 ,2
,2Shared functional modules for nasopharyngeal and oral squamous cell carcinoma identified by network analysis of transcriptomes
Yingjian Chen1,2, Yuanjun Liao1,2, Fan Lin1,2, Shengnan Sun1,2, Xiaolei Zhao2, Jiheng Qin2, Shaoqi Rao,2通讯作者:
编委: 周钢桥
收稿日期:2018-09-7修回日期:2018-10-26网络出版日期:2019-02-25
| 基金资助: |
Editorial board:
Received:2018-09-7Revised:2018-10-26Online:2019-02-25
| Fund supported: |
作者简介 About authors
陈应坚,硕士研究生,专业方向:公共卫生E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (751KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈应坚, 廖苑君, 林帆, 孙胜南, 赵小蕾, 覃继恒, 饶绍奇. 基于转录组数据的网络分析挖掘鼻咽癌与口腔鳞癌的共享功能模块[J]. 遗传, 2019, 41(2): 146-157 doi:10.16288/j.yczz.18-215
Yingjian Chen, Yuanjun Liao, Fan Lin, Shengnan Sun, Xiaolei Zhao, Jiheng Qin, Shaoqi Rao.
鼻咽癌(nasopharyngeal carcinoma, NPC)是一种起源于鼻咽上皮的鳞状细胞癌[1],在中国南方有很高的发病率。口腔鳞状细胞癌(oral squamous cell carcinoma, OSCC)是发生于口腔黏膜上皮的恶性肿瘤,是全球性第六大常见癌症[2]。NPC和OSCC在组织类型、流行病学、发病机制及治疗方法上具有较高的相似性和相关性,提示两者之间拥有共同的致病因素,而这个致病因素则可能来自共享的遗传因子—基因。这种一个基因座(locus)影响两个或多个性状(phenotypic trait)的现象称为基因多效性[3](pleiotropy),在癌症的遗传机制中普遍存在。例如,抑癌基因PTEN在细胞中具有磷酸酶依赖性活性和非磷酸酶依赖性活性,并控制着多种生物过程,包括维持基因组的稳定性、细胞存活、迁移、增殖和代谢,是已知的与多种癌症相关的多效性基因[4]。因此,挖掘NPC与OSCC的共享风险功能模块及其基因对寻找更有效的诊断方法,深度研究两者发病机制以及有效地预防NPC和OSCC具有重要意义。
近年来,基因表达谱数据迅速增加,利用生物信息学方法对基因表达谱数据进行深入研究已成为一个新的研究热点。目前对单一疾病的基因生物信息学的研究较多,然而对多种疾病的基因生物信息学的研究较少,因此,本研究利用大规模的转录数据来寻找NPC和OSCC的共享风险基因及其共享基因功能模块,以探索这两种疾病共享的分子机制。
1 材料与方法
1.1 数据来源
从GEO数据库(Gene Expression Omnibus,1.2 芯片数据的预处理
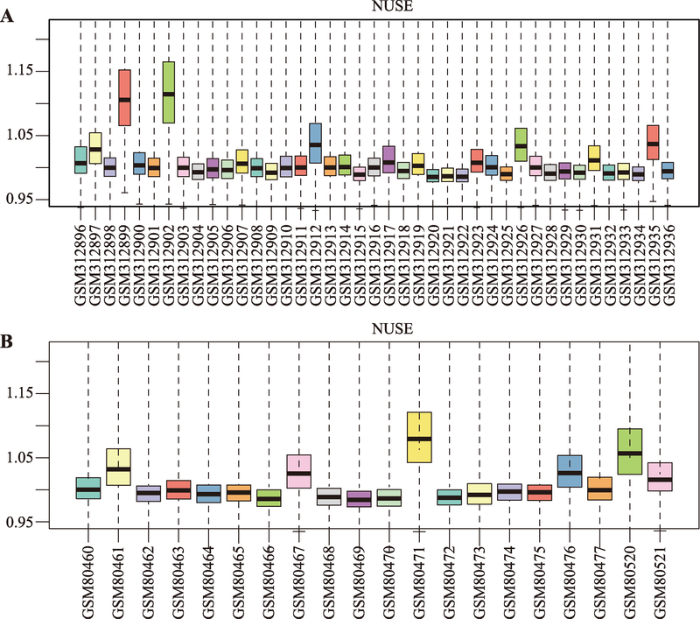
预处理通过质量控制,剔除不及格的芯片数据,只保留及格的进入下一步处理。然后通过标准化,将芯片数据中的基因表达值变换到一个可以比较的水平。本研究采用NUSE箱线图完成数据的质量控制。若所有芯片的质量都非常可靠,则NUSE值都在1附近;若NUSE值大于1.05,则芯片质量就有问题。本研究下载的基因芯片原始数据统一用RMA (Robust Multi-array Analysis)一体化算法对数据进行标准化和对数化。该过程利用R语言“affy”软件包完成。1.3 差异表达基因的筛选
采用倍数法(fold change, FC)和经验贝叶斯方法(empirical Bayes, EB)评估基因在病例对照中的差异表达程度和统计显著性。本研究采用相应实验平台的Affymetrix注释文件分别将两套芯片的探针编号注释为基因名称,若多个探针对应一个基因,则取所有探针的平均值作为基因的表达值。设定P值小于0.01且logFC的绝对值大于1,即FC>2作为阈值分别筛选NPC和OSCC的差异表达基因,然后对两种疾病的差异表达基因取交集,得到二者的共享风险基因。该过程利用R语言“limma”软件包完成。1.4 NPC与OSCC共享风险基因网络的构建
将筛选得到的NPC与OSCC共享风险基因作为初始种子基因(seed gene)。然后,将初始种子基因及其在PPI中的一级邻居基因,作为本研究构建NPC与OSCC共享风险基因网络的节点集合,对于集合中任意两个不同基因,如果其基因产物在PPI网络中存在互作,即视为网络中的连线,从而构建的NPC与OSCC共享风险基因网络,通过Cytoscape软件(版本3.6.1)进行PPI网络可视化。1.5 识别NPC与OSCC共享风险功能模块
功能模块在蛋白互作网络中的一个重要特征是功能模块内的基因间存在高度紧密的相互作用和联系,而模块内连接密度相比与其他功能模块间的联系要高很多。基于该研究假设,本研究拟对所构建的NPC与OSCC共享风险基因网络进行模块的划分。本研究采用Newman算法[5,6]对NPC与OSCC共享风险基因网络进行网络分解,挖掘网络中高度模块化的功能模块。该算法将网络分解问题转变成二次型优化问题,即求使目标函数Q最大的列向量s的取值:\[Q=\frac{1}{4m}{{s}^{T}}Bs\]
\[{{\mathbf{B}}_{\mathbf{ij}}}\mathbf{=}{{\mathbf{A}}_{\mathbf{ij}}}-\frac{{{\mathbf{k}}_{\mathbf{i}}}{{\mathbf{k}}_{\mathbf{j}}}}{2\mathbf{m}}\]
其中s是初始种子基因及其在PPI中的一级邻居基因组成的集合,A是集合s的邻接矩阵(adjacent matrix),用于表示网络中基因间的相互连接关系,Aij=1表示网络中基因i和基因j存在蛋白互作,反之Aij=0。m是网络中边的数目,ki和kj分别代表节点i和节点j的连通度。s是取值为1或-1的指示向量,si表示基因i所属模块的情况,即
\[{{s}_{i}}=\left\{ \begin{matrix} ^{{}}1{{,}^{{}}}i\in subnetwor{{k}^{{}}}1 \\ -1{{,}^{{}}}i\in subnetwor{{k}^{{}}}2 \\\end{matrix} \right.\]
根据柯西(Cauchy)不等式,当s取矩阵B的最大特征值对应的特征向量时,目标函数Q达到最大值。由于s只取1或-1,因此s只保留特征向量对应元素的正负号,从而根据特征向量的正负号将网络分解为两个子模块,重复这个过程,直到网络不能继续分解时,即可完成对网络的分解。最终,本研究将节点数大于30的子网络定义为网络模块。
1.6 网络拓扑属性分析与核心基因的识别
本研究对挖掘出的每个功能模块的拓扑属性进行评价,评价指标包括:(1)网络直径:指网络中任意两个连通节点间距离的最大值。网络直径代表了网络中任意两个节点间可能出现的最远距离,可从侧面反映网络的紧密性。
(2)特征路径长度:即模块中所有节点间的最短路径的平均值,反映了模块的紧密程度;
(3)连通度:即模块中直接与某一节点相连的边的数目,它不仅反映了单一节点在模块中的最基本的拓扑性质,而且是判断无标度网络属性的重要指标。无标度网络是网络中连通度的分布符合幂律分布,即p(k)∝k-a,其中k为接通度,α是指数参数,可通过极大似然法估计。通过Kolmogorov-Smirnov (KS)拟合优度检验评判网络的无标度性质。
无标度网络的属性很大程度上由少数的节点即核心基因(hub gene)决定的。虽然网络具有抗随机扰动的能力,但对中心节点的作用却很敏感。核心基因的识别一般通过检验其在网络中的连通度是否高出随机网络下的连通度期望值。该零假设可以通过泊松分布进行检验。那么,在一个节点数为N的随机网络中,某节点连通度等于t的概率为:
\[P(k=t)=\frac{{{\lambda }^{t}}{{e}^{-\lambda }}}{t!}\]
其中,λ=Np,表示随机网络中节点的期望连通度,p是网路中任意两个节点发生连接的概率。为控制假阳性率,FDR<0.01作为显著性水平。上述网络分析通过R软件实现,其中部分分析借助“igraph”软件包完成。
1.7 KEGG通路富集分析
KEGG (Kyoto Encyclopedia of Genes and Genomes)通路通过描述基因编码的生物大分子酶或者蛋白质间相互联系相互影响的情况以阐释基因及其产物的功能[7]。本研究利用在线注释网站DAVID软件(版本6.8)对每一个模块进行KEGG信号通路富集分析。为控制假阳性率,采用Fisher精确检验的方法,富集结果取FDR<0.001作为显著性基因富集。2 结果与分析
