 ,жаЙњХЉвЕПЦбЇдКББОЉаѓФСЪовНбаОПЫљ,ББОЉ 100193
,жаЙњХЉвЕПЦбЇдКББОЉаѓФСЪовНбаОПЫљ,ББОЉ 100193Applications of genome selection in sheep breeding
Zhida Zhao, Li Zhang,Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing 100193, ChinaЭЈбЖзїеп:
БрЮЏ: НЏЫМЮФ
ЪеИхШеЦк:2018-11-14аоЛиШеЦк:2019-02-5ЭјТчГіАцШеЦк:2019-04-20
| ЛљН№зЪжњ: |
Received:2018-11-14Revised:2019-02-5Online:2019-04-20
| Fund supported: |
зїепМђНщ About authors
еджОДя,ЫЖЪПбаОПЩњ,зЈвЕЗНЯђЃКбђЗжзгг§жжМАЩњВњE-mail:

еЊвЊ
ЙиМќДЪЃК
Abstract
KeywordsЃК
PDF (359KB)дЊЪ§ОнЖрЮЌЖШЦРМлЯрЙиЮФеТЕМГіEndNote|Ris|BibtexЪеВиБОЮФ
БОЮФв§гУИёЪН
еджОДя, еХРђ. ЛљвђзщбЁдёдкУрбђг§жжжаЕФгІгУ[J]. вХДЋ, 2019, 41(4): 293-303 doi:10.16288/j.yczz.18-251
Zhida Zhao, Li Zhang.
аѓЧнг§жжЭЈГЃЭЈЙ§ЙРЫуг§жжжЕ(estimated breeding values, EBVs)РДдЄВтКѓДњадзД,вдЦкЕУЕНЗћКЯШЫРрашЧѓЕФВњЦЗЁЃEBVЕФзМШЗЖШОіЖЈСЫг§жжЙЄзїаЇТЪЁЃЫцзХИпЭЈСПВтађММЪѕКЭЩњЮяаХЯЂбЇЕФЗЂеЙ,РћгУИїжжЛљвђзщбЇЪжЖЮЭкОђживЊОМУадзДЙІФмЛљвђвбГЩЮЊаѓЧнг§жжЕФбаОПШШЕуЁЃФПЧА,вбМјЖЈГівЛХњКЭаѓЧнОМУадзДОпгаЯджјЯрЙиЕФЪ§СПадзДЛљвђзљ(quantitative trait locus, QTL)ЁЃБъМЧИЈжњбЁдё(marker assisted selection, MAS)ОЭЪЧРћгУетаЉвбжЊЕФQTLНјаабЁг§,дквЛЖЈГЬЖШЩЯЬсИпСЫг§жжЙЄзїаЇТЪ,ЕЋФПЧАЫљЭкОђЕНЕФQTLЩаВЛзузмСПЕФ20%,ЧвMASФбвдНтЪЭИДдгЕФЪ§СПадзД,ДцдквЛЖЈЕФОжЯоадЁЃ2001Фъ,MeuwissenЕШ[1]ЪзДЮЬсГіЛљвђзщбЁдё(genome selection, GS),ИУЗНЗЈЪЙгУИВИЧШЋЛљвђзщЕФШЋВПвХДЋБъМЧНјааг§жжжЕЙРМЦ,ЯрБШДЋЭГЗНЗЈОпгаИќИпЕФзМШЗад,ВЛНіПЩвдЪЕЯждчЦкбЁжжЁЂЫѕЖЬЪРДњМфИє,ЛЙгаНЕЕЭНќНЛЁЂМгЫйвХДЋНјеЙЕФгХЕуЁЃ
ФПЧА,GSвбГЩЮЊЖЏжВЮяг§жжСьгђЕФбаОПШШЕуЁЃЮвЙњвбдкФЬХЃЁЂЩњжэКЭШтМІжаПЊеЙ,дкЦфЫћаѓЧнЩЯгІгУНЯЩйЁЃжаЙњзїЮЊбјбђДѓЙњ,ЖдбђМАЦфВњЦЗЕФашЧѓж№ФъдіГЄ,ШчКЮбЁг§КЭХрг§ВњСПИќИпЁЂЦЗжЪИќКУЕФгХСМЦЗжж,ЪЧЕБЧАбђВњвЕЗЂеЙиНашНтОіЕФЮЪЬтЁЃФПЧА,гЂЙњЁЂЗЈЙњЁЂАФДѓРћбЧКЭаТЮїРМЕШЙњМввбНЋGSММЪѕгІгУгкбђЕФг§жжжаВЂШЁЕУСЫСМКУЕФг§жжНјеЙ,ЖјЮвЙњдкетЗНУцШдЪЧПеАзЁЃБОЮФЖдУрбђ(Ovis aries)Лљвђзщг§жжММЪѕКЭGSЕФЗЂеЙМАгІгУНјааСЫзлЪі,вдЦкЮЊНёКѓдкУрбђЩЯПЊеЙGSбаОПЬсЙЉНшМјгыВЮПМЁЃ
1 Зжзгг§жжММЪѕЕФЗЂеЙЯжзД
ДЋЭГг§жжжївЊРћгУБэаЭаХЯЂКЭЯЕЦзаХЯЂ,ЭЈЙ§зюМбЯпадЮоЦЋЙРМЦ(best linear unbiased predication, BLUP)МЦЫуг§жжжЕ,ВЂвдДЫзїЮЊаѓЧнбЁг§ЕФЛљДЁДгЖјЛёЕУРэЯыЕФвХДЋНјеЙЁЃBLUPЗЈдкаѓЧнвЛаЉживЊОМУадзДЕФбЁдёЩЯШЁЕУСЫНЯКУЕФаЇЙћ,МЋДѓЕиЭЦЖЏСЫЮвЙњаѓЧнг§жжЙЄзїЁЃЕЋЪЧЭЈЙ§ЯЕЦзЕУГіЕФжЛЪЧЦкЭћЕФвХДЋЙиЯЕ,ЪЕМЪжаПЩФмвђУЯЕТЖћГщбљРыВюЖјЪЇзМ;ЭЌЪБ,ЖдгквЛаЉЕЭвХДЋСІЁЂЪмадБ№ЯожЦЁЂВЛФмдчЦкВтЖЈЁЂВтЖЈГЩБОИп,вдМАВтСПФбЖШДѓЕФадзД,ДЋЭГг§жжЗНЗЈДцдкНЯДѓЕФОжЯоадЁЃЫцзХЗжзгвХДЋбЇКЭЪ§СПвХДЋбЇЕФЗЂеЙ,аѓЧнг§жжж№НЅОлНЙЕНЗжзгЫЎЦНКЭЛљвђзщЫЎЦН,МДЛљвђзщг§жжММЪѕЁЃЯожЦадЦЌЖЮГЄЖШЖрЬЌадММЪѕ(restriction fragment length polymorphism, RFLP)гк1974ФъУцЪР,ИУММЪѕЭЦЖЏСЫШЫРрЖдDNAЖрЬЌадЕФбаОПЁЃMASЭЈЙ§RFLPБъМЧЛљвђзщВЂРћгУБъМЧЕФЛљвђаЭЙРМЦадзДЕФБэаЭ,вдДЫНјаабЁжжЁЃИУЗНЗЈЭкОђСЫгыаѓЧнживЊОМУадзДОпгаЯджјЯрЙиЕФQTLЛђжїаЇЛљвђВЂНјаабЁжжбЁг§,ЕБЦ№ЪМЛљвђЮЛЕужЎМфСЌЫјВЛЦНКт(linkage disequilibrium, LD)КмЧПЪБаЇЙћИќМб,дкЧхГ§вўадгаКІЛљвђЪБгШЮЊУїЯд,ВЂЧввХДЋБъМЧвХДЋЮШЖЈ,ВЛвзЪмЛЗОГгАЯь,ЮоадБ№ЁЂФъСфЯожЦ,ЭЈЙ§дчЦкбЁдёФмЙЛНкЪЁГЩИќЖрЕФГЩБО[2]ЁЃ
ФПЧА,вХДЋБъМЧЕФЭкОђЪжЖЮЯжвбгаЪЎМИжжГЩЪьЕФММЪѕ,ШчРЉдіЦЌЖЮГЄЖШЖрЬЌадБъМЧИЈжњбЁдё(amplified fragment length polymorphism, AFLP)ЁЂRFLPЁЂЫцЛњРЉдіЖрЬЌадБъМЧИЈжњбЁдё(random amplified polymorphic DNA, RAPD)ЁЂЮЂЮРаЧБъМЧИЈжњбЁдё(simple sequence repeat, SSR)КЭЕЅКЫмеЫсЖрЬЌадБъМЧИЈжњбЁдё(single nucleotide polymorphism, SNP)ЕШЁЃгЩгкSNPЪЧЛљгкЕЅКЫмеЫсЫЎЦНЕФЖрЬЌад,БШЦфЫћБъМЧвХДЋЮШЖЈадИќИп,ЖјЧввђЦфУмЖШИпЁЂЗжВМЙуЁЂМьГіТЪИп,ИќЪЪКЯПьЫйИпЭЈСПМьВтЁЃвђДЫ,SNPвбГЩЮЊЖЏжВЮяг§жжКЭЩњУќПЦбЇбаОПСьгђживЊЙЄОпжЎвЛ[3]ЁЃMASЭЈЙ§ЭкОђгыживЊОМУадзДЯджјЯрЙиЕФЗжзгБъМЧ,ВЂРћгУетаЉБъМЧЪЕЯжадзДЕФЖЈЯђбЁдёЁЃИУЗНЗЈашвЊевЕНгыадзДLDЕФЗжзгБъМЧ,ЕЋLDзДЬЌПЩФмвђжизщЖјДђЦЦ,дкВЛЭЌШКЬхжаLDГЬЖШврВЛЯрЭЌ,ЫљвдИУЗНЗЈгаЪБВЂВЛПЩППЁЃ
ЫцзХИпЭЈСПВтађММЪѕЕФЗЂеЙЁЂЩЬгУSNPаОЦЌЕФЮЪЪРМАЗжаЭММЪѕЕФЕЎЩњ,ВтађГЩБОДѓДѓНЕЕЭЁЃШЋЛљвђзщЙиСЊЗжЮі(genome-wide association study, GWAS)ЁЂПНБДЪ§Бфвь(copy number variation, CNV)ЕШЗНЗЈЯрМЬГіЯж,ВЂЧвгУгкаѓЧнОМУадзДжїаЇЛљвђЕФЭкОђгыМјЖЈЁЃ1996Фъ,RischЕШ[4]ЬсГіGWASЗНЗЈ,ИУЗНЗЈвдLDЮЊЛљДЁ,дкШЋЛљвђзщЫЎЦНЩЯЪЖБ№ИпУмЖШЕФSNPsБъМЧВЂгыИДдгадзДЕФБэаЭБфвьНјааЙиСЊЗжЮі,ДгЖјЗжЮіетаЉSNPsЖдБэаЭЕФвХДЋаЇгІ[5]ЁЃGWASзюдчгІгУгкШЫРрМВВЁбаОПЁЃ2005Фъ,ScienceдгжОЪзДЮБЈЕРСЫгыФъСфОпгаЯрЙиадЕФЛЦАпБфадGWASбаОП[6];жЎКѓ,гжга****ЖдЙкаФВЁ[7]ЁЂЗЪХж[8,9]ЕШМВВЁНјааGWASбаОПЁЃФПЧАGWASвбЙуЗКгІгУгкЭкОђаѓЧнживЊОМУадзДЕФвХДЋБъМЧбаОПжаЁЃеХРђЕШ[10]РћгУ50KаОЦЌЖд329жЛУрбђ(ЫеФсЬибђЁЂЕТЙњШтгУУРРћХЋбђКЭЖХВДбђ)ПЊеЙЬхжиадзДЕФGWASЗжЮі,МьВтЕНгыЖЯФЬКѓШедіжиЯрЙиВЂДяЕНЛљвђзщЯджјЫЎЦНЕФSNPsЮЛЕу10Иі,ДяЕНШОЩЋЬхЯджјЫЎЦНЕФЙиСЊЮЛЕу22Иі,ЛёЕУСЫMEF2BЁЂRFXANKКЭCAMKMTЕШгАЯьУрбђЬхжиадзДЕФживЊКђбЁЛљвђЁЃDemarsЕШ[11]ЖдGrvetteКЭOlkuskaСНИіУрбђЦЗжжНјааGWASЗжЮі,ЕУЕНгыУрбђВњИсЪ§ЯрЙиЕФживЊКђбЁЛљвђBMP15ЁЃJohnston[12]ЕШЖдSoayУрбђНјааGWASЗжЮі,ЕУЕНПижЦгаЮоНЧадзДЕФЛљвђRXFP2ЁЃDoreenЕШ[13]Жд23жЛЬиПЫШќЖћбђЕФМВВЁадзДНјааGWASЗжЮі,ЗЂЯжСЫгАЯьаЁблЛћаЮВЁЕФжївЊКђбЁЛљвђPITX3,ИУЛљвђЮЛгк22КХШОЩЋЬхЩЯЁЃДЫЭт,ЛЙгааэЖреыЖдУрбђЖрНЧЁЂУЋКЭУЋЩЋЁЂЩњВњЗЂг§ЁЂЗБжГЁЂШщгУадзДЁЂМВВЁЕШВЛЭЌОМУадзДЕФGWASбаОПЁЃ
CNVЪЧжИDNAЦЌЖЮдкkb~MbЗЖЮЇФкЕФЭЛБф,АќРЈгЩВхШыЁЂШБЪЇЁЂжиИДКЭИДКЯЖрЮЛЕуЕМжТЕФБфвьЁЃ2004Фъ,SebatЕШ[14]ЬсГіCNVsЖрЬЌаддкЗжЮігыХаЖЈШЫШКМфЕФвХДЋБфвьЩЯгаживЊзїгУЁЃ2006Фъ,RedonЕШ[15]ЗЂБэСЫШЫРрЛљвђзщЕквЛДњCNVЭМЦзЁЃгЩгкCNVгыФГаЉШЫРрМВВЁЕФжТВЁЛњРэДцдкУмЧаЯрЙиад,вђДЫвЛжБЪЧбаОПЕФШШЕуЁЃCNVЖдХрг§адФмгХауЁЂПЙВЁСІЧПЕФМвЧнвВОпгаживЊвтвх,СѕМбЩЕШ[16]РћгУ50KбђаОЦЌЖд71жЛЫеФсЬибђНјааЛљвђЗжаЭ,ЙВМьВтЕН134ИіПНБДЪ§БфвьЧјгђ(copy number variation regions, CNVR),ЖдCNVRИВИЧЕФЛљвђНјаазЂЪЭКЭЙІФмЗжЮі,НсЙћБэУїетаЉЛљвђгыасОѕИаЙйжЊОѕЁЂЛЏбЇДЬМЄИаЙйжЊОѕКЭЪЖБ№ЕШЛЗОГгІД№гаЙи,Жд5ИіCNVRНјааqRT-PCRбщжЄ,Цфжа3ИіCNVRЕУЕНбщжЄЁЃКюГЩСжЕШ[17]ВЩгУБШНЯЛљвђзщдгНЛаОЦЌММЪѕЖд3жжУрбђ(УЩЙХбђЁЂЙўШјПЫбђКЭВибђ)НјааCNVбаОП,ЙВМьВтЕН28ИіCNVR,ЭЈЙ§ЖдCNVRИВИЧЕФЛљвђНјаазЂЪЭКЭДњаЛЭЈТЗЗжЮі,ЗЂЯждкУЩЙХбђКЭВибђЕФбЊКьЕААзЛљвђДцдкCNVРЉеХ,ИУЯжЯѓПЩФмгыСНжжУрбђИпдЕЭбѕЕФЩњЛюЛЗОГгаЙиЁЃОЙ§бщжЄ,83.3%ЕФqRT-PCRНсЙћгыаОЦЌМьВтНсЙћвЛжТЁЃ
ОЁЙмGWASКЭCNVЕШЗНЗЈЪЧЭкОђаѓЧнОМУадзДжїаЇЛљвђЕФживЊЗНЗЈ,ЕЋгЩгкGWASЪЕбщНсЙћжаДцдкДѓСПМйбєадЮЪЬт,ВЂЧвФПЧАбаОПЫљЕУЕНЕФОпгаЯджјЯрЙиЕФQTLЪ§СПгаЯо,ЭЌЪБаѓЧнДѓЖрЪ§живЊЕФОМУадзДЖМЪЧЪмЖрЛљвђгыЛЗОГЙВЭЌЕїПиЕФИДдгЕФЪ§СПадзД,вђДЫНіНівРППвдЩЯЗНЗЈНјаабЁдёЛЙДцдквЛЖЈЕФВЛзуЁЃ
2 ЛљвђзщбЁдёММЪѕМАЦфгІгУ
ЫцзХаѓЧнЛљвђзщВтађЙЄзїЕФЯрМЬЭъГЩ,ВтађГЩБОдНРДдНЕЭ,МгжЎМЦЫуЛњдЫЫуФмСІВЛЖЯЬсЩ§,етЮЊШЋаТг§жжММЪѕЕФЗЂеЙДДдьСЫММЪѕЬѕМўЁЃGSжБНгРћгУШЋЛљвђзщЫљгаЕФБъМЧаЇгІШЅЙРМЦг§жжжЕ,ЮЊЯжДњг§жжЙЄзїЬсЙЉСЫаТЕФЫМТЗЁЃ2.1ЛљвђзщбЁдёЕФдРэ
аѓЧнадзДЪмвЛИіЛђЖрИіЛљвђПижЦ,ОЁЙмвбжЄЪЕДцдквЛаЉжїаЇЛљвђ,ЕЋКмЖрадзДЕФЙІФмЛљвђЩаВЛУїШЗ,ШддкЬНЫїжЎжаЁЃDNAЩЯДцдкДѓСПЕФвбжЊЮЛжУЕФSNPБъМЧ,ЫќУЧКЭФГаЉаѓЧнживЊадзДЕФЛљвђДцдквЛИіЛђЖрИіБъМЧLDЁЃгЩгкУПИіSNPгыжїаЇЛљвђМфДцдквЛИіЛђЖрИіLD,ЖрИіSNPОЭПЩФмдкВЛЭЌГЬЖШЩЯНтЪЭЭЌвЛИіЛљвђЕФаЇгІ,МДЭЈЙ§вбжЊSNPsЕФЖрЬЌадКЭБэаЭРДЙРМЦЦфаЇгІЁЃМйЖЈгАЯьЪ§СПадзДЕФУПвЛИіQTLКЭЛљвђаОЦЌжаЕФSNPsДцдкЖрИіLD[18],ЭЈЙ§ЖдШЋЛљвђзщЗЖЮЇФкЫљгагыQTLгаЙиЕФSNPЮЛЕуНјааСЌЫјМьВт,гЩВЮПМШКЕФБэаЭЙРМЦГіУПИіSNPsЕФаЇгІ,ВЂИљОнЦфЙРМЦаЇгІЖдКђбЁШКг§жжжЕНјааЙРМЦ,гЩДЫЕУЕНЕФг§жжжЕГЦЮЊЛљвђзщг§жжжЕ(genomic estimated breeding value, GEBV)[1]ЁЃИУЗНЗЈПЩвдзЗЫнЕНЫљгагАЯьФПБъадзДЕФQTL,НјвЛВНПЫЗўСЫДЋЭГMASжаБъМЧНвЪОвХДЋЗНВюНЯЩйЕФЮЪЬт,ЬсИпСЫг§жжжЕЕФзМШЗад[19],Ждг§жжЙЄзїЦ№ЕНжСЙиживЊЕФзїгУЁЃРћгУSNPЗжаЭКЭSNPаЇгІдЄВтг§жжжЕЙЋЪНЃК$GEBV={{m}_{1}}{{\hat{q}}_{1}}+{{m}_{2}}{{\hat{q}}_{2}}+\ldots +{{m}_{p}}{{\hat{q}}_{p}}=\underset{i=1}{\overset{p}{\mathop \sum }}\,{{m}_{i}}{{\hat{q}}_{i}}$
Цфжа,${{m}_{i}}$БэЪОЕкiЮЛЕуЕФБъМЧЛљвђаЭ,${{\hat{q}}_{i}}$БэЪОЕк$i$ЮЛЕуЕФБъМЧЙРМЦаЇгІжЕ,$p$БэЪОБъМЧЪ§СПЁЃ
MeuwissenЕШ[1]ЭЈЙ§ФЃФтЪЕбщЕУГі,НіЭЈЙ§БъМЧдЄВтг§жжжЕЕФзМШЗадОЭПЩвдДяЕН0.85ЁЃ2006Фъ,ХЃШЋЛљвђзщађСааХЯЂЙЋВМ,SchaefferЕШ[20]ЖдGSдкФЬХЃг§жжжаЕФгІгУНјааОМУбЇЗжЮі,ЕУГіGSММЪѕНЕЕЭСЫ92%ЕФФЬХЃг§жжГЩБОЁЃДЫЭт,ПЦбаШЫдБЖдGSдкжэ[21,22]ЁЂбђ[22,23]КЭМІ[24,25,26]ЕШаѓЧнжаЕФгІгУВпТдвВНјааСЫЬНЬжЁЃ
2.2 Лљвђзщг§жжжЕЕФГЃМћЫуЗЈ
2.2.1 МфНгЗЈМфНгЗЈЪЧЭЈЙ§ЙЙНЈВЮПМШК,РћгУВЮПМШКИіЬхЕФБэаЭКЭШЋЛљвђзщЕФЗжаЭаХЯЂ,ЙРМЦШЋЛљвђзщжаУПвЛИіSNPдкВЛЭЌадзДЕФаЇгІжЕЁЃжЎКѓЭЈЙ§ЖдКђбЁШКНјааSNPsЗжаЭ,РћгУВЮПМШКSNPsЕФБъМЧаЇгІ,РлМгЛёЕУКђбЁШКЕФGEBVs(ЭМ1)ЁЃ
ЭМ1
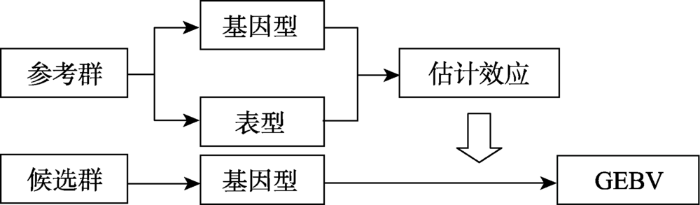 аТДАПкДђПЊ|ЯТдидЭМZIP|ЩњГЩPPT
аТДАПкДђПЊ|ЯТдидЭМZIP|ЩњГЩPPTЭМ1GSМфНгЗЈФЃЪНМЦЫуЭМ
