 ,西北农林科技大学生命科学学院,杨凌 712100
,西北农林科技大学生命科学学院,杨凌 712100Comparative analysis of gene family size provides insight into the adaptive evolution of vertebrates
Yu Meng, Ruolin Yang,College of Life Sciences, Northwest A&F University, Yangling 712100, China通讯作者:
编委: 于黎
收稿日期:2018-08-6修回日期:2018-12-13网络出版日期:2019-02-25
Editorial board:
Received:2018-08-6Revised:2018-12-13Online:2019-02-25
作者简介 About authors
孟玉,硕士研究生,专业方向:遗传学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1242KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孟玉, 杨若林. 基于基因家族大小的比较研究脊椎动物的适应性进化[J]. 遗传, 2019, 41(2): 158-174 doi:10.16288/j.yczz.18-225
Yu Meng, Ruolin Yang.
脊椎动物亚门是脊索动物门中物种数量最多、结构最复杂的一个亚门,大约在5~6亿年前从其他脊索动物(头索动物和尾索动物)中分歧出来[1,2],并演化出无颌类、鱼类、两栖类、爬行类、鸟类和哺乳类,经历了成功的演化革新和适应。鉴定出脊椎动物间表型差异背后潜在的遗传变化,并确定导致这种变化的进化动力虽然具有挑战性,但有着深刻的科学意义。
基因复制是新基因产生及基因家族扩增的主要机制之一[3],为生物体表型的创新及多样化等提供了遗传基础[4],并且与生物体基因组大小的进化和物种分化等紧密相关[5]。与基因复制相比,基因丢失曾被认为仅与冗余的基因拷贝的丢失有关,而不会产生明显的功能影响,因此常被忽视。然而,与日俱增的基因组学数据揭示了基因丢失作为遗传变异的普遍来源,其具有引起适应性表型多样性的巨大潜能,是一种非常重要的进化动力[6]。如虎尾海马(Hippocampus comes)基因组中的基因扩增和丢失与其特殊形态的演化密切相关。Lin等[7]对虎尾海马基因组进行了测序与分析,发现该物种基因组中Pastn (patristacin)基因家族(一种虾红素金属蛋白酶基因家族)经历了扩增,这与海马雄性孕育这一独特的繁殖方式密切相关。此外,虎尾海马基因组中P/Q-rich SCPP (proline/glutamine-rich secretory calcium- binding phosphoprotein)基因和tbx4基因的丢失分别是导致其没有牙齿和腹鳍的重要原因。
通过全基因组比较分析,已经揭示了不同物种间许多基因家族拷贝数发生了显著的数量变化[8,9,10,11],这种变化与基因得失速率息息相关,且受到自然选择与遗传漂变的共同作用[6,12,13]。物种间表型的差异与基因家族大小的差异关系密切,如抗冻糖蛋白(antifreeze glycoprotein, AFGP)基因在南极鱼亚目鱼类基因组中发生了大量扩增,在南极鱼类适应低温环境中发挥了非常重要的作用[14]。除AFGP基因外,铁调素、卵壳蛋白等100多个参与低温适应相关生物学途径的基因也在南极鱼类进化中发生了显著扩增[15],这体现出特定基因拷贝数的增加是南极鱼类适应持续寒冷环境的一种机制。研究表明,不同种系间基因家族大小的变化可能与物种形成或适应性有重要联系[16,17,18]。例如,Yu等[18]对非人灵长类高海拔适应机制的研究中发现,与恒河猴(Macaca mulatta)相比,生活在海拔高度为3500~4500米的滇金丝猴(Rhinopithecus bieti)基因组中有1187个基因家族发生了扩增,对其中231个显著扩增基因家族进行的功能富集分析表明,这些基因主要参与DNA修复和损伤应答以及氧化磷酸化过程。这一结果被认为可能与滇金丝猴暴露于高的紫外线辐射以及高海拔生存所需的能量代谢速率的增加有关。
植物和动物中基因家族大小的进化模式均已被广泛研究。然而,许多研究往往只涉及少数物种或只关注一个或某些基因家族的进化[19,20,21,22],缺乏全基因组水平的大规模分析。近年来,随着测序技术的发展,超过100种脊椎动物的全基因组已经被测序完成[23],这些数据的获得为人们揭示以下生物学问题提供了契机:(1)在脊椎动物中,物种间大规模的基因组差异,如基因家族大小的显著变化是否在物种的适应性进化中起到了重要的作用;(2)物种或种系特异性基因的特征(包括表达模式和功能等)能否在一定程度上反映出物种间表型的差异。为了回答上述问题,本文选取64个涵盖了脊椎动物几乎所有类群(无颌类、鱼类、两栖类、爬行类、鸟类和哺乳类)的物种作为研究对象,从大的进化时间跨度上揭示了脊椎动物基因家族的扩增、收缩模式;并结合表达数据和功能注释评估了物种或种系特异性基因对物种特有表型的影响。本研究为深入了解脊椎动物基因家族大小的进化、理解脊椎动物间的基因组差异和表型多样性提供了新的见解。
1 材料与方法
1.1 数据来源
64个脊椎动物物种及2个外群物种——玻璃海鞘(Ciona intestinalis)和萨氏海鞘(Ciona savignyi)完整的蛋白质组数据均下载自Ensembl v.84数据库。64个脊椎动物物种包含了1种无颌纲物种、12种鱼类、1种两栖动物、2种爬行动物、5种鸟类及43种哺乳动物。其中哺乳动物包括1种单孔目、3种有袋目、2种贫齿目、3种非洲兽总目、14种劳亚兽总目、2种兔形目、5种啮齿目、1种树鼩目和12种灵长目(表1)。从Ensembl网站(1.2 物种间直系同源基因的鉴定
为了获得高质量的蛋白质序列数据用以鉴定基因的同源关系,对上述66个物种的蛋白质组数据按以下两个条件进行过滤:(1)去除长度小于50个氨基酸的蛋白质;(2)对于由可变剪切产生的多个转录本所翻译的蛋白质,只保留每个基因最长转录本对应的蛋白质。过滤之后,66个物种共1 149 492条蛋白质序列作为输入数据提交至OrthoMCL v2.0.9[24]进行蛋白聚类。该软件运行中的两个关键步骤是:(1) All-against-all BLASTP,即使用BlastP v2.2.31将每个蛋白与所有其他蛋白进行比对(E-value < 1× 10-6),产生原始的blast输出;(2)使用马尔科夫聚类算法(Markov cluster algorithm, MCL)对解析的Blast结果构建马尔科夫矩阵,然后产生最终的基因家族[25]。MCL聚类的重要参数膨胀系数设为1.5。1.3 基因家族大小分析
将每个物种的所有基因家族按其拷贝数分为3类:(1)单拷贝基因家族,每个家族包含的基因数目为1,即通常所说的单拷贝基因;(2)包含两个拷贝的基因家族,即双拷贝基因家族;(3)包含3个及3个以上拷贝的多拷贝基因家族。本研究中每一个物种的Orphan基因家族(或基因)都是与其他65个物种进行比较得到的。例如,以人(Homo sapiens)为例,当某基因在除人以外的所有其他65个物种中都没有与之对应的同源基因时,就说明该基因是人的orphan基因。
1.4 基因得失的似然法分析
CAFE (computational analysis of gene family evolution, version 3.0)是研究基因家族大小进化的统计分析工具,使用生灭模型对基因家族大小在特定系统发生树上的进化过程进行建模,并确定出各个分支上基因家族的扩增和收缩模式[26]。由于上述66个物种基于分子水平的系统发生树与取自TimeTree[27]标有分歧时间的系统发生树不完全一致,为了便于分析和保证数据的可靠性,本研究从中选取57个脊椎动物物种进行后续分析。输入CAFE软件的树文件,为所选57个物种的Newick格式的有根系统发育树,且分支长度代表物种的分歧时间。数据文件是相应这些物种的各个基因家族大小的数据。使用的软件参数为:-p 0.05 -r 1000 -filter。最后通过lambda -s估算出所有基因家族总体的生灭参数λ。对于进化速率显著高于(P<0.0001)全基因组平均值的基因家族[28],该软件使用Viterbi法识别出相应的分支,即基因家族大小发生显著变化(P< 0.005)的分支[29]。
λ是基因家族大小进化分析中的一个重要参数,被用来度量单位时间(每百万年)内每个基因的得失概率。本研究中λ的估计值为0.0006,代表了所有基因家族整体水平的最有可能的生灭速率,或者说是基因家族随时间推移而扩增或收缩的速率。举例来说,所评估的基因得失速率意味着在特定基因组(如人类基因组)中,每百万年大约有13.467个新的拷贝和13.467个新的丢失被固定(0.0006得失/基因/百万年× 22 445基因)。
1.5 基因表达和GO (Gene Ontology)注释
从Expression Atlas数据库分别下载了人的16种组织(肝脏、淋巴结、甲状腺、骨骼肌、前列腺、大脑、睾丸、肾脏、肾上腺、肺脏、白细胞、卵巢、脂肪、乳腺、结肠和心脏)、鸡(Gallus gallus)的9种组织(大脑、心脏、肝脏、脾脏、肺脏、肾脏、结肠、睾丸和骨骼肌)的基因表达数据。斑马鱼的12种组织(骨、大脑、胚胎、卵巢、心脏、肠、肾脏、肝脏、肌肉、成熟卵泡、鳃和睾丸)的基因表达数据下载自Bgee数据库。基因表达的组织特异性参照文献[30,31]中描述的组织特异性指数τ来表示,计算公式如下:\[\tau =\frac{\sum\limits_{i=1}^{n}{(1-{{S}_{i}}/{{S}_{max}})}}{n-1}\]
其中,n是组织的数量,Si是基因在第i个组织中的表达量,Smax代表基因在所有组织中的最大表达量。本研究将τ≥0.85的基因视为组织特异性表达的基因,并关注这类基因最大表达值对应的组织;使用GOSlim对感兴趣的基因集进行功能富集分析。
2 结果与分析
2.1 基因家族大小的跨物种分布模式
为了鉴定脊椎动物间的直系同源基因,使用OrthoMCL[24]对涵盖了无颌类、鱼类、两栖类、爬行类、鸟类、哺乳类的64个脊椎动物物种和2个海鞘纲尾索动物物种(表1,图1A)共1 149 492个蛋白质序列进行了聚类分析,共产生32 498个直系同源基因家族。其中1648个基因家族是所有64个脊椎动物物种所共有,这可能代表了脊椎动物“核心”蛋白质组。Table 1
表1
表1 66个物种基因家族及成员基因数量
Table 1