2.1 芯片数据的预处理
从GEO数据库获取NPC与OSCC基因表达谱芯片原始数据后,对原始数据进行预处理。采用NUSE箱线图的数据处理结果分别见图1A和图1B。结果显示,NPC的正常样本GSM312899、GSM312902和OSCC的正常样本GSM80520、肿瘤样本GSM80471的NUSE 值均大于1.05,说明这4个样本的芯片存在质量问题。为保证后续分析计算结果的可信度,删除NPC的正常样本GSM312899、GSM312902和OSCC的正常样本GSM80520、肿瘤样本GSM80471,最终获得的有效样本包括NPC正常样本8个和肿瘤样本31个,OSCC正常样本3个和肿瘤样本15个。基于RMA一体化算法,使用R语言“affy”包对两种疾病的样本芯片数据分别进行标准化和对数化。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1NUSE箱线图
A:NPC基因表达谱芯片的NUSE箱线图;B:OSCC基因表达谱芯片的NUSE箱线图。
Fig. 1NUSE Box plot
2.2 差异表达基因的筛选
采用倍数法和经验贝叶斯检验方法,按照P值小于0.01且logFC的绝对值大于1为阈值,共筛选出NPC差异表达基因1279个,OSCC差异表达基因1293个,二者的共同差异表达基因278个。2.3 NPC与OSCC共享风险基因网络的构建
通过提取种子基因及其在PPI网络中的一级邻居,最终构建出38个大小不同的独立的基因互作网络,包含1412个节点和1851条边,其中节点表示蛋白(或称基因),边表示蛋白互作,见图2。最大子网络由1290个节点和1766条边组成。后面的分析均基于网络中的最大子网进行。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2NPC与OSCC共享的差异表达基因的PPI网络图
红色节点表示上调差异表达基因编码蛋白;绿色表示下调差异表达基因编码蛋白,蓝色表示其他蛋白。
Fig. 2PPI network of shared differentially expressed genes for NPC and OSCC
2.4 疾病网络模块与核心基因的识别
利用Newman 算法对最大子网进行分解,最终得15个节点数大于30的功能模块。表1列出了各模块的基本特征和拓扑属性。可以发现规模大的模块其包含的初始种子基因相对多,平均每个节点连接的边数也多。不同网络节点数相差也很大,最大的模块(M1)由196个基因(其中初始种子基因有18个)及270条边,平均每个节点含有1.4条边,然而最小的模块(M14和M15)由34个基因(其中初始种子基因分别有3个和9个)和34条边组成,平均每个节点含有1条边。这些模块的拓扑属性接近,网络直径变化范围在4~9,特征路径长度均小于6,符合“六度分离理论”[8],各模块的指数参数α估计值在2~3之间,符合生物学上无标度网络的常见表现。本研究对节点连通度是否符合幂律分布进行KS拟合优度检验,检验结果显示各模块的连通度均近似服从幂律分布(P>0.05),支持了各模块的无标度性,可认为是无标度网络。Table 1
表1
表1 各模块的基本特征和拓扑属性
Table 1
| 模块 | 初始种子基因个数 | 节点数 | 边数 | 网络直径 | 特征路径长度 | α值 | P值 |
|---|---|---|---|---|---|---|---|
| 模块1 | 18 | 196 | 270 | 7 | 2.980 | 2.215 | 1.000 |
| 模块2 | 16 | 130 | 173 | 8 | 4.022 | 2.123 | 1.000 |
| 模块3 | 16 | 128 | 189 | 7 | 3.061 | 2.054 | 0.817 |
| 模块4 | 10 | 88 | 96 | 9 | 3.111 | 2.487 | 1.000 |
| 模块5 | 9 | 77 | 84 | 7 | 3.389 | 2.458 | 0.999 |
| 模块6 | 7 | 70 | 72 | 6 | 3.041 | 2.623 | 1.000 |
| 模块7 | 6 | 67 | 68 | 5 | 3.395 | 2.665 | 0.927 |
| 模块8 | 7 | 56 | 60 | 7 | 3.285 | 2.429 | 0.997 |
| 模块9 | 6 | 55 | 56 | 7 | 3.277 | 2.613 | 0.999 |
| 模块10 | 8 | 48 | 48 | 8 | 3.097 | 2.459 | 1.000 |
| 模块11 | 6 | 46 | 47 | 7 | 4.074 | 2.422 | 0.981 |
| 模块12 | 4 | 41 | 41 | 6 | 3.149 | 2.534 | 0.990 |
| 模块13 | 5 | 38 | 43 | 9 | 4.058 | 2.172 | 1.000 |
| 模块14 | 3 | 34 | 34 | 4 | 2.929 | 2.555 | 0.961 |
| 模块15 | 9 | 34 | 34 | 9 | 4.447 | 2.222 | 1.000 |
新窗口打开|下载CSV
利用泊松检验,设定显著性水准为FDR<0.01,共得到58个核心基因。通过检索PubMed数据库,本研究发现共有34个核心基因被报道与NPC或OSCC相关,它们直接或间接参与了NPC和OSCC的发生发展,如增殖细胞核抗原(proliferating cell nuclear antigen, PCNA)、周期蛋白依赖激酶1 (cyclin- dependent kinase 1, CDK1)、信号转导与转录激活因子1 (signal transducer and activator of transcription 1, STAT1)、趋化因子CCL5 (C-C motif chemokine ligand 5, CCL5)等蛋白的编码基因。其余24个核心基因暂无文献报道与NPC或OSCC相关,这些基因可能对疾病的发生发展提供新的预测方向,如母体胚胎亮氨酸拉链激酶(maternal embryo leucine zipper kinase, MELK)、非转移性细胞蛋白1 (non-metastatic cell protein 1, NME1)、天冬氨酸特异性半胱氨酸蛋白酶-6 (caspase-6, CASP6)、RacGCT酶活性蛋白-1 (RACGAP1)、抑制素β A亚基基因(inhibin subunit beta A, INHBA)等蛋白的编码基因。核心基因数占模块中的初始种子基因数的44.6%。含有核心基因最多的模块是M2模块,共有10个核心基因。而含有核心基因最少的模块是M10模块,仅有含有1个核心基因(表2)。
Table 2
表2
表2 各个模块的核心基因
Table 2
| 模块 | 初始种子基因个数 | 核心基因个数 | 核心基因 |
|---|---|---|---|
| 模块1 | 18 | 6 | PCNA*、FEN1、CDK1*、CCNA2*、CCNB1*和TOP2A* |
| 模块2 | 16 | 10 | PSMA1、MCM6*、DBF4、MCM10、MCM2*、HOXA1*、 TAP1*、RAD54B、WDYHV1和RIBC2 |
| 模块3 | 16 | 8 | MMP1*、SPARC*、FN1*、COL1A2*、NID1、COL4A1*、COL1A1*和COL3A1* |
| 模块4 | 10 | 3 | CCL5*、STAT1*和MDK* |
| 模块5 | 9 | 4 | PRKDC*、CASP6*、MELK和GZMB* |
| 模块6 | 7 | 3 | SUMO1*、TOPBP1和PHB2 |
| 模块7 | 6 | 5 | ITGAV*、LGALS1*、SPP1*、CD14*和VCAM1* |
| 模块8 | 7 | 4 | BUB1B*、NDC80、AURKA*和MAD2L1* |
| 模块9 | 6 | 3 | COPS5、RRAD*和NME1 |
| 模块10 | 8 | 1 | KPNA2* |
| 模块11 | 6 | 2 | RACGAP1和INHBA |
| 模块12 | 4 | 3 | PLAT、SERPING1和CEP55 |
| 模块13 | 5 | 2 | CBX3和CBX1 |
| 模块14 | 3 | 2 | HSP90AB1和GMNN |
| 模块15 | 9 | 2 | NGFRAP1和CENPF* |
新窗口打开|下载CSV
2.5 模块的功能富集分析
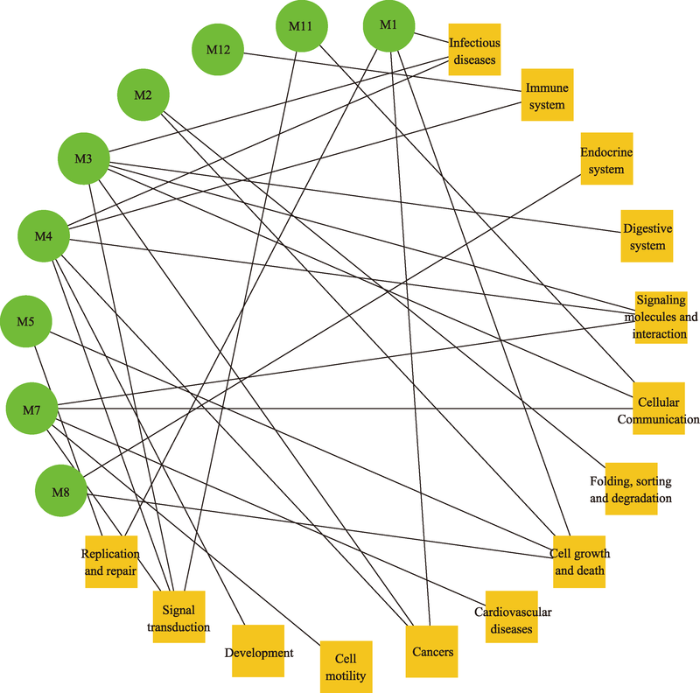
