flt3基因内部串联复制型的AML细胞系影响最为明显。研究发现,METTL3基因表达下调会促进AML细胞的分化,由于转录因子CEBPZ调控METTL3的表达上升,从而使癌基因SP1所转录的mRNA上的m6A甲基化水平明显上升,最终导致SP1蛋白表达量升高。同时发现,SP1蛋白可以持续影响造血干细胞向AML细胞分化。这一研究提示METTL3基因可能成为KMT2A-MLLT3基因和Flt3基因突变型AML的潜在治疗靶点。Vu等[48]也报道了METTL3与AML的发生发展有关。该研究发现,对CD34+的造血干细胞的METTL3基因下调后,该细胞的pAKT基因的表达上升,从而起到促进正常造血干细胞分化为AML细胞的作用,反之则会产生抑制作用。同时研究还发现,m6A甲基化会促进该细胞中c-MYC、BCL2和PTEN等基因的转录。Weng等[49]发现,METTL14基因在正常的造血干细胞和混合表形AML细胞(伴t(11q23)、t(15;17)或t(8;21))中呈高表达但在正常分化的骨髓细胞中则呈现低表达。METTL14基因编码写入蛋白,其升高会导致某些转录组中的m6A甲基化水平升高。该研究在AML细胞系中发现,敲低METTL14的表达会降低MYB和MYC所转录的mRNA的稳定性,使其加速降解。这说明,MYB和MYC受到了METTL14的调控。METTL14对造血干细胞分化为AML细胞有一定的作用,并在白血病的进展、维持和自我更新等方面都有一定作用。该研究还发现,沉默METTL14的表达可以有效地抑制AML细胞系的增殖。这一研究揭示了SPI1-METTL14-MYB/MYC通路对AML的影响,为AML的诊断和治疗提供了新的方向。Chen等[50]发现METTL3会改变抑癌基因SOCS2的m6A甲基化水平。并且METTL3过表达会促进肝细胞癌(HCC)的增殖和迁移,而降低METTL3则会抑制细胞的生长和转移。他们在进一步研究中发现,SOCS2在HCC中表现为抑癌基因,而METTL3通过调控m6A-YTHDF2通路,进而加速SOCS2的mRNA的降解,这项研究证明m6A高甲基化与HCC的发生发展也具有相关性。2.2 m6A低甲基化与肿瘤
Li等[51]发现在MLL基因重排的AML中高表达的FTO可以降低ASB2和 RARA基因的mRNA中m6A甲基化的水平从而引起AML的发生发展,同时发现高表达的FTO会抑制全反式维A酸介导AML细胞向正常血细胞分化的作用。这使得FTO这一去甲基化基因成为了MLL型AML的癌基因。Zhou等[52]发现在宫颈鳞状细胞癌(cervical squamous cell carcinoma,CSCC)患者的肿瘤组织中FTO表达显著升高,并发现这些患者对放化疗产生了耐受,这可能是由于FTO降低了某些基因的m6A甲基化水平进而激活β-catenin通路并影响到了ERCC1基因的表达而产生的作用。同时发现,CSCC患者中FTO和β-catenin表达同时升高相比较单独升高的患者,表现出了更差的预后(P = 0.041)。由此可见,FTO和β-catenin的表达对于CSCC的临床预后具有一定的评估价值。Wang等[53]研究了286例宫颈癌患者的大样本数据后发现,肿瘤组织中的m6A甲基化总体水平显著降低。同时发现METTL3和METTL14的表达量明显减少,而FTO和ALKBH5的表达量则明显升高。他们推测,m6A低甲基化可能在宫颈癌的发生发展中发挥了重要作用。通过对临床数据分析显示,m6A低甲基化的患者的无病生存率(disease free survival, DFS)和总生存率(overall survival, OS)显著降低,且低甲基化患者具有更高的复发率(P < 0.01)。随后作者通过功能实验发现,宫颈癌细胞中m6A总水平高甲基化会产生明显的增殖抑制作用。由于肿瘤中的m6A甲基化水平的高低往往取决于转甲基酶和去甲基酶的水平,所以研究肿瘤中这两种酶的基因表达水平有助于深入了解肿瘤的发生发展与m6A甲基化的相关性,帮助建立肿瘤早期诊断及预后分析的新方法(表2)。Table 2
表2
表2 m6A甲基化与肿瘤
Table 2
| m6A甲基化相关蛋白编码基因 | 表达趋势 | 肿瘤类型 | 调控基因 | 参考文献 |
|---|---|---|---|---|
| 写入基因(writers) | ||||
| METTL3 | ↑ | 白血病 | SP1、c-MYC、BCL2、PTEN | [47,48] |
| ↑ | 肝癌 | SOCS2 | [50] | |
| ↓ | 胶质母细胞瘤 | ADAM19、EPHA3、KLF4 CDKN2A、BRCA2、TP53 | [59] | |
| METTL14 | ↑ | 白血病 | MYB、MYC | [49] |
| ↓ | 胶质母细胞瘤 | ADAM19、EPHA3、KLF4 CDKN2A、BRCA2、TP53 | [59] | |
| 擦除基因(erasers) | ||||
| FTO | ↑ | 白血病 | ASB2、RARA、TP53 | [51] |
| ↑ | 宫颈癌 | β-catenin通路 | [53] | |
| ALKBH5 | ↑ | 乳腺癌 | KLF4、NANOG | [57] |
| 读取基因(readers) | ||||
| YTHDF1 | ↑ | 结直肠癌 | 未知 | [54] |
| ↑ | 肝癌 | 未知 | [56] | |
| YTHDF2 | ↑ | 肝癌 | SOCS2 | [50] |
新窗口打开|下载CSV
2.3 m6A甲基化读取基因与肿瘤
读取蛋白可以与RNA上的m6A位点特异性地结合,不同的读取蛋白与m6A位点结合后,会产生不同的生物学功能(表2)。如YTHDF2蛋白与mRNA中的m6A位点结合,起到加快mRNA降解的作用。Chen等[50]在研究中发现,HCC的抑癌基因SOCS2所表达的mRNA中m6A甲基化水平上调,这使得YTHDF2蛋白拥有了更多的m6A结合位点最终促进了mRNA降解。Nishizawa及其团队[54]发现YTHDF1基因在结直肠癌患者中处于高表达。通过临床病理数据分层研究中发现,YTHDF1基因的表达与肿瘤直径(P=0.009)、淋巴结转移(P=0.044)、远处转移(P=0.036)和临床分期(P=0.0226)具有相关性,但对于具体的调控机制未作研究。Yang等[55]收集了31例TNM三期的HCC患者样本,发现YTHDF2表达升高率高达83.9% (26/31),怀疑HCC晚期病人极可能与部分基因的m6A位点结合了YTHDF2蛋白相关。随后,作者在HepG2细胞系中过表达miR-145以降低YTHDF2的表达,发现HepG2细胞的增殖受到明显抑制。这提示miR-145可能成为HCC的潜在治疗手段。Zhao等[56]对TCGA中的肝癌数据进行研究发现,YTHDF1在TNM三、四期的HCC患者中相比于二期患者表达明显上调,且YTHDF1上调的患者具有更差预后;并发现受YTHDF1蛋白调控的潜在靶基因可能与肿瘤的细胞周期、多种氨基酸的降解以及各种脂类代谢有关,这些细胞生理功能的异常很可能与HCC的发生发展有关。最后作者通过分析TCGA和ChIP-atlas数据库数据预测并通过实验验证了c-Myc是调控YTHDF1的上游基因。2.4 m6A甲基化与肿瘤干细胞
