 ,, 仲永, 陈月婵, 张志威,石河子大学医学院,基础医学系,组织胚胎学教研室,石河子 832003
,, 仲永, 陈月婵, 张志威,石河子大学医学院,基础医学系,组织胚胎学教研室,石河子 832003Research progress on the roles of Krüppel-like factors in muscle tissues
Zhaohui Zhuang,, Yong Zhong, Yuechan Chen, Zhiwei Zhang,School of Medicine, Shihezi University, Shihezi 832003, China通讯作者:
编委: 杨中州
收稿日期:2018-04-10修回日期:2018-07-8网络出版日期:2018-09-20
| 基金资助: |
Editorial board:
Received:2018-04-10Revised:2018-07-8Online:2018-09-20
| Fund supported: |
作者简介 About authors
庄兆辉,本科生,专业方向:临床医学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1200KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
庄兆辉, 仲永, 陈月婵, 张志威. Krüppel样因子在肌肉组织中的功能研究进展[J]. 遗传, 2018, 40(9): 733-748 doi:10.16288/j.yczz.18-095
Zhaohui Zhuang, Yong Zhong, Yuechan Chen, Zhiwei Zhang.
Krüppel样因子(Krüppel-like factor, KLFs)得名于果蝇同源基因Krüppel,在动物体内一般作为转录因子发挥作用。KLFs C-末端是DNA结合域,由3个高度保守的C2H2锌指结构组成,相邻的锌指之间由保守序列TGEKP(Y/F)X链接[1, 2]。大多数KLFs通过该结构结合靶基因启动子或增强子区的CACCC模序或富含GC的顺式调控元件。KLFs N-末端是转录调控结构域,结构高度变异,能结合特异蛋白质,介导多种因子的转录调控作用。
目前人体内共发现了18种KLF因子。根据KLF因子的蛋白结构特征和转录调控作用,KLFs大致被分为3大类[3]:第1类包括KLF1、KLF2、KLF4、KLF5、KLF6和KLF7,这类KLFs的N-末端具有酸性蛋白模序,主要作为转录激活因子发挥作用;在一些特定情况下,这类KLFs也能够与转录抑制因子互作,发挥转录抑制作用[3]。第2类包括KLF3、KLF8和KLF12,N-末端具有PVDLT模序,可以与抑制辅助因子CtBP1互作,发挥转录抑制作用[3, 4]。第3类包括KLF9、KLF10、KLF11、KLF13、KLF14和KLF16,N-末端含有转录抑制结构域,主要发挥转录抑制作用;在一些特定情况下,这类KLFs的部分成员也能够与转录激活因子互作,发挥转录激活作用[5, 6]。目前,因为KLF15、KLF17和KLF18的蛋白互作模序目前还不完全清楚,所以尚未被划入上述分类之中[3, 7]。
在作用机制方面,大多数KLFs直接调控靶基因转录,少数KLFs则需要与其他因子形成复合物才能发挥调控作用[5]。不同的KLFs既能调控不同靶基因,也能调控相同靶基因,发挥相似或相反的调控作用[8]。此外,部分KLFs存在翻译后修饰调控,并且KLFs N-末端结合的辅助因子具有多样性,因此在不同生理条件下,同一种KLF分子可能表现出不同的作用[9]。KLFs参与多种细胞增殖、分化、表型转化和凋亡等生命过程的调控[10]。近年来,KLFs在肌肉组织中的功能正逐渐成为生命科学领域的一个研究热点。本文对KLFs在心肌、平滑肌和骨骼肌中的功能及其作用机制的研究进展进行了综述,并探讨了KLFs在3种肌肉组织的形成和疾病发生发展过程中的作用。
1 心肌中的KLFs
心肌(cardiac muscle)主要由心肌细胞和成纤维细胞构成。心脏的舒缩功能主要取决于心肌细胞的数量和形态。目前,心脏肥大和先天性心脏病已成为心血管相关疾病的研究热点。当心脏发生肥大时,心肌细胞体积增大、胚胎基因(fetal gene)重新表达、蛋白质合成增强[11]。此外,肌细胞增强因子2 (myocyte enhancer factor, MEF2)、GATA结合蛋白4 (GATA binding protein 4, GATA4)、核因子-κB (nuclear factor-κB, NF-κB)、活化T细胞核因子(nuclear factor of activated T cells, NFATs)、心肌素(myocardin, MYOCD)和心肌素相关转录因子A和B (myocardin- related transcription factors-A/B, MRTF-A/B)等在心肌肥大的发生、发展过程中具有重要作用[11]。内皮素-1 (endothelin 1, ET-1)和血管紧张素Ⅱ (angiotensin Ⅱ, Ang Ⅱ)处理或横向主动脉缩窄手术(transverse aortic constriction, TAC)能够诱导实验动物心肌肥大。对先天性心脏病的研究显示, 胚胎时期NK2同源框5 (NK2 homeobox 5, NKX2-5)、T盒蛋白5 (T-box 5, TBX5)和T盒蛋白20 (T-box 20, TBX20)等转录因子基因突变或缺失会导致心脏发育缺陷。已有的研究表明,KLFs对心肌细胞增殖[5, 6]、分化[12]和心脏成纤维细胞激活[13, 14]均有调控作用。研究KLFs在心肌中的作用对揭示心脏发育、心脏肥大、先天性心脏病和心脏纤维化等生理、病理进程具有重要参考价值。1.1 KLF4
KLF4参与对心肌肥大的负调控。在正常的生理状况下,KLF4-/-小鼠的心脏重量和A型钠尿肽(natriuretic peptide A, NPPA)基因的表达水平均高于对照组;在心脏高负荷情况下,多数KLF4-/-小鼠死亡,未死亡的KLF4-/-小鼠则出现心肌肥大和心力衰竭[15, 16];当静脉注射异丙肾上腺素后,KLF4-/-小鼠心脏中NPPA、B型钠尿肽(natriuretic peptide B, NPPB)和肌球蛋白重链7 (myosin heavy chain 7, MYHC7)基因表达的增幅均高于对照组[12]。研究显示,KLF4抑制心肌肥大的作用机制可能至少包括以下两种途径:(1)直接抑制胚胎基因的表达。KLF4因子被组蛋白脱乙酰酶抑制因子(histone deacetylase inhibitor, HDACI)诱导表达,并结合于小鼠NPPA启动子区KLF4结合位点,抑制NPPA基因的表达[12, 17];(2)间接抑制胚胎基因的表达。KLF4能通过下调小鼠MYOCD的基因表达水平,抑制胚胎基因表达[12] (图1)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1KLFs在心肌组织中的功能
在心肌组织中,KLF4、KLF11、KLF13和KLF15抑制心肌胚胎基因的表达,阻止心肌肥大的发生; KLF6调控心肌细胞与心脏成纤维细胞之间的信息交流,参与调控心脏纤维化进程。
Fig. 1Functions of KLFs in cardiac tissue
此外,KLF4影响心肌能量代谢,对维持心肌细胞线粒体数量和结构具有一定作用。KLF4与雌激素相关受体-ɑ (estrogen-related receptor ɑ, ERRɑ)、过氧化物酶体增殖物激活受体γ (peroxisome proliferator- activated receptor γ, PPARγ)共激活因子-1ɑ (PPARγ co-activators 1ɑ, PGC-1ɑ)共同形成KLF4-ERRɑ- PGC-1ɑ复合物,结合于核基因组中编码线粒体蛋白质的多个基因的启动子区,诱导线粒体相关蛋白的表达[16, 18](图1)。
1.2 KLF6
KLF6基因在心肌细胞和心脏成纤维细胞中表达。在ET-1诱导下,新生大鼠心肌细胞中KLF6的基因表达水平短暂上调,暗示其可能对心脏肥大具有调控作用[19]。KLF6是心肌细胞和心脏成纤维细胞的胞间交流信号分子。与野生型相比,KLF6+/-小鼠心脏纤维化程度降低[13]。AngⅡ能够特异性增加小鼠心肌细胞中KLF6基因的表达水平,并且,AngⅡ能够诱导心肌细胞中血小板反应蛋白-4 (thrombospondin 4, TSP-4)基因的表达,促进TSP4分泌到细胞间质,抑制成纤维细胞的激活[14];同时,AngⅡ增加心肌细胞中KLF6在TSP4启动子上的募集,而KLF6抑制TSP4启动子活性[13, 14]。因此,KLF6作为胞间信号分子在心脏纤维化过程中发挥了复杂的调控作用(图1)。1.3 KLF10