KEGG通路富集分析结果显示,除了模块M6、M9、M10、M13、M14、M15外,其余9个模块至少富集到一条生物学通路(表3)。这些模块的显著富集于ECM受体相互作用(hsa04512)、黏着斑(hsa04510)、细胞周期(hsa04110)、P53信号通路(hsa04115)、PI3K-Akt信号通路(hsa04151)、细胞因子—细胞因子受体相互作用(hsa04060)等45个有统计学意义的相关通路(FDR值<0.001)。本研究根据富集到的KEGG通路类别评估了这9个功能模块之间的相互作用。为了清楚地展示各模块之间的关系,本研究用Cytoscape软件画出模块和通路分类关系图(见图3)。图中的功能类是KEGG通路的分类:信号转导(signal transduction);信号分子和相互作用(signaling molecules and interaction);细胞运动(cell motility);细胞生长和死亡(cell growth and death);复制与修复(replication and repair);细胞通讯(cell communication);免疫系统(immune system);内分泌系统(endocrine system);癌症(cancer);心血管疾病(cardiovascular diseases);发育(development);消化系统(digestive system);折叠、分类和降解(folding, sorting and degradation);传染病(infectious diseases)。其中细胞生长和死亡(M1、M2、M5和M8)和信号转导(M3、M4、M7和M11)连通度最高,体现了这些功能与NPC和OSCC的发病机制有密切的联系。本研究的结果表明,各模块之间不是孤立的,功能相一致的模块之间相互作用,共同影响疾病的发生发展。Table 3
表3
表3 各个模块的KEGG通路分析结果
Table 3
| 模块 | KEGG通路 | FDR(<0.001) | 模块 | KEGG通路 | FDR (<0.001) |
|---|---|---|---|---|---|
| M1 | hsa04110:细胞周期 | 3.16E-23 | M4 | hsa04060:细胞因子—细胞因子受体相互作用 | 6.23E-14 |
| hsa03430:错配修复 | 5.75E-22 | hsa04062:趋化因子信号通路 | 4.03E-12 | ||
| hsa03410:碱基切除修复 | 2.44E-11 | hsa04630:Jak-STAT信号通路 | 4.08E-09 | ||
| hsa03030:DNA复制 | 9.30E-11 | hsa05162:麻疹 | 2.77E-07 | ||
| hsa03420:核苷酸切除修复 | 2.12E-10 | hsa04380:破骨细胞分化 | 3.13E-06 | ||
| hsa04115:p53信号通路 | 4.32E-08 | hsa05168:单纯疱疹感染 | 1.45E-05 | ||
| hsa05200:癌症相关通路 | 7.12E-07 | hsa05203:病毒致癌 | 5.65E-05 | ||
| hsa05222:小细胞肺癌 | 1.28E-05 | hsa05160:丙型肝炎 | 4.91E-04 | ||
| hsa05161:乙型肝炎 | 3.06E-05 | M5 | hsa03450:非同源末端连接 | 3.77E-05 | |
| hsa05166:HTLV-I感染 | 3.71E-05 | hsa04210:细胞凋亡 | 5.56E-04 | ||
| hsa03460:范可尼贫血通路 | 8.73E-05 | M7 | hsa04810:肌动蛋白细胞骨架调控 | 2.43E-10 | |
| hsa05215:前列腺癌 | 1.81E-04 | hsa04514:细胞黏附分子 | 2.00E-06 | ||
| hsa05169:EB病毒感染 | 2.48E-04 | hsa04512:ECM受体相互作用 | 5.55E-06 | ||
| M2 | hsa04110:细胞周期 | 2.64E-12 | hsa04510:黏着斑 | 8.62E-06 | |
| hsa03050:蛋白酶体 | 7.77E-08 | hsa04151:PI3K-Akt信号通路 | 4.04E-05 | ||
| M3 | hsa04512:ECM受体相互作用 | 1.73E-38 | hsa05410:肥厚型心肌病 | 8.97E-04 | |
| hsa04510:黏着斑 | 5.70E-28 | M8 | hsa04110:细胞周期 | 4.04E-09 | |
| hsa04151:PI3K-Akt信号通路 | 1.86E-21 | hsa04114:卵母细胞减数分裂 | 3.29E-08 | ||
| hsa04974:蛋白质的消化和吸收 | 5.20E-15 | hsa04914:孕激素介导的卵母细胞成熟 | 1.11E-04 | ||
| hsa05146:阿米巴病 | 2.02E-08 | M11 | hsa04350:TGF-β信号通路 | 1.22E-10 | |
| hsa05200:癌症相关通路 | 4.34E-06 | hsa04550:调节干细胞多能性信号通路 | 2.00E-05 | ||
| hsa05222:小细胞肺癌 | 4.37E-06 | M12 | hsa04610:补体和凝血级联 | 1.22E-13 | |
| hsa05205:癌症中的蛋白聚糖 | 9.25E-05 |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3共享功能模块与通路的关系图
Fig. 3The functional relationships between the identified shared modules in context of pathway involvements
3 讨论
基因多效性是癌症遗传机制中的普遍现象,大量全基因组关联研究(genome-wide association study, GWAS)的结果已经证实不同癌症表型可共享风险基因,即单个基因的突变可能引起不同的病理效应而参与多种癌症的发生发展[9]。基于基因多效性,本研究从GEO数据库分别下载NPC和OSCC基因芯片数据进行差异基因分析,以PPI知识为向导,两种癌症的共同差异表达基因为种子基因,构建NPC与OSCC共享风险基因网络。本研究利用Newman算法对NPC与OSCC共享风险基因网络进行网络分解,挖掘网络中高度模块化的共享功能模块和核心基因。通过阐明功能模块行使的生物学功能来挖掘NPC与OSCC的共享通路。在本研究中,共提取了15个NPC与OSCC相关的功能模块,并通过包括直径、特征路径长度、连通度等拓扑属性指标评价各模块的拓扑性质,结果显示各个模块都满足无标度性。模块中的核心基因担负着维系模块中各基因间相互联系和模块结构稳定性的作用,在模块行使特定生物学功能的过程中肩负着功能核心的作用,是功能模块必不可少的关键基因。通常,研究模块中核心基因的功能有助于阐明模块的生物学功能。
多数核心基因,如PCNA、CDK1、STAT1和CCL5等基因均被报道直接或间接与NPC和OSCC相关。它们可能直接或间接参与了肿瘤的生长和分化,如癌细胞的增殖、血管形成,侵袭和转移等。例如,PCNA基因的表达在真核细胞DNA复制的调控中起着至关重要的作用,是与多种肿瘤发生发展密切相关的基因。有研究表明,靶向PCNA基因的小干扰RNA (siRNA)通过沉默PCNA基因,可有效抑制PCNA对NPC细胞增殖的作用[10]。最近研究发现,高度侵袭性肿瘤表现出TP53突变和PCNA活性,从而提高OSCC侵袭前的增殖能力[11],可用于评估异型增生和不同级别口腔鳞癌的增殖能力和侵袭性[12]。还有研究称,PCNA可以成为区分正常口腔粘膜和口腔鳞癌组织的生物标志物[13]。CDK1通过调节细胞中心体周期和有丝分裂的发生,在真核细胞周期的调控中发挥关键作用。目前有文献报道,通过草酸盐对乳酸脱氢酶(lactate dehydrogenase, LDH)的抑制,调控CDK1/cyclinb1通路,从而诱导G2/M细胞周期阻滞。CDK1/cyclin B1通路有望成为NPC治疗的靶点[14]。Chen等[15]报道发现,CDK1在OSCC中高表达,可能与OSCC的恶性程度有关,可作为判断OSCC恶性程度和预测预后的有用指标。STAT1可能在抑制肿瘤细胞生长,促进免疫监视、凋亡和细胞周期阻滞方面发挥关键作用[16]。最近有研究报道,STAT1表达下调导致细胞周期S期减少,G2/M期增加,有助于提高NPC细胞的放射敏感性[17]。有研究表明,STAT1的活化主要见于分化程度较高的肿瘤,如果OSCC患者的肿瘤中保留了STAT1的表达,并表现出STAT1的活化,则对辅助化疗有较好的疗效[18]。近年来报道证实,CCL5能直接参与恶性肿瘤的发生发展,在前列腺癌[19]、结肠癌[20]、肺癌[21]、乳腺癌[22]中都有过表达,并被证明通过不同的信号途径促进多种癌症类型的肿瘤细胞的生长和(或)运动。CCL5通过PI3K/AKT和HIF-1a途径诱导肿瘤血管生成,与人鼻咽癌血管生成密切相关,是NPC潜在的治疗靶点[23]。在慢性炎症状态下CCL5能激活VEGF和(或) STAT信号通路,促进细胞增殖和血管生成,可通过激活STAT和NF信号通路促进肿瘤形成[24]。有研究证实,CCL5通过其特异性高亲和力受体CCR5的诱导,增加OSCC的移动力[25]。