Fig. 1Calculation diagram of GS indirect method
МфНгЗЈГЃгУЕФЙРМЦЗНЗЈАќРЈСыЛиЙщзюМбЯпадЮоЦЋдЄВтЗЈ(ridge regression best linear unbiased prediction, RRBLUP)КЭБДвЖЫЙЗЈ[1,27,28](BayesAЁЂBayesBЁЂBayesCЁЂBayesSКЭfBayesB)ЁЃ
RRBLUPЗЈМйЩшЫљгаБъМЧЖМгааЇгІ,ЧвЗНВюЯрЭЌ;BayesAМйЩшЫљгаБъМЧЖМгааЇгІ,ЧваЇгІЗНВюЗўДгФцПЈЗНЗжВМ(inversed chisquare distribution);BayesBМйЖЈБъМЧаЇгІЗНВювдІа(ІаЮЊв§ШыЕФвЛИіаТВЮЪ§)ЕФИХТЪжЕЮЊ0,вд(1-Іа)ЕФИХТЪЗўДгФцПЈЗНЗжВМ[1]ЁЃBayesSКЭBayesAЕФБъМЧаЇгІЗНВюЕФЗжВММйЩшЯрЭЌ,ЕЋдкаЇгІЙРМЦЪЧдЫгУбЙЫѕЫуЗЈ[27]ЁЃвђBayesBМЦЫуЪБМфГЄ,MeuwissenЕШ[29]ЭЈЙ§ЬѕМўЦкЭћЕќДњ(iteration condition expectation)ЕФЫуЗЈИФНјЕУЕНfBayesB(fast BayesB)ЁЃVerbylaЕШ[30]ЭЈЙ§ЫцЛњЫбЫїБфСПбЁдё(stochastic search variable selection, SSVS)ЖдІадкЙРМЦБъМЧаЇгІЪБНјааЧѓНтЁЃСэЭт,MeuwissenЕШ[31]НЋБъМЧаЇгІМйЖЈЮЊСНИіЗНВюВЛЕШЕФе§ЬЌЗжВМЕФЛьКЯЗжВМ,НЋЗНВюВЮЪ§КЭІазїЮЊФЃаЭБфСПЧѓНт,ЖдBayesBИФНјКѓЕФЫуЗЈГЦЮЊBayesC,ЯрНЯгкЧАеп,BayesCМЦЫуаЇТЪИќИпЁЃ
ДЫЭт,ЛЙгажїГЩЗжЗжЮі(principle component analysis)[32]ЁЂЛњЦїбЇЯА(machine learning)[33]ЁЂзюаЁЖўГЫЛиЙщ(least square regression)[1]ЁЂАыВЮЪ§ЗЈ(semiparametric)[34]ЁЂЗЧВЮЪ§ЗЈ(nonparametric)[35]КЭБДвЖЫЙLASSOЗЈ(least absolute shrinkage and selection operator)[36]ЕШЁЃ
2.2.2 жБНгЗЈ
жБНгЗЈЪЧЭЈЙ§SNPsЙЙНЈИіЬхМфЙиЯЕОиеѓ,НЋЙиЯЕОиеѓЗХШыЛьКЯФЃаЭЗНГЬзщ(mid model equations, MME)жБНгЛёЕУИіЬхЕФЛљвђзщЙРМЦг§жжжЕ(ЭМ2)ЁЃ
ЭМ2
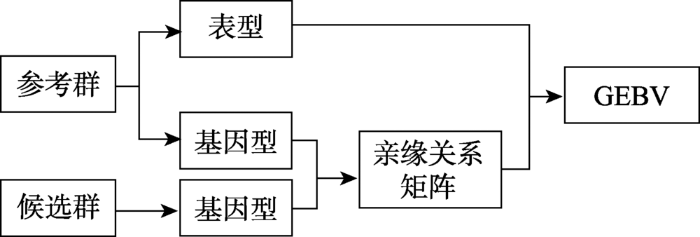 аТДАПкДђПЊ|ЯТдидЭМZIP|ЩњГЩPPT
аТДАПкДђПЊ|ЯТдидЭМZIP|ЩњГЩPPTЭМ2GSжБНгЗЈФЃЪНМЦЫуЭМ
Fig. 2Calculation diagram of GS direct method
жБНгЗЈГЃгУЕФЙРМЦЗНЗЈАќРЈЃКVanRadenЕШ[37]КЭHabierЕШ[38]ЬсГіЕФРћгУБъМЧЙЙНЈИіЬхМфЙиЯЕОиеѓGЕФЛљвђзщзюМбЯпадЮоЦЋЙРМЦЗЈ(genome best linear unbiased prediction, GBLUP);MisztalЕШ[39,40]ЁЂLegarraЕШ[41]КЭChristensenЕШ[42]ЯШКѓЬсГіЕФНЋЯЕЦзОиеѓAКЭЛљвђзщЙиЯЕОиеѓGЙЙНЈЮЊHОиеѓЕФвЛВНЗЈ(single step GBLUP, SSBLUP);ZhangЕШ[43]ЬсГіЕФИјгшВЛЭЌБъМЧВЛЭЌШЈжиЙЙНЈЙиЯЕОиеѓЕФTABLUPЗЈЁЃ
2.3 GEBVзМШЗадЕФгАЯьвђЫи
GEBVЕФзМШЗадЪЧGEBVКЭецЪЕг§жжжЕ(true breeding values, TBV)ЕФЯрЙиЯЕЪ§[1,44],МЦЫуЙЋЪНЮЊЃК$r=\frac{\text{GEBV}}{\text{TBV}}$
GEBVЕФзМШЗаджБНггАЯьг§жжЙЄзїаЇТЪЁЃМИжжгАЯьGEBVзМШЗадЕФГЃМћвђЫиШчЯТЁЃ
2.3.1 МЦЫуФЃаЭ
ЖдгкМфНгЗЈРДЫЕ,BayesBЗНЗЈдкЖрЪ§ЧщПіЯТгХгкЦфЫћЗНЗЈ[1,44],Calus[45]ШЯЮЊетПЩФмгыФЃФтЪ§ОнжагаЯоЕФQTLЪ§СПгаЙи,гыBayesBЗНЗЈЕФРэТлМйЩшБШНЯЮЧКЯЁЃMeuwissenЕШ[29]ЭЈЙ§ЖдBayesBНјааИФНјЕУЕНСЫМЦЫуаЇТЪИќКУЕФBayesC,fBayesBЕФзМШЗадБШBayesBТдЕЭ,ЕЋдЫЫуЪБМфМЋДѓЫѕЖЬЁЃ
ЖдгкжБНгЗЈРДЫЕ,GoddardЕШ[46]КЭHayesЕШ[47]ЭЈЙ§РэТлЗжЮі,ШЯЮЊGBLUPКЭRRBLUPЪЙгУЯрЭЌЕФаХЯЂЧвМйЩшЯрЭЌ,ВЂЕУГівдЩЯСНИіЗНЗЈЕШМлЕФНсТлЁЃZhangЕШ[43]ШЯЮЊ,ПМТЧЕНгАЯьг§жжжЕЦРЙРЪБадзДЕФЫљгаЛљвђЕФЮЛжУдкШЋЛљвђзщжаЗжВМВЛОљдШЁЂЛљвђаЇгІВЛЕШЕФЧщПі,гІЖдБъМЧЯШНјааМгШЈдйЙЙНЈИіЬхЙиЯЕОиеѓ,ЬсГіСЫЙЙНЈTAОиеѓ(trait-specific relationship matrix, TAОиеѓ)ЕФTABLUPЗЈЁЃИУЗНЗЈГфЗжПМТЧСЫФПБъадзДЕФвХДЋНсЙЙ,вђДЫгХгкRRBLUPКЭGBLUPЗЈ[48]ЁЃ
СэЭт,GarrickЕШ[49]ШЯЮЊ,ЖдгкгЩЩйЪ§QTLПижЦЕФадзД,ЛђРДздЖрЦЗжжЁЂдЖдЕЯрЙиЛљвђаЭЕФЪ§Он,Bayes LassoКЭMIXTUREЫуЗЈгааЇЕиРћгУСЫLDаХЯЂ,ЖдGEBVЕФзМШЗадгХгкGBLUPЫуЗЈЁЃOstersenЕШ[50]ШЯЮЊгУderegressed EBVДњЬцEBVзїЮЊЯьгІБфСП(response variable),ПЩЬсИпGEBVЕФзМШЗад,ЖјЫуЗЈгІАДееЪЕМЪЧщПіКЯРэбЁдёЁЃ
2.3.2 LDКЭБъМЧЪ§ФП
ПМТЧЕНGSЕФМйЩшЧАЬсЮЊгАЯьЪ§СПадзДЕФУПвЛИіQTLКЭЛљвђаОЦЌжаЕФSNPsДцдкЖрИіLD,МДSNPsгыQTLБиаывЊДцдкзуЙЛLD,МДБъМЧЪ§ФПдНЖр,зМШЗЖШдНИпЁЃMeuwissenЕШ[1]ЭЈЙ§РрБШМЦЫуЕУЕНЯрСкБъМЧжЎМфr2Ён0.2ВХФмНјааGS;CalusЕШ[45]дђШЯЮЊr2дНДѓ,зМШЗЖШдНДѓ,МДдіМгаОЦЌЕФУмЖШКЭШнСП,ПЩЬсЩ§зМШЗадЁЃЕЋаОЦЌУмЖШдНДѓ,ЛсдіМгГЩБОКЭМЦЫуЪБГЄ,вђДЫдкGSЪЕМЪЙЄзїжа,ашвЊГфЗжИљОнЫљбЁадзДвХДЋСІЕФИпЕЭКЭЪЕбщГЩБО,бЁдёКЯЪЪЕФаОЦЌЁЃErbeЕШ[51]баОПЗЂЯж,ПМТЧSNPМфЕФLDГЬЖШПЩЬсЩ§3.6%ЕФзМШЗад,ИУНсЙћгыдіМгаОЦЌУмЖШНсЙћРрЫЦ(аОЦЌЫљКЌSNPИіЪ§гЩ39 745діМгЕН624 214)ЁЃДЫЭт,РћгУЛљвђЬюГфММЪѕНЋЕЭУмЖШЬюГфЮЊжаИпУмЖШаОЦЌ[52,53]КЭЖЈжЦИіадЛЏаОЦЌ,дкГЩБОдЪаэЕФЧщПіЯТвВПЩвдЬсЩ§GEBVЕФзМШЗадЁЃ
2.3.3 ВЮПМШКЕФЙцФЃКЭНсЙЙ
дкЖдБъМЧаЇгІНјааЙРМЦЪБ,ашвЊЖдЙлВьЕНЕФБэаЭКЭЛљвђаЭНјааБШНЯ,ЪЕМЪЧщПіжаПЩФмЛсГіЯжИїжжЮѓВюЁЃдкИХТЪТлжа,ЪЕбщДЮЪ§дНЖр,ЦЕТЪдНЧїгкИХТЪЁЃвђДЫдкМѕЩйВтЖЈЮѓВюЕФЭЌЪБ,вЊОЁПЩФмМЧТМИќЖрИіЬхЕФБэаЭаХЯЂ,МДВЮПМШКЪ§СПдНДѓ,БъМЧаЇгІЙРМЦдНзМШЗЁЃMeuwissenЕШ[1]ЭЈЙ§ФЃФтЪЕбщЕУГіНсТл,дкЖдвХДЋСІЮЊ0.3ЕФадзДНјааг§жжжЕЙРМЦЪБ,бљБОЪ§Дѓгк2000ЪБзМШЗадНЯИпЁЃGoddardЕШ[54]баОПБэУї,адзДвХДЋСІдНЕЭ,ЫљашбљБОдНЖр,вХДЋСІЮЊ0.1ЕФадзДашвЊ10 000вдЩЯЕФШКЬхЪ§СПВХФмБЃжЄзМШЗЖШДяЕН0.4ЁЃJeremyЕШ[55]ШЯЮЊ,дкЗжЮіЛьКЯадБ№ФЃаЭЪБ,гІПМТЧадШОЩЋЬхЩЯЕФLD,АДадБ№ЙРМЦGEBVЁЃвђДЫ,ЮЊСЫЬсИпGEBVЕФзМШЗад,ашвЊПМТЧШКЬхНсЙЙ,ЩшМЦзуЙЛДѓЕФВЮПМШКЬхЛђбЁдёвХДЋСІНЯИпЕФФПБъадзДЁЃ
2.3.4 ЗЧМгадаЇгІ
ДѓВПЗжживЊадзДЖМгЩЖрИіЛљвђЙВЭЌЕїПи,ЛљвђМфЕФНЛЛЅзїгУЛсгАЯьБэаЭ,етжжЯжЯѓГЦЮЊЗЧМгадаЇгІ(non-additive allelic effect),жївЊАќРЈЯдадаЇгІЁЂЩЯЮЛаЇгІКЭЛЅзїаЇгІ,ВЛФмЮШЖЈвХДЋЁЃЖдгкЗЧМгадаЇгІЕФМЦЫу,XuЕШ[27,56]ЭЈЙ§в§ШыВЮЪ§,РћгУНќЫЦЗНЗЈМђЛЏМЦЫуВЂдкДѓТѓ(Hordeum vulgare)ЛиНЛЪЕбщжаЕУЕНгІгУЁЃGianolaЕШ[34]ЬсГіАыВЮЪ§ЗНЗЈВЂЪЙгУФЃФтЪ§ОнЖдвЛаЉЛЅзїаЇгІНјааСЫбаОП,ШЯЮЊИУЗНЗЈПЩФмЪЪгУгкИпУмЖШБъМЧЪ§ОнЙРМЦGEBVЁЃ
2.3.5 ЧздЕОрРы
LDдкЩњжГЯИАћМѕЪ§ЗжСбЙ§ГЬжа,ХфзгжаЕФDNAПЩФмЗЂЩњжизщЛђепЦЌЖЮЖЊЪЇЕШЭЛБф,ЪЙSNPsЪЇШЅгыдQTLЕФLDзДЬЌЁЃMeuwissenЕШ[1]ЭЈЙ§баОПЗЂЯж,ЫцЪРДњдіМг,GEBVЕФзМШЗадЛсж№НЅНЕЕЭ,дкОРњ5ИіЪРДњКѓгШЮЊУїЯд,вђДЫУПО3ИіЪРДњОЭашвЊжиаТЙРМЦБъМЧаЇгІЁЃHabierЕШ[38]ЗЂЯжЫцВЮПМШКгыбщжЄШКМфЪРДњдіМг,GEBVЕФзМШЗадШДгаЫљЯТНЕЁЃСэЭт,дкВЛЭЌШКЬхМф,QTLКЭSNPsМфЕФLDВЛОЁЯрЭЌ,вђДЫдкЖдЭЌвЛЦЗжжМфЖрГЁСЊКЯВтЖЈЪБ,БиаывЊПМТЧВЛЭЌГЁЧјЕФLDКЭЛЗОГаЇгІЁЃгЩДЫПЩМћ,дкУрбђЪЕМЪг§жжЙЄзїжа,ашвЊВЛЖЯЭъЩЦКЭгХЛЏВЮПМШКЬхЁЃУПФъПЩвдДгЛљвђЗжаЭЙ§ЕФКђбЁШКжабЁГівЛВПЗжНјааБэаЭВтЖЈ,дкгЕгаБэаЭКЭЛљвђЗжаЭЪБ,ИУШКЬхОЭПЩвдВЂШыВЮПМШКЁЃ
2.4 GSММЪѕдкбђг§жжжаЕФгІгУ
ФПЧА,ЙњФкЭтдкФЬХЃЩЯЪЕЪЉGSММЪѕвбШЁЕУУїЯдЕФвХДЋНјеЙ,ЕЋдкбђЩЯЕФгІгУКмЩйЁЃбђЪЧживЊЕФОМУМваѓ,вВЪЧЕБЧАЮвЙњЪЕЪЉЯчДхеёаЫеНТдКЭПЊеЙОЋзМЗіЦЖЕФЪЪвЫаѓжжЁЃИљОнжаЙњаѓФСвЕаХЯЂЭјКЭFAO (СЊКЯЙњСИЪГМАХЉвЕзщжЏ,http://www.fao.org)ЕФзюаТЭГМЦ,2009~2016ФъЮвЙњУрбђЕФЪ§СПДг1.28вкЭЗЩЯЩ§ЕН1.62вкЭЗ,бђШтЯћЗбСПвВгЩ433.7ЭђЖждіМгЕН468.0ЭђЖж,дЄМЦНёКѓЛЙЛсж№ФъЮШВНдіМгЁЃЫцзХбђШтЁЂбђУЋЁЂбђФЬЕШбђВњЦЗЕФЯћЗбдіГЄ,ПЊеЙбђИпаЇг§жжКЭШКЬхИФСМЦШдкУМНо,GSММЪѕЕФЪЕЪЉвВЪЧБиШЛЕФЧїЪЦЁЃбђГЃЙцг§жжЗжЮЊДПжжЗБг§КЭдгНЛг§жж,ЦфжавддгНЛг§жжОгЖрЁЃЙигкЛьКЯЦЗжжЕФGS,DoddsЕШ[57]ЭЈЙ§GBLUPМЦЫуЗНЗЈЖдRomneyЁЂCoopworthКЭPerendale 3жжШтУЋМцгУбђНјааGEBVЙРМЦгыбщжЄ,ШЯЮЊGSММЪѕПЩвддкЛьКЯЦЗжжЕФШКЬхжаНјааGEBVЙРМЦ,ИУбаОППМТЧСЫЦЗжжНсЙЙВЂЕУГігыдЄВтНсЙћЯрЫЦЕФНсТл,ВЂШЯЮЊПМТЧЦЗжжНсЙЙЕФаЇгІПЩвддіМгGEBVЙРМЦЕФзМШЗЖШ;СэЭт,ПМТЧЖЏЮяЕФЛљвђЗжаЭЛсМѕаЁЦЋВю,ЬсИпзМШЗадЁЃDaetwylerЕШ[58]ЪЙгУ50KаОЦЌдЫгУBayesAКЭGBLUPСНжжЫуЗЈ,вдДПжжУРРћХЋбђКЭгыжеЖЫдгНЛЕФУРРћХЋбђзїЮЊВЮПМШК,баОПСЫGSЖдШтжЪКЭбђУЋдЄВтЕФзМШЗад,ЦфжаGBLUPКЭBayesAНсТлВювьУїЯд,ЫЕУїЕБЧАSNPУмЖШВЛзувдПчЮяжжЪЙгУБъМЧаЇгІ,ПЩФмгЩВЛЭЌЦЗжжжаЯрЭЌQTLЕФЮЛжУВЛЭЌЕМжТ;ИУбаОПШЯЮЊЫцзХВЮПМШКбљБОКЌСПвдМАSNPаОЦЌУмЖШдіМг,НЋЛсНјвЛВНЬсИпGEBVЕФзМШЗадЁЃ
ЗБжГадзДжБНггАЯьбјбђЕФОМУаЇвц,ЪЧг§жжжаашвЊжиЕуЙизЂЕФОМУадзД,ЕЋгЩЖрЛљвђПижЦЧвЪмЛЗОГгАЯь,вХДЋСІМЋЕЭЁЃNewtonЕШ[59]РћгУЛљвђзщаХЯЂЦРЙРАФДѓРћбЧУрбђЗБжГадзДЕФвХДЋНјеЙ,баОПЗЂЯжВЛЭЌФъСфЕФЛљвђзщаХЯЂЛсгАЯьвХДЋдівц,ЙЋбђ1ЫъЪБЕФвХДЋдівцЯджјИпгк2ЫъЁЃPickeringЕШ[60]Жд4237жЛRomneyбђНјааGSдЄВт,ЗЂЯжЗБжГадзДЕФзМШЗаддк0.16~0.52жЎМфЁЃ
ыиЬхадзДКЭбђУЋадзДвВЪЧУрбђЕФживЊОМУадзДЁЃAuvrayЕШ[61]ЖдRomneyЁЂCoopworthЁЂPerendaleУрбђНјааGSЗжЮі,ВЂЕУГіGSММЪѕПЩвддквдЩЯ3жжбђжаНјааЖЯФЬжиЁЂыиЬхжиКЭЗрБуГцТбЪ§ЕФг§жжжЕдЄВт,дкRomneyжаПЩвдЖдВњИсЪ§НјаадЄВтЁЃSlack-SmithЕШ[62]баОПЗЂЯж,GSдкАФДѓРћбЧУрбђыиЬхЁЂЩњГЄадзДЁЂЖГКѓблМЁУцЛ§КЭМЁМфжЌЗОКЌСПЕФзМШЗадЗжБ№ЮЊ0.71ЁЂ0.82ЁЂ0.78КЭ0.61ЁЃBritoЕШ[63]дкаТЮїРМУрбђЛюЬхадзДзМШЗадЗЖЮЇЮЊ(0.18ЁР0.07)~(0.33ЁР0.10),ыиЬхадзДЮЊ(0.28ЁР0.09)~(0.55ЁР0.05),ШтжЪадзДЮЊ(0.21ЁР0.07)~(0.36ЁР0.08)ЁЃMoghaddarЕШ[64]ЖдPoll DorsetЁЂWhite SuffolkКЭBorder Leicester 3жжбђЕФЬхжиНјааGEBVЙРМЦ,Poll Dorset ЕФЙРМЦзМШЗадЮЊ0.11~0.27,КѓСНИіЦЗжжЕФзМШЗаддк0.25~0.63жЎМфЁЃShumbushoЕШ[65]ШЯЮЊ,дкгХЛЏг§жжЗНАИЛђЪЙгУШЋЛљвђзщаХЯЂОљБШДЋЭГЗНЗЈЕУЕНИќИпЕФУрбђВњШтадзДвХДЋЖШНјеЙЁЃDaetwylerЕШ[66]жИГі,НіеыЖдДПжжУРРћХЋбђРДЫЕ,1ЫъЪБЕФгЭадбђУЋжиСП(greasy fleece weigh, GFW)КЭ1ЫъЪБбђУЋЯЫЮЌжБОЖ(fibre diameter, FD)ЕФзМШЗЖШОљГЌЙ§0.70,ЖЬЯЫЮЌЧПЖШ(staple strength, SS)ЕФзМШЗадЕЭгк0.70,етПЩФмгыSSМЧТМЕФЪ§СПЩйЛђепSSБШGFWКЭFDЕФвХДЋСІИќЕЭЯрЙиЁЃMoghaddarЕШ[64]ЗЂЯжGSЖд1ЫъКЭГЩФъЕФMerinoбђУЋадзДЕФЙРМЦзМШЗадЮЊ0.33~0.75ЁЃBolormaaЕШ[67]Жд3жжбђУЋжЪСПадзДНјааGSЗжЮі,ЗЂЯжBayesRКЭGBLUPЫуЗЈЕФЦНОљGEBVОЋШЗЖШЯрЫЦ,дМЮЊ0.22,BayesRЖдбђУЋВњСПКЭFDзМШЗЖШОљДѓгк0.40,ЖјЖдбђЦЄжЪСПКЭЮлзЧГЬЖШЕФзМШЗЖШНЯВюЁЃ