KLF10也被称为转化生长因子-β (transforming growth factor-β, TGF-β)早期诱导基因-1 (TGF-β induces early gene-1, TIEG-1),对心肌肥大具有负 调控作用[20]。在心脏组织中KLF10基因低水平转 录[21]。与野生型相比,KLF10-/-小鼠出现心脏肥大,并且心脏组织中心肌肥厚相关基因垂体肿瘤转化基因-1 (pituitary tumor transforming gene-1, PTTG-1) 和组蛋白H3表达上调[20, 22]。研究显示,KLF10抑制小鼠PTTG-1启动子活性,因此,KLF10可能 通过抑制PTTG-1基因的表达,阻止心肌肥大[23] (图1)。1.4 KLF11
KLF11也被称为TGF-β诱导早期基因-2 (TGF-β induces early gene-2, TIEG-2),参与心肌肥大的负调控[20]。KLF11基因在心肌细胞中表达。与对照组相比,心力衰竭患者和心肌肥大小鼠的心脏组织中KLF11 mRNA水平显著下调[24]。ET-1刺激新生大鼠心肌细胞时,KLF11基因的表达水平下调,提示KLF11可能抑制心肌肥大的发生[25]。在正常情况下,KLF11转基因小鼠心脏无异常表现;在TAC模型中,未转基因的小鼠出现心肌肥大,而KLF11基因过表达的小鼠未见明显的心肌肥大和心脏纤维化,且与野生型小鼠相比,在过表达KLF11的小鼠心肌细胞中,胚胎基因的表达水平下调[24](图1)。1.5 KLF13
KLF13又称被为胚胎Krüppel样因子-2 (fetal Krüppel-like factor-2, FKLF-2)或基础转录元件结合蛋白3 (basal transcription element-binding protein 3, BTEB3),在红细胞、T淋巴细胞和心肌细胞等多种细胞中表达[26,27,28]。KLF13参与对心脏的早期发育调控:胚胎时期,小鼠心脏中KLF13基因的表达最早出现在E9.5,在心房和心室小梁中表达水平较高;出生后,心脏中KLF13基因的表达水平下调,在E15.5时主要在心房、房间隔、室间隔和心室小梁中表达[6]。敲低KLF13的非洲爪蟾胚胎出现房间隔缺损和心室小梁化程度低,并且可因GATA4基因的过表达而恢复正常[5]。单独缺失KLF13基因对心脏结构发育的影响微小,同时缺失KLF13与TBX5基因会导致非洲爪蟾房间隔缺损[6]。KLF13能够借助GATA4与其他因子如NKX2.5、TBX5或血清反应因子(serum response factor, SRF)等形成“KLF13-GATA4-其他因子”复合物,激活NPPA和NPPB等基因的转录,对心脏的早期发育 具有重要调控作用[5, 6](图1)。此外,KLF13能够保护小鼠心肌细胞免受外界刺激[如六水合氯化钴 (II) (CoCl2?6H2O)和多柔比星(doxorubicin)]导致的细胞死亡,对心脏毒性诱导的心力衰竭具有保护 作用[28]。
1.6 KLF15
KLF15参与对心肌肥大的负调控。在小鼠心脏发育过程中KLF15基因不表达或低水平表达;出生后KLF15基因的表达逐渐增加,约在3周龄时达到成年水平[29]。KLF15基因过表达能够阻止AngⅡ诱导的小鼠心脏肥大的发展[29, 30]。KLF15可通过调节胚胎基因转录和心肌能量代谢,抑制心肌肥大的发生:(1) KLF15能够与SRF竞争结合MYOCD和MRTF-A/B,抑制SRF-MYOCD和SRF-MRTF-A/B对胚胎基因的转录激活作用[29, 31, 32] (图1)。此外,KLF15抑制共激活物/乙酰酶p300的乙酰转移酶活性,抑制MEF2和GATA4分子的乙酰化,从而抑制MEF2和GATA4对小鼠NPPA和NPPB基因的转录激活作用[30, 33, 34](图1);(2)与野生型小鼠相比,KLF15-/-小鼠表现出对压力负荷异常敏感,可能与心肌细胞能量代谢能力的降低有关[30, 35]。研究显示,KLF15-/-小鼠的心肌细胞胞浆中出现巨 大线粒体、心肌细胞脂肪酸的转运和氧化能力降 低[36,37,38]。KLF15能够激活心肌脂质代谢、提高心肌细胞转运葡萄糖的能力。KLF15与p300结合,激活小鼠PDK4和FATP1等脂质代谢相关基因启动子活性,促进脂质代谢[37](图1)。此外,糖皮质激素能通过诱导大鼠KLF15基因的表达,激活支链氨基酸氨基转移酶2 (branched-chain amino acid transaminase 2, BCAT2)和葡萄糖转运蛋白4 (glucose transporter 4, GLUT4)基因的表达,降低心肌细胞支链氨基酸(branched-chain amino acid, BCAA)浓度和提高心肌细胞对葡萄糖的摄取能力[39](图1)。
此外,KLF15可通过调节心肌血管生成抑制AngⅡ诱导的心力衰竭[30]。KLF15通过抑制p300乙酰转移酶活性,抑制小鼠p53 Lys379乙酰化,进而抑制血小板反应蛋白-1(thrombospondin-1, TSP-1)等p53靶基因的表达,阻止p53累积导致的心力衰竭[30](图1)。
2 平滑肌中的KLFs
平滑肌(smooth muscle)广泛分布于消化道、呼吸道、血管和生殖管道等多种器官。体内平滑肌细胞存在2种表型:分化程度较高的收缩型,以及具有较高增殖和迁移能力的合成型。平滑肌表型的转化主要体现在平滑肌肌动蛋白(smooth muscle actin, SMA)、平滑肌肌球蛋白重链(smooth muscle myosin heavy chain, SMHC)、平滑肌22α (smooth muscle 22α, SM22α)、钙调蛋白和肌球蛋白轻链激酶(myosin light chain kinase, MLCK)等平滑肌标记基因和细胞周期基因表达水平的改变[40]。KLFs在多种平滑肌中具有调控作用,特别是KLFs在血管平滑肌细胞(vascular smooth muscle cells, VSMC)中的功能受到了研究者的广泛关注。2.1 KLF4
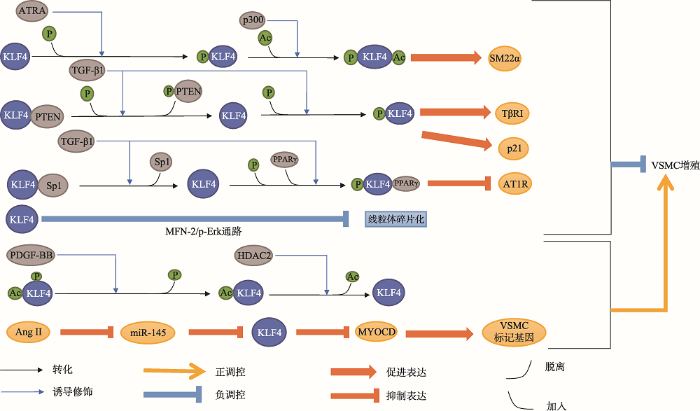
在非血管损伤条件下,大鼠VSMC中KLF4基因低水平表达;血管损伤后,大鼠VSMC中KLF4基因的表达量迅速上调。因此,过去认为KLF4主要发挥促VSMC增殖的作用[12]。近年来,多项研究显示,KLF4在血管平滑肌中具有促增殖和促分化的作用。在大鼠VSMC中,全反式维甲酸(all-trans retinoic acid, ATRA)和TGF-β1诱导KLF4分子发生磷酸化修饰,使其发挥促分化作用,而血小板衍生生长因子-BB (platelet derived growth factor-BB, PDGF- BB)诱导KLF4去磷酸化从而抑制其促VSMC分化的活性。此外,KLF4对线粒体碎片化和MYOCD基因的表达具有调控作用,间接影响VSMC的增殖和分化。因此,KLF4可能是VSMC表型转化的重要调控因子。磷酸化后的KLF4的促分化作用表现在:(1)诱导平滑肌标记基因表达。ATRA诱导KLF4磷酸化,募集p300,促使KLF4乙酰化。乙酰化的KLF4与SM22α启动子结合并激活SM22α基因的转录[8](图2);(2)抑制VSMC细胞周期。TGF-β1诱导KLF4磷酸化,磷酸化的KLF4一方面将p300募集至P21启动子区,乙酰化组蛋白H3,启动P21基因的表达;另一方面KLF4直接激活转化生长因子β受体I型(transforming growth factor β receptor I, TβRI)基因的表达,或与Smad2结合形成KLF4-Smad2复合物,协同激活TβRI基因的表达,抑制VSMC周期并促使其分化(图2);(3) 磷酸化的KLF4能与PPARγ结合形成KLF4-PPARγ复合物,占据AngⅡ1型受体(Ang Ⅱ type 1 receptor, AT1R)基因启动子区的TGF-β1调控元件(TGF-β1 control elements, TCE),抑制AT1R的启动子活性,抑制VSMC增殖[41,42,43,44](图2)。此外,KLF4能够调控线粒体融合蛋白-2/p-Erk (mitochondrial fusion protein - mitofusin-2, MFN-2/ p-Erk)信号通路,降低低氧诱导下大鼠肺动脉VSMC线粒体碎片化,抑制VSMC增殖[45](图2)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2血管平滑肌细胞中KLF4的翻译后修饰和相应功能
ATRA和TGF-β1诱导KLF4分子发生磷酸化修饰,磷酸化修饰后的KLF4诱导SM22α、TβRI和p21基因的表达,抑制AT1R基因的表达,从而抑制血管平滑肌细胞增殖;KLF4通过抑制线粒体碎片化,进而抑制血管平滑肌细胞增殖;PDGF-BB诱导下KLF4去磷酸化,抑制ATRA和TGF-β1诱导的血管平滑肌细胞分化;Ang II下调miRNA-145的表达水平,增加KLF4基因的表达;KLF4通过抑制MYOCD基因的表达,间接抑制血管平滑肌细胞标记基因的转录。
