 ,1,2, 宋晓峰,1
,1,2, 宋晓峰,1Molecular mechanisms of recursive splicing events in long introns of eukaryotes
Jinchuan Wei1, Tianyi Xu1, Jing Wu,1,2, Xiaofeng Song,1通讯作者:
编委: 张根发
收稿日期:2018-10-30修回日期:2018-12-24网络出版日期:2019-02-25
| 基金资助: |
Corresponding authors:
Editorial board:
Received:2018-10-30Revised:2018-12-24Online:2019-02-25
| Fund supported: |
作者简介 About authors
魏金川,硕士研究生,专业方向:生物信息学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (478KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
魏金川, 徐添翼, 吴静, 宋晓峰. 真核生物基因组长内含子递归剪接事件的分子机制[J]. 遗传, 2019, 41(2): 89-97 doi:10.16288/j.yczz.18-182
Jinchuan Wei, Tianyi Xu, Jing Wu, Xiaofeng Song.
真核生物基因组转录过程是以DNA双链中一条链为模板,经碱基互补配对得到前体mRNA,后通过对内含子与外显子的剪接加工得到成熟的mRNA。内含子是基因编码区中的非编码序列,在生物体内不编码蛋白,因此在前体mRNA转录加工过程中会被剪接体(spliceosome)识别并去除。起初研究者认为内含子不参与基因的表达调控,但近几年随着二代测序技术的发展和生物信息学工具的开发,人们发现内含子在转录及基因表达调控中具有重要的生物学功能,比如:有些内含子含有增强子、启动子或者其他的顺式作用元件,对基因的表达起调控作用;内含子的剪接增加了mRNA的稳定性;内含子还可能发生选择性剪接事件[1,2,3,4,5]。
前体mRNA的剪接过程中,内含子通过两个外显子的连接被完全切除,这个过程需要剪接体介导。前体mRNA的剪接需要两步实现:第一步,靠近内含子3′端分支位点的A残基的2′-OH对5′端的磷酸基团进行亲核攻击,形成2′→5′磷酸二酯键连接的套索结构;第二步,被剪接的外显子3′端核苷酸的-OH进攻内含子3′末端的磷酸基团,使得内含子3′端在剪接位点处断开,释放套索结构,两个外显子连接[6,7,8]。此过程是直接去除整个内含子的,属于一步剪接[9],但是有一些较长的内含子是经过了多步剪接来去除的。1999年,Hatton等[10]在黑腹果蝇(Drosophila melanogaster)Ubx基因中发现,其第1个内含子分3步去除,并将这一剪接过程命名为递归剪接(recursive splicing) (图1)。内含子中被剪接的核酸位点称为递归性剪接位点(RS-site)。近年来研究人员发现,除果蝇以外,递归剪接在其他真核生物基因组中也广泛存在,并与疾病的发生发展有重要关系。本文对真核生物基因组中长内含子递归剪接分子机制的国内外研究现状进行了综述,以期为RNA的剪接研究提供参考,同时为疾病的诊断和治疗提供新的 思路。
图1
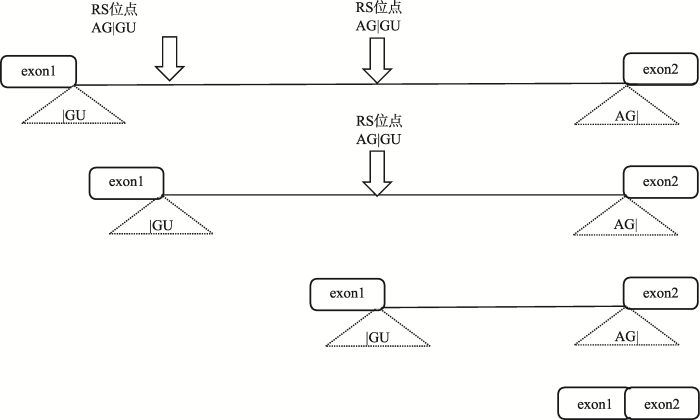 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1递归剪接示意图
含有两个剪接位点的内含子分步去除过程。exon1与exon2分别表示两个相邻外显子,中间内含子有两个递归剪接位点,灰色箭头指向剪接位点,一共经过了3步拼接在一起。
Fig. 1The schematic diagram of recursive splicing
1 递归剪接的特征
1.1 递归剪接的过程
通常内含子5′端剪接位点被称为供体位点(donor site),3′端剪接位点被称为受体位点(acceptor site)[11,12]。研究表明,内含子起始5′端两对碱基和结尾3′端的两对碱基最为保守,在所有的剪接位点中有99.24%的位点为GU-AG,0.7%的位点为GC-AG,0.05%的位点为AT-AC[13]。除了这两对保守碱基外,它们附近的碱基虽然在不同的物种之间存在差异,但在同一物种中通常具有保守性,如在脊椎动物中5′剪接端碱基通常为AG|GUAAGU[14]。由碱基序列可以看出供体与受体间不存在碱基互补配对的可能,所以它们并不是通过碱基互补的方式拼接到一起的。外显子转录是从5′端到3′端进行的,递归剪接就是把剪接位点处默认为含有0个核苷酸的外显子,所以递归剪接位点通常为AG|GU。递归剪接的具体过程大致如下[15,16,17]:(1)首先内含子3′端上游10~50 bp处的特定腺苷的2′羟基攻击5′剪接位点(图2A),形成5′-2′磷酸二酯键,从而构成套索结构,进而释放5′外显子(图2B);(2) 5′外显子的3′羟基攻击内含子3′端最近的递归剪接位点(RS sites),释放套索结构,与此同时,5′外显子的3′-羟基与下游外显子的5′端连接形成新的磷酸二酯键(图2C);(3)重复前面两个步骤,直到全部内含子去除。
1.2 递归剪接位点的特征
针对可变剪接的研究发现内含子5′和3′端碱基几乎都是GU和AG,并且内含子位点为AGGU的比例高达99.24%,所以人们对递归剪接位点的研究主要也主要针对AGGU型。此外,递归剪接在形成套索结构的过程中,在剪接位点上游第5和第6位置处的碱基基本上都为“U”,这也是寻找递归剪接位点的主要依据。而Kelly等[18]研究发现在人类(Homo sapiens)全基因组中并非所有递归剪接位点都是AGGU,而是AGMN(M、N分别表示A、G、C、U 4种碱基中的一种),其中AGGN的概率为44%,AGGU的概率仅为14% (表1)。因此,对于非经典(剪接位点不是AGGU)递归剪接位点的识别已成为当前研究的热点问题。图2
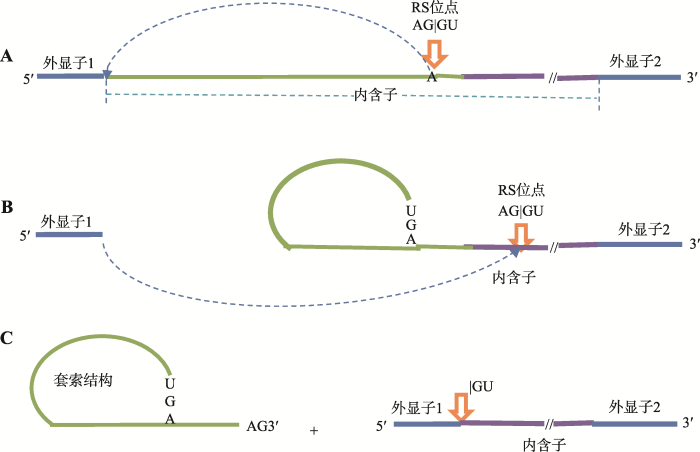 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2递归剪接套索结构形成与切除示意图
A:内含子3′端上游10~50 bp处的特定腺苷的2′羟基攻击5′剪接位点;B:构成套索结构,释放5′外显子;C:释放套索结构外显子与递归剪接位点相互连接。
Fig. 2The schematic diagram of recursive splicing lariat’ structure
Table 1
表1
表1 递归剪接位点AGMN中M、N为各碱基概率统计表
Table 1
| M | N | |||
|---|---|---|---|---|
| A | C | G | U | |
| A | 0.07 | 0.05 | 0.10 | 0.11 |
| C | 0.04 | 0.02 | 0.01 | 0.04 |
| G | 0.12 | 0.10 | 0.08 | 0.14 |
| U | 0.02 | 0.02 | 0.04 | 0.04 |
新窗口打开|下载CSV
1.3 递归剪接位点的识别