бђЕФУкШщадзДгыИсбђЕФГЩЛюТЪУмЧаЯрЙи,ЧвбђФЬвВОпгаживЊЕФОМУМлжЕ,вђДЫЖдбђУкШщадзДКЭШщЗПадзДЕФбаОПвВМЋЮЊживЊЁЃDucheminЕШ[68]ЖдЗЈЯЕLacauneУрбђШКЬх(дМ2500жЛ)ЕФУкШщадзДНјааСЫбаОП,ШЯЮЊGSММЪѕПЩвдЪЙЙЋИсЕФGEBVОЋШЗЖШЬсЩ§18%~25%,BayesCМЦЫуЪБМфЙ§ГЄ,sPLSЗЈЖдгкGEBVЕФЙРМЦОпгаЮШНЁад,ЦфНсЙћЗЧГЃНгНќGBLUPНсЙћЁЃMcLarenЕШ[69]ЭЈЙ§ЗжЮі29ИіШКЬх2957ЭЗДПжжTexelФИбђ,ШЯЮЊЦфШщЗПаЮЬЌКЭШщЗПбзгаНЯИпЕФЯрЙиад,ВЂгАЯьбђШтВњСП,ИУбаОПШЯЮЊПЩвдРћгУGSММЪѕЭЈЙ§ЗжЮігАЯьШщЗПаЮЬЌЕФЛљвђЬсИпбђШтВњСП,ВЂШЯЮЊетЪЧШтбђг§жжЕФвЛжжаТЫМТЗЁЃMuchaЕШ[70]Жд1960жЛгЂЙњФЬЩНбђЕФВњФЬСПКЭУкШщСПНјааGSЪдбщ,ЕУГіSSBLUPКЭGBLUPЕФдЄВтзМШЗадЗжБ№ЮЊ0.61КЭ0.32,SSBLUPОпгаИќИпЕФзМШЗадЁЃCarillierЕШ[71]ЖдЗЈЙњSaanenФЬЩНбђКЭAlpineбђНјааGSбЁдёг§жж,GEBVНЛВцбщжЄЕФзМШЗадЮЊ36%~53%ЁЃLarroqueЕШ[72]баОПЗЂЯж,GBLUPдкФЬЩНбђКЭФЬУрбђЩЯЕФзМШЗадБШКЩЫЙЬЙФЬХЃВю,ЕЋSSBLUPЕФзМШЗадБШGBLUPИп,ЭЈЙ§НЛВцбщжЄЗЂЯж,ЗЈЙњLacauneФЬУрбђGEBVзМШЗаддіМгСЫ0.47,LacauneФЬЩНбђдіМгСЫ0.43ЁЃBalocheЕШ[73]ЭЈЙ§ЖдБШBLUPКЭSSBLUPЖдLacauneФЬЩНбђЕФбЁдёаЇЙћ,ЗЂЯжGSБШДЋЭГг§жжЗНЗЈЕФзМШЗадИќИп,ДяЕН52%;ДЫЭт,SSBLUPБШBLUPзМШЗадИп58%ЁЃMolinaЕШ[74]ЪЙгУ55KаОЦЌЙРМЦЮїАрбРFloridaФЬЩНбђЕФGEBV,НсЙћЯдЪОSSBLUPЦНОљдіМг5.86%ЕФзМШЗадЁЃ
дкбщжЄВЛЭЌЩшМЦФЃаЭжаGSЕФаЇвцбаОПжа,Van der WerfЕШ[23]дкШтбђКЭЯИУЋбђШКЬхжаНјааGSФЃФтЪЕбщ,НсЙћБэУї,ЯрБШДЋЭГЗНЗЈ,GSЗНЗЈдкШтбђКЭЯИУЋбђжаОМУбЁдёжИЪ§ЗжБ№ЬсИпСЫ30%КЭ40%ЁЃRaoulЕШ[75]ВЩгУЕЭУмЖШSNPаОЦЌ(very low-density SNP panel)ЖдLacauneУрбђНјааGS,НсЙћБэУїЃКЖдаладВЮПМШКЪЙгУжаЕШУмЖШЛљвђаЭ(medium-density genotypes)ЪБ,ЛљвђзщЩшМЦ(genomic design)ЕФвХДЋаЇвцЬсЩ§26%,ЖдЭЌЪБАќКЌаладКЭДЦадЕФВЮПМШКЪЙгУжаЕШУмЖШЕФЛљвђаЭ,ЦфвХДЋаЇвцЬсЩ§54%;ЮоТлЛљвђзщЧщПіШчКЮ,ЛљвђзщЩшМЦЕФНќЧзЗБжГТЪЖМЛсБШДЋЭГЩшМЦЕЭЁЃЖдДЦадКЭаладКђбЁУрбђВЩгУМЋЕЭУмЖШЕФЛљвђаЭгыЙщМЏЙ§ГЬ(imputation process)ЯрНсКЯЕМжТаЁаЭЕФУрбђг§жжМЦЛЎЕФвХДЋаЇвцДѓЗљдіМгЁЃSantosЕШ[76]ЭЈЙ§БШНЯВЛЭЌЕФУрбђг§жжВпТдЕФзМШЗадМАОМУЪевц,ШЯЮЊЯдад+бЁдёадЛљвђзщбЁдё(pheno + selective GS)ПЩвддкзюЖЬЪБМфФкЪЕЯжгЏПїЦНКтЧваЇвцзюДѓ,ВЂгааЇНЕЕЭСЫвХДЋМфИєЁЃ
3 НсгягыеЙЭћ
GSг§жжБШДЋЭГг§жжММЪѕОпгаИќИпЕФзМШЗад,ПЩЪЕЯждчЦкбЁжжЁЂЫѕЖЬЪРДњМфИє,ЛЙОпгаНЕЕЭНќНЛЁЂМгЫйвХДЋНјеЙЕШгХЕу,ГЩЮЊЙњФкЭтаѓЧнг§жжЙЄзїЕФбаОПШШЕу[77]ЁЃдкЮвЙњ,GSвбдкФЬХЃЁЂЩњжэКЭШтМІЩЯДѓСПгІгУ,вЛаЉЦѓвЕКЭЪЕбщЪвПЊЪМЖдУрбђг§жжНјааGSЬНЫї,ЕЋжСНёЩаЮДгаЯЕЭГЕФбаОПКЭНсЙћЁЃЙњЭтGSЕФАИР§вбОжЄУїИУММЪѕФмЯджјЬсИпУрбђЕФЩњГЄадзДЁЂбђУЋадзДКЭУкШщадзДЕШбЁдёЕФзМШЗадЁЃвВгабаОПБэУїдкПМТЧЛљвђЗжаЭЁЂдіМгбљБОСПКЭШКЬхЙЙГЩЪБПЩЛёЕУИќИпЕФзМШЗад,ЖјВЩгУМЋЕЭУмЖШаОЦЌКЭЙщМЏЙ§ГЬЯрНсКЯЁЂЯдадКЭбЁдёадGSЯрНсКЯвВПЩвддіМгвХДЋаЇвцЁЃGSММЪѕЖдаѓЧнг§жжЕФЙБЯзКмДѓ,ЕЋдкРэТлбаОПКЭЪЕМЪВйзїжавВДцдквЛаЉЮЪЬт,жївЊБэЯждквдЯТ4ИіЗНУцЃК(1)GSашвЊжжбђГЁгаЭъећЕФЯЕЦзЁЂЩњВњадФмВтЖЈзЪСЯ,ЭЌЪБашвЊГжајЕФВтЖЈ,ВтЖЈЗбгУИп,ЛиБЈТ§;(2)GSЖдЕЭвХДЋСІадзДбЁдёаЇЙћНЯВю,ашвЊДѓЙцФЃЕФВЮПМШК;(3)GSвРОнВЮПМШКЕФSNPsаЇгІЙРМЦКђбЁШКЕФGEBV,ЕЋВЛЪЪгУгыаТГіЯжЕФОЋгЂИіЬхКЭдЖдЕИіЬх[55];(4)МЦЫуЪБМфНЯГЄ,ШдашНјвЛВНбаОПЫуЗЈЁЃ
ФПЧА,ЩњГЄЫйЖШТ§ЁЂЗБжГаЇТЪЕЭЪЧЮвЙњШтбђВњвЕЗЂеЙжаЕФЦПОБЮЪЬт,вђДЫ,дкжЦЖЈг§жжФПБъЪБгІНсКЯВњвЕЕФашЧѓНјааКЯРэЕФбЁг§ЁЃЩњВњДѓЪ§ОнЪЧПЊеЙGSбаОПЕФЛљДЁ,бђГЁБиаывЊГжајПЊеЙЩњВњадФмВтЖЈВЂНЈСЂКЫаФШКЩњВњаХЯЂЪ§ОнЦНЬЈЁЃЮЊНЕЕЭGSГЩБО,ПЩвдЯШВЩгУДЋЭГЗНЗЈНјаабЁдё,ШЛКѓдйНјааSSBLUPбЁдёЁЃвдФГбђГЁЮЊР§,МйЩшЖд3000жЛКђбЁбђНјааGSбЁг§,г§жжФПБъЪЧЬсИпЩњГЄЫйЖШКЭдіМгВњИсЪ§,ПЩвдЯШгУBLUPЖдКђбЁШКЬхНјааг§жжжЕЙРМЦВЂХХУћ,ШЛКѓЖдХХУћЧА30%(бЁдёЧПЖШЪгЪЕМЪЧщПі)ЕФИіЬхдйНјааGEBVЙРМЦЁЃЖдетХњбђНјааКѓајЩњГЄадФмВтЖЈ,ЕУЕНБэаЭ+ЯЕЦз+ЛљвђаЭаХЯЂ,ИУШКЬхгжПЩВЂШыВЮПМШКжа(НкЪЁВЮПМШКВтЖЈЗбгУ)ЁЃДЫЭтЭЈЙ§КЭЦфЫћЯрЭЌЦЗжжЕФбђГЁНјааСЊКЯг§жж[78],бђГЁжЎМфПЩвдЛЅЯрЕМбЊ,ЪЙПЊеЙСЊКЯг§жжЕФбђДцдквЛЖЈЕФЧздЕЙиЯЕ,МШПЩЪЙСЊКЯГЁЧјЕФВЮПМШКЕФаХЯЂЕУЕНЙВЯэ(СэашПМТЧГЁЧјаЇгІ),ЭЌЪБЛЙРЉДѓСЫВЮПМШКЕФЙцФЃ,ЬсИпСЫбЁдёЕФзМШЗадКЭаЇТЪ,ВЂМЋДѓГЬЖШЕиНЕЕЭСЫВтЖЈГЩБОЁЃЭЌЪБ,ЛЙгІНсКЯЖдКђбЁШКЬхЕФБэаЭбЁдё,вдМѕЩйОЋгЂИіЬхЕФвХТЉЁЃДЫЭт,ЮвЙњбђЦЗжжзЪдДЗсИЛ,зджїбаЗЂИВИЧгыУрбђживЊОМУадзДЯрЙиСЊЮЛЕуЕФжаЕЭУмЖШг§жжаОЦЌ,врФмЬсИпGBEVЕФзМШЗадЁЃ
BLUPКЭMASдкбђг§жжЙЄзїжаЫфШЁЕУвЛЖЈЕФГЩаЇ,ЕЋУцСйгЩЖрЛљвђПижЦЕФИДдгЪ§СПадзДЪБ,жЛФмдкКмаЁЕФВуУцШЅНтЪЭ,ЖјGSЭЈЙ§ЙиСЊЛљвђзщФкЫљгаБъМЧНјааЮЛЕуаЇгІЙРМЦ,КмКУЕиНтОіСЫДЋЭГЗНЗЈФбвдНтОіЕФЮЪЬт,ЪЧНёКѓаѓЧнг§жжЕФДѓЧїЪЦ,вВЪЧг§жжЗЂеЙЪЗЩЯЕФвЛИіживЊРяГЬБЎЁЃжаЙњХЉвЕПЦбЇдКББОЉаѓФСЪовНбаОПЫљЖдШтМІКЭШтХЃНјааСЫGSбаОПВЂЛёЕУНЯКУЕФГЩаЇ,ЭЌЪБзджїбаЗЂГіМІаОЦЌЁАIASCHICKЁБЁЃБОбаОПЭХЖгНЋЖдвбгаЕФУрбђг§жжНсЙћНјааЪсРэКЭзмНс,баЗЂИќМгЪЪКЯЮвЙњУрбђг§жжЕФЛљвђаОЦЌ,ВЂПЊеЙКўбђЕФGSг§жжбаОП,ЬНЫїGSММЪѕдкбђг§жжжаЕФЗНЗЈКЭаЇТЪЁЃ
ВЮПМЮФЯз дЮФЫГађ
ЮФЯзФъЖШЕЙађ
ЮФжав§гУДЮЪ§ЕЙађ
БЛв§ЦкПЏгАЯьвђзг
[БОЮФв§гУ: 11]
URLMagsci [БОЮФв§гУ: 1]

ЫцзХЗжзгЪ§СПвХДЋбЇМАЦфЯрЙибЇПЦЕФЗЂеЙЃЌгаЙиЖЏЮявХДЋБъМЧИЈжњбЁдёЗНУцЕФбаОПвВдкВЛЖЯЩюШыЃЌЧввбОдкЖЏЮявХДЋИФСМжагаСЫвЛаЉГЩЙІгІгУЕФЪОР§ЁЃОЭШчКЮзлКЯРћгУБэаЭЁЂЯЕЦзКЭвХДЋБъМЧаХЯЂНјааг§жжжЕЙРМЦЕФЭГМЦбЇЗНЗЈбаОПЗНУцЃЌФПЧАвбЛљБОаЮГЩСЫНЯЮЊЭъЩЦЕФЭГМЦбЇЗНЗЈЁЃЭЌЪБЃЌдкБъМЧИЈжњбЁдёЯрЖдаЇТЪМАЦфгАЯьвђЫиЃЌвдМАБъМЧИЈжњбЁдёЪЕЪЉЗНАИЕФбаОПЩЯвВШЁЕУСЫВЛЩйЯВШЫЕФГЩЙћЁЃБОЮФзлЪіСЫЖЏЮявХДЋБъМЧИЈжњбЁдёбаОПЕФвЛаЉНјеЙЃЌВЂЖдБъМЧИЈжњбЁдёдкЖЏЮявХДЋИФСМжагІгУЕФгаЙиЮЪЬтНјааСЫЬжТлЁЃ<br>Abstract:With the development of molecular and quantitative genetics and its related subjects,it made a great progress on the research about animal genetic marker-assisted selection (MAS).There were also some successful examples on the application of MAS to animal genetic improvement.The statistical method which using phenotypic,pedigree and genetic marker information to predict individual breeding values has already been developed.Many achievements were obtained from the researches,which carried on MAS relative efficiency and its affecting factors and selection schemes.The present paper reviewed some progresses of MAS research and discussed some problems about MAS application to animal breeding.
URLMagsci [БОЮФв§гУ: 1]
ЫцзХЗжзгЪ§СПвХДЋбЇМАЦфЯрЙибЇПЦЕФЗЂеЙЃЌгаЙиЖЏЮявХДЋБъМЧИЈжњбЁдёЗНУцЕФбаОПвВдкВЛЖЯЩюШыЃЌЧввбОдкЖЏЮявХДЋИФСМжагаСЫвЛаЉГЩЙІгІгУЕФЪОР§ЁЃОЭШчКЮзлКЯРћгУБэаЭЁЂЯЕЦзКЭвХДЋБъМЧаХЯЂНјааг§жжжЕЙРМЦЕФЭГМЦбЇЗНЗЈбаОПЗНУцЃЌФПЧАвбЛљБОаЮГЩСЫНЯЮЊЭъЩЦЕФЭГМЦбЇЗНЗЈЁЃЭЌЪБЃЌдкБъМЧИЈжњбЁдёЯрЖдаЇТЪМАЦфгАЯьвђЫиЃЌвдМАБъМЧИЈжњбЁдёЪЕЪЉЗНАИЕФбаОПЩЯвВШЁЕУСЫВЛЩйЯВШЫЕФГЩЙћЁЃБОЮФзлЪіСЫЖЏЮявХДЋБъМЧИЈжњбЁдёбаОПЕФвЛаЉНјеЙЃЌВЂЖдБъМЧИЈжњбЁдёдкЖЏЮявХДЋИФСМжагІгУЕФгаЙиЮЪЬтНјааСЫЬжТлЁЃ<br>Abstract:With the development of molecular and quantitative genetics and its related subjects,it made a great progress on the research about animal genetic marker-assisted selection (MAS).There were also some successful examples on the application of MAS to animal genetic improvement.The statistical method which using phenotypic,pedigree and genetic marker information to predict individual breeding values has already been developed.Many achievements were obtained from the researches,which carried on MAS relative efficiency and its affecting factors and selection schemes.The present paper reviewed some progresses of MAS research and discussed some problems about MAS application to animal breeding.
.
URL [БОЮФв§гУ: 1]
SNP (single nucleotide polymorphism) marker, as a molecular marker of the third generation, plays an important role in molecular genetics, pharmacogenetics, forensic medicine, diagnosis and treatment of diseases recently. In this paper, the definition and characteristics of SNP molecular marker were both introduced. And the application profiles of Bin map, high throughput SNP-chip classification and population evolution research on the basis of SNP molecular marker was presented respectively. Finally, the problems of SNP molecular marker at the present stage were proposed, and the prospects of SNP molecular marker was predicted as well.
URL [БОЮФв§гУ: 1]
