 ,福建省农业科学院水稻研究所,福州 350018
,福建省农业科学院水稻研究所,福州 350018Mapping and cloning of GAD1-2 for long awn using CSSLs in rice (Oryza sativa L.)
Dewei Yang, Xianghua Zheng, Chaoping Cheng, Ning Ye, Fenghuang Huang, Xinfu Ye,Institute of Rice, Fujian Academy of Agricultural Sciences, Fuzhou 350018, China通讯作者:
编委: 储成才
收稿日期:2018-02-13修回日期:2018-07-1网络出版日期:2018-12-20
| 基金资助: |
Received:2018-02-13Revised:2018-07-1Online:2018-12-20
| Fund supported: |
作者简介 About authors
杨德卫,博士研究生,研究方向:水稻遗传育种E-mail:dewei-y@163.com。

摘要
关键词:
Abstract
Keywords:
PDF (1221KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨德卫, 郑向华, 程朝平, 叶宁, 黄凤凰, 叶新福. 基于CSSLs群体定位和图位克隆水稻长芒基因GAD1-2[J]. 遗传, 2018, 40(12): 1101-1111 doi:10.16288/j.yczz.18-044
Dewei Yang, Xianghua Zheng, Chaoping Cheng, Ning Ye, Fenghuang Huang, Xinfu Ye.
水稻(Oryza sativa L.)是世界上最早驯化的重要粮食作物之一,栽培稻均是由野生稻驯化而来。在驯化过程中,许多农艺性状不仅受到自然选择的影响,同时最为重要的是受到人工选择的结果,如水稻芒的有无、种皮颜色、籽粒的落粒性、产量以及株高等性状[1]。驯化的主要目标是淘汰水稻中对生产不利的因素,同时增加对人类有利的因素。从基因与分子水平方面,驯化的主要方向就是改良与这些因素相关的基因,尤其是选择对人类有利的基 因[2]。因此,研究这些性状的基因功能和遗传机理,对进一步了解稻作起源具有十分重要的理论和实践意义。
植物芒是水稻、小麦(Triticum aestivum L.)、大麦(Hordeum vulgare L.)和高梁(Sorghum)等禾本科植物形态学特征之一,芒的形状、着生数量、长度在不同作物之间存在明显的差异[2]。研究发现,水稻无芒或短芒会遭受鸟的捕食,而有刺芒的水稻就免受鸟类的捕食[3]。芒是水稻最重要的驯化性状之一,目前大部分栽培稻都是短芒或无芒的,因此开展水稻长芒基因的鉴定和遗传机理等研究显得尤为重要。
近年来,科研人员已鉴定到一些与水稻芒性状相关的基因,有些基因已成功克隆。Kubo等[4]、Cai等[5]、Matsushita等[6]和Thomson等[7]利用不同的材料定位了18个与芒相关的QTL,分布于水稻第1、3、4、5、6、7、8、9、10、11和12号染色体上。2013年Luo等[8]从普通野生稻中分离了一个长芒基因An-1,这是水稻中第一个被克隆的长芒基因。 2015年Hua等[9]和Gu等[10]分别克隆了两个长芒基因—LABA1和An-2。2016年Jin等[11]从野生稻中分离出一个长芒基因GAD1。
目前调控水稻芒生长的遗传机制尚不清楚,而定位和克隆水稻芒长相关基因是研究水稻芒驯化遗传机制的前提和基础,对深入了解稻作起源具有极其重要的意义。因此,本研究以籼稻品种东南恢810为遗传背景、漳浦野生稻为染色体片段供体而构建的146个染色体片段置换系(chromosome segment substitution lines, CSSLs)为材料,通过检测获得了4份稳定遗传具有长芒性状的CSSLs,并对GAD1-2基因进行定位和克隆。本研究为进一步理解水稻起源演化和水稻芒长发育基因的遗传调控机制奠定了 基础。
1 材料和方法
1.1 供试材料
以籼稻品种东南恢810为遗传背景、漳浦野生稻为染色体片段供体而构建的 146个CSSLs及两个亲本(受体亲本东南恢810和供体亲本漳浦野生稻)为供试材料,这些材料均为本实验室构建的永久性群体。在146个CSSLs中,其中145个株系只携带一个置换片段,1个株系携带两个置换片段,分布于水稻的12条染色体,平均长度为7.79 Mb,置换片段对水稻基因组的覆盖总长度为1683.75 Mb,平均覆盖率为100% [12]。1.2 材料种植和性状调查
供试材料于2015年7月种植于福建省农业科学院水稻研究所实验农场。146个CSSLs及两个亲本分别种植1个小区,每个小区设置3个重复,且每个小区3行,每行20株,小区种植按置换系编号顺序排列,常规管理。146个CSSLs及两个亲本齐穗后,以整穗完全无芒作为无芒单株;定位群体齐穗后,调查群体中有芒和无芒单株的分离比例。
1.3 水稻粒长和芒长的测定
水稻成熟时单株收取种子,稻谷粒长的测定参照Tan等[13]方法进行;每个水稻株系随机取10粒进行芒长测定,然后取其平均值。1.4 水稻基因组DNA的提取及电泳检测
水稻成熟期叶片基因组DNA的提取、PCR扩增及扩增产物的电泳检测均参照杨德卫等[14]方法。1.5 重叠代换作图法对控制水稻长芒的QTL进行鉴定
重叠代换作图法是鉴定作物QTL的重要方法之一[15]。本文采用重叠代换作图法对控制水稻长芒QTL进行鉴定,参照何风华[16]的方法并作适当修改。如果在相互重叠的两个或两个以上的CSSL的置换片段上都鉴定有长芒QTL,则认为控制该长芒的QTL位于这些置换片段的重叠区段上;如果在一个CSSL的置换片段上检测出长芒QTL,在与置换片段的某一区段相互重叠的另一个或多个CSSLs中未检测出,则认为控制该长芒的QTL位于非重叠的区段上。1.6 水稻长芒遗传分析
将CSSL108和CSSL109分别与东南恢810杂交,根据F1的表型和调查F2代分离群体中长芒单株与无芒单株比例,来确定该长芒性状是遗传特性。1.7 水稻长芒基因GAD1-2初步定位
GAD1-2基因的置换片段长度的计算按Young等[17]方法进行。当染色体某一区段的两端的标记基因型均为供体基因型时,则认为这一区段为100%的供体片段;当染色体某一区段的两端的标记基因型均为受体基因型时,则认为这一区段为0%的供体片段;当染色体某一区段的一端的标记基因型为受体基因型,另一端的标记基因型为供体基因型时,则认为这一区段为50%的供体片段。1.8 水稻长芒基因GAD1-2的精确定位
为了进一步定位GAD1-2基因,分别构建东南恢810与CSSL108、CSSL109、CSSL110和CSSL111次级分离群体,获得2726单株,其中1836个为显性单株。利用1836个显性单株,并通过已经公布的水稻数据库引物及水稻基因组序列,在目标基因附近区域筛选并合成新的SSR引物,经检测获得25对多态性较好的引物。如果在目标区域筛选不到多态性SSR引物时,进一步开发并合成新的Indel标记,经检测获得4对多态性较好的引物,进而完成精细定位。Indel标记的开发以及两个分子标记间物理距离的确定均参照杨德卫等[14]方法。用于精细定位的GAD1-2基因具体引物见表1。