Duff等[15]对黑腹果蝇进行了转录组测序,并将得到的Total RNA-seq数据用Tophat[19]进行了处理。从得到的reads与全基因组匹配结果中,他们发现基因的转录是多个RNA聚合酶同时进行,所以靠近5′外显子处的转录酶更多(图3A)。存在递归剪接现象的内含子在reads的分布上存在着波峰与波谷,并且整体呈现锯齿状,这种分布模式体现了递归剪接去除内含子的机理,其理想的reads密度图为倒三角状(图3B)。递归剪接通常存在于大于10 kb的内含子中,人类基因组中此类内含子较多,长度大于24 kb的内含子有8000个以上;大于50 kb的内含子有3400多个;而超过100 kb的内含子都有1200个以上[20,21]。Sibley等[22]对人脑组织Total RNA-seq数据进行了分析,从中发现了8个递归剪接位点,这些位点所在的内含子长度都超过150 kb。Duff等[15]用Tophat将果蝇Total RNA-seq数据与果蝇全基因组(modENCODE注释文件)相匹配,发现含有剪接位点的内含子长度都大于2 kb。近来,递归剪接位点的识别主要基于Total RNA-Seq数据,辅以对应物种的基因组注释数据及基因组序列信息。通常,递归剪接位点的识别主要分为以下4步(图4):(1) 通过FastQC、Fastx-toolkit分别对其质量检测、控制;(2)利用Tophat进行reads映射,得到Bam文件与Junction文件;(3)依据内含子长度、剪接位点保守性对Junction位点进行筛选;(4)将Bam文件转换成Sam文件,并与Junction文件相结合,进而通过生物信息学方法识别递归剪接位点。目前,常将周边reads分布呈锯齿状的位点判定为潜在的递归剪接位点。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Total RNA-seq数据匹配全基因组统计得到的结果图
A:含递归剪接的基因组转录示意图。AG/GU为递归剪接位点,曲线表示RNA转录情况,椭圆形表示RNA聚合酶Ⅱ,代表一条DNA链在同时进行多次转录。B:Total RNA-seq的reads密度图。
Fig. 3Total RNA-seq data mapping results
图4
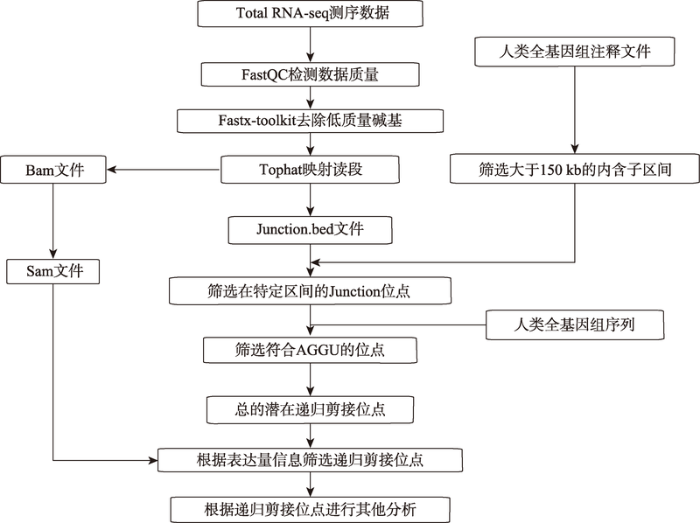 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4递归剪接位点识别流程图
Fig. 4Flow chart for identifying recursive splice sites
矩形框中的是数据处理过程、用到的文件、得到的结果等。
2 递归剪接研究的进展
2.1 果蝇基因组中递归剪接的研究
Hatton等[10]于1998年在黑腹果蝇Ubx基因中首次发现了递归性剪接现象,在Ubx基因的第1个外显子和第2个外显子之间发现了新的剪接位点,并且在3′剪接位点的下游的碱基序列为GTAAGA。研究者对Ubx基因进行了深入研究,发现该基因第1个内含子长73 kb,该内含子的去除经历了3次递归剪接[24]。Conklin等[25]于2005年通过套索结构识别递归剪接位点,并于Ubx基因的第一个内含子中成功识别出Hatton等[10]找到的两个递归剪接位点。针对套索结构RNA分子半衰期通常较短,且转录的过程不能准确把握等问题,他们通过控制PCR技术对分支连接处2′→5′磷酸二酯键的特异性扩增,提高了RNA套索结构分析的灵敏度。
2015年,Duff 等[15]以黑腹果蝇为材料,对其进行实验处理,通过二代测序技术得到对应的Total RNA-seq数据,然后通过Tophat/Bowtie软件比对得到Junction位点信息,结合reads在Junction位点附近的分布信息,最终筛选出197个潜在的递归剪接位点。这些递归剪接位点分布于115个基因的130个内含子中,他们挑选了其中14个基因用作实验验证,证实这些基因含有24个递归剪接位点。对存在递归性剪接的基因(115 个)和内含子统计分析发现,其中有100个基因只存在一个递归性剪接内含子,剩下的15个基因含有两个递归性剪接内含子。此外,这130个内含子的剪接位点个数都在1~6之间。递归性剪接内含子的碱基长度为11 341~132 736 bp,内含子的平均长度为45 164 bp。进一步分析发现,存在递归剪接的内含子长度都相对较长,但是并不是所有的较长内含子都存在递归剪接,在较长的内含子中只有大约6%存在递归性剪接。他们还发现U2AF蛋白减少会导致含有递归剪接的Pre-mRNA加工速率降低。
Pre-mRNA在内含子转录完成几秒后就会剪切掉内含子,递归剪接是内含子中间片段的剪接。所以,2018年Pai等[26]利用4sU对果蝇S2细胞进行标记,分别获取转录5 min、10 min、20 min的Total RNA-seq数据,从中发现了541个递归剪接位点,其中实验证明的有119个,这些位点所属内含子长度大部分都超过40 kb。
2.2 脊椎动物基因组中递归剪接的研究
除果蝇以外,研究者还对多种脊柱动物进行了递归剪接的研究,包括人、大鼠(Rattus norvegicus)、斑马鱼(Danio rerio)等。2012年,Taggart 等[27]开发了一个基于RNA套索结构识别内含子3′剪接位点的生物信息学方法,其假定套索结构都位于3′剪接位点附近。利用该方法,他们对人类12个组织的转录组测序数据进行了处理,共筛选出861个内含子3′剪接位点。其中,2118个reads经匹配映射到这些剪接位点附近的套索结构中,而属于递归性剪接套索结构的reads有70个(3%)。
2015年,Kelly等[18]对SAMD4A基因转录过程中的内含子递归剪接事件进行了探究。SAMD4A基因全长221 kb,其中第1个内含子长134 kb。研究人员用人脐静脉内皮细胞(human umbilical vein endothelial cells, HUVEC)作为实验材料,利用肿瘤坏死因子(TNFα)对HUVEC进行刺激,并通过二代测序技术(high-throughput sequencing)得到相关的Total RNA- seq数据。接着,将reads匹配到全基因组(hg19),发现reads匹配的位置主要分为3类情况:第一类为直接整体匹配到单一外显子上;第二类匹配到exon-exon上,即分别匹配到上游外显子的3′ 端部分碱基和下游外显子的5′端部分碱基;第三类为exon-intron,即一部分匹配到上游外显子上,而另一部分匹配到相邻内含子的中间部分。其中第三类reads能反映出内含子存在递归剪接与可变剪接的可能。最后研究者用生物信息学的方法对套索结构进行筛选,从中发现SAMD4A基因的第1个内含子存在递归性剪接现象。SAMD4A基因在人脐静脉内皮细胞中基本不表达,而TNFα对SAMD4A基因的转录有着很强的刺激作用,所以研究人员通过荧光原位杂交技术(fluorescence in situ hybridization, FISH)对SAMD4A基因表达情况进行检测,发现较长的内含子并不是转录结束后直接切除的,而是边转录边剪接的。对于SAMD4A基因的荧光标记发现,剪切部分的水解时间占整个内含子转录时间的1/15;它每次剪切大约经历了15 min,其平均半衰期大约为4.2 min。研究表明,在SAMD4A基因递归剪接过程中,内含子片段的水解过程与形成套索切除的过程是同步进行的;在较长的内含子中,转录的速度大约3 kb/min。研究人员使用CRISPR-Cas9技术[28]对递归剪接的分支点(branch point)进行突变与删除,发现这样会大大降低Pre-mRNA到成熟mRNA的加工速率。并且,对任意一个递归剪接位点(RS-site)进行基因突变或基因敲除处理,SAMD4A基因的表达量都会降低35%~50%,表明递归剪接的分支点和RS位点在对应基因的表达过程中有着重要的作用。同时研究人员还发现,在人体静脉细胞中含有多个剪接位点的基因在某一个剪接位点失效后,会通过下一个剪接位点或者其他的补偿机制进行处理,虽然效率降低,但是还可以表达[18]。因此,递归剪接位点突变后,应同样存在某种补偿机制,使得SAMD4A基因仍能正常转录。
2015年,Sibley等[22]对脊椎动物神经元细胞进行了RNA深度测序,分析发现在脊椎动物神经元中存在大量的递归剪接现象。由此,他们基于对人类脑组织中递归剪接的研究建立了一个双重剪接的模型,该模型大体对AGGU为剪接位点的现象进行了研究,发现递归剪接位点上游内含子去除后,供体部分的外显子与剪接后的受体首端的GU相互连接起来,形成新的外显子-内含子-外显子复合物,在进行下次剪接过程中,有可能本次外显子-内含子会保持连在一起,最终的产物会有两种,一种是外显子-外显子(exon-exon),另外一种为外显子-递归剪接中间小段内含子(exon-intron)这两种产物。虽然该模型还不够完善,但也说明了人类细胞中mRNA亚型具有多样性。Emmett[29]发现在人类脑组织中,存在递归剪接现象的7个基因与神经系统疾病密切有关。