SNP (single nucleotide polymorphism) marker, as a molecular marker of the third generation, plays an important role in molecular genetics, pharmacogenetics, forensic medicine, diagnosis and treatment of diseases recently. In this paper, the definition and characteristics of SNP molecular marker were both introduced. And the application profiles of Bin map, high throughput SNP-chip classification and population evolution research on the basis of SNP molecular marker was presented respectively. Finally, the problems of SNP molecular marker at the present stage were proposed, and the prospects of SNP molecular marker was predicted as well.
URLPMID:9583430 [БОЮФв§гУ: 1]
The identification of the genetic basis of complex human diseases such as schizophrenia and diabetes has proven difficult. In their Perspective, Risch and Merikangas propose that we can best accomplish this goal by combining the power of the human genome project with association studies, a method for determining the basis of a genetic disease.
URLPMID:15716906 [БОЮФв§гУ: 1]
Abstract Genetic factors strongly affect susceptibility to common diseases and also influence disease-related quantitative traits. Identifying the relevant genes has been difficult, in part because each causal gene only makes a small contribution to overall heritability. Genetic association studies offer a potentially powerful approach for mapping causal genes with modest effects, but are limited because only a small number of genes can be studied at a time. Genome-wide association studies will soon become possible, and could open new frontiers in our understanding and treatment of disease. However, the execution and analysis of such studies will require great care.
URL [БОЮФв§гУ: 1]
URL [БОЮФв§гУ: 1]
URL [БОЮФв§гУ: 1]
URLPMID:17434869 [БОЮФв§гУ: 1]
Obesity is a serious international health problem that increases the risk of several common diseases. The genetic factors predisposing to obesity are poorly understood. A genome-wide search for type 2 diabetes-susceptibility genes identified a common variant in the FTO (fat mass and obesity associated) gene that predisposes to diabetes through an effect on body mass index (BMI). An additive association of the variant with BMI was replicated in 13 cohorts with 38,759 participants. The 16% of adults who are homozygous for the risk allele weighed about 3 kilograms more and had 1.67-fold increased odds of obesity when compared with those not inheriting a risk allele. This association was observed from age 7 years upward and reflects a specific increase in fat mass.
[БОЮФв§гУ: 1]
Motivated by mining major candidate genes across Ovine genome, the present study is to perform genome-wide association studies(GWAS) to detect genes associated with body weight traits. Using Illumina OvineSNP50 BeadChip, we performed a GWA study in 329 purebred sheep phenotyped for 6 body weight traits(birth weight, weaning weight, 6-month weight, pre-weaning gain, post-weaning gain, daily weight gain). Statistics and data analysis were based on TASSEL programЃЌmixed linear model and the latest Ovis_aries_v3.1 genome sequence (released October 2012). The results indicated that 10 SNPs consistently reached genome-wise significant level for post-weaning gain and 22 SNPs reached chromosome-wise significant level for other body weight traits. The SNPs were within (MEF2B,RFXANKЃЌet al) or close to some ovine genes, which were thought to be the most important candidate genes associated with body weight traits. The results will contribute to identify candidate genes for ovine body weight traits, and facilitate the potential utilization of genes involved production traits in sheep in future.
[БОЮФв§гУ: 1]
Motivated by mining major candidate genes across Ovine genome, the present study is to perform genome-wide association studies(GWAS) to detect genes associated with body weight traits. Using Illumina OvineSNP50 BeadChip, we performed a GWA study in 329 purebred sheep phenotyped for 6 body weight traits(birth weight, weaning weight, 6-month weight, pre-weaning gain, post-weaning gain, daily weight gain). Statistics and data analysis were based on TASSEL programЃЌmixed linear model and the latest Ovis_aries_v3.1 genome sequence (released October 2012). The results indicated that 10 SNPs consistently reached genome-wise significant level for post-weaning gain and 22 SNPs reached chromosome-wise significant level for other body weight traits. The SNPs were within (MEF2B,RFXANKЃЌet al) or close to some ovine genes, which were thought to be the most important candidate genes associated with body weight traits. The results will contribute to identify candidate genes for ovine body weight traits, and facilitate the potential utilization of genes involved production traits in sheep in future.
URL [БОЮФв§гУ: 1]
URLPMID:21651634 [БОЮФв§гУ: 1]
Understanding the genetic architecture of phenotypic variation in natural populations is a fundamental goal of evolutionary genetics. Wild Soay sheep (Ovis aries) have an inherited polymorphism for horn morphology in both sexes, controlled by a single autosomal locus, Horns. The majority of males have large normal horns, but a small number have vestigial, deformed horns, known as scurs; females have either normal horns, scurs or no horns (polled). Given that scurred males and polled females have reduced fitness within each sex, it is counterintuitive that the polymorphism persists within the population. Therefore, identifying the genetic basis of horn type will provide a vital foundation for understanding why the different morphs are maintained in the face of natural selection. We conducted a genome-wide association study using т36 000 single nucleotide polymorphisms (SNPs) and determined the main candidate for Horns as RXFP2, an autosomal gene with a known involvement in determining primary sex characters in humans and mice. Evidence from additional SNPs in and around RXFP2 supports a new model of horn-type inheritance in Soay sheep, and for the first time, sheep with the same horn phenotype but different underlying genotypes can be identified. In addition, RXFP2 was shown to be an additive quantitative trait locus (QTL) for horn size in normal-horned males, accounting for up to 76% of additive genetic variation in this trait. This finding contrasts markedly from genome-wide association studies of quantitative traits in humans and some model species, where it is often observed that mapped loci only explain a modest proportion of the overall genetic variation.
URLPMID:2805710 [БОЮФв§гУ: 1]
Microphthalmia in sheep is an autosomal recessive inherited congenital anomaly found within the Texel breed. It is characterized by extremely small or absent eyes and affected lambs are absolutely blind. For the first time, we use a genome-wide ovine SNP array for positional cloning of a Mendelian trait in sheep. Genotyping 23 cases and 23 controls using Illumina's OvineSNP50 BeadChip allowed us to localize the causative mutation for microphthalmia to a 2.4 Mb interval on sheep chromosome 22 by association and homozygosity mapping. ThePITX3gene is located within this interval and encodes a homeodomain-containing transcription factor involved in vertebrate lens formation. An abnormal development of the lens vesicle was shown to be the primary event in ovine microphthalmia. Therefore, we consideredPITX3a positional and functional candidate gene. An ovine BAC clone was sequenced, and after full-length cDNA cloning thePITX3gene was annotated. Here we show that the ovine microphthalmia phenotype is perfectly associated with a missense mutation (c.338G>C, p.R113P) in the evolutionary conserved homeodomain ofPITX3. Selection against this candidate causative mutation can now be used to eliminate microphthalmia from Texel sheep in production systems. Furthermore, the identification of a naturally occurringPITX3mutation offers the opportunity to use the Texel as a genetically characterized large animal model for human microphthalmia.