| 物种名称 | 单拷贝 基因家族 | 双拷贝 基因家族 | 多拷贝 基因家族 | 基因 家族总数 | 最大基因 家族的大小 |
|---|---|---|---|---|---|
| 萨氏海鞘(Ciona savignyi) | 9396 | 552 (1104) | 223 (949) | 10 171 (11 449) | 45 |
| 玻璃海鞘(Ciona intestinalis) | 12 962 | 825 (1650) | 288 (1173) | 14 075 (15 785) | 20 |
| 海七鳃鳗(Petromyzon marinus) | 7017 | 902 (1804) | 291 (1319) | 8210 (10 140) | 52 |
| 眼斑雀鳝(Lepisosteus oculatus) | 12 458 | 1390 (2780) | 713 (3088) | 14 561 (18 326) | 50 |
| 墨西哥丽脂鲤(Astyanax mexicanus) | 13 454 | 2269 (4538) | 1139 (5032) | 16 862 (23 024) | 82 |
| 斑马鱼(Danio rerio) | 12 204 | 2311 (4622) | 1414 (8745) | 15 929 (25 571) | 601 |
| 大西洋鳕鱼(Gadus morhua) | 12 261 | 1760 (3520) | 938 (4206) | 14 959 (19 987) | 79 |
| 红鳍东方鲀(Takifugu rubripes) | 10 221 | 1845 (3690) | 1053 (4560) | 13 119 (18 471) | 23 |
| 绿河鲀(Tetraodon nigroviridis) | 11 306 | 1948 (3896) | 1021 (4354) | 14 275 (19 556) | 20 |
| 尼罗罗非鱼(Oreochromis niloticus) | 11 069 | 2085 (4170) | 1253 (6192) | 14 407 (21 431) | 164 |
| 三刺鱼(Gasterosteus aculeatus) | 11 897 | 1913 (3826) | 1058 (5046) | 14 868 (20 769) | 152 |
| 青鱂(Oryzias latipes) | 11 453 | 1823 (3646) | 993 (4480) | 14 269 (19 579) | 45 |
| 月光鱼(Xiphophorus maculatus) | 12 020 | 1976 (3952) | 1034 (4384) | 15 030 (20 356) | 18 |
| 花帆鱂(Poecilia formosa) | 12 233 | 2458 (4916) | 1403 (6458) | 16 094 (23 607) | 95 |
| 腔棘鱼(Latimeria chalumnae) | 11 917 | 1491 (2982) | 855 (4650) | 14 263 (19 549) | 147 |
| 热带爪蟾(Xenopus tropicalis) | 10 810 | 1277 (2554) | 782 (5069) | 12 869 (18 433) | 174 |
| 绿安乐蜥(Anolis carolinensis) | 11 653 | 1336 (2672) | 717 (4125) | 13 706 (18 450) | 185 |
| 中华鳖(Pelodiscus sinensis) | 11 692 | 1254 (2508) | 656 (3948) | 13 602 (18 148) | 508 |
| 白领姬鹟(Ficedula albicollis) | 11 123 | 1130 (2260) | 446 (1710) | 12 699 (15 093) | 20 |
| 斑胸草雀(Taeniopygia guttata) | 11 461 | 1570 (3140) | 543 (2695) | 13 574 (17 296) | 151 |
| 绿头鸭(Anas platyrhynchos) | 11 050 | 1213 (2426) | 443 (1684) | 12 706 (15 160) | 12 |
| 鸡(Gallus gallus) | 11 108 | 1125 (2250) | 496 (2115) | 12 729 (15 473) | 111 |
| 火鸡(Meleagris gallopavo) | 10 126 | 1112 (2224) | 448 (1695) | 11 686 (14 045) | 11 |
| 鸭嘴兽(Ornithorhynchus anatinus) | 14 068 | 1787 (3574) | 788 (3896) | 16 643 (21 538) | 470 |
| 家短尾负鼠(Monodelphis domestica) | 13 268 | 1470 (2940) | 851 (5017) | 15 589 (21 225) | 482 |
| 袋獾(Sarcophilus harrisii) | 12 961 | 1384 (2768) | 700 (3037) | 15 045 (18 766) | 80 |
| 尤金袋鼠(Macropus eugenii) | 11 281 | 1038 (2076) | 488 (1882) | 12 807 (15 239) | 14 |
| 九带犰狳(Dasypus novemcinctus) | 14 393 | 1650 (3300) | 960 (4962) | 17 003 (22 655) | 99 |
| 霍氏树懒(Choloepus hoffmanni) | 9570 | 871 (1742) | 256 (977) | 10 697 (12 289) | 18 |
| 小马岛猬(Echinops telfairi) | 12 173 | 1164 (2328) | 482 (1976) | 13 819 (16 477) | 36 |
| 非洲草原象(Loxodonta africana) | 12 959 | 1384 (2768) | 863 (4245) | 15 206 (19 972) | 66 |
| 非洲蹄兔(Procavia capensis) | 12 105 | 1028 (2056) | 474 (1840) | 13 607 (16 001) | 27 |
| 刺猬(Erinaceus europaeus) | 10 914 | 1058 (2116) | 385 (1483) | 12 357 (14 513) | 13 |
| 鼩鼱(Sorex araneus) | 9772 | 901 (1802) | 341 (1528) | 11 014 (13 102) | 119 |
| 野猪(Sus scrofa) | 12 841 | 2270 (4540) | 990 (4154) | 16 101 (21 535) | 24 |
| 羊驼(Vicugna pacos) | 9370 | 744 (1488) | 223 (847) | 10 337 (11 705) | 16 |
| 宽吻海豚(Tursiops truncates) | 12 387 | 1052 (2104) | 514 (1990) | 13 953 (16 481) | 20 |
| 物种名称 | 单拷贝 基因家族 | 双拷贝 基因家族 | 多拷贝 基因家族 | 基因 家族总数 | 最大基因 家族的大小 |
| 绵羊(Ovis aries) | 14 376 | 1498 (2996) | 801 (3419) | 16 675 (20 791) | 27 |
| 牛(Bos taurus) | 13 565 | 1400 (2800) | 829 (3590) | 15 794 (19 955) | 32 |
| 小棕蝠(Myotis lucifugus) | 12 393 | 1570 (3140) | 863 (4103) | 14 826 (19 636) | 65 |
| 大狐蝠(Pteropus vampyrus) | 12 761 | 1050 (2100) | 523 (2065) | 14 334 (16 926) | 18 |
| 马(Equus caballus) | 13 080 | 1334 (2668) | 813 (4628) | 15 227 (20 376) | 514 |
| 家猫(Felis catus) | 13 922 | 1320 (2640) | 704 (2889) | 15 946 (19 451) | 40 |
| 狗(Canis lupus familiaris) | 14 076 | 1365 (2730) | 727 (3013) | 16 168 (19 819) | 45 |
| 大熊猫(Ailuropoda melanoleuca) | 13 853 | 1308 (2616) | 695 (2781) | 15 856 (19 250) | 22 |
| 雪貂(Mustela putorius furo) | 14 474 | 1306 (2612) | 690 (2794) | 16 470 (19 880) | 21 |
| 北美鼠兔(Ochotona princeps) | 11 811 | 1120 (2240) | 459 (1857) | 13 390 (15 908) | 66 |
| 穴兔(Oryctolagus cuniculus) | 12 584 | 1431 (2862) | 808 (3793) | 14 823 (19 239) | 56 |
| 豚鼠(Cavia porcellus) | 12 813 | 1365 (2730) | 724 (3021) | 14 902 (18 564) | 37 |
| 斑纹地松鼠(Ictidomys tridecemlineatus) | 12 836 | 1389 (2778) | 755 (3158) | 14 980 (18 772) | 25 |
| 奥氏更格卢鼠(Dipodomys ordii) | 11 789 | 1001 (2002) | 497 (1945) | 13 287 (15 736) | 24 |
| 褐家鼠(Rattus norvegicus) | 13 739 | 1762 (3524) | 1015 (4966) | 16 516 (22 229) | 78 |