以上研究都是以组织的Total RNA-seq数据为材料进行分析实验。2018年Hayashi等[30]开发了一种对单细胞Total RNA-seq进行测序的方法,即RamDA-seq。与其他方法相比,这种测序方法对non- poly(A) RNA敏感性更高。Hayashi等[30]用RamDA- seq对不同时期的小鼠胚胎干细胞进行实验分析,在长度大于150 kb的内含子中发现了207个潜在的递归剪接位点。2018年Pai等[31]对果蝇的30 000个内含子进行研究,发现存在递归剪接的内含子也具有半衰期。此外,Srndic 等[32]在人类多个组织中检测到12 000个递归剪接事件,并发现递归剪接对无义介导的RNA降解(nonsense-mediated decay)可能起着反作用。
3 递归剪接研究的意义
随着RNA测序技术的发展,越来越多的内含子被发现在基因表达调控中发挥着重要的作用,因此对递归剪接的深入研究有助于理解内含子调控功能的分子机制[33]。全基因组研究发现机体可以通过改变RNA聚合酶Ⅱ的合成速度来控制转录的速率[34],碱基突变或缺失容易造成各类疾病[35,36]。人类基因组除了必要的编码信息外,还有许多必要的剪接序列,其内部位点的突变或者缺失经常会导致疾病的发生[37,38,39]。研究人员还发现在剪接位点发生突变或者删除时,成熟mRNA的生成速率会大大降低。在人类基因组中,递归剪接位点比其他位置更具有保守性,并且广泛存在[18,22,29,40,41]。研究发现,人体内的突变主要来自于不编码蛋白的区域,占总突变事件的90%以上,且超过40%的突变来自于内含子区域[35]。剪接过程中的拼接错误是导致疾病的重要原因,递归剪接位点突变可能对某些疾病的研究具有重要意义[42]。目前,人们已对神经元[29]及内皮细胞[18]中的递归剪接事件进行了研究,发现剪接位点与神经系统疾病如帕金森综合征、循环系统疾病如视网膜硬化或者高血压等病变的关联[43]。
递归性剪接对生物体的生长发育具有重要的调节作用。例如,剪接位点的选择通过顺式作用元件[44]和反式作用因子共同控制,例如SR蛋白、hnRPNs等蛋白质。这些因素通过影响U1 snRNP,U2辅助因子和U2 snRNP剪接体的装配来调控mRNA的序列,从而控制生物体的生长发育[45]。
递归性剪接还有待进一步深入研究,目前尚待解决的问题有:(1)内含子递归剪接的机制,递归剪接位点间的片段是按前后顺序依次切除,还是另有其他调控方式;(2)递归剪接事件的发生是否与生物发育的不同阶段有关;(3)递归剪接位点突变与疾病间的关系。以上问题的研究必将有助于进一步理解RNA分子剪接机制,为疾病相关研究提供有价值的科研线索。
4 结语与展望
随着RNA测序技术的发展与研究的深入,起初认为内含子是单步剪接,现在已发现部分长内含子存在多步递归剪接。目前,研究者已在人类、果蝇等多个真核生物基因组中发现了递归剪接现象,找到了多个递归剪接位点。虽然近年来对递归剪接的研究发展迅速,但是递归剪接位点突变或缺失造成的影响尚未可知,仍需要人们更进一步的探索。随着递归剪接位点的不断发现,递归剪接位点突变或缺失在人类疾病发生发展中的功能作用将越来越清楚,为疾病的预防和治疗提供新的科学线索。三代测序技术的迅猛发展对真核生物递归剪接的研究具有重要的推动作用。与二代测序技术相比,三代测序技术具有测序速度更快、测序长度激增、可直接测RNA序列等特点[46]。三代测序技术的测序长度由二代测序的上百碱基到现在的几千碱基,因此其对基因长内含子的递归剪接事件的识别分析提供了更好的技术手段,相信未来会有更多有价值的递归剪接的生物学机制被发现。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:9671053 [本文引用: 1]

Abstract Generally, mRNAs that prematurely terminate translation are abnormally low in abundance. In the case of mammalian cells, nonsense codons most often mediate a reduction in the abundance of newly synthesized, nucleus-associated mRNA by a mechanism that is not well understood. With the aim of defining cis-acting sequences that are important to the reduction process, the effects of particular beta-globin gene rearrangements on the metabolism of beta-globin mRNAs harboring one of a series of nonsense codons have been assessed. Results indicate that nonsense codons located 54 bp or more upstream of the 3'-most intron, intron 2, reduce the abundance of nucleus-associated mRNA to 10-15% of normal without altering the level of either of the two introns within pre-mRNA. The level of cytoplasmic mRNA is also reduced to 10-15% of normal, indicating that decay does not take place once the mRNA is released from an association with nuclei into the cytoplasm. A nonsense codon within exon 2 that does not reduce mRNA abundance can be converted to the type that does by (1) inserting a sufficiently large in-frame sequence immediately upstream of intron 2 or (2) deleting and reinserting intron 2 a sufficient distance downstream of its usual position. These findings indicate that only those nonsense codons located more than 54 bp upstream of the 3'-most intron reduce beta-globin mRNA abundance, which is remarkably consistent with which nonsense codons within the triosephosphate isomerase (TPI) gene reduce TPI mRNA abundance. We propose that the 3'-most exon-exon junction of beta-globin mRNA and, possibly, most mRNAs is marked by the removal of the 3'-most intron during pre-mRNA splicing and that the "mark" accompanies mRNA during transport to the cytoplasm. When cytoplasmic ribosomes terminate translation more than 54 nt upstream of the mark during or immediately after transport, the mRNA is subjected to nonsense-mediated decay. The finding that deletion of beta-globin intron 2 does not appreciably alter the effect of any nonsense codon on beta-globin mRNA abundance suggests that another cis-acting sequence functions in nonsense-mediated decay comparably to intron 2, at least in the absence of intron 2, possibly as a fail-safe mechanism. The analysis of deletions and insertions indicates that this sequence resides within the coding region and can be functionally substituted by intron 2.