Table 1
表1
表1 本研究开发的SSR和Indel分子标记引物序列
Table 1
| 标记 | 引物序列(5′→3′) | 位点 | 扩增片段大小(bp) |
|---|---|---|---|
| CM-2 | F: TGGATGCGGGAGAGGTTGTCG | P0461F06 | 196 |
| R: TTCTATCACTTTGCCGGCCTAATCG | |||
| CM-4 | F: GGAGGAGAGCCAAGCGATGG | P0013B04 | 111 |
| R: ACCGTCTTGACGCTGAGAGTGC | |||
| CM-5 | F: CGACGAGCAAGTAGAGCACACG | P0451G12 | 157 |
| R: AGAATCTGACGACTGTGGGAACC | |||
| CM-9 | F: CATCCTACCCTATGACATGAGACC | OJ1111_E05 | 110 |
| R: CTCTAAGATTAGCGTTCCAGAGAGC | |||
| CM-11 | F: GCTTCGATTTAGCTATTGGTACGG | OJ1666_A04 | 94 |
| R: GTGCCAGCATGAAGAGGTATGG | |||
| CM-61 | F: CAGCTAGTGCCGGAAAGATTCG | OJ1113_A10 | 200 |
| R: CCCATGCAGTATATTTCGGTTGG | |||
| CM-65 | F: GTTCTTGAGGCACTCCCGATACC | OJ1113_A10 | 292 |
| R: CTTAAGCTCGCCAAGAACACACC | |||
| CM-14 | F: GCAGGTGCAATTTACTCATAGGG | OSJNBb0092C08 | 249 |
| R: GGATGATGATGACGACGATTACG | |||
| CM-17 | F: CGTCGTCCTCCGATCAAATCC | OJ1111_H02 | 375 |
| R: CTGGCTCGGTGTTCCAGTCG | |||
| CM-21 | F: CAGTCCAACGGGTTCGTCATCG | P0481F05 | 392 |
| R: TACCTGCAGTGCCTCCTCCAGTCC | |||
| CM-26 | F: ATGATTGCATCTGCATCACTGC | P0481F05 | 379 |
| R: ATACCTGTTTCCAATGCGTAGCC | |||
| CM-29 | F: CGGTCGGAGGTGTACACGAAGC | P0028A08 | 236 |
| R: GAGGAGATCCGGTGGGATTGC | |||
| CM-68 | F: CGATCAGATCTCCGCAAGTTGG | P0605H02 | 361 |
| R: GGCGTGGTTGATGGTGAACG | |||
| CM-71 | F: TGTCAGTGAGGTAGGTCAATAGC | P0605H02 | 200 |
| R: AGGCCCTAAGTCATATCTTCC | |||
| CM-32 | F: GATGGAGCCAAGACACGAAAGC | P0686H11 | 127 |
| R: TGACTTCGTCCGTATGCTACTGC | |||
| CM-75 | F: TTGCTACACCTTAGCTGCTGTGC | P0686H11 | 300 |
| R: GGTGTTCTTGTTGCTAGGAGATGC | |||
| CM-35 | F: TCCTCTCGTCATCTCACCTCACC | B1142B04 | 178 |
| R: CTCACCCACTCCTGTGTGACTCC | |||
| CM-39 | F: CTCTCCTCCTCACCTACGCATCC | OJ1118_A06 | 289 |
| R: GAAGTCCGGCTGGGAGTAGTGC | |||
| CM-42 | F: TGCAGAAGAACTACTGAGAGAAGACG | OJ1345_D02 | 135 |
| R: CATCTCCTTCAACCTGCCTTCC | |||
| CM-77 | F: AGGTTGACCTGTGTGAGTAGCAAGG | OJ1345_D02 | 294 |
| R: ACATCGCCAACCATCTCAAGG | |||
| 标记 | 引物序列(5′→3′) | 位点 | 扩增片段大小(bp) |
| CM-46 | F: CAATAAATCTCGCCCTCGTTGC | OJ1134_H03 | 333 |
| R: GTGAGGTCGGCCTTGAAGTACC | |||
| CM-47 | F: CCATCTCAACTCCTTCGTTTACTGC | OSJNBa0025J22 | 297 |
| R: TCGACTGTTTGCTTGGAATAGGC | |||
| CM-51 | F: GTCGGTCACGAAGTTCAGATCC | B1168A08 | 186 |
| R: TCAGGCAAAGTTGAAGATGGTAGC | |||
| CM-52 | F: CGGCGAGGTAGAAGGTGACG | OSJNBa0016N23 | 196 |
| R: CAGTGATTGTGTGACAGTGTGAGAGG | |||
| CM-58 | F: TCGATGGAGGAGGAGGAGTACG | P0711H09 | 100 |
| R: AAATTGATCGCTCCTCCACTGC | |||
| Indel-3 | F: CAGATGGCGAGTTGTCAGTTGC | P0419H09 | 232 |
| R: CATCTCGCCGATCCAAGTAAGC | |||
| Indel-5 | F: ACAGCCTATAGCTCACACCAAACC | P0419H09 | 224 |
| R: GAACACCTCCGTCTCCATTGC | |||
| Indel-8 | F: GACGCTGTAGAGACACAGATACATGG | P0481F05 | 397 |
| R: CTGTTGGTGATGGCCTTAGTGC | |||
| Indel-11 | F: AAGCTGCAGTTGGGTGAGAAGC | P0481F05 | 484 |
| R: GCCCAAGATACAGCAAGTTCTCG |
新窗口打开|下载CSV
1.9 候选基因的预测
在精细定位的基础上,利用水稻数据库RAD (rice automatic interpretation database, http://ricegaas. dna.affrc.go.jp/rgadb/)和RAP-DB (rice annotation project database, http://rapdb.dna.affrc.go.jp/),分析定位区间内所有的基因信息。同时与Gramene (http:// www.gramene.org/rice_mutant/)中基因信息进行分析和比对,对候选基因进行预测,从而确定候选基因。2 结果与分析