肿瘤干细胞具有自我更新和无限增值的能力,在肿瘤的发生发展、转移及复发中起到重要作用。Zhang等[57]发现缺氧的状态下乳腺癌干细胞(breast cancer stem cells, BCSCs)会产生富集。并发现在低氧状态下,乳腺癌细胞会依赖HIF通路并影响ZNF217和ALKBH5的表达,从而影响KLF4和NANOG的mRNA表达。ALKBH5蛋白是一种擦除蛋白,可以降低RNA上的m6A甲基化水平,而KLF4和NANOG则是与多能干细胞相关的转化基因。由此推测,m6A甲基化可能与低氧状态下乳腺癌干细胞的生成有一定联系。并在分子病理检查中发现,雌、孕激素受体(ER、PR)阳性和人表皮生长因子受体2 (HER2)阳性的患者具有更高的ALKBH5或HIF-1α阳性率。同时发现,低水平的ALKBH5表达与乳腺癌的发生以及肺转移具有一定的相关性。众所周知,肿瘤细胞对于能量和氧的消耗极高,这一研究发现低氧条件会升高ALKBH5表达可能影响了KLF4和NANOG的表达上调,使乳腺癌细胞向肿瘤干细胞转化,这为肿瘤干细胞的研究提供了新的思路。TAKETO等[58]发现下调胰腺癌细胞系的METTL3表达可以使细胞成球能力明显降低,这表明METTL3可以促进肿瘤干细胞的形成。进一步研究发现在METTL3表达降低的胰腺癌细胞中,抗癌药物对其具有更明显的作用,也对放射性外照射更加敏感。Cui等[59]在m6A甲基化与胶质母细胞瘤干细胞(CSC)的研究中发现,m6A甲基化水平能影响SCS的分化。METTL3或METTL14基因高表达可以抑制CSC的生长和自我更新。同时发现下调METTL3和(或) METTL14基因会促进肿瘤的进展并缩短接种GSC动物模型的生存时间。其后,作者发现使用FTO抑制剂可以明显地抑制GSC细胞的生长并减少GSC细胞的转化为肿瘤干细胞的频率。在METTL3和(或)METTL14下调的CSC细胞中发现,癌基因ADAM19、EPHA3和KLF4出现上调,而抑癌基因CDKN2A、BRCA2和TP53则出现下调。为验证这些癌基因表达的改变是由于METTL3和(或)METTL14下调导致m6A甲基化水平下降而引起的。作者进一步通过上调METTL3基因的表达和使用FTO抑制剂使m6A甲基化水平上升,发现ADAM19、EPHA3和KLF4这3个癌基因表达出现下降,这表明METTL3蛋白可能通过影响m6A甲基化而提高了这3个基因的表达并最终引起SCS的发生。2.5 m6A甲基化与抗肿瘤药物
TAKETO等[58]发现,使用吉西他滨(GEM)可以使METTL3低表达的胰腺癌细胞出现凋亡。Nishizawa等[54]则发现将 YTHDF1基因表达降低后,肿瘤细胞的增殖得到了明显的抑制,且对5-氟脲嘧啶(5-FU)和奥沙利铂(L-OHP)这两种化疗药更为敏感。常规的化疗药物在恶性肿瘤的治疗中一直扮演着重要的角色,除了具有较强的副作用外,其最大的缺点就是容易产生耐药,所以在临床中往往会选择联合用药,以上两项m6A甲基化与化疗药物的研究为在肿瘤的临床用药提供了新的思路。Lai等[60]发现鼻咽癌也与m6A甲基化相关,并发现黄芩苷可以通过上调鼻咽癌中的m6A甲基化水平来影响mRNA的剪接,这会对鼻咽癌细胞的凋亡和细胞周期产生影响。黄芩苷是一种有名的中药成分,该研究为人们在中药中寻找合适的抗癌药物提供了指引。大黄素(Rhein)是首个发现的FTO抑制剂,可以有效增加细胞内的m6A甲基化水平。虽然大黄素对于FTO的抑制效果很强,但也发现大黄素的专一性却很差[61]。随后Huang等[17]发现MA(meclofenamic acid)类抗炎药物可以作为FTO抑制剂,它可以在RNA中的m6A位点与FTO蛋白竞争性结合,从而起到抑制FTO的去甲基作用。同时,他们发现MA的乙酯化异构体MA2,该药物具有更好的细胞穿透性且能显著地提高细胞内的m6A甲基化水平。而Cui等[59]发现,对胶质母细胞瘤使用MA2可以有效抑制FTO的表达并抑制肿瘤的进展。这也为人们寻找新的靶向药物提供了指引。同时也有文献报道IOX3可以抑制细胞内的FTO基因的表达,但是否能改变细胞内的m6A甲基化水平尚未得出明确结论[62]。柠檬酸盐结晶也被发现可以抑制细胞内ALKBH5基因的表达[63]。另有两篇文献报道了N-CDPCB和CHTB药物可以通过与FTO转录的mRNA特异性结合而可以作为FTO抑制剂[64,65]。目前,虽然已经文献报道了多种m6A去甲基酶的小分子药物的存在(表3),但无论是其特异性或有效性都不能很好的达到精准治疗的目的,精准、有效的m6A靶向药物依然有待进一步研发。Table 3
表3
表3 去甲基酶抑制剂
Table 3
| 药物种类 | 是否作用于FTO | 是否作用于ALKBH5 | 参考文献 |
|---|---|---|---|
| Rhein | 是 | 未知 | [61] |
| MA | 是 | 否 | [17] |
| IOX3 | 是 | 是 | [62] |
| Citrate | 是 | 是 | [63] |
| N-CDPCB | 是 | 未知 | [64] |
| CHTB | 是 | 未知 | [65] |
新窗口打开|下载CSV
3 结语与展望
m6A甲基化作为众多RNA表观遗传修饰家族中的一员,基于目前对其与肿瘤的认识,其本身并无“好坏”之分,主要通过调控相关的癌基因或抑癌基因的mRNA表达量起到对肿瘤细胞的促进或抑制作用。随着人们对m6A甲基化机制的研究逐渐深入,发现与其相关的RNA水平的调控变得更加复杂和多样。有趣的是,读取蛋白结合m6A甲基化位点后,既可能促进相关mRNA的表达,也可能降低相关mRNA的稳定性而加速降解。此外,非编码RNA可以调控目的基因的表达水平,而m6A甲基化出现在非编码RNA中,又会调控非编码RNA本身的表达水平。m6A甲基化这种双面调控行为特别是对于非编码RNA的调控,也许能够帮助人们从新角度去阐明一些原本无法解释的问题。肿瘤干细胞是一种获得了多能性的肿瘤细胞,其恶性程度极高,拥有自我更新能力使其能更快地突变以产生耐药能力或适应微环境的改变。研究发现,目前有一定数量的m6A甲基化与肿瘤的研究都与肿瘤干细胞有关,这为人们对于肿瘤干细胞的认识提供了一个新的视角,并对于更全面地理解肿瘤细胞的发生发展和突变提供了很好的帮助,也为对肿瘤干细胞相关肿瘤的治疗指引了新的方向。如今,肿瘤的诊断和治疗已步入了分子时代,肿瘤的分子诊断现已广泛开展,分子靶向药物的研制也取得了一定的突破。对于不同肿瘤中m6A甲基化位点的研究可以帮助医疗工作者们寻找新的分子诊断位点。另外,RNA中的m6A甲基化水平与细胞内写入、擦除基因的表达量密切相关,而读取基因表达的蛋白分子则与m6A甲基化位点相结合从而执行一系列生物学功能。所以在肿瘤中,无论是m6A相关的基因还是蛋白的表达水平改变都将有可能成为肿瘤分子诊断的潜在标志物,同时也将为临床分子靶向治疗药物的研发提供新的 靶点。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URL [本文引用: 1]
URLPMID:25171402 [本文引用: 1]

Although more than 100 types of RNA modification have been described thus far, most of them were thought to be rare in mRNAs and in regulatory noncoding RNAs. Recent developments have unveiled that at least some of the modifications are considerably abundant and widely conserved. This Minireview summarizes the molecular machineries and biological functions of methylation (N6-methyladenosine, m6A) and uridylation (U-tail).