Fig. 2The post-translational modifications of KLF4 and the corresponding functions in vascular smooth muscle cells (VSMC)
在血管损伤情况下,PDGF-BB诱导KLF4去磷酸化,促使KLF4与HDAC2相互作用使得KLF4去乙酰化,抑制KLF4与SM22α基因启动子的结合和对SM22α转录的激活[8](图2)。PDGF-BB还可促使KLF4与磷酸酶/张力蛋白同源物(phosphatase and tensin homolog, PTEN)形成KLF4-PTEN复合物,抑制TGF-β1诱导下KLF4的促分化作用[41, 43]。此外,Ang II能够通过下调miRNA-145的表达水平,增加KLF4基因的表达,进而下调MYOCD基因的表达水平,降低MYOCD对VSMC标记基因转录的诱导作用,促进VSMC增殖和迁移[9, 46~48](图2)。VSMC中存在一个围绕KLF4的庞大调节网络,遗传和环境因素通过改变KLF4的表达、分子修饰和复合物状态,转换KLF4在VSMC中的功能,以应对内外环境(如血管损伤等)的改变。
2.2 KLF5
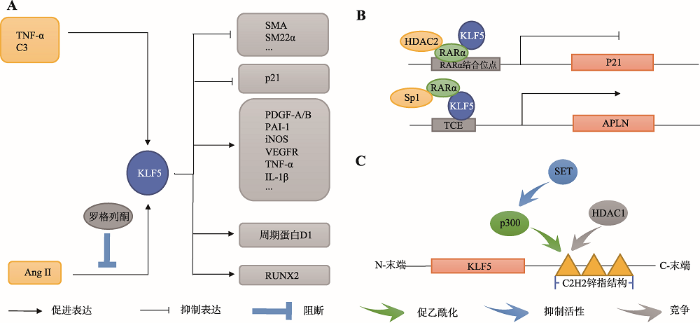
KLF5参与调控VSMC增殖和血管重塑过程。胚胎期时,人和兔等多物种的VSMC中KLF5基因呈现高水平表达,而在成年期时KLF5基因却转变为低水平表达状态[49]。在血管损伤或AngⅡ输注条件下,大鼠主动脉VSMC中KLF5基因的表达增加[50]。KLF5至少可通过以下4条途径引起VSMC增殖和血管钙化:(1) KLF5上调PDGF-A/B、纤溶酶原激活物抑制因子-1 (plasminogen activator inhibitor-1,PAI-1)、诱导型一氧化氮合酶(inducible nitric oxide synthase, iNOS)、血管内皮生长因子受体(vascular endothelial growth factor receptor, VEGFR)、肿瘤坏死因子-α (tumor necrosis factor-α, TNF-α)和白细胞介素-1β (interleukin-1β, IL-1β)等炎性反应因子基因的表达水平,发挥促VSMC增殖的作用[49, 51](图3A);(2) KLF5抑制SMA和SM22α等VSMC标记基因的启动子活性[40](图3A);(3) KLF5上调细胞周期蛋白D1基因的表达和抑制P21基因的表达[42, 50](图3A);(4) KLF5能够激活参与成骨细胞和软骨细胞分化调控的关键转录因子RUNX2基因的表达,诱导大鼠VSMC向成骨样细胞转化,引起血管钙化[52](图3A)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3血管平滑肌细胞中的KLF5功能
A:诱导KLF5基因表达的分子和被KLF5所诱导表达的基因;B:KLF5与其他因子形成不同复合物调控靶基因P21和APLN的表达;C:SET、p300和HDACI影响KLF5功能的作用机制。
Fig. 3The functions of KLF5 in vascular smooth muscle cells (VSMC)
多种因子或药物可通过以下几种方式调控KLF5基因的表达或分子状态,进而在VSMC中发挥促增殖或促分化功能:(1)调控KLF5基因的转录。TNF-α、补体C3(complement C3, C3)和AngⅡ等可上调KLF5基因的表达水平,促进VSMC增殖;在大鼠中AngⅡ诱导的KLF5基因的表达可被罗格列酮阻断[8, 50, 53, 54](图3A);(2)与KLF5形成复合物。当视黄酸受体-α(retinoic acid receptor-α, RARα)与HDAC2、KLF5在大鼠P21基因启动子的RARα结合位点上形成HDAC2-RARα-KLF5复合物时,抑制P21启动子活性,间接促进VSMC增殖[42](图3B)。此外,RARα能与KLF5、Sp1在大鼠爱帕琳肽(apelin, APLN)基因启动子的TCE上形成Sp1-RARα-KLF5复合物,诱导APLN表达,促进VSMC增殖[55](图3B)。值得注意的是,在RARα特异性激动剂Am80的作用下,大鼠VSMC分化活性增强[42, 55]。Am80能够磷酸化HDAC2并使其与RARα解离,磷酸化的HDAC2使KLF5脱乙酰化,促使KLF5从P21基因的启动子上解离,解除对P21转录的抑制。但是,Am80可促使Sp1-RARα-KLF5复合物形成进而诱导APLN基因的表达,因此,Am80对VSMC中RARα的靶基因的调控机制尚不清楚[42, 55];(3)影响KLF5翻译后修饰。致癌调节因子/组蛋白伴侣SET可抑制p300介导的KLF5锌指结构乙酰化,HDAC1可通过N-末端与p300竞争KLF5 C-末端第一锌指,通过作用于锌指结构,发挥抑制KLF5结合靶基因启动子的作用[49, 56](图3C)。
2.3 KLF8
KLF8参与维持VSMC的收缩型状态。收缩型VSMC中KLF8基因高水平表达,在TNF-α诱导大鼠VSMC去分化过程中,VSMC标记基因和KLF8基因的表达下调[40]。KLF8可通过激活SMA等VSMC标记基因启动子活性和抑制KLF5基因启动子活性,维持VSMC的收缩型,但对KLF4基因的表达没有影响[40]。此外,大鼠KLF8基因启动子上存在C- Ets-1、CAAT/增强结合蛋白β(CAAT/enhance binding protein β, C/EBPβ)、RARɑ、Ap1、Sp1、KLF4和NF-κB等转录因子结合位点,MYOCD可通过上调KLF4或NF-κB基因的表达水平,间接激活KLF8基因的启动子活性[40]。2.4 KLF15
KLF15参与对VSMC增殖的负调控。在正常生理状况下,小鼠VSMC中KLF15基因高表达;在促增殖和促炎因子刺激下,KLF15基因的表达水平显著降低[57]。与野生型小鼠相比,血管损伤时,KLF15-/-小鼠新生血管内膜中的VSMC表现为增殖及迁移增强[58]。KLF15可通过抑制促增殖信号分子,抑制VSMC增殖:KLF15基因过表达会抑制PDGF- BB诱导的小鼠VSMC增殖,但是具体机制还不清楚[58];此外,KLF15可通过其转录激活结构域以浓度依赖的方式与p65竞争p300结合位点,改变NF-κB乙酰化状态并抑制其活性,进而抑制VSMC增殖[59]。3 骨骼肌中的KLFs
骨骼肌(skeletal muscle)肌纤维数目在胚胎期时已基本固定。骨骼肌特异性基因表达受生肌决定基因(myogenic determination gene, MyoD)家族、MEF2家族和细胞外信号调节激酶5 (extracellular signal- regulated kinases 5, ERK5)等的调节[60],此外,良好的血管发生和能量代谢促进骨骼肌发育[61]。KLFs参与对骨骼肌细胞增殖、融合、肌小管形成、能量代谢和血管发生的调控。3.1 KLF2和KLF4
KLF2和KLF4促进骨骼肌细胞融合。在小鼠骨骼肌细胞分化过程中,KLF2和KLF4基因的表达上调,且可被ERK5抑制剂阻断[60]。他汀类药物可诱导人脐带内皮细胞ERK5磷酸化,诱导KLF4基因的表达,但其对KLF2基因的诱导作用仍然缺少证据[62]。ERK5信号通路能通过Sp1通路上调小鼠骨骼肌细胞KLF2和KLF4基因的表达,进而上调NPNT基因的表达水平,通过促进细胞-基质粘附,促进细胞融合。因此,骨骼肌中可能存在MEK5-ERK5- KLF2/4-NPNT途径调控着骨骼肌细胞融合[60]。3.2 KLF3
在横纹肌终末分化时期,KLF3基因的表达水平增加,并在内源性促肌肉形成基因的顺式调控元件上富集,但是与野生型相比,KLF3-/-小鼠没有表 现出明显的肌肉缺陷,这可能是由于分子冗余造成的[63, 64]。本课题组前期在鸡前脂肪细胞中发现,KLF3基因过表达能够调控PPARγ和C/EBPα等脂肪细胞分化标记基因的表达,并且该调控作用部分依赖KLF3 N-末端的抑制结构域(repression domain, RD)中的PVDLT模序[65]。与此一致的是,在多数组织中,KLF3发挥转录抑制作用依赖其N-末端的RD序列与转录抑制因子的结合[66]。但是,在小鼠骨骼肌中,KLF3对肌肉特异基因表达的调节机制是:KLF3的C-末端到RD间的序列与SRF结合,引起KLF3的RD构象改变,导致其不再募集转录抑制因子,转而募集转录激活因子,激活肌酸激酶(muscle creatine kinase, MCK)等基因的转录[64]。3.3 KLF10