此外,本研究还发现了一些鲜有报道的与NPC和OSCC相关的核心基因,例如MELK、NME1、RACGAP1和INHBA等基因。这些核心基因在其他肿瘤的发生发展中起到重要作用,可能与NPC和OSCC也存在密切相关。例如,MELK高表达,与肝癌[26]、肾上腺皮质癌[27]、乳腺癌[28]、胃癌[29]和脑癌[30]患者预后不良相关。MELK在肝癌中高表达,可通过FOXM1/b-catenin信号通路调控细胞周期相关基因和有丝分裂进程,沉默MELK可以降低肝癌细胞的活力和增殖,诱导细胞凋亡和有丝分裂,MELK的稳定沉默能抑制肝癌细胞的侵袭、干性和致瘤性[26]。有报道指出,NME1基因的编码蛋白被认为是潜在的典型转录因子,通过其DNA结合活性来调节基因转录与表达,与肿瘤细胞分化和转移抑制功能等高度相关。此外,NME1也参与DNA修复,这表明NME蛋白的表达减少可能导致基因组不稳定,从而导致癌症的发展[31]。RACGAP1具有调节细胞质分裂和细胞分化的作用。有文献报道,RACGAP1能激活RhoA和ERK蛋白,增强卵巢上皮癌细胞的迁移和侵袭能力[32]。RACGAP1的过表达与直肠癌的进展和预后密切相关,其生物学和临床意义为判断淋巴结转移和预后不良提供了充分的证据[33]。INHBA编码TGF-β超家族蛋白,在不同类型的肿瘤中发挥着重要作用。INHBA在食管腺癌中高表达,通过启动子去甲基化和组蛋白乙酰化可促进细胞增殖[34],也与淋巴结转移有关[35]。
为了阐明每个模块的生物学功能,本研究利用DAVID在线软件作了基于KEGG信号通路的富集分析。这些模块显著富集于ECM受体相互作用(has04512)、黏着斑(hsa04510)、细胞周期(hsa04110)、p53信号通路(hsa04115)、PI3K-Akt信号通路(hsa04151)、细胞因子—细胞因子受体相互作用(hsa04060)等。其中最显著相关的信号通路是ECM受体相互作用(hsa04512, M3)。ECM在细胞形态、增殖、迁移、分化、凋亡和癌变等多个生物学过程中发挥着重要的作用[36]。黏着斑是介导细胞对ECM粘附的调节效应的亚细胞结构[37]。根据KEEG信号通路富集分析显示,本研究富集到的ECM受体相互作用和粘着斑通路有许多基因重叠,例如编码胶原蛋白、整合素和层粘连蛋白等基因,有密切的相互作用。这些相互作用有利于癌细胞的增殖、运动、分化和ECM代谢,同时抑制细胞死亡、平稳的极化生长和ECM的稳定性[38]。ECM受体相互作用和粘着斑信号通路对癌细胞有明显调控的作用。在p53信号通路中,抑癌基因TP53通过诱导细胞周期阻滞、凋亡或衰老,在细胞对各种应激信号的反应中起着至关重要的作用[39]。TP53的失活能使TP53调控的靶基因异常增殖和(或)表达缺失,可使细胞周期G1/S检查点失控,细胞凋亡失控,导致细胞癌变[40]。另外,模块与模块之间不是孤立的,功能相一致的模块之间相互作用,共同影响疾病的发生发展。模块M1、M2、M5和M8与细胞生长与死亡有关,共同调控细胞周期。模块M3和M7主要富集于ECM受体相互作用与黏着斑信号通路,共同协调参与癌细胞的增殖与迁移。M4模块主要富集于细胞因子—细胞因子受体相互作用、趋化因子信号通路和Jak-STAT信号通路,模块M11主要富集于TGF-β信号通路,M12主要富集于补体与凝血级联信号通路,这些信号通路都参与癌症的炎症反应,说明模块M4、M11和M12之间有密切联系。
总而言之,本研究通过对NPC和OSCC芯片数据的挖掘以及生物信息学分析,筛选出15个共享功能模块,58个密切相关的核心基因(如PCNA、CDK1、STAT1和CCL5等)以及p53信号通路、ECM受体相互作用、黏着斑等在NPC和OSCC的发生发展、侵袭、转移可能起着重要作用的信号通路,为研究NPC与OSCC的共享发病机制、肿瘤标志物及治疗靶点的筛选方面提供了更多的信息,有助于解释多效性基因在致癌过程中所起的作用。除此之外,本研究还发现了一些鲜有报道的核心基因,如MELK、NME1、RACGAP1和INHBA等,这些新发现为探索NPC与OSCC的共同发生发展机制提供了新线索。综上所述,本研究表明鼻咽癌和口腔鳞癌具有相似的致癌机制,所挖掘的共享模块可能是这两种疾病演化的核心分子相互作用机制。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
URLPMID:28416242 [本文引用: 1]

Abstract Oral squamous cell carcinoma (OSCC) is a common cancer worldwide. Besides tobacco use and alcohol consumption, human papillomavirus (HPV) infection has also been identified as a risk factor for OSCC recently. The OSCC incidence has increased in recent years, especially among younger women. The purpose of this article is to review clinical and epidemiological studies on the association between HPV infection and OSCCs, and the efficacy of HPV vaccine, so as to provide possible policy implications for preventing HPV-positive OSCC. It is necessary to review the present related body of knowledge to determine whether the association between HPV infection and OSCC has been thoroughly studied. The study was based on literature review. Studies were identified using electronic databases including MEDLINE, PubMed, EMBASE, etc. The inclusion and exclusion criteria were based on consultation from a panel of experts in this area and carefully designed. Based on a systematic review of literatures, HPV infection is a possible cause for the incidence of HPV-positive OSCCs. The prevalence of HPV infection possibly contributed to the increasing trends of HPV-positive OSCCs. Oral HPV infection is a form of HPV transmission. Oral sex behaviors and open-mouthed kissing are probably reasons for oral HPV infection. We also have some epidemiological evidences proving that HPV vaccine provides a possible solution for preventing oral HPV infection. Increased awareness of HPV-positive OSCCs is essential due to the severity of this problem. Biological and epidemiological data regarding the link between sexual behavior and HPV-associated cancers indicate a probable connection, although definitive data are needed. Future studies are needed to investigate the mechanisms of how HPV infection causes HPV-positive OSCCs, whether HPV vaccine provides a prevention for OSCCs, and other important issues. Copyright 2017 Elsevier Inc. All rights reserved.
URLPMID:21062962 [本文引用: 1]
No abstract available.