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLMagsci [本文引用: 1]
<p>RNA酶促共价修饰研究, 尤其是m<sup>6</sup>A(6-甲基腺嘌呤), 是RNA生物学研究的一个新兴领域。m<sup>6</sup>A是真核生物mRNA内部序列中最常见的一种转录后修饰形式, 由包含3个独立组分的复合物mRNA: m<sup>6</sup>A甲基转移酶催化生成。最新研究发现肥胖相关蛋白FTO可以脱掉m<sup>6</sup>A上的甲基, 表明该甲基化过程是可逆的。抑制或敲除m<sup>6</sup>A甲基转移酶会引起重要的表型变化, 但是由于过去的检测方法受限, m<sup>6</sup>A确切的作用机制目前为止还不甚清楚。二代测序技术结合免疫沉淀方法为大规模检测m<sup>6</sup>A修饰并研究其作用机制提供了可能。文章主要综述了m<sup>6</sup>A的发现史、生成机制、组织和基因组分布、检测方法、生物学功能等及其最新研究进展, 并通过比较3种IP-seq技术和数据分析的异同及优缺点, 对m<sup>6</sup>A这种RNA表观修饰研究中尚未解决的问题进行了讨论。</p>
URLMagsci [本文引用: 1]
<p>RNA酶促共价修饰研究, 尤其是m<sup>6</sup>A(6-甲基腺嘌呤), 是RNA生物学研究的一个新兴领域。m<sup>6</sup>A是真核生物mRNA内部序列中最常见的一种转录后修饰形式, 由包含3个独立组分的复合物mRNA: m<sup>6</sup>A甲基转移酶催化生成。最新研究发现肥胖相关蛋白FTO可以脱掉m<sup>6</sup>A上的甲基, 表明该甲基化过程是可逆的。抑制或敲除m<sup>6</sup>A甲基转移酶会引起重要的表型变化, 但是由于过去的检测方法受限, m<sup>6</sup>A确切的作用机制目前为止还不甚清楚。二代测序技术结合免疫沉淀方法为大规模检测m<sup>6</sup>A修饰并研究其作用机制提供了可能。文章主要综述了m<sup>6</sup>A的发现史、生成机制、组织和基因组分布、检测方法、生物学功能等及其最新研究进展, 并通过比较3种IP-seq技术和数据分析的异同及优缺点, 对m<sup>6</sup>A这种RNA表观修饰研究中尚未解决的问题进行了讨论。</p>
.
URLPMID:29205437 [本文引用: 1]
Abstract Messenger RNA is a flexible tool box that plays a key role in the dynamic regulation of gene expression. RNA modifications variegate the message conveyed by the mRNA. Similar to DNA and histone modifications, mRNA modifications are reversible and play a key role in the regulation of molecular events. Our understanding about the landscape of RNA modifications is still rudimentary in contrast to DNA and histone modifications. The major obstacle has been the lack of sensitive detection methods since they are non-editing events. However, with the advent of next-generation sequencing techniques, RNA modifications are being identified precisely at single nucleotide resolution. In recent years, methylation at the N6 position of adenine (m 6 A) has gained the attention of RNA biologists. The m 6 A modification has a set of writers (methylases), erasers (demethylases), and readers. Here, we provide a summary of interesting facts, conflicting findings, and recent advances in the technical and functional aspects of the m 6 A epitranscriptome.
.
URLPMID:29125541 [本文引用: 1]
The broad application of next-generation sequencing technologies in conjunction with improved bioinformatics has helped to illuminate the complexity of the transcriptome, both in terms of quantity and variety. In humans, 70 90% of the genome is transcribed, but only ~2% carries the blueprint for proteins. Hence, there is a huge class of non-translated transcripts, called long non-coding RNAs (lncRNAs), which have received much attention in the past decade. Several studies have shown that lncRNAs are involved in a plethora of cellular signaling pathways and actively regulate gene expression via a broad selection of molecular mechanisms. Only recently, sequencing-based, transcriptome-wide studies have characterized different types of post-transcriptional chemical modifications of RNAs. These modifications have been shown to affect the fate of RNA and further expand the variety of the transcriptome. However, our understanding of their biological function, especially in the context of lncRNAs, is still in its infancy. In this review, we will focus on three epitranscriptomic marks, namely pseudouridine ( ),N6-methyladenosine (m6A) and 5-methylcytosine (m5C). We will introduce writers, readers, and erasers of these modifications, and we will present methods for their detection. Finally, we will provide insights into the distribution and function of these chemical modifications in selected, cancer-related lncRNAs.
.
URLPMID:25799998 [本文引用: 1]
Abstract The first step in the biogenesis of microRNAs is the processing of primary microRNAs (pri-miRNAs) by the microprocessor complex, composed of the RNA-binding protein DGCR8 and the type III RNase DROSHA. This initial event requires recognition of the junction between the stem and the flanking single-stranded RNA of the pri-miRNA hairpin by DGCR8 followed by recruitment of DROSHA, which cleaves the RNA duplex to yield the pre-miRNA product. While the mechanisms underlying pri-miRNA processing have been determined, the mechanism by which DGCR8 recognizes and binds pri-miRNAs, as opposed to other secondary structures present in transcripts, is not understood. Here we find in mammalian cells that methyltransferase-like 3 (METTL3) methylates pri-miRNAs, marking them for recognition and processing by DGCR8. Consistent with this, METTL3 depletion reduced the binding of DGCR8 to pri-miRNAs and resulted in the global reduction of mature miRNAs and concomitant accumulation of unprocessed pri-miRNAs. In vitro processing reactions confirmed the sufficiency of the N(6)-methyladenosine (m(6)A) mark in promoting pri-miRNA processing. Finally, gain-of-function experiments revealed that METTL3 is sufficient to enhance miRNA maturation in a global and non-cell-type-specific manner. Our findings reveal that the m(6)A mark acts as a key post-transcriptional modification that promotes the initiation of miRNA biogenesis.
.
URLPMID:4344304 [本文引用: 1]
Methylation of N6-adenosine (m6A) has been observed in many different classes of RNA, but its prevalence in microRNAs (miRNAs) has not yet been studied. Here we show that a knockdown of the m6A demethylase FTO affects the steady-state levels of several miRNAs. Moreover, RNA immunoprecipitation with an anti-m6A-antibody followed by RNA-seq revealed that a significant fraction of miRNAs contains m6A. By motif searches we have discovered consensus sequences discriminating between methylated and unmethylated miRNAs. The epigenetic modification of an epigenetic modifier as described here adds a new layer to the complexity of the posttranscriptional regulation of gene expression.
.
URLPMID:9409616 [本文引用: 1]
The methylation of internal adenosine residues in eukaryotic mRNA, forming N6-methyladenosine (m6A), is catalyzed by a complex multicomponent enzyme. Previous studies suggested that m6A affects the efficiency of mRNA processing or transport, although the mechanism by which this occurs is not known. As a step toward better understanding the mechanism and function of this ubiquitous posttranscriptional modification, we have shown that HeLa mRNA (N6-adenosine)-methyltransferase requires at least two separate protein factors, MT-A and MT-B, and MT-A contains the AdoMet binding site on a 70-kDa subunit (MT-A70). MT-A70 was purified by conventional chromatography and electrophoresis, and was microsequenced. The peptide sequence was used to design a degenerate oligodeoxynucleotide that in turn was used to isolate the cDNA clone coding for MT-A70 from a HeLa cDNA library. Recombinant MT-A70 was expressed as a fusion protein in bacteria and was used to generate anti-MT-A70 antisera in rabbits. These antisera recognize MT-A70 in HeLa nuclear extracts by western blot and are capable of depleting (N6-adenosine)-methyltransferase activity from HeLa nuclear extract, confirming that MT-A70 is a critical subunit of (N6-adenosine)-methyltransferase. Northern blot analysis reveals that MT-A70 mRNA is present in a wide variety of human tissues and may undergo alternative splicing. MT-A70 cDNA probe hybridizes to a 2.0-kilobase (kb) polyadenylated RNA isolated from HeLa cells, whereas it hybridizes to two predominant RNA species (approximately 2.0 kb and 3.0 kb) using mRNA isolated from six different human tissues. Analysis of the cDNA sequence indicates that it codes for a 580-amino acid protein with a predicted MW = 65 kDa. The predicted protein contains sequences similar to consensus methylation motifs I and II identified in prokaryotic DNA (N6-adenosine)-methyltransferases, suggesting the functional conservation of peptide motifs. MT-A70 also contains a long region of homology to the yeast protein SPO8, which is involved in induction of sporulation by an unknown mechanism.
.