KLF10抑制骨骼肌形成,因此,理论上KLF10基因在骨骼肌形成过程中表达下调;但是,在小鼠骨骼肌细胞分化过程中,KLF10基因的表达增加;并且在鸡成肌细胞和肌管中能够检测到KLF10基因的表达,提示了KLF10在骨骼肌组织中的功能可能比较复杂[67, 68]。过表达KLF10基因的小鼠骨骼肌表现为细胞数量减少和肌管形成受损;与野生型相比,KLF10-/-小鼠表现出比目鱼肌和趾长伸肌酵解性肥大(glycolytic hypertrophy)和增生[68, 69]。KLF10通过抑制促增殖信号分子功能和周期蛋白表达抑制成肌细胞增殖:(1)在鸡成肌细胞中,KLF10与成纤维细胞生长因子受体1(fibroblast growth factors, FGFR1)基因的启动子近端的Sp1结合位点结合,抑制FGFR1启动子活性[67];(2)在敲低KLF10的小鼠成肌细胞中,细胞周期蛋白CCNA2、CCNB2和BIRC5基因的表达增加,暗示KLF10可能对其转录具有抑制作用[68]。3.4 KLF15
KLF15参与骨骼肌分化的正调控。骨骼肌分化过程中KLF15基因的表达上调,但敲低KLF15对小鼠骨骼肌分化没有影响,这可能是由于分子冗余造成的[70]。功能研究显示,KLF15可以通过NFATc1信号通路诱导小鼠成肌细胞肌球蛋白重链-β/慢速(myosin heavy chain-β/slow, MHC-β/slow)基因的表达,促进骨骼肌形成[70]。此外,KLF15对骨骼肌能量代谢也有调控作用。耐力运动实验显示,与野生型小鼠相比,KLF15-/-小鼠骨骼肌过多依赖碳水化合物,不能很好地利用脂肪[71]。功能研究显示,KLF15对骨骼肌中蛋白质、糖和脂质转化和代谢具有调控作用:(1)在蛋白质代谢方面,在糖皮质激素刺激下,KLF15通过激活BCAT2基因的表达,加速BCAA降解,抑制哺乳动物雷帕霉素靶向基因(mammalian target of rapamycin, mTOR)信号通路活性,抑制肌肉蛋白的合成[72]。此外,KLF15与叉头框蛋白O1 (forhead box O1, FoxO1)协同激活大鼠成肌细胞中Atrogin-1和MuRF-1基因的表达,作用于MyoD和MYHC等蛋白底物,加速肌肉蛋白分解[72];(2)在糖代谢方面,KLF15能够直接结合GLUT4基因近端启动子区的MEF2A结合位点,激活GLUT4的转录[73];此外,KLF15能协同转录因子Sp1激活果蝇乙酰CoA合成酶2 (acetyl-CoA synthetase 2, ACECS2)基因的转录,加速糖代谢[74];(3)在脂质代谢方面,在骨骼肌中KLF15能诱导小鼠FATP1等脂质转运、代谢等相关基因的表达[71];此外KLF15能结合于牛骨骼肌长链酰基辅酶A合成酶1 (long-chain acyl-CoA synthetase 1, ACSL1)基因的启动子,激活其转录。因此,KLF15在骨骼肌能量代谢方面具有调控作用[75]。
4 结语和展望
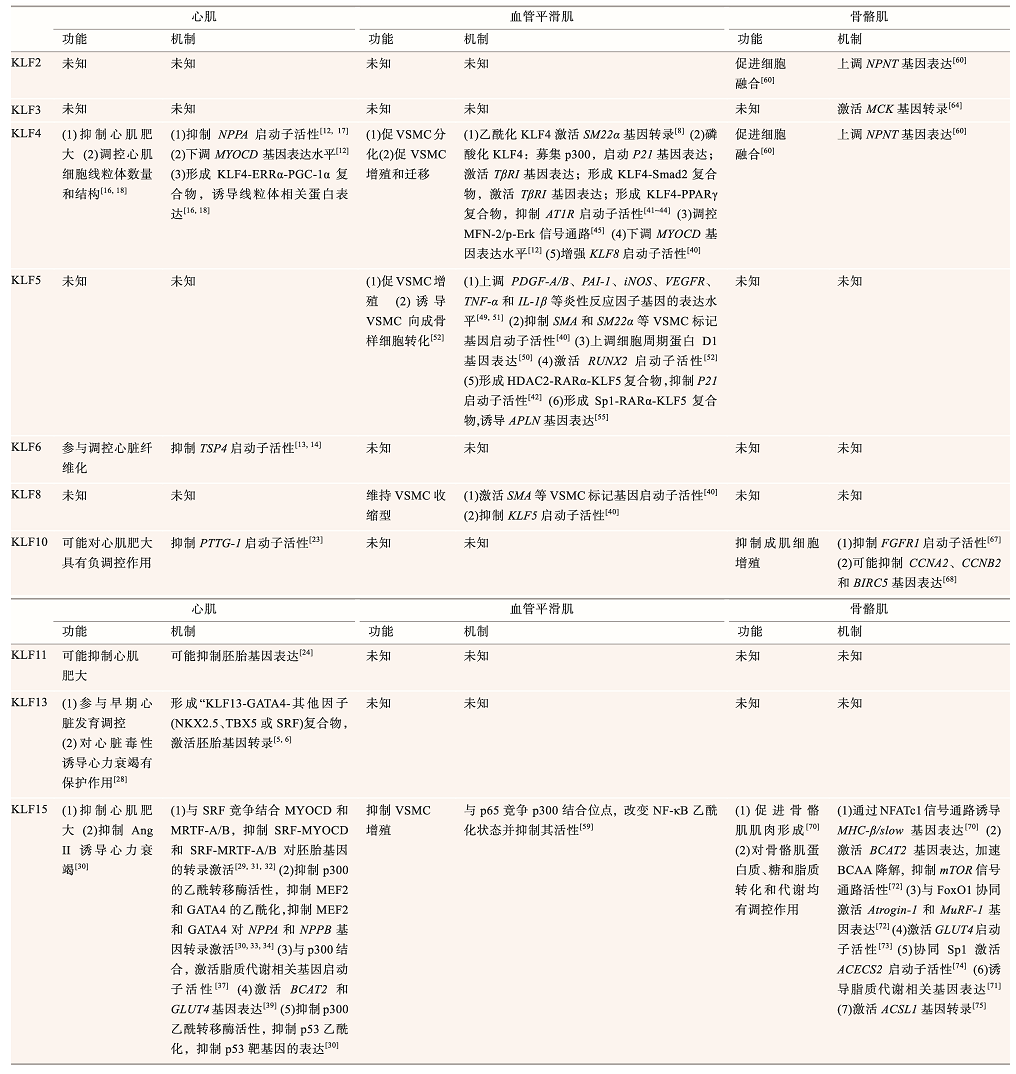
KLFs在3种肌肉组织的发育和功能维持中均具有重要调控作用。在心肌组织中,KLF4、KLF10、KLF11和KLF15参与心肌肥大的负调控,KLF6参与调控心脏纤维化,KLF13参与调控胚胎时期的心肌发育。在血管平滑肌中,随着分子修饰和复合物组成的变化,KLF4发挥着促增殖或促分化作用,KLF5促进血管平滑肌增殖,KLF8和KLF15抑制血管平滑肌增殖。在骨骼肌中,KLF2、KLF3、KLF4、KLF10和KLF15参与对骨骼肌发育的调控。另外,KLF15是3种肌肉组织能量代谢的重要调节因子。对比同种KLF在3种不同肌肉组织中的功能显示,同种KLF可在2种或2种以上的肌肉组织中通过相似的作用机制发挥相似作用,如在心肌和骨骼肌中,KLF15均对线粒体脂质代谢相关基因的表达有调控作用(表1)。此外,同种KLF也可以在2种或2种以上的肌肉组织中通过不同的作用机制发挥不同或相似的调控作用,如KLF4在3种肌肉组织中通过3种完全不同的机制发挥了不同的调控作用,而KLF10在心肌和骨骼肌中则通过不同的机制均发挥了抑制肌肉组织形成的作用(表1)。
Table 1
表1
表1 KLFs在心肌、平滑肌和骨骼肌中的功能机制
Table 1
 |
新窗口打开|下载CSV
此外,不同KLF因子在同种肌肉组织中可能具有相似或相反作用,如KLF4、KLF10、KLF11和KLF15均参与对心肌肥大的负调控,在心肌中KLF4和KLF15对MYOCD基因的表达或活性具有调控作用[12, 31],在血管平滑肌细胞中KLF4和KLF5对SM22α基因的表达具有调控作用[8, 40]。
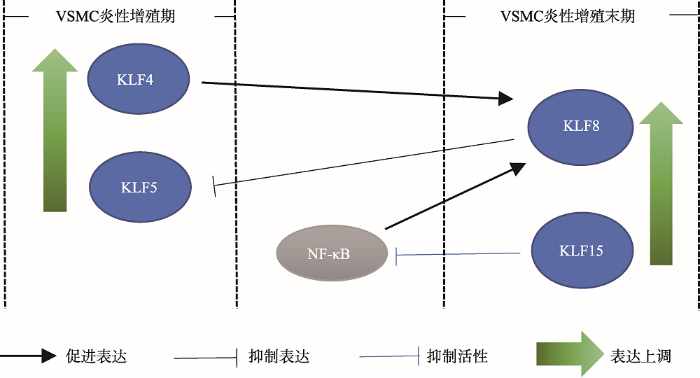
值得注意的是,在血管损伤条件下,KLF4和KLF5基因的表达上调,参与促进血管平滑肌细胞增殖[12, 49, 51];在血管平滑肌细胞的炎性增殖末期,KLF4和NF-κB能够上调KLF8基因的表达,被诱导表达的KLF8因子会抑制KLF5基因的启动子活性,进而抑制KLF5诱导的血管平滑肌细胞增殖[40];KLF15能够抑制NF-κB分子活性[59],在血管平滑肌细胞的炎性增殖末期,KLF8和KLF15可能参与抑制血管平滑肌细胞的过度增殖(图4,表1)。在血管平滑肌细胞的炎性增殖过程中,KLF家族成员的顺序调控可能发挥了重要作用。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4血管平滑肌细胞中KLF4、KLF5、KLF8和KLF15的表达模式及分子间的相互作用
在血管平滑肌细胞炎性增殖期,KLF4和KLF5基因的表达上调;在血管平滑肌细胞炎性增殖末期,KLF8和KLF15基因的表达上调。在血管平滑肌细胞中,KLF4和NF-κB上调KLF8基因的表达,KLF8抑制KLF5基因的启动子活性,KLF15又可以抑制NF-κB的分子活性;上述KLFs共同调控了血管平滑肌细胞的炎性增殖。
Fig. 4Expression patterns and intermolecular interactions among KLF4, KLF5, KLF8, and KLF15 in vascular smooth muscle cells (VSMC)
除此之外,在不同的肌肉组织中的研究显示,不同的KLF因子发挥着类似的细胞周期调控作用,如心肌中的KLF15、血管平滑肌中的KLF4、KLF5和骨骼肌中的KLF10都可通过调控细胞周期基因进而调控细胞增殖(表1)。鉴于细胞周期的保守性,有可能KLF因子在同种肌肉组织的细胞增殖中具有协同或顺序调控现象。
此外,骨骼肌中的研究报道显示,KLF2和KLF4在骨骼肌中均对NPNT基因具有调控作用(表1)。KLF2、KLF3、KLF4和KLF15基因均在骨骼肌分化期间表达上调,并促进成肌细胞分化和肌肉组织形成;KLF10作为骨骼肌形成的负调控因子在分化期间其基因的表达上调,可能参与抑制骨骼肌的过度增殖。因此,KLF家族成员可能在肌细胞分化过程中发挥了协同或顺序调控,从而精细调控肌细胞的增殖和分化。
目前还有很多KLF家族成员在肌肉组织中的功能不明确,如心肌细胞中KLF2、KLF3、KLF5的作用尚不明确。进一步研究KLFs在肌肉组织中的作用及其调控的靶基因将有助于揭示肌肉组织发育及相关疾病的发生机制。借助过表达、siRNA干扰和荧光素酶报告基因分析等分子生物学技术研究调控KLFs表达的上游信号分子和基因,可能为开发心肌肥大、动脉粥样硬化、糖尿病等疾病的靶向药物提供新思路。此外,对KLF因子的分子结构的研究可能有助于发现KLF家族成员在物种间的功能共性,为进一步揭示锌指样转录因子的功能提供参考。
(责任编委: 杨中州)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 4]
[本文引用: 1]
[本文引用: 1]
URLPMID:17053787 [本文引用: 5]