2.1 亲本及置换系的芒长和粒长性状比较
对双亲的粒长和芒长进行表型观察和调查,结果表明,亲本东南恢810表现无芒,而漳浦野生稻表现长芒,差异达到显著水平。在146个CSSLs 中,CSSL108、CSSL109、CSSL110和CSSL111这4个株系与受体亲本东南恢810相比表现长芒,差异达到显著水平(图1,表2);东南恢810与漳浦野生稻粒长差异不显著,而在146个CSSLs 中,CSSL108、CSSL109、CSSL110和CSSL111这4个株系与受体亲本东南恢810相比表现粒型变长,差异达到极显著水平(表2,图2C)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1亲本芒长性状差异比较
Fig. 1Comparison of the differences in parents
Table 2
表2
表2 双亲和置换系材料芒长、粒长性状比较
Table 2
| 性状 | 株系名称 | ||||||
|---|---|---|---|---|---|---|---|
| 漳浦野生稻 | 东南恢810 | 漳浦野生稻/东南恢810 | CSSL108 | CSSL109 | CSSL110 | CSSL111 | |
| 芒长(mm) | 34.6** | 0 | 34.2** | 28.4** | 28.6** | 29.1** | 28.6** |
| 粒长(mm) | 98.1 | 96.8 | 98.3 | 103.2* | 103.6* | 104.5* | 103.6* |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2亲本东南恢810和CSSL108的穗部性状比较
Fig. 2Comparison of the panicle traits between DongNanHui 810 and CSSL108
2.2 利用重叠代换作图法对控制水稻长芒的QTL进行鉴定
通过比较146个CSSLs的芒长与东南恢810的差异,发现有4个CSSLs的芒长显著大于东南恢810,分别是CSSL108、CSSL109、CSSL110和CSSL111,差异均达到极显著水平;而余下置换系的芒长与东南恢810之间未检测到显著差异(图2,表2)。这4个CSSL的置换片段来源于同一个亲本—漳浦野生稻[12](图3),表明这4个CSSL中存在控制芒长性状的QTL。图3
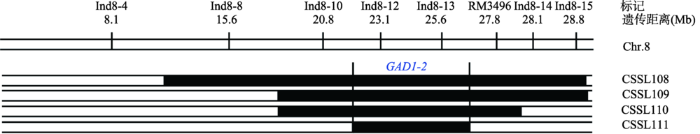 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3水稻长芒基因GAD1-2的初步定位
Fig. 3Primary mapping of the GAD1-2 gene
2.3 长芒性状的遗传分析
为了进一步对该长芒性状进行遗传分析,本文从4个长芒CSSLs中随机选择CSSL108和CSSL109分别与东南恢810杂交,F1代均表现长芒的表型。将F1自交种子全部种下,田间调查F2代分离比,经卡平方(χ2)检验(χ2c<χ20.05=3.84),长芒单株与无芒单株的分离符合孟德尔遗传3:1比例(表3),表明该长芒性状是由1对显性单基因控制的。Table 3
表3
表3 长芒性状的遗传分析
Table 3
| 杂交组合 | F1表型 | F2群体 | χ2(3:1) | P | ||
|---|---|---|---|---|---|---|
| 长芒株数 | 无芒株数 | 总株数 | ||||
| CSSL108/东南恢810 | 长芒 | 580 | 182 | 762 | 0.302* | 0.5~0.75 |
| CSSL109/东南恢810 | 长芒 | 492 | 183 | 675 | 0.486* | 0.25~0.5 |
新窗口打开|下载CSV
2.4 水稻长芒基因初步定位
分析发现,CSSL108、CSSL109、CSSL110和CSSL111 共4个CSSLs株系控制水稻长芒基因在同一个区域,命名为GAD1-2。由于GAD1-2在CSSL108、CSSL109、CSSL110和CSSL111这4个CSSLs株系中均能检测到,表明GAD1-2位于这4个置换片段的重叠区段上。按Young等[17]方法进行置换片段长度计算,将GAD1-2定位在2个标记Ind8-10和RM4936之间,遗传距离约为4.75 Mb (图3)。2.5 长芒基因GAD1-2的精确定位
为了将GAD1-2基因界定在较小的区域内,本文将东南恢810分别与CSSL108、CSSL109、CSSL110和CSSL111杂交,构建了4个分离群体。2017年6月8日将这4个分离群体种植在福建省农业科学院水稻研究所福州市仓山区城门镇水稻实验基地,共获得2726单株。由于GAD1-2是受1对显性单基因控制,显性纯合和杂合体均表现长芒的表型,显性纯合和杂合的单株均含有GAD1-2基因。显性纯合单株合基因型为GAD1-2/GAD1-2,杂合体单株基因型为GAD1-2/gad1-2,隐性纯合单株合基因型为gad1-2/gad1-2,而隐性纯合单株不含有GAD1-2基因,同时显性纯合单株和杂合单株总数量接近是隐性纯合单株数量3倍(AA:Aa:aa=1:2:1)。为了更有效和准确的定位GAD1-2基因,本文选择显性单株进行定位,4个分离群体中共获得1836个显性单株。本文利用Ind8-10、Ind8-12、Ind8-13和RM4936标记对1836个显性单株进行检测,将GAD1-2基因界定在标记Ind8-12和Ind8-13之间,遗传距离约为2.5 Mb (图4 A)。
图4
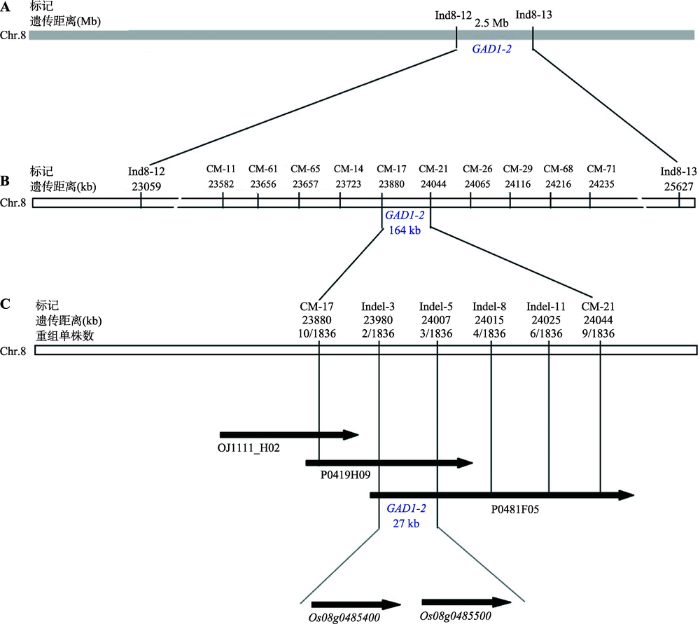 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4水稻长芒基因GAD1-2物理图谱的构建
A:GAD1-2基因的初步定位;B:GAD1-2基因的精细定位;C:GAD1-2候选基因的确定。
Fig. 4Genetic and physical maps of the GAD1-2 gene
为了将GAD1-2基因界定在更小的区域内,本文在标记Ind8-12和Ind8-13之间合成78对引物,经检测获得25对多态性较好的引物(引物信息见表1)。利用这25对引物分别对1836个显性单株进行检测,最终将GAD1-2基因定位在标记CM-17和CM-21之间,物理距离约164 kb (图4 B)。
为了进一步图位克隆GAD1-2基因,本文在标记CM-17和CM-21之间开发12对Indel引物,经检测获得4对多态性引物,分别是Indel-3、Indel-5、Indel-8和Indel-11 (引物信息见表1)。利用这4对引物对1836个显性单株进行分析,结果表明,Indel-3检测到的所有显性单株均与CM-17检测到的显性单株重叠,而Indel-5、Indel-8和Indel-11检测到的所有显性单株均与CM-21检测到的显性单株重叠,且Indel-11检测到的显性单株包含所有Indel-8检测到的显性单株,Indel-8检测到的显性单株包含所有Indel-5检测到的显性单株。因此GAD1-2基因位于标记Indel-3和Indel-5之间,物理距离约27 kb (图4C)。