URLPMID:20202062020202020202020 [本文引用: 1]
N(6)-methyladenosine (m(6)A) has been identified as the most abundant internal modification of messenger RNA in eukaryotes. m(6)A modification is involved in cell fate determination in yeast and embryo development in plants. Its mammalian function remains unknown but thousands of mammalian mRNAs and long non-coding RNAs (lncRNAs) show m(6)A modification and m(6)A demethylases are required for mammalian energy homeostasis and fertility. We identify two proteins, the putative m(6)A MTase, methyltransferase-like 3 (Mettl3; ref. ), and a related but uncharacterized protein Mettl14, that function synergistically to control m(6)A formation in mammalian cells. Knockdown of Mettl3 and Mettl14 in mouse embryonic stem cells (mESCs) led to similar phenotypes, characterized by lack of m(6)A RNA methylation and lost self-renewal capability. A large number of transcripts, including many encoding developmental regulators, exhibit m(6)A methylation inversely correlated with mRNA stability and gene expression. The human antigen R (HuR) and microRNA pathways were linked to these effects. This gene regulatory mechanism operating in mESCs through m(6)A methylation is required to keep mESCs at their ground state and may be relevant to thousands of mRNAs and lncRNAs in various cell types.
.
URLPMID:3915904 [本文引用: 1]
The methyltransferase like 3 (METTL3)-containing methyltransferase complex catalyzes the N6-methyladenosine (m6A) formation, a novel epitranscriptomic marker; however, the nature of this complex remains largely unknown. Here we report two new components of the human m6A methyltransferase complex, Wilms' tumor 1-associating protein (WTAP) and methyltransferase like 14 (METTL14). WTAP interacts with METTL3 and METTL14, and is required for their localization into nuclear speckles enriched with pre-mRNA processing factors and for catalytic activity of the m6A methyltransferase in vivo. The majority of RNAs bound by WTAP and METTL3 in vivo represent mRNAs containing the consensus m6A motif. In the absence of WTAP, the RNA-binding capability of METTL3 is strongly reduced, suggesting that WTAP may function to regulate recruitment of the m6A methyltransferase complex to mRNA targets. Furthermore, transcriptomic analyses in combination with photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) illustrate that WTAP and METTL3 regulate expression and alternative splicing of genes involved in transcription and RNA processing. Morpholino-mediated knockdown targeting WTAP and/or METTL3 in zebrafish embryos caused tissue differentiation defects and increased apoptosis. These findings provide strong evidence that WTAP may function as a regulatory subunit in the m6A methyltransferase complex and play a critical role in epitranscriptomic regulation of RNA metabolism.
.
URLPMID:24100041 [本文引用: 1]
Background: WTAP is a ubiquitously expressed nuclear protein that is required for mammalian early embryo development and cell cycle progression. Results: WTAP forms a complex with several splicing regulators. Conclusion: WTAP regulates both the cell cycle and alternative splicing by the formation of a protein complex. Significance: Characterization of this protein complex will help to elucidate the critically important function of WTAP in alternative splicing and cell proliferation.Wilms' tumor 1-associating protein (WTAP) is a putative splicing regulator that is thought to be required for cell cycle progression through the stabilization of cyclin A2 mRNA and mammalian early embryo development. To further understand how WTAP acts in the context of the cellular machinery, we identified its interacting proteins in human umbilical vein endothelial cells and HeLa cells using shotgun proteomics. Here we show that WTAP forms a novel protein complex including Hakai, Virilizer homolog, KIAA0853, RBM15, the arginine/serine-rich domain-containing proteins BCLAF1 and THRAP3, and certain general splicing regulators, most of which have reported roles in post-transcriptional regulation. The depletion of these respective components of the complex resulted in reduced cell proliferation along with G(2)/M accumulation. Double knockdown of the serine/arginine-rich (SR)-like proteins BCLAF1 and THRAP3 by siRNA resulted in a decrease in the nuclear speckle localization of WTAP, whereas the nuclear speckles were intact. Furthermore, we found that the WTAP complex regulates alternative splicing of the WTAP pre-mRNA by promoting the production of a truncated isoform, leading to a change in WTAP protein expression. Collectively, these findings show that the WTAP complex is a novel component of the RNA processing machinery, implying an important role in both posttranscriptional control and cell cycle regulation.
.
URL [本文引用: 3]
.
URLPMID:28525753 [本文引用: 1]
Maintenance of proper levels of the methyl donor S-adenosylmethionine (SAM) is critical for a wide variety of biological processes. We demonstrate that the N 6 -adenosine methyltransferase METTL16 regulates expression of human MAT2A, which encodes the SAM synthetase expressed in most cells. Upon SAM depletion by methionine starvation, cells induce MAT2A expression by enhanced splicing of a retained intron. Induction requires METTL16 and its methylation substrate, a vertebrate conserved hairpin (hp1) in the MAT2A 3′ UTR. Increasing METTL16 occupancy on the MAT2A 3′ UTR is sufficient to induce efficient splicing. We propose that, under SAM-limiting conditions, METTL16 occupancy on hp1 increases due to inefficient enzymatic turnover, which promotes MAT2A splicing. We further show that METTL16 is the long-unknown methyltransferase for the U6 spliceosomal small nuclear RNA (snRNA). These observations suggest that the conserved U6 snRNA methyltransferase evolved an additional function in vertebrates to regulate SAM homeostasis.
URLPMID:24981863 [本文引用: 1]
N6-methyladenosine (m6A) is a highly abundant modification of mRNA. Schwartz et al. identify and validate a network of proteins required for mRNA methylation in mammalian cells. They define two distinct classes of methylation sites. The majority of sites depend on the identified proteins, are located at internal positions in transcripts, and inversely correlate with mRNA stability. Sites independent of these proteins form at the first transcribed base as part of the cap structure, forming a previously unappreciated layer of transcriptome complexity.
.
URLPMID:25452335 [本文引用: 2]
Abstract Two human demethylases, the fat mass and obesity-associated (FTO) enzyme and ALKBH5, oxidatively demethylate abundant N(6)-methyladenosine (m(6)A) residues in mRNA. Achieving a method for selective inhibition of FTO over ALKBH5 remains a challenge, however. Here, we have identified meclofenamic acid (MA) as a highly selective inhibitor of FTO. MA is a non-steroidal, anti-inflammatory drug that mechanistic studies indicate competes with FTO binding for the m(6)A-containing nucleic acid. The structure of FTO/MA has revealed much about the inhibitory function of FTO. Our newfound understanding, revealed herein, of the part of the nucleotide recognition lid (NRL) in FTO, for example, has helped elucidate the principles behind the selectivity of FTO over ALKBH5. Treatment of HeLa cells with the ethyl ester form of MA (MA2) has led to elevated levels of m(6)A modification in mRNA. Our collective results highlight the development of functional probes of the FTO enzyme that will (i) enable future biological studies and (ii) pave the way for the rational design of potent and specific inhibitors of FTO for use in medicine. The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
.
URL [本文引用: 2]
URLPMID:24284625 [本文引用: 1]
Abstract N(6)-methyladenosine (m(6)A) is the most prevalent internal (non-cap) modification present in the messenger RNA of all higher eukaryotes. Although essential to cell viability and development, the exact role of m(6)A modification remains to be determined. The recent discovery of two m(6)A demethylases in mammalian cells highlighted the importance of m(6)A in basic biological functions and disease. Here we show that m(6)A is selectively recognized by the human YTH domain family 2 (YTHDF2) 'reader' protein to regulate mRNA degradation. We identified over 3,000 cellular RNA targets of YTHDF2, most of which are mRNAs, but which also include non-coding RNAs, with a conserved core motif of G(m(6)A)C. We further establish the role of YTHDF2 in RNA metabolism, showing that binding of YTHDF2 results in the localization of bound mRNA from the translatable pool to mRNA decay sites, such as processing bodies. The carboxy-terminal domain of YTHDF2 selectively binds to m(6)A-containing mRNA, whereas the amino-terminal domain is responsible for the localization of the YTHDF2-mRNA complex to cellular RNA decay sites. Our results indicate that the dynamic m(6)A modification is recognized by selectively binding proteins to affect the translation status and lifetime of mRNA.
URLPMID:4673968 [本文引用: 2]
The RNA-binding protein HNRNPA2B1 is a nuclear “reader” of the m6A mark, acting as an adaptor that recruits the Microprocessor complex to a subset of precursor miRNAs, facilitating their processing into mature miRNAs.