URLPMID:15273396 [БОЮФв§гУ: 1]
The extent to which large duplications and deletions contribute to human genetic variation and diversity is unknown. Here, we show that large-scale copy number polymorphisms (CNPs) (about 100 kilobases and greater) contribute substantially to genomic variation between normal humans. Representational oligonucleotide microarray analysis of 20 individuals revealed a total of 221 copy number differences representing 76 unique CNPs. On average, individuals differed by 11 CNPs, and the average length of a CNP interval was 465 kilobases. We observed copy number variation of 70 different genes within CNP intervals, including genes involved in neurological function, regulation of cell growth, regulation of metabolism, and several genes known to be associated with disease.
[БОЮФв§гУ: 1]
URL [БОЮФв§гУ: 1]
In this study, 71 Sunite sheep were genotyped using Illumina OvineSNP50 BeadChip array, a total of 134 CNV regions (CNVR) across the genome were identified, which covered 25.95 Mb of the sheep genome.CNVR size on autosomes range from 29.48 kb to 1.30 Mb. Functional indicated that the most genes overrepresented in environmental responses, which involved in sensory perception of smell, sensory perception of chemical stimulus, sensory perception, cognition. To confirm these findings, quantitative PCR (qPCR) was performed for 5 CNVR and 3 of them were successfully validated. Our findings would be of help to understand the sheep genome and provide preliminary foundation for investigating the association between important economic traits and CNV.
URL [БОЮФв§гУ: 1]
In this study, 71 Sunite sheep were genotyped using Illumina OvineSNP50 BeadChip array, a total of 134 CNV regions (CNVR) across the genome were identified, which covered 25.95 Mb of the sheep genome.CNVR size on autosomes range from 29.48 kb to 1.30 Mb. Functional indicated that the most genes overrepresented in environmental responses, which involved in sensory perception of smell, sensory perception of chemical stimulus, sensory perception, cognition. To confirm these findings, quantitative PCR (qPCR) was performed for 5 CNVR and 3 of them were successfully validated. Our findings would be of help to understand the sheep genome and provide preliminary foundation for investigating the association between important economic traits and CNV.
URLMagsci [БОЮФв§гУ: 1]
ЮЊбАевУрбђЛљвђзщжаПЩФмЕФвХДЋадзДЯрЙиБъМЧ,БОЪдбщВЩгУБШНЯЛљвђзщдгНЛ(comparative genomic hybridization,CGH)аОЦЌММЪѕ,ЙЙНЈСЫУЩЙХбђЁЂЙўШјПЫбђЁЂВибђЕФПНБДЪ§Бфвь(copy number variation,CNV)ЖрбљадЭМЦзЁЃЪдбщНсЙћЯдЪО,ЙВМьВтГі28ИіCNVЧјгђ(CNV region,CNVRs),АќРЈ11ИіРЉдіаЭЁЂ15ИіШБЪЇаЭКЭ2ИіРЉдіЁЊШБЪЇаЭЁЃЭЈЙ§ЙІФмзЂЪЭКЭДњаЛЭЈТЗЗжЮіЗЂЯж,дкУЩЙХбђКЭВибђЛљвђзщжабЊКьЕААзЛљвђДцдкПНБДЪ§РЉеХ,ПЩФмгыСНжжУрбђГЄЦкЩњЛюдкИпдЕЭбѕЛЗОГжаВњЩњЕФЪЪгІадгаЙиЁЃЖдCNVRsКЭCNVЯрЙиЛљвђНјааЪЕЪБгЋЙтЖЈСПPCRМьбщ,83.3%ЕФЪЕЪБгЋЙтЖЈСПPCRНсЙћгыаОЦЌМьВтНсЙћвЛжТЁЃЭЈЙ§ЖджаЙњББЗН3жжУрбђЕФЛљвђзщCNVЕФбаОП,ЮЊВЛЭЌУрбђЦЗжжМфвХДЋБфвьЕФбаОПЕьЖЈСЫЛљДЁЁЃ
URLMagsci [БОЮФв§гУ: 1]
ЮЊбАевУрбђЛљвђзщжаПЩФмЕФвХДЋадзДЯрЙиБъМЧ,БОЪдбщВЩгУБШНЯЛљвђзщдгНЛ(comparative genomic hybridization,CGH)аОЦЌММЪѕ,ЙЙНЈСЫУЩЙХбђЁЂЙўШјПЫбђЁЂВибђЕФПНБДЪ§Бфвь(copy number variation,CNV)ЖрбљадЭМЦзЁЃЪдбщНсЙћЯдЪО,ЙВМьВтГі28ИіCNVЧјгђ(CNV region,CNVRs),АќРЈ11ИіРЉдіаЭЁЂ15ИіШБЪЇаЭКЭ2ИіРЉдіЁЊШБЪЇаЭЁЃЭЈЙ§ЙІФмзЂЪЭКЭДњаЛЭЈТЗЗжЮіЗЂЯж,дкУЩЙХбђКЭВибђЛљвђзщжабЊКьЕААзЛљвђДцдкПНБДЪ§РЉеХ,ПЩФмгыСНжжУрбђГЄЦкЩњЛюдкИпдЕЭбѕЛЗОГжаВњЩњЕФЪЪгІадгаЙиЁЃЖдCNVRsКЭCNVЯрЙиЛљвђНјааЪЕЪБгЋЙтЖЈСПPCRМьбщ,83.3%ЕФЪЕЪБгЋЙтЖЈСПPCRНсЙћгыаОЦЌМьВтНсЙћвЛжТЁЃЭЈЙ§ЖджаЙњББЗН3жжУрбђЕФЛљвђзщCNVЕФбаОП,ЮЊВЛЭЌУрбђЦЗжжМфвХДЋБфвьЕФбаОПЕьЖЈСЫЛљДЁЁЃ
[БОЮФв§гУ: 1]
URLPMID:19109259 [БОЮФв§гУ: 1]
Genetic progress will increase when breeders examine genotypes in addition to pedigrees and phenotypes. Genotypes for 38,416 markers and August 2003 genetic evaluations for 3,576 Holstein bulls born before 1999 were used to predict January 2008 daughter deviations for 1,759 bulls born from 1999 through 2002. Genotypes were generated using the Illumina BovineSNP50 BeadChip and DNA from semen contributed by US and Canadian artificial-insemination organizations to the Cooperative Dairy DNA Repository. Genomic predictions for 5 yield traits, 5 fitness traits, 16 conformation traits, and net merit were computed using a linear model with an assumed normal distribution for marker effects and also using a nonlinear model with a heavier tailed prior distribution to account for major genes. The official parent average from 2003 and a 2003 parent average computed from only the subset of genotyped ancestors were combined with genomic predictions using a selection index. Combined predictions were more accurate than official parent averages for all 27 traits. The coefficients of determination (R2) were 0.05 to 0.38 greater with nonlinear genomic predictions included compared with those from parent average alone. Linear genomic predictions had R2 values similar to those from nonlinear predictions but averaged just 0.01 lower. The greatest benefits of genomic prediction were for fat percentage because of a known gene with a large effect. The R2 values were converted to realized reliabilities by dividing by mean reliability of 2008 daughter deviations and then adding the difference between published and observed reliabilities of 2003 parent averages. When averaged across all traits, combined genomic predictions had realized reliabilities that were 23% greater than reliabilities of parent averages (50 vs. 27%), and gains in information were equivalent to 11 additional daughter records. Reliability increased more by doubling the number of bulls genotyped than the number of markers genotyped. Genomic prediction improves reliability by tracing the inheritance of genes even with small effects.
URLPMID:16882088 [БОЮФв§гУ: 1]
Summary Animals can be genotyped for thousands of single nucleotide polymorphisms (SNPs) at one time, where the SNPs are located at roughly 1-cM intervals throughout the genome. For each contiguous pair of SNPs there are four possible haplotypes that could be inherited from the sire. The effects of each interval on a trait can be estimated for all intervals simultaneously in a model where interval effects are random factors. Given the estimated effects of each haplotype for every interval in the genome, and given an animal's genotype, a ЁЎgenomicЁЏ estimated breeding value is obtained by summing the estimated effects for that genotype. The accuracy of that estimator of breeding values is around 80%. Because the genomic estimated breeding values can be calculated at birth, and because it has a high accuracy, a strategy that utilizes these advantages was compared with a traditional progeny testing strategy under a typical Canadian-like dairy cattle situation. Costs of proving bulls were reduced by 92% and genetic change was increased by a factor of 2. Genome-wide selection may become a popular tool for genetic improvement in livestock.
URL [БОЮФв§гУ: 1]
This article describes the current status of sequencing of the pig genome and how the findings will benefit pig production in terms of genetic improvement through selection for better performance traits.
[БОЮФв§гУ: 2]
[БОЮФв§гУ: 2]
URLPMID:21255418 [БОЮФв§гУ: 1]
Background Genomic selection involves breeding value estimation of selection candidates based on high-density SNP genotypes. To quantify the potential benefit of genomic selection, accuracies of estimated breeding values (EBV) obtained with different methods using pedigree or high-density SNP genotypes were evaluated and compared in a commercial layer chicken breeding line. Methods The following traits were analyzed: egg production, egg weight, egg color, shell strength, age at sexual maturity, body weight, albumen height, and yolk weight. Predictions appropriate for early or late selection were compared. A total of 2,708 birds were genotyped for 23,356 segregating SNP, including 1,563 females with records. Phenotypes on relatives without genotypes were incorporated in the analysis (in total 13,049 production records). The data were analyzed with a Reduced Animal Model using a relationship matrix based on pedigree data or on marker genotypes and with a Bayesian method using model averaging. Using a validation set that consisted of individuals from the generation following training, these methods were compared by correlating EBV with phenotypes corrected for fixed effects, selecting the top 30 individuals based on EBV and evaluating their mean phenotype, and by regressing phenotypes on EBV. Results Using high-density SNP genotypes increased accuracies of EBV up to two-fold for selection at an early age and by up to 88% for selection at a later age. Accuracy increases at an early age can be mostly attributed to improved estimates of parental EBV for shell quality and egg production, while for other egg quality traits it is mostly due to improved estimates of Mendelian sampling effects. A relatively small number of markers was sufficient to explain most of the genetic variation for egg weight and body weight.
[БОЮФв§гУ: 1]
URLPMID:21406356 [БОЮФв§гУ: 1]
Salmonella propagation by apparently healthy chickens could be decreased by the selection and use of chicken lines that are more resistant to carrier state. Using a reduced set of markers, this study investigates, for the first time to the authors' knowledge, the feasibility of a genomic selection approach for resistance to carrier state in hen lines. In this study, commercial laying hen lines were divergently selected for resistance to Salmonella carrier state at 2 different ages: young chicks and adults at the peak of lay. A total of 600 birds were typed with 831 informative SNP markers and artificially infected with Salmonella Enteritidis. Phenotypes were collected 28 d (389 young animals) or 38 d (208 adults) after infection. Two types of variance component analyses, including SNP data or not, were performed and compared. The set of SNP used was efficient in capturing a large part of the genetic variation. Average accuracies from mixed model equations did not change between analyses, showing that using SNP data does not increase information in this data set. These results confirm that genomic selection for Salmonella carrier state resistance in laying hens is promising. Nevertheless, a denser SNP coverage of the genome on a greater number of animals is still needed to assess its feasibility and efficiency.
URLPMID:12618414 [БОЮФв§гУ: 3]
Molecular markers have been used to map quantitative trait loci. However, they are rarely used to evaluate effects of chromosome segments of the entire genome. The original interval-mapping approach and various modified versions of it may have limited use in evaluating the genetic effects of the entire genome because they require evaluation of multiple models and model selection. Here we present a Bayesian regression method to simultaneously estimate genetic effects associated with markers of the entire genome. With the Bayesian method, we were able to handle situations in which the number of effects is even larger than the number of observations. The key to the success is that we allow each marker effect to have its own variance parameter, which in turn has its own prior distribution so that the variance can be estimated from the data. Under this hierarchical model, we were able to handle a large number of markers and most of the markers may have negligible effects. As a result, it is possible to evaluate the distribution of the marker effects. Using data from the North American Barley Genome Mapping Project in double-haploid barley, we found that the distribution of gene effects follows closely an L-shaped Gamma distribution, which is in contrast to the bell-shaped Gamma distribution when the gene effects were estimated from interval mapping. In addition, we show that the Bayesian method serves as an alternative or even better QTL mapping method because it produces clearer signals for QTL. Similar results were found from simulated data sets of F2 and backcross (BC) families.
URLMagsci [БОЮФв§гУ: 1]
<p>Лљвђзщг§жжжЕЙРМЦЪЧЛљвђзщбЁдёЕФживЊЛЗНк, Лљвђзщг§жжжЕЕФзМШЗадЪЧЛљвђзщбЁдёГЩЙІгІгУЕФЙиМќ, ЖјЦфзМШЗаддкКмДѓГЬЖШЩЯШЁОігкЙРМЦЗНЗЈЁЃФПЧАбаОПКЭгІгУзюЖрЕФЛљвђзщг§жжжЕЙРМЦЗНЗЈЪЧБДвЖЫЙ(Bayes)КЭзюМбЯпадЮоЦЋдЄВт(BLUP)СНДѓРрЗНЗЈЁЃЮФеТЯЕЭГНщЩмСЫФПЧАвбЬсГіЕФИїжжBayesЗНЗЈ, ВЂзмНсСЫИУРрЗНЗЈЕФЙРМЦаЇЙћКЭИїЗНУцЕФИФНјЁЃФЃФтЪ§ОнКЭЪЕМЪЪ§ОнбаОПНсЙћЖМБэУї, BayesРрЗНЗЈЙРМЦЛљвђзщг§жжжЕЕФзМШЗадгХгкBLUPРрЗНЗЈ, ЬиБ№ЖдгкДцдкНЯДѓаЇгІQTLЕФадзДЦфгХЪЦИќУїЯдЁЃгЩгкBayesЗНЗЈЕФРэТлКЭМЦЫуЙ§ГЬЯрЖдИДдг, ФПЧАЦфдкЪЕМЪг§жжжаЕФдЫгУВЛШчBLUPРрЗНЗЈЦеБщ, ЕЋЫцзХПьЫйЫуЗЈЕФПЊЗЂКЭМЦЫуЛњгВМўЕФИФНј, МЦЫуЮЪЬтгаЭћЕУЕННтОі; СэЭт, ЫцзХЖдЛљвђзщКЭадзДвХДЋНсЙЙбаОПЕФЩюШыПЊеЙ, ФмЮЊBayesЗНЗЈЬсЙЉИќЮЊзМШЗЕФЯШбщаХЯЂ, ДгЖјЪЙBayesЗНЗЈЙРМЦЛљвђзщг§жжжЕзМШЗадЕФгХЪЦИќМгЭЛГі, гІгУНЋЛсИќМгЙуЗКЁЃ</p>
URLMagsci [БОЮФв§гУ: 1]
<p>Лљвђзщг§жжжЕЙРМЦЪЧЛљвђзщбЁдёЕФживЊЛЗНк, Лљвђзщг§жжжЕЕФзМШЗадЪЧЛљвђзщбЁдёГЩЙІгІгУЕФЙиМќ, ЖјЦфзМШЗаддкКмДѓГЬЖШЩЯШЁОігкЙРМЦЗНЗЈЁЃФПЧАбаОПКЭгІгУзюЖрЕФЛљвђзщг§жжжЕЙРМЦЗНЗЈЪЧБДвЖЫЙ(Bayes)КЭзюМбЯпадЮоЦЋдЄВт(BLUP)СНДѓРрЗНЗЈЁЃЮФеТЯЕЭГНщЩмСЫФПЧАвбЬсГіЕФИїжжBayesЗНЗЈ, ВЂзмНсСЫИУРрЗНЗЈЕФЙРМЦаЇЙћКЭИїЗНУцЕФИФНјЁЃФЃФтЪ§ОнКЭЪЕМЪЪ§ОнбаОПНсЙћЖМБэУї, BayesРрЗНЗЈЙРМЦЛљвђзщг§жжжЕЕФзМШЗадгХгкBLUPРрЗНЗЈ, ЬиБ№ЖдгкДцдкНЯДѓаЇгІQTLЕФадзДЦфгХЪЦИќУїЯдЁЃгЩгкBayesЗНЗЈЕФРэТлКЭМЦЫуЙ§ГЬЯрЖдИДдг, ФПЧАЦфдкЪЕМЪг§жжжаЕФдЫгУВЛШчBLUPРрЗНЗЈЦеБщ, ЕЋЫцзХПьЫйЫуЗЈЕФПЊЗЂКЭМЦЫуЛњгВМўЕФИФНј, МЦЫуЮЪЬтгаЭћЕУЕННтОі; СэЭт, ЫцзХЖдЛљвђзщКЭадзДвХДЋНсЙЙбаОПЕФЩюШыПЊеЙ, ФмЮЊBayesЗНЗЈЬсЙЉИќЮЊзМШЗЕФЯШбщаХЯЂ, ДгЖјЪЙBayesЗНЗЈЙРМЦЛљвђзщг§жжжЕзМШЗадЕФгХЪЦИќМгЭЛГі, гІгУНЋЛсИќМгЙуЗКЁЃ</p>
PMID:2637029 [БОЮФв§гУ: 2]
Genomic selection uses genome-wide dense SNP marker genotyping for the prediction of genetic values, and consists of two steps: (1) estimation of SNP effects, and (2) prediction of genetic value based on SNP genotypes and estimates of their effects. For the former step, BayesB type of estimators have been proposed, which assume a priori that many markers have no effects, and some have an effect coming from a gamma or exponential distribution, i.e. a fat-tailed distribution. Whilst such estimators have been developed using Monte Carlo Markov chain (MCMC), here we derive a much faster non-MCMC based estimator by analytically performing the required integrations. The accuracy of the genome-wide breeding value estimates was 0.011 (s.e. 0.005) lower than that of the MCMC based BayesB predictor, which may be because the integrations were performed one-by-one instead of for all SNPs simultaneously. The bias of the new method was opposite to that of the MCMC based BayesB, in that the new method underestimates the breeding values of the best selection candidates, whereas MCMC-BayesB overestimated their breeding values. The new method was computationally several orders of magnitude faster than MCMC based BayesB, which will mainly be advantageous in computer simulations of entire breeding schemes, in cross-validation testing, and practical schemes with frequent re-estimation of breeding values.