| 小鼠(Mus musculus) | 13 837 | 1523 (3046) | 1016 (5627) | 16 376 (22 510) | 122 |
| 树鼩(Tupaia belangeri) | 11 437 | 1037 (2074) | 423 (1855) | 12 897 (15 366) | 78 |
| 小耳大婴猴(Otolemur garnettii) | 13 278 | 1480 (2960) | 774 (3210) | 15 532 (19 448) | 48 |
| 倭狐猴(Microcebus marinus) | 12 209 | 1050 (2100) | 496 (1902) | 13 755 (16 211) | 16 |
| 菲律宾眼镜猴(Tarsius syrichta) | 10 438 | 872 (1744) | 337 (1360) | 11 647 (13 542) | 22 |
| 狨猴(Callithrix jacchus) | 14 436 | 1567 (3134) | 761 (3255) | 16 764 (20 825) | 44 |
| 绿猴(Chlorocebus sabaeus) | 13 710 | 1299 (2598) | 664 (2781) | 15 673 (19 089) | 41 |
| 恒河猴(Macaca mulatta) | 14 794 | 1633 (3266) | 831 (3626) | 17 258 (21 686) | 44 |
| 东非狒狒(Papio anubis) | 13 570 | 1316 (2632) | 698 (2940) | 15 584 (19 142) | 38 |
| 白颊长臂猿(Nomascus leucogenys) | 13 543 | 1272 (2544) | 588 (2452) | 15 403 (18 539) | 41 |
| 苏门答腊猩猩(Pongo abelii) | 14 409 | 1429 (2858) | 671 (2773) | 16 509 (20 040) | 34 |
| 西非低地大猩猩(Gorllia gorilla gorilla) | 14 595 | 1512 (3024) | 713 (2929) | 16 820 (20 548) | 27 |
| 黑猩猩(Pan troglodytes) | 13 502 | 1277 (2554) | 615 (2560) | 15 394 (18 616) | 42 |
| 人(Homo sapiens) | 13 037 | 1799 (3598) | 1077 (5810) | 15 913 (22 445) | 200 |
新窗口打开|下载CSV
本研究首先对每个物种基因组中3类基因家族及其成员基因的数量进行了统计。在所研究的物种中,基因家族总数从8210 (海七鳃鳗,Petromyzon marinus)至17 258 (恒河猴)不等(表1)。每个物种最大的基因家族由11 (火鸡,Meleagris gallopavo)至601 (斑马鱼)个基因组成(表1),这显示基因家族大小有着较大的跨物种变异程度。
进一步统计显示,除斑马鱼外,脊椎动物各物种基因组中半数以上的基因都以单拷贝的形式存在(图1B)。与双拷贝基因相比,单拷贝和多拷贝基因在各物种基因组中所占比例有更大差异。具体而言,双拷贝基因家族中的基因数占各物种总基因数的比例从12.4% (大狐蝠)至21.1% (野猪)不等,斑马鱼基因组中有最多的多拷贝基因(34.2%)和最少的单拷贝基因(47.7%),而羊驼基因组中有最少的多拷贝基因(7.2%)和最多的单拷贝基因(80.1%) (图1B)。
图1
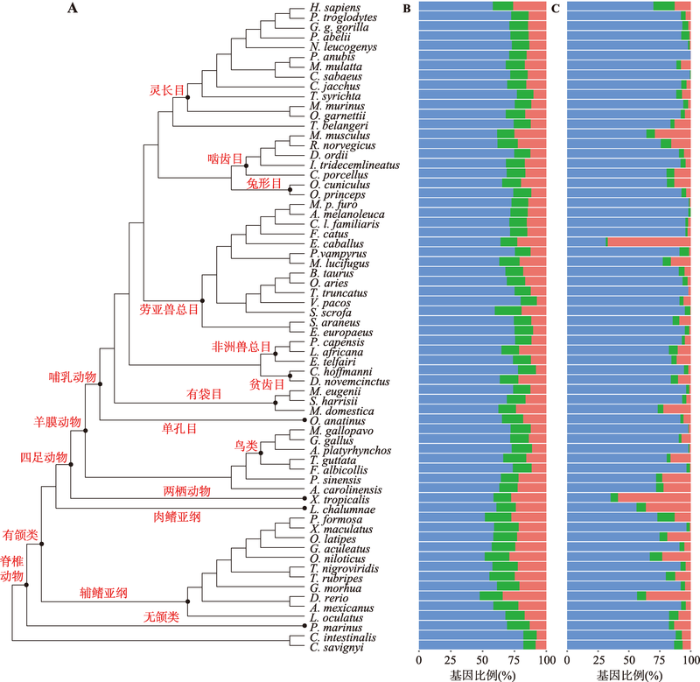 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1脊椎动物系统发生关系及基因家族大小分布
A:66个物种的系统发育树(数据来自Ensembl v.84 数据库,图中黑色节点及相应的红色文字表示物种分类);B:各物种全基因组水平的基因家族大小分布;C:各物种Orphan基因家族大小分布(条形图中的蓝、绿、红分别表示单拷贝、双拷贝及多拷贝基因)。
Fig. 1Phylogeny and gene family size distribution of vertebrates
2.2 基因家族的扩增与收缩
基因得失的似然法分析中,需要假定所分析的基因家族在所有物种最近共同祖先中至少含有一个基因。在57个脊椎动物物种包含的28 084个基因家族中,只有6857个基因家族符合这一要求,因此本研究只对这些基因家族的扩增与收缩模式进行分析(图2)。脊椎动物最近共同祖先处6857个基因家族中有6712个都在至少一个种系中发生了扩增或收缩。在57个脊椎动物物种组成的系统发育树的不同分支上基因家族扩增和收缩的模式来看,脊椎动物基因家族在大部分种系中都是收缩的,其中霍氏树懒中有最大程度的收缩(发生扩增和收缩的基因家族分别有74个和2151个),而斑马鱼中有最大程度的扩增(发生扩增和收缩的基因家族分别有912个和343个) (图2)。在鸟类中,除了斑胸草雀这一末端分支上发生了相对多的基因家族扩增以外,其他鸟类的基因家族均发生了较大收缩,这与鸟类基因组进化过程中整体的基因组变小现象一致[32]。已知鸟类基因组是羊膜动物中最小的,研究表明广泛的基因丢失比转座子活性降低对维持鸟类较小的基因组有更重要的贡献[33]。在辐鳍鱼中,真骨附类进化早期有大量的基因家族发生扩增,随后又有较多的基因家族呈现出收缩的模式(图2),这与真骨附类祖先物种发生了特有的全基因组复制以及复制后往往伴随着大量的基因丢失现象基本吻合[34,35]。
图2
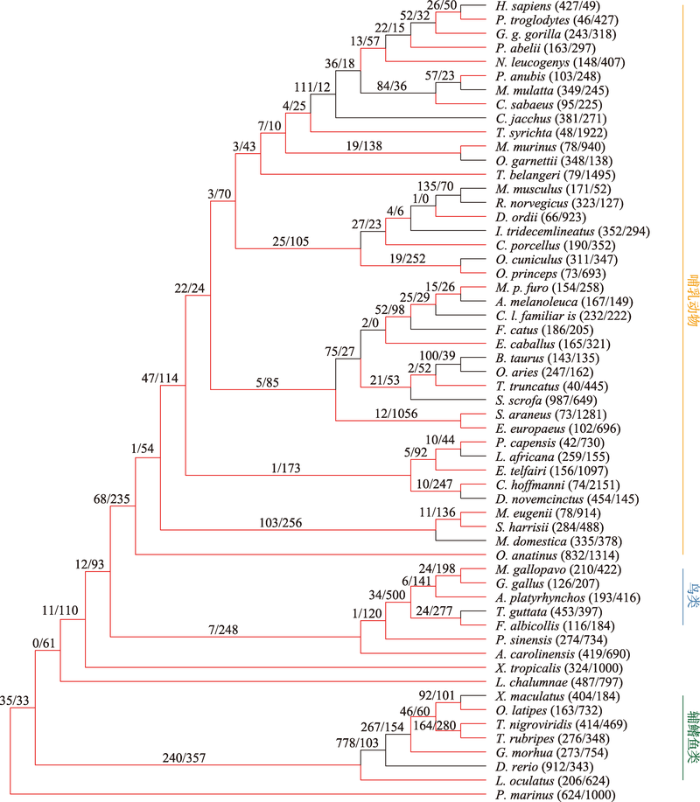 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2脊椎动物中基因家族的扩增和收缩
Fig. 2Expansions and contractions of gene families in vertebrates
分支上“/”线左右两侧的数字分别表示该分支上发生扩增及收缩的基因家族的数量;物种名称之后的数字表示相应物种基因组中发生扩增及收缩的基因家族的数量。黑色和红色分支分别表示从整体来说基因家族在特定分支上是扩增或收缩的。右侧橘色、蓝色和绿色的竖线分别标注了哺乳动物、鸟类及辐鳍鱼类在系统发育树中的位置。
似然法分析能够识别基因家族大小的进化速率显著高于全基因组平均值的基因家族[28]。在所分析的6857个基因家族中,有148个是快速进化的基因家族(FDR<0.01%),其中22个快速进化的基因家族在人这一末端分支上发生了显著扩增。例如,CT抗原中CTAGE (cutaneous T-cell-lymphoma-associated antigen)基因家族是一类由生殖细胞系基因编码的肿瘤/睾丸抗原,在人类的很多肿瘤中CT抗原会异常表达[36]。本研究的数据显示,该基因家族在人类基因组中有10个拷贝,而在黑猩猩中的拷贝数为2,用CAFE软件所推断的人与黑猩猩最近共同祖先中的该基因家族拷贝数为2。之前有研究发现CTAGE基因家族在灵长类的进化中发生了快速的扩增,人类基因组中的CTAGE基因家族包含了多个单外显子基因拷贝,这些单外显子拷贝基因受到明显的正选择作用,有可能对人类早期进化中适应性表型的产生有贡献[37]。
2.3 Orphan基因家族的大小分布、特征及起源进化
2.3.1 Orphan基因家族大小的跨物种分布模式特定物种基因组中的orphan基因指的是在其他物种基因组中找不到其同源基因的一类基因[38],它们被认为与相应物种具有的特异的发育模式,适应特定的环境紧密相关[39]。本研究统计了上述66个物种基因组中各物种特异的基因家族及其成员基因的数量。结果表明,Orphan基因家族的数目和成员基因总数在这些物种中变异很大,如宽吻海豚基因组中仅有223个Orphan基因家族和相应的226个基因;而玻璃海鞘则具有最多的4956个Orphan基因家族,共包含5383个成员基因。各物种基因组中orphan
基因所占比例从1.4% (宽吻海豚)到19.4% (鸭嘴兽)不等(表1,表2)。
Table 2
表2
表2 物种特异性Orphan基因家族及成员基因的数量
Table 2
| 物种名称 | 单拷贝 基因家族 | 双拷贝 基因家族 | 多拷贝 基因家族 | Orphan基因 家族总数 | 最大Orphan 基因家族的大小 |
|---|---|---|---|---|---|
| 萨氏海鞘(C. savignyi) | 1937 | 76 (152) | 42 (145) | 2055 (2234) | 6 |
| 玻璃海鞘(C. intestinalis) | 4733 | 136 (272) | 87 (378) | 4956 (5383) | 20 |
| 海七鳃鳗(P. marinus) | 1243 | 32 (64) | 34 (201) | 1309 (1508) | 22 |
| 眼斑雀鳝(L. oculatus) | 892 | 41 (82) | 20 (107) | 953 (1081) | 17 |
| 墨西哥丽脂鲤(A. mexicanus) | 2502 | 50 (100) | 26 (106) | 2578 (2708) | 9 |
| 斑马鱼(D. rerio) | 1343 | 86 (172) | 95 (853) | 1524 (2368) | 67 |
| 大西洋鳕鱼(G. morhua) | 1443 | 27 (54) | 16 (70) | 1486 (1567) | 10 |
| 红鳍东方鲀(T. rubripes) | 423 | 20 (40) | 11 (66) | 454 (529) | 14 |
| 绿河鲀(T. nigroviridis) | 1366 | 31 (62) | 14 (59) | 1411 (1487) | 7 |
| 尼罗罗非鱼(O. niloticus) | 599 | 45 (90) | 25 (206) | 669 (895) | 71 |
| 三刺鱼(G. aculeatus) | 940 | 20 (40) | 11 (52) | 971 (1032) | 12 |
| 青鱂(O. latipes) | 1218 | 51 (102) | 49 (308) | 1318 (1628) | 23 |