利用CM-17、Indel-3、Indel-5、Indel-8、Indel-11和CM-21分子标记,对Line 143、Line 207、Line 746、Line 960和Line 1088共5个显性单株进行基因型分析。结果表明,只有Line 207单株表现纯合基因型,其他4个单株均表现杂合基因型。其中,Line 143在CM-17和Indel-3标记位点表现杂合基因型,Line 746在CM-17、Indel-3、Indel-5、Indel-8、Indel-11和CM-21标记位点均表现杂合基因型,Line 960和Line 1088在Indel-5、Indel-8、Indel-11和CM-21标记位点表现杂合基因型(图5)。
图5
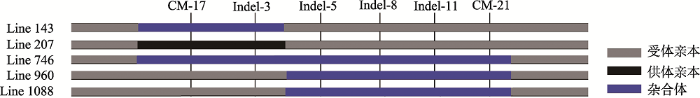 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5精细定位中5个显性单株的基因型分析
Fig. 5Genotype analysis of five dominant plants
2.6 长芒基因GAD1-2候选基因的确定
为了确定GAD1-2的候选基因,本文对27 kb区域进行候选基因预测和分析,发现该27 kb区域内只有Os08g0485400和Os08g0485500这2个候选基因(图4C)。本文对这2个候选基因分别进行测序,发现东南恢810、CSSL108和漳浦野生稻在Os08g0485400基因内的序列没有任何差异,而在Os08g0485500基因序列,东南恢810、CSSL108和漳浦野生稻均有差异,CSSL108和漳浦野生稻在差异位置缺失6个碱基(图6A)。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6漳浦野生稻、CSSL108与受体亲本东南恢810中Os08g0485400的序列比对分析
A:cDNA比对分析;B:氨基酸序列的比对分析。
Fig. 6Sequence comparison and analysis of Os08g0485400 during DongNanHui 810, ZhangPu wild rice and CSSL108
本课题组前期研究发现,水稻长芒发育相关基因GAD1也在Os08g0485500基因内[12],因此,本文推测GAD1-2与GAD1是等位基因。本文进一步对东南恢810、CSSL108和漳浦野生稻Os08g0485400基因进行测序,通过比对和分析可知,GAD1-2中6个碱基缺失的位置在Os08g0485400保守的ORF区域,本文推测这6个碱基的缺失导致丝氨酸和半胱氨酸缺失(图6B),从而引起水稻长芒的表型。
3 讨 论
3.1 水稻芒长性状的驯化和遗传研究
在水稻驯化过程中,也伴随着人类对水稻性状选择的过程。因此,对水稻驯化过程中相关基因的深入研究,将为水稻起源演化过程提供有利的证据。水稻芒性状是一个复杂的数量性状,受到光照、温度、逆境和海拔等影响,而出现不同表型,如出现芒长的有无和芒的长短等现象。亲本均无芒的籼粳材料杂交后,后代分离群体中会出现有芒的个体,显然水稻芒是一个不稳定的性状[1]。研究表明,水稻芒性状是由显示基因控制,并且可能是同时由几对基因控制,基因之间还存在上位性[18]。由于芒的产生可能存在多基因互作和遗传的复杂性,因此需要更为精细的材料才能进行深入研究。CSSLs与其受体亲本之间除了在置换片段上存在差异外,其余部分均与受体亲本相同,遗传背景较单一。通过CSSLs与其受体亲本之间以及CSSLs之间的性状比较,可以检测出置换片段上的QTL,进一步通过重叠代换作图就可以对检测的基因进行鉴定和分离[12,19]。本研究利用重叠代换作图方法鉴定了1个控制水稻长芒的基因GAD1-2,遗传分析表明该长芒性状是由1对显性单基因控制的。
3.2 已鉴定的芒基因比较和分析
近年来,随着测序技术的快速发展,科研人员已鉴定出多个与水稻芒性状相关的基因,分布在水稻不同的染色体上[3]。本研究鉴定的水稻长芒基因GAD1-2,经分析发现与Jin等[12]从野生稻中分离的长芒基因GAD1是等位基因。GAD1基因使水稻表现长芒、籽粒变长和每穗粒数减少等性状[12],本研究从漳浦野生稻鉴定了一个控制长芒的基因GAD1-2,含有该基因的CSSL108株系与受体亲本赣优810相比表现长芒和粒型变长(表2) (图2,B和C),与GAD1基因控制的性状一致。但是,本研究发现含有GAD1-2基因的CSSL108株系与受体亲本赣优810相比,每穗粒数并没有减少(图2 A),这可能是由于插入片段大小不同,影响基因之间互作和效应,从而影响其每穗粒数。3.3 利用显性单株鉴定和分离水稻显性基因
在定位由显性性状控制的基因时,一般均是利用分离群体中的隐性单株。本研究利用CSSLs群体鉴定了一个显性的水稻长芒基因GAD1-2,利用显性单株将该基因定位在物理距离约27 kb区域内(图 4C),该区域只有2个候选基因。本研究在分离群体中共获得2726单株,其中显性单株是1836个,依据遗传学中的单基因控制基因型分离比例(AA:Aa:aa=1:2:1),隐性单株不会超过890个。如果利用890个隐性单株很难将该长芒基因GAD1-2定位在27 kb区域内,当然会有个例情况。本研究利用分离群体中的2726单株,将该基因定位在较小的区间,证明该方法可以有效分离和定位由显性性状控制的基因。
同时,本文对定位区间中5个显性单株的基因型进行了分析,结果表明,5个与GAD1-2基因紧密连锁的单株中,只有1个单株Line 207在这两个标记上表现纯合基因型,而其余4个单株均表现杂合的基因型(图5)。显然,4个杂合基因型单株在精细定位GAD1-2基因时发挥了重要作用。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 2]

Long-awn trait was preserved from wild rice (F population which segregation ratio of awn accorded with 3∶1 to develop a large mapping population with heterozygous BCF plants.Using bulked segregant analysis (BSA),from 1 512 SSR makers evenly distributed on rice genome,we rough mapped the awn gene between RM6283 and RM5685 on chromosome 3,named AWN3-1.After developed and screened new markers,AWN3-1 was fine-mapped between marker Y5 and marker Y9,which genetic distance was 0.5 and 0.4 cM,respectively.This work provided a useful help for cloning AWN3-1.