URLPMID:19922694 [БОЮФв§гУ: 1]
Genomic selection describes a selection strategy based on genomic breeding values predicted from dense single nucleotide polymorphism (SNP) data. Multiple methods have been proposed but the critical issue is how to decide whether an SNP should be included in the predictive set to estimate breeding values. One major disadvantage of the traditional Bayes B approach is its high computational demands caused by the changing dimensionality of the models. The use of stochastic search variable selection (SSVS) retains the same assumptions about the distribution of SNP effects as Bayes B, while maintaining constant dimensionality. When Bayesian SSVS was used to predict genomic breeding values for real dairy data over a range of traits it produced accuracies higher or equivalent to other genomic selection methods with significantly decreased computational and time demands than Bayes B.
URLPMID:2708128 [БОЮФв§гУ: 1]
Background Recent developments in SNP discovery and high throughput genotyping technology have made the use of high-density SNP markers to predict breeding values feasible. This involves estimation of the SNP effects in a training data set, and use of these estimates to evaluate the breeding values of other 'evaluation' individuals. Simulation studies have shown that these predictions of breeding values can be accurate, when training and evaluation individuals are (closely) related. However, many general applications of genomic selection require the prediction of breeding values of 'unrelated' individuals, i.e. individuals from the same population, but not particularly closely related to the training individuals. Methods Accuracy of selection was investigated by computer simulation of small populations. Using scaling arguments, the results were extended to different populations, training data sets and genome sizes, and different trait heritabilities. Results Prediction of breeding values of unrelated individuals required a substantially higher marker density and number of training records than when prediction individuals were offspring of training individuals. However, when the number of records was 2*Ne*L and the number of markers was 10*Ne*L, the breeding values of unrelated individuals could be predicted with accuracies of 0.88 ??? 0.93, where Ne is the effective population size and L the genome size in Morgan. Reducing this requirement to 1*Ne*L individuals, reduced prediction accuracies to 0.73???0.83. Conclusion For livestock populations, 1NeL requires about ~30,000 training records, but this may be reduced if training and evaluation animals are related. A prediction equation is presented, that predicts accuracy when training and evaluation individuals are related. For humans, 1NeL requires ~350,000 individuals, which means that human disease risk prediction is possible only for diseases that are determined by a limited number of genes. Otherwise, genotyping and phenotypic recording need to become very common in the future.
URLPMID:2671482 [БОЮФв§гУ: 1]
Partial least square regression (PLSR) and principal component regression (PCR) are methods designed for situations where the number of predictors is larger than the number of records. The aim was to compare the accuracy of genome-wide breeding values (EBV) produced using PLSR and PCR with a Bayesian method, 'BayesB'. Marker densities of 1, 2, 4 and 8 Ne markers/Morgan were evaluated when the effective population size (Ne) was 100. The correlation between true breeding value and estimated breeding value increased with density from 0.611 to 0.681 and 0.604 to 0.658 using PLSR and PCR respectively, with an overall advantage to PLSR of 0.016 (s.e = 0.008). Both methods gave a lower accuracy compared to the 'BayesB', for which accuracy increased from 0.690 to 0.860. PLSR and PCR appeared less responsive to increased marker density with the advantage of 'BayesB' increasing by 17% from a marker density of 1 to 8Ne/M. PCR and PLSR showed greater bias than 'BayesB' in predicting breeding values at all densities. Although, the PLSR and PCR were computationally faster and simpler, these advantages do not outweigh the reduction in accuracy, and there is a benefit in obtaining relevant prior information from the distribution of gene effects.
URLPMID:18076475 [БОЮФв§гУ: 1]
Summary Genome-wide association studies using single nucleotide polymorphisms (SNPs) can identify genetic variants related to complex traits. Typically thousands of SNPs are genotyped, whereas the number of phenotypes for which there is genomic information may be smaller. When predicting phenotypes, options for statistical model building range from incorporating all possible markers into the specification to including only sets of relevant SNPs (features). In the latter case, an efficient method of selecting influential features is required. A two-step feature selection method for binary traits was developed, which consisted of filtering (using information gain), and wrapping (using na07ve Bayesian classification). The filter reduces the large number of SNPs to a much smaller size, to facilitate the wrapper step. As the procedure is tailored for discrete outcomes, an approach based on discretization of phenotypic values was developed, to enable feature selection in a classification framework. The method was applied to chick mortality rates (0ЈC1402days of age) on progeny from 201 sires in a commercial broiler line, with the goal of identifying SNPs (over 5000) related to progeny mortality. To mimic a caseЈCcontrol study, sires were clustered into two groups, low and high, according to two arbitrarily chosen mortality rate cut points. By varying these thresholds, 11 different ЁЎcaseЈCcontrolЁЏ samples were formed, and the SNP selection procedure was applied to each sample. To compare the 11 sets of chosen SNPs, predicted residual sum of squares (PRESS) from a linear model was used. The two-step method improved na07ve Bayesian classification accuracy over the case without feature selection (from around 50 to above 90% without and with feature selection in each caseЈCcontrol sample). The best caseЈCcontrol group (63 sires above or below the thresholds) had the smallest PRESS statistic among groups with model p-values below 0.003. The 17 SNPs selected using this group accounted for 31% of the variation in raw mortality rates between sire families.
URL [БОЮФв§гУ: 2]
[БОЮФв§гУ: 1]
URLPMID:19293140 [БОЮФв§гУ: 1]
Abstract The availability of genomewide dense markers brings opportunities and challenges to breeding programs. An important question concerns the ways in which dense markers and pedigrees, together with phenotypic records, should be used to arrive at predictions of genetic values for complex traits. If a large number of markers are included in a regression model, marker-specific shrinkage of regression coefficients may be needed. For this reason, the Bayesian least absolute shrinkage and selection operator (LASSO) (BL) appears to be an interesting approach for fitting marker effects in a regression model. This article adapts the BL to arrive at a regression model where markers, pedigrees, and covariates other than markers are considered jointly. Connections between BL and other marker-based regression models are discussed, and the sensitivity of BL with respect to the choice of prior distributions assigned to key parameters is evaluated using simulation. The proposed model was fitted to two data sets from wheat and mouse populations, and evaluated using cross-validation methods. Results indicate that inclusion of markers in the regression further improved the predictive ability of models. An R program that implements the proposed model is freely available.
URLPMID:18946147 [БОЮФв§гУ: 1]
Efficient methods for processing genomic data were developed to increase reliability of estimated breeding values and to estimate thousands of marker effects simultaneously. Algorithms were derived and computer programs tested with simulated data for 2,967 bulls and 50,000 markers distributed randomly across 30 chromosomes. Estimation of genomic inbreeding coefficients required accurate estimates of allele frequencies in the base population. Linear model predictions of breeding values were computed by 3 equivalent methods: 1) iteration for individual allele effects followed by summation across loci to obtain estimated breeding values, 2) selection index including a genomic relationship matrix, and 3) mixed model equations including the inverse of genomic relationships. A blend of first- and second-order Jacobi iteration using 2 separate relaxation factors converged well for allele frequencies and effects. Reliability of predicted net merit for young bulls was 63% compared with 32% using the traditional relationship matrix. Nonlinear predictions were also computed using iteration on data and nonlinear regression on marker deviations; an additional (about 3%) gain in reliability for young bulls increased average reliability to 66%. Computing times increased linearly with number of genotypes. Estimation of allele frequencies required 2 processor days, and genomic predictions required <1 d per trait, and traits were processed in parallel. Information from genotyping was equivalent to about 20 daughters with phenotypic records. Actual gains may differ because the simulation did not account for linkage disequilibrium in the base population or selection in subsequent generations.
URLPMID:20170500 [БОЮФв§гУ: 2]
Background The impact of additive-genetic relationships captured by single nucleotide polymorphisms (SNPs) on the accuracy of genomic breeding values (GEBVs) has been demonstrated, but recent studies...
URL [БОЮФв§гУ: 1]
URLPMID:20105546 [БОЮФв§гУ: 1]
The first national single-step, full-information (phenotype, pedigree, and marker genotype) genetic evaluation was developed for final score of US Holsteins. Data included final scores recorded from 1955 to 2009 for 6,232,548 Holsteins cows. BovineSNP50 (Illumina, San Diego, CA) genotypes from the Cooperative Dairy DNA Repository (Beltsville, MD) were available for 6,508 bulls. Three analyses used a repeatability animal model as currently used for the national US evaluation. The first 2 analyses used final scores recorded up to 2004. The first analysis used only a pedigree-based relationship matrix. The second analysis used a relationship matrix based on both pedigree and genomic information (single-step approach). The third analysis used the complete data set and only the pedigree-based relationship matrix. The fourth analysis used predictions from the first analysis (final scores up to 2004 and only a pedigree-based relationship matrix) and prediction using a genomic based matrix to obtain genetic evaluation (multiple-step approach). Different allele frequencies were tested in construction of the genomic relationship matrix. Coefficients of determination between predictions of young bulls from parent average, single-step, and multiple-step approaches and their 2009 daughter deviations were 0.24, 0.37 to 0.41, and 0.40, respectively. The highest coefficient of determination for a single-step approach was observed when using a genomic relationship matrix with assumed allele frequencies of 0.5. Coefficients for regression of 2009 daughter deviations on parent-average, single-step, and multiple-step predictions were 0.76, 0.68 to 0.79, and 0.86, respectively, which indicated some inflation of predictions. The single-step regression coefficient could be increased up to 0.92 by scaling differences between the genomic and pedigree-based relationship matrices with little loss in accuracy of prediction. One complete evaluation took about 2h of computing time and 2.7 gigabytes of memory. Computing times for single-step analyses were slightly longer (2%) than for pedigree-based analysis. A national single-step genetic evaluation with the pedigree relationship matrix augmented with genomic information provided genomic predictions with accuracy and bias comparable to multiple-step procedures and could account for any population or data structure. Advantages of single-step evaluations should increase in the future when animals are pre-selected on genotypes.
URLPMID:22818478 [БОЮФв§гУ: 1]
The single-step genomic BLUP (SSGBLUP) is a method that can integrate pedigree and genotypes at molecular markers in an optimal way. However, its present form (regular SSGBLUP) has a high computational cost (cubic in the number of genotyped animals) and may need extensive rewriting of genetic evaluation software. In this work, we propose several strategies to implement the single step in a simpler manner. The first one expands the single-step mixed-model equations to obtain equivalent equations from which the regular (including pedigree and records only) mixed-model equations are a subset. These new equations (unsymmetric extended SSGBLUP) have low computational cost, but require a nonsymmetric solver such as the biconjugate gradient stabilized method or successive underrelaxation, which is a variant of successive overrelaxation, with a relaxation factor lower than 1. In addition, we show a new derivation of the single-step method, which includes, as an extra effect, deviations from strictly polygenic breeding values. As a result, the same set of equations as above is obtained. We show that, whereas the new derivation shows apparent problems of nonpositive definiteness for certain covariance matrices, a proper equivalent model including imaginary effects always exists, leading always to the regular SSGBLUP mixed model equations. The system of equations can be solved (iterative SSGBLUP) by iterating between a pedigree and records evaluation and a genomic evaluation (each one solved by any iterative or direct method), whereas global iteration can use a block version of successive underrelaxation, which ensures convergence. The genomic evaluation can explicitly include marker or haplotype effects and possibly involve nonlinear (e.g., Bayesian by Markov chain Monte Carlo) methods. In a simulated example with 28,800 individuals and 1,800 genotyped individuals, all methods converged quickly to the same solutions. Using existing efficient methods with limited memory requirements to compute the products Gt and A22t for any t (where G and A22 are genomic and pedigree relationships for genotyped animals, and t is a vector), all strategies can be converted to iteration on data procedures for which the total number of operations is linear in the number of animals + number of genotyped animals number of markers.
URLPMID:2834608 [БОЮФв§гУ: 1]
Background The use of genomic selection in breeding programs may increase the rate of genetic improvement, reduce the generation time, and provide higher accuracy of estimated breeding values (EBVs). A number of different methods have been developed for genomic prediction of breeding values, but many of them assume that all animals have been genotyped. In practice, not all animals are genotyped, and the methods have to be adapted to this situation. Results In this paper we provide an extension of a linear mixed model method for genomic prediction to the situation with non-genotyped animals. The model specifies that a breeding value is the sum of a genomic and a polygenic genetic random effect, where genomic genetic random effects are correlated with a genomic relationship matrix constructed from markers and the polygenic genetic random effects are correlated with the usual relationship matrix. The extension of the model to non-genotyped animals is made by using the pedigree to derive an extension of the genomic relationship matrix to non-genotyped animals. As a result, in the extended model the estimated breeding values are obtained by blending the information used to compute traditional EBVs and the information used to compute purely genomic EBVs. Parameters in the model are estimated using average information REML and estimated breeding values are best linear unbiased predictions (BLUPs). The method is illustrated using a simulated data set. Conclusions The extension of the method to non-genotyped animals presented in this paper makes it possible to integrate all the genomic, pedigree and phenotype information into a one-step procedure for genomic prediction. Such a one-step procedure results in more accurate estimated breeding values and has the potential to become the standard tool for genomic prediction of breeding values in future practical evaluations in pig and cattle breeding.
[БОЮФв§гУ: 2]
URL [БОЮФв§гУ: 2]
Genomic selection (GS) is a marker-assisted selection method, in which high density markers covering the whole genome are used simultaneously for individual genetic evaluation via genomic estimated breeding values (GEBVs). GS can increase the accuracy of selection, shorten the generation interval by selecting individuals at the early stage of life, and accelerate genetic progress. With the availability of high density whole genome SNP (single nucleotide polymorphism) chips for livestock, GS is reshaping the conventional animal breeding systems. In many countries, GS is becoming the major genetic evaluation method for bull selection in dairy cattle and GS may soon completely replace the traditional genetic evaluation system. In recent years, GS has become an important research topic in animal, plant and aquiculture breeding and many exciting results have been reported. In this paper, the methods for obtaining GEBVs, factors affecting the accuracy of GEBVs, and the current status of implementation of GS in livestock are reviewed. Some unresolved issues related to GS in livestock are also discussed.
URLPMID:22443868 [БОЮФв§гУ: 2]
Animal breeding faces one of the most significant changes of the past decades the implementation of genomic selection. Genomic selection uses dense marker maps to predict the breeding value of animals with reported accuracies that are up to 0.31 higher than those of pedigree indexes, without the need to phenotype the animals themselves, or close relatives thereof. The basic principle is that because of the high marker density, each quantitative trait loci (QTL) is in linkage disequilibrium (LD) with at least one nearby marker. The process involves putting a reference population together of animals with known phenotypes and genotypes to estimate the marker effects. Marker effects have been estimated with several different methods that generally aim at reducing the dimensions of the marker data. Nearly all reported models only included additive effects. Once the marker effects are estimated, breeding values of young selection candidates can be predicted with reported accuracies up to 0.85. Although results from simulation studies suggest that different models may yield more accurate genomic estimated breeding values (GEBVs) for different traits, depending on the underlying QTL distribution of the trait, there is so far only little evidence from studies based on real data to support this. The accuracy of genomic predictions strongly depends on characteristics of the reference populations, such as number of animals, number of markers, and the heritability of the recorded phenotype. Another important factor is the relationship between animals in the reference population and the evaluated animals. The breakup of LD between markers and QTL across generations advocates frequent re-estimation of marker effects to maintain the accuracy of GEBVs at an acceptable level. Therefore, at low frequencies of re-estimating marker effects, it becomes more important that the model that estimates the marker effects capitalizes on LD information that is persistent across generations.