| 月光鱼(X. maculatus) | 431 | 6 (12) | 1 (3) | 438 (446) | 3 |
| 美帆鱂(P. formosa) | 693 | 66 (132) | 22 (122) | 781 (947) | 30 |
| 腔棘鱼(L. chalumnae) | 866 | 58 (116) | 69 (557) | 993 (1539) | 77 |
| 热带爪蟾(X. tropicalis) | 632 | 53 (106) | 95 (1052) | 780 (1790) | 141 |
| 绿安乐蜥(A. carolinensis) | 943 | 39 (78) | 36 (286) | 1018 (1307) | 45 |
| 中华鳖(P. sinensis) | 884 | 31 (62) | 31 (281) | 946 (1227) | 45 |
| 白领姬鹟(F. albicollis) | 501 | 7 (14) | 1 (3) | 509 (518) | 3 |
| 斑胸草雀(T. guttata) | 1660 | 31 (62) | 26 (334) | 1717 (2056) | 52 |
| 绿头鸭(A. platyrhynchos) | 695 | 3 (6) | 2 (7) | 700 (708) | 4 |
| 鸡(G. gallus) | 759 | 9 (18) | 13 (62) | 781 (839) | 11 |
| 火鸡(M. gallopavo) | 397 | 1 (2) | 2 (6) | 400 (405) | 3 |
| 鸭嘴兽(O. anatinus) | 3834 | 52 (104) | 40 (241) | 3926 (4179) | 22 |
| 家短尾负鼠(M. domestica) | 1428 | 43 (86) | 37 (430) | 1508 (1944) | 236 |
| 袋獾(S. harrisii) | 1294 | 24 (48) | 11 (47) | 1329 (1389) | 9 |
| 尤金袋鼠(M. eugenii) | 709 | 9 (18) | 2 (8) | 720 (735) | 5 |
| 九带犰狳(D. novemcinctus) | 1978 | 68 (136) | 51 (238) | 2097 (2352) | 16 |
| 霍氏树懒(C. hoffmanni) | 652 | 12 (24) | 4 (13) | 668 (689) | 4 |
| 小马岛猬(E. telfairi) | 1343 | 33 (66) | 29 (180) | 1405 (1589) | 23 |
| 非洲草原象(L. africana) | 561 | 24 (48) | 14 (71) | 599 (680) | 14 |
| 非洲蹄兔(P. capensis) | 543 | 7 (14) | 4 (27) | 554 (584) | 14 |
| 刺猬(E. europaeus) | 694 | 13 (26) | 2 (8) | 709 (728) | 5 |
| 鼩鼱(S. araneus) | 699 | 22 (44) | 15 (75) | 736 (818) | 11 |
| 野猪(S. scrofa) | 1619 | 37 (74) | 1 (4) | 1657 (1697) | 4 |
| 羊驼(V. pacos) | 399 | 7 (14) | 3 (25) | 409 (438) | 16 |
| 宽吻海豚(T. truncatus) | 222 | 0 (0) | 1 (4) | 223 (226) | 4 |
| 绵羊(O. aries) | 1222 | 27 (54) | 6 (31) | 1255 (1307) | 9 |
| 牛(B. taurus) | 611 | 16 (32) | 6 (33) | 633 (676) | 13 |
| 物种名称 | 单拷贝 基因家族 | 双拷贝 基因家族 | 多拷贝 基因家族 | Orphan基因 家族总数 | 最大Orphan 基因家族的大小 |
| 小棕蝠(M. lucifugus) | 976 | 41 (82) | 25 (202) | 1042 (1260) | 65 |
| 大狐蝠(P. vampyrus) | 282 | 12 (24) | 1 (4) | 295 (310) | 4 |
| 马(E. caballus) | 492 | 12 (24) | 28 (1055) | 532 (1571) | 514 |
| 家猫(F. catus) | 1049 | 10 (20) | 6 (26) | 1065 (1095) | 8 |
| 狗(C. l. familiaris) | 1334 | 16 (32) | 4 (27) | 1354 (1393) | 15 |
| 大熊猫(A. melanoleuca) | 761 | 7 (14) | 0 (0) | 768 (775) | 2 |
| 雪貂(M. p. furo) | 2011 | 8 (16) | 4 (14) | 2023 (2041) | 4 |
| 北美鼠兔(O. princeps) | 538 | 11 (22) | 6 (21) | 555 (581) | 4 |
| 穴兔(O. cuniculus) | 841 | 31 (62) | 24 (137) | 896 (1040) | 17 |
| 豚鼠(C. porcellus) | 657 | 25 (50) | 21 (108) | 703 (815) | 12 |
| 斑纹地松鼠(I. tridecemlineatus) | 455 | 10 (20) | 6 (20) | 471 (495) | 4 |
| 奥氏更格卢鼠(D. ordii) | 516 | 12 (24) | 6 (30) | 534 (570) | 9 |
| 褐家鼠(R. norvegicus) | 1054 | 59 (118) | 36 (218) | 1149 (1390) | 26 |
| 小鼠(M. musculus) | 752 | 40 (80) | 39 (339) | 831 (1171) | 104 |
| 树鼩(T. belangeri) | 835 | 16 (32) | 13 (130) | 864 (997) | 78 |
| 小耳大婴猴(O. garnettii) | 805 | 15 (30) | 5 (40) | 825 (875) | 20 |
| 倭狐猴(M. murinus) | 576 | 12 (24) | 3 (12) | 591 (612) | 5 |
| 菲律宾眼镜猴(T. syrichta) | 574 | 15 (30) | 8 (45) | 597 (649) | 16 |
| 狨猴(C. jacchus) | 1591 | 36 (72) | 12 (57) | 1639 (1720) | 8 |
| 绿猴(C. sabaeus) | 359 | 1 (2) | 0 (0) | 360 (361) | 2 |
| 恒河猴(M. mulatta) | 1699 | 36 (72) | 26 (149) | 1761 (1920) | 12 |
| 东非狒狒(P. anubis) | 359 | 0 (0) | 0 (0) | 359 (359) | 0 |
| 白颊长臂猿(N. leucogenys) | 421 | 3 (6) | 1 (3) | 425 (430) | 3 |
| 苏门答腊猩猩(P. abelii) | 819 | 29 (58) | 1 (9) | 849 (886) | 9 |
| 西非低地大猩猩(G. g. gorilla) | 950 | 25 (50) | 4 (18) | 979 (1018) | 6 |
| 黑猩猩(P. troglodytes) | 297 | 6 (12) | 4 (13) | 307 (322) | 4 |
| 人(H. sapiens) | 332 | 41 (82) | 15 (61) | 388 (475) | 12 |
新窗口打开|下载CSV
进一步统计显示,orphan基因在绝大部分物种中主要以单拷贝的形式存在,而斑马鱼、腔棘鱼、热带爪蟾和马中有相对较多的多拷贝的orphan基因(图1C)。例如,马基因组有最高比例的多拷贝orphan基因,这些基因分布在28个多拷贝的Orphan基因家族中,包含1055个基因,占该物种所有orphan基因的67%。其中有两个家族分别含有514和271个基因,GO功能注释信息显示这些基因富集于RNA介导的转座这一功能类别。也就是说,马中多拷贝orphan基因的高比例很可能是由逆转录转座产生了个别较大的基因家族而导致的。
2.3.2 orphan基因特征
以人类基因组中鉴定到的475个orphan基因为例,分别从序列属性、表达水平、基因表达的组织特异性、功能注释等方面探究了orphan基因的部分特征。
由图3A可知,orphan基因编码的蛋白其序列长度显著低于非orphan基因编码的蛋白(曼-惠特尼U检验,P<2.20×10-16)。对该基因在16种人类组织的表达水平进行分析,发现这475个orphan基因中只有292个基因有可利用的表达谱数据。与非orphan基因相比,这些orphan基因的表达水平较低(曼-惠特尼U检验,P<2.20×10-16) (图3B);约60%的orphan基因都是组织特异性表达(图3C),且主要倾向于在淋巴结中特异性表达(图3D),这暗示orphan基因可能与免疫响应密切相关。
图3
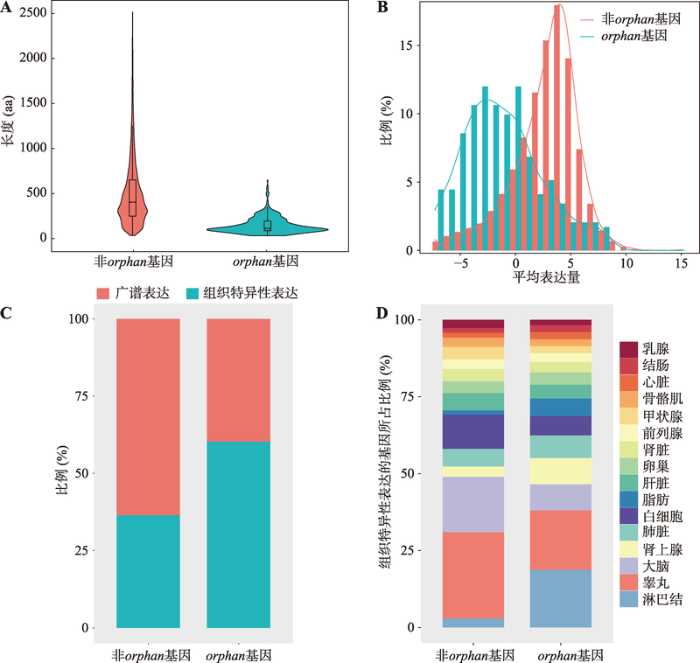 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3orphan基因的序列长度与表达模式
Fig. 3Sequence length and expression pattern of orphan genes
A:人类基因组中orphan基因、非orphan基因编码的氨基酸序列长度;B:orphan基因及非orphan基因的表达水平(该图反映了特定表达水平(x轴)对应的基因所占的比例(y轴),每个基因的表达水平以所有样本中该基因表达水平的平均值取log来表示);C:orphan基因与非orphan基因中广谱表达基因及组织特异性表达基因所占的比例;D:组织特异性表达的orphan基因和非orphan基因在各组织中的分布。