URLPMID:17122369 [本文引用: 1]
Abstract Evolutionary biologists frequently rely on estimates of the neutral rate of evolution when characterizing the selective pressure on protein-coding genes. We introduce a new method to estimate this value based on intron nucleotide substitutions. The new method uses a metascript model that considers alternative splicing forms and an algorithm to pair orthologous introns, which we call Introndeuce. We compare the intron method with a widely used method that uses observed substitutions in synonymous coding nucleotides, by using both methods to estimate the neutral rate for human-dog and mouse-rat comparisons. The estimates of the 2 methods correlate strongly (r(S) = 0.75), but cannot be considered directly equivalent. We also investigate the effect of alignment error and G + C content on the variance in the intron method: in both cases there is an effect, and it is species-pair specific. Although the intron method may be more useful for shorter evolutionary distances, it is less useful at longer distances due to the poor alignment of less-conserved positions.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:15285897 [本文引用: 1]
Abstract We propose a framework for modeling sequence motifs based on the maximum entropy principle (MEP). We recommend approximating short sequence motif distributions with the maximum entropy distribution (MED) consistent with low-order marginal constraints estimated from available data, which may include dependencies between nonadjacent as well as adjacent positions. Many maximum entropy models (MEMs) are specified by simply changing the set of constraints. Such models can be utilized to discriminate between signals and decoys. Classification performance using different MEMs gives insight into the relative importance of dependencies between different positions. We apply our framework to large datasets of RNA splicing signals. Our best models out-perform previous probabilistic models in the discrimination of human 5' (donor) and 3' (acceptor) splice sites from decoys. Finally, we discuss mechanistically motivated ways of comparing models.
URLPMID:5182069 [本文引用: 1]
Abstract Noncoding variants play a central role in the genetics of complex traits, but we still lack a full understanding of the molecular pathways through which they act. We quantified the contribution of cis-acting genetic effects at all major stages of gene regulation from chromatin to proteins, in Yoruba lymphoblastoid cell lines (LCLs). About ~65% of expression quantitative trait loci (eQTLs) have primary effects on chromatin, whereas the remaining eQTLs are enriched in transcribed regions. Using a novel method, we also detected 2893 splicing QTLs, most of which have little or no effect on gene-level expression. These splicing QTLs are major contributors to complex traits, roughly on a par with variants that affect gene expression levels. Our study provides a comprehensive view of the mechanisms linking genetic variation to variation in human gene regulation. Copyright 2016, American Association for the Advancement of Science.