.
URLPMID:25719671 [本文引用: 2]
Abstract RNA-binding proteins control many aspects of cellular biology through binding single-stranded RNA binding motifs (RBMs). However, RBMs can be buried within their local RNA structures, thus inhibiting RNA-protein interactions. N(6)-methyladenosine (m(6)A), the most abundant and dynamic internal modification in eukaryotic messenger RNA, can be selectively recognized by the YTHDF2 protein to affect the stability of cytoplasmic mRNAs, but how m(6)A achieves its wide-ranging physiological role needs further exploration. Here we show in human cells that m(6)A controls the RNA-structure-dependent accessibility of RBMs to affect RNA-protein interactions for biological regulation; we term this mechanism 'the m(6)A-switch'. We found that m(6)A alters the local structure in mRNA and long non-coding RNA (lncRNA) to facilitate binding of heterogeneous nuclear ribonucleoprotein C (HNRNPC), an abundant nuclear RNA-binding protein responsible for pre-mRNA processing. Combining photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) and anti-m(6)A immunoprecipitation (MeRIP) approaches enabled us to identify 39,060 m(6)A-switches among HNRNPC-binding sites; and global m(6)A reduction decreased HNRNPC binding at 2,798 high-confidence m(6)A-switches. We determined that these m(6)A-switch-regulated HNRNPC-binding activities affect the abundance as well as alternative splicing of target mRNAs, demonstrating the regulatory role of m(6)A-switches on gene expression and RNA maturation. Our results illustrate how RNA-binding proteins gain regulated access to their RBMs through m(6)A-dependent RNA structural remodelling, and provide a new direction for investigating RNA-modification-coded cellular biology.
.
URLPMID:26593424 [本文引用: 1]
N6-methyladenosine (m6A) residues within the 5′ UTR of mRNAs promote translation initiation through a mechanism that does not require the 5′ cap or cap-binding proteins. Diverse cellular stresses selectively increase the levels of m6A within 5′ UTRs, suggesting that 5′ UTR m6A is important for mediating stress-induced translational responses.
.
URLPMID:28106076 [本文引用: 1]
Cell death and differentiation is a monthly research journal focused on the exciting field of programmed cell death and apoptosis. It provides a single accessible source of information for both scientists and clinicians, keeping them up-to-date with advances in the field. It encompasses programmed cell death, cell death induced by toxic agents, differentiation and the interrelation of these with cell proliferation.
.
URLPMID:26046440 [本文引用: 1]
Human YTHDF1 binds m6A-modified mRNAs and through interactions with initiation factors and ribosomes increases translational output from those messages.
.
URLPMID:4851248 [本文引用: 1]
The most abundant mRNA post-transcriptional modification isN6-methyladenosine (m6A) that has broad roles in RNA biology1-5. In mammalian cells, the asymmetric distribution of m6A along mRNAs leaves relatively less methylation in the 5′ untranslated region (5′UTR) compared to other regions6,7. However, whether and how 5′UTR methylation is regulated is poorly understood. Despite the crucial role of the 5′UTR in translation initiation, very little is known whether m6A modification influences mRNA translation. Here we show that in response to heat shock stress, m6A is preferentially deposited to the 5′UTR of newly transcribed mRNAs. We found that the dynamic 5′UTR methylation is a result of stress-induced nuclear localization of YTHDF2, a well characterized m6A “reader”. Upon heat shock stress, the nuclear YTHDF2 preserves 5′UTR methylation of stress-induced transcripts by limiting the m6A “eraser” FTO from demethylation. Remarkably, the increased 5′UTR methylation in the form of m6A promotes cap-independent translation initiation, providing a mechanism for selective mRNA translation under heat shock stress. Using Hsp70 mRNA as an example, we demonstrate that a single site m6A modification in the 5′UTR enables translation initiation independent of the 5′ end m7G cap. The elucidation of the dynamic feature of 5′UTR methylation and its critical role in cap-independent translation not only expands the breadth of physiological roles of m6A, but also uncovers a previously unappreciated translational control mechanism in heat shock response.
URLPMID:26876937 [本文引用: 1]
Xiao et02al. show that m6A reader YTHDC1 promotes exon inclusion of targeted mRNAs through facilitating SRSF3 while blocking SRSF10 mRNA binding, demonstrating how m6A reader YTHDC1 directly regulates mRNA splicing by bridging interactions oftrans- andcis-regulatory elements.
URL [本文引用: 1]
First page of article
URLPMID:8373778 [本文引用: 1]
Abstract A new technique has been developed for the facile location of pseudouridylate (psi) residues in any RNA molecule. The method uses two known modification procedures which in combination uniquely identify U residues which have been converted into psi. The first procedure involves reaction of all U-like and G-like residues with N-cyclohexyl-N'-beta-(4-methylmorpholinium)ethylcarbodiimide p-tosylate (CMC), followed by alkaline removal of all CMC groups except those linked to the N3 of psi. This stops reverse transcription, resulting in a gel band which identifies the U residue. The second procedure is uridine-specific hydrazinolysis which cleaves the RNA chain at all U residues and produces a gel band upon reverse transcription. psi residues, being resistant to hydrazinolysis, are not cleaved and do not stop reverse transcription. This leads to the absence of a band at psi residues. The combined method can also distinguish psi from 5-methyluridine, 4-thiouridine, uridine-5-oxyacetic acid, and 2-thio-5-methylaminomethyluridine as shown by treating rRNA and tRNA species known to contain these modified bases at defined sites. By this procedure, four new sites for psi in Escherichia coli 23S RNA were discovered, and one was disproven. The four new sites are at positions 2457, 2504, 2580, and 2605. The erroneous site is at position 2555. These four new psi residues, which are all in or within 2-3 residues of the peptidyltransferase ring, are thus in a position to play a functional and/or structural role at the peptidyltransferase center.(ABSTRACT TRUNCATED AT 250 WORDS)
.
URL [本文引用: 1]
URLPMID:22608085 [本文引用: 1]
Adenosine methylation (m6A) is a widespread RNA modification found in >7,000 mammalian genes, encoding both mRNAs and noncoding RNAs, with an especially high prevalence in the developing brain. In both mouse and human mRNAs, m6A is enriched within 3′ UTRs that also contain miRNA-binding sites.
URLPMID:28002401 [本文引用: 1]
Abstract Internal bases in mRNA can be subjected to modifications that influence the fate of mRNA in cells. One of the most prevalent modified bases is found at the 5' end of mRNA, at the first encoded nucleotide adjacent to the 7-methylguanosine cap. Here we show that this nucleotide, N 6 ,2'-O-dimethyladenosine (m 6 A m ), is a reversible modification that influences cellular mRNA fate. Using a transcriptome-wide map of m 6 A m we find that m 6 A m -initiated transcripts are markedly more stable than mRNAs that begin with other nucleotides. We show that the enhanced stability of m 6 A m -initiated transcripts is due to resistance to the mRNA-decapping enzyme DCP2. Moreover, we find that m 6 A m is selectively demethylated by fat mass and obesity-associated protein (FTO). FTO preferentially demethylates m 6 A m rather than N 6 -methyladenosine (m 6 A), and reduces the stability of m 6 A m mRNAs. Together, these findings show that the methylation status of m 6 A m in the 5' cap is a dynamic and reversible epitranscriptomic modification that determines mRNA stability.
.
URLPMID:4487409 [本文引用: 1]
N6-methyladenosine (m6A) is the most abundant modified base in eukaryotic mRNA and has been linked to diverse effects on mRNA fate. Current m6A mapping approaches localize m6A residues to 100–200 nt-long regions of transcripts. The precise position of m6A in mRNAs cannot be identified on a transcriptome-wide level because there are no chemical methods to distinguish between m6A and adenosine. Here we show that anti-m6A antibodies can induce specific mutational signatures at m6A residues after ultraviolet light-induced antibody-RNA crosslinking and reverse transcription. We find these antibodies similarly induce mutational signatures atN6,2′-O-dimethyladenosine (m6Am), a nucleotide found at the first encoded position of certain mRNAs. Using these mutational signatures, we map m6A and m6Am at single-nucleotide resolution in human and mouse mRNA and identify snoRNAs as a novel class of m6A-containing ncRNAs.