In humans, congenital heart defects occur in 10900092% of live birth, but the molecular mechanisms and causative genes remain unidentified in the majority of cases. We have uncovered a novel transcription pathway important for heart morphogenesis. We report that KLF13, a member of the Kr0104ppel-like family of zinc-finger proteins, is expressed predominantly in the heart, binds evolutionarily conserved regulatory elements on cardiac promoters and activates cardiac transcription. KLF13 is conserved across species and knockdown of KLF13 in Xenopus embryos leads to atrial septal defects and hypotrabeculation similar to those observed in humans or mice with hypomorphic GATA-4 alleles. Physical and functional interaction with GATA-4, a dosage-sensitive cardiac regulator, provides a mechanistic explanation for KLF13 action in the heart. The data demonstrate that KLF13 is an important component of the transcription network required for heart development and suggest that KLF13 is a GATA-4 modifier; by analogy to other GATA-4 collaborators, mutations in KLF13 may be causative for congenital human heart disease.
URLPMID:28164238 [本文引用: 5]
Abstract TBX5, a member of the T-box family of transcription factors, is a dosage sensitive regulator of heart development. Mutations in TBX5 are responsible for Holt-Oram Syndrome, an autosomal dominant disease with variable and partially penetrant cardiac defects suggestive of the existence of genetic and environmental modifiers. KLF13, a member of the Kr ppel-like family of zinc finger proteins is co-expressed with TBX5 in several cardiac cells including atrial cardiomyocytes and cells of the interatrial septum. We report that KLF13 interacts physically and functionally with TBX5 to synergistically activate transcription of cardiac genes. We show that TBX5 contacts KLF13 via its T-domain and find that several disease-causing mutations therein have decreased KLF13 interaction. Whereas Klf13 heterozygote mice have no detectable cardiac defects, loss of a Klf13 allele in Tbx5 heterozygote mice significantly increases the penetrance of TBX5-dependent cardiac abnormalities including atrial, atrial-ventricular and ventricular septal defects. The results reveal for the first time combinatorial interaction between a T-box protein and a KLF family member and its importance for heart and possibly other organ development. The data also suggest that, in human, KLF13 may be a genetic modifier of the Holt-Oram Syndrome gene TBX5. The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Magsci [本文引用: 1]
<p>Sp1/Krüppel样因子(Sp1-like and Krüppel-like factors, Sp1/KLFs)是一组与真核细胞转录调控密切相关的锌指蛋白。Sp1/KLFs的羧基末端高度保守, 含有3个串联的Cys<sub>2</sub>His<sub>2</sub>锌指结构, 用于结合DNA; 其氨基末端在不同的家族成员间存在较大差异, 主要是通过结合辅助因子发挥转录调控作用。Sp1/KLFs的表达具有组织、细胞分布以及发育时期的特异性, 它们通过调控多种富含GC或CACCC的启动子的基因的表达, 参与细胞增殖、分化、凋亡和肿瘤发生、发展等多种生理、病理过程。文章综述了Sp1/KLFs的结构特征、作用机制及生物学功能。</p>
Magsci [本文引用: 1]
<p>Sp1/Krüppel样因子(Sp1-like and Krüppel-like factors, Sp1/KLFs)是一组与真核细胞转录调控密切相关的锌指蛋白。Sp1/KLFs的羧基末端高度保守, 含有3个串联的Cys<sub>2</sub>His<sub>2</sub>锌指结构, 用于结合DNA; 其氨基末端在不同的家族成员间存在较大差异, 主要是通过结合辅助因子发挥转录调控作用。Sp1/KLFs的表达具有组织、细胞分布以及发育时期的特异性, 它们通过调控多种富含GC或CACCC的启动子的基因的表达, 参与细胞增殖、分化、凋亡和肿瘤发生、发展等多种生理、病理过程。文章综述了Sp1/KLFs的结构特征、作用机制及生物学功能。</p>
URL [本文引用: 5]
[本文引用: 2]
URLPMID:28366734 [本文引用: 1]
Specificity proteins (SPs) and Kr ppel-like factors (KLFs) belong to the family of transcription factors that contain conserved zinc finger domains involved in binding to target DNA sequences. Many of these proteins are expressed in different tissues and have distinct tissue-specific activities and functions. Studies have shown that SPs and KLFs regulate not only physiological processes such as growth, development, differentiation, proliferation, and embryogenesis, but pathogenesis of many diseases, including cancer and inflammatory disorders. Consistently, these proteins have been shown to regulate normal functions and pathobiology in the digestive system. We review recent findings on the tissue- and organ-specific functions of SPs and KLFs in the digestive system including the oral cavity, esophagus, stomach, small and large intestines, pancreas, and liver. We provide a list of agents under development to target these proteins.