URL [本文引用: 1]
[本文引用: 1]
URLPMID:15697438 [本文引用: 1]
The discovery and analysis of community structure in networks is a topic of considerable recent interest within the physics community, but most methods proposed so far are unsuitable for very large networks because of their computational cost. Here we present a hierarchical agglomeration algorithm for detecting community structure which is faster than many competing algorithms: its running time on a network with n vertices and m edges is O (md log n) where d is the depth of the dendrogram describing the community structure. Many real-world networks are sparse and hierarchical, with m approximately n and d approximately log n, in which case our algorithm runs in essentially linear time, O (n log(2) n). As an example of the application of this algorithm we use it to analyze a network of items for sale on the web site of a large on-line retailer, items in the network being linked if they are frequently purchased by the same buyer. The network has more than 400 000 vertices and 2 x 10(6) edges. We show that our algorithm can extract meaningful communities from this network, revealing large-scale patterns present in the purchasing habits of customers.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
Arbitrarily selected individuals (N=296) in Nebraska and Boston are asked to generate acquaintance chains to a target person in Massachusetts, employing "the small world method" (Milgram, 1967). Sixty-four chains reach the target person. Within this group the mean number of intermediaries between starters and targets is 5.2. Boston starting chains reach the target person with fewer intermediaries than those starting in Nebraska; subpopulations in the Nebraska group do not differ among themselves. The funneling of chains through sociometric "stars" is noted, with 48 per cent of the chains passing through three persons before reaching the target. Applications of the method to studies of large scale social structure are discussed.
URLPMID:4104202 [本文引用: 1]
Abstract Genome-wide association studies have identified many variants that each affects multiple traits, particularly across autoimmune diseases, cancers and neuropsychiatric disorders, suggesting that pleiotropic effects on human complex traits may be widespread. However, systematic detection of such effects is challenging and requires new methodologies and frameworks for interpreting cross-phenotype results. In this Review, we discuss the evidence for pleiotropy in contemporary genetic mapping studies, new and established analytical approaches to identifying pleiotropic effects, sources of spurious cross-phenotype effects and study design considerations. We also outline the molecular and clinical implications of such findings and discuss future directions of research.
URLPMID:27698802 [本文引用: 1]
Nasopharyngeal carcinoma (NPC) is the most common cancer originating from the nasopharynx, and can be induced by infection with Epstein-Barr virus (EBV). To study the mechanisms of EBV-associated NPC, a microarray of the GSE12452 dataset was analyzed. GSE12452 was downloaded from Gene Expression Omnibus and consisted of 31 NPC samples and 10 normal healthy nasopharyngeal tissue samples. The differentially-expressed genes (DEGs) were screened using the linear models for microarray data package in R. Using Database for Annotation, Visualization and Integrated Discovery software, potential functions of the DEGs were predicted by Gene Ontology and pathway enrichment analyses. With the information from the Search Tool for the Retrieval of Interacting Genes/Proteins database, the protein-protein interaction (PPI) network was visualized by Cytoscape. Furthermore, modules of the PPI network were searched using ClusterONE in Cytoscape. A total of 951 DEGs were screened in the NPC samples compared with the normal healthy nasopharyngeal tissue samples. Function enrichment indicated that the upregulated genes were associated with the cell cycle, cytoskeleton organization and DNA metabolism. Meanwhile, the downregulated genes were mainly associated with cell differentiation, hormone metabolism, inflammatory response and immune response. PPI networks for the DEGs suggested that upregulated mitotic arrest deficient 2-like 1 (MAD2L1; degree=133), proliferating cell nuclear antigen (PCNA; degree=125) and cyclin B1 (CCNB1; degree=115), and downregulated member A1 of aldehyde dehydrogenase 1 (ALDH1A1; degree=15) may be of great importance as they exhibited higher degrees on interaction. Mucin 1 (MUC1) was a key node of module 4. Overall, the study indicated thatMAD2L1,CCNB1,PCNA,ALDH1A1andMUC1may have a correlation with EBV-associated NPC.
URLPMID:21501231Magsci [本文引用: 1]
J Oral Pathol Med (2011) 40: 693-698Background: Abnormalities in cell-cycle-controlling genes are important in the malignant transformation and proliferation of tumors. Among these genes, the tumor suppressor gene p53 is the most notable, and its mutations provide an indicator of tumor progression and prognosis. Proliferating cell nuclear antigen (PCNA) is a highly conserved nuclear protein that is expressed during cell replication and DNA repair. This study examined the expression of p53 and PCNA at the invasive front of oral squamous cell carcinomas (OSCC) by immunohistochemical staining, and investigated the relationship of these proteins to clinicopathological findings and prognosis.Methods: Fifty-nine biopsy cases of OSCC were examined by immunohistochemical staining. Clinicopathological data were gathered and patient survival was analyzed.Results: The p53 labeling index (p53-LI) and PCNA labeling index (PCNA-LI) were examined at the invasive front of the tumors. A high p53-LI (p53+) was observed in 17 of the 59 cases (28.8%) and a high PCNA-LI (PCNA+) was observed in 28 of the 59 cases (47.5%). Among the modes of cancer invasion, many of the p53+/PCNA+ cases could be confirmed as highly invasive cancer (P < 0.05). In addition, the p53+/PCNA+ cases showed a high risk of tumor recurrence compared with the other expression forms, and patients with p53+/PCNA+ had a worse prognosis than those with the other expression forms. High labeling indices of p53 and PCNA are associated with poor prognosis in patients with OSCC.Conclusion: We suggest that it is important to investigate the expression of p53 and PCNA at the invasive front of OSCC.
URLPMID:26266215 [本文引用: 1]
Abstract INTRODUCTION: Cancer has multifactorial aetiology and is a multistep process involving initiation, promotion and tumour progression. Cellular proliferation is one of the important indicators for the biologic aggressiveness of a malignant lesion. The dysregulated proliferation may be a significant change to determine the potential prognosis of various malignant tumours. AIM: The aim of this study was to evaluate the expression of proliferating cell nuclear antigen (PCNA) as an indicator for clinical aggressiveness in oral premalignancy and squamous cell carcinoma. MATERIALS AND METHODS: A total of 50 blocks were taken from the Department of Oral Pathology which was diagnosed previously histopathologically. It comprised of normal oral mucosa (10), dysplasia (10) and grades of oral squamous cell carcinoma (30) of patients between the age group of 40-60 years. From each block, sections of 4 micro metre thicknesses were prepared and placed on poly- L lysine coated slides. These sections were immunohistochemically stained with monoclonal proliferating cell antibody (PC10). The stained slides were evaluated by a single examiner for cell count. RESULTS: A comparison between study groups and controls showed a probability value (p-value) < 0.05. Significant increase in the proliferative index from the normal to oral squamous cell carcinoma was noticed. Poorly differentiated squamous cell carcinoma showed maximum proliferative index followed by moderately differentiated, well differentiated squamous cell carcinoma, dysplasia and normal mucosa. CONCLUSION: Present study concluded that PCNA index can be used to assess the proliferation and aggressiveness in dysplasia and different grades oral squamous cell carcinoma.
URLPMID:4774281 [本文引用: 1]
To evaluate the cell proliferation rate by the expression of proliferating cell nuclear antigen (PCNA) and argyrophilic nucleolar organizing region (AgNOR) counts and to assess its usefulness as a marker for malignant potential in oral epithelial lesions. The study group included 30 cases of leukoplakia, 15 nondysplastic (NDL), 15 dysplastic (DL), 15 cases of oral squamous cell carcinoma (OSCC) and 5 cases of normal oral mucosa. Formalin fixed paraffin embedded tissues were subjected to immunohistochemical staining for PCNA and AgNOR technique. The PCNA labeling index (LI) and the AgNOR dots were evaluated for the entire sample. ANOVA, Tukey honestly significant difference, Pearson's correlation. In this study, the AgNOR count of OSCC was lower than the DL lesions moreover the AgNOR counts were found to be higher in normal mucosa as compared to the DL and the NDL epithelium. The study results also showed that the mean AgNOR count failed to distinguish between DL and NDL lesions. Overall we observed increased PCNA expression from normal epithelium to NDL to DL lesion. Based on the findings of the present study on oral epithelial precancerous and cancerous lesions we conclude that mean AgNOR count alone cannot be a valuable parameter to distinguish between the normal, NDL, DL epithelium and OSCC but, on the other hand, we found out that PCNA can be a useful biomarker for delineating normal epithelium from DL epithelium and OSCC.
URLPMID:24064966Magsci [本文引用: 1]
An elevated rate of glucose consumption and the dependency on aerobic glycolysis for ATP generation have long been observed in cancer cells, a phenomenon known as the Warburg effect. The altered energy metabolism in cancer cells provides an attractive opportunity for developing novel cancer therapeutic strategies. Lactate dehydrogenase (LDH), which catalyzes the transformation of pyruvate to lactate, plays a vital role in the process of glycolysis. It has been reported that the level of LDH-A expression is increased both in head and neck cancer cells and in the blood serum of nasopharyngeal carcinoma (NPC) patients, and is associated with poor prognosis. However, the effect of LDH-A inhibition on NPC cells remains unknown. Here, in the present study, we found that oxamate, a classical inhibitor of LDH-A, suppressed cell proliferation in a dose- and time-dependent manner both in CNE-1 and CNE-2 cells, two NPC cancer cell lines. LDH inhibition by oxamate induced G(2)/M cell cycle arrest via downregulation of the CDK1/cyclin B1 pathway and promoted apoptosis through enhancement of mitochondrial ROS generation. N-acetylcysteine, a specific scavenger of ROS, significantly blocked the growth inhibition effect induced by oxamate. We also identified that oxamate increased sensitivity to ionizing radiation in the two NPC cancer cell lines. Furthermore, we verified similar results in tumor xenograft models. Collectively, these results suggest that LDH-A may serve as a promising therapeutic target for NPC treatment.