[本文引用: 1]
.
URLPMID:26404942 [本文引用: 1]
Abstract We adapted UV CLIP (cross-linking immunoprecipitation) to accurately locate tens of thousands of m(6)A residues in mammalian mRNA with single-nucleotide resolution. More than 70% of these residues are present in the 3'-most (last) exons, with a very sharp rise (sixfold) within 150-400 nucleotides of the start of the last exon. Two-thirds of last exon m(6)A and >40% of all m(6)A in mRNA are present in 3' untranslated regions (UTRs); contrary to earlier suggestions, there is no preference for location of m(6)A sites around stop codons. Moreover, m(6)A is significantly higher in noncoding last exons than in next-to-last exons harboring stop codons. We found that m(6)A density peaks early in the 3' UTR and that, among transcripts with alternative polyA (APA) usage in both the brain and the liver, brain transcripts preferentially use distal polyA sites, as reported, and also show higher proximal m(6)A density in the last exons. Furthermore, when we reduced m6A methylation by knocking down components of the methylase complex and then examined 661 transcripts with proximal m6A peaks in last exons, we identified a set of 111 transcripts with altered (approximately two-thirds increased proximal) APA use. Taken together, these observations suggest a role of m(6)A modification in regulating proximal alternative polyA choice. 2015 Ke et al.; Published by Cold Spring Harbor Laboratory Press.
.
URLPMID:27376769 [本文引用: 1]
Abstract N(6)-Methyladenosine (m(6)A) is a widespread, reversible chemical modification of RNA molecules, implicated in many aspects of RNA metabolism. Little quantitative information exists as to either how many transcript copies of particular genes are m(6)A modified ('m(6)A levels') or the relationship of m(6)A modification(s) to alternative RNA isoforms. To deconvolute the m(6)A epitranscriptome, we developed m(6)A-level and isoform-characterization sequencing (m(6)A-LAIC-seq). We found that cells exhibit a broad range of nonstoichiometric m(6)A levels with cell-type specificity. At the level of isoform characterization, we discovered widespread differences in the use of tandem alternative polyadenylation (APA) sites by methylated and nonmethylated transcript isoforms of individual genes. Strikingly, there is a strong bias for methylated transcripts to be coupled with proximal APA sites, resulting in shortened 3' untranslated regions, while nonmethylated transcript isoforms tend to use distal APA sites. m(6)A-LAIC-seq yields a new perspective on transcriptome complexity and links APA usage to m(6)A modifications.
.
URLPMID:24141618 [本文引用: 1]
N-6-methyladenosine (m(6)A) is the most abundant modification in mammalian mRNA and long noncoding RNA (lncRNA). Recent discoveries of two m(6)A demethylases and cell-type and cell-state-dependent m(6)A patterns indicate that m(6)A modifications are highly dynamic and likely play important biological roles for RNA akin to DNA methylation or histone modification. Proposed functions for m(6)A modification include mRNA splicing, export, stability, and immune tolerance; but m(6)A studies have been hindered by the lack of methods for its identification at single nucleotide resolution. Here, we develop a method that accurately determines m(6)A status at any site in mRNA/lncRNA, termed site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET). The method determines the precise location of the m(6)A residue and its modification fraction, which are crucial parameters in probing the cellular dynamics of m(6)A modification. We applied the method to determine the m(6)A status at several sites in two human lncRNAs and three human mRNAs and found that m(6)A fraction varies between 6% and 80% among these sites. We also found that many m(6)A candidate sites in these RNAs are however not modified. The precise determination of m(6)A status in a long noncoding RNA also enables the identification of an m(6)A-containing RNA structural motif.
URLPMID:26075521 [本文引用: 1]
Pseudouridine (02¨) is the most abundant post-transcriptional RNA modification, yet little is known about its prevalence, mechanism and function in mRNA. Here, we performed quantitative MS analysis and show that 02¨ is much more prevalent (02¨/U ratio 0903040.2-0.6%) in mammalian mRNA than previously believed. We developed N3-CMC-enriched pseudouridine sequencing (CeU-Seq), a selective chemical labeling and pulldown method, to identify 2,084 02¨ sites within 1,929 human transcripts, of which four (in ribosomal RNA and EEF1A1 mRNA) are biochemically verified. We show that hPUS1, a known 02¨ synthase, acts on human mRNA; under stress, CeU-Seq demonstrates inducible and stress-specific mRNA pseudouridylation. Applying CeU-Seq to the mouse transcriptome revealed conserved and tissue-specific pseudouridylation. Collectively, our approaches allow comprehensive analysis of transcriptome-wide pseudouridylation and provide tools for functional studies of 02¨-mediated epigenetic regulation.
.
URLPMID:3936739 [本文引用: 1]
Abstract Chemical landscape of natural RNA species is decorated with the large number of modified nucleosides. Some of those could easily be detected by reverse transcription, while others permit only high-performance liquid chromatography or mass-spectrometry detection. Presence of m(6)A nucleoside at a particular position of long RNA molecule is challenging to observe. Here we report an easy and high-throughput method for detection of m(6)A nucleosides in RNA based on high-resolution melting analysis. The method relies on the previous knowledge of the modified nucleoside position at a particular place of RNA and allows rapid screening for conditions or genes necessary for formation of that modification.
.
URLPMID:4537718 [本文引用: 1]
With the development of new sequencing technology, the entire N6-methyl-adenosine (m6A) RNA methylome can now be unbiased profiled with methylated RNA immune-precipitation sequencing technique (MeRIP-Seq), making it possible to detect differential methylation states of RNA between two conditions, for example, between normal and cancerous tissue. However, as an affinity-based method, MeRIP-Seq has yet provided base-pair resolution; that is, a single methylation site determined from MeRIP-Seq data can in practice contain multiple RNA methylation residuals, some of which can be regulated by different enzymes and thus differentially methylated between two conditions. Since existing peak-based methods could not effectively differentiate multiple methylation residuals located within a single methylation site, we propose a hidden Markov model (HMM) based approach to address this issue. Specifically, the detected RNA methylation site is further divided into multiple adjacent small bins and then scanned with higher resolution using a hidden Markov model to model the dependency between spatially adjacent bins for improved accuracy. We tested the proposed algorithm on both simulated data and real data. Result suggests that the proposed algorithm clearly outperforms existing peak-based approach on simulated systems and detects differential methylation regions with higher statistical significance on real dataset.
URLPMID:26748145 [本文引用: 1]
Just like PTM or PTLM (post-translational modification) in proteins, PTCM (post-transcriptional modification) in RNA plays very important roles in biological processes. Occurring at adenine (A) with the genetic code motif (GAC), N6-methyldenosine (m6A) is one of the most common and abundant PTCMs in RNA found in viruses and most eukaryotes. Given an uncharacterized RNA sequence containing many GAC motifs, which of them can be methylated, and which cannot? It is important for both basic research and drug development to address this problem. Particularly with the avalanche of RNA sequences generated in the postgenomic age, it is highly demanded to develop computational methods for timely identifying the N6-methyldenosine sites in RNA. Here we propose a new predictor called pRNAm-PC, in which RNA sequence samples are expressed by a novel mode of pseudo dinucleotide composition (PseDNC) whose components were derived from a physical hemical matrix via a series of auto-covariance and cross covariance transformations. It was observed via a rigorous jackknife test that, in comparison with the existing predictor for the same purpose, pRNAm-PC achieved remarkably higher success rates in both overall accuracy and stability, indicating that the new predictor will become a useful high-throughput tool for identifying methylation sites in RNA, and that the novel approach can also be used to study many other RNA-related problems and conduct genome analysis. A user-friendly Web server for pRNAm-PC has been established athttp://www.jci-bioinfo.cn/pRNAm-PC, by which users can easily get their desired results without needing to go through the mathematical details.