为了揭示出这些基因可能的生物学功能,本研究对orphan基因进行功能富集分析。结果表明,尽管orphan基因与非orphan基因相比具有更高比例的未知功能基因(图4A),但已知功能的orphan基因主要参与角质化、皮肤发育、上皮细胞分化、免疫响应等生物学过程(图4B)。
图4
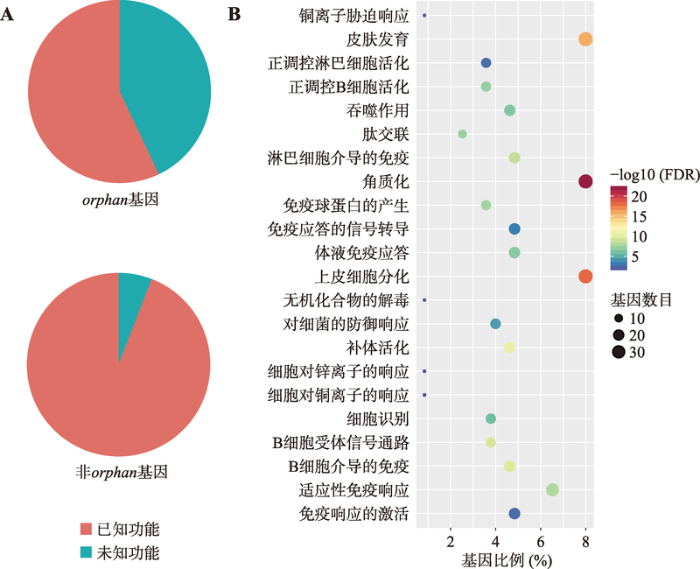 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4orphan基因的功能注释
A:orphan基因和非orphan基因中有GO注释的基因所占的比例;B:orphan基因的功能富集。
Fig. 4Functional annotation of orphan genes
2.3.3 种系特异性基因家族的起源和进化
上述分析只涉及单个物种的特异性基因,而种系特异性基因对于理解特定分类学阶元的物种的基因组和表型进化也具有重要的意义。因此本研究进一步对种系特异性基因家族进行了鉴定。参考文献[40]中的方法,对于系统发育树上感兴趣的内部节点,当某基因家族包含了该节点下半数以上物种的基因时,该基因家族即被认为是相应节点起源的种系特异性基因家族。按照此原则,共有9488个种系特异性基因家族分布到脊椎动物主要类群系统发育树的节点上(图5)。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5脊椎动物不同种系中基因家族的数量

Fig. 5The number of gene families in different lineages of vertebrates
数据显示,从64个脊椎动物物种共同祖先起源的基因家族有1854个,脊椎动物在进化过程中,自有颌纲祖先物种起源的基因家族数量最多,为3839个。
鱼类在早期进化中发生了一次该类群特异性的全基因组复制事件。数据显示辐鳍鱼特有的基因家族高达453个,推测这可能与鱼类特异的全基因组复制事件有关(图5)。当把腔棘鱼考虑在内时,硬骨鱼特有的基因家族则有183个,根据简约法原理,这些基因家族很可能是四足动物进化早期丢失的基因家族。为了调查这些基因是否对脊椎动物由水生到陆生的进化方式有贡献,本研究利用ZFIN (The Zebrafish Information Network)数据库中的基因表达、基因敲除、基因敲低数据对这些基因进行了分析。结果发现有84个基因其功能与鱼类特有的发育过程关系密切:分别有9个基因与鳍的发育有关,11个基因与躯干、体节、尾巴发育有关,7个基因与耳石及耳朵的发育有关,15个基因与肾脏发育有关,24个基因与眼睛及27个基因与大脑发育相关。这暗示脊椎动物从水生到陆生转变中某些关键特征的形成,如鳍到肢的转变、耳的重塑以及排氮形式的改变等与四足动物中特定基因的缺失有着密切联系。Amemiya等[41]鉴定到的55个在四足动物早期进化中丢失的基因中,有20个基因在本研究分析中得到了进一步证实。
为了探究硬骨鱼特有基因的表达特征,本研究以斑马鱼中的硬骨鱼特有基因为对象进行了分析。结果表明,斑马鱼中硬骨鱼特有的基因通常比非特有基因的表达水平低(图6A,曼-惠特尼U检验,P < 2.20×10-16),但这些基因表达的组织特异性较高,且主要集中在鳃中特异性表达(图6B),这反映了硬骨鱼特有基因在硬骨鱼特异的发育过程中发挥了至关重要的作用。
本研究还调查了鸟类特有的基因家族。在199个鸟类特有的基因家族中,有134个家族含有共151个鸡的直系同源基因,GO功能富集分析(FDR<0.05)显示其中许多基因参与了对细菌的防御响应以及与细胞骨架的结构成分有关。这些基因中有7个注释为羽毛角蛋白基因,分别是LOC426913、LOC426914、LOC431325、LOC427060、F-KER、LOC429492和LOC769486。与鱼类中观察结果相似,鸟类特有的基因相对非鸟类特有的基因通常表达水平更低(图6C,曼-惠特尼U检验,P = 1.65×10-8)、表达的组织特异性更高,但无显著的组织偏好性(图6D)。
图6
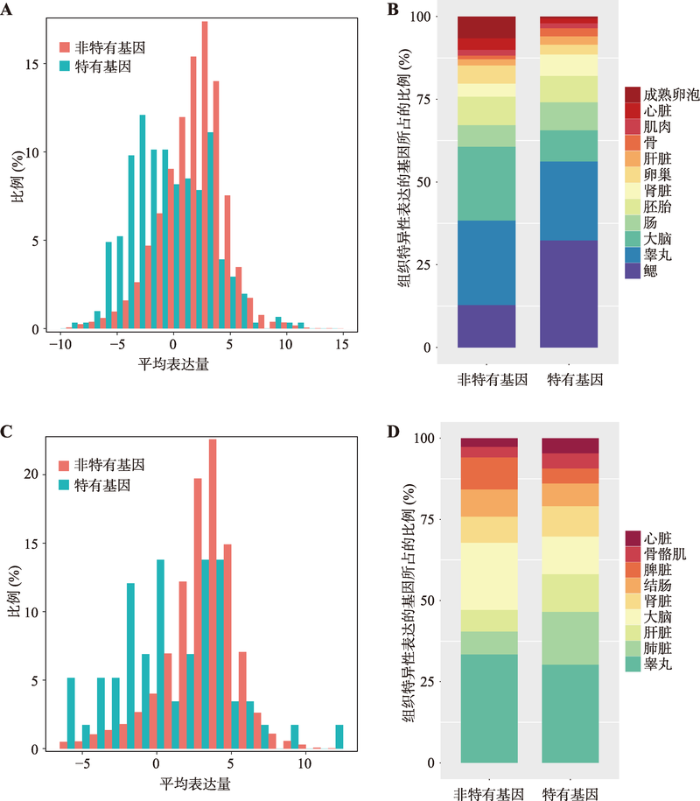 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6硬骨鱼、鸟类特有基因的表达分析
A:斑马鱼基因组中所包含的硬骨鱼特有的基因及非特有基因的表达水平;B:组织特异性表达的硬骨鱼特有基因和非特有基因在斑马鱼各组织中的分布;C:鸡基因组中所含有的鸟类特有基因及非特有基因的表达水平;D:组织特异性表达的鸟类特有基因和非特有基因在鸡不同组织中的分布。
Fig. 6Expression analysis of bony fish- and birds-specific genes
3 讨论
本研究对跨约6亿年进化时间的64个脊椎动物物种及2个海鞘纲外群物种进行了基因家族的鉴定和初步分析,揭示了脊椎动物基因家族大小的动态进化,并对部分基因家族拷贝数变异与特定分类群的宏进化之间的联系进行了推测。从全基因组水平来看,脊椎动物中的基因主要以单拷贝的形式存在,这与植物中观察到的现象不同。植物基因组中的基因大都以多基因家族的形式存在[20],这主要是由于植物中除了小规模复制外,还发生了非常广泛的全基因组复制事件。而脊椎动物中除了进化早期发生的两轮全基因组复制及真骨鱼类中额外的全基因组复制外,只在两栖类和辐鳍鱼部分物种中发现独立的全基因组复制事件[42,43]。Demuth等[22]对人、黑猩猩、小鼠、大鼠和狗基因组中基因家族的扩增与收缩的研究发现,在灵长类动物中,人的基因组中有最少的基因丢失,而且相比之下,黑猩猩在相同时期内却丢失了更多基因。本研究也得到一致的结果,这在一定程度上揭示了这两个物种间表型差异背后的遗传变化。基因家族的大小受到各种因素的影响。基因复制、基因的de novo起源等会增加基因家族的大小;而基因缺失(包括单个基因或染色体片段中几个基因的缺失)会使基因家族的大小减小[20]。除此之外,研究表明基因的功能也是决定基因家族大小的一个主要因素[22]。例如,脊椎动物中参与调控、信号转导、转录、蛋白质运输和蛋白质修饰的基因家族趋向于扩增,而参与新陈代谢过程的基因家族倾向于收缩。随机过程与自然选择是基因家族大小进化的驱动力[22]。有研究表明不同真核生物中基因家族的大小与选择压力的关系有所差异,如在单细胞真核生物酵母中,选择约束与基因家族的大小有很强的正相关关系,然而在多细胞真核生物中则呈现出弱的负相关[44]。
物种或种系特异性orphan基因与其他物种中的基因序列不具有同源性,常被认为可能对物种的适应性进化有重要贡献[45,46,47]。本研究发现脊椎动物中的orphan基因只在极个别物种中有较多的多拷贝,绝大多数仍以单拷贝的形式存在。与全基因组水平的基因家族大小分布相比,orphan基因中单拷贝基因所占的比例高于全基因组中单拷贝基因的比例。这可能是由于脊椎动物中基因的复制能力低,或者是这些基因太“年轻”而没有足够的时间进化出额外的拷贝。此外,orphan基因的产生机制比较特殊,该基因的形成贯穿整个进化历程并且是一个持续不断的过程,它不但可以通过复制和重排过程产生,也可以从基因组中的非编码区de novo起源[48]。基因表达数据与功能注释等的结合进一步揭示了脊椎动物中物种或种系特异性基因的一般属性及其对物种适应性的影响。详细而言,这类基因通常编码的蛋白质序列长度较短、表达水平低、而表达的组织特异性高;硬骨鱼特有的基因中包含了对鱼类适应水生环境的重要基因,鸟类特异性基因中富集了羽毛角蛋白基因,这些分析证实了该类基因对物种或种系特异性表型创新的贡献。其中,对鸟类特异性基因的研究中,增加鸟类样本大小可能更有利于评估这类基因对鸟类特异性适应的影响。
综上所述,本研究系统地阐述了脊椎动物进化过程中动态的基因得失过程导致的不同种系间基因家族大小的差异及其蕴含的生物学意义,对物种或种系特异的基因的分析为理解脊椎动物间表型的多样性提供了理论基础。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URLPMID:25523484 [本文引用: 1]

Vertebrates diverged from other chordates ~500-yr ago and experienced successful innovations and adaptations, but the genomic basis underlying vertebrate origins are not fully understood. Here we suggest, through comparison with multiple lancelet (amphioxus) genomes, that ancient vertebrates experienced high rates of protein evolution, genome rearrangement and domain shuffling and that these rates greatly slowed down after the divergence of jawed and jawless vertebrates. Compared with lancelets, modern vertebrates retain, at least relatively, less protein diversity, fewer nucleotide polymorphisms, domain combinations and conserved non-coding elements (CNE). Modern vertebrates also lost substantial transposable element (TE) diversity, whereas lancelets preserve high TE diversity that includes even the long-sought RAG transposon. Lancelets also exhibit rapid gene turnover, pervasive transcription, fastest exon shuffling in metazoans and substantial TE methylation not observed in other invertebrates. These new lancelet genome sequences provide new insights into the chordate ancestral state and the vertebrate evolution.