URLPMID:26292705 [本文引用: 1]
Abstract Splicing of precursor messenger RNA is performed by the spliceosome. In the cryogenic electron microscopy structure of the yeast spliceosome, U5 small nuclear ribonucleoprotein acts as a central scaffold onto which U6 and U2 small nuclear RNAs (snRNAs) are intertwined to form a catalytic center next to Loop I of U5 snRNA. Magnesium ions are coordinated by conserved nucleotides in U6 snRNA. The intron lariat is held in place through base-pairing interactions with both U2 and U6 snRNAs, leaving the variable-length middle portion on the solvent-accessible surface of the catalytic center. The protein components of the spliceosome anchor both 5' and 3' ends of the U2 and U6 snRNAs away from the active site, direct the RNA sequences, and allow sufficient flexibility between the ends and the catalytic center. Thus, the spliceosome is in essence a protein-directed ribozyme, with the protein components essential for the delivery of critical RNA molecules into close proximity of one another at the right time for the splicing reaction. Copyright 2015, American Association for the Advancement of Science.
URLPMID:9885566 [本文引用: 3]
Little is known about mechanisms that regulate and ensure accurate processing of complex transcription units with long introns. We investigate this in the Ultrabithorax gene of Drosophila . A consensus 5- splice site is regenerated at the junction between the first exon and a small internal exon (mI); this splice site is used in a developmentally regulated manner to remove mI during subsequent processing of the downstream intron. Conserved elements within mI and an interaction with exon mII modulate use of the regenerated splice site. Structural similarities predict the same process for mII. This resplicing mechanism avoids competition between distant splice sites for control of exon inclusion and allows removal of a 74 kb intron as a series of smaller fragments.
URLPMID:16386465 [本文引用: 1]
Previously, Patterson et al. showed that mRNA structure information aids splice site prediction in human genes [Patterson, D.J., Yasuhara, K., Ruzzo, W.L., 2002. Pre-mRNA secondary structure prediction aids splice site prediction. Pac. Symp. Biocomput. 7, 223–234]. Here, we have attempted to predict splice sites in selected genes of Saccharomyces cerevisiae using the information obtained from the secondary structures of corresponding mRNAs. From Ares database, 154 genes were selected and their structures were predicted by Mfold. We selected a 20-nucleotide window around each site, each containing 4 nucleotides in the exon region. Based on whether the nucleotide is in a stem or not, the conventional four-letter nucleotide alphabet was translated into an eight-letter alphabet. Two different three-layer-based perceptron neural networks were devised to predict the 5′ and 3′ splice sites. In case of 5′ site determination, a network with 3 neurons at the hidden layer was chosen, while in case of 3′ site 20 neurons acted more efficiently. Both neural nets were trained applying Levenberg–Marquardt backpropagation method, using half of the available genes as training inputs and the other half for testing and cross-validations. Sequences with GUs and AGs non-sites were used as negative controls. The correlation coefficients in the predictions of 5′ and 3′ splice sites using eight-letter alphabet were 98.0% and 69.6%, respectively, while these values were 89.3% and 57.1% when four-letter alphabet is applied. Our results suggest that considering the secondary structure of mRNA molecules positively affects both donor and acceptor site predictions by increasing the capacity of neural networks in learning the patterns.
URLPMID:6489319 [本文引用: 1]
Abstract We have synthesised a 32-bp oligonucleotide containing sequences conforming to the consensus sequences for donor and acceptor splice sites. The oligonucleotide has been inserted into an RNA polymerase B (II) transcription unit and the resulting recombinant used to study the splicing mechanism. Our findings are as follows: (i) the synthetic sites function when separated by several different prokaryotic or eukaryotic DNA fragments providing bulk intron sequence, (ii) intron size need not be greater than 29 bp, (iii) an AG dinucleotide 11 bp upstream from the invariant AG of an acceptor splice site renders the latter non-functional, and (iv) sequence changes distant from splice sites can affect the efficiency of their utilisation.
URLPMID:11058137 [本文引用: 1]
Abstract A set of 43 337 splice junction pairs was extracted from mammalian GenBank annotated genes. Expressed sequence tag (EST) sequences support 22 489 of them. Of these, 98.71% contain canonical dinucleotides GT and AG for donor and acceptor sites, respectively; 0.56% hold non-canonical GC-AG splice site pairs; and the remaining 0.73% occurs in a lot of small groups (with a maximum size of 0.05%). Studying these groups we observe that many of them contain splicing dinucleotides shifted from the annotated splice junction by one position. After close examination of such cases we present a new classification consisting of only eight observed types of splice site pairs (out of 256 a priori possible combinations). EST alignments allow us to verify the exonic part of the splice sites, but many non-canonical cases may be due to intron sequencing errors. This idea is given substantial support when we compare the sequences of human genes having non-canonical splice sites deposited in GenBank by high throughput genome sequencing projects (HTG). A high proportion (156 out of 171) of the human non-canonical and EST-supported splice site sequences had a clear match in the human HTG. They can be classified after corrections as: 79 GC-AG pairs (of which one was an error that corrected to GC-AG), 61 errors that were corrected to GT-AG canonical pairs, six AT-AC pairs (of which two were errors that corrected to AT-AC), one case was produced from non-existent intron, seven cases were found in HTG that were deposited to GenBank and finally there were only two cases left of supported non-canonical splice sites. If we assume that approximately the same situation is true for the whole set of annotated mammalian non-canonical splice sites, then the 99.24% of splice site pairs should be GT-AG, 0.69% GC-AG, 0.05% AT-AC and finally only 0.02% could consist of other types of non-canonical splice sites. We analyze several characteristics of EST-verified splice sites and build weight matrices for the major groups, which can be incorporated into gene prediction programs. We also present a set of EST-verified canonical splice sites larger by two orders of magnitude than the current one (22 199 entries versus approximately 600) and finally, a set of 290 EST-supported non-canonical splice sites. Both sets should be significant for future investigations of the splicing mechanism.
[本文引用: 1]
URLPMID:4529404 [本文引用: 4]
Recursive splicing is a process in which large introns are removed in multiple steps by re-splicing at ratchet points--5' splice sites recreated after splicing. Recursive splicing was first identified in the Drosophila Ultrabithorax (Ubx) gene and only three additional Drosophila genes have since been experimentally shown to undergo recursive splicing. Here we identify 197 zero nucleotide exon ratchet points in 130 introns of 115 Drosophila genes from total RNA sequencing data generated from developmental time points, dissected tissues and cultured cells. The sequential nature of recursive splicing was confirmed by identification of lariat introns generated by splicing to and from the ratchet points. We also show that recursive splicing is a constitutive process, that depletion of U2AF inhibits recursive splicing, and that the sequence and function of ratchet points are evolutionarily conserved in Drosophila. Finally, we identify four recursively spliced human genes, one of which is also recursively spliced in Drosophila. Together, these results indicate that recursive splicing is commonly used in Drosophila, occurs in humans, and provides insight into the mechanisms by which some large introns are removed.