URLPMID:26314792 [本文引用: 1]
Occurring at adenine (A) with the consensus motif GAC,N6-methyladenosine (m6A) is one of the most abundant modifications in RNA, which plays very important roles in many biological processes. The nonuniform distribution of m6A sites across the genome implies that, for better understanding the regulatory mechanism of m6A, it is indispensable to characterize its sites in a genome-wide scope. Although a series of experimental technologies have been developed in this regard, they are both time-consuming and expensive. With the avalanche of RNA sequences generated in the postgenomic age, it is highly desired to develop computational methods to timely identify their m6A sites. In view of this, a predictor called “iRNA-Methyl” is proposed by formulating RNA sequences with the “pseudo dinucleotide composition” into which three RNA physiochemical properties were incorporated. Rigorous cross-validation tests have indicated that iRNA-Methyl holds very high potential to become a useful tool for genome analysis. For the convenience of most experimental scientists, a web-server for iRNA-Methyl has been established athttp://lin.uestc.edu.cn/server/iRNA-Methylby which users can easily get their desired results without needing to go through the mathematical details.
.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:29040692 [本文引用: 1]
react-text: 539 A hallmark of aberrant activation of the Wnt/β-catenin signaling pathway has been observed in most colorectal cancers (CRC), but little is known about the role of non-coding RNAs regulated by this pathway. Here, we found that miR-150 was the most significantly upregulated microRNA responsive to elevated of Wnt/β-catenin signaling activity in both HCT116 and HEK293T cells. Mechanistically, the... /react-text react-text: 540 /react-text [Show full abstract]
URL [本文引用: 1]
@@
URLPMID:29186125 [本文引用: 1]
Abstract N 6 -methyladenosine (m 6 A) is an abundant internal RNA modification in both coding and non-coding RNAs that is catalysed by the METTL3-METTL14 methyltransferase complex. However, the specific role of these enzymes in cancer is still largely unknown. Here we define a pathway that is specific for METTL3 and is implicated in the maintenance of a leukaemic state. We identify METTL3 as an essential gene for growth of acute myeloid leukaemia cells in two distinct genetic screens. Downregulation of METTL3 results in cell cycle arrest, differentiation of leukaemic cells and failure to establish leukaemia in immunodeficient mice. We show that METTL3, independently of METTL14, associates with chromatin and localizes to the transcriptional start sites of active genes. The vast majority of these genes have the CAATT-box binding protein CEBPZ present at the transcriptional start site, and this is required for recruitment of METTL3 to chromatin. Promoter-bound METTL3 induces m 6 A modification within the coding region of the associated mRNA transcript, and enhances its translation by relieving ribosome stalling. We show that genes regulated by METTL3 in this way are necessary for acute myeloid leukaemia. Together, these data define METTL3 as a regulator of a chromatin-based pathway that is necessary for maintenance of the leukaemic state and identify this enzyme as a potential therapeutic target for acute myeloid leukaemia.
.
[本文引用: 1]
URLPMID:29290617 [本文引用: 1]
Abstract N 6 -methyladenosine (m 6 A), the most prevalent internal modification in eukaryotic messenger RNAs (mRNAs), plays critical roles in many bioprocesses. However, its functions in normal and malignant hematopoiesis remain elusive. Here, we report that METTL14, a key component of the m 6 A methyltransferase complex, is highly expressed in normal hematopoietic stem/progenitor cells (HSPCs) and acute myeloid leukemia (AML) cells carrying t(11q23), t(15;17), or t(8;21) and is downregulated during myeloid differentiation. Silencing of METTL14 promotes terminal myeloid differentiation of normal HSPCs and AML cells and inhibits AML cell survival/proliferation. METTL14 is required for development and maintenance of AML and self-renewal of leukemia stem/initiation cells (LSCs/LICs). Mechanistically, METTL14 exerts its oncogenic role by regulating its mRNA targets (e.g., MYB and MYC) through m 6 A modification, while the protein itself is negatively regulated by SPI1. Collectively, our results reveal the SPI1-METTL14-MYB/MYC signaling axis in myelopoiesis and leukemogenesis and highlight the critical roles of METTL14 and m 6 A modification in normal and malignant hematopoiesis.
URLPMID:29171881 [本文引用: 2]
Abstract Epigenetic alterations have contributed greatly to human carcinogenesis. Conventional epigenetic studies have predominantly focused on DNA methylation, histone modifications, and chromatin remodeling. Recently, diverse and reversible chemical modifications of RNAs have emerged as a new layer of epigenetic regulation. N6-methyladenosine (m6A) is the most abundant chemical modification of eukaryotic messenger RNA (mRNA) and is important for the regulation of mRNA stability, splicing, and translation. Using transcriptome sequencing, we discovered that methyltransferase-like 3 (METTL3), a major RNA N6-adenosine methyltransferase, was significantly up-regulated in human hepatocellular carcinoma (HCC) and multiple solid tumors. Clinically, overexpression of METTL3 is associated with poor prognosis of patients with HCC. Functionally, we proved that knockdown of METTL3 drastically reduced HCC cell proliferation, migration, and colony formation in vitro. Knockout of METTL3 remarkably suppressed HCC tumorigenicity and lung metastasis in vivo. On the other hand, using the CRISPR/dCas9-VP64 activation system, we demonstrated that overexpression of METTL3 significantly promoted HCC growth both in vitro and in vivo. Through transcriptome sequencing, m6A sequencing, and m6A methylated RNA immuno-precipitation quantitative reverse-transcription polymerase chain reaction, we identified suppressor of cytokine signaling 2 (SOCS2) as a target of METTL3-mediated m6A modification. Knockdown of METTL3 substantially abolished SOCS2 mRNA m6A modification and augmented SOCS2 mRNA expression. We also showed that m6A-mediated SOCS2 mRNA degradation relied on the m6A reader protein YTHDF2-dependent pathway. CONCLUSION: METTL3 is frequently up-regulated in human HCC and contributes to HCC progression. METTL3 represses SOCS2 expression in HCC through an m6A-YTHDF2-dependent mechanism. Our findings suggest an important mechanism of epigenetic alteration in liver carcinogenesis. (Hepatology 2017). 2017 by the American Association for the Study of Liver Diseases.
URL [本文引用: 1]
URLPMID:29315835 [本文引用: 1]
Abstract The role of N 6 -methyladenosine (m 6 A) demethylase fat mass and obesity-associated protein (FTO) in the regulation of chemo-radiotherapy resistance remains largely unknown. Here, we show that the mRNA level of FTO is elevated in cervical squamous cell carcinoma (CSCC) tissues when compared with respective adjacent normal tissues. FTO enhances the chemo-radiotherapy resistance both in vitro and in vivo through regulating expression of -catenin by reducing m 6 A levels in its mRNA transcripts and in turn increases excision repair cross-complementation group 1 (ERCC1) activity. Clinically, the prognostic value of FTO for overall survival is found to be dependent on -catenin expression in human CSCC samples. Taken together, these findings uncover a critical function for FTO and its substrate m 6 A in the regulation of chemo-radiotherapy resistance, which may bear potential clinical implications for CSCC treatment.
URLPMID:5716777 [本文引用: 1]
The m6A mRNA methylation involves in mRNA splicing, degradation and translation. Recent studies have revealed that reduced m6A mRNA methylation might promote cancer development. However, the role of m6A mRNA methylation in cervical cancer development remains unknown. Therefore, we investigated the role of m6A methylation in cervical cancer in the current study. We first evaluated the m6A mRNA methylation level in 286 pairs of cervical cancer samples and their adjacent normal tissues by dot blot assay. Then the role of m6A on patient survival rates and cervical cancer progression were assessed. The m6A level was significantly reduced in the cervical cancer when comparing with the adjacent normal tissue. The m6A level reduction was significantly correlated with the FIGO stage, tumor size, differentiation, lymph invasion and cancer recurrence. It was also shown to be an independent prognostic indicator of disease-free survival and overall survival for patients with cervical cancer. Reducing m6A level via manipulating the m6A regulators expression promoted cervical cancer cell proliferation. And increasing m6A level significantly suppressed tumor development bothin vitroandin vivo. Our results showed that the reduced m6A level is tightly associated with cervical cancer development and m6A mRNA methylation might be a potential therapeutic target in cervical cancer.