URLPMID:1779523 [本文引用: 1]
We show that: first, the majority of duplicated genes in extant vertebrate genomes are ancient and were created at times that coincide with proposed whole genome duplication events; second, there exist significant differences in gene retention for different functional categories of genes between fishes and land vertebrates; third, there seems to be a considerable bias in gene retention of regulatory genes towards the mode of gene duplication (whole genome duplication events compared to smaller-scale events), which is in accordance with the so-called gene balance hypothesis; and fourth, that ancient duplicates that have survived for many hundreds of millions of years can still be lost.Based on phylogenetic analyses, we show that both the mode of duplication and the functional class the duplicated genes belong to have been of major importance for the evolution of the vertebrates. In particular, we provide evidence that massive gene duplication (probably as a consequence of entire genome duplications) at the dawn of vertebrate evolution might have been particularly important for the evolution of complex vertebrates.The sequencing of vertebrate genomes occurs at an ever-increasing pace. Currently, the genome sequences, or at least first drafts thereof, are available for more than 14 different vertebrate species, while many more are underway. These vertebrate genome sequences cover a phylogenetic distance of more than 450 million years of evolution, dating back as far as the split between fishes and land vertebrates. Unfortunately, genome sequences of cartilaginous fish such as sharks, rays or skates, or of jawless vertebrates such as lampreys and hagfish, which diverged well before that time, are not available yet.Based on rather inaccurate indicators such as genome size and isozyme complexity, Ohno already suggested in 1970 that the genomes of (early) vertebrates have been shaped by two whole genome duplications (WGDs) [1]. More than 20 years later, important indicatio
.
URLPMID:17684299 [本文引用: 1]
Abstract Genomic plasticity of human chromosome 8p23.1 region is highly influenced by two groups of complex segmental duplications (SDs), termed REPD and REPP, that mediate different kinds of rearrangements. Part of the difficulty to explain the wide range of phenotypes associated with 8p23.1 rearrangements is that REPP and REPD are not yet well characterized, probably due to their polymorphic status. Here, we describe a novel primate-specific gene family, named FAM90A (family with sequence similarity 90), found within these SDs. According to the current human reference sequence assembly, the FAM90A family includes 24 members along 8p23.1 region plus a single member on chromosome 12p13.31, showing copy number variation (CNV) between individuals. These genes can be classified into subfamilies I and II, which differ in their upstream and 5'-untranslated region sequences, but both share the same open reading frame and are ubiquitously expressed. Sequence analysis and comparative fluorescence in situ hybridization studies showed that FAM90A subfamily II suffered a big expansion in the hominoid lineage, whereas subfamily I members were likely generated sometime around the divergence of orangutan and African great apes by a fusion process. In addition, the analysis of the Ka/Ks ratios provides evidence of functional constraint of some FAM90A genes in all species. The characterization of the FAM90A gene family contributes to a better understanding of the structural polymorphism of the human 8p23.1 region and constitutes a good example of how SDs, CNVs and rearrangements within themselves can promote the formation of new gene sequences with potential functional consequences.
.
URL [本文引用: 1]
The importance of gene duplication in supplying raw genetic material to biological evolution has been recognized since the 1930s. Recent genomic sequence data provide substantial evidence for the abundance of duplicated genes in all organisms surveyed. But how do newly duplicated genes survive and acquire novel functions, and what role does gene duplication play in the evolution of genomes and organisms? Detailed molecular characterization of individual gene families, computational analysis of genomic sequences and population genetic modeling can all be used to help us uncover the mechanisms behind the evolution by gene duplication.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 2]
.
URLPMID:27974754 [本文引用: 1]
Abstract Seahorses have a specialized morphology that includes a toothless tubular mouth, a body covered with bony plates, a male brood pouch, and the absence of caudal and pelvic fins. Here we report the sequencing and de novo assembly of the genome of the tiger tail seahorse, Hippocampus comes. Comparative genomic analysis identifies higher protein and nucleotide evolutionary rates in H. comes compared with other teleost fish genomes. We identified an astacin metalloprotease gene family that has undergone expansion and is highly expressed in the male brood pouch. We also find that the H. comes genome lacks enamel matrix protein-coding proline/glutamine-rich secretory calcium-binding phosphoprotein genes, which might have led to the loss of mineralized teeth. tbx4, a regulator of hindlimb development, is also not found in H. comes genome. Knockout of tbx4 in zebrafish showed a 'pelvic fin-loss' phenotype similar to that of seahorses.
.
[本文引用: 1]
.
[本文引用: 1]
.
URLPMID:11861885 [本文引用: 1]
Abstract We conducted a detailed analysis of duplicate genes in three complete genomes: yeast, Drosophila, and Caenorhabditis elegans. For two proteins belonging to the same family we used the criteria: (1) their similarity is > or =I (I = 30% if L > or = 150 a.a. and I = 0.01n + 4.8L(-0.32(1 + exp(-L/1000))) if L or = 80% of the longer protein. We found it very important to delete isoforms (caused by alternative splicing), same genes with different names, and proteins derived from repetitive elements. We estimated that there were 530, 674, and 1,219 protein families in yeast, Drosophila, and C. elegans, respectively, so, as expected, yeast has the smallest number of duplicate genes. However, for the duplicate pairs with the number of substitutions per synonymous site (K(S)) < 0.01, Drosophila has only seven pairs, whereas yeast has 58 pairs and nematode has 153 pairs. After considering the possible effects of codon usage bias and gene conversion, these numbers became 6, 55, and 147, respectively. Thus, Drosophila appears to have much fewer young duplicate genes than do yeast and nematode. The larger numbers of duplicate pairs with K(S) < 0.01 in yeast and C. elegans were probably largely caused by block duplications. At any rate, it is clear that the genome of Drosophila melanogaster has undergone few gene duplications in the recent past and has much fewer gene families than C. elegans.
.
[本文引用: 1]
.
URLPMID:11586358 [本文引用: 1]
Gene duplication followed by adaptive evolution is one of the primary forces for the emergence of new gene function. Here we describe the recent proliferation, transposition and selection of a 20-kilobase (kb) duplicated segment throughout 15 Mb of the short arm of human chromosome 16. The dispersal of this segment was accompanied by considerable variation in chromosomal-map location and copy number among hominoid species. In humans, we identified a gene family (morpheus) within the duplicated segment. Comparison of putative protein-encoding exons revealed the most extreme case of positive selection among hominoids. The major episode of enhanced amino-acid replacement occurred after the separation of human and great-ape lineages from the orangutan. Positive selection continued to alter amino-acid composition after the divergence of human and chimpanzee lineages. The rapidity and bias for amino-acid-altering nucleotide changes suggest adaptive evolution of the morpheus gene family during the emergence of humans and African apes. Moreover, some genes emerge and evolve very rapidly, generating copies that bear little similarity to their ancestral precursors. Consequently, a small fraction of human genes may not possess discernible orthologues within the genomes of model organisms.
.
URLPMID:14660798 [本文引用: 1]
Previous studies of genome evolution usually have involved one or two genomes and have thus been limited in their ability to detect the direction and rate of evolutionary change. Here, we use complete genome data from 20 poxvirus genomes to build a robust phylogeny of the Poxviridae and to study patterns of genome evolution. We show that, although there has been little gene order evolution, there are substantial differences between poxviruses in terms of genome content. Furthermore, we show that the rate of gene acquisition is not constant over time and that it has increased in the orthopox lineage (which includes smallpox and vaccinia). We also tested for positive selection on 204 groups of genes and show that a disproportionately high proportion of genes in the orthopox clade are under positive selection. The association of an increased rate of gene gain and positive selection is indicative of adaptive genome evolution. Many of the genes involved in these processes are likely to be associated with host-parasite coevolution.
.
URLPMID:12885956 [本文引用: 1]
The fish fauna of the Antarctic Ocean is dominated by five endemic families of the Perciform suborder Notothenioidei, thought to have arisen in situ within the Antarctic through adaptive radiation of an ancestral stock that evolved antifreeze glycoproteins (AFGPs) enabling survival as the ocean chilled to subzero temperatures. The endemism results from geographic confinement imposed by a massive oceanographic barrier, the Antarctic Circumpolar Current, which also thermally isolated Antarctica over geologic time, leading to its current frigid condition. Despite this voluminous barrier to fish dispersal, a number of species from the Antarctic family Nototheniidae now inhabit the nonfreezing cool temperate coasts of the southern continents. The origin of these temperate-water nototheniids is not completely understood. Since the AFGP gene apparently evolved only once, before the Antarctic notothenioid radiation, the presence of AFGP genes in extant temperate-water nototheniids can be used to infer an Antarctic evolutionary origin. Genomic Southern analysis, PCR amplification of AFGP genes, and sequencing showed that Notothenia angustata and Notothenia microlepidota endemic to southern New Zealand have two to three AFGP genes, structurally the same as those of the Antarctic nototheniids. At least one of these genes is still functional, as AFGP cDNAs were obtained and low levels of mature AFGPs were detected in the blood. A phylogenetic tree based on complete ND2 coding sequences showed monophyly of these two New Zealand nototheniids and their inclusion in the monophyletic Nototheniidae consisted of mostly AFGP-bearing taxa. These analyses support an Antarctic ancestry for the New Zealand nototheniids. A divergence time of approximately 11 Myr was estimated for the two New Zealand nototheniids, approximating the upper Miocene northern advance of the Antarctic Convergence over New Zealand, which might have served as the vicariant event that lead to the northward dispersal of their most recent common ancestor. Similar secondary northward dispersal likely applies to the South American nototheniid Paranotothenia magellanica, which has four AFGP genes in its DNA, but not to the sympatric nototheniid Patagonotothen tessellata, which does not appear to have any AFGP sequences in its genome at all.
.
URLPMID:18753634 [本文引用: 1]
The antifreeze glycoprotein-fortified Antarctic notothenioid fishes comprise the predominant fish suborder in the isolated frigid Southern Ocean. Their ecological success undoubtedly entailed evolutionary acquisition of a full suite of cold-stable functions besides antifreeze protection. Prior studies of adaptive changes in these teleost fishes generally examined a single genotype or phenotype. We report here the genome-wide investigations of transcriptional and genomic changes associated with Antarctic notothenioid cold adaptation. We sequenced and characterized 33,560 ESTs from four tissues of the Antarctic notothenioid Dissostichus mawsoni and derived 3,114 nonredundant protein gene families and their expression profiles. Through comparative analyses of same-tissue transcriptome profiles of D. mawsoni and temperate/tropical teleost fishes, we identified 177 notothenioid protein families that were expressed many fold over the latter, indicating cold-related up-regulation. These up-regulated gene families operate in protein biosynthesis, protein folding and degradation, lipid metabolism, antioxidation, antiapoptosis, innate immunity, choriongenesis, and others, all of recognizable functional importance in mitigating stresses in freezing temperatures during notothenioid life histories. We further examined the genomic and evolutionary bases for this expressional up-regulation by comparative genomic hybridization of DNA from four pairs of Antarctic and basal non-Antarctic notothenioids to 10,700 D. mawsoni cDNA probes and discovered significant to astounding (3- to >300-fold, P < 0.05) Antarctic-specific duplications of 118 protein-coding genes, many of which correspond to the up-regulated gene families. Results of our integrative tripartite study strongly suggest that evolution under constant cold has resulted in dramatic genomic expansions of specific protein gene families, augmenting gene expression and gene functions contributing to physiological fitness of Antarctic notothenioids in freezing polar conditions.