URLPMID:16936699 [本文引用: 2]
Abstract The mammalian heart is a dynamic organ that can grow and change to accommodate alterations in its workload. During development and in response to physiological stimuli or pathological insults, the heart undergoes hypertrophic enlargement, which is characterized by an increase in the size of individual cardiac myocytes. Recent findings in genetically modified animal models implicate important intermediate signal-transduction pathways in the coordination of heart growth following physiological and pathological stimulation.
URL [本文引用: 7]
URL [本文引用: 3]
[本文引用: 3]
URLPMID:2885477 [本文引用: 1]
Kr眉ppel-like factors (KLF) are a subfamily of the zinc-finger class of transcriptional regulators that play important roles in diverse cellular processes. While a number of KLFs are expressed in cardiomyocytes, little is known about their specific roles in the heart in vivo. Here, we demonstrate that KLF4 is induced by hypertrophic stimuli in cultured cardiomyocytes and in the mouse heart. Overexpression of KLF4 in neonatal rat ventricular myocytes inhibits three cardinal features of cardiomyocyte hypertrophy: fetal gene expression, protein synthesis, and cell enlargement. Conversely, mice with cardiomyocyte-specific deletion of KLF4 (CM-K4KO) are highly sensitized to transverse aortic constriction (TAC) and exhibit high rates of mortality. CM-K4KO mice that survive TAC display severe pathologic cardiac hypertrophy characterized by increased cardiac mass, depressed LV systolic function, pulmonary congestion, cavity dilation and attenuated LV wall thickening when compared to control genotypes. In addition, CM-K4KO mice develop increased myocardial fibrosis and apoptotic cell death after TAC. Collectively, these studies implicate KLF4 as a novel transcriptional regulator that is indispensible for the heart's response to stress in vivo.
[本文引用: 2]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URLPMID:3104724 [本文引用: 3]
Since the discovery by this laboratory of the zinc finger transcription factor, KLF10, a member of the Kr ppel-like family of transcription factors, there have been multiple publications regarding its functions and its immediate family members, in numerous cell types. KLF10 has been shown to be rapidly induced by TGF1, 2, 3, E2, epidermal growth factor, and bone morphogenetic protein-2. TGF inducible early gene-1 activates the TGF-Smad signaling pathway via repression of Smad 7 expression and activation of Smad 2 expression and activity. Overall, KLF10 has been implicated in cell differentiation, as a target gene for a variety of signaling pathways, and in serving as a potential marker for human diseases such as breast cancer, cardiac hypertrophy, and osteoporosis. Like other KLF members, KLF10 is expressed in specific cell types in numerous tissues and is known to be involved in repressing cell proliferation and inflammation as well as inducing apoptosis similar to that of TGF. KLF10 binds to Sp-1-GC rich DNA sequences and can activate or repress the transcription of a number of genes. Overall, KLF10 has been shown to play a major role in the TGF inhibition of cell proliferation and inflammation and induction of apoptosis, and its overexpression in human osteoblasts and pancreatic carcinoma cells mimics the actions of TGF.
URL [本文引用: 1]
URLPMID:22234868 [本文引用: 1]
Hypertrophic cardiomyopathy (HCM) is the most common heritable cardiovascular disease. A recent study showed that male KLF10-encoded TGF0205 Inducible Early Gene-1 knock-out mice (TIEG090808/090808) develop HCM with 13-fold up-regulation of PTTG1-encoded pituitary tumor-transforming gene 1. We hypothesized TIEG1 could be a novel candidate gene in the pathogenesis of genotype negative HCM in humans, possibly through a loss of its repression on PTTG1 expression. A cohort of 923 unrelated patients from two independent HCM centers was analyzed for mutations in TIEG's four translated exons using DHPLC and direct DNA-sequencing. Site directed mutagenesis was performed to clone novel variants. The effect of TIEG1 mutations on SMAD7 and PTTG1 promoters was studied using transient transfection and luciferase-assays. Altered expression of PTTG1 in cardiac tissue was studied by immunohistochemistry (IHC) to determine levels of PTTG1 protein in hypertrophic diseases. Six novel TIEG1 missense mutations were discovered in six patients (two males/four females, mean age at diagnosis 56.209000900±09000923 years, MLVWT 20.809000900±0900094090009mm). Compared to WT TIEG1, five TIEG1 mutants significantly increased PTTG1 promoter function similar to TIEG1090808/090808-mice. By IHC, PTTG1-protein expression was significantly increased in multiple models of hypertrophic cardiac disease, including TIEG1-mutation positive HCM compared to normal hearts. This is the first article to associate mutations in TIEG1 to human disease with the discovery of six novel, HCM-associated variants. Functional assays suggest a role for PTTG1 in the pathogenesis of TIEG1-mediated HCM. Up-regulation of PTTG1 seems to be a common pathway in hypertrophic heart disease, including TIEG1-mediated HCM. J. Cell. Biochem. 113: 18960900091903, 2012. 0008 2012 Wiley Periodicals, Inc.
URL [本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 1]
URLPMID:132020 [本文引用: 1]
To study the function of the Kr ppel-like factor in vivo, we generated with a disrupted allele. Although (-/-) are viable, fewer were present at 3 weeks than predicted by Mendelian inheritance. Viable (-/-) had reduced numbers of circulating erythrocytes and a larger spleen. The spleen contained an increased number of Ter119()CD71(hi), Ter119(hi)CD71(hi), and Ter119(hi)CD71() cells but not Ter119(hi)CD71(-) cells, indicating an increase in less mature erythroblasts. A higher proportion of the Ter119()CD71(hi) cells were proliferating, indicating that the were under a degree of erythropoietic stress. These data indicate that is involved in the normal control of erythropoiesis.
URLPMID:248219701344 [本文引用: 1]
Kruppel-like factor (KLF) 13 is a transcription factor that positively regulates expression of the chemokine RANTES 3-5 d after activation of T cells. In this study, we document a key role for KLF13 in the expression of IL-4 in CD4(+) T cells. Gene expression analysis in activated T cells from Klf13(-/-) mice showed that IL-4, along with other Th2 cytokine genes, was downregulated when compared with cells from wild-type mice. The decreased levels of IL-4 were not associated with changes in expression of the Th2-inducing transcription factors GATA3 or c-Maf. Additional analysis revealed that KLF13 directly binds to IL-4 promoter regions and synergizes with c-Maf to positively regulate IL-4 expression. These results indicate that KLF13 is a positive regulator for differentiation of Th2 cells, as part of the transcriptional machinery that regulates IL-4 production in Th2 cells.
URL [本文引用: 2]
URLPMID:22586493 [本文引用: 3]
The Kruppel-like factor (KLF) family of transcription factors regulates diverse cell biological processes including proliferation, differentiation, survival and growth. Previous studies have shown that KLF15 inhibits cardiac hypertrophy by repressing the activity of pivotal cardiac transcription factors such as GATA4, MEF2 and myocardin. We set out this study to characterize the interaction of KLF15 with putative other transcription factors. We first show that KLF15 interacts with myocardin-related transcription factors (MRTFs) and strongly represses the transcriptional activity of MRTF-A and MRTF-B. Second, we identified a region within the C-terminal zinc fingers of KLF15 that contains the nuclear localization signal. Third, we investigated whether overexpression of KLF15 in the heart would have therapeutic potential. Using recombinant adeno-associated viruses (rAAV) we have overexpressed KLF15 specifically in the mouse heart and provide the first evidence that elevation of cardiac KLF15 levels prevents the development of cardiac hypertrophy in a model of Angiotensin II induced hypertrophy.