URL [本文引用: 2]
Long-awn trait was preserved from wild rice (F population which segregation ratio of awn accorded with 3∶1 to develop a large mapping population with heterozygous BCF plants.Using bulked segregant analysis (BSA),from 1 512 SSR makers evenly distributed on rice genome,we rough mapped the awn gene between RM6283 and RM5685 on chromosome 3,named AWN3-1.After developed and screened new markers,AWN3-1 was fine-mapped between marker Y5 and marker Y9,which genetic distance was 0.5 and 0.4 cM,respectively.This work provided a useful help for cloning AWN3-1.
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 2]
亚洲栽培稻(Oryza sativa L.)是从普通野生稻(Oryza rufipogon Griff.)驯化而来的。与野生祖先相比,栽培稻在很多形态和生理性状上发生了巨大的改变。野生稻不仅具有长芒,而且芒表面布满长约200μm的锋利的芒刺。野生稻的长刺芒能够粘附在动物皮毛上,帮助种子借助动物进行传播;平衡自由下落的种子使种子带有胚的一端埋入土壤,有利于萌发和幼苗建成;还能够保护种子不被鸟兽掠食,所以长刺芒对野生稻的野外生存和扩散至关重要。相反,栽培稻一般无芒或仅有少量光滑的短芒,有利于种子的收获、储藏和加工,提高稻米产量。因此,野生稻的长刺芒到栽培稻的短光芒的转变是水稻驯化过程中一个重要事件。本研究在云南元江普通野生稻与籼稻品种93-11构建的渗入系中,筛选到一个具有长刺芒的渗入系(9YIL304),用该系与轮回亲本93,11构建分离群体,将控制野生稻长刺芒的基因(LONG AND BARBED AWN1,LABA1)定位在第4染色体长臂的一个约35-kb的区间内。通过双亲序列比对,发现93-11在一个编码细胞分裂素激活酶基因的第1个外显子中存在单碱基缺失,引起编码蛋白提前终止。该单碱基的缺失与栽培稻的芒长和芒刺有无高度相关。遗传互补实验表明该基因就是控制野生稻长刺芒的基因LABA1。重组LABA1蛋白能够将细胞分裂素核糖5’-单磷酸(iPRMP)催化成为具有生物活性的细胞分裂素iP。细胞分裂素含量测定表明互补转基因植株幼穗内活性细胞分裂素含量(iP,tZ)显著高于转基因对照,验证了LABA1在体内也具有细胞分裂素激活酶功能。LABA1特异地在芒原基的表皮细胞中表达,组织学分析和OsHistone H4的原位杂交表明,ABA1能够通过提高芒原基的细胞分裂活性从而增加芒长和芒的着生比例。栽培稻在LABAl位点的核苷酸多样性相比于野生稻发生了严重下降,中性测验表明labal基因很可能受到了定向选择。通过多位点核苷酸多样性分析,在栽培稻中鉴定到一个包含LABAl的约800-kb的基因组选择性清除区域,表明LABA1是水稻驯化过程中一个重要的人工选择靶点。LABA1基因的克隆不仅为揭示水稻驯化的分子机理提供了新的见解,而且为研究水稻芒的发育机制提供了重要线索。
URL [本文引用: 2]
亚洲栽培稻(Oryza sativa L.)是从普通野生稻(Oryza rufipogon Griff.)驯化而来的。与野生祖先相比,栽培稻在很多形态和生理性状上发生了巨大的改变。野生稻不仅具有长芒,而且芒表面布满长约200μm的锋利的芒刺。野生稻的长刺芒能够粘附在动物皮毛上,帮助种子借助动物进行传播;平衡自由下落的种子使种子带有胚的一端埋入土壤,有利于萌发和幼苗建成;还能够保护种子不被鸟兽掠食,所以长刺芒对野生稻的野外生存和扩散至关重要。相反,栽培稻一般无芒或仅有少量光滑的短芒,有利于种子的收获、储藏和加工,提高稻米产量。因此,野生稻的长刺芒到栽培稻的短光芒的转变是水稻驯化过程中一个重要事件。本研究在云南元江普通野生稻与籼稻品种93-11构建的渗入系中,筛选到一个具有长刺芒的渗入系(9YIL304),用该系与轮回亲本93,11构建分离群体,将控制野生稻长刺芒的基因(LONG AND BARBED AWN1,LABA1)定位在第4染色体长臂的一个约35-kb的区间内。通过双亲序列比对,发现93-11在一个编码细胞分裂素激活酶基因的第1个外显子中存在单碱基缺失,引起编码蛋白提前终止。该单碱基的缺失与栽培稻的芒长和芒刺有无高度相关。遗传互补实验表明该基因就是控制野生稻长刺芒的基因LABA1。重组LABA1蛋白能够将细胞分裂素核糖5’-单磷酸(iPRMP)催化成为具有生物活性的细胞分裂素iP。细胞分裂素含量测定表明互补转基因植株幼穗内活性细胞分裂素含量(iP,tZ)显著高于转基因对照,验证了LABA1在体内也具有细胞分裂素激活酶功能。LABA1特异地在芒原基的表皮细胞中表达,组织学分析和OsHistone H4的原位杂交表明,ABA1能够通过提高芒原基的细胞分裂活性从而增加芒长和芒的着生比例。栽培稻在LABAl位点的核苷酸多样性相比于野生稻发生了严重下降,中性测验表明labal基因很可能受到了定向选择。通过多位点核苷酸多样性分析,在栽培稻中鉴定到一个包含LABAl的约800-kb的基因组选择性清除区域,表明LABA1是水稻驯化过程中一个重要的人工选择靶点。LABA1基因的克隆不仅为揭示水稻驯化的分子机理提供了新的见解,而且为研究水稻芒的发育机制提供了重要线索。
[本文引用: 1]
URLPMID:12582574 [本文引用: 1]
The genetic basis of character association related to differentiation found in the primary gene pool of rice was investigated based on the genomic distribution of quantitative trait loci (QTLs). Major evolutionary trends in cultivated rice of Asiatic origin ( Oryza sativa ) and its wild progenitor ( O. rufipogon ) are: (1) differentiation from wild to domesticated types (domestication), (2) ecotype differentiation between the perennial and annual types in wild races, and (3) the Indica versus Japonica type differentiation in cultivated races. Using 125 recombinant inbred lines (RILs) derived from a cross between an Indica cultivar of O. sativa and a strain of O. rufipogon carrying some Japonica-like characteristics, we mapped 147 markers, mostly RFLPs, on 12 chromosomes. Thirty-seven morphological and physiological quantitative traits were evaluated, and QTLs for 24 traits were detected. The mapped loci showed a tendency to form clusters that are composed of QTLs of the domestication-related traits as well as Indica/Japonica diagnostic traits. QTLs for perennial/annual type differences did not cluster. This cluster phenomenon could be considered "multifactorial linkages" followed by natural selection favoring co-adapted traits. Further, it is possible that the clustering phenomenon is partly due to pleiotropy of some unknown key factor(s) controlling various traits through diverse metabolic pathways. Chromosomal regions where QTL clusters were found coincided with the regions harboring genes or gene blocks where the frequency of cultivar-derived alleles in RILs is higher than expected. This distortion may be partly due to unconscious selection favoring cultivated plant type during the establishment of RILs.
[本文引用: 1]
URLPMID:12736777 [本文引用: 1]