.
[本文引用: 1]
.
URLPMID:15252450 [本文引用: 1]
pGiven that gene duplication is a major driving force of evolutionary change and the key mechanism underlying the emergence of new genes and biological processes, this study sought to use a novel genome-wide approach to identify genes that have undergone lineage-specific duplications or contractions among several hominoid lineages. Interspecies cDNA array-based comparative genomic hybridization was used to individually compare copy number variation for 39,711 cDNAs, representing 29,619 human genes, across five hominoid species, including human. We identified 1,005 genes, either as isolated genes or in clusters positionally biased toward rearrangement-prone genomic regions, that produced relative hybridization signals unique to one or more of the hominoid lineages. Measured as a function of the evolutionary age of each lineage, genes showing copy number expansions were most pronounced in human (134) and include a number of genes thought to be involved in the structure and function of the brain. This work represents, to our knowledge, the first genome-wide gene-based survey of gene duplication across hominoid species. The genes identified here likely represent a significant majority of the major gene copy number changes that have occurred over the past 15 million years of human and great ape evolution and are likely to underlie some of the key phenotypic characteristics that distinguish these species./p
URLPMID:27399969 [本文引用: 2]
Nature Genetics 48, 947 (2016). doi:10.1038/ng.3615Authors: Li Yu, Guo-Dong Wang, Jue Ruan, Yong-Bin Chen, Cui-Ping Yang, Xue Cao, Hong Wu, Yan-Hu Liu, Zheng-Lin Du, Xiao-Ping Wa ...
URLPMID:24357323 [本文引用: 1]
Amborella trichopoda is strongly supported as the single living species of the sister lineage to all other extant flowering plants, providing a unique reference for inferring the genome content and structure of the most recent common ancestor (MRCA) of living angiosperms. Sequencing the Amborella genome, we identified an ancient genome duplication predating angiosperm diversification, without evidence of subsequent, lineage-specific genome duplications. Comparisons between Amborella and other angiosperms facilitated reconstruction of the ancestral angiosperm gene content and gene order in the MRCA of core eudicots. We identify new gene families, gene duplications, and floral protein-protein interactions that first appeared in the ancestral angiosperm. Transposable elements in Amborella are ancient and highly divergent, with no recent transposon radiations. Population genomic analysis across Amborella's native range in New Caledonia reveals a recent genetic bottleneck and geographic structure with conservation implications.
URLPMID:23216999 [本文引用: 3]
Gene family size variation is an important mechanism that shapes the natural variation for adaptation in various species. Despite its importance, the pattern of gene family size variation in green plants is still not well understood. In particular, the evolutionary pattern of genes and gene families remains unknown in the model plant Arabidopsis thaliana in the context of green plants. In this study, eight representative genomes of green plants are sampled to study gene family evolution and characterize the origination of A. thaliana genes, respectively. Four important insights gained are that: (i) the rate of gene gains and losses is about 0.001359 per gene every million years, similar to the rate in yeast, Drosophila, and mammals; (ii) some gene families evolved rapidly with extreme expansions or contractions, and 2745 gene families present in all the eight species represent the -ore- proteome of green plants; (iii) 70% of A. thaliana genes could be traced back to 450 million years ago; and (iv) intriguingly, A. thaliana genes with early origination are under stronger purifying selection and more conserved. In summary, the present study provides genome-wide insights into evolutionary history and mechanisms of genes and gene families in green plants and especially in A. thaliana.
.
URLPMID:17183716 [本文引用: 1]
Gene families are groups of homologous genes that are likely to have highly similar functions. Differences in family size due to lineage-specific gene duplication and gene loss may provide clues to the evolutionary forces that have shaped mammalian genomes. Here we analyze the gene families contained within the whole genomes of human, chimpanzee, mouse, rat, and dog. In total we find that more than half of the 9,990 families present in the mammalian common ancestor have either expanded or contracted along at least one lineage. Additionally, we find that a large number of families are completely lost from one or more mammalian genomes, and a similar number of gene families have arisen subsequent to the mammalian common ancestor. Along the lineage leading to modern humans we infer the gain of 689 genes and the loss of 86 genes since the split from chimpanzees, including changes likely driven by adaptive natural selection. Our results imply that humans and chimpanzees differ by at least 6% (1,418 of 22,000 genes) in their complement of genes, which stands in stark contrast to the oft-cited 1.5% difference between orthologous nucleotide sequences. This genomic -渞evolving door- of gene gain and loss represents a large number of genetic differences separating humans from our closest relatives.
.
[本文引用: 4]
.
URLPMID:28736437 [本文引用: 1]
With the generation of more than 100 sequenced vertebrate genomes in less than 25 years, the key question arises of how these resources can be used to inform new or ongoing projects. In the past, this diverse collection of sequences from human as well as model and non-model organisms has been used to annotate the human genome and to increase the understanding of human disease. In the future, comparative vertebrate genomics in conjunction with additional genomic resources will yield insights into the processes of genome function, evolution, speciation, selection and adaptation, as well as the quantification of species diversity. In this Review, we discuss how the genomics of non-human organisms can provide insights into vertebrate biology and how this can contribute to the understanding of human physiology and health.
.
URLPMID:12952885 [本文引用: 2]
Abstract The identification of orthologous groups is useful for genome annotation, studies on gene/protein evolution, comparative genomics, and the identification of taxonomically restricted sequences. Methods successfully exploited for prokaryotic genome analysis have proved difficult to apply to eukaryotes, however, as larger genomes may contain multiple paralogous genes, and sequence information is often incomplete. OrthoMCL provides a scalable method for constructing orthologous groups across multiple eukaryotic taxa, using a Markov Cluster algorithm to group (putative) orthologs and paralogs. This method performs similarly to the INPARANOID algorithm when applied to two genomes, but can be extended to cluster orthologs from multiple species. OrthoMCL clusters are coherent with groups identified by EGO, but improved recognition of "recent" paralogs permits overlapping EGO groups representing the same gene to be merged. Comparison with previously assigned EC annotations suggests a high degree of reliability, implying utility for automated eukaryotic genome annotation. OrthoMCL has been applied to the proteome data set from seven publicly available genomes (human, fly, worm, yeast, Arabidopsis, the malaria parasite Plasmodium falciparum, and Escherichia coli). A Web interface allows queries based on individual genes or user-defined phylogenetic patterns (http://www.cbil.upenn.edu/gene-family). Analysis of clusters incorporating P. falciparum genes identifies numerous enzymes that were incompletely annotated in first-pass annotation of the parasite genome.
.
[本文引用: 1]
.
[本文引用: 1]
.
URLPMID:17021158 [本文引用: 1]
Biologists and other scientists routinely need to know times of divergence between species and to construct phylogenies calibrated to time (timetrees). Published studies reporting time estimates from molecular data have been increasing rapidly, but the data have been largely inaccessible to the greater community of scientists because of their complexity. TimeTree brings these data together in a consistent format and uses a hierarchical structure, corresponding to the tree of life, to maximize their utility. Results are presented and summarized, allowing users to quickly determine the range and robustness of time estimates and the degree of consensus from the published literature.
.
URLPMID:16077014 [本文引用: 2]
Abstract Comparison of whole genomes has revealed that changes in the size of gene families among organisms is quite common. However, there are as yet no models of gene family evolution that make it possible to estimate ancestral states or to infer upon which lineages gene families have contracted or expanded. In addition, large differences in family size have generally been attributed to the effects of natural selection, without a strong statistical basis for these conclusions. Here we use a model of stochastic birth and death for gene family evolution and show that it can be efficiently applied to multispecies genome comparisons. This model takes into account the lengths of branches on phylogenetic trees, as well as duplication and deletion rates, and hence provides expectations for divergence in gene family size among lineages. The model offers both the opportunity to identify large-scale patterns in genome evolution and the ability to make stronger inferences regarding the role of natural selection in gene family expansion or contraction. We apply our method to data from the genomes of five yeast species to show its applicability.
[本文引用: 1]
.
URLPMID:15388519 [本文引用: 1]
Genes are often characterized dichotomously as either housekeeping or single-tissue specific. We conjectured that crucial functional information resides in genes with midrange profiles of expression.
[本文引用: 1]
.
[本文引用: 1]
.
URLPMID:28179571 [本文引用: 1]
Abstract Genome size in mammals and birds shows remarkably little interspecific variation compared with other taxa. However, genome sequencing has revealed that many mammal and bird lineages have experienced differential rates of transposable element (TE) accumulation, which would be predicted to cause substantial variation in genome size between species. Thus, we hypothesize that there has been covariation between the amount of DNA gained by transposition and lost by deletion during mammal and avian evolution, resulting in genome size equilibrium. To test this model, we develop computational methods to quantify the amount of DNA gained by TE expansion and lost by deletion over the last 100 My in the lineages of 10 species of eutherian mammals and 24 species of birds. The results reveal extensive variation in the amount of DNA gained via lineage-specific transposition, but that DNA loss counteracted this expansion to various extents across lineages. Our analysis of the rate and size spectrum of deletion events implies that DNA removal in both mammals and birds has proceeded mostly through large segmental deletions (>10 kb). These findings support a unified "accordion" model of genome size evolution in eukaryotes whereby DNA loss counteracting TE expansion is a major determinant of genome size. Furthermore, we propose that extensive DNA loss, and not necessarily a dearth of TE activity, has been the primary force maintaining the greater genomic compaction of flying birds and bats relative to their flightless relatives.
.