URLPMID:20375365 [本文引用: 5]
Current therapies for diseases of heart muscle (cardiomyopathy) and aorta (aortopathy) include inhibitors of the renin-angiotensin system, beta-adrenergic antagonists, and the statin class of cholesterol-lowering agents. These therapies have limited efficacy, as adverse cardiovascular events continue to occur with some frequency in patients taking these drugs. Although cardiomyopathy and aortopathy can coexist in a number of conditions (for example, Marfan's syndrome, acromegaly, pregnancy, and aging), pathogenetic molecular links between the two diseases remain poorly understood. We reasoned that identification of common molecular perturbations in these two tissues could point to therapies for both conditions. Here, we show that deficiency of the transcriptional regulator Kruppel-like factor 15 (Klf15) in mice leads to both heart failure and aortic aneurysm formation through a shared molecular mechanism. Klf15 concentrations are markedly reduced in failing human hearts and in human aortic aneurysm tissues. Mice deficient in Klf15 develop heart failure and aortic aneurysms in a p53-dependent and p300 acetyltransferase-dependent fashion. KLF15 activation inhibits p300-mediated acetylation of p53. Conversely, Klf15 deficiency leads to hyperacetylation of p53 in the heart and aorta, a finding that is recapitulated in human tissues. Finally, Klf15-deficient mice are rescued by p53 deletion or p300 inhibition. These findings highlight a molecular perturbation common to the pathobiology of heart failure and aortic aneurysm formation and suggest that manipulation of KLF15 function may be a productive approach to treat these morbid diseases.
URLPMID:20566642 [本文引用: 2]
Abstract Pathological forms of left ventricular hypertrophy (LVH) often progress to heart failure. Specific transcription factors have been identified that activate the gene program to induce pathological forms of LVH. It is likely that apart from activating transcriptional inducers of LVH, constitutive transcriptional repressors need to be removed during the development of cardiac hypertrophy. Here, we report that the constitutive presence of Kr ppel-like factor 15 (KLF15) is lost in pathological hypertrophy and that this loss precedes progression toward heart failure. We show that transforming growth factor-beta-mediated activation of p38 MAPK is necessary and sufficient to decrease KLF15 expression. We further show that KLF15 robustly inhibits myocardin, a potent transcriptional activator. Loss of KLF15 during pathological LVH relieves the inhibitory effects on myocardin and stimulates the expression of serum response factor target genes, such as atrial natriuretic factor. This uncovers a novel mechanism where activated p38 MAPK decreases KLF15, an important constitutive transcriptional repressor whose removal seems a vital step to allow the induction of pathological LVH.
URL [本文引用: 1]
URLPMID:17438289 [本文引用: 1]
Abstract Cardiac hypertrophy is a common response to injury and hemodynamic stress and an important harbinger of heart failure and death. Herein, we identify the Kruppel-like factor 15 (KLF15) as an inhibitor of cardiac hypertrophy. Myocardial expression of KLF15 is reduced in rodent models of hypertrophy and in biopsy samples from patients with pressure-overload induced by chronic valvular aortic stenosis. Overexpression of KLF15 in neonatal rat ventricular cardiomyocytes inhibits cell size, protein synthesis and hypertrophic gene expression. KLF15-null mice are viable but, in response to pressure overload, develop an eccentric form of cardiac hypertrophy characterized by increased heart weight, exaggerated expression of hypertrophic genes, left ventricular cavity dilatation with increased myocyte size, and reduced left ventricular systolic function. Mechanistically, a combination of promoter analyses and gel-shift studies suggest that KLF15 can inhibit GATA4 and myocyte enhancer factor 2 function. These studies identify KLF15 as part of a heretofore unrecognized pathway regulating the cardiac response to hemodynamic stress.
URLPMID:2726266 [本文引用: 1]
Acetyltransferase p300 is essential for cardiac development and is thought to be involved in cardiac myocyte growth through MEF2- and GATA4-dependent transcription. However, the importance of p300 in the modulation of cardiac growth in vivo is unknown.Pressure overload induced by transverse aortic coarctation, postnatal physiological growth, and human heart failure were associated with large increases in p300. Minimal transgenic overexpression of p300 (1.5- to 3.5-fold) induced striking myocyte and cardiac hypertrophy. Both mortality and cardiac mass were directly related to p300 protein dosage. Heterozygous loss of a single p300 allele reduced pressure overload-induced hypertrophy by approximately 50% and rescued the hypertrophic phenotype of p300 overexpressers. Increased p300 expression had no effect on total histone deacetylase activity but was associated with proportional increases in p300 acetyltransferase activity and acetylation of the p300 substrates histone 3 and GATA-4. Remarkably, a doubling of p300 levels was associated with the de novo acetylation of MEF2. Consistent with this, genes specifically upregulated in p300 transgenic hearts were highly enriched for MEF2 binding sites.Small increments in p300 are necessary and sufficient to drive myocardial hypertrophy, possibly through acetylation of MEF2 and upstream of signals promoting phosphorylation or nuclear export of histone deacetylases. We propose that induction of myocardial p300 content is a primary rate-limiting event in the response to hemodynamic loading in vivo and that p300 availability drives and constrains adaptive myocardial growth. Specific reduction of p300 content or activity may diminish stress-induced hypertrophy and forestall the development of heart failure.
URLPMID:26686628 [本文引用: 1]
Zhang et02al. identify the transcription factor KLF15 as a regulator of the bimodal circadian gene expression landscape in the heart, with a distinct catabolic phase and a remodeling and repair phase. They further find that KLF15 promotes and represses oscillation in a different subset of genes.
[本文引用: 1]
URLPMID:3937660 [本文引用: 2]
The mammalian heart, the body's largest energy consumer, has evolved robust mechanisms to tightly couple fuel supply with energy demand across a wide range of physiologic and pathophysiologic states, yet, when compared with other organs, relatively little is known about the molecular machinery that directly governs metabolic plasticity in the heart. Although previous studies have defined Kruppel-like factor 15 (KLF15) as a transcriptional repressor of pathologic cardiac hypertrophy, a direct role for the KLF family in cardiac metabolism has not been previously established. We show in human heart samples that KLF15 is induced after birth and reduced in heart failure, a myocardial expression pattern that parallels reliance on lipid oxidation. Isolated working heart studies and unbiased transcriptomic profiling in Klf15-deficient hearts demonstrate that KLF15 is an essential regulator of lipid flux and metabolic homeostasis in the adult myocardium. An important mechanism by which KLF15 regulates its direct transcriptional targets is via interaction with p300 and recruitment of this critical co-activator to promoters. This study establishes KLF15 as a key regulator of myocardial lipid utilization and is the first to implicate the KLF transcription factor family in cardiac metabolism.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:28066139 [本文引用: 7]
Regulation of vascular smooth muscle cell (VSMC) phenotype plays an essential role in many cardiovascular diseases. In the present study, we provide evidence that krüppel-like factor 8 (KLF8) is essential for tumor necrosis factor α (TNFα)-induced phenotypic conversion of VSMC obtained from thoracic aorta from 4-week-old SD rats. Stimulation of the contractile phenotype of VSMCs with TNFα significantly reduced the VSMC marker gene expression and KLF8. The gene expression of KLF8 was blocked by TNFα stimulation in an ERK-dependent manner. The promoter region of KLF8 contained putative Sp1, KLF4, and NFκB binding sites. Myocardin significantly enhanced the promoter activity of KLF4 and KLF8. The ectopic expression of KLF4 strongly enhanced the promoter activity of KLF8. Moreover, silencing of Akt1 significantly attenuated the promoter activity of KLF8; conversely, the overexpression of Akt1 significantly enhanced the promoter activity of KLF8. The promoter activity of SMA, SM22α, and KLF8 was significantly elevated in the contractile phenotype of VSMCs. The ectopic expression of KLF8 markedly enhanced the expression of SMA and SM22α concomitant with morphological changes. The overexpression of KLF8 stimulated the promoter activity of SMA. Stimulation of VSMCs with TNFα enhanced the expression of KLF5, and the promoter activity of KLF5 was markedly suppressed by KLF8 ectopic expression. Finally, the overexpression of KLF5 suppressed the promoter activity of SMA and SM22α, thereby reduced the contractility in response to the stimulation of angiotensin II. These results suggest that cross-regulation of KLF family of transcription factors plays an essential role in the VSMC phenotype.
URL [本文引用: 2]
URL [本文引用: 5]
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URLPMID:16102021 [本文引用: 4]
Summary. Structural remodeling of the heart and blood vessels is an important pathologic process in the development of many cardiovascular diseases. However, transcriptional regulation of altered gene expression during cardiovascular remodeling is not well understood. We previously isolated KLF5/basic transcription element-binding (BTEB)2, a Krüppel-like factor, as a transcription factor that binds the promoter of the embryonic smooth muscle myosin heavy chain gene (SMemb). KLF5 activates many genes inducible during cardiovascular remodeling, such as platelet-derived growth factor (PDGF)-A/B, Egr-1, plasminogen activator inhibitor-1 (PAI-1), inducible nitric oxide synthase (iNOS), and vascular endothelial growth factor (VEGF) receptors. KLF5 is abundantly expressed in embryonic smooth muscles and is down-regulated with vascular development, but reinduced in proliferative neointimal smooth muscles in response to vascular injury. In KLF5 gene-targeted mice, homozygotes die at an early embryonic stage whereas heterozygotes are apparently normal. However, in response to external stress, arteries of heterozygotes exhibit diminished levels of smooth muscle and adventitial cell activation. Furthermore, angiotensin II-induced cardiac hypertrophy and fibrosis are attenuated in heterozygotes. KLF5 activities are regulated by many transcriptional regulators and nuclear receptors, such as retinoic acid receptor- α (RAR α ), NF- κ B, PPAR γ , p300, and SET. Interestingly, RAR α agonist suppresses KLF5 and cardiovascular remodeling, whereas RAR α antagonist activates KLF5 and induces angiogenesis. These results indicate that KLF5 is an essential transcription factor in cardiovascular remodeling and a potential therapeutic target for cardiovascular disease.