URL [БОЮФв§гУ: 1]
URLPMID:19220931 [БОЮФв§гУ: 1]
Abstract Dense marker genotypes allow the construction of the realized relationship matrix between individuals, with elements the realized proportion of the genome that is identical by descent (IBD) between pairs of individuals. In this paper, we demonstrate that by replacing the average relationship matrix derived from pedigree with the realized relationship matrix in best linear unbiased prediction (BLUP) of breeding values, the accuracy of the breeding values can be substantially increased, especially for individuals with no phenotype of their own. We further demonstrate that this method of predicting breeding values is exactly equivalent to the genomic selection methodology where the effects of quantitative trait loci (QTLs) contributing to variation in the trait are assumed to be normally distributed. The accuracy of breeding values predicted using the realized relationship matrix in the BLUP equations can be deterministically predicted for known family relationships, for example half sibs. The deterministic method uses the effective number of independently segregating loci controlling the phenotype that depends on the type of family relationship and the length of the genome. The accuracy of predicted breeding values depends on this number of effective loci, the family relationship and the number of phenotypic records. The deterministic prediction demonstrates that the accuracy of breeding values can approach unity if enough relatives are genotyped and phenotyped. For example, when 1000 full sibs per family were genotyped and phenotyped, and the heritability of the trait was 0.5, the reliability of predicted genomic breeding values (GEBVs) for individuals in the same full sib family without phenotypes was 0.82. These results were verified by simulation. A deterministic prediction was also derived for random mating populations, where the effective population size is the key parameter determining the effective number of independently segregating loci. If the effective population size is large, a very large number of individuals must be genotyped and phenotyped in order to accurately predict breeding values for unphenotyped individuals from the same population. If the heritability of the trait is 0.3, and N(e)=100, approximately 12474 individuals with genotypes and phenotypes are required in order to predict GEBVs of un-phenotyped individuals in the same population with an accuracy of 0.7 [corrected].
URL [БОЮФв§гУ: 1]
URLPMID:20043827 [БОЮФв§гУ: 1]
Background Genomic prediction of breeding values involves a so-called training analysis that predicts the influence of small genomic regions by regression of observed information on marker genotypes for a given population of individuals. Available observations may take the form of individual phenotypes, repeated observations, records on close family members such as progeny, estimated breeding values (EBV) or their deregressed counterparts from genetic evaluations. The literature indicates that researchers are inconsistent in their approach to using EBV or deregressed data, and as to using the appropriate methods for weighting some data sources to account for heterogeneous variance. Methods A logical approach to using information for genomic prediction is introduced, which demonstrates the appropriate weights for analyzing observations with heterogeneous variance and explains the need for and the manner in which EBV should have parent average effects removed, be deregressed and weighted. Results An appropriate deregression for genomic regression analyses is EBV/r2 where EBV excludes parent information and r2 is the reliability of that EBV. The appropriate weights for deregressed breeding values are neither the reliability nor the prediction error variance, two alternatives that have been used in published studies, but the ratio (1 - h2)/[(c + (1 - r2)/r2)h2] where c > 0 is the fraction of genetic variance not explained by markers. Conclusions Phenotypic information on some individuals and deregressed data on others can be combined in genomic analyses using appropriate weighting.
[БОЮФв§гУ: 1]
URL [БОЮФв§гУ: 1]
URLPMID:20412938 [БОЮФв§гУ: 1]
The availability of dense single nucleotide polymorphism (SNP) genotypes for dairy cattle has created exciting research opportunities and revolutionized practical breeding programs. Broader application of this technology will lead to situations in which genotypes from different low-, medium-, or high-density platforms must be combined. In this case, missing SNP genotypes can be imputed using family- or population-based algorithms. Our objective was to evaluate the accuracy of imputation in Jersey cattle, using reference panels comprising 2,542 animals with 43,385 SNP genotypes and study samples of 604 animals for which genotypes were available for 1, 2, 5, 10, 20, 40, or 80% of loci. Two population-based algorithms, fastPHASE 1.2 (P. Scheet and M. Stevens; University of Washington TechTransfer Digital Ventures Program, Seattle, WA) and IMPUTE 2.0 (B. Howie and J. Marchini; Department of Statistics, University of Oxford, UK), were used to impute genotypes on Bos taurus autosomes 1, 15, and 28. The mean proportion of genotypes imputed correctly ranged from 0.659 to 0.801 when 1 to 2% of genotypes were available in the study samples, from 0.733 to 0.964 when 5 to 20% of genotypes were available, and from 0.896 to 0.995 when 40 to 80% of genotypes were available. In the absence of pedigrees or genotypes of close relatives, the accuracy of imputation may be modest (generally 40,000 SNP) from a reference population. Accurate imputation of high-density genotypes from inexpensive low- or medium-density platforms could greatly enhance the efficiency of whole-genome selection programs in dairy cattle.
URLPMID:20965364 [БОЮФв§гУ: 1]
The availability of high-density bovine genotyping arrays made implementation of genomic selection possible in dairy cattle. Development of low-density single nucleotide polymorphism (SNP) panels will allow the extension of genomic selection to a larger portion of the population. Prediction of ungenotyped markers, called imputation, is a strategy that allows using the same low-density chips for all traits (and for different breeds). In the present study, we evaluated the accuracy of imputation with low-density genotyping arrays in the Dutch Holstein population. Five different sizes of genotyping arrays were tested, from 384 to 6,000 SNP. According to marker density, the overall allelic imputation error rate obtained with the program DAGPHASE, which relies on linkage disequilibrium and linkage, ranged from 11.7 to 2.0%, and that obtained with the program CHROMIBD, which relies on linkage and the set of all genotyped ancestors, ranged from 10.7 to 3.3%. However, imputation efficiency was influenced by the relationship between low-density and high-density genotyped animals. Animals with both parents genotyped had particularly low imputation error rates:
URLPMID:19448663 [БОЮФв§гУ: 1]
Genome-wide panels of SNPs have recently been used in domestic animal species to map and identify genes for many traits and to select genetically desirable livestock. This has led to the discovery of the causal genes and mutations for several single-gene traits but not for complex traits. However, the genetic merit of animals can still be estimated by genomic selection, which uses genome-wide SNP panels as markers and statistical methods that capture the effects of large numbers of SNPs simultaneously. This approach is expected to double the rate of genetic improvement per year in many livestock systems.
URL [БОЮФв§гУ: 2]
78 Equivalent models for Genomic Selection (GS) using GBLUP are demonstrated. 78 SNP effects are estimated by regression on genomic breeding values. 78 Genomic relationship matrices are biased due to the use of only common SNP. 78 Sex-linked loci are incorporated into GS and GWAS analyses. 78 Accuracy of GS depends on relatedness of individuals to training population members.
URL [БОЮФв§гУ: 1]
The doubled-haploid (DH) barley population (Harrington x TR306) developed by the North American Barley Genome Mapping Project (NABGMP) for QTL mapping consisted of 145 lines and 127 markers covering a total genome length of 1270 cM. These DH lines were evaluated in approximately 25 environments for seven quantitative traits: heading, height, kernel weight, lodging, maturity, test weight, and yield. We applied an empirical Bayes method that simultaneously estimates 127 main effects for all markers and 127(127-1)/2=8001 interaction effects for all marker pairs in a single model. We found that the largest main-effect QTL (single marker) and the largest epistatic effect (single pair of markers) explained approximately 18 and 2.6% of the phenotypic variance, respectively. On average, the sum of all significant main effects and the sum of all significant epistatic effects contributed 35 and 6% of the total phenotypic variance, respectively. Epistasis seems to be negligible for all the seven traits. We also found that whether two loci interact does not depend on whether or not the loci have individual main effects. This invalidates the common practice of epistatic analysis in which epistatic effects are estimated only for pairs of loci of which both have main effects.
In:
[БОЮФв§гУ: 1]
URLPMID:22585798 [БОЮФв§гУ: 1]
In genome-wide association studies, failure to remove variation due to population structure results in spurious associations. In contrast, for predictions of future phenotypes or estimated breeding values from dense SNP data, exploiting population structure arising from relatedness can actually increase the accuracy of prediction in some cases, for example, when the selection candidates are offspring of the reference population where the prediction equation was derived. In populations with large effective population size or with multiple breeds and strains, it has not been demonstrated whether and when accounting for or removing variation due to population structure will affect the accuracy of genomic prediction. Our aim in this study was to determine whether accounting for population structure would increase the accuracy of genomic predictions, both within and across breeds. First, we have attempted to decompose the accuracy of genomic prediction into contributions from population structure or linkage disequilibrium (LD) between markers and QTL using a diverse multi-breed () data set, genotyped for 48,640 SNP. We demonstrate that SNP from a single can achieve up to 86% of the accuracy for genomic predictions using all SNP. This result suggests that most of the prediction accuracy is due to population structure, because a single is expected to capture relationships but is unlikely to contain all QTL. We then explored principal component analysis () as an approach to disentangle the respective contributions of population structure and LD between SNP and QTL to the accuracy of genomic predictions. Results showed that fitting an increasing number of principle components (PC; as covariates) decreased within breed accuracy until a lower plateau was reached. We speculate that this plateau is a measure of the accuracy due to LD. In conclusion, a large proportion of the accuracy for genomic predictions in our data was due to variation associated with population structure. Surprisingly, accounting for this structure generally decreased the accuracy of across breed genomic predictions.
URL [БОЮФв§гУ: 1]
Genomic selection could be useful in sheep-breeding programs, especially if rams and ewes are first mated at an earlier age than is the current industry practice. However, young-ewe (1 year old) fertility rates are known to be lower and more variable than those of mature ewes. The aim of the present study was to evaluate how young-ewe fertility rate affects risk and expected genetic gain in Australian sheep-breeding programs that use genomic information and select ewes and rams at different ages. The study used stochastic simulation to model different flock age structures and young-ewe fertility levels with and without genomic information for Merino and maternal sheep-breeding programs. The results from 10 years of selection were used to compare breeding programs on the basis of the mean and variation in genetic gain. Ram and ewe age, availability of genomic information on males and young-ewe fertility level all significantly (P < 0.05) affected expected genetic gain. Higher young-ewe fertility rates significantly increased expected genetic gain. Low fertility rate of young ewes (10%) resulted in net genetic gain similar to not selecting ewes until they were 19 months old and did not increase breeding-program risk, as the likelihood of genetic gain being lower than the range of possible solutions from a breeding program with late selection of both sexes was zero. Genomic information was of significantly (P < 0.05) more value for 1-year-old rams than for 2-year-old rams. Unless genomic information was available, early mating of rams offered no greater gain in Merino breeding programs and increased breeding-program risk. It is concluded that genomic information decreases the risk associated with selecting replacements at 7 months of age. Genetic progress is unlikely to be adversely affected if fertility levels above 10% can be achieved. Whether the joining of young ewes is a viable management decision for a breeder will depend on the fertility level that can be achieved in their young ewes and on other costs associated with the early mating of ewes.
In:
URL [БОЮФв§гУ: 1]
ABSTRACT To identify the impact of using molecular breeding values (mBVs) on the New Zealand sheep dual-purpose (DP) index, genomic selection (GS) accuracies were estimated using a training and validation data set consisting of 4,237 genotyped and pedigree recorded Romney animals. Molecular BVs and their accuracies for a range of DP production traits including live weight, fleece weight, faecal egg count, dagginess, reproduction and survival were estimated. The Romney mBV accuracies ranged from 0.16 to 0.52. For the majority of production traits the accuracies of the mBVs contributed information equivalent to having 1 to 8 measured progeny. For the traits: number of lambs born, lamb survival and lamb survival maternal the mBVs contributed between 11 to 145 measured progeny, albeit lamb survival maternal had a large error estimate. Combined with reducing the generation interval of rams used, from 2 years to 1 year, the potential increase in genetic gain in using mBVs in a New Zealand DP index was estimated to be 84%.
URLPMID:25149326 [БОЮФв§гУ: 1]
The aim of genomic prediction is to predict breeding value from genomic data. We describe the development of genomic prediction equations and accuracies for molecular breeding values (MBV) for industry use, focusing on the methodology used to deal with predictions for the New Zealand sheep population structure. This is made up of a mixture of pure and crossbred animals, but principally Romney based. In particular, we used pedigree-based EBV for 8 traits (weaning weight as a direct effect, weaning weight as a maternal effect, live weight at 8 mo, live weight at 12 mo, greasy fleece weight at 12 mo, lamb fleece weight, adult fleece weight, and number of lambs born) and Illumina OvineSNP50 BeadChip genotypes from 13,420 animals to investigate BLUP with different genomic relationship matrices (GRM) based on SNP markers and to investigate varying sets of older animals (training sets) to predict the MBV of younger animals (validation sets). The GRM tested included modifications to account for allele frequency differences between breeds, rescaling so that the mean GRM is equal to the mean of the traditional pedigree numerator relationship matrix A: , and combining of the GRM with A: using a convex combination with a weight estimated by maximizing a conditional restricted likelihood. We found that these modifications were beneficial and recommend using a breed-adjusted GRM combined with A: . Training data sets with Romney, Coopworth, and Perendale animals all together usually predicted better than using just a pure breed training data set for all traits. But predictions for the breed Perendale were more accurate with a Perendale training set for 3 of the 8 traits. We concluded that using a mixed-breed training set for all combinations of traits and breeds was best but advise that increasing the number of Perendale animals genotyped should be a priority to increase the MBV accuracies obtained for that breed.
[БОЮФв§гУ: 1]
URLPMID:5267438 [БОЮФв§гУ: 1]
New Zealand has some unique Terminal Sire composite sheep breeds, which were developed in the last three decades to meet commercial needs. These composite breeds were developed based on crossing various Terminal Sire and Maternal breeds and, therefore, present high genetic diversity compared to other sheep breeds. Their breeding programs are focused on improving carcass and meat quality traits. There is an interest from the industry to implement genomic selection in this population to increase the rates of genetic gain. Therefore, the main objectives of this study were to determine the accuracy of predicted genomic breeding values for various growth, carcass and meat quality traits using a HD SNP chip and to evaluate alternative genomic relationship matrices, validation designs and genomic prediction scenarios. A large multi-breed population (n=6514,845) was genotyped with the HD SNP chip (60002K) and phenotypes were collected for a variety of traits. The average observed accuracies (ЁР SD) for traits measured in the live animal, carcass, and, meat quality traits ranged from 0.1865ЁР650.07 to 0.3365ЁР650.10, 0.2865ЁР650.09 to 0.5565ЁР650.05 and 0.2165ЁР650.07 to 0.3665ЁР650.08, respectively, depending on the scenario/method used in the genomic predictions. When accounting for population stratification by adjusting for 2, 4 or 6 principal components (PCs) the observed accuracies of molecular breeding values (mBVs) decreased or kept constant for all traits. The mBVs observed accuracies when fitting bothGandAmatrices were similar to fitting onlyGmatrix. The lowest accuracies were observed for k-means cross-validation and forward validation performed within each k-means cluster. The accuracies observed in this study support the feasibility of genomic selection for growth, carcass and meat quality traits in New Zealand Terminal Sire breeds using the Ovine HD SNP chip. There was a clear advantage on using a mixed training population instead of performing analyzes per genomic clusters. In order to perform genomic predictions per breed group, genotyping more animals is recommended to increase the size of the training population within each group and the genetic relationship between training and validation populations. The different scenarios evaluated in this study will help geneticists and breeders to make wiser decisions in their breeding programs. The online version of this article (doi:10.1186/s12863-017-0476-8) contains supplementary material, which is available to authorized users.
URL [БОЮФв§гУ: 2]
URLPMID:23736059 [БОЮФв§гУ: 1]
In conventional small ruminant breeding programs, only pedigree and phenotype records are used to make selection decisions but prospects of including genomic information are now under consideration. The objective of this study was to assess the potential benefits of genomic selection on the genetic gain in French sheep and goat breeding designs of today. Traditional and genomic scenarios were modeled with deterministic methods for 3 breeding programs. The models included decisional variables related to male selection candidates, progeny testing capacity, and economic weights that were optimized to maximize annual genetic gain (AGG) of i) a meat sheep breeding program that improved a meat trait of heritability (h(2)) = 0.30 and a maternal trait of h(2) = 0.09 and ii) dairy sheep and goat breeding programs that improved a milk trait of h(2) = 0.30. Values of +/- 0.20 of genetic correlation between meat and maternal traits were considered to study their effects on AGG. The Bulmer effect was accounted for and the results presented here are the averages of AGG after 10 generations of selection. Results showed that current traditional breeding programs provide an AGG of 0.095 genetic standard deviation (sigma(a)) for meat and 0.061 sigma(a) for maternal trait in meat breed and 0.147 sigma(a) and 0.120 sigma(a) in sheep and goat dairy breeds, respectively. By optimizing decisional variables, the AGG with traditional selection methods increased to 0.139 sigma(a) for meat and 0.096 sigma(a) for maternal traits in meat breeding programs and to 0.174 sigma(a) and 0.183 sigma(a) in dairy sheep and goat breeding programs, respectively. With a medium-sized reference population (nref) of 2,000 individuals, the best genomic scenarios gave an AGG that was 17.9% greater than with traditional selection methods with optimized values of decisional variables for combined meat and maternal traits in meat sheep, 51.7% in dairy sheep, and 26.2% in dairy goats. The superiority of genomic schemes increased with the size of the reference population and genomic selection gave the best results when nref > 1,000 individuals for dairy breeds and nref > 2,000 individuals for meat breed. Genetic correlation between meat and maternal traits had a large impact on the genetic gain of both traits. Changes in AGG due to correlation were greatest for low heritable maternal traits. As a general rule, AGG was increased both by optimizing selection designs and including genomic information.
URL [БОЮФв§гУ: 1]
Estimated breeding values for the selection of more profitable sheep for the sheep meat and wool industries are currently based on pedigree and phenotypic records. With the advent of a medium-density DNA marker array, which genotypes ~50 000 ovine single nucleotide polymorphisms, a third source of information has become available. The aim of this paper was to determine whether this genomic information can be used to predict estimated breeding values for wool and meat traits. The effects of all single nucleotide polymorphism markers in a multi-breed sheep reference population of 7180 individuals with phenotypic records were estimated to derive prediction equations for genomic estimated breeding values (GEBV) for greasy fleece weight, fibre diameter, staple strength, breech wrinkle score, weight at ultrasound scanning, scanned eye muscle depth and scanned fat depth. Five hundred and forty industry sires with very accurate Australian sheep breeding values were used as a validation population and the accuracies of GEBV were assessed according to correlations between GEBV and Australian sheep breeding values . The accuracies of GEBV ranged from 0.15 to 0.79 for wool traits in Merino sheep and from 0.07 to 0.57 for meat traits in all breeds studied. Merino industry sires tended to have more accurate GEBV than terminal and maternal breeds because the reference population consisted mainly of Merino haplotypes. The lower accuracy for terminal and maternal breeds suggests that the density of genetic markers used was not high enough for accurate across-breed prediction of marker effects. Our results indicate that an increase in the size of the reference population will increase the accuracy of GEBV.