URLPMID:11269499 [本文引用: 1]
Abstract Top of page Abstract Introduction Results Discussion Methods Acknowledgements References Pre-mRNA splicing has to be coordinated with other processes occurring in the nucleus including transcription, mRNA 3′ end formation and mRNA export. To analyze the relationship between transcription and splicing, we constructed a network of nested introns. Introns were inserted in the 5′ splice site and/or branchpoint of a synthetic yeast intron interrupting a reporter gene. The inserted introns mask the recipient intron from the cellular machinery until they are removed by splicing. Production of functional mRNA from these constructs therefore requires recognition of a spliced RNA as a splicing substrate. We show that recurrent splicing occurs in a sequential and ordered fashion in vivo . Thus, in Saccharomyces cerevisiae , intron recognition and pre-spliceosome assembly is not tightly coupled to transcription.
URLPMID:23395799Magsci [本文引用: 1]
The mechanisms by which huge human introns are spliced out precisely are poorly understood. We analyzed large intron 7 (110 199 nucleotides) generated from the human dystrophin (DMD) pre-mRNA by RT-PCR. We identified branching between the authentic 5' splice site and the branch point; however, the sequences far from the branch site were not detectable. This RT-PCR product was resistant to exoribonuclease (RNase R) digestion, suggesting that the detected lariat intron has a closed loop structure but contains gaps in its sequence. Transient and concomitant generation of at least two branched fragments from nested introns within large intron 7 suggests internal nested splicing events before the ultimate splicing at the authentic 5' and 3' splice sites. Nested splicing events, which bring the authentic 5' and 3' splice sites into close proximity, could be one of the splicing mechanisms for the extremely large introns. (C) 2013 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
URLPMID:25897131 [本文引用: 6]
The conventional model for splicing involves excision of each intron in one piece; we demonstrate this inaccurately describes splicing in many human genes. First, after switching on transcription ofSAMD4A, a gene with a 134 kb-long first intron, splicing joins the 3′ end of exon 1 to successive points within intron 1 well before the acceptor site at exon 2 is made. Second, genome-wide analysis shows that >60% of active genes yield products generated by such intermediate intron splicing. These products are present at 6515% the levels of primary transcripts, are encoded by conserved sequences similar to those found at canonical acceptors, and marked by distinctive structural and epigenetic features. Finally, using targeted genome editing, we demonstrate that inhibiting the formation of these splicing intermediates affects efficient exon–exon splicing. These findings greatly expand the functional and regulatory complexity of the human transcriptome.
URLPMID:19289445 [本文引用: 1]
Abstract MOTIVATION: A new protocol for sequencing the messenger RNA in a cell, known as RNA-Seq, generates millions of short sequence fragments in a single run. These fragments, or 'reads', can be used to measure levels of gene expression and to identify novel splice variants of genes. However, current software for aligning RNA-Seq data to a genome relies on known splice junctions and cannot identify novel ones. TopHat is an efficient read-mapping algorithm designed to align reads from an RNA-Seq experiment to a reference genome without relying on known splice sites. RESULTS: We mapped the RNA-Seq reads from a recent mammalian RNA-Seq experiment and recovered more than 72% of the splice junctions reported by the annotation-based software from that study, along with nearly 20,000 previously unreported junctions. The TopHat pipeline is much faster than previous systems, mapping nearly 2.2 million reads per CPU hour, which is sufficient to process an entire RNA-Seq experiment in less than a day on a standard desktop computer. We describe several challenges unique to ab initio splice site discovery from RNA-Seq reads that will require further algorithm development. AVAILABILITY: TopHat is free, open-source software available from http://tophat.cbcb.umd.edu. SUPPLEMENTARY INFORMATION: Supplementary data are available at Bioinformatics online.
URLPMID:18769727 [本文引用: 1]
Abstract While many properties of eukaryotic gene structure are well characterized, differences in the form and function of introns that occur at different positions within a transcript are less well understood. In particular, the dynamics of intron length variation with respect to intron position has received relatively little attention. This study analyzes all available data on intron lengths in GenBank and finds a significant trend of increased length in first introns throughout a wide range of species. This trend was found to be even stronger when using high-confidence gene annotation data for three model organisms (Arabidopsis thaliana, Caenorhabditis elegans, and Drosophila melanogaster) which show that the first intron in the 5' UTR is--on average--significantly longer than all downstream introns within a gene. A partial explanation for increased first intron length in A. thaliana is suggested by the increased frequency of certain motifs that are present in first introns. The phenomenon of longer first introns can potentially be used to improve gene prediction software and also to detect errors in existing gene annotations.
URLPMID:19924226 [本文引用: 1]
Abstract In mammals a considerable 92% of genes contain introns, with hundreds and hundreds of these introns reaching the incredible size of over 50,000 nucleotides. These "large introns" must be spliced out of the pre-mRNA in a timely fashion, which involves bringing together distant 5' and 3' acceptor and donor splice sites. In invertebrates, especially Drosophila, it has been shown that larger introns can be spliced efficiently through a process known as recursive splicing-a consecutive splicing from the 5'-end at a series of combined donor-acceptor splice sites called RP-sites. Using a computational analysis of the genomic sequences, we show that vertebrates lack the proper enrichment of RP-sites in their large introns, and, therefore, require some other method to aid splicing. We analyzed over 15,000 non-redundant, large introns from six mammals, 1,600 from chicken and zebrafish, and 560 non-redundant large introns from five invertebrates. Our bioinformatic investigation demonstrates that, unlike the studied invertebrates, the studied vertebrate genomes contain consistently abundant amounts of direct and complementary strand interspersed repetitive elements (mainly SINEs and LINEs) that may form stems with each other in large introns. This examination showed that predicted stems are indeed abundant and stable in the large introns of mammals. We hypothesize that such stems with long loops within large introns allow intron splice sites to find each other more quickly by folding the intronic RNA upon itself at smaller intervals and, thus, reducing the distance between donor and acceptor sites.
URLPMID:4471124 [本文引用: 3]
It is generally believed that splicing removes introns as single units from precursor messenger RNA transcripts. However, some long Drosophila melanogaster introns contain a cryptic site, known as a recursive splice site (RS-site), that enables a multi-step process of intron removal termed recursive splicing. The extent to which recursive splicing occurs in other species and its mechanistic basis have not been examined. Here we identify highly conserved RS-sites in genes expressed in the mammalian brain that encode proteins functioning in neuronal development. Moreover, the RS-sites are found in some of the longest introns across vertebrates. We find that vertebrate recursive splicing requires initial definition of an `RS-exon' that follows the RS-site. The RS-exon is then excluded from the dominant mRNA isoform owing to competition with a reconstituted 5' splice site formed at the RS-site after the first splicing step. Conversely, the RS-exon is included when preceded by cryptic promoters or exons that fail to reconstitute an efficient 5' splice site. Most RS-exons contain a premature stop codon such that their inclusion can decrease mRNA stability. Thus, by establishing a binary splicing switch, RS-sites demarcate different mRNA isoforms emerging from long genes by coupling cryptic elements with inclusion of RS-exons.