URL [本文引用: 2]
Recent studies that have emerged on the diversity of RNA modification in tumors suggest their eligibility as bona fide targets in diagnosis and drug discovery. N6-methyladenosine (m6A) was first reported and is most common in epitranscriptome modification of various RNAs. The YT521-B homology (YTH) domain family are representative m6A-binding proteins, but how the YTH domain family is involved in cancer remains to be clearly understood. Given that clinical sequence data in colorectal cancer indicate that overexpression of YTHDF1 is outstanding among other family members, we studied the role of Ythdf1 and the transcriptional control of YTHDF1. Immunostaining of Ythdf1 showed that its expression was associated with various malignant tumor behaviors, such as depth, lymph node metastasis, and poorer cancer stages. The study of patient survival indicated that patients with high Ythdf1 expression had significantly poorer overall survival. The results indicated that Ythdf1 expression is an independent prognostic factor of patients. Thein vitrostudy showed that the knockdown of YTHDF1 resulted in the suppression of cancer proliferation and sensitization to the exposure of anticancer drugs such as fluorouracil and oxaliplatin. Importantly, the study upstream of the YTHDF1 gene indicated that an oncogenic transcription factor c-Myc was associated with YTHDF1 in both expression and chromatin immunoprecipitation data. Moreover, the knockdown experiments of c-Myc showed the inhibition of YTHDF1, supporting a notion of c-Myc-driven YTHDF1 axis significance. These data suggest that m6A reader Ythdf1 plays a significant role in colorectal cancer progression.
[本文引用: 1]
URLPMID:29439311 [本文引用: 1]
Abstract In China, hepatocellular carcinoma (HCC) is the most commonly diagnosed cancer and the leading cause of cancer death in men, followed by lung and stomach cancer. There was an urgent need to identify novel prognostic biomarkers for HCC. We explored the expression pattern of m6A related proteins in HCC tissues by using TCGA in this study. We found that the m6A 'reader' YTHDF1 was significantly upregulated in HCC and was positive correlated with pathology stage. Kaplan-Meier analysis showed that Lower YTHDF1 expression level was associated with better survival of HCC patients. Furthermore, we performed GO and KEGG pathway analysis of YTHDF1 co-expressed genes and found YTHDF1 played an important role in regulating HCC cell cycle progression and metabolism. We believed that this study will provide a potential new therapeutic and prognostic target for HCC.
URLPMID:27590511 [本文引用: 1]
Exposure of breast cancer cells to hypoxia increases the percentage of breast cancer stem cells (BCSCs), which are required for tumor initiation and metastasis, and this response is dependent on the activity of hypoxia-inducible factors (HIFs). We previously reported that exposure of breast cancer cells to hypoxia induces the ALKBH5-mediated demethylation of N6-methyladenosine (m6A) in NANOG mRNA leading to increased expression of NANOG, which is a pluripotency factor that promotes BCSC specification. Here we report that exposure of breast cancer cells to hypoxia also induces ZNF217-dependent inhibition of m6A methylation of mRNAs encoding NANOG and KLF4, which is another pluripotency factor that mediates BCSC specification. Although hypoxia induced the BCSC phenotype in all breast-cancer cell lines analyzed, it did so through variable induction of pluripotency factors and ALKBH5 or ZNF217. However, in every breast cancer line, the hypoxic induction of pluripotency factor and ALKBH5 or ZNF217 expression was HIF-dependent. Immunohistochemistry revealed that expression of HIF-1 and ALKBH5 was concordant in all human breast cancer biopsies analyzed. ALKBH5 knockdown in MDA-MB-231 breast cancer cells significantly decreased metastasis from breast to lungs in immunodeficient mice. Thus, HIFs stimulate pluripotency factor expression and BCSC specification by negative regulation of RNA methylation.
URLPMID:29345285 [本文引用: 2]
Abstract N6-methyladenosine (m6A) is the most abundant epitranscriptome modification in mammalian mRNA. Recent years have seen substantial progress in m6A epitranscriptomics, indicating its crucial roles in the initiation and progression of cancer through regulation of RNA stabilities, mRNA splicing, microRNA processing and mRNA translation. However, by what means m6A is dynamically regulated or written by enzymatic components represented by methyltransferase-like 3 (METTL3) and how m6A is significant for each of the numerous genes remain unclear. We focused on METTL3 in pancreatic cancer, the prognosis of which is not satisfactory despite the development of multidisciplinary therapies. We established METTL3-knockdown pancreatic cancer cell line using short hairpin RNA. Although morphologic and proliferative changes were unaffected, METTL3-depleted cells showed higher sensitivity to anticancer reagents such as gemcitabine, 5-fluorouracil, cisplatin and irradiation. Our data suggest that METTL3 is a potent target for enhancing therapeutic efficacy in patients with pancreatic cancer. In addition, we performed cDNA expression analysis followed by gene ontology and protein-protein interaction analysis using the Database for Annotation, Visualization, and Integrated Discovery and Search Tool for the Retrieval of Interacting Genes/Proteins databases, respectively. The results demonstrate that METTL3 was associated with mitogen-activated protein kinase cascades, ubiquitin-dependent process and RNA splicing and regulation of cellular process, suggesting functional roles and targets of METTL3.
URLPMID:28297667 [本文引用: 2]
RNA modifications play critical roles in important biological processes. However, the functions of N 6 -methyladenosine (m 6 A) mRNA modification in cancer biology and cancer stem cells remain largely unknown. Here, we show that m 6 A mRNA modification is critical for glioblastoma stem cell (GSC) self-renewal and tumorigenesis. Knockdown of METTL3 or METTL14, key components of the RNA methyltransferase complex, dramatically promotes human GSC growth, self-renewal, and tumorigenesis. In contrast, overexpression of METTL3 or inhibition of the RNA demethylase FTO suppresses GSC growth and self-renewal. Moreover, inhibition of FTO suppresses tumor progression and prolongs lifespan of GSC-grafted mice substantially. m 6 A sequencing reveals that knockdown of METTL3 or METTL14 induced changes in mRNA m 6 A enrichment and altered mRNA expression of genes (e.g., ADAM19 ) with critical biological functions in GSCs. In summary, this study identifies the m 6 A mRNA methylation machinery as promising therapeutic targets for glioblastoma.
URL [本文引用: 1]
Baicalin hydrate (BH), a natural compound, has been investigated for many years because of its traditional medicinal properties. However, the anti-tumor activities of BH and its epigenetic role in NPC have not been elucidated. In this study, we identified that BH inhibits NPC cell growthin vivoandin vitroby inducing apoptosis and cell cycle arrest. BH epigenetically regulated genome instability by up-regulating the expression of satellite 2 (Sat2), alpha satellite ( -Sat), and major satellite (Major-Sat). BH also increased the level of IKK , Suv39H1, and H3K9me3 and decreased LSH expression. Interestingly, BH promoted the splicing of Suv39H1 via the enhancement of m6A RNA methylation, rather than DNA methylation. Taken together, our results demonstrated that BH has an anti-tumor role in NPC and revealed a unique role of BH in genome instability and splicing in response to DNA damage.
URL [本文引用: 1]
.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:26314339 [本文引用: 1]
Abstract N-(5-Chloro-2,4-dihydroxyphenyl)-1-phenylcyclobutanecarboxamide (N-CDPCB, 1a) is found to be an inhibitor of the fat mass and obesity associated protein (FTO). The crystal structure of human FTO with 1a reveals a novel binding site for the FTO inhibitor and defines the molecular basis for recognition by FTO of the inhibitor. The identification of the new binding site offers new opportunities for further development of selective and potent inhibitors of FTO, which is expected to provide information concerning novel therapeutic targets for treatment of obesity or obesity-associated diseases.
URLPMID:26915401 [本文引用: 1]
FeII and α-ketoglutarate-dependent fat mass and obesity associated protein (FTO)-dependent demethylation of m6A is important for regulation of mRNA splicing and adipogenesis. Developing FTO-specific inhibitors can help probe the biology of FTO and unravel novel therapeutic targets for treatment of obesity or obesity-associated diseases. In the present paper, we have identified that 4-chloro-6-(6′-chloro-7′-hydroxy-2′,4′,4′-trimethyl-chroman-2′-yl)benzene-1,3-diol (CHTB) is an inhibitor of FTO. The crystal structure of CHTB complexed with human FTO reveals that the novel small molecule binds to FTO in a specific manner. The identification of the novel small molecule offers opportunities for further development of more selective and potent FTO inhibitors.

{kind=link}
{kind=link}