URLPMID:25129248 [本文引用: 1]
To evaluate the clinical significance of cyclin-dependent kinase 1 (CDK1) in 77 oral squamous cell carcinomas (OSCC) using immunohistochemical methods.Immunohistochemical expression of CDK1 was compared with various clinicopathological features in 77 OSCC and 60 controlled epithelia adjacent to the tumours. In addition, correlation of CDK1 expression and prognostic and the 5-year accumulative survival rate of OSCC were investigated.The CDK1 protein was expressed in 52 cases of 77 tumor tissues (67.5%), compared with 21 cases of 60 controlled (35.0%). The expression of CDK1 was significantly correlated with the histological grade of OSCC (P<0.05). The CDK1 protein was over-expressed in recurrent tumors or in those with lymph node metastasis. Statistical analysis showed a significant reduction in the 5-year accumulative survival rate in CDK1 positive cases compared with CDK1 negative cases (P<0.05). Namely, the CDK1 positive patients had poor prognosis.The expression of CDK1 might serve as malignant degree and prognostic markers for the survival of OSCC.
URLPMID:28950072 [本文引用: 1]
Abstract The first STAT family member, STAT1, is an essential component of interferon (IFN)-signaling, which mediates several cellular functions in response to stimulation by cytokines, growth factors, and hormones, such as the IFNs and IL-6. The role and significance of STAT1 in cancer biology have been studied for a decade. The majority of evidence shows that activating STAT1 plays a tumor suppressor role in cancer cells. Nevertheless, results from some experiments and clinical studies suggest that STAT1 also exerts tumor promoter effects under specific conditions. In some malignant phenotypes, STAT1 can function either as an oncoprotein or tumor suppressor in the same cell type, depending on the specific genetic background. Thus, the function of STAT1 in cancer biology remains a mystery. In this review, we discuss both the "friend" and "foe" features of STAT1 by summarizing its tumor suppressor or oncogenic functions and mechanisms. To explain how STAT1 may mediate its tumor suppressor effects, we discuss several possible mechanisms, one of which is linked to the role of STAT1, an isoform of STAT1.
URL [本文引用: 1]
Radioresistance remains a major obstacle for clinicians in the treatment of nasopharyngeal carcinoma (NPC). Others and we have reported that signal transducer and activator of transcription 1 (STAT1) may be as an important gene for resistance to radiation. However, the relationship between STAT1 and radioresistance is still elusive. In this study, by constitutive silencing STAT1 in human radioresistant nasopharyngeal carcinoma CNE-2R cell line, we showed that inhibition of STAT1 enhanced radiosensitivity of CNE-2R. Furthermore, knockdown of STAT1 led to growth suppression and apoptosis promotionin vitroandin vivo. Moreover, cells with low STAT1 expression increased G2/M phase and decreased S phase at 2Gy. These result revealed that knockdown of stat1 expression could sensitizes the CNE-2R to radiotherapy, But the exact mechanism needs to be further clarified.
[本文引用: 1]
URL [本文引用: 1]
Chemokines and their receptors have key roles in cancer progression. The present study investigated chemokine activity in the prostate cancer bone metastasis microenvironment. Growth and migration of human prostate cancer cells were assayed in cocultures with bone stromal cells. The migration ofLNCaP cells significantly increased when co‐cultured with bone stromal cells isolated from prostate cancer bone metastases. Cytokine array analysis of conditioned medium from bone stromal cell cultures identifiedCCL5 as a concentration‐dependent promoter ofLNCaP cell migration. The migration ofLNCaP cells was suppressed when C‐C motif ligand 5 (CCL5) neutralizing antibody was added to cocultures with bone stromal cells. Knockdown of androgen receptor with small interferingRNAincreased the migration ofLNCaP cells compared with control cells, andCCL5 did not promote the migration of androgen receptor knockdownLNCaP. ElevatedCCL5 secretion in bone stromal cells from metastatic lesions induced prostate cancer cell migration by a mechanism consistent withCCL5 activity upstream of androgen receptor signaling.
URLPMID:25546229 [本文引用: 1]
Tumor micro-environment is a critical factor in the development of cancer. The aim of this study was to investigate the inflammatory cytokines secreted by tumor-associated dendritic cells (TADCs) that contribute to enhanced migration, invasion, and epithelial-to-mesenchymal transition (EMT) in colon cancer. The administration of recombinant human chemokine (C-C motif) ligand 5 (CCL5), which is largely expressed by colon cancer surrounding TADCs, mimicked the stimulation of TADC-conditioned medium on migration, invasion, and EMT in colon cancer cells. Blocking CCL5 by neutralizing antibodies or siRNA transfection diminished the promotion of cancer progression by TADCs. Tumor-infiltrating CD11c(+) DCs in human colon cancer specimens were shown to produce CCL5. The stimulation of colon cancer progression by TADC-derived CCL5 was associated with the up-regulation of non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT-1), which subsequently increased the expression of Snail. Blocking MALAT-1 significantly decreased the TADC-conditioned medium and CCL5-mediated migration and invasion by decreasing the enhancement of Snail, suggesting that the MALAT-1/Snail pathway plays a critical role in TADC-mediated cancer progression. In conclusion, the inhibition of CCL5 or CCL5-related signaling may be an attractive therapeutic target in colon cancer patients.
URLPMID:19073147Magsci [本文引用: 1]
CCL5 (previously called RANTES) is in the CC-chemokine family and plays a crucial role in the migration and metastasis of human cancer cells. Besides, integrins are the major adhesive molecules in mammalian cells. Here we found CCL5 increased the migration and cell surface expression of αvβ3 integrin in human lung cancer cells (A549 cells). CCL5 stimulation increased phosphorylation of the p85α subunit of phosphatidylinositol 3-kinase (PI3K) and serine 473 of Akt. Also, we found that PI3K inhibitor (Ly294002) or Akt inhibitor suppressed CCL5-induced migration activities and integrin expression of A549 cells. Transfection of cells with p85 or Akt mutant also reduced CCL5-mediated cancer migration. In addition, treatment of A549 cells with CCL5 induced IκB kinase α/β (IKK α/β) phosphorylation, IκB phosphorylation, p65 Ser phosphorylation, and κB-luciferase activity. Furthermore, the CCL5-mediated increases in p65 Ser phosphorylation were inhibited by Ly294002 and Akt inhibitor. Taken together, our results suggest that CCL5 acts through PI3K/Akt, which in turn activates IKKα/β and NF-κB, resulting in the activation of αvβ3 integrin and contributing to the migration of human lung cancer cells.
URLPMID:4968951 [本文引用: 1]
A variety of therapeutic strategies are currently under investigation to inhibit factors that promote tumor invasion, as metastasis is the most common cause of mortality for cancer patients. Notably, considerable emphasis has been placed on studying metastasis as a dynamic process that is highly dependent on the tumor microenvironment. In regards to breast cancer, chemokine C-C motif ligand 5 (CCL5), which is produced by tumor-associated stromal cells, has been established as an important contributor to metastatic disease. This review summarizes recent discoveries uncovering the role of this chemokine in breast cancer metastasis, including conditions that increase the generation of CCL5 and effects induced by this signaling pathway. In particular, CCL-5-mediated cancer cell migration and invasion are discussed in the context of intertwined feedback loops between breast cancer cells and stromal cells. Moreover, the potential use of CCL5 and its receptor chemokine C-C motif receptor 5 (CCR5) as targets for preventing breast cancer metastasis is also reviewed.
URL [本文引用: 1]
Nasopharyngeal carcinoma (NPC) is etiologically associated with Epstein–Barr virus (EBV) infection and is known to be highly vascularized. Previous studies have suggested thatEBVoncoproteins contribute toNPCangiogenesis. However, the regulatory network ofEBVin angiogenesis still remains elusive. Herein, we reveal a novel mechanism ofEBV‐induced angiogenesis inNPC. First, we showed thatEBV‐infectedNPCcell lines generated larger tumors with more microvessels in mouse xenograft models. Subsequent proteomic analysis revealed thatEBVinfection increased the expression of a series of angiogenic factors, including chemokine (C‐C motif) ligand 5 (CCL5). We then proved thatCCL5 was a target ofEBVin inducing tumor angiogenesis and growth. Further investigation through transcriptome analysis indicated that the pro‐angiogenic function ofCCL5 might be mediated by thePI3K/AKTpathway. Furthermore, we confirmed that activation of thePI3K/AKTand hypoxia‐inducible factor‐1α pathways was essential forCCL5‐promoted angiogenesis. Finally, the immunohistochemical analysis of humanNPCspecimens also showed thatCCL5 was correlated with angiogenesis. Taken together, our study identifiesCCL5 as a keyEBV‐regulated molecular driver that promotesNPCangiogenesis, suggesting it as a potential therapeutic target.