[本文引用: 3]
[本文引用: 2]
[本文引用: 1]
URL [本文引用: 1]
URLPMID:18178156 [本文引用: 1]
We have shown that spontaneously hypertensive rat (SHR)-derived vascular smooth muscle cells (VSMCs) change to the synthetic phenotype and show increased expression of complement 3 (C3) and that C3 plays a role in the change to the synthetic phenotype. To determine the mechanisms underlying the effects of C3 on this phenotypic change, we examined the effects of C3a on transcription factors involved in VSMC phenotype and found that C3a increased the expression of Kr眉ppel-like zinc-finger transcription factor 5 (KLF5) mRNA. C3a increased KLF5 promoter activity in a concentration-dependent manner. Deletion analysis of the promoter region of the KLF5 gene revealed that the region between nucleotides-991 and -699 contains the transcriptional regulatory element stimulated by C3a. C3a induced extracellular signal-regulated kinase (ERK) phosphorylation, and C3a-increased KLF5 promoter activity was completely inhibited by the MEK inhibitor U0126. These findings suggest that C3 increases KLF5 promoter activity and gene expression via ERK signaling.
URL [本文引用: 3]
URL [本文引用: 1]
URLPMID:4410857 [本文引用: 1]
Alaiti MA, Orasanu G, Tugal D, Lu Y, Jain MK.
URLPMID:20508206 [本文引用: 2]
Abstract To determine the role of Kruppel-like factor (KLF) 15, a zinc finger transcriptional factor that is expressed in vascular smooth muscle cells (VSMCs) in vascular biology. VSMCs respond to mechanical injury via a tightly orchestrated series of gene regulatory events. KLF15 is broadly expressed in both arterial and venous vascular beds in a VSMC restricted fashion. KLF15 expression is markedly reduced by both pharmacological and mechanical stimuli. To examine the specific role of KLF15 in the vascular response to injury, we performed femoral artery wire injury in KLF15(-/-) and wild-type mice. KLF15(-/-) mice develop exaggerated neointimal growth, with evidence of increased SMC proliferation and migration within the neointima. In concordance, gain and loss of function studies in isolated VSMCs demonstrate that KLF15 can directly inhibit SMC proliferation and migration. To our knowledge, these data are the first to identify KLF15 as a novel inhibitor of VSMC proliferation and migration and to implicate this factor as a critical regulator of the vascular response to injury.
URLPMID:3785338 [本文引用: 2]
Activation of cells intrinsic to the vessel wall is central to the initiation and progression of vascular inflammation. As the dominant cellular constituent of the vessel wall, vascular smooth muscle cells (VSMCs) and their functions are critical determinants of vascular disease. While factors that regulate VSMC proliferation and migration have been identified, the endogenous regulators of VSMC proinflammatory activation remain incompletely defined. The Kruppel-like family of transcription factors (KLFs) are important regulators of inflammation. In this study, we identified Kruppel-like factor 15 (KLF15) as an essential regulator of VSMC proinflammatory activation. KLF15 levels were markedly reduced in human atherosclerotic tissues. Mice with systemic and smooth muscle-specific deficiency of KLF15 exhibited an aggressive inflammatory vasculopathy in two distinct models of vascular disease: orthotopic carotid artery transplantation and diet-induced atherosclerosis. We demonstrated that KLF15 alters the acetylation status and activity of the proinflammatory factor NF-kappa B through direct interaction with the histone acetyltransferase p300. These studies identify a previously unrecognized KLF15-dependent pathway that regulates VSMC proinflammatory activation.
URLPMID:21316587 [本文引用: 3]
78 The ERK5 MAP kinase pathway regulates muscle cell fusion 78 Klf2 and Klf4 function downstream of ERK5 as key regulators of muscle cell fusion 78 Sp1 transcription factor links ERK5 to Klf2/4 78 Nephronectin, a target of the ERK5/Klf module, is involved in muscle cell fusion
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 2]
URLPMID:25036958 [本文引用: 1]
Studies in mammalian species showed that Krüppel-like factor 3 (KLF3) regulated adipose tissue development. However, it was not reported in chicken. In the current study, we found that during the growth and development of abdominal fat tissue, chicken KLF3 (Gallus gallus KLF3, gKLF3) was consecutively expressed, and its transcripts were higher at 765weeks of age and lower at 1065weeks of age in lean broilers than in fat broilers. In addition, gKLF3 overexpression suppressed chicken CCAAT/enhancer binding protein alpha (C/EBPα), fatty acid binding protein 4 (FABP4), fatty acid synthase (FASN), and lipoprotein lipase (LPL) promoter activities, but increased chicken peroxisome proliferator-activated receptor gamma (PPARγ) promoter activity. Additionally, point mutagenesis analysis showed that the substitution of Asp by Gly within the Pro-Val-Asp-Leu-Thr (PVDLT) motif of gKLF3 significantly reduced the ability of gKLF3 to regulate the promoter activities of FABP4, FASN, LPL, C/EBPα, and PPARγ. Effects of Overexpression of KLF3 and Its Mutant on the Promoter Activities of Chicken FABP4, FASN, LPL, C/EBPα, and PPARγ in DF1 Cells.
[本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 3]
[本文引用: 1]
URLPMID:24680826 [本文引用: 2]
A comprehensive understanding of genetic and environmental factors that control skeletal muscle fiber type specification and transformation is essential not only in sports science, but also in myopathy and metabolic disorders. Kr ppel-like factors (KLFs) are a subfamily of the zinc-finger class of transcription factors, which are involved in the development, homeostasis, and pathology of cardiovascular systems. Compared to cardiac and smooth muscles, the role of KLFs in skeletal muscle is much less understood. In this study, the endogenous expression of KLF15 was analyzed in differentiating C2C12 muscle cells and mouse skeletal muscle. Our data indicated that Klf15 was upregulated during myogenic differentiation and higher levels of Klf15 mRNA were detected in mouse slow, oxidative soleus muscle (SL) compared to that in fast, glycolytic tibialis anterior muscle (TA), indicating that KLF15 may play a role in myogenesis or myofiber typing. Additional studies revealed that KLF15 regulated the expression of MHC-/slow rather than muscle cell differentiation. Gene silencing, overexpression, and luciferase reporter assay showed that KLF15 regulated MHC-/slow by binding to Nfatc1 promoter, inducing its activity, therefore mediating calcineurin/NFAT signaling. Our study contributed to the current knowledge on KLFs in skeletal muscle, and it indicated a need for further intensive studies on the redundant and divergent functions of KLFs.
URLPMID:22493257 [本文引用: 2]
The ability of skeletal muscle to enhance lipid utilization during exercise is a form of metabolic plasticity essential for survival. Conversely, metabolic inflexibility in muscle can cause organ dysfunction and disease. Although the transcription factor Kruppel-like factor 15 (KLF15) is an important regulator of glucose and amino acid metabolism, its endogenous role in lipid homeostasis and muscle physiology is unknown. Here we demonstrate that KLF15 is essential for skeletal muscle lipid utilization and physiologic performance. KLF15 directly regulates a broad transcriptional program spanning all major segments of the lipid-flux pathway in muscle. Consequently, Klf15-deficient mice have abnormal lipid and energy flux, excessive reliance on carbohydrate fuels, exaggerated muscle fatigue, and impaired endurance exercise capacity. Elucidation of this heretofore unrecognized role for KLF15 now implicates this factor as a central component of the transcriptional circuitry that coordinates physiologic flux of all three basic cellular nutrients: glucose, amino acids, and lipids.
URLPMID:21284984 [本文引用: 2]
Maintenance of skeletal muscle mass relies on the dynamic balance between anabolic and catabolic processes and is important for motility, systemic energy homeostasis, and viability. We identified direct target genes of the glucocorticoid receptor (GR) in skeletal muscle, i.e., REDD1 and KLF15. As well as REDD1, KLF15 inhibits mTOR activity, but via a distinct mechanism involving BCAT2 gene activation. Moreover, KLF15 upregulates the expression of the E3 ubiquitin ligases atrogin-1 and MuRF1 genes and negatively modulates myofiber size. Thus, GR is a liaison involving a variety of downstream molecular cascades toward muscle atrophy. Notably, mTOR activation inhibits GR transcription function and efficiently counteracts the catabolic processes provoked by glucocorticoids. This mutually exclusive crosstalk between GR and mTOR, a highly coordinated interaction between the catabolic hormone signal and the anabolic machinery, may be a rational mechanism for fine-tuning of muscle volume and a potential therapeutic target for muscle wasting.Graphical AbstractView high quality image (416K)
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}