URLPMID:15486693Magsci [本文引用: 1]
For many genes, ray-finned fish (Actinopterygii) have two paralogous copies, where only one ortholog is present in tetrapods. The discovery of an additional, almost-complete set of Hox clusters in teleosts (zebrafish, pufferfish, medaka, and cichlid) but not in basal actinopterygian lineages ( Polypterus) led to the formulation of the fish-specific genome duplication hypothesis. The phylogenetic timing of this genome duplication during the evolution of ray-finned fish is unknown, since only a few species of basal fish lineages have been investigated so far. In this study, three nuclear genes ( fzd8, sox11, tyrosinase) were sequenced from sturgeons (Acipenseriformes), gars (Semionotiformes), bony tongues (Osteoglossomorpha), and a tenpounder (Elopomorpha). For these three genes, two copies have been described previously teleosts (e.g., zebrafish, pufferfish), but only one orthologous copy is found in tetrapods. Individual gene trees for these three genes and a concatenated dataset support the hypothesis that the fish-specific genome duplication event took place after the split of the Acipenseriformes and the Semionotiformes from the lineage leading to teleost fish but before the divergence of Osteoglossiformes. If these three genes were duplicated during the proposed fish-specific genome duplication event, then this event separates the species-poor early-branching lineages from the species-rich teleost lineage. The additional number of genes resulting from this event might have facilitated the evolutionary radiation and the phenotypic diversification of the teleost fish.
.
URLPMID:19652647 [本文引用: 1]
Abstract Many organisms are currently polyploid, or have a polyploid ancestry and now have secondarily 'diploidized' genomes. This finding is surprising because retained whole-genome duplications (WGDs) are exceedingly rare, suggesting that polyploidy is usually an evolutionary dead end. We argue that ancient genome doublings could probably have survived only under very specific conditions, but that, whenever established, they might have had a pronounced impact on species diversification, and led to an increase in biological complexity and the origin of evolutionary novelties.
.
URLPMID:16034368 [本文引用: 1]
Nat Rev Cancer. 2005 Aug;5(8):615-25. Review
.
URLPMID:24916032Magsci [本文引用: 1]
Cancer/testis (CT) antigens are encoded by germline genes and are aberrantly expressed in a number of human cancers. Interestingly, CT antigens are frequently involved in gene families that are highly expressed in germ cells. Here, we presented an evolutionary analysis of the CTAGE (cutaneous T-cell-lymphoma-associated antigen) gene family to delineate its molecular history and functional significance during primate evolution. Comparisons among human, chimpanzee, gorilla, orangutan, macaque, marmoset, and other mammals show a rapid and primate specific expansion of CTAGE family, which starts with an ancestral retroposition in the haplorhini ancestor. Subsequent DNA-based duplications lead to the prosperity of single-exon CTAGE copies in catarrhines, especially in humans. Positive selection was identified on the single-exon copies in comparison with functional constraint on the multiexon copies. Further sequence analysis suggests that the newly derived CTAGE genes may obtain regulatory elements from long terminal repeats. Our result indicates the dynamic evolution of primate genomes, and the recent expansion of this CT antigen family in humans may confer advantageous phenotypic traits during early human evolution.
.
URLPMID:10498776 [本文引用: 1]
Author information: (1)Faculty of Natural Science, Department of Math and Computer Science, Ben Gurion University, Beer-Sheva 84015, Israel. dfischer@cs.bgu.ac.il
.
URLPMID:14634634 [本文引用: 1]
Genome data have revealed great variation in the numbers of genes in different organisms, which indicates that there is a fundamental process of genome evolution: the origin of new genes. However, there has been little opportunity to explore how genes with new functions originate and evolve. The study of ancient genes has highlighted the antiquity and general importance of some mechanisms of gene origination, and recent observations of young genes at early stages in their evolution have unveiled unexpected molecular and evolutionary processes.
.
URLPMID:5554394 [本文引用: 1]
The birth of genes that encode new protein sequences is a major source of evolutionary innovation. However, we still understand relatively little about how these genes come into being and which functions they are selected for. To address these questions, we have obtained a large collection of mammalian-specific gene families that lack homologues in other eukaryotic groups. We have combined gene annotations and de novo transcript assemblies from 30 different mammalian species, obtaining 6,000 gene families. In general, the proteins in mammalian-specific gene families tend to be short and depleted in aromatic and negatively charged residues. Proteins which arose early in mammalian evolution include milk and skin polypeptides, immune response components, and proteins involved in reproduction. In contrast, the functions of proteins which have a more recent origin remain largely unknown, despite the fact that these proteins also have extensive proteomics support. We identify several previously described cases of genes originated de novo from noncoding genomic regions, supporting the idea that this mechanism frequently underlies the evolution of new protein-coding genes in mammals. Finally, we show that most young mammalian genes are preferentially expressed in testis, suggesting that sexual selection plays an important role in the emergence of new functional genes.
.
URLPMID:23598338Magsci [本文引用: 1]
The discovery of a living coelacanth specimen in 1938 was remarkable, as this lineage of lobe-finned fish was thought to have become extinct 70 million years ago. The modern coelacanth looks remarkably similar to many of its ancient relatives, and its evolutionary proximity to our own fish ancestors provides a glimpse of the fish that first walked on land. Here we report the genome sequence of the African coelacanth, Latimeria chalumnae. Through a phylogenomic analysis, we conclude that the lungfish, and not the coelacanth, is the closest living relative of tetrapods. Coelacanth protein-coding genes are significantly more slowly evolving than those of tetrapods, unlike other genomic features. Analyses of changes in genes and regulatory elements during the vertebrate adaptation to land highlight genes involved in immunity, nitrogen excretion and the development of fins, tail, ear, eye, brain and olfaction. Functional assays of enhancers involved in the fin-to-limb transition and in the emergence of extra-embryonic tissues show the importance of the coelacanth genome as a blueprint for understanding tetrapod evolution.
.
URLMagsci [本文引用: 1]
Whole genome duplication (leading to polyploidy) is widely accepted as an important evolutionary force in plants, but it is less recognized as a driver of animal diversification. Nevertheless, it occurs across a wide range of animals; this review investigates why it is particularly common in fish and amphibians, while rare among other vertebrates. We review the current geographic, ecological and phylogenetic distributions of sexually reproducing polyploid taxa before focusing more specifically on what factors drive polyploid formation and establishment. In summary, (1) polyploidy is phylogenetically restricted in both amphibians and fishes, although entire fish, but not amphibian, lineages are derived from polyploid ancestors. (2) Although mechanisms such as polyspermy are feasible, polyploid formation appears to occur principally through unreduced gamete formation, which can be experimentally induced by temperature or pressure shock in both groups. (3) External reproduction and fertilization in primarily temperate freshwater environments potentially exposes zygotes to temperature stress, which can promote increased production of unreduced gametes. (4) Large numbers of gametes and group breeding in relatively confined areas could increase the probability of compatible gamete combinations in both groups. (5) Both fish and amphibians have a propensity to form reproductively successful hybrids; although the relative frequency of autopolyploidy versus allopolyploidy is difficult to ascertain, multiple origins involving hybridization have been confirmed for a number of species in both groups. (6) Problems with establishment of polyploid lineages associated with minority cytotype exclusion could be overcome in amphibians via assortative mating by acoustic recognition of the same ploidy level, but less attention has been given to chemical or acoustic mechanisms that might operate in fish. (7) There is no strong evidence that polyploid fish or amphibians currently exist in more extreme environments than their diploid progenitors or have broader ecological ranges. (8) Although pathogens could play a role in the relative fitness of polyploid species, particularly given duplication of genes involved in immunity, this remains an understudied field in both fish and amphibians. (9) As in plants, many duplicate copies of genes are retained for long periods of time, indicative of selective maintenance of the duplicate copies, but we find no physiological or other reasons that could explain an advantage for allelic or genetic complexity. (10) Extant polyploid species do not appear to be more or less prone to extinction than related diploids in either group. We conclude that, while polyploid fish and amphibians share a number of attributes facilitating polyploidy, clear drivers of genome duplication do not emerge from the comparison. The lack of a clear association of sexually reproducing polyploids with range expansion, harsh environments, or risk of extinction could suggest that stronger correlations in plants may be driven by shifts in mating system more than ploidy. However, insufficient data currently exist to provide rigorous tests of these hypotheses and we make a plea for zoologists to also consider polyploidy as a possibility in continuing taxonomic surveys.
[本文引用: 1]
.
URLPMID:12140322 [本文引用: 1]
We present a publicly available software tool (http://www.unm.edu/~compbio/software/GenomeHistory) that identifies all pairs of duplicate genes in a genome and then determines the degree of synonymous and non-synonymous divergence between each duplicate pair. Using this tool, we analyze the relations between (i) gene function and the propensity of a gene to duplicate and (ii) the number of genes in a gene family and the family's rate of sequence evolution. We do so for the complete genomes of four eukaryotes (fission and budding yeast, fruit fly and nematode) and one prokaryote (Escherichia coli). For some classes of genes we observe a strong relationship between gene function and a gene's propensity to undergo duplication. Most notably, ribosomal genes and transcription factors appear less likely to undergo gene duplication than other genes. In both fission and budding yeast, we see a strong positive correlation between the selective constraint on a gene and the size of the gene family of which this gene is a member. In contrast, a weakly negative such correlation is seen in multicellular eukaryotes.
.
URLPMID:16079329 [本文引用: 1]
Author information: (1)Molecular Evolution and Bioinformatics Section, CEH-Oxford, UK.
.
URLPMID:4631527 [本文引用: 1]
New genes in human genomes have been found relevant in evolution and biology of humans. It was conservatively estimated that the human genome encodes more than 300 human-specific genes and 1000 primate-specific genes. These new arrivals appear to be implicated in brain function and male reproduction. Surprisingly, increasing evidence indicates that they may also bring negative pleiotropic effects, while assuming various possible biological functions as sources of phenotypic novelties, suggesting a non-progressive route for functional evolution. Similar to these fixed new genes, polymorphic new genes were found to contribute to functional evolution within species, for example, with respect to digestion or disease resistance, revealing that new genes can acquire new or diverged functions in its initial stage as prototypic genes. These progresses have provided new opportunities to explore the genetic basis of human biology and human evolutionary history in a new dimension.
.
URLPMID:19064677 [本文引用: 1]
Abstract Genomes contain a large number of genes that do not have recognizable homologues in other species and that are likely to be involved in important species-specific adaptive processes. The origin of many such "orphan" genes remains unknown. Here we present the first systematic study of the characteristics and mechanisms of formation of primate-specific orphan genes. We determine that codon usage values for most orphan genes fall within the bulk of the codon usage distribution of bona fide human proteins, supporting their current protein-coding annotation. We also show that primate orphan genes display distinctive features in relation to genes of wider phylogenetic distribution: higher tissue specificity, more rapid evolution, and shorter peptide size. We estimate that around 24% are highly divergent members of mammalian protein families. Interestingly, around 53% of the orphan genes contain sequences derived from transposable elements (TEs) and are mostly located in primate-specific genomic regions. This indicates frequent recruitment of TEs as part of novel genes. Finally, we also obtain evidence that a small fraction of primate orphan genes, around 5.5%, might have originated de novo from mammalian noncoding genomic regions.
.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}