URLPMID:20702412 [本文引用: 1]
Oral squamous cell carcinoma (OSCC) is a major health problem worldwide, and patients have a particularly poor 5-year survival rate. Thus, identification of the molecular targets in OSCC and subsequent innovative therapies are greatly needed. Prolonged exposure to alcohol, tobacco, and pathogenic agents are known risk factors and have suggested that chronic inflammation may represent a potential common denominator in the development of OSCC. Microarray analysis of gene expression in OSCC cell lines with high basal NF-B activity and OSCC patient samples identified dysregulation of many genes involved in inflammation, wound healing, angiogenesis, and growth regulation. In particular , , , and gene expression was up-regulated in OSCC. Moreover, IL-8 protein levels were significantly higher in OSCC cell lines as compared with normal human oral keratinocytes. Targeting IL-8 expression by siRNA significantly reduced the survival of OSCC cells, indicating that it plays an important role in OSCC development and/or progression. Inhibiting the inflammatory pathway by aspirin and the proteasome/NF-B pathway by bortezomib resulted in marked reduction in cell viability in OSCC lines. Taken together our studies indicate a strong link between inflammation and OSCC development and reveal IL-8 as a potential mediator. Treatment based on prevention of general inflammation and/or the NF-B pathway shows promise in OSCCs.
URLPMID:19334035 [本文引用: 1]
CCL5 (previously called RANTES) is in the CC-chemokine family and plays a crucial role in the migration and metastasis of human cancer cells. On the other hand, the effect of CCL5 is mediated via CCR receptor. RT-PCR and flow cytometry studies demonstrated CCR5 but not CCR1 and CCR3 mRNA in oral cancer cell lines, especially higher in those with high invasiveness (SCC4) as compared with lower levels in HSC3 cells and SCC9 cells. Stimulation of oral cancer cells with CCL5 directly increased the migration and metalloproteinase-9 (MMP-9) production. MMP-9 small interfering RNA inhibited the CCL5-induced MMP-9 expression and thereby significantly inhibited the CCL5-induced cell migration. Activations of phospholipase C (PLC), protein kinase C (PKC), and NF-B pathways after CCL5 treatment was demonstrated, and CCL5-induced expression of MMP-9 and migration activity was inhibited by the specific inhibitor of PLC, PKC, and NF-B cascades. In addition, migration-prone sublines demonstrate that cells with increasing migration ability had more expression of MMP-9, CCL5, and CCR5. Taken together, these results indicate that CCL5/CCR5 axis enhanced migration of oral cancer cells through the increase of MMP-9 production. J. Cell. Physiol. 220: 418-426, 2009. 2009 Wiley-Liss, Inc.
URLPMID:27693640 [本文引用: 2]
61MELK is overexpressed in HCC and correlates with early recurrence and survival.61Silencing MELK inhibits growth, invasion, stemness and tumorigenicity of HCC cells.61MELK regulates cell cycle progression and mitosis-related genes through targeting FOXM1.
URL [本文引用: 1]
Abstract Adrenocortical carcinoma (ACC) is an aggressive cancer with a five-year survival less than 35%. Mortality remains high due to lack of targeted therapies. Using bioinformatic analyses, we identified Maternal Embryonic Leucine Zipper Kinase (MELK) as 4.1-fold overexpressed in ACC compared to normal adrenal samples. High MELK expression in human tumors correlated with shorter survival and with increased expression of genes involved in cell division and growth. We investigated the functional effects of MELK inhibition using newly developed ACC cell lines with variable MELK expression, CU-ACC1 and CU-ACC2, compared with H295R cells. In vitro treatment with the MELK inhibitor, OTSSP167, resulted in a dose dependent decrease in rates of cell proliferation, colony formation and cell survival, with relative sensitivity of each ACC cell line based upon the level of MELK overexpression. To confirm a MELK specific anti-tumorigenic effect, MELK was inhibited in H295R cells via multiple shRNAs. MELK silencing resulted in 1.9-fold decrease in proliferation, and 3 to 10-fold decrease in colony formation in soft agar and clonogenicity assays, respectively. In addition, while MELK silencing had no effect on survival in normoxia, exposure to a hypoxia resulted in a 6- and 8-fold increase in apoptosis as assessed by caspase-3 activation and TUNEL, respectively. Together these data suggest that MELK is a modulator of tumor cell growth and survival in a hypoxic microenvironment in adrenal cancer cells, and support future investigation of its role as a novel therapeutic kinase target in patients with adrenocortical carcinoma.
URLPMID:27225691 [本文引用: 1]
Abstract PURPOSE: While effective targeted therapies exist for estrogen receptor-positive and HER2-positive breast cancer, no such effective therapies exist for triple-negative breast cancer (TNBC), thus it is clear that additional targets for radiosensitization and treatment are critically needed. EXPERIMENTAL DESIGN: Expression microarrays, qRT-PCR, and western blotting were used to assess MELK RNA and protein expression levels. Clonogenic survival assays were used to quantitate the radiosensitivity of cell lines at baseline and after MELK inhibition. The effect of MELK knockdown on DNA damage repair kinetics was determined using H2AX staining. The in vivo effect of MELK knockdown on radiosensitivity was performed using mouse xenograft models. Kaplan-Meier analysis was used to estimate local control and survival information and a Cox proportional hazards model was constructed to identify potential factors impacting local recurrence-free survival. RESULTS: MELK expression is significantly elevated in breast cancer tissues compared to normal tissue as well as in TNBC compared to non-TNBC. MELK RNA and protein expression is significantly correlated with radioresistance in breast cancer cell lines. Inhibition of MELK (genetically and pharmacologically) induces radiation sensitivity in vitro and significantly delayed tumor growth in vivo in multiple models. Kaplan-Meier survival and multivariable analyses identify increasing MELK expression as being the strongest predictor of radioresistance and increased local recurrence in multiple independent datasets. CONCLUSIONS: Here, we identify MELK as a potential biomarker of radioresistance and target for radiosensitization in TNBC. Our results support the rationale for developing clinical strategies to inhibit MELK as a novel target in TNBC. Copyright 2016, American Association for Cancer Research.
URLPMID:26701722 [本文引用: 1]
Maternal embryonic leucine zipper kinase (MELK) is upregulated in a variety of human tumors, and is considered an attractive molecular target for cancer treatment. We characterized the expression of MELK in gastric cancer (GC) and measured the effects of reducing MELK mRNA levels and protein activity on GC growth. MELK was frequently overexpressed in primary GCs, and higher MELK levels correlated with worse clinical outcomes. Reducing MELK expression or inhibiting kinase activity resulted in growth inhibition, G2/M arrest, apoptosis and suppression of invasive capability of GC cellsin vitroandin vivo. MELK knockdown led to alteration of epithelial mesenchymal transition (EMT)-associated proteins. Furthermore, targeting treatment with OTSSP167 in GC patient-derived xenograft (PDX) models had anticancer effects. Thus, MELK promotes cell growth and invasiveness by inhibiting apoptosis and promoting G2/M transition and EMT in GC. These results suggest that MELK may be a promising target for GC treatment.
URLPMID:28536141 [本文引用: 1]
Abstract Medulloblastoma is the most common malignant brain tumor in children. Although accumulated research has suggested that cancer stem-like cells play a key role in medulloblastoma tumorigenesis, the specific molecular mechanism regarding proliferation remains elusive. Here, we reported that more abundant expression of maternal embryonic leucine-zipper kinase (MELK) and enhancer of zeste homolog 2 (EZH2) in medulloblastoma stem-like cells than in neural stem cells and the interaction between the two proteins could mediate the self-renewal of sonic hedgehog (SHH)-subtype medulloblastoma. In human medulloblastoma, extensive nodularity and large cell/anaplastic subgroups differed according to the staining levels of MELK and EZH2 from the other two subgroups. The proportion of MELK- or EZH2-positive staining status could be considered as the potential indicator for survival. Mechanistically, MELK bound to and phosphorylated EZH2 and its methylation was induced by EZH2 in medulloblastoma, which could regulate the proliferation of cancer stem-like cells. In xenografts, loss of MELK or EZH2 attenuated medulloblastoma stem-like cell-derived tumor growth and promoted differentiation. These findings indicate that MELK-induced phosphorylation and EZH2-mediated methylation in MELK/EZH2 pathway are essential for medulloblastoma stem-like cell-derived tumor proliferation, thereby identifying a potential therapeutic strategy for these patients. IMPLICATIONS: This study demonstrates that the interaction occurring between MELK and EZH2 promotes self-proliferation and stemness; thus, representing an attractive therapeutic target and potential candidate for diagnosis of medulloblastoma. Copyright 2017, American Association for Cancer Research.