URLPMID:4678815
Abstract Alternative splicing is a powerful mechanism present in eukaryotic cells to obtain a wide range of transcripts and protein isoforms from a relatively small number of genes. The mechanisms regulating (alternative) splicing and the paradigm of consecutive splicing have recently been challenged, especially for genes with a large number of introns. RNA-Seq, a powerful technology using deep sequencing in order to determine transcript structure and expression levels, is usually performed on mature mRNA, therefore not allowing detailed analysis of splicing progression. Sequencing pre-mRNA at different stages of splicing potentially provides insight into mRNA maturation. Although the number of tools that analyze total and cytoplasmic RNA in order to elucidate the transcriptome composition is rapidly growing, there are no tools specifically designed for the analysis of nuclear RNA (which contains mixtures of pre- and mature mRNA). We developed dedicated algorithms to investigate the splicing process. In this paper, we present a new classification of RNA-Seq reads based on three major stages of splicing: pre-, intermediate- and post-splicing. Applying this novel classification we demonstrate the possibility to analyze the order of splicing. Furthermore, we uncover the potential to investigate the multi-step nature of splicing, assessing various types of recursive splicing events. We provide the data that gives biological insight into the order of splicing, show that non-sequential splicing of certain introns is reproducible and coinciding in multiple cell lines. We validated our observations with independent experimental technologies and showed the reliability of our method. The pipeline, named SplicePie, is freely available at: https://github.com/pulyakhina/splicing_analysis_pipeline. The example data can be found at: https://barmsijs.lumc.nl/HG/irina/example_data.tar.gz. The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
[本文引用: 1]
URLPMID:16314266 [本文引用: 1]
The analysis of lariats produced in vivo during pre-mRNA splicing is a powerful tool for elucidation of regulatory mechanisms and identification of natural recursive splicing events. Nevertheless, this analysis is technically challenging because lariats normally have short half-lives. With appropriate controls, RT-PCR amplification and sequencing of the region spanning the 2′–5′ phosophodiester bond at the branch junction can be a sensitive and versatile method for lariat analysis. This approach can be facilitated and enhanced by reducing the activity of debranching enzyme (DBR) in order to stabilize lariats. We have generated a set of plasmids for dsRNA-mediated knockdown of DBR under diverse conditions in transgenic Drosophila and in cultured cells. We describe the use of these plasmids and protocols for lariat analysis. We have generated transgenic Drosophila strains carrying a GAL4-regulated RNAi construct that allows selective knockdown of DBR in specific tissues or developmental stages, using the large collection of available GAL4 expression lines. These strains should prove useful for detailed developmental analyses of alternative and recursive splicing and for genetic analyses of splicing factors. Similar approaches should be readily adaptable to other organisms.
[本文引用: 1]
URLPMID:3465671 [本文引用: 1]
Nat Struct Mol Biol. 2012 Jun 17;19(7):719-21. doi: 10.1038/nsmb.2327. Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov't; Research Support, U.S. Gov't, Non-P.H.S.
Magsci [本文引用: 1]
Targeted nucleases are powerful tools for mediating genome alteration with high precision. The RNA-guided Cas9 nuclease from the microbial clustered regularly interspaced short palindromic repeats (CRISPR) adaptive immune system can be used to facilitate efficient genome engineering in eukaryotic cells by simply specifying a 20-nt targeting sequence within its guide RNA. Here we describe a set of tools for Cas9-mediated genome editing via nonhomologous end joining (NHEJ) or homology-directed repair (HDR) in mammalian cells, as well as generation of modified cell lines for downstream functional studies. To minimize off-target cleavage, we further describe a double-nicking strategy using the Cas9 nickase mutant with paired guide RNAs. This protocol provides experimentally derived guidelines for the selection of target sites, evaluation of cleavage efficiency and analysis of off-target activity. Beginning with target design, gene modifications can be achieved within as little as 1-2 weeks, and modified clonal cell lines can be derived within 2-3 weeks.
[本文引用: 3]
URLPMID:5809388 [本文引用: 2]
Total RNA sequencing has been used to reveal poly(A) and non-poly(A) RNA expression, RNA processing and enhancer activity. To date, no method for full-length total RNA sequencing of single cells has been developed despite the potential of this technology for single-cell biology. Here we describe random displacement amplification sequencing (RamDA-seq), the first full-length total RNA-sequencing method for single cells. Compared with other methods, RamDA-seq shows high sensitivity to non-poly(A) RNA and near-complete full-length transcript coverage. Using RamDA-seq with differentiation time course samples of mouse embryonic stem cells, we reveal hundreds of dynamically regulated non-poly(A) transcripts, including histone transcripts and long noncoding RNANeat1. Moreover, RamDA-seq profiles recursive splicing in >300-kb introns. RamDA-seq also detects enhancer RNAs and their cell type-specific activity in single cells. Taken together, we demonstrate that RamDA-seq could help investigate the dynamics of gene expression, RNA-processing events and transcriptional regulation in single cells. TotalRNA sequencing has been used to profile poly(A) and non-poly(A) RNA expression, processing and the activity of enhancers. Here the authors develop RamDA-seq, a method for full-length total RNA sequencing in single cells.
,
URL [本文引用: 1]
[本文引用: 1]
URLPMID:22465326Magsci [本文引用: 1]
Alternative splicing (AS) is the process by which splice sites in precursor (pre)-mRNA are differentially selected to produce multiple mRNA and protein isoforms. During the past few years the application of genome-wide profiling technologies coupled with bioinformatic approaches has transformed our understanding of AS complexity and regulation. These studies are further driving research directed at elucidating the functions of networks of regulated AS events in the context of normal physiology and disease. Major strides have also been made in understanding how AS is functionally integrated with- and coupled to- gene regulation at the level of chromatin and transcription. Particularly intriguing is the discovery of new AS ‘switches’ that control transcriptional networks required for animal development and behavior.
Magsci [本文引用: 1]
Most eukaryotic protein-coding transcripts contain introns, which vary in number and position along the transcript body. Intron removal through pre-mRNA splicing is tightly linked to transcription by RNA polymerase II as it translocates along each gene. Here, we review recent evidence that transcription and splicing are functionally coupled. We focus on how RNA polymerase II elongation rates impact splicing through local regulation and transcriptional pausing within genes. Emerging concepts of how splicing-related changes in elongation might be achieved are highlighted. We place the interplay between transcription and splicing in the context of chromatin where nucleosome positioning influences elongation, and histone modifications participate directly in the recruitment of splicing regulators to nascent transcripts.