An advanced backcross population between an accession of Oryza rufipogon (IRGC 105491) and the U.S. cultivar Jefferson ( Oryza sativa ssp. japonica ) was developed to identify quantitative trait loci (QTLs) for yield, yield components and morphological traits. The genetic linkage map generated for this population consisted of 153 SSR and RFLP markers with an average interval size of 10.3 cM. Thirteen traits were examined, nine of which were measured in multiple environments. Seventy-six QTLs above an experiment-wise significance threshold of P 3.6 or a composite interval mapping LOD > 3.9) were identified. For the traits measured in multiple environments, 47% of the QTLs were detected in at least two environments. The O. rufipogon allele was favorable for 53% of the yield and yield component QTLs, including loci for yield, grains per panicle, panicle length, and grain weight. Morphological traits related to the domestication process and/or weedy characteristics, including plant height, shattering, tiller type and awns, were found clustered on chromosomes 1 and 4. Comparisons to previous studies involving wild cultivated crosses revealed O. rufipogon alleles with stable effects in multiple genetic backgrounds and environments, several of which have not been detected in studies between Oryza sativa cultivars, indicating potentially novel alleles from O. rufipogon . Some O. rufipogon -derived QTLs, however, were in similar regions as previously reported QTLs from Oryza sativa cultivars, providing evidence for conservation of these QTLs across the Oryza genus. In addition, several QTLs for grain weight, plant height, and flowering time were localized to putative homeologous regions in maize where QTLs for these traits have been previously reported, supporting the hypothesis of functional conservation of QTLs across the grasses.
URLPMID:24076974 [本文引用: 1]
Long awns are important for seed dispersal in wild rice (Oryza rufipogon), but are absent in cultivated rice (Oryza sativa). The genetic mechanism involved in loss-of-awn in cultivated rice remains unknown. We report here the molecular cloning of a major quantitative trait locus, An-1, which regulates long awn formation in O. rufipogon. An-1 encodes a basic helix-loophelix protein, which regulates cell division. The nearly-isogenic line (NIL-An-1) carrying a wild allele An-1 in the genetic background of the awnless indica Guangluai4 produces long awns and longer grains, but significantly fewer grains per panicle compared with Guangluai4. Transgenic studies confirmed that An-1 positively regulates awn elongation, but negatively regulates grain number per panicle. Genetic variations in the An-1 locus were found to be associated with awn loss in cultivated rice. Population genetic analysis of wild and cultivated rice showed a significant reduction in nucleotide diversity of the An-1 locus in rice cultivars, suggesting that the An-1 locus was a major target for artificial selection. Thus, we propose that awn loss was favored and strongly selected by humans, as genetic variations at the An-1 locus that cause awn loss would increase grain numbers and subsequently improve grain yield in cultivated rice.
.
URLPMID:26283047 [本文引用: 1]
Many morphological and physiological traits have changed between cultivated riceOryza sativaand wild riceOryza rufipogonunder domestication. We reveal that a genetic variation in theAn-2locus has a large impact on reducing awn length and increasing tiller and grain numbers in domesticated rice. Nucleotide diversity of theAn-2locus in cultivated rice was found to be significantly reduced compared with that of wild rice, suggesting that theAn-2locus was subjected to artificial selection. We propose that the selection of genetic variation inAn-2was due to reduced awn length and increased grain yield in cultivated rice.
URLPMID:26082172 [本文引用: 1]
Common wild rice (Oryza rufipogon), the wild relative of Asian cultivated rice (Oryza sativa), flaunts long, barbed awns, which are necessary for efficient propagation and dissemination of seeds. By contrast, O. sativa cultivars have been selected to be awnless or to harbor short, barbless awns, which facilitate seed processing and storage. The transition from long, barbed awns to short, barbless awns was a crucial event in rice domestication. Here, we show that the presence of long, barbed awns in wild rice is controlled by a major gene on chromosome 4, LONG AND BARBED AWN1 (LABA1), which encodes a cytokinin-activating enzyme. A frame-shift deletion in LABA1 of cultivated rice reduces the cytokinin concentration in awn primordia, disrupting barb formation and awn elongation. Sequencing analysis demonstrated low nucleotide diversity and a selective sweep encompassing an 800-kb region around the derived laba1 allele in cultivated rice. Haplotype analysis revealed that the laba1 allele originated in the japonica subspecies and moved into the indica gene pool via introgression, suggesting that humans selected for this locus in early rice domestication. Identification of LABA1 provides new insights into rice domestication and also sheds light on the molecular mechanism underlying awn development.