URLPMID:5558709 [БОЮФв§гУ: 1]
The application of genomic selection to sheep breeding could lead to substantial increases in profitability of wool production due to the availability of accurate breeding values from single nucleotide polymorphism (SNP) data. Several key traits determine the value of wool and influence a sheep susceptibility to fleece rot and fly strike. Our aim was to predict genomic estimated breeding values (GEBV) and to compare three methods of combining information across traits to map polymorphisms that affect these traits. GEBV for 5726 Merino and Merino crossbred sheep were calculated using BayesR and genomic best linear unbiased prediction (GBLUP) with real and imputed 510,174 SNPs for 22 traits (at yearling and adult ages) including wool production and quality, and breech conformation traits that are associated with susceptibility to fly strike. Accuracies of these GEBV were assessed using fivefold cross-validation. We also devised and compared three approximate multi-trait analyses to map pleiotropic quantitative trait loci (QTL): a multi-trait genome-wide association study and two multi-trait methods that use the output from BayesR analyses. One BayesR method used local GEBV for each trait, while the other used the posterior probabilities that a SNP had an effect on each trait. BayesR and GBLUP resulted in similar average GEBV accuracies across traits (~0.22). BayesR accuracies were highest for wool yield and fibre diameter (>0.40) and lowest for skin quality and dag score (<0.10). Generally, accuracy was higher for traits with larger reference populations and higher heritability. In total, the three multi-trait analyses identified 206 putative QTL, of which 20 were common to the three analyses. The two BayesR multi-trait approaches mapped QTL in a more defined manner than the multi-trait GWAS. We identified genes with known effects on hair growth (i.e.FGF5,STAT3,KRT86, andALX4) near SNPs with pleiotropic effects on wool traits. The mean accuracy of genomic prediction across wool traits was around 0.22. The three multi-trait analyses identified 206 putative QTL across the ovine genome. Detailed phenotypic information helped to identify likely candidate genes. The online version of this article (doi:10.1186/s12711-017-0337-y) contains supplementary material, which is available to authorized users.
URLPMID:22541502 [БОЮФв§гУ: 1]
Genomic selection aims to increase accuracy and to decrease generation intervals, thus increasing genetic gains in animal breeding. Using real data of the French Lacaune dairy sheep breed, the purpose of this study was to compare the observed accuracies of genomic estimated breeding values using different models (infinitesimal only, markers only, and joint estimation of infinitesimal and marker effects) and methods [BLUP, Bayes C , partial least squares (PLS), and sparse PLS]. The training data set included results of progeny tests of 1,886 rams born from 1998 to 2006, whereas the validation set had results of 681 rams born in 2007 and 2008. The 3 lactation traits studied (milk yield, fat content, and somatic cell scores) had heritabilities varying from 0.14 to 0.41. The inclusion of molecular information, as compared with traditional schemes, increased accuracies of estimated breeding values of young males at birth from 18 up to 25%, according to the trait. Accuracies of genomic methods varied from 0.4 to 0.6, according to the traits, with minor differences among genomic approaches. In Bayes C , the joint estimation of marker and infinitesimal effects had a slightly favorable effect on the accuracies of genomic estimated breeding values, and were especially beneficial for somatic cell counts, the less heritable trait. Inclusion of infinitesimal effects also improved slopes of predictive regression equations. Methods that select markers implicitly (Bayes C and sparse PLS) were advantageous for some models and traits, and are of interest for further quantitative trait loci studies.
URL [БОЮФв§гУ: 1]
Mastitis can prove expensive in sheep reared for meat production due to costs associated with treatment methods, poor lamb growth and premature culling of ewes. The most commonly used method to detect mastitis, in dairy systems, is somatic cell counts. However, in many meat-producing sheep flocks ewes are not routinely handled, thus regular milk sampling is not always possible. It is therefore worthwhile to investigate alternative phenotypes, such as those associated with udder conformation and methods of evaluating somatic cell counts in the milk, such as the California Mastitis Test. The main objectives of this study were therefore, a) to estimate genetic parameters of traits relating to mastitis and udder conformation in a meat sheep breed; b) estimate the level of association between somatic cell counts and the California Mastitis Test and c) assess the relationships between mastitis and both udder conformation and lamb live weights. Data were collected from Texel ewes based on 29 flocks, throughout the UK, during 2015 and 2016. The ewes were scored twice each year, at mid- and late-lactation. Eight different conformation traits, relating to udder and teat characteristics, and milk samples were recorded. The data set comprised of data available for 2 957 ewes. The pedigree file used contained sire and dam information for 31 775 individuals. The animal models used fitted relevant fixed and random effects. Heritability estimates for traits relating to mastitis (somatic cell score and the California Mastitis Test), ranged from 0.08 to 0.11 and 0.07 to 0.11 respectively. High genetic correlations were observed between somatic cell score and the California Mastitis Test (0.76 to 0.98), indicating the California Mastitis Test to be worthwhile for assessing infection levels, particularly at mid-lactation. The strongest correlations observed between the mastitis traits and the udder conformation traits were associated with udder depth (0.61 to 0.75) also at mid-lactation. Moderately negative correlations were also observed between the mastitis traits and teat angle, with those estimated at mid-lactation ranging from -0.41 to -0.55. Negative phenotypic correlations were estimated between mastitis and the weight of lamb reared by the ewe (-0.15 to -0.23), suggesting that lamb weights fell as infection levels rose. Genetic correlations were not significantly different from zero. Reducing mastitis will lead to improvements in flock productivity and the health and welfare of the animals. It will also improve the efficiency of production and the resilience to disease challenge. The economic benefits, therefore, of these results combined could be substantial not only in this breed but also in the overall meat sheep industry.
URLPMID:26342984 [БОЮФв§гУ: 1]
The objective of this study was to estimate genomic breeding values for milk yield in crossbred dairy goats. The research was based on data provided by 2 commercial goat farms in the UK comprising 590,409 milk yield records on 14,453 dairy goats kidding between 1987 and 2013. The population was created by crossing 3 breeds: Alpine, Saanen, and Toggenburg. In each generation the best performing animals were selected for breeding, and as a result, a synthetic breed was created. The pedigree file contained 30,139 individuals, of which 2,799 were founders. The data set contained test-day records of milk yield, lactation number, farm, age at kidding, and year and season of kidding. Data on milk composition was unavailable. In total 1,960 animals were genotyped with the Illumina 50K caprine chip. Two methods for estimation of genomic breeding value were compared LUP at the single nucleotide polymorphism level (BLUP-SNP) and single-step BLUP. The highest accuracy of 0.61 was obtained with single-step BLUP, and the lowest (0.36) with BLUP-SNP. Linkage disequilibrium (r2, the squared correlation of the alleles at 2 loci) at 50 kb (distance between 2 SNP) was 0.18. This is the first attempt to implement genomic selection in UK dairy goats. Results indicate that the single-step method provides the highest accuracy for populations with a small number of genotyped individuals, where the number of genotyped males is low and females are predominant in the reference population.
URL [БОЮФв§гУ: 1]
In:
URL [БОЮФв§гУ: 1]
ABSTRACT Abstract Text: Genomic selection opens new perspectives for breeding programs of dairy ruminants. In France, several projects have enabled the creation of reference populations in dairy sheep and goats. Early studies have shown that the reliability of genomic evaluations using a GBLUP method in these species is lower than in Holstein dairy cattle breed. The single-step approach gives best predictions for candidates at birth (genomic evaluation accuracy obtained by cross validation for milk yield of 0.47 in Lacaune dairy sheep and 0.43 in goats breeds). The multi-breed approach is effective in goats by blending Alpine and Saanen breeds, but not in sheep. Finally, switching to genomic selection is planned in Lacaune dairy sheep and is under consideration for other sheep breeds. In goats, inclusion of major genes in genomic evaluations should be explored before switching to genomic breeding programs. Keywords: Dairy goats Dairy sheep Genomic selection
In:
URL [БОЮФв§гУ: 1]
Abstract: Genomic selection experiment in Lacaune dairy sheep: progeny test results of rams initially selected either on parent average or on genomic prediction (10th World Congress on Genetics Applied to Livestock Production)
URL [БОЮФв§гУ: 1]
URLPMID:29065868 [БОЮФв§гУ: 1]
Building an efficient reference population for genomic selection is an issue when the recorded population is small and phenotypes are poorly informed, which is often the case in sheep breeding programs. Using stochastic simulation, we evaluated a genomic design based on a reference population with medium-density genotypes [around 4502K single nucleotide polymorphisms (SNPs)] of dams that were imputed from very low-density genotypes (Ём021000 SNPs). A population under selection for a maternal trait was simulated using real genotypes. Genetic gains realized from classical selection and genomic selection designs were compared. Genomic selection scenarios that differed in reference population structure (whether or not dams were included in the reference) and genotype quality (medium-density or imputed to medium-density from very low-density) were evaluated. The genomic design increased genetic gain by 26% when the reference population was based on sire medium-density genotypes and by 54% when the reference population included both sire and dam medium-density genotypes. When medium-density genotypes of male candidates and dams were replaced by imputed genotypes from very low-density SNP genotypes (1000 SNPs), the increase in gain was 22% for the sire reference population and 42% for the sire and dam reference population. The rate of increase in inbreeding was lower (from 61 20 to 61 34%) for the genomic design than for the classical design regardless of the genomic scenario. We show that very low-density genotypes of male candidates and dams combined with an imputation process result in a substantial increase in genetic gain for small sheep breeding programs.
URL [БОЮФв§гУ: 1]
[БОЮФв§гУ: 1]
[БОЮФв§гУ: 1]
URLMagsci [БОЮФв§гУ: 1]
ЦЗжжбЁг§дкХЉвЕЩњВњжаеМгаЪЎЗжживЊЕФЕиЮЛ, г§жжжЕЙРМЦЪЧЦЗжжбЁг§ЕФКЫаФЁЃЫцзХвХДЋБъМЧЕФЗЂеЙ, гШЦфЪЧИпЭЈСПЕФЛљвђЗжаЭММЪѕ, ЪЙЕУДгЛљвђзщЫЎЦНЙРМЦг§жжжЕГЩЮЊПЩФм, МДЛљвђзщбЁдёЁЃЮФеТНЋЛљвђзщбЁдёЕФЗНЗЈЗжЮЊСНРрЃКвЛЪЧЛљгкЙРМЦЕШЮЛЛљвђаЇгІРДдЄВтЛљвђзщЙРМЦг§жжжЕ(GEBV), ШчзюаЁЖўГЫЗЈ, ЫцЛњЛиЙщ-зюМбЯпадЮоЦЋдЄВт(RR-BLUP)ЁЂBayesЁЂжїГЩЗжЗжЮіЕШЗНЗЈ; ЖўЪЧЛљгквХДЋЙиЯЕОиеѓРДдЄВтGEBV, ЭЈЙ§ВЩгУИпЭЈСПБъМЧЙЙНЈИіЬхМфЕФвХДЋЙиЯЕОиеѓ, ШЛКѓгУЯпадЛьКЯФЃаЭРДдЄВтг§жжжЕ, МДGBLUPЗЈ, ВЂвдетСНжжЗжРрМђвЊНщЩмСЫЛљвђзщбЁдёИїжжЗНЗЈЕФДѓжТдРэЁЃгАЯьЛљвђзщбЁдёзМШЗадЕФвђЫижївЊгаБъМЧРраЭКЭУмЖШЁЂЕЅБЖаЭГЄЖШЁЂВЮПМШКЬхДѓаЁКЭБъМЧ-Ъ§СПадзДЛљвђзљ(QTL)СЌЫјВЛЦНКт(LD)ДѓаЁЕШ; дкЛљвђзщбЁдёЕФИїжжЗНЗЈжа, вЛАуЫЕРДBayesЗНЗЈКЭGBLUPЗНЗЈОпгаНЯИпЕФзМШЗад, зюаЁЖўГЫЗЈзюВю; GBLUPМЦЫуЫйЖШПь, ФмЙЛНЋБъМЧКЭЯЕЦзНсКЯЦ№РД, вђЖјБШЦфЫћЗНЗЈИќОпгХЪЦЁЃОЁЙмЛљвђзщбЁдёШЁЕУСЫКмДѓНјеЙ, ЕЋдкРэТлЗНУцЛЙУцСйзХвЛаЉЬєеН, ШчСЊКЯг§жжЁЂГЄЦкбЁдёЕФвХДЋНјеЙМАШчКЮНтЮігыадзДгаЙиКЭЮоЙиЕФБъМЧЕШЁЃЛљвђзщбЁдёдквЛаЉЖЏжВЮяг§жжЩЯвбОПЊЪМгІгУ, дкШЫРрвХДЋЧуЯђдЄВтКЭНјЛЏЖЏСІбЇбаОПжавВгаЧБдкЕФгІгУЧАОАЁЃЛљвђзщбЁдёдкИіЬхМфЧздЕЙиЯЕЕФСПЛЏЩЯгаСЫЭЛЦЦ, БШДЋЭГЗНЗЈИќМгОЋШЗ, вђДЫ, ЛљвђзщбЁдёНЋЛсЪЧЖЏжВЮяг§жжЪЗЩЯИяУќадЕФЪТМўЁЃ
URLMagsci [БОЮФв§гУ: 1]
ЦЗжжбЁг§дкХЉвЕЩњВњжаеМгаЪЎЗжживЊЕФЕиЮЛ, г§жжжЕЙРМЦЪЧЦЗжжбЁг§ЕФКЫаФЁЃЫцзХвХДЋБъМЧЕФЗЂеЙ, гШЦфЪЧИпЭЈСПЕФЛљвђЗжаЭММЪѕ, ЪЙЕУДгЛљвђзщЫЎЦНЙРМЦг§жжжЕГЩЮЊПЩФм, МДЛљвђзщбЁдёЁЃЮФеТНЋЛљвђзщбЁдёЕФЗНЗЈЗжЮЊСНРрЃКвЛЪЧЛљгкЙРМЦЕШЮЛЛљвђаЇгІРДдЄВтЛљвђзщЙРМЦг§жжжЕ(GEBV), ШчзюаЁЖўГЫЗЈ, ЫцЛњЛиЙщ-зюМбЯпадЮоЦЋдЄВт(RR-BLUP)ЁЂBayesЁЂжїГЩЗжЗжЮіЕШЗНЗЈ; ЖўЪЧЛљгквХДЋЙиЯЕОиеѓРДдЄВтGEBV, ЭЈЙ§ВЩгУИпЭЈСПБъМЧЙЙНЈИіЬхМфЕФвХДЋЙиЯЕОиеѓ, ШЛКѓгУЯпадЛьКЯФЃаЭРДдЄВтг§жжжЕ, МДGBLUPЗЈ, ВЂвдетСНжжЗжРрМђвЊНщЩмСЫЛљвђзщбЁдёИїжжЗНЗЈЕФДѓжТдРэЁЃгАЯьЛљвђзщбЁдёзМШЗадЕФвђЫижївЊгаБъМЧРраЭКЭУмЖШЁЂЕЅБЖаЭГЄЖШЁЂВЮПМШКЬхДѓаЁКЭБъМЧ-Ъ§СПадзДЛљвђзљ(QTL)СЌЫјВЛЦНКт(LD)ДѓаЁЕШ; дкЛљвђзщбЁдёЕФИїжжЗНЗЈжа, вЛАуЫЕРДBayesЗНЗЈКЭGBLUPЗНЗЈОпгаНЯИпЕФзМШЗад, зюаЁЖўГЫЗЈзюВю; GBLUPМЦЫуЫйЖШПь, ФмЙЛНЋБъМЧКЭЯЕЦзНсКЯЦ№РД, вђЖјБШЦфЫћЗНЗЈИќОпгХЪЦЁЃОЁЙмЛљвђзщбЁдёШЁЕУСЫКмДѓНјеЙ, ЕЋдкРэТлЗНУцЛЙУцСйзХвЛаЉЬєеН, ШчСЊКЯг§жжЁЂГЄЦкбЁдёЕФвХДЋНјеЙМАШчКЮНтЮігыадзДгаЙиКЭЮоЙиЕФБъМЧЕШЁЃЛљвђзщбЁдёдквЛаЉЖЏжВЮяг§жжЩЯвбОПЊЪМгІгУ, дкШЫРрвХДЋЧуЯђдЄВтКЭНјЛЏЖЏСІбЇбаОПжавВгаЧБдкЕФгІгУЧАОАЁЃЛљвђзщбЁдёдкИіЬхМфЧздЕЙиЯЕЕФСПЛЏЩЯгаСЫЭЛЦЦ, БШДЋЭГЗНЗЈИќМгОЋШЗ, вђДЫ, ЛљвђзщбЁдёНЋЛсЪЧЖЏжВЮяг§жжЪЗЩЯИяУќадЕФЪТМўЁЃ

{kind=link}
{kind=link}
{kind=link}
{kind=link}