URL [本文引用: 1]
Nuclear functions of NME proteinsLaboratory Investigation, Published online: 23 October 2017; doi:10.1038/labinvest.2017.109
URLPMID:29095547 [本文引用: 1]
Rac GTPase activating protein 1 (RacGAP1) can regulate cytokinesis and cell differentiation. The oncogenic role of RacGAP1 has been partially studied in gastric cancer, colorectal cancer, and breast cancer. In the present study, we endeavor to evaluate its expression and functions in epithelial ovarian cancer (EOC). We retrospectively collected the clinicopathological information of 117 patients who underwent curative surgery for EOC. Expression of RacGAP1 protein in primary tumor tissues was evaluated by immunohistochemistry, which was significantly associated with tumor pathological grade, tumor stage, and lymph node metastasis. Patients with lower RacGAP1 level had a longer survival time and lower recurrence risk. Multivariate results identified the independent prognostic role of RacGAP1 for both recurrence and survival in EOC patients. Cellular studies showed that RacGAP1 can positively regulate the activation of RhoA and Erk proteins. In addition, wound healing assay and Transwell assay found that RacGAP1 can up egulate the migration and invasion process of EOC cells, respectively. In all, our results not only confirmed the prognostic role of RacGAP1 for recurrence and survival in EOC patients, but also highlighted its possible potency for drug development.
URLPMID:25568185 [本文引用: 1]
Rac GTPase-activating protein (RacGAP) 1 plays a key role in controlling various cellular phenomena including cytokinesis, transformation, invasive migration and metastasis. This study investigated the function and clinical significance of RacGAP1 expression in colorectal cancer (CRC). The intrinsic functions of RacGAP1 in CRC cells were analyzed using small interfering RNA (siRNA). We analyzed RacGAP1 mRNA expression in surgical specimens from 193 CRC patients (Cohort 1) by real-time PCR. Finally, we validated RacGAP1 protein expression using formalin-fixed paraffin-embedded samples from 298 CRC patients (Cohort 2) by immunohistochemistry. Reduced RacGAP1 expression by siRNA in CRC cell lines showed significantly decreased cellular proliferation, migration and invasion. In Cohort 1, RacGAP1 expression in CRC was significantly higher than in adjacent normal mucosa and increased according to tumor node metastasis stage progression. High RacGAP1 expression in tumors was significantly associated with progression and prognosis. In Cohort 2, RacGAP1 protein was overexpressed mainly in the nuclei of CRC cells; however, its expression was scarcely observed in normal colorectal mucosa. RacGAP1 protein expression was significantly higher in CRC patients with higher T stage, vessel invasion and lymph node and distant metastasis. Increased expression of RacGAP1 protein was significantly associated with poor disease-free and overall survival. Multivariate analyses revealed that high RacGAP1 expression was an independent predictive marker for lymph node metastasis, recurrence and poor prognosis in CRC. Our data provide novel evidence for the biological and clinical significance of RacGAP1 as a potential biomarker for identifying patients with lymph node metastasis and poor prognosis in CRC.
URLPMID:19240652 [本文引用: 1]
The expression, mechanisms of regulation, and functional impact of activin (INHBA) in esophageal adenocarcinoma (EAC) have not been fully defined. INHBAexpression was examined in 46 esophageal samples (nine Barrett's metaplasia (BM); seven BM/low-grade dysplasia; eight low-grade dysplasia; seven high-grade dysplasia; 15 EAC) using oligonucleotide microarrays and real-time reverse transcription-polymerase chain reaction (RT-PCR) and in 90 tissue samples (79 EAC; 8 dysplastic; 3 BM) using immunohistochemistry (IHC). The proliferation of EAC cell lines FLO and OE-33 was examined after treatment with exogenous activin. The proliferation of OE-33 was also examined after treatment with the activin inhibitor follistatin andINHBA-targeting siRNA. OE-33 and FLO cells were treated with 5-aza-2 eoxycytidine (5-AZA) and trichostatin A to investigate the role of epigenetic regulation inINHBAexpression. Primary EACs expressed 5.7-times moreINHBAmRNA than BM samples on oligonucleotide microarray. Transcript overexpression in EAC relative to BM was confirmed on real-time RT-PCR. IHC suggested higherINHBAprotein expression in EAC (69.6%) than in the dysplastic (37.5%) and BM samples (33.3%). FLO and OE-33 treated with activin demonstrated increased proliferation, and OE-33 cells treated with follistatin andINHBA-targeting siRNA demonstrated reduced proliferation, relative to untreated controls. Treatment of FLO cells with trichostatin A and 5-AZA up-regulatedINHBAmRNA and protein production by real time RT-PCR and IHC. INHBAis overexpressed in EAC relative to dysplastic and BM tissue.INHBAoverexpression may promote cell proliferation and may be affected by promoter demethylation and histone acetylation in EAC cell lines.
URL [本文引用: 1]
Although extranodal NK/T cell lymphoma (ENKTCL) is consistently associated with Epstein-Barr virus (EBV) infection, the manifestation and prognostic value of serum EBV antibodies still remain unknown. One hundred and forty-one patients with ENKTCL were evaluated for serum EBV EA-IgA and VCA-IgA antibodies levels in the past 24 years in our institution. Their correlation with... [Show full abstract]
[本文引用: 1]
URLPMID:19262093 [本文引用: 1]
Abstract Cell-extracellular matrix (ECM) is an important property of virtually all cells in multi-cellular organisms. Cell-ECM adhesion research, therefore, has broad impact on biology and medicine. Studies over the past three decades have resulted in tremendous advance in our understanding of the molecular basis and functions of cell-ECM adhesion. Here, I focus on some of the general lessons that we have learned from recent studies on cell-ECM adhesion. In addition, I highlight several topics in this rapidly advancing research area. These topics, which include assembly, disassembly and regulation of cell-ECM adhesion structures, the molecular mechanisms of bi-directional signaling through cell-ECM adhesions, and the tissue and organ pathobiology of cell-ECM adhesion, are pertinent to our understanding of cell-ECM adhesion and signaling.
URLPMID:4706424 [本文引用: 1]
Oral squamous cell carcinoma (OSCC) has been reported as the most prevalent cancer of the head and neck region, while early diagnosis remains challenging. Here we took a comprehensive bioinformatics study on microarray data of 326 OSCC clinical samples with control of 165 normal tissues. The cell interaction pathways of ECM-receptor interaction and focal adhesion were found to be significantly regulated in OSCC samples. Further analysis of the topological properties and expression consistency identified that three hub genes in the gene interaction network, MMP9, PDIA3 and BGH3, were consistently up-expressed in OSCC samples. When being validated on additional microarray datasets of 41 OSCC samples, the validation rate of over-expressed BGH3, MMP9, and PDIA3 reached 90%, 90% and 84% respectively. At last, immuno-histochemical assays were done to test the protein expression of the three genes on newly collected clinical samples of 35 OSCC, 20 samples of pre-OSCC stage, and 12 normal oral mucosa specimens. Their protein expression levels were also found to progressively increase from normal mucosa to pre-OSCC stage and further to OSCC (ANOVA p = 0.000), suggesting their key roles in OSCC pathogenesis. Based on above solid validation, we propose BGH3, MMP9 and PDIA3 might be further explored as potential biomarkers to aid OSCC diagnosis.
[本文引用: 1]
URLPMID:26462838 [本文引用: 1]
It has been reported that p53 dysfunction is closely related to the carcinogenesis of nasopharyngeal carcinoma (NPC). Recently, an increasing body of evidence has indicated that microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) participate in p53-associated signaling pathways and, in addition to mRNAs, form a complex regulation network to promote tumor occurrence and progression. The aim of this study was to elucidate the p53-regulated miRNAs, mRNAs, and lncRNAs and their regulating networks in NPC. Firstly, we overexpressed p53 in the NPC cell line HNE2 and performed transcriptomic gene expression profiling (GEP) analysis, which included miRNAs, mRNAs, and lncRNAs, using microarray technology at 0, 12, 24, and 48 h after transfection. There were 38 miRNAs (33 upregulated and 5 downregulated), 2107 mRNAs (296 upregulated and 1811 downregulated), and 1190 lncRNAs (133 upregulated and 1057 downregulated) that were significantly dysregulated by p53. Some of the dysregulated molecules were confirmed by quantitative real-time polymerase chain reaction (qRT-PCR). Then, we integrated previously published miRNAs, mRNAs, and lncRNAs GEP datasets from NPC biopsies to investigate the expression of these p53 regulated molecules and found that 7 miRNAs, 218 mRNAs, and 101 lncRNAs regulated by p53 were also differentially expressed in NPC tissues. Finally, p53-regulated miRNA, mRNA, and lncRNA networks were constructed using bioinformatics methods. These miRNAs, mRNAs, and lncRNAs may participate in p53 downstream signaling pathways and play important roles in the carcinogenesis of NPC. Thorough investigations of their biological functions and regulating relationships will provide a novel view of the p53 signaling pathway, and the restoration of p53 functioning or its downstream gene regulating network is potentially of great value in treating NPC patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}