URLPMID:27634315 [本文引用: 1]
Abstract Cultivated rice (Oryza sativa) was domesticated from wild rice (Oryza rufipogon), which typically displays fewer grains per panicle and longer grains than cultivated rice. In addition, wild rice has long awns, whereas cultivated rice has short awns or lacks them altogether. These changes represent critical events in rice domestication. Here, we identified a major gene, GRAIN NUMBER, GRAIN LENGTH AND AWN DEVELOPMENT1 (GAD1), that regulates those critical changes during rice domestication. GAD1 is located on chromosome 8 and is predicted to encode a small secretary signal peptide belonging to the EPIDERMAL PATTERNING FACTOR-LIKE family. A frame-shift insertion in gad1 destroyed the conserved cysteine residues of the peptide, resulting in a loss of function, and causing the increased number of grains per panicle, shorter grains, and awnless phenotype characteristic of cultivated rice. Our findings provide a useful paradigm for revealing functions of peptide signal molecules in plant development and helps elucidate the molecular basis of rice domestication. 2016 American Society of Plant Biologists. All rights reserved.
URLPMID:5121215 [本文引用: 6]
Common wild rice (Oryza rufipogonGriff.) represents an important resource for rice improvement. Genetic populations provide the basis for a wide range of genetic and genomic studies. In particular, chromosome segment substitution lines (CSSLs) are most powerful tools for the detection and precise mapping of quantitative trait loci (QTLs). In this study, 146 CSSLs were produced; they were derived from the crossing and back-crossing of two rice cultivars: Dongnanihui 810 (Oryza sativaL.), anindicarice cultivar as the recipient, and ZhangPu wild rice, a wild rice cultivar as the donor. First, a physical map of the 146 CSSLs was constructed using 149 molecular markers. Based on this map, the total size of the 147 substituted segments in the population was 1145.65 Mb, or 3.04 times that of the rice genome. To further facilitate gene mapping, heterozygous chromosome segment substitution lines (HCSSLs) were also produced, which were heterozygous in the target regions. Second, a physical map of the 244 HCSSLs was produced using 149 molecular markers. Based on this map, the total length of substituted segments in the HCSSLs was 1683.75 Mb, or 4.47 times the total length of the rice genome. Third, using the 146 CSSLs, two QTLs for plant height, and one major QTL for apiculus coloration were identified. Using the two populations of HCSSLs, theqPa-6-2gene was precisely mapped to an 88 kb region. These CSSLs and HCSSLs may, therefore, provide powerful tools for future whole genome large-scale gene discovery in wild rice, providing a foundation enabling the development of new rice varieties. This research will also facilitate fine mapping and cloning of quantitative trait genes, providing for the development of superior rice varieties.
URL [本文引用: 1]
Appearance quality of the rice grain represents a major problem of rice production in many rice-producing areas of the world, especially in hybrid rice production in China. In this study, we conducted a molecular marker-based genetic analysis of the traits that are determinants of the appearance quality of rice grains, including traits specifying grain shape and endosperm opacity. The materials used in the analysis included an F 2:3 population and an F 10 recombinant inbred line population from a cross between the parents of Shanyou 63, the most widely grown rice hybrid in China. Molecular marker-based QTL (quantitative trait locus) analyses revealed that grain length and grain width were each controlled by a major QTL accounting for a very large proportion of the genetic variation, plus one or two minor QTLs each explaining a small proportion of the genetic variation. The major QTLs can be detected in both the F 2:3 and recombinant inbred line population using both paddy rice and brown rice, whereas the minor QTLs were detected only occasionally. The QTL located in the interval of RG393-C1087 on chromosome 3 is the major locus for grain length, and the one in the interval RG360-C734a on chromosome 5 plays a major role in determining grain width. Similarly, white belly, which largely determines the opacity of the endosperm, is almost entirely controlled by a major locus on chromosome 5, located in the same genomic region as the major QTL for grain width. The implications of the results with respect to hybrid rice improvement were discussed.
Magsci [本文引用: 2]
水稻产量和品质受花器官发育的直接影响, 因此对水稻颖花发育机理的研究将有助于水稻产量提高和品质的改良。文章利用<sup>60</sup>Coγ射线辐照亲本8PW33 (籼稻背景) 获得一个性状能稳定遗传的内颖退化突变体(编号:MU102), 并对其农艺性状和花器官进行了观察和分析。结果显示, 相对于野生型, 该突变体的株高、每穗总粒数及剑叶宽均显著增加, 而结实率则显著降低, 差异均达显著水平。解剖镜下观察表明, 该突变体内颖退化, 外颖弯曲呈现镰刀状, 其余器官与野生型表型基本一致。扫描电镜观察显示, 突变体与野生型叶片维管束的结构组成以及外颖表皮细胞组成、排列均正常, 没有明显差异; 与野生型相比, 突变体内颖表皮细胞排列较为紧密, 推测可能是内颖收缩退化导致的。遗传分析显示该突变性状是由隐性单基因控制, 并命名为<em>pd2</em>。利用实验室现有的SSR分子标记将<em>PD2</em>基因定位于水稻第9号染色体上, 通过进一步扩大群体和开发新的Indel标记, 将<em>PD2</em>基因定位在2个Indel标记之间, 两者间的物理距离大约是82 kb。在该物理区间内有一个已经克隆的内颖发育基因<em>REP1</em>, 经过测序和比对分析, 推测<em>REP1</em>与<em>PD2</em>为等位基因。
Magsci [本文引用: 2]
水稻产量和品质受花器官发育的直接影响, 因此对水稻颖花发育机理的研究将有助于水稻产量提高和品质的改良。文章利用<sup>60</sup>Coγ射线辐照亲本8PW33 (籼稻背景) 获得一个性状能稳定遗传的内颖退化突变体(编号:MU102), 并对其农艺性状和花器官进行了观察和分析。结果显示, 相对于野生型, 该突变体的株高、每穗总粒数及剑叶宽均显著增加, 而结实率则显著降低, 差异均达显著水平。解剖镜下观察表明, 该突变体内颖退化, 外颖弯曲呈现镰刀状, 其余器官与野生型表型基本一致。扫描电镜观察显示, 突变体与野生型叶片维管束的结构组成以及外颖表皮细胞组成、排列均正常, 没有明显差异; 与野生型相比, 突变体内颖表皮细胞排列较为紧密, 推测可能是内颖收缩退化导致的。遗传分析显示该突变性状是由隐性单基因控制, 并命名为<em>pd2</em>。利用实验室现有的SSR分子标记将<em>PD2</em>基因定位于水稻第9号染色体上, 通过进一步扩大群体和开发新的Indel标记, 将<em>PD2</em>基因定位在2个Indel标记之间, 两者间的物理距离大约是82 kb。在该物理区间内有一个已经克隆的内颖发育基因<em>REP1</em>, 经过测序和比对分析, 推测<em>REP1</em>与<em>PD2</em>为等位基因。
URLPMID:12582914 [本文引用: 1]
A major QTL for P uptake had previously been mapped to a 13-cM marker interval on the long arm of chromosome 12. To map that major QTL with higher precision and certainty, a secondary mapping population was developed by backcrossing a near-isogenic line containing the QTL from the donor parent to the recurrent parent of low P uptake. Two different mapping strategies have been followed in this study. A conventional QTL mapping approach was based on individual F 2 RFLP data and the phenotypic evaluation of family means in the F 3 . The second strategy employed a substitution-mapping approach. Phenotypic and marker data were obtained for 160 F 3 individuals of six highly informative families that differed in the size of donor chromosomal segments in the region of the putative QTL. QTL mapping showed that close to 80% of the variation between families was due to a single QTL, hereafter referred to as Pup1 (Phosphorus uptake 1). Pup1 was placed in a 3-cM interval flanked by markers S14025 and S13126, which is within 1 cM of the position identified in the original QTL mapping experiment. Other chromosomal regions and epistatic effects were not significant. Substitution mapping revealed that Pup1 co-segregated with marker S13126 and that the flanking markers, S14025 and S13752, were outside the interval containing Pup1 . The two mapping strategies therefore yielded almost identical results and, in combining the advantages of both, Pup1 could be mapped with high certainty. The QTL mapping appoach showed that the phenotypic variation between families was due to only one QTL without any additional epistacic interactions, whereas the advantage of substitution mapping was to place clearly defined borders around the QTL.