URLPMID:3771521 [本文引用: 2]
Genome-wide association studies have identified many noncoding variants associated with common diseases and traits. We show that these variants are concentrated in regulatory DNA marked by deoxyribonuclease I (DNase I) hypersensitive sites (DHSs). Eighty-eight percent of such DHSs are active during fetal development and are enriched in variants associated with gestational exposure-related phenotypes. We identified distant gene targets for hundreds of variant-containing DHSs that may explain phenotype associations. Disease-associated variants systematically perturb transcription factor recognition sequences, frequently alter allelic chromatin states, and form regulatory networks. We also demonstrated tissue-selective enrichment of more weakly disease-associated variants within DHSs and the de novo identification of pathogenic cell types for Crohn disease, multiple sclerosis, and an electrocardiogram trait, without prior knowledge of physiological mechanisms. Our results suggest pervasive involvement of regulatory DNA variation in common human disease and provide pathogenic insights into diverse disorders.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
In addition to coding information, human exons contain sequences necessary for correct splicing. These elements are known to be under purifying selection and their disruption can cause disease. However, the density of functional exonic splicing information remains profoundly uncertain. Several groups have experimentally investigated how mutations at different exonic positions affect splicing. They have found splice information to be distributed widely in exons, with one estimate putting the proportion of splicing-relevant nucleotides at >90%. These results suggest that splicing could place a major pressure on exon evolution. However, analyses of sequence conservation have concluded that the need to preserve splice regulatory signals only slightly constrains exon evolution, with a resulting decrease in the average human rate of synonymous evolution of only 1-4%. Why do these two lines of research come to such different conclusions? Among other reasons, we suggest that the methods are measuring different things: one assays the density of sites that affect splicing, the other the density of sites whose effects on splicing are visible to selection. In addition, the experimental methods typically consider short exons, thereby enriching for nucleotides close to the splice junction, such sites being enriched for splice-control elements. By contrast, in part owing to correction for nucleotide composition biases and to the assumption that constraint only operates on exon ends, the conservation-based methods can be overly conservative. The online version of this article (doi:10.1007/s00439-017-1798-3) contains supplementary material, which is available to authorized users.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
URLPMID:29727661 [本文引用: 1]
Tools to understand how the spliceosome functions invivo have lagged behind advances in the structural biology of the spliceosome. Here, methods are described to globally profile spliceosome-bound pre-mRNA, intermediates, and spliced mRNA at nucleotide resolution. These tools are applied to three yeast species that span 600 million years of evolution. The sensitivity of the approach enables the detection of canonical and non-canonical events, including interrupted, recursive, and nested splicing. This application of statistical modeling uncovers independent roles for the size and position of theintron and the number of introns per transcript in substrate progression through the two catalytic stages. These include species-specific inputs suggestive of spliceosome-transcriptome coevolution. Further investigations reveal the ATP-dependent discard of numerous endogenous substrates afterspliceosome assembly invivo and connect this discard to intron retention, a form of splicing regulation. Spliceosome profiling is a quantitative, generalizable global technology used to investigate an RNP central to eukaryotic gene expression.
URLPMID:27658249 [本文引用: 1]
Abstract The Candida CTG clade is a monophyletic group of fungal species that translates CTG as serine, and includes the pathogens Candida albicans and Candida parapsilosis. Research has typically focused on identifying protein-coding genes in these species. Here, we use bioinformatic and experimental approaches to annotate known classes of non-coding RNAs in three CTG-clade species, Candida parapsilosis, Candida orthopsilosis and Lodderomyces elongisporus. We also update the annotation of ncRNAs in the C. albicans genome. The majority of ncRNAs identified were snoRNAs. Approximately 50% of snoRNAs (including most of the C/D box class) are encoded in introns. Most are within mono- and polycistronic transcripts with no protein coding potential. Five polycistronic clusters of snoRNAs are highly conserved in fungi. In polycistronic regions, splicing occurs via the classical pathway, as well as by nested and recursive splicing. We identified spliceosomal small nuclear RNAs, the telomerase RNA component, signal recognition particle, RNase P RNA component and the related RNase MRP RNA component in all three genomes. Stem loop IV of the U2 spliceosomal RNA and the associated binding proteins were lost from the ancestor of C. parapsilosis and C. orthopsilosis, following the divergence from L. elongisporus. The RNA component of the MRP is longer in C. parapsilosis, C. orthopsilosis and L. elongisporus than in S. cerevisiae, but is substantially shorter than in C. albicans.
URLPMID:26728853 [本文引用: 1]
Abstract Examples of associations between human disease and defects in pre-messenger RNA splicing/alternative splicing are accumulating. Although many alterations are caused by mutations in splicing signals or regulatory sequence elements, recent studies have noted the disruptive impact of mutated generic spliceosome components and splicing regulatory proteins. This review highlights recent progress in our understanding of how the altered splicing function of RNA-binding proteins contributes to myelodysplastic syndromes, cancer, and neuropathologies. 0008 2016 Chabot and Shkreta.
URLPMID:5126111 [本文引用: 1]
Over time eukaryotic genomes have evolved to host genes carrying multiple exons separated by increasingly larger intronic, mostly non-protein-coding, sequences. Initially, little attention was paid to these intronic sequences, as they were considered not to contain regulatory information. However, advances in molecular biology, sequencing, and computational tools uncovered that numerous segments within these genomic elements do contribute to the regulation of gene expression. Introns are differentially removed in a cell type-specific manner to produce a range of alternatively-spliced transcripts, and many span tens to hundreds of kilobases. Recent work in human and fruitfly tissues revealed that long introns are extensively processed cotranscriptionally and in a stepwise manner, before their two flanking exons are spliced together. This process, called ecursive splicing,- often involves non-canonical splicing elements positioned deep within introns, and different mechanisms for its deployment have been proposed. Still, the very existence and widespread nature of recursive splicing offers a new regulatory layer in the transcript maturation pathway, which may also have implications in human disease.
[本文引用: 1]
URLPMID:27507607 [本文引用: 1]
The splicing rates of very long mammalian introns and of short ones are similar. It is therefore baffling how spatially distant splice sites (SSs) are rapidly brought into proximityin vivowhen introns are thousands of nucleotides long. There is much evidence concerning the functional associations between splicing factors and RNA polymerase II (RNAPII) as well as between splicing factors and chromatin. Since splicing factors involved in the identification of the 5′ and 3′ SSs associate with RNAPII, SS pairing could potentially occur closely following the synthesis of long introns as they are still attached to chromatin via RNAPII. Functional associations between splicing factors and chromatin would later promote splicing factor recruitment to the RNA to advance the splicing reaction.
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}