URLMagsci [本文引用: 1]
抽穗期是水稻品种的重要农艺性状之一,对抽穗期QTL进行定位并研究其遗传效应在水稻育种中是至关重要的。本研究利用以6个水稻品种为供体的52个单片段代换系为试验材料,通过t测验比较单片段代换系与受体亲本华粳籼74之间抽穗期的差异,对代换片段上的抽穗期QTL进行了鉴定。以P≤0.001为阈值共鉴定出20个抽穗期QTL,这些QTL分布于水稻的10条染色体。QTL加性效应值为-5.9~1.1,加性效应百分率为-7.4%~1.4%。有8个QTL被定位在小于10.0 cM的区段内。利用1个单片段代换系与华粳籼74杂交发展的F2群体对qHD-3-1进行了定位。在作图群体中,早抽穗和迟抽穗植株数符合3:1的分离比,早抽穗表现为显性。利用微卫星标记将qHD-3-1定位于3号染色体短臂,PSM304和RM569分别位于其两侧,遗传距离分别为2.4 cM和5.1 cM。
URLMagsci [本文引用: 1]
抽穗期是水稻品种的重要农艺性状之一,对抽穗期QTL进行定位并研究其遗传效应在水稻育种中是至关重要的。本研究利用以6个水稻品种为供体的52个单片段代换系为试验材料,通过t测验比较单片段代换系与受体亲本华粳籼74之间抽穗期的差异,对代换片段上的抽穗期QTL进行了鉴定。以P≤0.001为阈值共鉴定出20个抽穗期QTL,这些QTL分布于水稻的10条染色体。QTL加性效应值为-5.9~1.1,加性效应百分率为-7.4%~1.4%。有8个QTL被定位在小于10.0 cM的区段内。利用1个单片段代换系与华粳籼74杂交发展的F2群体对qHD-3-1进行了定位。在作图群体中,早抽穗和迟抽穗植株数符合3:1的分离比,早抽穗表现为显性。利用微卫星标记将qHD-3-1定位于3号染色体短臂,PSM304和RM569分别位于其两侧,遗传距离分别为2.4 cM和5.1 cM。
URL [本文引用: 2]
URL [本文引用: 1]
水稻芒的性状与驯化过程密切相关,具体表现为芒长短和芒的分布 等.利用一个无芒品种越光(轮回亲本)和一个有芒品种Kasalath杂交产生的回交重组自交系群体,在上海、海南两地对芒长和芒的分布(芒出现比例)进 行了相关分析及其相关基因(QTL)定位,结果发现这两个性状间存在显著的正相关.共检测到影响这两个性状的12个QTL,分布在6条染色体上的7个区 域,贡献率介于2.90%-53.24%,其正效应大多来自有芒亲本Kasalath,但也有3个QTL正效应来自无芒亲本越光,并对这个现象产生的原因 进行了讨论.本研究检测到的QTL及其两侧的分子标记可以用于水稻驯化的研究和理想型无芒品种分子辅助育种.
URL [本文引用: 1]
水稻芒的性状与驯化过程密切相关,具体表现为芒长短和芒的分布 等.利用一个无芒品种越光(轮回亲本)和一个有芒品种Kasalath杂交产生的回交重组自交系群体,在上海、海南两地对芒长和芒的分布(芒出现比例)进 行了相关分析及其相关基因(QTL)定位,结果发现这两个性状间存在显著的正相关.共检测到影响这两个性状的12个QTL,分布在6条染色体上的7个区 域,贡献率介于2.90%-53.24%,其正效应大多来自有芒亲本Kasalath,但也有3个QTL正效应来自无芒亲本越光,并对这个现象产生的原因 进行了讨论.本研究检测到的QTL及其两侧的分子标记可以用于水稻驯化的研究和理想型无芒品种分子辅助育种.
URL [本文引用: 1]
Chromosome segment substitution lines (CSSLs) are powerful tools for detecting and precisely mapping quantitative trait loci (QTLs) and evaluating gene action as a single factor. In this study, 103 CSSLs were produced using two sequenced rice cultivars: 93-11, an elite restorer indica cultivar as recipient, and Nipponbare, a japonica cultivar, as donor. Each CSSL carried a single chromosome substituted segment. The total length of the substituted segments in the CSSLs was 2,590.6 cM, which was 1.7 times of the rice genome. To evaluate the potential application of these CSSLs for QTL detection, phenotypic variations of seed shattering, grain length and grain width in 10 CSSLs were observed. Two QTLs for seed shattering and three for grain length and grain width were identified and mapped on rice chromosomes. The results demonstrate that CSSLs are excellent genetic materials for dissecting complex traits into a set of monogenic loci. These CSSLs are of great potential value for QTL mapping and plant marker-assisted breeding (MAB).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}